

Summary
Much of current molecular and cell biology research relies on the ability to purify cell types by fluorescence-activated cell sorting (FACS). FACS typically relies on the ability to label cell types of interest with antibodies or fluorescent transgenic constructs. However, antibody availability is often limited, and genetic manipulation is labor intensive or impossible in the case of primary human tissue. To date, no systematic method exists to enrich for cell types without a priori knowledge of cell-type markers. Here, we propose GateID, a computational method that combines single-cell transcriptomics with FACS index sorting to purify cell types of choice using only native cellular properties such as cell size, granularity, and mitochondrial content. We validate GateID by purifying various cell types from zebrafish kidney marrow and the human pancreas to high purity without resorting to specific antibodies or transgenes.
Keywords: flow cytometry, single-cell transcriptomics, FACS gate prediction and normalization, optimization algorithm, machine learning, cell type purification, zebrafish hematopoiesis, human pancreas, bisulphite sequencing
Graphical Abstract
Highlights
-
•
GateID predicts non-intuitive FACS gates to purify cell types based on scRNA-seq data
-
•
Predicted gates can be normalized and used in an unlimited amount of experiments
-
•
Zebrafish hematopoietic and human pancreatic cell types can be enriched up to 100%
-
•
Live GateID-purified populations can be used for downstream analyses
By integrating single-cell transcriptomics data with FACS index sorting data, GateID can be used to set nonintuitive sorting gates to efficiently isolate pure, live populations of desired cell types from heterogenous mixtures without the need for identifying labels, such as antibodies or transgenic reporters.
Introduction
Purification of cell types present in heterogenous tissues is a central goal for biologists. Fluorescence-activated cell sorting (FACS) is the method of choice to isolate up to millions of single cells based on many cellular parameters. Over the years, flow cytometry became an important tool to study the cellular heterogeneity of many complex tissues, especially in the field of cellular immunology (reviewed by O’Donnell et al., 2013). In a flow cytometer, light scatter parameters can be used to segregate single cells based on general cellular properties such as size and granularity. For example, hematopoietic cell types are known to occupy distinct areas in forward and side scatter space, which allows their isolation (Balla et al., 2010, De Rosa et al., 2001, Freel et al., 2010, Ost et al., 1998, Salzman et al., 1975). Additionally, FACS can be used to separate single cells based on the expression of a specific protein when cells are labeled with fluorescent transgenic constructs or fluorescently coupled antibodies raised against that protein (Rodriguez et al., 2017). Although demonstrated to be powerful, these purification strategies require a priori knowledge of a cell-specific marker and depend on the availability of antibodies and/or transgenic constructs. For example, purification of hematopoietic stem and progenitor cells (HSPCs) is crucial to study and treat blood-related disorders. However, no HSPC-specific marker is currently available, and HSPCs can only be enriched using elaborate sorting strategies that achieve imperfect purities (Balazs et al., 2006, Bertrand et al., 2008, Iwasaki et al., 2010, Kiel et al., 2005, Ma et al., 2011, Osawa et al., 1996, Spangrude et al., 1988). Similarly, the isolation of α and β cells from the human pancreas is essential for diabetes research. Despite efforts, antibody discovery has been hampered by trial-and-error methods that do not deliver pure populations (Banerjee and Otonkoski, 2009, Dorrell et al., 2011, Dorrell et al., 2016). Recently, intelligent image-activated cell sorting (IACS) demonstrated the ability to perform real-time high-throughput cell microscopy analysis prior to cell sorting (Nitta et al., 2018). IACS reported high specificity and sensitivity in identifying targeted populations based on parameters such as intracellular protein localization and cell-cell interaction. Although a significant instrument innovation, the use of IACS remains limited because of the need to engineer a highly complex instrument. Additionally, IACS does not eliminate the need for prior knowledge of the targeted population and reports sorting purities below 80%. Overall, no universal sorting strategy applicable in many tissues and model organisms exists, making purification of many cell types imperfect or impossible.
Sorting decisions are taken based on gate combinations that select the desired population based on the scatter and fluorescence intensity values of choice. Gate placement happens manually and is therefore highly variable between samples and error prone. Several methods have been developed with the aim to automate the gating process, such as CCAST or SPADE (reviewed by Anchang and Plevritis, 2016). Although these methods bring an automated step to the gate design, they are limited to datasets with prior knowledge of the tissue cellular composition and rely on potential markers for the cell type of choice, ignoring all other available FACS parameters.
Single-cell RNA-sequencing (scRNA-seq) has become the method of choice to study cellular heterogeneity within complex tissues (reviewed by Choi and Kim, 2019, Grün and van Oudenaarden, 2015, Kiselev et al., 2019, Svensson et al., 2018)). Importantly, scRNA-seq datasets have been used to find new and more specific markers for cell types composing heterogenous tissues. For instance, human pancreatic tissue has been widely studied to discover novel markers for all cell types present in the pancreas (Baron et al., 2016, Enge et al., 2017, Muraro et al., 2016, Segerstolpe et al., 2016, Tritschler et al., 2017). These datasets have also been mined for novel cell surface markers to enrich α cells to 85% purity (Muraro et al., 2016). In a similar manner, new surface markers for human blood cell types were uncovered (Björklund et al., 2016, Villani et al., 2017). Furthermore, CITE-Seq (cellular indexing of transcriptomes and epitopes by sequencing), a method allowing transcriptome and epitope profiling in single cells, was developed to link mRNA and protein expression (Stoeckius et al., 2017). Overall, the large diversity of available scRNA-seq datasets and the need for better solutions to purify cell types led to the creation of CellMarker, a resource for cell type markers in many mouse and human tissues (Zhang et al., 2019). Although a valuable resource, cell type purification is still limited to cell types for which commercial antibodies are available or for which transgene construction is an option.
To solve this challenge, we set out to devise a method that could purify cell types of choice without a priori knowledge of cell type markers and would rely on general cellular properties. To achieve this, we combined high-throughput measurements of many cellular properties by FACS with unbiased cell type identification by scRNA-seq. This allowed us to find the best cellular parameters to purify any cell type, as identified by scRNA-seq, of our tissues of interest. To achieve this goal, we developed GateID, a computational method that predicts unintuitive combinations of FACS gates to purify cell types of choice. GateID (1) uses scRNA-seq for unbiased cell type identification rather than on a limited set of known cell markers; (2) is antibody and transgene free and solely relies on general cellular properties such as, but not restricted to, cell refractive index, granularity, nuclear staining, cellular proliferation, and mitochondrial activity; and (3) offers automated gate design and placement, which eliminates manual gating strategies and corrects for biological and technical variability between experiments. Using GateID, we purified various cell types from zebrafish kidney marrow, including HSPCs, solely based on their general cellular properties. Additionally, we isolated live α and β cells from the human pancreas up to 100% purity and demonstrated that GateID-sorted cells can be used for downstream experiments, such as methylome profiling.
Results
GateID Design
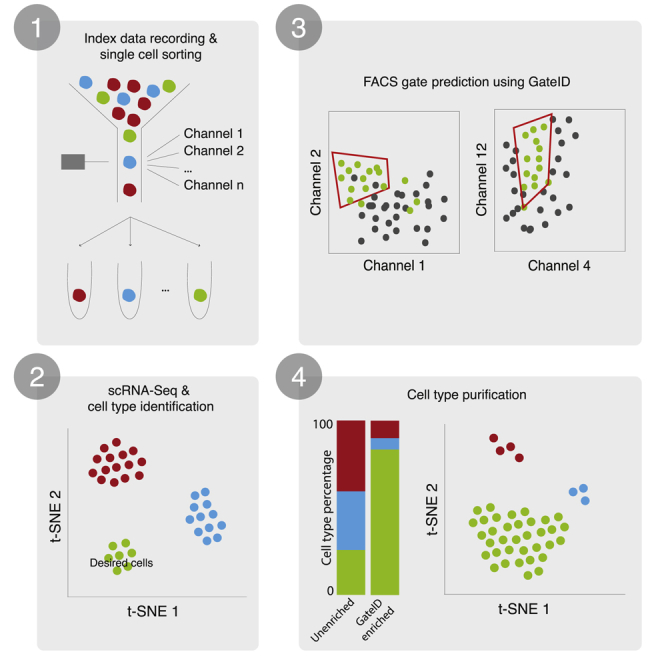
GateID is an optimization algorithm that combines the FACS index and transcriptome information of many single cells to predict non-intuitive combinations of FACS gates capable of purifying a transcriptionally distinct cell type. Importantly, gate prediction using GateID is solely data driven and does not require a priori information about FACS gates, cell types, and cellular markers. The GateID workflow starts with generating a training dataset (TD) of the organ or tissue of interest (Figure 1, step 1). To this end, single live cells are sorted while recording index data in all available scatter and fluorescence channels (Figure 1, step 1A). Next, the transcriptome of all sorted single cells is sequenced using SORT-Seq (sorting and robot-assisted transcriptome sequencing), and the cell type composition of the organ/tissue of interest is determined (Figure 1, step 1B; Muraro et al., 2016). The TD is generated by merging the index sorting parameters with the cell type information obtained by scRNA-seq for each cell (Figure 1, step 1C). After defining the desired cell type, the computational gate design occurs (Figure 1, step 2). At the core of GateID is an optimization algorithm that attempts to predict gates to obtain the maximum number of desired cells while minimizing the number of undesired cells. It iterates this procedure through all combinations of FACS channels and, subsequently, through combinations of gates to predict the best gates in terms of purity and yield (Figure 1, steps 2D and 2E; STAR Methods). Finally, the GateID-predicted gates are experimentally validated using a new sample of the organ or tissue of interest (Figure 1, step 3). Predicted gates are normalized to the new experimental dataset to correct for biological inter-individual variability and FACS technical variability (Figure 1, step 3F; STAR Methods). Single cells passing through normalized GateID gates are sorted and sequenced using scRNA-seq (Figure 1, steps 3G and 3H). The cell type composition of the GateID-enriched library is determined by clustering all cells purified by GateID gates as well as an unenriched set of cells. Using this complete dataset, the experimental purity of each GateID-enriched library is calculated.
Figure 1.

GateID Workflow
In step 1, the GateID TD is generated.
(A) Live single cells from the organ of interest are sorted in an unbiased manner, and index data for all available channels are recorded.
(B) Single cells sorted in (A) are sequenced to determine the cell type composition of the organ.
(C) The TD is generated after merging the FACS index data and the cell type information for each single cell.
In step 2, the gates are computationally designed for the desired cell type.
(D) Gates are computed for each possible combination of channels.
(E) The best combination of gates is chosen to maximize the yield and purity of the desired cell type.
In step 3, GateID-predicted gates are tested experimentally.
(F) The predicted gates are normalized to the new experimental dataset.
(G) Single cells in GateID gates are sorted.
(H) After scRNA-seq, cell types present in the GateID-enriched library are determined, and the experimental purity is calculated by comparison with the unenriched library.
See also Figure S7H and STAR Methods.
GateID Allows Purification of Zebrafish Eosinophils
First, we focused on zebrafish whole-kidney marrow (WKM), the primary site of production of hematopoietic cells in zebrafish (Murayama et al., 2006). Their isolation relies on a limited number of antibodies, transgenic lines, or manual gating subject to high variability (Traver et al., 2003, Wittamer et al., 2011). To assess whether GateID could be a more attractive method, we generated a TD1 of single live WKM hematopoietic cells (DAPI−) by merging FACS index data in 12 dimensions (BD FACSJazz) and cell type information for 1,252 cells from 3 zebrafish. Using cell clustering and known markers, we identified 7 hematopoietic cell types (Carmona et al., 2017, Grün et al., 2016, Kobayashi et al., 2010, Macaulay et al., 2016, Moore et al., 2016; Figures S1A–S1C; STAR Methods) and first aimed to enrich for eosinophils. GateID predicted a purity of 79.3% and a yield of 46.9% to isolate eosinophils using a combination of two gates (Figures 2A and S1D; Table S1). We define the purity of a set of GateID gates as the number of desired cells in the gates divided by the total number of cells in the gates. Additionally, the yield of GateID is the number of desired cells in the gates divided by the number of desired cells in our tissue of interest. We verified that 46.9% of eosinophils selected by GateID-predicted gates were not representing a transcriptionally distinct population of eosinophils (Figure S1E). To experimentally validate the predicted gates, we sorted GateID-enriched and unenriched single cells from three independent WKMs (Figure 2B). Predicted gates were normalized to each new WKM to correct for inter-individual and technical variability (Figure 2B; STAR Methods). After scRNA-seq, to ensure high confidence in our cell type identification and purity estimates, we clustered all of our zebrafish GateID experiments together (WKM 1–15, TD1–3), resulting in 15,984 single cells (Figures 2C and S1F; STAR Methods). We calculated the experimental purity of all of our WKM experiments based on this full dataset. The above-mentioned eosinophil enrichment experiments achieved an experimental purity of between 68.9% and 78%, even with eosinophil content as low as 0.6% in the unenriched population (Figure 2D, barplots; n = 3). The reason why experimental purity can vary from predicted values lies in the individual variation that can be observed in both cell type composition and FACS measurements per experiment (Figures 2 and S2A). Interestingly, we observed that contaminating cells intermingled with enriched eosinophils in FACS space and were difficult to eliminate (Figure S2B). The contaminating population in all experiments consisted mainly of monocytes. This is not surprising because eosinophils and myeloid cells occupy partly overlapping FACS regions (Balla et al., 2010). Importantly, enriched eosinophils from each experiment clustered with eosinophils in the unenriched population (Figure 2D, t-SNE [t-distributed stochastic neighbor embedding] maps), showing that GateID-enriched cells capture the existing transcriptional variance in eosinophils from the unenriched library. Finally, to compare GateID with manual gating, we isolated eosinophils as described previously (Balla et al., 2010; Figure S2C). This manual gating yielded lower enrichment compared with GateID and revealed stronger myeloid contamination (Figures S2D and S2E).
Figure S1.

Generation of the Zebrafish WKM Unstained TD, Related to Figure 2
(A) Contour plots of sorted live WKM cells to generate WKM TD1. The left panel show the DAPI- gate used to select live cells and the right panel shows the gate used to exclude erythrocytes that are low in FSC Height space.
(B) t-SNE map of zebrafish WKM TD1 generated on BD FACSJazz. Single cells are colored based on cell type.
(C) Heatmap showing marker genes for all hematopoietic cell types identified in the WKM full dataset.
(D) GateID predicted gates to isolate eosinophils from unstained WKM TD1. Grey points are undesired cells in TD1. Orange points are eosinophils outside both GateID predicted gates (excluded by GateID). Red points are eosinophils inside both GateID predicted gates (sorted by GateID).
(E) t-SNE map of zebrafish WKM TD1 where eosinophils inside and outside of GateID gates are colored in red and blue respectively.
(F) t-SNE map of experimental contributions to the zebrafish WKM full dataset. Single cells are colored based on experiment number.
Figure 2.

Proof of Principle: Enrichment of Zebrafish Eosinophils Using GateID
(A) GateID-predicted gates to isolate eosinophils from unstained WKM on BD FACSJazz. Gates were predicted on unstained WKM TD1. Red points show desired cells (eosinophils) present in TD1, and blue points show undesired cells present in the other gate. A blue undesired cell in a gate denotes an impure cell that will be sorted.
(B) Contour plots of unstained WKM cells showing experimental sorting gates for eosinophils for the WKM 2 experiment (representative example for WKM 1–3 eosinophil enrichment experiments) on BD FACSJazz. Gates in black represent GateID-predicted gates prior to normalization, whereas red gates show GateID-normalized sorting gates. Sorted cells passed through normalized gate 1 and gate 2. Percentages of events within each gate are indicated.
(C) t-SNE map of the complete zebrafish WKM dataset (all WKM TDs and enrichment experiment datasets of this study, n = 15,984 cells). Single cells are colored based on cell type.
(D) Barplots and t-SNE maps showing the outcome of GateID eosinophil enrichments for three independent experiments (WKM 1–3) on BD FACSJazz. Gates were predicted on unstained TD1. In the barplots, numbers within the bars indicate the percentage of eosinophils in the corresponding library, and numbers above the bars indicate the cell type fold enrichment between the unenriched and GateID enriched library. On the t-SNE maps, gray points represent all cells from the WKM dataset. For each experiment, black dots represent single cells in the unenriched library for a given experiment, whereas colored dots represent single cells in the GateID-enriched library for the same experiment.
(E) Left panel: principal-component analysis (PCA) of zebrafish WKM TD1 (unstained, BD FACSJazz). Each point represents a single cell, and single cells are colored based on cell type identification from scRNA-seq. The ellipses represent normal contour lines that contain 50% of the data points for each cell type. Right panel: PC1 and PC2 loadings. Each point represents a FACS channel measured by the BD FACSJazz.
(F) Curves showing the trade-off between yield and purity of GateID solutions for HSPCs, lymphocytes, monocytes, and eosinophils on stained (solid line) and unstained (dashed line) cells from the same zebrafish WKM (WKM 7).
See also Figures S1 and S2 and Tables S1 and S2.
Figure S2.

Eosinophil Enrichments with Unstained WKM Cells on BD FACSJazz, Related to Figure 2
(A) FSC Height and SSC Height contour plots of all WKM cells analyzed for eosinophils enrichment experiments WKM 1 to 3. Histograms on each plot show population density is FSC and SSC Height channels.
(B) Plots showing sorted unenriched and GateID enriched cells for eosinophil experiments WKM 1 to 3 in FSC and SSC Height. Grey points are cells from the unenriched library and colored points are cells from the GateID enriched library. Sorted eosinophils in the GateID enriched library are highlighted in orange and sorted non-eosinophil contaminating cells in the GateID enriched library are represented in black.
(C) FSC Height and SSC Height contour plot of all WKM cells for WKM 2. The eosinophil manual gate used in WKM 2 experiment is represented in red (representative for WKM 2 and 3 manual enrichment experiments).
(D and E) Barplots and t-SNE maps showing the outcome of eosinophil enrichments using manual gating for two independent experiments: (D) WKM 2 and (E) WKM 3 on BD FACSJazz. In the barplots, numbers in the bars indicate the percentage of eosinophils in the corresponding library and numbers above the bars indicate the cell type fold enrichment between unenriched and manually enriched library. On the t-SNE maps, gray points represent all cells from the WKM dataset. For each experiment, black dots are single cells in the unenriched library for a given experiment, while colored dots are single cells in the manually enriched library for the same experiment.
(F) Curves showing trade-off between yield and purity of GateID solutions for eosinophils, monocytes, lymphocytes and HSPCS for the unstained TD1. All gates for a given cell type with lower purity or yield are internal to these curves and are not shown. Dashed lines represent our thresholds for acceptable yield (0.3) and purity (0.8).
General Cellular Properties Can Be Used to Further Segregate Cell Types in FACS Space
We next aimed to isolate additional hematopoietic cell types from WKM. Our TD1 was obtained using WKM cells with a limited number of cells per individual, and GateID was unable to predict gates with satisfying purity and yield for HSPCs, lymphocytes, or monocytes (Figure S2F). Principal-component analysis (PCA) of the FACS index data showed that most of the cell types intermingle in PCA space (Figure 2E, left panel). When plotting the principal-component loadings of the dataset, we observed that only forward scatter (FSC) and side scatter (SCC) parameters contribute to significant variability in PC space and that fluorescent channels have a contribution close to zero (Figure 2E, right panel). We hypothesized that enhancing cell type separation in FACS space was necessary to purify additional hematopoietic cell types. We aimed to keep GateID antibody- or transgene-free and therefore chose to stain WKM cells with generic, easy-to-use cellular dyes. We chose MitoTracker, a fluorescent dye that reflects mitochondrial abundance and activity, and carboxyfluorescein succinimidyl ester (CFSE), which binds to cytoplasmic proteins. Neither dye stains any one cell type specifically. As validation, we used a new WKM and split it in two parts, one of which was stained with MitoTracker, CFSE, and DAPI (hereafter referred to as “stained”), whereas the other was stained only with DAPI (hereafter referred to as “unstained”). We sorted and performed scRNA-seq on both libraries and evaluated all two-gate combinations for HSPCs, lymphocytes, monocytes, and eosinophils (Figure 2F). We observed that unstained samples resulted in gates with lower yield and purity compared with stained samples.
GateID Allows Purification of Zebrafish HSPCs on Distinct FACS Machines
We generated a new TD2 containing 1,202 stained WKM cells on a BD FACSJazz (Figure S3A). PCA showed that cell types were more segregated, and we found that MitoTracker and CFSE fluorescence channels ([488] 670/long pass [LP] and [488] 530/40, respectively) highly contributed to this segregation (Figure 3A). Additionally, we generated another stained WKM TD on a BD FACSInflux. This TD3 contained 1,036 cells for which index data were recorded in 27 dimensions (Figure S3B). We used TD2 and TD3 to design gates to enrich multiple hematopoietic cell types and demonstrated that GateID performance would be independent of the FACS machine of use. First, we repeated the eosinophil enrichments using stained WKM cells and sorting with a BD FACSJazz (Figure S3C). Notably, although GateID-predicted gates are unconventional and humanly unintuitive, we show that our gated and enriched population is transcriptionally unbiased compared with unenriched cells and maps back in the same region as the classical manual FACS gate (Balla et al., 2010; Figures S3D and S3E). We obtained higher purities (85.4% on average) compared with unstained cells (73.9% on average) (Figure 3B; Figures S3F and S3G; n = 3). Next we used GateID to predict gates to enrich for HSPCs. GateID predicted a yield of 20% and a purity of 90.5% to isolate HSPCs on a BD FACSJazz using a combination of two gates, one of them using the MitoTracker fluorescence channel (Figure 3C). Not surprisingly, the projection of GateID-enriched HSPCs on the classical dimensions of FSC height and SSC height was similar to what is published (Figure 3D; Balla et al., 2010). We validated that the GateID-selected HSPCs clustered with the ones excluded by GateID, proving unbiased gate prediction (Figure 3E). Experimentally, we were able to enrich HSPCs to an average purity of 89% (Figures 3F–3H; n = 3). Additionally, GateID predicted a yield of 30% and purity of 98.6% to isolate HSPCs on a BD FACSInflux (Figures S4A–S4C). We obtained purities averaging 67% and observed no bias toward a subset of HSPCs upon GateID enrichment (Figures S4D–S4F; n = 3). Importantly, we compared our transcriptomics method for cell type calling with manual histological classification to calculate purities. We found high correlation between both methods to calculate HSPC purities after enrichment using GateID (Figure S4G). Finally, to benchmark GateID, we compared it with a classical method of enriching HSPCs based on their low expression of CD41 (Figure S3H; Bertrand et al., 2008, Ma et al., 2011). Enriched HSPCs from the CD41low fraction from CD41-eGFP transgenic zebrafish yielded inferior purity compared with GateID-predicted gates (Figures S3I and S3J). Surprisingly, the enriched HSPCs were contaminated by neutrophils. This result suggested that neutrophils reside partially in the cd41low WKM fraction, an observation that would have gone undetected without the combination of single-cell FACS and transcriptome information.
Figure S3.

General Dyes Enhance Hematopoietic Cell Type Segregation in FACS Space and Allow Purification of Eosinophils and HSPCs on BD FACSJazz, Related to Figure 3
(A) t-SNE map of zebrafish stained WKM TD2 generated on BD FACSJazz. Single cells are colored based on cell type.
(B) t-SNE map of zebrafish stained WKM TD3 generated on BD FACSInflux. Single cells are colored based on cell type.
(C) Contour plots of stained WKM cells showing normalized sorting gates for eosinophils for WKM 8 experiment (representative example for WKM 4, WKM 8 and WKM 9 eosinophil enrichments) on BD FACSJazz. Sorted cells passed through gate 1 and gate 2. Percentages of events within each gate are indicated.
(D) t-SNE map of zebrafish WKM TD2 where eosinophils inside and outside of GateID gates are colored in red and blue respectively.
(E) Projection of the sorted GateID eosinophils in WKM 8 (representative example for WKM 4, 8 and 9 eosinophil enrichment experiments) in FSC Height versus SSC Height.
(F and G) Barplots and t-SNE maps showing the outcome of eosinophil enrichments for (F) WKM 8 and (G) WKM 9 on BD FACSJazz.
(H) Left panel: FSC Height versus CD41-EGFP dot plot of live singlet WKM cells. The CD41low gate is represented in red. Right panel: projection of the CD41low sorted cells in FSC Height versus SSC Height.
(I) Barplot indicating cell type percentages for sorted CD41low cells. Percentage in the barplot indicates HSPC percentage in the sorted library.
(J) t-SNE map showing sorted CD41low cells. Non HSPCs are represented in gray and HSPCs in red.
Figure 3.

General Dyes Enhance Hematopoietic Cell Type Segregation in FACS Space to Allow HSPC Purification with GateID
(A) Left panel: PCA of zebrafish WKM TD2 (stained, BD FACSJazz). Each point represents a single cell, and single cells are colored based on cell type identification from scRNA-seq. The ellipses represent normal contour lines that contain 50% of the data points for each cell type. Right panel: PC1 and PC2 loadings. Each point represents a FACS channel measured by the BD FACSJazz.
(B) Barplots and t-SNE map showing the outcome of GateID enrichments of eosinophils (WKM 4) on BD FACSJazz. Gates were predicted on stained TD2.
(C) Contour plots of stained WKM cells showing experimental sorting gates for HSPCs for the WKM 10 experiment (representative example for WKM 5, 10, and 15 HSPC enrichment experiments) on BD FACSJazz. Sorted cells passed through gate 1 and gate 2. Percentages of events within each gate are indicated.
(D) Projection of the sorted GateID HSPCs for WKM 10 in FSC height versus SSC height (representative example for WKM 5, 10, and 15 HSPC enrichment experiments).
(E) t-SNE map of zebrafish WKM TD2, where HSPCs inside and outside of GateID gates are colored red and blue, respectively.
(F–H) Barplots and t-SNE maps showing the outcome of HSPC enrichments for (F) WKM 5, (G) WKM 10, and (H) WKM 15 on BD FACSJazz.
See also Figures S3 and S4 and Table S1.
Figure S4.

HSPC Enrichments with Stained WKM Cells on BD FACSInflux, Related to Figures 3 and 4
(A) Contour plots of stained WKM cells showing experimental sorting gates for HSPC for the WKM 11 experiment (representative example for WKM 11, 12 and 13 HSPC enrichment experiments) on BD FACSInflux. Sorted cells passed through gate 1 and gate 2. Percentages of events within each gate are indicated.
(B) t-SNE map of zebrafish WKM TD3 where HSPCs inside and outside of GateID gates are colored in red and blue respectively.
(C) Projection of the sorted GateID HSPCs for WKM 10 in FSC Height versus SSC Height (representative example for WKM 11, 12 and 13 HSPC enrichment experiments).
(D–F) Barplots and t-SNE maps showing the outcome of HSPC enrichments for (D) WKM 11, (E) WKM 12 and (F) WKM 13 on BD FACSInflux.
(G) Scatterplot showing experimental purities of GateID predicted gates determined by scRNA-seq (x axis) and histological analysis (y axis) for HSPCs (dark blue) and monocytes (light blue) on BD FACSJazz (triangle) and BD FACSInflux (circle).
(H) Design of in silico reconstruction of the manual gate for lymphocyte enrichment. Cells from WKM TD2 are represented in gray and manual gate is drawn in red.
(I) Barplots indicating cell type percentages for the lymphocyte in silico manual gate. Percentage in the barplot indicates lymphocyte percentage in the in silico manual gate.
GateID Allows Purification of Zebrafish Lymphocytes and Monocytes
Next we used GateID to isolate lymphocytes (Figures 4A–4C). Experimentally, with BD FACSJazz, we obtained unbiased enrichment between 77% and 91.7% (Figures 4D–4G; n = 4). In silico, we tested the efficiency of lymphocyte manual gating because lymphocytes are characterized by their small FSC height and SSC height properties (Figure S4H; Balla et al., 2010). The manual gate yielded 60.9% purity and exhibited HSPC contamination (Figure S4I). We then challenged GateID to isolate a subset of myeloid cells on both BD FACSJazz and BD FACSInflux. Neutrophils and monocytes are strongly intermingled in side scatter height versus forward scatter height (Balla et al., 2010). However, GateID made use of the CFSE or MitoTracker dimensions to design gates to purify monocytes (see Figures 5A–5C for BD FACSInflux and Figures S5A–S5C for BD FACSJazz). We succeeded in enriching monocytes to average purities of 87.1% on BD FACSInflux and 79.7% on BD FACSJazz (Figures 5D–5F; Figures S5D–S5F). We found the enriched populations to overlap with the one present in the live population in t-SNE space for all experiments and found neutrophils to be the highest source of contamination. Importantly, we show that the performance of GateID does not depend on the proportion of the desired cell type in the tissue of interest because no correlation (Pearson’s r = 0.07) was found between the achieved experimental purity and the abundance of the cell type of interest in all 22 of our WKM enrichment experiments (Figure 5G).
Figure 4.

GateID Allows Purification of Zebrafish Lymphocytes
(A) Contour plots of stained WKM cells showing experimental sorting gates for lymphocytes for the WKM 10 experiment (representative example for WKM 5, 6, 8, and 10 lymphocyte enrichment experiments) on BD FACSJazz. Sorted cells passed through gate 1 and gate 2. Percentages of events within each gate are indicated.
(B) Projection of the sorted GateID lymphocytes for WKM10 in FSC height versus SSC height (representative example for WKM 5, 6, 8, and 10 lymphocyte enrichment experiments).
(C) t-SNE map of zebrafish WKM TD2 where lymphocytes inside and outside of GateID gates are colored red and blue, respectively.
(D–G) Barplots and t-SNE maps showing the outcome of lymphocyte enrichments for (D) WKM 5, (E) WKM 6, (F) WKM 8, and (G) WKM 10 on BD FACSJazz.
See also Table S1.
Figure 5.

GateID Allows Purification of Zebrafish Monocytes
(A) Contour plots of stained WKM cells, showing experimental sorting gates for monocytes for the WKM 11 experiment (representative example for WKM 11, 12, and 14 monocyte enrichment experiments) on BD FACSInflux. Sorted cells passed through gate 1 and gate 2. Percentages of events within each gate are indicated.
(B) Projection of the sorted GateID monocytes for WKM 11 in FSC height versus SSC height (representative example for WKM 11, 12, and 14 monocyte enrichment experiments).
(C) t-SNE map of zebrafish WKM TD3, where monocytes inside and outside of GateID gates are colored red and blue, respectively.
(D–F) Barplots and t-SNE maps showing the outcome of monocyte enrichments for (D) WKM 11, (E) WKM 12, and (F) WKM 14 on BD FACSInflux.
(G) Scatterplot showing the percentage of the desired cell type in the unenriched library versus the achieved GateID purity in the enriched library for all WKM enrichment experiments. Points are colored based on the cell type enriched (orange for eosinophils, dark blue for HSPCs, light blue for monocytes, and green for lymphocytes) and shaped based on the FACS machine used for isolation (triangles for BD FACSJazz and circles for BD FACSInflux).
Figure S5.

Monocyte Enrichments with Stained WKM Cells on BD FACSJazz, Related to Figure 5
(A) Contour plots of stained WKM cells showing experimental sorting gates for monocytes for WKM 8 experiment (representative example for WKM 4, 7 and 8 monocyte enrichment experiments) on BD FACJazz. Sorted cells passed through gate 1 and gate 2. Percentages of events within each gate are indicated.
(B) Projection of the sorted GateID monocytes for WKM 8 in FSC Height versus SSC Height (representative example for WKM 4, 7 and 8 monocyte enrichment experiments).
(C) t-SNE map of zebrafish WKM TD2 where monocytes inside and outside of GateID gates are colored in red and blue respectively.
(D–F) Barplots and t-SNE maps showing the outcome of monocyte enrichments for (D) WKM 4, (E) WKM 7, and (F) WKM 8 BD FACJazz.
GateID Allows Purification of α and β Cells from the Human Pancreas
Next we used GateID on primary human tissue with clinical relevance. We and others have previously sequenced single cells from islets of Langerhans obtained from human cadaveric material to describe the transcriptomes of the major pancreatic cell types (α, β, δ, PP [pancreatic polypeptide], acinar, and ductal cells) (reviewed in Carrano et al., 2017). Unfortunately, isolating live α and β cells to high purity remains a challenge. Although a screen for novel markers for human pancreatic cell types resulted in purified populations of several pancreatic cell types, δ cell markers were found in the enriched β cell population compared with other cell types, indicating contamination from delta cells (see Table 1 in Dorrell et al., 2011). We thus set out to use GateID to enrich α and β cells to high purity from human pancreas without resorting to any antibodies. First we used one of the donors from our previous dataset (donor 30) as a TD (Muraro et al., 2016). We merged the BD FACSJazz index parameters with the cell type information for 664 DAPI− single cells (Figure S6A, TD1). GateID predicted 43% yield and 100% purity for α cells and 52% yield and 100% purity for β cells (Figures S6B and S6D). To experimentally validate the GateID-predicted gates, we used a new donor (donor 1) to sort enriched and unenriched cells (Figures S6C and S6E). We clustered all scRNA-seq data from our pancreas experiments to call cell types and calculate the experimental purities. This combined dataset resulted in 10,176 cells representing 8 distinct pancreatic cell types (Figures 6A and S6F). For donor 1, we obtained 97.2% α cell purity and a 78.3% pure β cell population (Figure S6G, barplot). Importantly, GateID-enriched α and β cells clustered together with the unenriched population, revealing unbiased enrichment of both cell types (Figure S6G, t-SNE maps). We observed that contamination in donor 1 when enriching for β cells originated from all other pancreatic cell types, indicating overall inefficient exclusion of undesired cell types. We hypothesized that TD1 did not contain enough information about the undesired cells that would be present in an experimental sort or a larger dataset, leading to inefficient exclusion of undesired cells. To test this hypothesis, we built a larger TD2 of 2,255 cells by sorting DAPI-stained single cells on a BD FACSJazz and performing scRNA-seq to identify the main pancreatic cell types (Figure S6H). PCA showed that the cell types present in TD2 were robustly segregated in PCA space (Figure 6B). First, we repeated the α cell enrichment with new predicted gates designed on TD2 (yield 51% and purity 97%; Figures 6C and 6D) and obtained 89% experimental purity (Figure 6H). GateID predicted gates of 26% yield and 98% purity for β cells (Figures 6E and 6F). We experimentally validated these gates with three independent donors (donors 2–4) and achieved an average purity of 95% (Figures 6G and 6H; Figure S6I). In t-SNE space, GateID-enriched β cells did not separate from the ones in the unenriched fraction showing unbiased enrichment. Driven by these experimental results and to more precisely estimate the adequate size of a TD, we performed β cell gate design using GateID on various datasets computationally generated from TD1 (Figure S7A; STAR Methods). We computationally changed the ratio of contaminating cells in the enlarged datasets to visualize the effect of the proportion of non-β cells on the performance of GateID gates. We observed that gates designed on a smaller dataset (1× TD1, 664 cells) fare poorly in comparison with gates designed on a larger dataset (2× and 3× TD1; 1,328 and 1,992 cells, respectively). Overall, in line with our results with WKM TDs, we found TDs ranging from 1,000 to 1,300 to allow robust gate design using GateID.
Figure S6.

Gates for Enrichments of α and β Cells from Unstained Pancreatic Tissue on BD FACSJazz, Related to Figure 6
(A) t-SNE map of human pancreas TD1 generated on on BD FACSJazz. Single cells are colored based on cell type.
(B) GateID predicted gates to isolate alpha cells from human pancreas. Gates were predicted on TD1. Red points show desired cells (alpha cells) present in TD and the blue points show undesired cells falling in the other gate.
(C) Contour plots of unstained human pancreas cells showing experimental gates used to sort alpha cells from donor 1. Sorted cells passed through gate 1 and gate 2. Percentages of events within each gate are indicated.
(D) GateID predicted gates to isolate beta cells from human pancreas. Gates were predicted on TD1. Red points show desired cells (beta cells) present in TD and the blue points show undesired cells falling in the other gate.
(E) Contour plots of unstained human pancreas cells showing experimental gates used to sort beta cells from donor 1. Sorted cells passed through gate 1 and gate 2. Percentages of events within each gate are indicated.
(F) t-SNE map of human pancreas full dataset. Single cells are colored based on experiment number.
(G) Barplots and t-SNE map showing the outcome of GateID alpha and beta cell enrichments for donor 1 on BD FACSJazz. Gates for were predicted on unstained TD1.
(H) t-SNE map of human pancreas TD2 generated on BD FACSJazz. Single cells are colored based on cell type.
(I) Barplots and t-SNE map showing the outcome of GateID beta cell enrichment for donor 2 on BD FACSJazz. Gates for were predicted on unstained TD2.
Figure 6.

GateID Allows Enrichment of α and β Cells from Human Pancreatic Islets
(A) t-SNE map of the complete pancreas dataset (all pancreas TDs and unstained enrichment experiment datasets, n = 10,176 cells). Single cells are colored based on cell type.
(B) Left panel: PCA of human pancreas TD2 (unstained, BD FACSJazz). Each point represents a single cell, and single cells are colored based on cell type identification from scRNA-seq. The ellipses represent normal contour lines that contain 50% of the data points for each cell type. Right panel: PC1 and PC2 loadings. Each point represents a FACS channel measured by the BD FACSJazz.
(C) Contour plots of unstained human pancreas cells showing experimental gates used to sort α cells from donor 4. Sorted cells passed through gate 1 and gate 2. Percentages of events within each gate are indicated.
(D) t-SNE map of human pancreas TD2, where α cells inside and outside of GateID gates are colored red and blue, respectively.
(E) Contour plots of unstained human pancreas cells showing experimental gates used to sort β cells from donor 3. Sorted cells passed through gate 1 and gate 2. Percentages of events within each gate are indicated.
(F) t-SNE map of human pancreas TD2, where β cells inside and outside of GateID gates are colored red and blue, respectively.
(G and H) Barplots and t-SNE maps showing the outcome of GateID α and β cell enrichments for (G) donor and (H) donor 4 on BD FACSJazz. Gates were predicted on unstained TD2.
See also Figures S6 and S7 and Table S1.
Figure S7.

α and β Cells Purified with GateID Can Be Used for Methylome Analysis, Related to Figure 6
(A) Average beta cell purity depending on TD size and proportion of contaminating cells in the TD. The y axis denotes the average GateID purity and its standard deviation. The x axis represents the fold change of the proportion of the contaminating cells in the TD. The curves represent different datasets: 1x is the original pancreas TD1 (678 cells), while 2x and 3x datasets are enlarged by two (1356 cells) or three (2034 cells) fold, respectively.
(B) Hierarchical clustering of mean methylation values for differentially methylated bins from the most variable bins, wherein methylation is shown in a gradient from blue (low) to red (high). Methylation in pancreatic alpha and beta cells cluster by cell type instead of donor of origin (indicated in columns). Bins with annotated genes of interest (rows) are shown on the right.
(C) Bins used in (D) were annotated and grouped by their genomic features for donor 4, wherein each point represents an average methylation value for a certain bin. Average methylation from alpha cells is shown on the x axis while y axis represents beta cells.
(D and E) Histogram of UMI counts and number of detected genes per cell for (D) zebrafish WKM full dataset and (E) human pancreas full dataset.
(F) Purity estimate for 100 samples of gate optimization for a pair of gates using different optimization algorithms. The figure shows that MA-LS-Chains shows the best purity in comparison to 8 different optimization algorithms used here.
(G) Time (in seconds) 100 samples of gate optimization for a pair of gates using different optimization algorithms. NMK and BOQA algorithms are fast but at the cost of substandard solution for the gate prediction problem.
(H) Workflow of the normalization of GateID predicted gates to a new experimental dataset. In step a, the data of 10000 events is exported live from the FACS machine to a laptop. In step b, the GateID gates are normalized leading to normalized gate coordinates (for each gate vertex (rows) the x and y gate coordinates are printed). Finally, in step c, the normalized gate coordinated are imported back into the FACS instrument via a software interface or the XML file of the workspace).
GateID Improves Cell Type Purification Using Antibodies
Finally, we tested whether GateID could improve manual gating of antibody-stained cells. To this end, we performed a new set of experiments for which we created an antibody-stained human pancreas dataset (Figure 7A). This dataset contained (1) our previously published pancreas dataset stained with CD24-fluorescein isothiocyanate (FITC) (an endocrine marker) and TM4SF4-APC (an α cell marker) antibodies (Muraro et al., 2016; termed TD3 here) and (2) two new GateID experiments (donors 5 and 6) where gates were predicted on the abovementioned TD3. To obtain TD3, TM4SF4+ cells within the CD24− fraction were gated to purify α cells (Figure 7B). Additionally, CD24− and NOT(TM4SF4+) cells were sorted to enrich for β cells. Finally, unenriched cells were isolated to obtain the cell type composition of the pancreatic tissue. We clustered the cells from TD3, donor 5, and donor 6 and calculated that manual gating with antibodies resulted in 80.8% purity for α cells and 76.8% for β cells (Figure 7C). Next we used GateID to predict gates on TD3 for α and β cells. PCA revealed that the antibody channels ([640] 660/20 for TM4SF4 and [488] 530/40 for CD24) contributed to α and β cell separation (Figure 7D). GateID predicted combinations of two gates for α cells and three gates for β cells (Figures 7E and 7F, respectively). Using two new pancreatic samples (donors 5 and 6) to test GateID-predicted gates and calculate experimental purities, we obtained 96.1% and 100% purity for α cells and 85.9% purity for β cells (Figures 7G and 7H).
Figure 7.

Gates for Enrichments of α and β Cells from Antibody-Stained Pancreatic Tissue on BD FACSJazz
(A) t-SNE map of the human pancreas antibody-stained dataset (TD3, donors 5 and 6). Single cells are colored based on cell type.
(B) FACS plot of TD3, showing the manual sorting gate for α cells. Displayed cells are live singlets.
(C) Barplot and t-SNE map showing the outcome of manual gating enrichments for TD3 on BD FACSJazz.
(D) Upper panel: PCA of TD3 (antibody-stained, BD FACSJazz). Each point represents a single cell, and single cells are colored based on cell type identification from scRNA-seq. The ellipses represent normal contour lines that contain 50% of the data points for each cell type. Bottom panel: PC1 and PC2 loadings. Each point represents a FACS channel measured by the BD FACSJazz.
(E and F) Contour plots of antibody-stained human pancreas cells showing experimental sorting gates for (E) α cells and (F) β cells for donor 6 (representative example for donor 5 and 6 α cell enrichment experiments) on BD FACSJazz. Sorted cells passed through normalized gate 1 and gate 2. Percentages of events within each gate are indicated.
(G and H) Barplots and t-SNE maps showing the outcome of GateID enrichments for (G) donor 5 and (H) donor 6 on BD FACSJazz.
See also Table S1.
Purified GateID Live Cells Can Be Used for Downstream Methylome Profiling
To demonstrate that live cells purified by GateID can be used as input for downstream experiments, we set out to obtain the methylomes from GateID-enriched α and β cells from the human pancreas. Despite efforts, an unbiased genome-wide characterization based on bisulfite sequencing (BS-seq) has been lacking (Neiman et al., 2017). We used BS-seq on GateID-purified populations of α and β cells isolated from two donors (donors 4 and 5, 250 cells each; Clark et al., 2017). As described in Neiman et al. (2017), we identified many differentially methylated loci. Within the top 1,000 bins ordered by the variance of their average methylation values per bin, we found 183 bins to be significantly differentially methylated (Benjamini and Hochberg-adjusted p < 0.05; Table S2). Figure S7B shows the output of hierarchical clustering using these bins, with cell types from different donors clustering together, whereas Figure S7C shows the same bins annotated for different genomic features. We found promoter and 5′ or 3′ UTRs with significant differential methylation between α and β cells, some of which are highlighted in Figure S7B. These include the insulin (INS2) promoter, which is methylated in α cells but significantly lowly methylated in β cells. Other examples of genes include SIX2, which we and others found to be differentially upregulated in β cells and which shows age-related expression in humans (Arda et al., 2016, Muraro et al., 2016). Interestingly, WNT5B, a gene previously associated with type 2 diabetes but for which α cell-specific expression has never been reported, appears to display β cell hypermethylation that would require further validation (Dorrell et al., 2011). Other genes in this analysis include genes with known pancreatic cell-type-specific function, like INS-IGF2 and PDX1, but also many others that have no reported pancreatic function, which yields a valuable resource for further research regarding the link between methylome and pancreatic cell function.
Discussion
We have described a novel computational method that combines single-cell transcriptomics and single-cell FACS to predict FACS gates that allow cell type enrichment without the aid of transgenes or antibodies. To demonstrate the effectiveness of GateID, we enriched four major hematopoietic cell types from the zebrafish WKM. Our approach proves sufficiently robust to enrich for hematopoietic cell types ranging from 0.5% (eosinophils) to 35% (monocytes) of the total WKM cell composition (Figures 2, 3, 4, and 5). The performance of GateID is also independent of age and gender because we did not control for these parameters when choosing our zebrafish or human samples. Additionally, our approach allows purification of more than one cell type from one animal, as shown by purifying eosinophils, lymphocytes, and monocytes from WKM 8 (Figures 4G, S3F, and S5F).
We also showed that GateID could enrich for human α and β cells from the islets of Langerhans up to 99% purity (Figures 6 and 7). With our pancreatic datasets, we demonstrate the importance of the composition of the TD and we find that three factors play a role in the performance of a TD to design gates, in order of importance: (1) the distinction between desired and undesired cell types in FACS space, (2) the proportion of undesired cells (potential contamination) in the TD, and (3) the number of desired cells in the TD. Although generating such a TD can be a limitation because of the costs of scRNA-seq, we note that TDs can be used to generate gates for all cell types present in the organ of choice. Additionally, because of GateID’s robust normalization strategy, the user is able to enrich for a desired cell type in an unlimited number of experiments on different samples. Overall, our pancreatic enrichment experiments demonstrate that GateID can be used to purify human cell types from primary tissues without resorting to antibodies often limited in their availability.
Overall, with the increase in applications using machine learning in everyday and scientific areas, we believe that FACS machines could benefit from innovations such as GateID. For example, models capable of identifying cell types from FACS readouts could be trained prior to a sort and deployed as part of the FACS software. Computations achieved in real time as the cell passes through the FACS nozzle could allow gate-free cell type purification. We anticipate that GateID will pave the way for implementing machine learning-based antibody and/or transgene-free automated cell type purification as a routine FACS tool.
Limitations
In the WKM, we demonstrate that separation of the desired cell type compared with other cell types in FACS space can be an important variable in the performance of GateID. A limitation of GateID is that it may not always perform well when cell types largely overlap in FACS space. That is, certain cell types might prove difficult to segregate based on their scatter and autofluorescence properties alone. Although these properties were sufficient to predict GateID gates for zebrafish eosinophils and human pancreatic α and β cells, they were not sufficient for zebrafish lymphocytes, HSPCs, and monocytes (Figure S2F). Although remaining antibody- and transgene-free, we demonstrated that general dyes (MitoTracker and CFSE) allow segregation of hematopoietic cell types in FACS space and allow successful gate prediction and validation (Figures 3, 4, and 5). General dyes are very diverse, can be coupled to various fluorochromes, and are inexpensive and very easy to use. We therefore believe that implementing general dyes when generating a TD is relatively simple and will generate many additional gate combinations, allowing gating for the desired cell type.
In our pancreas experiments, we demonstrated the effect of biological variability between training and experimental datasets on GateID’s performance. Variability mainly springs from variable proportions and statistical properties of cell types in different datasets. GateID offers a normalization strategy to correct for such variability. However, GateID normalization performs adequately only when the TD captures sufficient diversity from the chosen sample. Therefore, the TD will need to contain a sufficient number of cells and cover the cellular diversity of the tissue of interest. In light of these observations, GateID’s performance will be limited in biological systems where new cell states arise, such as a differentiating tissue or different time points during embryonic development. In such systems, a new TD will be required to capture the FACS index and transcriptome features of the new cell types or states and design GateID gates.
Finally, GateID requires a scRNA-seq method that is compatible with the recording of index data (Figure 1). Here we demonstrate GateID’s performance using SORT-Seq to generate transcriptomics data of single cells sorted on a BD FACSJazz or FACSInflux to record index information. We envision that any flow cytometer that allows importing of gates from external sources or a manual input of gate coordinates can be used in combination with any plate-based scRNA-seq method. However, microfluidics-based scRNA-seq methods that take single-cell suspensions as input are not compatible with GateID. Despite this technical limitation, and as sequencing costs continue to decline, we expect broad application of GateID to make purification of any given cell type easier and to allow enrichment of cell types never before isolated.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| FITC-Mouse anti Human CD24 Clone ML5 | BD | Cat #560992; RRID: AB_10562033 |
| TM4SF4-APC | BD | Cat #FAB7998A |
| MitoTracker Deep Red | Molecular Probes | Cat #M22426 |
| CellTrace CFSE Cell Proliferation Kit, for flow cytometry | Invitrogen | Cat #C34554 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Accutase | StemCell Technologies | Cat #07920 |
| CMRL 1066 medium | Mediatech | Cat #99663-CV |
| DAPI Solution 1 mg/mL | Thermofisher | Cat #62248 |
| Paraformaldehyde Aqueous Solution −16% | EMS | Cat #15710 |
| May-Grunwald Stain | Sigma | Cat #MG1L-1L |
| 10XPBS Buffer pH7.4 | Ambion | Cat #AM9625 |
| Giemsa Stain, Modified | Sigma | Cat #GS1l-1L |
| Deposited Data | ||
| scRNA-seq data | This paper | GEO: GSE112438 |
| Experimental Models: Organisms/Strains | ||
| Zebrafish Danio-Rerio AB strain | ZIRC | N/A |
| Zebrafish Danio-Rerio CD41:eGFP strain | Bertrand et al., 2008 | N/A |
| Software and Algorithms | ||
| GateID algorithm | This paper | https://github.com/chlbaron/GateID |
| Rstudio software Version 1.1.463 | N/A | https://www.rstudio.com/products/rstudio/download/ |
| Burrows-Wheeler Aligner (BWA) | Li and Durbin, 2009 | http://bio-bwa.sourceforge.net/ |
| Hclust function | Scialdone et al., 2016 | https://www.rdocumentation.org/packages/stats/versions/3.6.1/topics/hclust |
| DynamicTreeCut R package | Langfelder and Horvath, 2008 | https://cran.r-project.org/web/packages/dynamicTreeCut/index.html |
| mnnCorrect function | Haghverdi et al., 2018 | https://rdrr.io/bioc/scran/man/mnnCorrect.html |
| Rmalschains R package | Bergmeir et al., 2016 | https://cran.r-project.org/web/packages/Rmalschains/index.html |
| Controlled random search (CRS) and bound optimization with quadratic approximation (BOQA) functions (R package nloptr) | Price, 1983 | https://cran.r-project.org/web/packages/nloptr/index.html |
| Kaelo and Ali, 2006 | ||
| Continous genetic algorithm (R package: GA) | Scrucca, 2013 | https://cran.r-project.org/web/packages/GA/index.html |
| HJK (R package: dfoptim) | Kelley, 1999 | https://cran.r-project.org/web/packages/dfoptim/index.html |
| SA (R package: GenSA) | Xiang et al., 2013 | https://cran.r-project.org/web/packages/GenSA/index.html |
| DEoptim (R package: RcppDE) | Mullen et al., 2011 | https://cran.r-project.org/web/packages/RcppDE/index.html |
| R package tmvtnorm | Wilhelm and Manjunath, 2010 | https://cran.r-project.org/web/packages/tmvtnorm/index.html |
| Other | ||
| BD FACSJazz | BD | N/A |
| BD FACSInflux | BD | N/A |
| SORT-Seq reagents and equipment for scRNA-seq (CEL-Seq 2 based) | Muraro et al., 2016 | N/A |
| Nanodrop II liquid handling platform | GCBiotech | N/A |
| 40um cell strainer | VWR | Cat #10054-462 |
| 70um cell strainer | VWR | Cat #10054-456 |
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Alexander van Oudenaarden, Hubrecht Institute (a.vanoudenaarden@hubrecht.eu).
Experimental Model and Subject Details
Human cadaveric donor pancreata were procured through a multi-organ donor program. Pancreatic tissue was only used if the pancreas could not be used for clinical pancreas or islet transplantation, according to national laws, and if research consent was available. Age and sex of donors were not controlled. In total, 6 human donor pancreata were procured.
Zebrafish experiments using AB and CD41:GFP transgenic lines were performed in accordance with institutional and governmental regulations and were approved by the Dier Experimenten Commissie of the Royal Netherlands Academy of Arts and Science and performed according to the guidelines. Age and sex of zebrafish were not controlled.
Method Details
Tissue isolation
The WKM of WT and CD41-GFP zebrafish were isolated as described previously (Stachura and Traver, 2011). Briefly, after a ventral midline incision the internal organs were removed. The kidney was carefully dissected and collected in PBS supplemented with 5% FCS. To mechanically dissociate single hematopoietic cells, the tissue was passed multiple times through a 1 mL low-bind pipet tip. The cells were filtered (70um and 40um cell strainers (VWR)) and washed. The pellet of hematopoietic cells was resuspended in PBS/FCS supplemented with DAPI (dilution 1/2000, Thermo Fisher) to assess cell viability. In case of staining, the pellet of hematopoietic cells was resuspended in PBS/FCS supplemented with both MitoTracker and CFSE (dilution 1/4000) and incubated at room temperature for 10 min. Cells were washed and resuspended in PBS/FCS supplemented with DAPI as described above. For TD generation, DAPI- single cells were sorted (BD FACSJazz or BD FACSInflux) and erythrocytes with low forward and side scatter were excluded as described in Figure S1A. For GateID enrichment experiments, cells passing through all gates were sorted. For histology, pools of 10.000 cells were sorted in PBS supplemented with FCS and fixed 10 min in 4% PFA. After washing, cytospins were performed as described in ref. 5. Cells were post-fixed on slide and May-Grunwald-Giemsa staining was performed following manufacturer’s instructions.
Human pancreas isolation and staining with APC-TM4SF4 and FITC-CD24 was done as described previously (Muraro et al., 2016). Briefly, islets were cultured in CMRL 1066 medium (5.5 mM glucose) (Mediatech) supplemented with 10% human serum, 20 mg/ml ciprofloxacin, 50 mg/ml gentamycin, 2 mM L-gluta- min, 0.25 mg/ml fungizone, 10 mM HEPES and 1.2 mg/ml nicotinamide for 3-6 days. Islets were maintained in culture at 37C in a 5% CO2 humidified atmosphere. Medium was refreshed the day after isolation and every 2-3 days thereafter until cell sorting. The islets were cultured for 3-5 days after islet isolation. Culture time depended on the decision time needed for considering islets for trans- plantation and FACS. For cells sorted on cell surface markers; filtered, dispersed cells were incubated with FITC-CD24 (BD, 560992) and APC-TM4SF4 (BD, FAB7998A) antibodies for 30 min post dispersion on ice, followed by brief washing and sorting as above.
scRNA-Seq
We used SORT-seq to sequence the transcriptome from single cells and store FACS information from single cells (index files) (Muraro et al., 2016). All sorts were carried out using BD FACSJazz or BD FACSInflux. Unless mentioned otherwise, we used the following protocol for both model systems mentioned in this study. We lysed cells by incubating them at 65°C for 5 min, and then used Nanodrop II liquid handling platform (GC biotech) to dispense RT and second strand mixes. The aqueous phase was separated from the oil phase after pooling all cells into one library, followed by IVT transcription. The CEL-Seq2 protocol was used for library prep (Muraro et al., 2016). Primers consisted of a 24 bp polyT stretch, a 4 or 6bp random molecular barcode (UMI), a cell-specific 8bp barcode, the 5′ Illumina TruSeq small RNA kit adaptor and a T7 promoter. We used TruSeq small RNA primers (Illumina) for preparation of Illumina sequencing libraries and then paired-end sequenced them at 75 bp read length using Illumina NextSeq at approximately 45 million and 30 million reads for zebrafish kidney marrow and human pancreatic libraries respectively.
GateID algorithm
The goal of GateID is to predict gates toward sorting a desired cell type from a mixture of multiple cell types. In other words, we want to purify a specific cell type to maximum purity while sorting a sufficient fraction of the desired cells. Recent advances in flow cytometry allow users to index sort, which is to save and associate flow cytometry readouts pertinent to each sorted cell. After performing single-cell mRNA sequencing, one can then merge this information with the cell type annotation (Figure 1 – Step 1) for each cell. Such a merged dataset forms the starting point for GateID, and we refer to it as training data.
Gate prediction
We treat gate prediction as an optimization problem, wherein predicted gates allow a minimal number of undesired cells while maximizing the number of desired cells. The algorithm takes as input a matrix with FACS measurements and cell type annotation for each cell. It requires the desired cell type and the minimum yield to be input by the user. Yield is defined as a percentage of desired cells (of the total number of desired cells) that are predicted to pass through the gates. GateID first predicts a gate for each pair of flow cytometer channels, comprising scatter and fluorescence channels, where each gate is represented as a polygon with four vertices. The starting gate is computed by setting its vertices to represent the 2nd and 98th percentile in each of the x and y axis and functions as the starting point for the optimization algorithm. We use a two-step optimization as follows for the prediction of a gate: 1- The first step finds a gate that contains at least the user-specified minimum yield for desired cells while minimizing the number of undesired cells in the gate. Fitness of each solution is thus defined by the number of undesired cells in the gate. The highest fitness is the complete absence of undesired cells within the gate. The requirement of minimum yield is enforced by assigning the worst fitness (equivalent to the total number of undesired cells in the dataset) to a solution not adhering to this constraint. 2- The second step takes as input the solution (gate) of the first step and tries to maximize the yield while disallowing an increase in undesired cells. Fitness in this step is thus defined as the number of desired cells within the gate. Best fitness is achieved when all desired cells are sorted by the gate. The requirement of maximum number of undesired cells is enforced by assigning the worst fitness of zero yield to a solution not adhering to the constraint.
By default, each step is run for 20000 iterations. While evaluating fitness at each iteration, we only allow solutions involving convex polygons thereby dismissing non-convex shapes that may result in over-fitting on the training data. Once gates for each pair of FACS channels are predicted, gate combinations can then be evaluated in logical conjunction (AND combination) such as all combinations of two gates, all combinations of three gates or a higher order. For example, many of the experiments in this study were carried out on BD FACSJazz, which records cytometry readouts in twelve channels, six scatter and six fluorescence channels. There are thus C(12,2) = 66 channel pairs and 66 gates. 66 gates can be further combined to yield 2145 pairwise gate combinations (C(66,2)) evaluated in an AND configuration, meaning a cell has to pass through both gates to be sorted. As a general rule, the more gates are added in AND configuration, the slower the sorting during enrichment will be and the more cells will be lost during the sort. This is especially crucial to keep in mind when enriching from limited input material.
A possibly better strategy could be to optimize a pair of gates in AND combination together, because optimization together may allow an increase in yield while reducing impurity in a coordinated fashion. One can thus optimize all 2145 combination of pairwise gates together. In all examples we tested, two gates were enough to achieve high purity. These include stained samples of HSPCs, lymphocytes, eosinophils, and monocytes. While optimizing all 2145 pairwise gates is possible, the number quickly explodes thereafter to 45760 (combinations of 3 gates) and 720720 for 4 gate combinations and thus may become intractable. This leads us to the third intuitive approach - that of recursive gating: once gates for each combination of FACS channels are predicted (66 gates in this study), the best gate in terms of purity is selected. This gate is paired with each other gate and re-optimized together. This process is repeated until 100% purity is reached, no overall improvement is observed in the subsequent iteration or if the number of gates exceeds a user-defined preset limit. Even if there are differences in methods mentioned above, different approaches predict gates that are comparable in yield and purity, demonstrated by experiments enriching eosinophils from the unstained sample (Figure 2), wherein the first method was used versus experiments enriching eosinophils from the stained sample, where each pair of gates was optimized together (Figures 3, 4, and 5). Both experiments predicted similar purities for enrichment of eosinophils. Gates for pancreatic cell types were predicted by using the first method of optimizing gates separately, which predicted and achieved high purities experimentally (Figures 6 and 7; Figure S6).
As stated above, the objective function of the optimization procedure is to predict gates that allow a minimal number of undesired cells while maximizing the number of desired cells. This presents a discrete problem for optimization, the objective function for which is not smooth. In addition, scRNA-seq along with flow cytometry results in a limited number of cells, wherein the complete variance of each cell type population may not be captured sufficiently, especially for rarer cell populations. To address these problems, we chose a derivative-free, fast, and robust optimization algorithm called MA-LS-Chains, which combines an evolutionary algorithm along with a local search and is available as an R package (Rmalschains) (Bergmeir et al., 2016). Such algorithms are known to converge faster and more reliably without being trapped in local optima (references within Bergmeir et al., 2016). While theoretically any robust global optimization algorithm may suffice, a comparison with other algorithms (Figures S7F and S7G, and see below) shows that MA-LS-Chains is both fast and optimizes to the best purity. This is not surprising in the light of the “no free lunch” theorems, which state that certain optimization algorithms may do better than others for a certain kind of problem (Wolpert and Macready, 1997).
Gate normalization
The procedure above states in brief how gates are predicted. However, every sorted biological sample is different owing to multiple sources of variability. For example, variability is introduced during tissue isolation and subsequent sorting. An added layer of variability springs from fluctuating proportion of each cell type per isolation and variability in the statistical properties for each cell type in FACS space. For instance, the inconsistency in the proportion of each cell type can be readily observed by comparing the unenriched barplots in Figures 2, 3, 4, 5, S2, S3, S4, and S5 for the zebrafish, and Figures 6, 7, and S6 for human pancreas. Such inconsistency is further exacerbated by an overall shift in the distribution of all points demonstrated in Figure S2A (WKM1-3). For example, the distribution of side scatter height changes from a maximum of ∼500 (WKM1) to ∼100 (WKM2 and WKM3). Such variability requires that GateID predicted gates also change with respect to the current experiment in real-time (Figure 1 – Step 3F).
The first approach, normalization method 1, is to deal with such variability by standardizing the values for the vertices of gates to the unenriched population of cells of the TD using z-normalization. During FACS enrichment, one can analyze sufficient events (∼10000) and use the mean and the standard deviation of the population of the current sort to normalize gates using the reverse of z-normalization procedure. Another method for gate normalization is elaborate and requires machine learning, and we refer to it as normalization method 2. Briefly, one first trains a machine learning classifier to classify the desired cell type based on the training data. The current sort, however, could have a different overall distribution of points, different cell type proportion therefore changing the statistical variance in different dimensions and is known to create a problem for classifiers. Thus, data from the current sort needs to be normalized to the target distribution of the training data for each FACS channel. To do this, one can use methods such as non-linear qspline normalization used to compare different microarray chips to each other (Workman et al., 2002). Once the new data are normalized in this fashion, we classify the cells therein as desired and undesired cells using the trained classifier. We next z-normalize predicted gates to the desired cells from our training data and renormalize them to the predicted desired cells in the new data from the current sort. We again use z-normalization, but instead of normalizing the gates to the complete dataset, we normalize them using only the predicted desired population.
More practically, for both methods, the initial step is to acquire data in all FACS channels for 10000 events of the new experimental dataset (Figure S7H, step a). This data, which we refer to as “pre-sort data” is exported from the FACS machine (as an FCS file) and loaded into R to perform the normalization. This step can be done live at the FACS in a few minutes. The exported pre-sort data loaded into R is used for gate normalization (Figure S7H step b): 1- Method 1 performs reverse z-normalization of the GateID predicted gates using the mean and standard deviation of all the cells in the pre-sort data. Computationally, method 1 will take no longer than a few minutes. 2- Method 2 first performs a non-linear q-spline normalization of the pre-sort data to match the target distribution of the TD (Workman et al., 2002). Then, the identity of the cells in the normalized pre-sort data are classified as desired or undesired using the trained classifier, which is trained prior to the sort. This classifier needs to be trained only once on the training data and can be used as is for every subsequent sort. Finally, the GateID predicted gates are z-normalized to the desired cells (classified by the trained classifier). Computationally, method 2 can take up to 15 min if the classifier was not trained prior to the sorting experiment. However, one can train the machine learning classifier on the TD in advance, making normalization method 2 no longer than a few minutes. This approach accounts for high variability in cell type proportions from experiment to experiment, as opposed to the reverse z-normalization strategy on the complete set of points, which accounts for overall variability in the distribution of the whole dataset. We note that one can also use normalization method 2 without the use of qspline normalization but with machine learning included to train and then predict desired cells from the current FACS enrichment experiment. The output of both methods is normalized gate coordinates (x and y values for each vertex of each gate) that can be imported to the FACS machine through a software interface or the XML file of the software workspace (Figure S7H step c).
Gate prediction and normalization for zebrafish WKM
To predict gates for eosinophils from the unstained zebrafish WKM, we used GateID to optimize gates on each of the pairs of FACS channels (66 gates) and then computed the best combination of two gates in an AND combination. Gates were normalized using the normalization method 1 for each of the eosinophil sorts from the unstained WKM. For experiments concerning hematopoietic cell types in the stained WKM on BD FACSJazz (HSPCs, lymphocytes, monocytes, and eosinophils), we optimized all 2145 gate combinations together in a pairwise fashion. For experiments on BD FACSInflux, we optimized 61425 gate combinations together in a pairwise fashion. As one can observe from the eosinophil enrichment, both methods yielded experimentally similar results (Figures 2D, 3B, and S3). Gates were normalized for each sort using normalization method 2.
Gate prediction and normalization for human pancreatic alpha and beta cells
For alpha and beta cells from the human islets of Langerhans, we optimized gates for each of the pairs of FACS channels (66 gates) and computed the best combination of gates in AND configuration. Gates for alpha cell and beta cells predicted from the smaller training data (TD1, Figure S6A) were normalized using the mean normalization method 1 relying on the whole population of cells, as were the beta cell gates for the second donor, based on the second TD (Figure S6H). To compare normalization methods, beta cells from the third donor were normalized using both normalization method 1 and 2 that yielded similar results. We display the results from method 2 in Figure 6.
Comparison of different optimization algorithms
Different optimization algorithms may perform variably for different optimization tasks. To check if our choice of using MA-LS-Chains was indeed the best, we evaluated eight different optimization algorithms (Figures S7F and S7G). These were controlled random search (CRS, R package: nloptr (Price, 1983) (Kaelo and Ali, 2006)), continuous genetic algorithm (GA, R package: GA (Scrucca, 2013)), MA-LS-Chains (R package: Rmalschains (Bergmeir et al., 2016)), bounded Hooke-Jeeves (HJK, R package: dfoptim (Kelley, 1999)), bounded Nelder-Mead (NMK, R package: dfoptim (Kelley, 1999)), simulated annealing (SA, R package: GenSA (Xiang et al., 2013)), DEoptim (R package: RcppDE (Mullen et al., 2011)), bound optimization with quadratic approximation (BOQA, R package: nloptr (Powell, 2009)). We randomly chose two gates to optimize together using the stained WKM and HSPCs as the desired cells. For each optimization algorithm, we optimized those gates for maximum purity with at least a 20% yield. We repeated this process 100 times while choosing two random gates to optimize every iteration and recorded the purity of each optimization algorithm.
Computational generation of inflated dataset(s) for understanding the size of training data
It is important to understand the size of TD required for the generation of robust gates. Here, we wish to make a distinction between the feasibility of designing a gate and its efficacy during a real experiment. GateID can design gates with little number of cells, as in the case of eosinophils from the unstained training data 1 for the zebrafish WKM. In this particular case, there are 48 eosinophils in the TD (3.8% of total cell composition). Eosinophils are relatively distinct in FACS space allowing GateID to predict gates with high-predicted purity. However, an enrichment experiment involves many more cells with higher variance in their FACS readouts that may not always be represented in the TD. If this is the case, GateID cannot take into account FACS profiles for possible contaminating cell types that are not visible to the algorithm while predicting gates. Thus, in practice, GateID predicted gates may not perform as predicted using a smaller dataset. We believe this is the reason behind the fact that the predicted beta cell gates designed on the first limited TD (664 cells) of the pancreas did not yield the predicted purity in the enrichment experiment (Figure 6G, donor 1 – 78.3% purity). To further check if our hypothesis was true, we did the following computational experiment. We used our limited TD from the pancreas (TD1, 664 cells) to generate two larger datasets using truncated multivariate sampling. Briefly, we sampled random instances from a normal distribution parameterized by the mean and variance of each cell cluster in the dataset, for each FACS channel. We used this method to increase the size of our dataset twofold and then threefold. We took care that the random deviates resided within the bounds of zero and a maximum of the particular FACS channel, similar to data from a FACS experiment. This method also ensured that the artificial datasets would have identical proportion of cell clusters in comparison to the training data. We then used GateID to design gates on both these artificial datasets.
To evaluate the performance of the gates generated above, it is important to take into account that contaminating cells may be higher in number in another experiment. We therefore listed the most common cell type(s) that contaminates beta cell gates (alpha cells and delta cells for our dataset) and increased its proportion in stepwise fashion while generating a dataset of a larger size. Here, we used a size of 20000 cells, which is in the same range as an actual experiment involving sorting of live cells. We then evaluated the gates predicted by GateID on our actual TD 1 (664 cells) and two artificial TDs on this test dataset. We repeated this evaluation test 50 times for each of the three sets of gates. We observed that gates designed on a smaller dataset (1x) fare poorly in comparison to gates designed on a larger dataset (2x and 3x) (Figure S7A). Specifically, increasing the TD two-fold to 1328 cells ensures higher mean purity even in the case of twice the number of contaminating cells and may ensure higher robustness to fluctuations in cell proportions.
Truncated multivariate sampling was carried out using package ‘tmvtnorm’ in R.
Quantification and Statistical Analysis
scRNA-Seq data analysis
Zebrafish WKM and human pancreas were analyzed separately as follows. For each model system we analyzed, paired-end reads were aligned to the transcriptome of that model system using BWA (Li and Durbin, 2009). We used Read 1 for assigning reads to correct cells and libraries, while read 2 was mapped to gene models. Only reads mapping to unique locations were kept. We corrected read counts for UMI barcodes by removing duplicate reads that had identical combinations of library, cellular, and molecular barcodes and were mapped to the same gene. Transcripts were counted using 256 UMI barcodes for the human pancreas (TD3 and donors 1, 5 and 6) and 4096 UMI barcodes for the other human donors and the zebrafish kidney. The counts were then adjusted using Poissonian counting statistics to yield the number of UMIs detected per cell as described in (Figures S7D and S7E).
Data were normalized by median normalization to a minimum number of 500 transcripts and genes expressing at least three transcripts in at least two cells were retained for zebrafish WKM. Pancreatic data were median normalized to 3000 transcripts and only genes expressing 5 transcripts in at least 3 cells were retained for downstream analysis. We then computed the Pearson’s distance (1 - p) between cells. To cluster cells, we used a method previously published in (Scialdone et al., 2016). Briefly, we used hierarchical clustering (‘hclust’ R function with ‘ward.D2′ method) to cluster cells. To identify the number of clusters, we used ‘cutreeDynamic’ along with the ‘hybrid’ method which allows the user to specify a ‘deepSplit’ parameter controlling the sensitivity of clustering. We evaluated 100 subsamples of our data by randomly selecting 90% of the genes in the dataset, specifying the ‘deepSplit’ parameter as an integer from 0 to 4 and evaluating the average silhouette width of the number of clusters. This procedure resulted in identifying the correct cell types for both datasets of the zebrafish WKM data and the pancreatic data.
While evaluating the results of our enrichment experiments, we clustered all data together to ensure maximum confidence in resulting purity estimates. For zebrafish, this involved clustering both TDs and enrichment experiments (WKM 1-15) resulting in 15984 cells in all. For this clustering, we used the mnnCorrect function for batch correction (Haghverdi et al., 2018). For the pancreas data, clustering both TDs and data from four donors resulted in a total of 10176 cells.
Differentially expressed genes between two subgroups of cells were identified similar to a previously published method (Grün et al., 2014). Briefly, we started by modeling the background expected transcript count variability. We then identified genes in each subgroup that were variably expressed by representing gene expression of each gene as a negative binomial distribution. We then computed Benjamini-Hochberg corrected p-values for the observed difference in transcript counts between the two subgroups as described earlier (Anders and Huber, 2010) and identified differentially expressed genes (adjusted p-value < 0.01). Such genes were then used to annotate specific cell types within each model system based on known published literature.
For the zebrafish WKM data, we selected the topmost ten genes for each cell type ordered by their log fold change in expression when comparing the gene’s expression in a specific cell cluster compared to other cell clusters taken together (Figure S1C). Some known marker genes, especially for HSPCs and lymphocytes do not make the top ten list. We manually added them to our list of differentially expressed genes. We then used hierarchical clustering to cluster genes in seven clusters (one for each cell type). We found that our manually added genes, namely, meis1b, myb (denoting HSPCs) and pax5, cd79b (denoting lymphocytes) clustered in the appropriate clusters and do not show expression elsewhere (Figure S1C) (Tang et al., 2017). Marker gene lists for all hematopoietic cell types are appended to this manuscript as Table S3.
Data and Code Availability
The accession numbers for the scRNA-seq datasets reported in this study are available on GEO: GSE112438. The R code is available on Github: https://github.com/chlbaron/GateID.
Acknowledgments
This work was supported by a European Research Council advanced grant (ERC-AdG 742225-IntScOmics) and a Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO) TOP award (NWO-CW 714.016.001). Financial support was also provided by the Dutch Diabetes Research Foundation and the DON Foundation. This work is part of the Oncode Institute, which is partly financed by the Dutch Cancer Society. We especially thank Dr. Jake Yeung, Josi Peterson-Maduro, Dr. Lennart Kester, and all other members of the A.v.O. laboratory for discussions and input. In addition, we thank the Hubrecht Sorting Facility and the Utrecht Sequencing Facility, subsidized by the University Medical Center Utrecht; the Hubrecht Institute, and Utrecht University.
Author Contributions
A.v.O. and A.B. conceived and designed the project. A.B. developed the GateID algorithm. A.B., C.S.B., M.J.M., and A.v.O. further refined the algorithm. A.B. performed the gate design and normalization for BD FACSJazz WKM and pancreas experiments. C.S.B. performed the gate design and normalization for BD FACSInflux WKM experiments. R.v.d.L. operated both FACS machines used in this study and assisted with gate normalization. C.S.B. performed zebrafish WKM scRNA-seq experiments. A.B. and C.S.B. analyzed the zebrafish WKM scRNA-seq data. M.J.M. and G.D. performed human pancreas scRNA-seq experiments. M.J.M. analyzed the human pancreas scRNA-seq data. G.D. and E.J.P.d.K. provided human pancreatic tissue. All authors discussed and interpreted the results. C.S.B., A.B., M.J.M., and A.v.O. wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: October 3, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2019.08.006.
Supplemental Information
Table S1 shows several sorting parameters for all zebrafish WKM and human pancreas enrichment experiments. The TD used for the gate design is indicated as well as the fraction of desired cells in the corresponding TD. The GateID predicted yield and purity are indicated for each experiment. The experimentally sorted fraction of each cell type is shown next to the experimental purity. The experimental purity column is equivalent to the cell type percentage shown in Figures 2, 3, 4, 5, 6, and 7, and the barplots in Figures S1, S2, S3, S4, S5, S6 and S7.
Table S2 shows mean methylation fraction for bins of size 2 kb for alpha and beta cells in donors 4 and 5 along with gene annotation corresponding to that bin. Only significantly differentially methylated bins (Benjamini & Hochberg adjusted p value < 0.05) are included in this table.
Each tab contains the differentially expressed genes expressed in the different cell types identified in our zebrafish WKM full dataset (Figure 2C). Columns indicate, in order, mean expression in the non-cell type, mean expression in the cell type, fold change between the non-cell type and cell type, p value and adjusted p value.
References
- Anchang B., Plevritis S.K. Automated population identification and sorting algorithms for high-dimensional single-cell data. bioRxiv. 2016 [Google Scholar]
- Anders S., Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arda H.E., Li L., Tsai J., Torre E.A., Rosli Y., Peiris H., Spitale R.C., Dai C., Gu X., Qu K. Age-Dependent Pancreatic Gene Regulation Reveals Mechanisms Governing Human β Cell Function. Cell Metab. 2016;23:909–920. doi: 10.1016/j.cmet.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balazs A.B., Fabian A.J., Esmon C.T., Mulligan R.C. Endothelial protein C receptor (CD201) explicitly identifies hematopoietic stem cells in murine bone marrow. Blood. 2006;107:2317–2321. doi: 10.1182/blood-2005-06-2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balla K.M., Lugo-Villarino G., Spitsbergen J.M., Stachura D.L., Hu Y., Bañuelos K., Romo-Fewell O., Aroian R.V., Traver D. Eosinophils in the zebrafish: prospective isolation, characterization, and eosinophilia induction by helminth determinants. Blood. 2010;116:3944–3954. doi: 10.1182/blood-2010-03-267419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee M., Otonkoski T. A simple two-step protocol for the purification of human pancreatic beta cells. Diabetologia. 2009;52:621–625. doi: 10.1007/s00125-009-1259-1. [DOI] [PubMed] [Google Scholar]
- Baron M., Veres A., Wolock S.L., Faust A.L., Gaujoux R., Vetere A., Ryu J.H., Wagner B.K., Shen-Orr S.S., Klein A.M. A Single-Cell Transcriptomic Map of the Human and Mouse Pancreas Reveals Inter- and Intra-cell Population Structure. Cell Syst. 2016;3:346–360.e4. doi: 10.1016/j.cels.2016.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmeir C., Molina D., Benítez J.M. Memetic Algorithms with Local Search Chains in R : The Rmalschains Package. J. Stat. Softw. 2016;75:1–33. [Google Scholar]
- Bertrand J.Y., Kim A.D., Teng S., Traver D. CD41+ cmyb+ precursors colonize the zebrafish pronephros by a novel migration route to initiate adult hematopoiesis. Development. 2008;135:1853–1862. doi: 10.1242/dev.015297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björklund A.K., Forkel M., Picelli S., Konya V., Theorell J., Friberg D., Sandberg R., Mjösberg J. The heterogeneity of human CD127(+) innate lymphoid cells revealed by single-cell RNA sequencing. Nat. Immunol. 2016;17:451–460. doi: 10.1038/ni.3368. [DOI] [PubMed] [Google Scholar]
- Carmona S.J., Teichmann S.A., Ferreira L., Macaulay I.C., Stubbington M.J., Cvejic A., Gfeller D. Single-cell transcriptome analysis of fish immune cells provides insight into the evolution of vertebrate immune cell types. Genome Res. 2017;27:451–461. doi: 10.1101/gr.207704.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrano A.C., Mulas F., Zeng C., Sander M. Interrogating islets in health and disease with single-cell technologies. Mol. Metab. 2017;6:991–1001. doi: 10.1016/j.molmet.2017.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y.H., Kim J.K. Dissecting Cellular Heterogeneity Using Single-Cell RNA Sequencing. Mol. Cells. 2019;42:189–199. doi: 10.14348/molcells.2019.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark S.J., Smallwood S.A., Lee H.J., Krueger F., Reik W., Kelsey G. Genome-wide base-resolution mapping of DNA methylation in single cells using single-cell bisulfite sequencing (scBS-seq) Nat. Protoc. 2017;12:534–547. doi: 10.1038/nprot.2016.187. [DOI] [PubMed] [Google Scholar]
- De Rosa S.C., Herzenberg L.A., Herzenberg L.A., Roederer M. 11-color, 13-parameter flow cytometry: identification of human naive T cells by phenotype, function, and T-cell receptor diversity. Nat. Med. 2001;7:245–248. doi: 10.1038/84701. [DOI] [PubMed] [Google Scholar]
- Dorrell C., Schug J., Lin C.F., Canaday P.S., Fox A.J., Smirnova O., Bonnah R., Streeter P.R., Stoeckert C.J., Jr., Kaestner K.H., Grompe M. Transcriptomes of the major human pancreatic cell types. Diabetologia. 2011;54:2832–2844. doi: 10.1007/s00125-011-2283-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorrell C., Schug J., Canaday P.S., Russ H.A., Tarlow B.D., Grompe M.T., Horton T., Hebrok M., Streeter P.R., Kaestner K.H., Grompe M. Human islets contain four distinct subtypes of β cells. Nat. Commun. 2016;7:11756. doi: 10.1038/ncomms11756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enge M., Arda H.E., Mignardi M., Beausang J., Bottino R., Kim S.K., Quake S.R. Single-Cell Analysis of Human Pancreas Reveals Transcriptional Signatures of Aging and Somatic Mutation Patterns. Cell. 2017;171:321–330.e14. doi: 10.1016/j.cell.2017.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freel S.A., Lamoreaux L., Chattopadhyay P.K., Saunders K., Zarkowsky D., Overman R.G., Ochsenbauer C., Edmonds T.G., Kappes J.C., Cunningham C.K. Phenotypic and functional profile of HIV-inhibitory CD8 T cells elicited by natural infection and heterologous prime/boost vaccination. J. Virol. 2010;84:4998–5006. doi: 10.1128/JVI.00138-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grün D., van Oudenaarden A. Design and Analysis of Single-Cell Sequencing Experiments. Cell. 2015;163:799–810. doi: 10.1016/j.cell.2015.10.039. [DOI] [PubMed] [Google Scholar]
- Grün D., Kester L., van Oudenaarden A. Validation of noise models for single-cell transcriptomics. Nat. Methods. 2014;11:637–640. doi: 10.1038/nmeth.2930. [DOI] [PubMed] [Google Scholar]
- Grün D., Muraro M.J., Boisset J.C., Wiebrands K., Lyubimova A., Dharmadhikari G., van den Born M., van Es J., Jansen E., Clevers H. De Novo Prediction of Stem Cell Identity using Single-Cell Transcriptome Data. Cell Stem Cell. 2016;19:266–277. doi: 10.1016/j.stem.2016.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghverdi L., Lun A.T.L., Morgan M.D., Marioni J.C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 2018;36:421–427. doi: 10.1038/nbt.4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki H., Arai F., Kubota Y., Dahl M., Suda T. Endothelial protein C receptor-expressing hematopoietic stem cells reside in the perisinusoidal niche in fetal liver. Blood. 2010;116:544–553. doi: 10.1182/blood-2009-08-240903. [DOI] [PubMed] [Google Scholar]
- Kaelo P.A., Ali M.M. Some Variants of the Controlled Random Search Algorithm for Global Optimization. J. Optim. Theory Appl. 2006;130:253–264. [Google Scholar]
- Kelley C.T. Society for Industrial and Applied Mathematics; 1999. Iterative Methods for Optimization. [Google Scholar]
- Kiel M.J., Yilmaz O.H., Iwashita T., Yilmaz O.H., Terhorst C., Morrison S.J. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121:1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- Kiselev V.Y., Andrews T.S., Hemberg M. Challenges in unsupervised clustering of single-cell RNA-seq data. Nat. Rev. Genet. 2019;20:273–282. doi: 10.1038/s41576-018-0088-9. [DOI] [PubMed] [Google Scholar]
- Kobayashi I., Ono H., Moritomo T., Kano K., Nakanishi T., Suda T. Comparative gene expression analysis of zebrafish and mammals identifies common regulators in hematopoietic stem cells. Blood. 2010;115:e1–e9. doi: 10.1182/blood-2009-07-232322. [DOI] [PubMed] [Google Scholar]
- Langfelder P., Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D., Zhang J., Lin H.F., Italiano J., Handin R.I. The identification and characterization of zebrafish hematopoietic stem cells. Blood. 2011;118:289–297. doi: 10.1182/blood-2010-12-327403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macaulay I.C., Svensson V., Labalette C., Ferreira L., Hamey F., Voet T., Teichmann S.A., Cvejic A. Single-Cell RNA-Sequencing Reveals a Continuous Spectrum of Differentiation in Hematopoietic Cells. Cell Rep. 2016;14:966–977. doi: 10.1016/j.celrep.2015.12.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore F.E., Garcia E.G., Lobbardi R., Jain E., Tang Q., Moore J.C., Cortes M., Molodtsov A., Kasheta M., Luo C.C. Single-cell transcriptional analysis of normal, aberrant, and malignant hematopoiesis in zebrafish. J. Exp. Med. 2016;213:979–992. doi: 10.1084/jem.20152013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen K., Ardia D., Gil D., Windover D., Cline J. DEoptim: An R package for Global Optimization by Differential Evolution. J. Stat. Softw. 2011;40:1–26. [Google Scholar]
- Muraro M.J., Dharmadhikari G., Grun D., Groen N., Dielen T., Jansen E., van Gurp L., Engelse M.A., Carlotti F., de Koning E.J. A Single-Cell Transcriptome Atlas of the Human Pancreas. Cell Syst. 2016;3:385–394.e3. doi: 10.1016/j.cels.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murayama E., Kissa K., Zapata A., Mordelet E., Briolat V., Lin H.F., Handin R.I., Herbomel P. Tracing hematopoietic precursor migration to successive hematopoietic organs during zebrafish development. Immunity. 2006;25:963–975. doi: 10.1016/j.immuni.2006.10.015. [DOI] [PubMed] [Google Scholar]
- Neiman D., Moss J., Hecht M., Magenheim J., Piyanzin S., Shapiro A.M.J., de Koning E.J.P., Razin A., Cedar H., Shemer R., Dor Y. Islet cells share promoter hypomethylation independently of expression, but exhibit cell-type-specific methylation in enhancers. Proc. Natl. Acad. Sci. USA. 2017;114:13525–13530. doi: 10.1073/pnas.1713736114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitta N., Sugimura T., Isozaki A., Mikami H., Hiraki K., Sakuma S., Iino T., Arai F., Endo T., Fujiwaki Y. Intelligent Image-Activated Cell Sorting. Cell. 2018;175:266–276.e13. doi: 10.1016/j.cell.2018.08.028. [DOI] [PubMed] [Google Scholar]
- O’Donnell E.A., Ernst D.N., Hingorani R. Multiparameter flow cytometry: advances in high resolution analysis. Immune Netw. 2013;13:43–54. doi: 10.4110/in.2013.13.2.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osawa M., Hanada K., Hamada H., Nakauchi H. Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science. 1996;273:242–245. doi: 10.1126/science.273.5272.242. [DOI] [PubMed] [Google Scholar]
- Ost V., Neukammer J., Rinneberg H. Flow cytometric differentiation of erythrocytes and leukocytes in dilute whole blood by light scattering. Cytometry. 1998;32:191–197. [PubMed] [Google Scholar]
- Powell M.J.D. 2009. The BOBYQA algorithm for bound constrained optimization without derivatives.http://www.damtp.cam.ac.uk/user/na/NA_papers/NA2009_06.pdf [Google Scholar]
- Price W.L. Global optimization by controlled random search. J. Optim. Theory Appl. 1983;40:333–348. [Google Scholar]
- Rodriguez E.A., Campbell R.E., Lin J.Y., Lin M.Z., Miyawaki A., Palmer A.E., Shu X., Zhang J., Tsien R.Y. The Growing and Glowing Toolbox of Fluorescent and Photoactive Proteins. Trends Biochem. Sci. 2017;42:111–129. doi: 10.1016/j.tibs.2016.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzman G.C., Crowell J.M., Martin J.C., Trujillo T.T., Romero A., Mullaney P.F., LaBauve P.M. Cell classification by laser light scattering: identification and separation of unstained leukocytes. Acta Cytol. 1975;19:374–377. [PubMed] [Google Scholar]
- Scialdone A., Tanaka Y., Jawaid W., Moignard V., Wilson N.K., Macaulay I.C., Marioni J.C., Göttgens B. Resolving early mesoderm diversification through single-cell expression profiling. Nature. 2016;535:289–293. doi: 10.1038/nature18633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scrucca L.G. A Package for Genetic Algorithms in R. J. Stat. Softw. 2013;53:1–37. [Google Scholar]
- Segerstolpe Å., Palasantza A., Eliasson P., Andersson E.M., Andréasson A.C., Sun X., Picelli S., Sabirsh A., Clausen M., Bjursell M.K. Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes. Cell Metab. 2016;24:593–607. doi: 10.1016/j.cmet.2016.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangrude G.J., Heimfeld S., Weissman I.L. Purification and characterization of mouse hematopoietic stem cells. Science. 1988;241:58–62. doi: 10.1126/science.2898810. [DOI] [PubMed] [Google Scholar]
- Stachura D.L., Traver D. Cellular dissection of zebrafish hematopoiesis. Methods Cell Biol. 2011;101:75–110. doi: 10.1016/B978-0-12-387036-0.00004-9. [DOI] [PubMed] [Google Scholar]
- Stoeckius M., Hafemeister C., Stephenson W., Houck-Loomis B., Chattopadhyay P.K., Swerdlow H., Satija R., Smibert P. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods. 2017;14:865–868. doi: 10.1038/nmeth.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson V., Vento-Tormo R., Teichmann S.A. Exponential scaling of single-cell RNA-seq in the past decade. Nat. Protoc. 2018;13:599–604. doi: 10.1038/nprot.2017.149. [DOI] [PubMed] [Google Scholar]
- Tang Q., Iyer S., Lobbardi R., Moore J.C., Chen H., Lareau C., Hebert C., Shaw M.L., Neftel C., Suva M.L. Dissecting hematopoietic and renal cell heterogeneity in adult zebrafish at single-cell resolution using RNA sequencing. J. Exp. Med. 2017;214:2875–2887. doi: 10.1084/jem.20170976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traver D., Paw B.H., Poss K.D., Penberthy W.T., Lin S., Zon L.I. Transplantation and in vivo imaging of multilineage engraftment in zebrafish bloodless mutants. Nat. Immunol. 2003;4:1238–1246. doi: 10.1038/ni1007. [DOI] [PubMed] [Google Scholar]
- Tritschler S., Theis F.J., Lickert H., Böttcher A. Systematic single-cell analysis provides new insights into heterogeneity and plasticity of the pancreas. Mol. Metab. 2017;6:974–990. doi: 10.1016/j.molmet.2017.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villani A.C., Satija R., Reynolds G., Sarkizova S., Shekhar K., Fletcher J., Griesbeck M., Butler A., Zheng S., Lazo S. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science. 2017;356 doi: 10.1126/science.aah4573. eaah4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm S., Manjunath B. tmvtnorm: A Package for the Truncated Multivariate Normal Distribution. The R Journal. 2010;2 [Google Scholar]
- Wittamer V., Bertrand J.Y., Gutschow P.W., Traver D. Characterization of the mononuclear phagocyte system in zebrafish. Blood. 2011;117:7126–7135. doi: 10.1182/blood-2010-11-321448. [DOI] [PubMed] [Google Scholar]
- Wolpert D.H., Macready W.G. No free lunch theorems for optimization. IEEE Trans. Evol. Comput. 1997;1:67–82. [Google Scholar]
- Workman C., Jensen L.J., Jarmer H., Berka R., Gautier L., Nielser H.B., Saxild H.H., Nielsen C., Brunak S., Knudsen S. A new non-linear normalization method for reducing variability in DNA microarray experiments. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-9-research0048. research0048–research0048.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y., Gubian S., Suomela B., Hoeng J. Generalized Simulated Annealing for Global Optimization: The GenSA Package: An Application to Non-Convex Optimization in Finance and Physics. R Journal. 2013;5:13–38. [Google Scholar]
- Zhang X., Lan Y., Xu J., Quan F., Zhao E., Deng C., Luo T., Xu L., Liao G., Yan M. CellMarker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 2019;47(D1):D721–D728. doi: 10.1093/nar/gky900. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 shows several sorting parameters for all zebrafish WKM and human pancreas enrichment experiments. The TD used for the gate design is indicated as well as the fraction of desired cells in the corresponding TD. The GateID predicted yield and purity are indicated for each experiment. The experimentally sorted fraction of each cell type is shown next to the experimental purity. The experimental purity column is equivalent to the cell type percentage shown in Figures 2, 3, 4, 5, 6, and 7, and the barplots in Figures S1, S2, S3, S4, S5, S6 and S7.
Table S2 shows mean methylation fraction for bins of size 2 kb for alpha and beta cells in donors 4 and 5 along with gene annotation corresponding to that bin. Only significantly differentially methylated bins (Benjamini & Hochberg adjusted p value < 0.05) are included in this table.
Each tab contains the differentially expressed genes expressed in the different cell types identified in our zebrafish WKM full dataset (Figure 2C). Columns indicate, in order, mean expression in the non-cell type, mean expression in the cell type, fold change between the non-cell type and cell type, p value and adjusted p value.
Data Availability Statement
The accession numbers for the scRNA-seq datasets reported in this study are available on GEO: GSE112438. The R code is available on Github: https://github.com/chlbaron/GateID.