

SUMMARY
Break-induced replication (BIR) is a pathway of homology-directed repair that repairs one-ended DNA breaks, such as those formed at broken replication forks or uncapped telomeres. In contrast to conventional S phase DNA synthesis, BIR proceeds by a migrating D-loop and results in conservative synthesis of the nascent strands. DNA polymerase delta (Pol δ) initiates BIR; however, it is not known whether synthesis of the invading strand switches to a different polymerase or how the complementary strand is synthesized. By using alleles of the replicative DNA polymerases that are permissive for ribonucleotide incorporation, thus generating a signature of their action in the genome that can be identified by hydrolytic end sequencing, we show that Pol δ replicates both the invading and the complementary strand during BIR. In support of this conclusion, we show that depletion of Pol δ from cells reduces BIR, whereas depletion of Pol ε has no effect.
Graphical Abstract
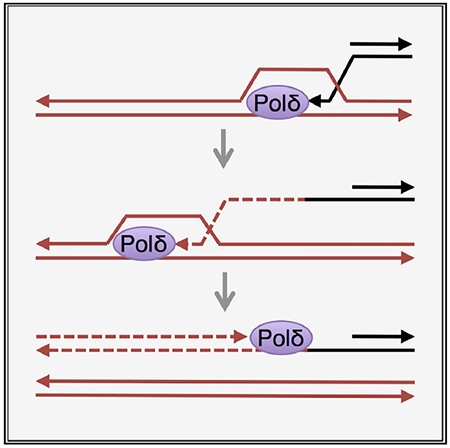
In Brief
Donnianni et al. elucidate DNA polymerase usage during break-induced replication (BIR). In contrast to conventional S phase DNA replication, in which DNA Pol ε synthesizes the leading strand and Pols α and δ synthesize the lagging strand, the authors show that Pol δ synthesizes both strands during BIR.
INTRODUCTION
DNA double-strand breaks (DSBs) are toxic lesions that must be healed to preserve genome integrity. DSBs resulting from endonuclease cleavage or ionizing irradiation have two ends (two-end DSBs) and can be repaired through either non-homologous end joining (NHEJ) or homologous recombination (HR). By contrast, broken or reversed replication forks, or eroded telomeres, present only one free end (single-end DSB) and must be repaired by HR to avoid formation of rearranged chromosomes by NHEJ. The invading end of a single-end DSB can prime extensive DNA synthesis in a process referred to as break-induced replication (BIR) (Sakofsky and Malkova, 2017). This process has been modeled in yeast by creating an endonuclease-induced DSB between sequence homologous to a donor duplex and heterologous downstream sequence that will be lost after DSB formation (Donnianni and Symington, 2013; Lydeard et al., 2007; Malkova et al., 2005; Morrow et al., 1997). As a result, extensive DNA synthesis (up to several hundred kb) from the site of strand invasion to the telomere is required to restore chromosome integrity. BIR can result in loss of heterozygosity when it occurs between non-sister chromatids in diploid cells, and it can result in non-reciprocal translocation or copy number variation when dispersed repeats are involved (Costantino et al., 2014; Payen et al., 2008; Sakofsky and Malkova, 2017; Smith et al., 2007).
Like other HR processes, BIR initiates by resection of the DSB ends to generate 3′ overhangs. Rad51 catalyzes pairing of a 3′ overhang with an intact homologous duplex and promotes strand invasion to form a displacement loop (D-loop) intermediate (Figure 1A; Davis and Symington, 2004; Malkova et al., 2005). The 3′ end of the invading end within the D-loop is extended by DNA synthesis, templated by the donor duplex. To complete repair of a two-end DSB by the simplest mechanism, the extended invading strand is released by a DNA helicase and anneals to the other resected DSB end, and the captured 3′ end then primes DNA synthesis to fill the gap. During repair of a single-end DSB, there is no second end to anneal with the extended invading end (Figure 1A), and DNA synthesis continues to the telomere or until merging with a converging replication fork (Mayle et al., 2015; Saini et al., 2013). The frequency of BIR increases in the absence of the Mph1 helicase, which normally displaces the invading strand of D-loop intermediates for second end capture (Prakash et al., 2009), consistent with the view that BIR intermediates are similar to those formed during two-end break repair (Luke-Glaser and Luke, 2012; Mehta et al., 2017; Stafa et al., 2014). Furthermore, BIR DNA synthesis is conservative, and synthesis of the invading and complementary (second) strands is uncoupled, resulting in a long-lived single-stranded DNA (ssDNA) intermediate, in agreement with BIR synthesis occurring by a migrating D-loop from the site of invasion to the telomere (Donnianni and Symington, 2013; Saini et al., 2013; Wilson et al., 2013). A conservative mode of replication has also been reported for mitotic DNA synthesis and alternative lengthening of telomeres in human cells, processes thought to occur by BIR (Bhowmick et al., 2016; Roumelioti et al., 2016). The ssDNA generated during BIR is subjected to clustered mutations and can invade other homologous templates (Deem et al., 2011; Sakofsky et al., 2014; Smith et al., 2007), leading to speculation that BIR contributes to kataegis and complex rearrangements associated with a variety of human diseases (Hastings et al., 2009).
Figure 1. The pol3-L612G Mutant Is Defective for BIR.
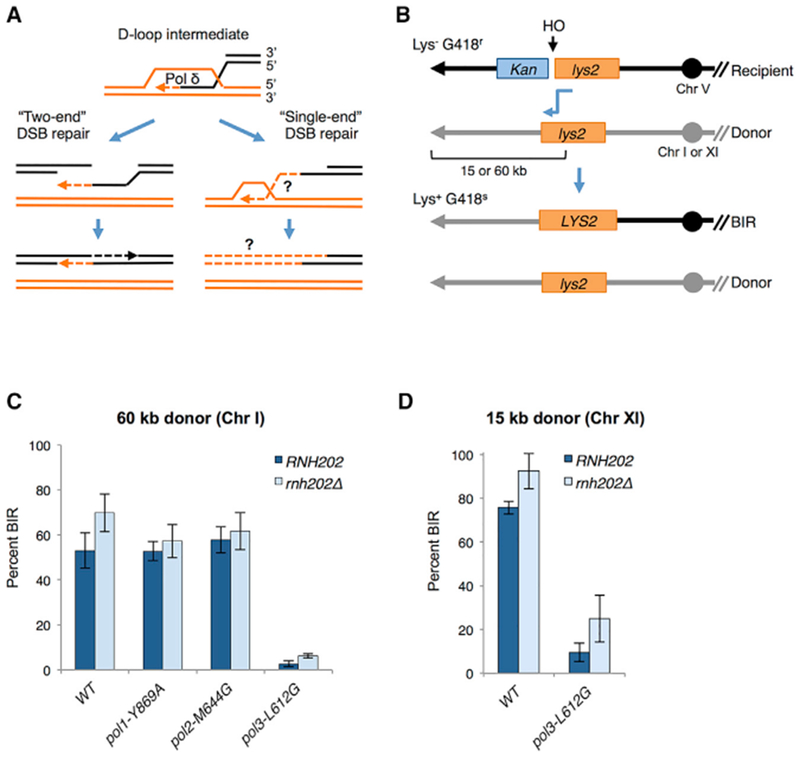
(A) Homology-dependent repair initiates by invasion of a homologous duplex by a 3′ overhang formed by end resection. The invading 3′ end is extended by DNA synthesis catalyzed by DNA Pol δ. For repair of a two-end DSB by the simplest mechanism, the extended invading end displaced by a DNA helicase pairs with the other resected end of the broken duplex. Gap-filling DNA synthesis initiated at the captured 3′ end, followed by ligation, completes repair. Because there is no second end to pair with, extension of the invading end at a single-end DSB can continue to the end of the chromosome as a migrating D-loop. Synthesis of the second (top) strand must rely on de novo priming on the extended invading (bottom) strand because there is no 3′ end capture. The DNA polymerase(s) responsible for completing first-strand synthesis and synthesis of the second strand is unknown (indicated by ?).
(B) Schematic of the ectopic BIR assay (see text for details). After HO cleavage, the truncated lys2 sequence on the recipient cassette invades the donor lys2 cassette, generating a functional LYS2 gene (shown by blue connecting arrow). Continued synthesis to the end of the donor chromosome generates a non-reciprocal translocation. The non-invading end produced by HO cleavage is degraded. The donor cassette is placed 60 kb from the left telomere of Chr I or 15 kb from the left telomere of Chr XI. Only the left arms of the recipient (R) and donor (D) chromosomes are shown; centromeres are indicated by solid circles and telomeres as arrowheads. Note that the native LYS2 gene on Chr II is deleted in all BIR strains, and the lys2 fragments within the D and R cassettes are encoded on the Crick strand.
(C) BIR frequency of polymerase mutants in strains with the 60-kb donor. BIR frequencies were determined by colony-forming units (CFUs) Lys+ G418S YPGal/CFU YPD for each of the indicated strains from at least three independent trials; error bars show SDs.
(D) BIR frequency of WT and pol3-L612G strains with the 15-kb donor from three independent trials; error bars show SD. BIR frequencies were determined as in (C).
During normal S phase replication of undamaged DNA, DNA polymerase (Pol) ε primarily synthesizes the leading strand, and Pol α and Pol δ synthesize the discontinuous lagging strand (Lujan et al., 2016). Assignment of DNA polymerase usage genome-wide has been greatly facilitated by the development of next-generation sequencing methods to detect ribonucleotides inserted during DNA synthesis. By using a set of yeast strains, each expressing a variant of yeast Pol α, δ, or ε that increases the probability of ribonucleotide incorporation into DNA, coupled to a mutation in RNaseH2 to prevent their removal, the locations of ribonucleotides can be identified genome-wide, generating a signature for polymerase usage (Clausen et al., 2015; Daigaku et al., 2015; Koh et al., 2015; Reijns et al., 2015). In vitro and in vivo studies have shown that Pol δ can also synthesize regions of the leading strand under some circumstances but that Pol ε is more effective at leading strand replication as a result of its direct interaction with the MCM replicative helicase (Garbacz et al., 2018; Georgescu et al., 2014; Yeeles et al., 2017).
By contrast, the division of labor between DNA polymerases during BIR is poorly understood. Elimination of the translesion DNA polymerases, Pol ζ and Pol η, does not reduce the frequency of BIR, indicating that the replicative polymerases must carry out the bulk of DNA synthesis (Lydeard et al., 2010). Several studies support the involvement of Pol δ in extension of the 3′ end of strand invasion intermediates (Costantino et al., 2014; Deem et al., 2008; Dilley et al., 2016; Li et al., 2009; Lydeard et al., 2007; Maloisel et al., 2008; Smith et al., 2009; Wilson et al., 2013). Notably, the Pol32/POLD3 subunit of the Pol δ complex, which is not essential for S phase synthesis in budding yeast, is required for BIR (Costantino et al., 2014; Dilley et al., 2016; Lydeard et al., 2007). More puzzling is the mechanism for synthesis of the second strand. In contrast to canonical two-end DSB repair, where capture of the other break end provides a 3′ end to prime second-strand synthesis, there is no second end capture in BIR and hence no primer (Figure 1A). Pol α-primase is the only polymerase capable of de novo priming in yeast. Earlier studies to address the role of the replicative DNA polymerases during BIR utilized conditional alleles and measured BIR by direct physical assays following depletion of each polymerase in G2-arrested cells (Lydeard et al., 2007). These studies support the involvement of Pols α and δ to extend the invading end within the D-loop intermediate. However, because BIR fails to initiate in Pol δ-depleted cells, it is not known whether Pol δ completes synthesis of the first strand and whether it also catalyzes second-strand synthesis. Lydeard et al. (2007) reported that DNA Pol ε is required to complete BIR synthesis, suggesting the possibility of a polymerase switch.
In this study, we used two complementary approaches to address DNA polymerase usage during BIR. First, by using the signature of ribonucleotides incorporated during BIR, we show that Pol δ replicates both the invading and second strands during BIR, with little or no contribution from Pol ε. Second, by utilizing auxin-inducible degron (AID) alleles of each replicative DNA polymerase, we find that depletion of Pol α or Pol δ from cells reduces BIR, whereas depletion of Pol ε has no effect.
RESULTS
Increased Ribonucleotide Incorporation by Pol δ Impairs BIR
The BIR assay used here is comprised of an HO endonuclease cleavage site adjacent to a 3′ truncated lys2 gene on recipient chromosome V (Chr V) that shares 2.2 kb of homology on only one side of the DSB to a 5′ truncated lys2 gene inserted, with the same polarity, at variable distances from the telomere of the donor chromosome (Figure 1B; Donnianni and Symington, 2013). Because genes that lie between the recipient chromosome telomere and the DSB are not essential, the non-homologous end can be lost from haploid cells without loss of viability. In these strains, HO endonuclease is expressed from a galactose-inducible promoter. Additionally, the strains have the MATa-inc allele to prevent HO cleavage at the endogenous MAT locus. After DSB formation, the 3′ truncated lys2 cassette invades homologous lys2 sequence on the donor chromosome and DNA synthesis continues until the end of the donor chromosome, generating a non-reciprocal translocation (Figure 1B). The terminal fragment generated by HO cleavage is unable to engage in HR and is degraded. The BIR frequency is determined by the plating efficiency on galactose-containing medium (YPGal) relative to glucose-containing medium. In wild-type (WT) cells, >99% of the colonies formed on YPGal are Lys+ and G418s, indicating reconstitution of the LYS2 gene by BIR and loss of the Kan marker. Additionally, physical analysis of genomic DNA isolated at different time points after HO induction can be used to detect BIR intermediates and the BIR translocation chromosome can be monitored by pulsed-field gel electrophoresis (PFGE) of intact chromosomes (Donnianni and Symington, 2013).
To evaluate DNA polymerase usage during BIR, we employed alleles of yeast Pols α, δ, and ε that increase the probability of ribonucleotide incorporation into DNA (pol1-Y869A, pol3-L612G, and pol2-M644G, respectively; Clausen et al., 2015; Nick McElhinny et al., 2010). To prevent the removal of ribonucleotides incorporated during DNA synthesis by ribonucleotide excision repair (RER), we constructed rnh202Δ (RNH202 encodes one of the three subunits of the RNaseH2 complex) derivatives of each strain. Ribonucleotides incorporated into genomic DNA provide markers of polymerization reactions in vivo. The sites of ribonucleotide incorporation can be identified by alkaline hydrolysis or by RNaseHII digestion of genomic DNA, followed by sequencing of libraries prepared from the resulting ssDNA fragments and identification of 5′ hydrolytic DNA ends (Clausen et al., 2015; Daigaku et al., 2015; Koh et al., 2015; Reijns et al., 2015). In this study, we used a modified HydEn-seq procedure (STAR Methods) that was improved on quantitation, specificity, and signal-to-noise ratio upon the published protocol (Clausen et al., 2015).
First, we assessed BIR proficiency of the DNA polymerase mutants by formation of Lys+ recombinants after HO induction with a donor cassette located 60 kb from the left telomere of Chr I. BIR is reduced by 20-fold in thepol3-L612G mutant relative to WT, whereas BIR is unaffected by the pol1-Y869A or pol2-M644G mutation (Figure 1C). Although this result strongly supports Pol δ involvement in BIR, the low efficiency would hinder our ability to conduct HydEn-seq analysis. Because we have shown previously that BIR frequency is inversely proportional to the distance of the donor cassette from the telomere (Donnianni and Symington, 2013), we used a variation of the BIR system that requires copying of only 15 kb from the donor chromosome. The BIR frequency of the pol3-L612G strain with a donor cassette located 15 kb from the telomere of Chr XI is improved compared to the 60-kb donor strain but is still 8-fold lower than WT (Figure 1D). Loss of RNase H2 activity (rnh202Δ) increases the BIR efficiency of the pol3-L612G strain to a level where it is only 3.7-fold lower than the rnh202Δ single mutant. A strain with a different POL3 substitution, pol3-L612M, which is less permissive for ribonucleotide incorporation than pol3-L612G (Figure S1A), shows a higher BIR frequency than pol3-L612G, but this is still lower than WT (p < 0.005; Figure S1B).
There are two possible explanations for the pol3-L612G BIR defect. First, the activity of Pol δL61213 may be impaired, making the extensive DNA synthesis required for BIR more challenging. The primary determinant of Pol δ processivity is through interaction with PCNA, which should not be affected by the L612G mutation because it is located far away from the PCNA interacting motif (Acharya et al., 2011). We suspect that Pol δL612G is not less processive but may have a lower synthesis rate, perhaps due to slow extension from its many misincorporated ribonucleotides (Watt et al., 2011). During S phase synthesis, the template for Pol3L612G would be a strand synthesized mostly by WT Pol ε in the previous S phase. By contrast, if Pol3L612G synthesizes both strands during BIR, it would have to synthesize on templates with ribonucleotides incorporated from the previous S phase and from first-strand synthesis, lowering the efficiency of extension, particularly on long templates. Indeed, mating type interconversion, which requires only ~700 bp DNA synthesis, is unaffected by the pol3-L612G mutation (Figures S1C and S1D). Second, ribonucleotides in the ssDNA intermediate formed during BIR or completed products may be susceptible to enzymatic or spontaneous hydrolysis. The three-fold higher frequency of BIR in the rnh202Δ derivative of pol3-L612G supports the idea that ribonucleotides present in BIR intermediates are labile. The elevated mutation rate of pol3-L612G is unlikely to cause the BIR defect because the pol3-01 mutant, defective for Pol δ proofreading (Venkatesan et al., 2006), shows close to WT BIR frequency (Figure S1B).
DNA Pol δ Synthesizes Both Strands during BIR
By decreasing the amount of DNA to be synthesized to 15 kb, we were able to partially overcome the pol3-L612G BIR defect. In order to further optimize the system, we modified the BIR assay to prevent cells from initiating a new cell cycle after completing repair, which would erase BIR-specific ribonucleotide incorporation. To ensure that only the BIR signature is detected, we placed the recipient cassette on Chr X adjacent to CDC6, a gene required for S phase synthesis, but not for BIR (Lydeard et al., 2010). After DSB formation, resection through the CDC6 locus on the side of the break unable to engage in repair would impede CDC6 transcription (Manfrini et al., 2015) and thus entry into a new S phase (Figure 2A). In this strain, the donor cassette was placed 21 kb from the telomere of Chr VI to enable clear separation of the BIR product from the donor chromosome by PFGE (Figure 2B). We verified that BIR in this system results in loss of cell viability and causes cell cycle arrest after BIR completion (Figures S2A and S2B). BIR efficiency was measured by physical analysis of genomic DNA isolated before HO induction (0 h) and at 2, 6, and 12 h after HO induction. PCR using one primer up-stream of the break on the recipient chromosome and a reverse primer downstream of the homologous sequence on the donor chromosome detects extension of the invading end and completed BIR products (Figures 2C and 2D). BIR was also monitored by EcoRV digestion of genomic DNA and Southern blot hybridization (Figures 2C and 2D). Consistent with the genetic assay, pol1-Y869A and pol2-M644G strains show WT levels of BIR products, and repair is delayed and reaches a lower final level in the pol3-L612G strain (Figure 2D). When attempting HydEn-seq with total genomic DNA isolated 6 or 24 h after HO induction, reads from the newly synthesized 21-kb BIR region were not clearly distinguishable from those of the donor chromosome. Therefore, the der(X)t(VI:X) translocation chromosome produced by BIR (697 kb) was separated from recipient (Chr X; 746 kb) and donor (Chr VI; 270 kb) chromosomes by PFGE. DNA extracted from the BIR chromosome band excised from the gel for library preparation was shown to be free from Chr VI contamination by PCR analysis (Figure S2C).
Figure 2. Physical Analysis of BIR in the Ribonucleotide-Permissive DNA Polymerase Mutants.
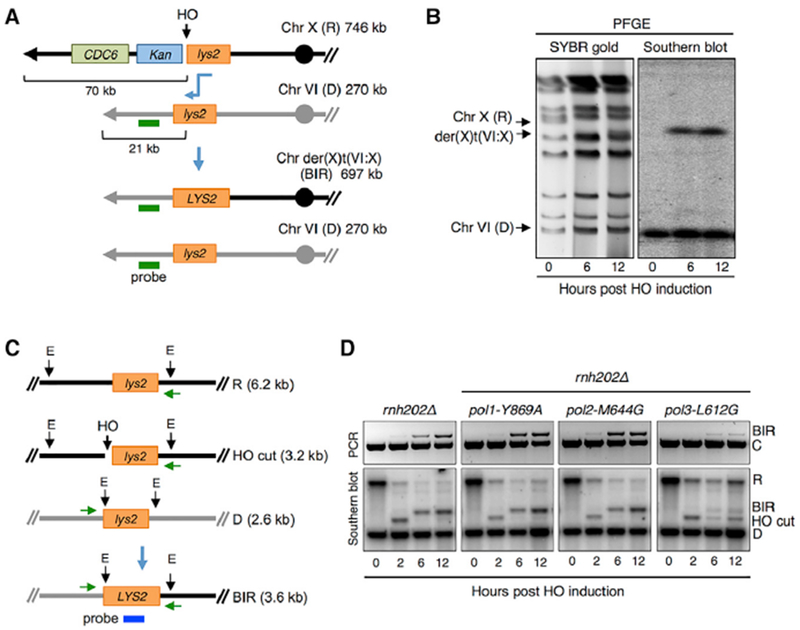
(A) Schematic of the BIR assay with the recipient cassette inserted telomere distal to the CDC6 locus on Chr X and donor cassette 21 kb from the telomere of Chr VI. The sizes of the R, D, and BIR product are indicated to the right; the horizontal green line indicates the position of the hybridization probe used to detect the BIR translocation product by PFGE.
(B) Final BIR translocation product (der(X)t(VI:X)) analyzed by PFGE and SYBR gold stain for all chromosomes or Southern blot hybridization using a probe specific to a duplicated region of Chr VI.
(C) Schematic of physical assays to detect BIR. Green horizontal arrows indicate the locations of primers used to monitor BIR product by PCR. Vertical black arrows show the location of EcoRV (E) sites, and sizes of digestion products for the recipient chromosome before and after HO cutting, the donor chromosome, and BIR product are indicated. The probe used for Southern blot analysis hybridizes to LYS2 sequence shared by R and D cassettes and final BIR product.
(D) PCR (top panel) and Southern blot (bottom panel) of the indicated strains showing initiation and completion of BIR after HO induction. Representative images from three independent trials are shown. Control primers (“C”) were designed to amplify sequences 66-kb centromere proximal to the DSB and were included in the same reaction with primers to detect BIR.
We performed HydEn-seq analysis of purified Chr X or der(X) t(VI:X) from RER-defective strains encoding pol1-Y869A, pol2-M644G, or pol3-L612G at different time points before and after HO induction. Following HO induction, reads mapping to the normal Chr X telomere-proximal region from the DSB site were barely detectable in non-hydrolyzed control samples (data not shown), consistent with removal of this region of the chromosome. In contrast, after HO induction, there was a gradual increase in sequence reads from the 21-kb donor region of Chr VI (Figure S2D). The increase was slower in the pol3-L612G strain as a consequence of the BIR defect. Because cutting therefore appeared efficient, sequencing reads from samples before HO induction (time point 0) were mapped to the original Chr X, and reads from samples after HO induction were mapped to der(X)t(VI:X) reference chromosome.
We determined the relative contributions of Pols α, ε, and δ to DNA synthesis across Chr X before HO induction (Figure 3A) and across der(X)t(VI:X) after HO induction (Figure 3B). As described previously (Garbacz et al., 2018), the ribonucleotide density yi,j,k) at position i on strand j in strain K was set equal to a polymerase-independent noise factor (w) times the sum of the products of the fractional contributions (f) of each polymerase (k) and the ribonucleotide insertion rate (s) of that polymerase (). Using HydEn-seq data from RER-defective strains encoding the variant polymerases (pol1-Y869A, pol2-M644G, and pol3-L612G) and assuming constant polymerase usage among these strains (i.e., fi,j,wild type pol = fi,j,variant pol), a system of simultaneous equations was solved (i.e., yi,j,pol1-Y869A versus yi,j,pol2-M644G and yi,j,pol3-L612G) for the fractional polymerase contributions. Interestingly, the data from later time points are noisier (Figures 3 and S3), suggesting either a progressive loss of embedded ribonucleotides (e.g., by additional inefficient ribonucleotide removal mechanisms) or an increase in background noise in non-cycling cells (i.e., DNA strand breaks or emergence of additional hydrolytic targets).
Figure 3. DNA Polymerase Enzymology on Chromosome X before and after HO Induction.
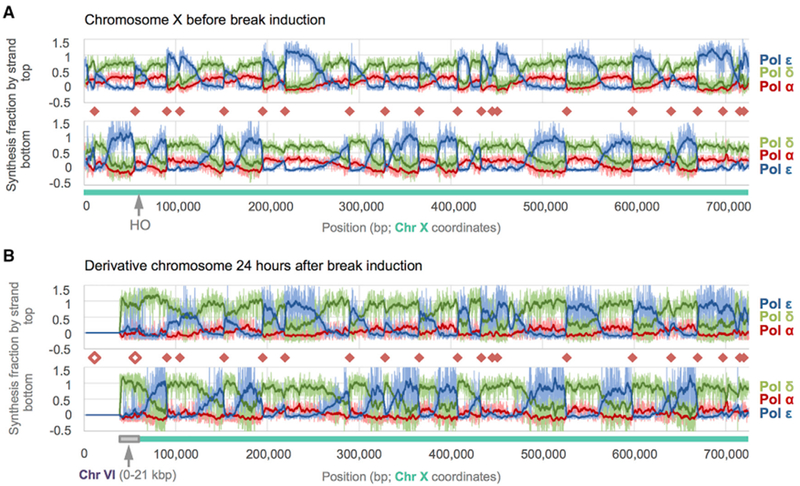
Fraction of synthesis across S. cerevisiae Chr X due to each replicative polymerase as calculated from HydEn-seq end densities. Chr-X- and Chr-VI-derived sequences are indicated beneath each graph by blue-green and gray bars, respectively. Fractional synthesis contributions due to Pols α, δ, and ε data are shown in shades of red, green, and blue, respectively. Pale lines are raw fractions (100-bp bins). Dark curves are 25-bin moving averages (2.5 kb). Origins (orange diamonds) coincide with abrupt reversals in Pol α + δ versus Pol ε trends. A greater reversal on one strand indicates an origin that is often overrun by forks proceeding from a neighboring origin.
(A) Synthesis fractions before double-strand break (DSB) induction mapped to the Chr X reference.
(B) Synthesis fractions 24 h post-DSB are mapped to the derivative chromosome, der(X)t(VI;X). Noise increases with incubation time (compare A to B), but polymerase usage patterns remain constant beyond the DSB +60 kb. The Pol α contribution is lower (indistinguishable from noise) on both strands in the donor region than in lagging strand regions elsewhere. The unfilled orange diamonds indicate origins that were deleted during BIR.
See also Figures S2–S4.
The division of labor among the three replicative polymerases remains largely unchanged in Chr X centromere proximal to the DSB (Figures 3A and 3B). However, the contributions are very different in the translocation region. At 24 h after break induction, the HydEn-seq data reveal that Pol δ is the major polymerase in the translocation region, and the Pol ε footprint is minimal. Pol α contribution in the BIR region 24 h post-HO induction is negligible on both strands and is indistinguishable from zero. This is much less than in lagging-strand-specific regions in the rest of the chromosome (average usage >10%; higher than expected), suggesting that neither BIR strand is synthesized like a canonical lagging strand. However, it is important to note that the resolution does not allow us to discern individual priming events. In particular, the repetitive nature of the telomere and sub-telomeric regions further prevents us from assessing Pol α priming near chromosome ends. For simplicity, we plot the combined fraction of Pols α and δ synthesis in the BIR region at different time points (Figure 4A). On the invading strand (Figure 4A, bottom panel), this fraction reaches 100% by about 12 h, consistent with the idea that Pol δ synthesized most, and perhaps all, of this strand. Similarly, Pols α and δ perform nearly all synthesis of the second strand by about 12 h post-HO induction (Figure 4A, top panel). The predominant use of Pol δ is highly reproducible in a biological replica (Figure S3). Thus, within the error of the measurement, the data suggest that Pol δ performs the vast majority of synthesis on both strands during BIR (Figure 4B).
Figure 4. Polymerase Contributions and BIR Mechanism.
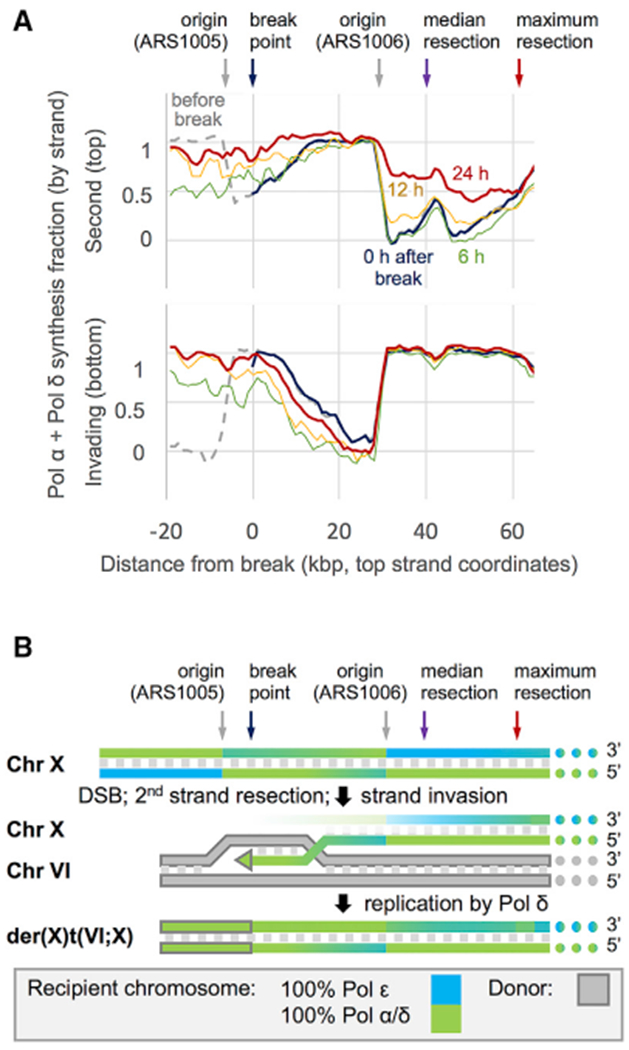
(A) 3-kb moving averages Pol α + δ synthesis fractions before (dashed gray) and immediately after break induction (blue); polymerase usage reflects the previous round of synthesis. Sequencing depth in the donated region is low (relative to the rest of Chr X and particularly for pol3-L612G strain) through at least the first 6 h (green), and the HydEn-seq signal therein results from a mix of new synthesis and low-level residual donor DNA. By 12 h after break induction (yellow), as BIR products increase and the signal is dominated by new synthesis, the Pol α + δ contribution in the donated regions approaches 100% and begins to increase up to 50 kb outside of the donated region on the second or top strand. 5′-to-3′ resection and re-synthesis are expected on this strand. By 24 h after induction (red), the Pol α + δ contribution beyond the DSB indicates continuing resection and re-synthesis up to 60 kb from the break. The degree of the Pol α + δ contribution beyond the DSB is consistent with a median resection distance of over 40 kb.
(B) A model of BIR derived from polymerase usage as measured by HydEn-seq. The scale is the same as in (A). Gradients indicate polymerase usage (full blue, 100% Pol ε; full green, 100% Pol α + δ; gray fill, donor chromosome; gray outline, donor sequence).
See also Figures S3 and S4.
One concern with this interpretation is the low efficiency of BIR observed for the pol3-L612G strain, which could potentially be due to an altered mode of DNA synthesis. Therefore, we also determined the fractional polymerase contributions using the pol3-L612M strain. Although the signal-to-noise ratio from the pol3-L612M mutant was lower than found for pol3-L612G, due to the lower incorporation rate of ribonucleotides, the contribution of Pol δ to synthesis within the translocation region was still 90% or above (Figure S4).
Interestingly, both the length and magnitude of Pol δ second-strand synthesis centromere proximal to the DSB site increases over time, up to 60 kb by 24 h after DSB induction (Figures 4A, top panel, and S3). This suggests that 5′ to 3′ end resection proceeds until replacement synthesis “catches up” with the receding 5′ end (Figure 4B). Previous studies have shown that end resection is more extensive during BIR than two-end break repair (Chung et al., 2010). The slower BIR kinetics in pol3-L612G exacerbate the extent of DSB end resection and result in under-estimation of the Pol δ contribution in the donor region, particularly at 6 h post-HO induction.
BIR Requires DNA Pols α and δ
The HydEn-seq data provide compelling evidence for Pol δ-catalyzed synthesis of both strands during BIR, but it was unclear whether Pol α did not contribute or whether its contributions were simply below the compounded noise level. Thus, we employed conditional alleles of relevant genes and measured BIR by PCR and Southern blot analyses using the Chr V recipient and Chr XI 15-kb donor strain (Figure 5). Initially, we tested the BIR efficiency of the temperature-sensitive pri2-1 (one of the subunits of the Pol α-primase complex) mutant grown at permissive (25°C) or restrictive temperatures (37°C). Cells were grown at 25°C and then arrested with nocodazole prior to temperature shift and HO induction to ensure that S phase replication had completed before inducing BIR (Figure 5A; Wang et al., 2004). At the permissive temperature, PCR and Southern blot products appear with similar kinetics to WT (Figures 5B and 5C). By contrast, product formation is greatly decreased in the pri2-1 mutant at the restrictive temperature, consistent with a previous study (Lydeard et al., 2007). If DNA synthesis involves one or more priming events, we would predict a requirement for DNA ligase I to seal the fragments. We find a similar reduction in BIR by inactivation of DNA ligase I (encoded by CDC9) as observed for the pri2-1 mutant (Figures 5B and 5C). In principle, the PCR assay requires extension of the invading strand by 278 nt for product detection. However, the PCR extension product is barely detectable in pri2-1 and cdc9-1 mutants, suggesting either that Pol α and DNA ligase are needed to extend the invading 3′ end or that the ssDNA intermediate is highly unstable if second-strand synthesis is prevented.
Figure 5. DNA Pol ε Complex Is Dispensable for BIR.
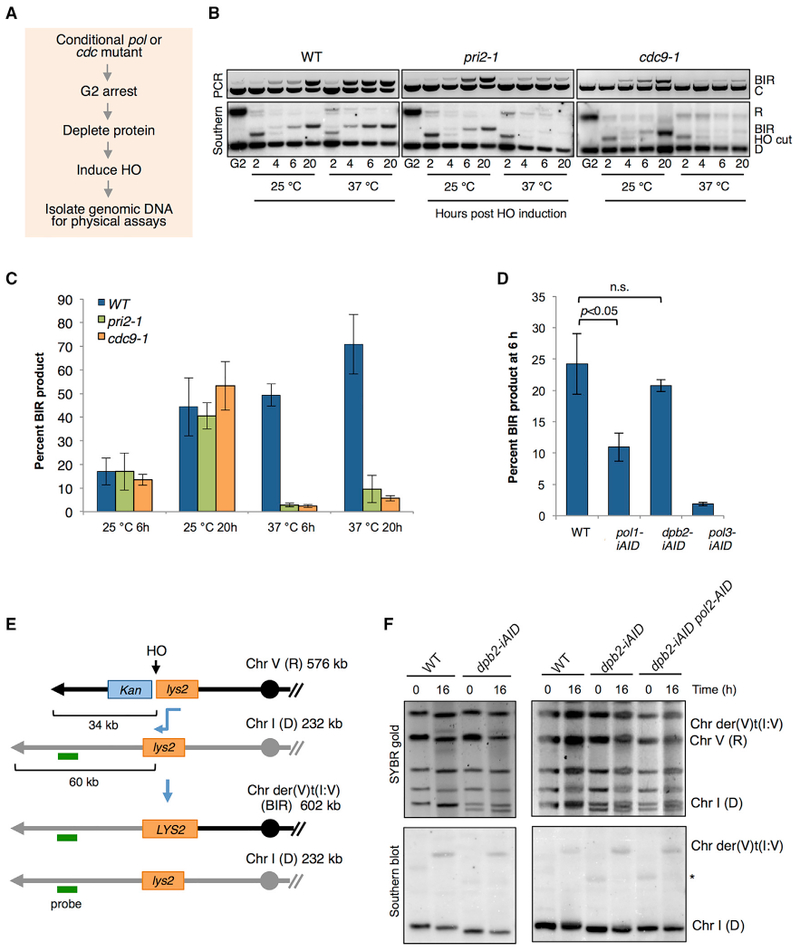
(A) Schematic of protocol used to analyze conditional mutants.
(B) Analysis of BIR kinetics in nocodazole-arrested cells at permissive (25°C) and upon inactivation of Pol α/primase or DNA ligase I by pri2-1 or cdc9-1 mutations, respectively, at non-permissive temperature (37°C). Primer extension PCR is shown in the top panel, and Southern blot analysis is shown in the lower panel (see Figure 2C for locations of primers and restriction endonuclease sites used to monitor BIR).
(C) Quantification of BIR product detected by Southern blot hybridization at 6 h and 24 h is plotted. Error bars show SD from three trials.
(D) Cells were arrested in nocodazole, and the relevant polymerase was depleted by addition of 2.5 mM IAA and 50 μg/mL Dox prior to HO induction. Graph shows quantification of BIR product by Southern blot 6 h after DSB induction. Error bars show SD (n = 3); n.s. denotes not significant.
(E) Schematic of the BIR assay with the recipient cassette on Chr V and donor cassette 60 kb from the telomere of Chr I. The sizes of the R, D, and BIR product are indicated; the horizontal green line indicates the position of the hybridization probe used to detect the BIR translocation product by PFGE and Southern blot.
(F) Final BIR translocation product (der(V)t(I:V)) of the indicated strains analyzed by PFGE and SYBR gold stain for all chromosomes and Southern blot hybridization using a probe specific to a duplicated region of Chr I. Representative images from three independent trials are shown. The WT and dpb2-iAID strains have a slightly different-sized Chr I because they were derived from a cross. Smaller chromosomes often show size polymorphism due to variable numbers of sub-telomeric repeats. * indicates a non-specific hybridization signal observed in some Southern blots.
Next, we employed Tet-OFF auxin-inducible degron (iAID) alleles encoding subunits of each DNA polymerase complex (pol1-iAID, dpb2-iAID, and pol3-iAID), which were previously shown to be defective for S phase progression under restrictive conditions (Tanaka et al., 2015). DPB2 encodes the second largest subunit of the Pol ε complex. In these strains, a Tet-OFF promoter controls expression of the relevant gene and degradation of the encoded protein is induced by addition of auxin (IAA) to the growth medium. In the absence of IAA and doxycycline (Dox), all the strains exhibit similar BIR frequencies to WT (Figure S5A). We verified efficient depletion of the polymerases by the low plating efficiency of the BIR reporter strains expressing pol1-iAID, dpb2-iAID, or pol3-iAID on medium containing Dox and IAA and by western blot (Figures S5B and S5C). For physical monitoring of BIR, cells were arrested with nocodazole prior to polymerase depletion and HO induction. As anticipated, BIR products were reduced by >10-fold following depletion of Pol3, whereas no defect was found for Dpb2-depleted cells (Figure 5D). The lack of a requirement for Dpb2 is consistent with the HydEn-seq data, showing little or no synthesis by Pol ε in the translocation region. In contrast to the pri2-1 mutant, we observed only a 50% reduction in BIR when DNA Pol α was depleted using the degron system. It is possible that sufficient DNA Pol α remains after depletion for the few priming events necessary for BIR, but not for S phase synthesis using the degron system. Alternatively, the mutant protein encoded by pri2-1 might have a negative effect on DNA synthesis at the restrictive temperature.
A previous study reported a four-fold decrease in the production of full-length BIR products in Pol ε-depleted cells, and the authors suggested that a switch between Pol δ and Pol ε was needed to complete BIR repair in a system where cells were required to copy 30 kb (Lydeard et al., 2007). Therefore, one possible explanation for the failure to observe a BIR defect in our assay is that there is no switch from Pol δ to Pol ε when replicating only 15 kb by BIR. To address this possibility, we generated the dpb2-iAID allele in a strain with the donor cassette located 60 kb from the left telomere of Chr I and monitored completion of BIR by PFGE 16 h after HO induction (Figures 5E and 5F). In dpb2-iAID cells, BIR final products are detected at comparable frequencies without or with addition of Dox and IAA to deplete Pol ε, and the percent BIR is not significantly different from WT (DPB2) cells (Figures 5F, S5D, and S5E). Note that the BIR frequency is lower for the 60-kb donor than observed for the 21-kb donor (Figure 2B); furthermore, BIR is much less efficient in G2-arrested cells than cycling cells (Donnianni and Symington, 2013). To rule out the possibility that depletion of Pol ε is incomplete and sufficient polymerase remains for BIR, we measured BIR in a strain containing a pol2-AID allele in addition to dpb2-iAID. Although pol2-AID exacerbates the growth defect of the dpb2-iAID mutant on auxin-containing medium, the double mutant remains proficient for BIR (Figures 5F, S5B, and S5E).
DISCUSSION
In this study, we used two different experimental approaches to address DNA polymerase usage during BIR: HydEn-seq and conditional depletion of essential replication proteins. Collectively, our data provide compelling evidence that Pol δ synthesizes both strands during BIR. These findings are in agreement with previous studies showing that Pol δ is required to initiate BIR and is able to synthesize both leading and lagging strands after homologous-recombination-dependent fork restart at a protein barrier in Schizosaccharomyces pombe (Costantino et al., 2014; Deem et al., 2008; Dilley et al., 2016; Lydeard et al., 2007; Maloisel et al., 2008; Miyabe et al., 2015; Smith et al., 2009). In contrast to fork restart in S. pombe, which involves semi-conservative replication (Miyabe et al., 2015), BIR results from an uncoupled and conservative DNA synthesis of the two nascent strands.
In principle, a single priming event by Pol α-primase within the telomere tract would be sufficient for synthesis of the second strand by Pol δ. The non-unique nature of telomeric sequences hinders read mapping in these regions, rendering Hyden-seq blind to this possibility. Due to the limitation of resolution, we cannot conclusively assign a role for DNA Pol α by HydEn-seq, determine whether there are few or multiple priming events, or exclude the possibility of Pol α involvement in synthesis of the invading strand. The use of conditional alleles of the Pol α complex supports its role in BIR, and the need for DNA ligase I to detect products suggests that synthesis is discontinuous. More surprising is the failure to detect the BIR strand invasion intermediate when Pol α or ligase I is depleted from cells. It is not obvious why a priming event or ligation would be required to extend the 3′ invading end; thus, we favor the hypothesis that the ssDNA intermediate is highly unstable and can only be detected when converted to the double-stranded DNA (dsDNA) form by synthesis of the second strand. The mechanism for any recruitment of Pol α complex to the nascent leading strand is unclear. Pol α is known to interact with RPA, which would presumably be bound to the ssDNA intermediate formed by migrating D-loop synthesis (Ruff et al., 2016). A recent study reported a direct interaction between Xenopus laevis Rad51 and the Pol α complex (Kolinjivadi et al., 2017), and if conserved in yeast, this could potentially direct Pol α to recombination intermediates. Additionally, Pol α interacts with Cdc13 and the Stn1 at telomeres, raising the possibility of priming events within the telomeric repeat sequence via Cdc13 and/or Stn1 interaction (Grossi et al., 2004; Qi and Zakian, 2000).
In contrast to a previous study (Lydeard et al., 2007), we find no evidence for Pol ε involvement during BIR. Although we do not have a simple explanation for the 3- to 4-fold reduction in BIR previously reported for Pol ε-depleted cells 24 h after HO induction, one possibility is that a fraction of cells escaped nocodazole arrest after completing BIR repair. WT cells would then have resumed cell division, amplifying the BIR signal, whereas the Pol ε-depleted cells would have become permanently arrested even if they had completed repair and attempted to enter the next cell cycle. The lack of Pol ε requirement observed here is consistent with studies showing that BIR synthesis occurs by a migrating D-loop with Pif1 helicase facilitating strand separation to create a template for BIR rather than re-establishment of a replication fork driven by the MCM complex (Saini et al., 2013; Wilson et al., 2013). Our results suggest that Pol δ carries out most, if not all, synthesis during BIR with Pif1 facilitating strand separation for synthesis of the invading strand in the context of the D-loop (Wilson et al., 2013).
The HydEn-seq data reveal a more extensive footprint for DNA Pol δ on the top (non-invading) strand than the bottom (invading) strand, with synthesis continuing for up to 60 kb centromere proximal to the initiating DSB. End resection at the invading end is expected to continue until synthesis of both strands is complete. A previous study reported that more than 50% of cells in a population undergoing ectopic BIR have resection tracts of >15 kb (Chung et al., 2010). Because of the delay in BIR kinetics in thepol3-L612G mutant, resection might continue much further than in cells with WT Pol δ. We suggest that fill-in synthesis of the top strand eventually catches up with the 5′ strand being degraded by end resection. If Pol α and Pol δ are capable of filling in resected tracts, it raises the question of why this is only observed during BIR and not at resected breaks engaged in two-end repair. Interestingly, the mammalian CTC1-STN1-TEN1 (CST) complex, analogous to Cdc13-Stn1-Ten1 of yeast, recruits Pol α to DSBs in addition to telomeres and is suggested to counteract extensive resection by Pol α-dependent fill-in synthesis (Mirman et al., 2018). CST and Pol α recruitment to non-telomeric breaks requires the recently described shieldin complex, which acts with 53BP1 to limit end resection (Greenberg, 2018). Although Shieldin is not conserved in yeast, Cdc13 can be detected at DSBs, suggesting that fill-in synthesis by Pol α and Pol δ might act at all DSBs at low frequency, particularly when repair is delayed.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Lorraine Symington (lss5@cumc.columbia.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
S. cerevisiae W303 background strains were used for all experiments (see Table S1 for genotypes of all the strains used in this study). Media and growth conditions were as described previously (Amberg et al., 2005). Experiments were carried out with log-phase cells, unless otherwise indicated. Cells were grown at 30°C for all the experiments except for those employing temperature sensitive alleles, in which cells were grown at 23°C or 37°C.
METHOD DETAILS
Construction of yeast strains
All strains were derived from W303 (leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15 RAD5) and are listed in Table S1. BIR strains with 15, 21 and 60 kb donors were described previously (Donnianni and Symington, 2013). To construct the BIR strain with the recipient cassette adjacent to CDC6, PCR products were designed with 40-bp flanking homology to direct homologous integration 400 bp downstream of the CDC6 ORF. All targeted chromosomes have the recipient and donor lys2 fragments with the same polarity on the left chromosome arms. W303 derivatives with rnh202Δ, pol2-M664G and pol3-L612M mutations were gifts from H. Klein (NYU School of Medicine) (Epshtein et al., 2016), and S. Jinks-Robertson (Duke University) provided the pol3-01 strain (Guo et al., 2017). These strains were crossed to each of the BIR reporter strains to obtain haploid progeny with the BIR reporter and relevant mutations. The pol1-Y869A, and pol3-L612G mutations were created in W303 using plasmids YIAL31-URA and p170-URA, respectively, by two-step gene replacement, and the resulting strains crossed to strains with the BIR reporters. W303 derivatives with pri2-1, cdc9-1 or iAID polymerase alleles (Tanaka et al., 2015) were crossed to the 15 kb donor BIR strain, and pol2-AID and dpb2-iAID were also crossed to the 60 kb donor strain. Oligonucleotides used for genotyping and for BIR physical assays are listed in Table S2.
Determination of BIR Frequencies
The genetic and physical methods used to determine the BIR fraction after HO induction were performed as described previously (Donnianni and Symington, 2013). Briefly, cells were grown to exponential phase in 1% yeast extract, 2% peptone, 2% raffinose (YPR), and then plated on YP medium containing 2% glucose (YPD) or 2% galactose (YPGal). After 3 days growth, YPG plates were replicated to medium lacking lysine and to plates containing G418 to confirm BIR. The BIR frequency was determined by the ratio of colony-forming units (CFU) on YPG (Lys+) to CFU on YPD. For physical assays, cells were grown in YPR to OD600 0.5 and aliquots were removed before galactose addition (0 h) or at different time points after galactose addition to 2% final concentration. DNA was extracted from cells and analyzed by PCR or Southern blot as described previously (Donnianni and Symington, 2013). For the strains with pri2-1 or cdc9-1 mutations, cells were grown at 25°C until early log phase, treated with 20 μg/ml nocodazole to induce a G2-M arrest and after two hours were shifted to 37°C for one hour before addition of galactose. The iAID strains were grown to early log phase, treated with 20 μg/ml nocodazole to induce a G2-M arrest in the presence of 0.1 ug/ml doxycycline, and after two hours 2.5 mM 3-indoleacetic acid (IAA) and 50 μg/ml doxycycline were added to the medium. One hour later, galactose was added to 2% final concentration for HO induction.
Analysis of BIR by PFGE
Samples for PFGE were obtained from 30 mL aliquots of cultures (OD600 0.5). Cell pellets were resuspended with 800 μL of 0.5% Certified™ low melt agarose in 100 mM EDTA pH 7.5 and 80 μL of Zymolyase 20T (25 mg/ml in 10 mM KPO4, pH 7.5) and incubated for 20 minutes at 4°C. Solidified agarose embedded-cells are incubated overnight at 37°C in 1 mL of 500 mM EDTA, 10 mM Tris, pH 7.5, then treated with 5 mg/ml proteinase K in 5% sarcosyl, 500 mM EDTA pH 7.5 for 5 h at 50°C. Plugs are washed five times with 2 mM Tris, 1 mM EDTA, pH 8.0. Chromosomes were separated by electrophoresis through 1% agarose at 6 V in 0.5 X Tris-borate-EDTA at 14°C for 24 h (initial time = 45 s, final time = 95 s) using a CHEF-DR II Pulsed-Field Electrophoresis system. Gels were stained with SYBR gold and the chromosomes were then transferred to nylon membranes. To assay for the completion of BIR, membranes were probed with a PCR product generated by amplification of sequence corresponding to the BIR-duplicated region of the donor chromosome.
Chromosome purification from PFGE
Chromosomes were separated by electrophoresis through 1% Certified™ low melt agarose at 6 V in 4 l of 0.5 X Tris-borate-EDTA at 14°C for 96 h (initial time = 45 s, final time = 95 s) using a CHEF-DR II Pulsed-Field Electrophoresis system. After 48 h, half of exhausted buffer was replaced with fresh 0.5X Tris-borate-EDTA. Bands corresponding to Ch X before (time point 0 h) and after BIR (time points 6, 12, 24 h) were excised from the gel and melted at 60°C for 10 minutes. Molten agarose was equilibrated with β-Agarase I buffer (1X final), cooled 10 minutes at 42°C and then incubated with 6 U of β-Agarase I overnight at 42°C. Salt concentration of the β-Agarase I treated solution containing ChrX was adjusted with 0.3 M NaOAc, chilled on ice for 15 min and centrifuged at 15,000 X g for 15 minutes at 4°C. DNA from the supernatant was precipitated with 2 volumes of 100% isopropanol and 2 μL of 20 μg/ml glycogen. After washing the pellet with 70% isopropanol, DNA was resuspended with 25 μL of 1 X TE. Purity of the extracted DNA was evaluated by PCR using primers specific for Chr X, VI or the BIR translocation product.
Flow Cytometry
DNA content of nocodazole-treated cells was analyzed using LSR Fortessa (Becton Dickinson) cell analyzer and FlowJo software.
Immunoblotting
Protein extracts for western blot analysis were prepared from cells grown under the same conditions as for the BIR assay. Pelleted cells from 10 mL of culture were resuspended in 1 mL 2 M LiAc, incubated for 5 min at room temperature and then pelleted. Cell pellets were resuspended in 1 mL 0.4 M NaOH, incubated on ice for 5 min, pelleted and then resuspended in 0.2 mL SDS-PAGE sample buffer. Anti-AID-tag was used for western blot analysis with anti-PSTAIR (anti-CDK1) as a loading control.
HydEn-seq library construction and data analysis
Ribonucleotide footprints in purified chromosomal DNA were mapped as originally described with a few modifications (Clausen et al., 2015). Briefly, 10-20 ng of gel-purified chromosome X or BIR translocation chromosome DNA was mixed with 200 ng of genomic DNA from HeLa or HEK293T cell lines. The mixed DNA was treated by Shrimp Alkaline Phosphatase to reduce ligatable 5′ DNA ends, and followed by restriction digestion by PmeI, creating an internal control of known DNA ends, and then split in two. One half was treated with bacterial RNase HII to cleave at single genomic ribonucleotides. Then, the DNA was denatured by incubation at 90°C for 2 min and ligated to adaptor ARC140 by T4 RNA ligase 1 overnight at 25°C (Clausen et al., 2015). The DNA was denatured again and annealed to the duplex adaptor ARC76/77 (Clausen et al., 2015). The second strand synthesis was carried out by T7 DNA polymerase. Finally, the DNA was amplified for 20 cycles using KAPA HiFi HotStart Ready Mix. 0.8 volumes of MagBio HighPrep PCR beads were used to purify DNA in between all treatments that involves changing buffers and in the final cleanup. The library was sequenced on the Illumina HiSeq 2500 platform for paired end 50 bp reads.
HydEn-seq reads were aligned to reference sequence with Bowtie 2 and uniquely mapped pair and single reads with 1 mismatch were retained. 5′ ends that map to PmeI sites were counted. The geometric mean of the end counts of each PmeI site across all samples was determined. Normalization factor for each sample was determined by the median of the ratios of all PmeI sites to their respective geometric means. The reads with perfect or 1 mismatch to PmeI were excluded for subsequent analysis. After PmeI normalization, the end count within a given genomic bin (usually 50 bp) was further normalized against the internal control to get an end density that can be compared across samples. The untreated density was subtracted from the treated sample density. The resulting background-subtracted densities were compared and those from strains with mutator polymerases were adjusted until regions replicated with wild-type polymerases match the equivalent regions in the strain with only wild-type polymerases.
The fraction of DNA synthesized by each polymerase was calculated as previously described (Garbacz et al., 2018) except that instead of meta-analysis of all active origins with regression to determine polymerase-specific ribonucleotide incorporation parameters, improved library construction allowed direct calculation of polymerase synthesis fractions across the chromosome. Specifically, a system of equations is constructed assuming that each mutant polymerase inserts more ribonucleotides than its wild-type equivalent without changing the fraction of DNA synthesis for which it is responsible:
where the ribonucleotide density (yi,j,K) at position i on strand j in strain K is equal to a polymerase-independent noise factor (w) times the sum of the products of the fractional contributions (f) of each polymerase (k) and the ribonucleotide insertion rate (s) of that polymerase.
The noise scalar factors out and each insertion rate is approximated with the corrected end density on the appropriate strand in the appropriate strain in regions where Pol ε synthesizes 100% of the nascent leading strand. The resulting simultaneous equations are solved for each fractional polymerase contribution. The ribonucleotide incorporation rate approximation for Pol α included a correction factor. Results were stable when this factor ranged from 0.01 to 0.25 (default 0.05), which contains the range of accepted Pol α fractional contributions to the mature lagging strand.
QUANTIFICATION AND STATISTICAL ANALYSIS
PRISM (GraphPad) was used for statistical analysis of BIR assays. For the BIR plating assays, significance was determined by an unpaired Student’s t test from at least three independent trials of each genotype. For the Southern blot assays of digested genomic DNA, recipient, donor, DSB and BIR band intensities were first quantified by ImageJ. The average percent BIR for each mutant was calculated by the percent BIR product normalized to the percent DSB formation, where BIR product = (BIR signal/donor)time point/(recipient/donor)t0*100; and percent DSB formation = 100-(recipient/donor)time point/(recipient/donor)t0*100. For Southern blot analysis of PFGE, BIR and donor bands were quantified by ImageJ and the percent BIR was determined by BIR/donor*100 at 16 hours. Significance was determined by a t test using the mean values from at least three independent trials, and representative gels are shown.
DATA AND CODE AVAILABILITY
DNA sequencing data was analyzed as described in Method Details; the data files are available in the Gene Expression Omnibus accession number GEO: GSE133558. Unprocessed agarose gels and Southern blot images are available at http://doi.org/10.17632/44hk5n646g.3.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-AID-tag | MBL | Cat# M214-3 |
| Anti-PSTAIR | Sigma-Aldrich | Cat# P7962; RRID:AB_261183 |
| Bacterial Strains and Plasmids | ||
| NEB 5-alpha Competent E. coli (High Efficiency) | New England Biolabs (NEB) | Cat# C29871 |
| YIAL3-URA | (Pavlov et al., 2001) | N/A |
| P170-URA | (Clausen et al., 2015) | N/A |
| Biological Samples | ||
| Genomic DNA isolated from HeLa and HEK293T cells | Björn Rabe | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Nocodazole | Sigma-Aldrich | Cat# M1404 |
| Doxycycline hyclate | Sigma-Aldrich | Cat# D9891 |
| 3-indoleacetic acid (IAA) | Abcam | Cat# ab146403 |
| Certified low melt agarose | Bio-rad | Cat# 1613111 |
| SYBR Gold | Invitrogen | Cat# S11494 |
| dCTP-alpha-P32 6000Ci/mmol | Perkin Elmer | Cat# BLU513H250 |
| β Agarase I | NEB | Cat# M0392 |
| Shrimp Alkaline Phosphatase | NEB | Cat# M0371 |
| RNase HII | NEB | Cat# M0288 |
| T4 RNA Ligase I | NEB | Cat# M0204 |
| T7 DNA Polymerase | NEB | Cat# M0274 |
| Zymolyase 20T | USBiological | Cat# 37340-57-1 |
| Proteinase K | Roche | Cat# 03115852001 |
| Critical Commercial Assays | ||
| MasterPure Yeast DNA Purification Kit | Lucigen | Cat# MPY80200 |
| KAPA HiFi HotStart Ready Mix | Kapa Biosystems | Cat# KK2602 |
| HighPrep PCR Clean-Up System | MagBio Genomics | Cat# AC-60050 |
| Deposited Data | ||
| Agarose gels and Southern blot data | This paper | https://doi.org/10.17632/44hk5n646g.3 |
| Raw and analyzed DNA sequencing data | This paper | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE133558 |
| Experimental Models: Organisms/Strains | ||
| S. cerevisiae: Strain background: W303 (Table S1) | R. Rothstein | N/A |
| Oligonucleotides (Table S2) | ||
| Software and Algorithms | ||
| Flow Jo | Flow Jo | https://www.flowjo.com/ |
| ImageJ | ImageJ | https://imagej.nih.gov/ij/ |
| Bowtie 2 | (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Custom algorithm for HydEn-seq analysis | This paper | |
| Other | ||
| CHEF-DR II Pulsed-Field Gel Electrophoresis system | Bio-rad | N/A |
Highlights.
Pol δ synthesizes both strands during BIR
Pol α-primase and DNA ligase I are required for BIR
DNA synthesis during BIR is independent of Pol ε
HydEn-seq technology is applied to DNA repair synthesis
ACKNOWLEDGMENTS
We thank S. Jinks-Robertson, H. Klein, S. Tanaka, I. Whitehouse, and X. Zhao for yeast strains; L. Argueso for sharing PFGE protocols; and K. Bebenek, R. Gnügge, W.K. Holloman, L. Langston, N. Saini, and C. Sakofsky for critical reading of and thoughtful comments on the manuscript. We thank P. Mieczkowski and others from the High Throughput Sequencing Facility of UNC Chapel Hill for performing deep sequencing. This study was supported by NIH grant R35 GM126997 to L.S.S. and by project Z01 ES065070 to T.A.K. from the Division of Intramural Research of the NIH, NIEHS.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.molcel.2019.07.033.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Acharya N, Klassen R, Johnson RE, Prakash L, and Prakash S (2011). PCNA binding domains in all three subunits of yeast DNA polymerase δ modulate its function in DNA replication. Proc. Natl. Acad. Sci. USA 108, 17927–17932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amberg DC, Burke DJ, and Strathern JN (2005). Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual (Cold Spring Harbor Laboratory Press; ). [Google Scholar]
- Bhowmick R, Minocherhomji S, and Hickson ID (2016). RAD52 facilitates mitotic DNA synthesis following replication stress. Mol. Cell 64, 1117–1126. [DOI] [PubMed] [Google Scholar]
- Chung WH, Zhu Z, Papusha A, Malkova A, and Ira G (2010). Defective resection at DNA double-strand breaks leads to de novo telomere formation and enhances gene targeting. PLoS Genet. 6, e1000948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen AR, Lujan SA, Burkholder AB, Orebaugh CD, Williams JS, Clausen MF, Malc EP, Mieczkowski PA, Fargo DC, Smith DJ, and Kunkel TA (2015). Tracking replication enzymology in vivo by genomewide mapping of ribonucleotide incorporation. Nat. Struct. Mol. Biol 22, 185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantino L, Sotiriou SK, Rantala JK, Magin S, Mladenov E, Helleday T, Haber JE, Iliakis G, Kallioniemi OP, and Halazonetis TD (2014). Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 343, 88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigaku Y, Keszthelyi A, Müller CA, Miyabe I, Brooks T, Retkute R, Hubank M, Nieduszynski CA, and Carr AM (2015). A global profile of replicative polymerase usage. Nat. Struct. Mol. Biol 22, 192–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis AP, and Symington LS (2004). RAD51-dependent break-induced replication in yeast. Mol. Cell. Biol 24, 2344–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deem A, Barker K, Vanhulle K, Downing B, Vayl A, and Malkova A (2008). Defective break-induced replication leads to half-crossovers in Saccharomyces cerevisiae. Genetics 179, 1845–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deem A, Keszthelyi A, Blackgrove T, Vayl A, Coffey B, Mathur R, Chabes A, and Malkova A (2011). Break-induced replication is highly inaccurate. PLoS Biol. 9, e1000594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilley RL, Verma P, Cho NW, Winters HD, Wondisford AR, and Greenberg RA (2016). Break-induced telomere synthesis underlies alternative telomere maintenance. Nature 539, 54–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnianni RA, and Symington LS (2013). Break-induced replication occurs by conservative DNA synthesis. Proc. Natl. Acad. Sci. USA 110, 13475–13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epshtein A, Potenski CJ, and Klein HL (2016). Increased spontaneous recombination in RNase H2-deficient cells arises from multiple contiguous rNMPs and not from single rNMP residues incorporated by DNA polymerase epsilon. Microb. Cell 3, 248–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbacz MA, Lujan SA, Burkholder AB, Cox PB, Wu Q, Zhou ZX, Haber JE, and Kunkel TA (2018). Evidence that DNA polymerase δ contributes to initiating leading strand DNA replication in Saccharomyces cerevisiae. Nat. Commun 9, 858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgescu RE, Langston L, Yao NY, Yurieva O, Zhang D, Finkelstein J, Agarwal T, and O’Donnell ME (2014). Mechanism of asymmetric polymerase assembly at the eukaryotic replication fork. Nat. Struct. Mol. Biol 21, 664–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg RA (2018). Assembling a protective shield. Nat. Cell Biol 20, 862–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossi S, Puglisi A, Dmitriev PV, Lopes M, and Shore D (2004). Pol12, the B subunit of DNA polymerase alpha, functions in both telomere capping and length regulation. Genes Dev. 18, 992–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Hum YF, Lehner K, and Jinks-Robertson S (2017). Regulation of hetDNA length during mitotic double-strand break repair in yeast. Mol. Cell 67, 539–549.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings PJ, Ira G, and Lupski JR (2009). A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 5, e1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh KD, Balachander S, Hesselberth JR, and Storici F (2015). Ribose-seq: global mapping of ribonucleotides embedded in genomic DNA. Nat. Methods 12, 251–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolinjivadi AM, Sannino V, De Antoni A, Zadorozhny K, Kilkenny M, Techer H, Baldi G, Shen R, Ciccia A, Pellegrini L, et al. (2017). Smarcal1-mediated fork reversal triggers Mre11-dependent degradation of nascent DNA in the absence of Brca2 and Stable Rad51 nucleofilaments. Mol. Cell 67, 867–881.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Stith CM, Burgers PM, and Heyer WD (2009). PCNA is required for initiation of recombination-associated DNA synthesis by DNA polymerase delta. Mol. Cell 36, 704–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan SA, Williams JS, and Kunkel TA (2016). DNA polymerases divide the labor of genome replication. Trends Cell Biol. 26, 640–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luke-Glaser S, and Luke B (2012). The Mph1 helicase can promote telomere uncapping and premature senescence in budding yeast. PLoS ONE 7, e42028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lydeard JR, Jain S, Yamaguchi M, and Haber JE (2007). Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature 448, 820–823. [DOI] [PubMed] [Google Scholar]
- Lydeard JR, Lipkin-Moore Z, Sheu YJ, Stillman B, Burgers PM, and Haber JE (2010). Break-induced replication requires all essential DNA replication factors except those specific for pre-RC assembly. Genes Dev. 24, 1133–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkova A, Naylor ML, Yamaguchi M, Ira G, and Haber JE (2005). RAD51-dependent break-induced replication differs in kinetics and checkpoint responses from RAD51-mediated gene conversion. Mol. Cell. Biol 25, 933–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloisel L, Fabre F, and Gangloff S (2008). DNA polymerase delta is preferentially recruited during homologous recombination to promote heteroduplex DNA extension. Mol. Cell. Biol 28, 1373–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfrini N, Clerici M, Wery M, Colombo CV, Descrimes M, Morillon A, d’Adda di Fagagna F, and Longhese MP (2015). Resection is responsible for loss of transcription around a double-strand break in Saccharomyces cerevisiae. eLife 4, e08942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayle R, Campbell IM, Beck CR, Yu Y, Wilson M, Shaw CA, Bjergbaek L, Lupski JR, and Ira G (2015). DNA REPAIR. Mus81 and converging forks limit the mutagenicity of replication fork breakage. Science 349, 742–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta A, Beach A, and Haber JE (2017). Homology requirements and competition between gene conversion and break-induced replication during double-strand break repair. Mol. Cell 65, 515–526.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirman Z, Lottersberger F, Takai H, Kibe T, Gong Y, Takai K, Bianchi A, Zimmermann M, Durocher D, and de Lange T (2018). 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polα-dependent fill-in. Nature 560, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyabe I, Mizuno K, Keszthelyi A, Daigaku Y, Skouteri M, Mohebi S, Kunkel TA, Murray JM, and Carr AM (2015). Polymerase δ replicates both strands after homologous recombination-dependent fork restart. Nat. Struct. Mol. Biol 22, 932–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow DM, Connelly C, and Hieter P (1997). “Break copy” duplication: a model for chromosome fragment formation in Saccharomyces cerevisiae. Genetics 147, 371–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nick McElhinny SA, Kumar D, Clark AB, Watt DL, Watts BE, Lundström EB, Johansson E, Chabes A, and Kunkel TA (2010). Genome instability due to ribonucleotide incorporation into DNA. Nat. Chem. Biol 6, 774–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov YI, Shcherbakova PV, and Kunkel TA (2001). In vivo consequences of putative active site mutations in yeast DNA polymerases alpha, epsilon, delta, and zeta. Genetics 159, 47–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payen C, Koszul R, Dujon B, and Fischer G (2008). Segmental duplications arise from Pol32-dependent repair of broken forks through two alternative replication-based mechanisms. PLoS Genet. 4, e1000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash R, Satory D, Dray E, Papusha A, Scheller J, Kramer W, Krejci L, Klein H, Haber JE, Sung P, and Ira G (2009). Yeast Mph1 helicase dissociates Rad51-made D-loops: implications for crossover control in mitotic recombination. Genes Dev. 23, 67–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi H, and Zakian VA (2000). The Saccharomyces telomere-binding protein Cdc13p interacts with both the catalytic subunit of DNA polymerase alpha and the telomerase-associated est1 protein. Genes Dev. 14, 1777–1788. [PMC free article] [PubMed] [Google Scholar]
- Reijns MAM, Kemp H, Ding J, de Procé SM, Jackson AP, and Taylor MS (2015). Lagging-strand replication shapes the mutational landscape of the genome. Nature 518, 502–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roumelioti FM, Sotiriou SK, Katsini V, Chiourea M, Halazonetis TD, and Gagos S (2016). Alternative lengthening of human telomeres is a conservative DNA replication process with features of break-induced replication. EMBO Rep. 17, 1731–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruff P, Donnianni RA, Glancy E, Oh J, and Symington LS (2016). RPA stabilization of single-stranded DNA is critical for break-induced replication. Cell Rep. 17, 3359–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini N, Ramakrishnan S, Elango R, Ayyar S, Zhang Y, Deem A, Ira G, Haber JE, Lobachev KS, and Malkova A (2013). Migrating bubble during break-induced replication drives conservative DNA synthesis. Nature 502, 389–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakofsky CJ, and Malkova A (2017). Break induced replication in eukaryotes: mechanisms, functions, and consequences. Crit. Rev. Biochem. Mol. Biol 52, 395–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakofsky CJ, Roberts SA, Malc E, Mieczkowski PA, Resnick MA, Gordenin DA, and Malkova A (2014). Break-induced replication is a source of mutation clusters underlying kataegis. Cell Rep. 7, 1640–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CE, Llorente B, and Symington LS (2007). Template switching during break-induced replication. Nature 447, 102–105. [DOI] [PubMed] [Google Scholar]
- Smith CE, Lam AF, and Symington LS (2009). Aberrant double-strand break repair resulting in half crossovers in mutants defective for Rad51 or the DNA polymerase delta complex. Mol. Cell. Biol 29, 1432–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafa A, Donnianni RA, Timashev LA, Lam AF, and Symington LS (2014). Template switching during break-induced replication is promoted by the Mph1 helicase in Saccharomyces cerevisiae. Genetics 196, 1017–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Miyazawa-Onami M, Iida T, and Araki H (2015). iAID: an improved auxin-inducible degron system for the construction of a ‘tight’ conditional mutant in the budding yeast Saccharomyces cerevisiae. Yeast 32, 567–581. [DOI] [PubMed] [Google Scholar]
- Venkatesan RN, Hsu JJ, Lawrence NA, Preston BD, and Loeb LA (2006). Mutator phenotypes caused by substitution at a conserved motif A residue in eukaryotic DNA polymerase delta. J. Biol. Chem 281, 4486–4494. [DOI] [PubMed] [Google Scholar]
- Wang X, Ira G, Tercero JA, Holmes AM, Diffley JF, and Haber JE (2004). Role of DNA replication proteins in double-strand break-induced recombination in Saccharomyces cerevisiae. Mol. Cell. Biol 24, 6891–6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt DL, Johansson E, Burgers PM, and Kunkel TA (2011). Replication of ribonucleotide-containing DNA templates by yeast replicative polymerases. DNA Repair (Amst.) 10, 897–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MA, Kwon Y, Xu Y, Chung WH, Chi P, Niu H, Mayle R, Chen X, Malkova A, Sung P, and Ira G (2013). Pif1 helicase and Polδ promote recombination-coupled DNA synthesis via bubble migration. Nature 502, 393–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeeles JTP, Janska A, Early A, and Diffley JFX (2017). How the eukaryotic replisome achieves rapid and efficient DNA replication. Mol. Cell 65, 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
DNA sequencing data was analyzed as described in Method Details; the data files are available in the Gene Expression Omnibus accession number GEO: GSE133558. Unprocessed agarose gels and Southern blot images are available at http://doi.org/10.17632/44hk5n646g.3.