

Abstract
Human acid ceramidase (AC) is a lysosomal cysteine amidase, which has received a great deal of interest in recent years as a potential target for the development of new therapeutics against melanoma and glioblastoma tumors. Despite the strong interest in obtaining structural information, only the structures of the apo-AC enzyme in its zymogen and activated conformations are available. In this work, the crystal structure of AC in complex with the covalent carmofur inhibitor is presented. Carmofur is an antineoplastic drug containing an electrophilic carbonyl reactive group that targets the catalytic cysteine. This novel structural data explains the basis of the AC inhibition, provides insights into the enzymatic properties of the protein, and is a great aid toward the structure-based drug design of potent inhibitors for AC, providing the detailed mechanism, which has eluded the scientific community for more than 30 years, of carmofur’s mysterious 5-fluorouracil-independent anti-tumor activity.
Keywords: glioblastoma, acid ceramidase, acid ceramidase inhibitors, benzoxazolone carboxamides, carmofur, radioresistance, radiation, sphingosine, sphingosine-1-phosphate, S1P
Graphical Abstract
INTRODUCTION
The pyrimidine analogue 5-fluorouracil (5-FU) inhibits thymidylate synthase, an enzyme responsible for the synthesis of thymidine that contributes to DNA replication1. The compound has been effective in the treatment of several solid tumors, particularly head, neck, and gastrointestinal tumors2. In fact, the compound is the third most common agent employed against solid cancers globally2. Efforts to discover derivatives of 5-FU that were less toxic to the host and more effective against malignancies led to the discovery of carmofur (1-hexylcarbamoyl-5-fluorouracil), which has been used to treat patients with colorectal cancers for more than 30 years3,4. Early studies suggested that the hexylcarbamoyl structure (more lipophilic) eases the transport of carmofur through the cell membrane, providing a higher therapeutic ratio4. For more than 30 years, carmofur has been considered a masked compound of 5-FU, and believed to functions similarly to 5-FU5, 6. However, some researchers have questioned this mechanism as carmofur was also as effective in targeting 5-FU resistant cells compared to control6. The exact mechanism of the 5-FU independent anti-tumor activity of carmofur has remained a mystery. Studies have implicated carmofur’s involvement in multiple pathways7,8. Carmofur, which has seen clinical use since 1981, significantly augments overall survival and disease-free survival in colon cancers9. In addition, carmofur appears effective against breast, gastric, bladder cancer, and colorectal cancer cancers7, 10, 11.
Human AC (EC 3.5.1.23), an intracellular two-subunit cysteine amidase catalyzes hydrolysis of ceramides into sphingosine and free fatty acids12–14. Ceramides favor cell-cycle arrest and apoptosis; on the other hand, sphingososine-1-phospate (S1P), a direct derivative of sphingosine, encourages angiogenesis, cell survival, and cell proliferation15. AC levels are raised in several malignancies, including breast16, prostate17, colorectal18, melanoma19, and brain20, 21. Moreover, downregulation or inhibition of AC has been associated with anti-cancer activity. The deletion of AC gene in melanoma cells has been shown to hinder the formation of cancer-initiating cells22. We have shown that inhibition of AC effectively combats several brain cancer cell lines, including glioblastoma, medulloblastoma, and atypical teratoid / rhaboid tumors13, 20, 21, 23–26. Thus, recent data has made AC an attractive target for designing selective and potent inhibitors, which can modulate the balance between cell proliferation and death with the influence on both growth and survival of normal and neoplastic cells. Currently, there is no AC inhibitor clinically available to treat patients.
Recently, colleagues discovered that carmofur inhibits the activity of AC and using liquid chromatography mass spectrometry, they revealed a derivative of carmofur inhibited AC through the covalent modification of the catalytic cysteine residue27–29. However, the exact mechanism of inhibition between actual carmofur and AC has not been elaborated. These results prompted us to study the interaction between the carmofur inhibitor and human AC at the structural level.
The inactive zymogen form of AC is secreted as a single-chain polypeptide of 374 residues with molecular weight of 50–55 kDa, depending on the extent of glycosylation12–14. Upon auto-activation, the AC proenzyme is cleaved at the Thr142-Cys143 peptide bond, generating an α-subunit consisting of a 13 kDa chain and a 37–42 kDa β-subunit chain held together by the Cys31-Cys340 disulfide bond30, 31. In recent work, a catalytic mechanism for AC auto-activation was described based on structural data obtained for the apo-form of the enzyme in its zymogen and activated states30.
In this work, we crystallized and solved the structure of human active AC in complex with carmofur at 2.7 Å resolution. The mature form of AC in the complex adopts the same fold as the apo protein. The fatty acid moiety of the carmofur molecule after catalytic cleavage of the inhibitor at its carbonyl group covalently modifies the active site Cys143 and occupies one of the hydrophobic cavities close to the enzyme active site. Simple modeling data reveals the plausible mechanism of substrate degradation. Also, the structure of the AC-inhibitor complex provides important information about the geometry of the binding site, which is a critical asset for understanding the determinants for selectivity and specificity in AC-ligand interactions.
Via crystal structural analysis, this study is the first to definitively demonstrate that carmofur directly binds to AC, and prompts its inhibition, providing the detailed mechanism, which has eluded the scientific community for more than 30 years, of its mysterious 5-FU-independent anti-tumor activity. This provides the impetus to now engage in clinical trial with carmofur for treatment of various types of cancers targeting AC as a new class of anticancer drugs.
RESULTS AND DISCUSSION
Overall structure of the AC-carmofur complex
The structure of human AC in covalent complex with the free fatty acid tail of the small carmofur inhibitor (5-fluoro-N-hexyl-2,4-dioxo-pyrimidine-1-carboxamide, supplemental Figure 1) was solved at a resolution of 2.7 Å (Table 1). For the final model, 114 residues (residues 27–140 excluding the signal peptide) of the α-subunit and all 253 residues (residues 143–395) of the β-subunit, with the exception of a few side chains, were traceable in the electron density map (Figure 1). The heterodimeric structure of AC displays strong structural similarity with that of other members of the Ntn-hydrolase family, consisting of two β sheets forming the substrate-binding site and the catalytic residues, Cys143, Asp162, Glu225, and Asn320, located at the cleft between these two sheets, which are flanked by six α helices (Figure 1A). The main body of the α-subunit is positioned on the molecular surface of the β–subunit far away from the active site entrance with its N-terminal segment wrapping around the β-subunit to reach a distance of 12.5 Å between Cα atoms of the α–chain N-terminal Trp27 and the α-chain C-terminal Trp396, and is covalently linked to the β-domain through a disulfide bridge between Cys31–Cys340. The α-chain also makes contacts from both the backbone and side chains of its C-terminus to the residues in the β-domain located close to the active site cleft (Figure 1A). These include a weak hydrogen bond from the backbone carbonyl of Glu138 to the backbone amino group of Gly232, a hydrogen bond from the side chain of Glu138 to the backbone amino group of Ile249, and a hydrogen bond from the side chain of Asn134 to the backbone amino group of Val208. Finally, the side chains of Phe140, Leu139, Tyr137, and Ile135 of α–subunit form extensive hydrophobic interactions with residues located on the helix (230–241) and the loop (244–249) of the β–subunit.
Table 1.
Data collection and refinement statistics.
| Data collection | ||
|---|---|---|
| Wavelength (Å) | 0.9793 | |
| Resolution range (Å) | 50.00 – 2.74 (2.79 – 2.74)a | |
| Space group | C 2 | |
| (α, β, γ) (°) | 90.000 120.727 90.000 | |
| Total reflections | 114,299 | |
| Unique reflections | 23,259 (1,186) | |
| Multiplicity | 4.9 (4.0) | |
| Completeness (%) | 100.00 (99.9) | |
| <I>/(I) | 17.86 (1.89) | |
| Rmergeb (%) | 11.1 (68.0) | |
| Wilson B-factor (Å2) | 53.95 | |
| Refinement | ||
| Resolution range (Å) | 48.65 – 2.74 (2.84 – 2.74) | |
| Rwork/Rfreec (%) | 21.41 (29.57)/226.16 (36.72) | |
| Number of non-hydrogen atomsd | 6208 | |
| protein | 5864 | |
| ligands | 265 | |
| waters | 79 | |
| RMS (bonds, Å) | 0.006 | |
| RMS (angles, °) | 1.006 | |
| Ramachandran favored (%) | 97.25 | |
| Ramachadran allowed (%) | 2.75 | |
| Ramachandran outliers (%) | 0.00 | |
| Average B-factor (Å2) | 51.13 | |
| macromolecules | 50.36 | |
| ligands | 69.90 | |
| waters | 45.25 | |
RMS, Root Mean Square deviation from ideal values (crystallography).
Statistics for the highest-resolution shell are shown in parentheses.
Rmerge = 100Σ(h)Σ(i)|I(i)-<I>|/Σ(h)Σ(i)I(i), where I(i) is the ith intensity measurement of reflection h, and <I> is the average intensity from multiple observations.
Rfactor = Σ||Fobs|-|Fcalc||/Σ|Fobs|. Where Fobs and Fcalc are the structure factor amplitudes from the data and the model, respectively. 8.6 % reflections were used to calculate Rfree values.
Per asymmetric unit.
Figure 1. Crystal Structure of Human AC in Covalent Complex with the Carmofur Fatty Acid Moiety.

(A) Ribbon diagram (α-subunit in magenta, β-subunit in cyan) of the covalent complex. The inhibitor fatty acid moiety is shown in yellow sticks. The side chains of N-glycosylated Asn173, Asn259, Asn286, and Asn342 are shown in cyan sticks and modeled sugar units are shown in orange sticks. One disulfide bond, Cys31-Cys340, is shown in yellow. The N- and C-termini are labeled. (B) Superposition of the covalent complex (cyan) and apo-form of AC (magenta) crystal structures. The two structures are superimposed by aligning all Cα atoms of β-subunits and are presented as cartoon diagrams.
The AC protein has 6 cysteine residues. Four of them form two disulfide bridges, Cys31-Cys340, Cys388-Cys392, whereas two other are not oxidized: Cys143 is the active site cysteine, and Cys292 is buried in the body of the protein molecule. There are 6 potential N-glycosylation sites in the molecule of human AC and only four of them, Asn173, Asn259, Asn286, and Asn342, could be modeled in the final electron density map. The sugar residues of N-glycosylation sites at Asn173 and Asn259 located on the surface loops of the β–subunit make extensive hydrogen bonds with both subunits.
The structure of the complex is very similar to that of the apo-form in its active conformation with an overall root mean square deviation (RMSD) of 0.41 Å for all Cα pairs (Figure 1B). The main conformational differences between the two structures are observed in the areas of the N-terminal part of helix (230–241) and the surface loop (319–322) of the β–subunit with the maximum distances between the corresponding Cα atoms of 3.1 Å. These results suggest that the covalent modification of the active site cysteine during the inhibition of the enzyme by the potent carmofur inhibitor does not induce major conformation changes in the body of both the α– and β–subunits.
Inhibitor binding
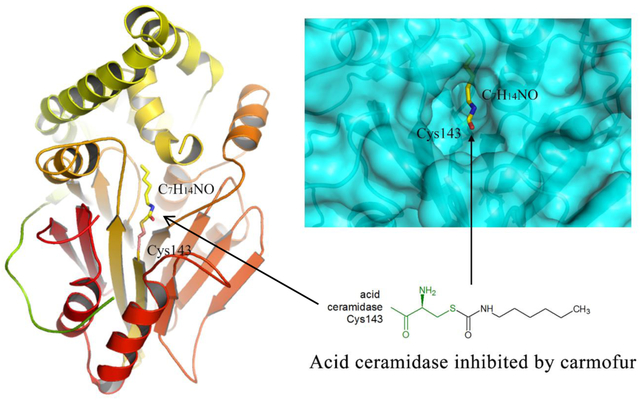
In a recent work, the small compound 2-oxo-N—(4-phenylbutyl)-1,3-benzoxazole-3-carboxamide (supplemental Figure S1), a close structural homologue of carmofur, was shown to be an effective noncompetitive inhibitor of the enzyme with an inhibition constant of 64 nM for the human AC27, 29. The authors proposed that the catalytic Cys143 attacks the electrophilic carbonyl group of the inhibitor, which results in the covalent modification of the cysteine residue by the C11H14NO moiety of the compound29. We revealed a similar mechanism of inhibition of human AC by carmofur, which is a close structural homologous of the above mentioned 2,4-dioxopyrimidine-1-carboxamide and a potent inhibitor of acid ceramidases with an IC50 value of 29 nM for the rat ortholog27. The fatty acid moiety of the carmofur inhibitor (C7H14NO, supplemental Figure S1) in the crystal structure is clearly defined by electron density in the enzyme substrate-binding site, with Cys143 positioned at the bottom of the pocket (Figure 2A). Continuous electron density links the inhibitor C7 atom to the Sγ of Cys143, indicating the presence of a covalent linkage between the inhibitor moiety and the enzyme. In addition, other electrostatic interactions include hydrogen bonds between the backbone carbonyl oxygen of Asp162, the backbone amide of Glu225, and the Nδ of Asn320 with the carbonyl and amide groups of the free fatty acid tail. An extended conformation of the fatty acid moiety of the inhibitor molecule in this complex is sandwiched between the side chains of Phe136, Phe163, Met161, Leu211, and Leu223 (Figure 2A). This binding mode is similar to the covalent inhibition of the related enzyme N-acylethanolamine acid amidase (NAAA) by the beta-lactam inhibitor ARN72632.
Figure 2. Binding interactions of the inhibitor fatty acid to AC active site.

(A) Close up view of the binding area in the complex (magenta and cyan ribbons for α– and β–subunits, respectively). The inhibitor moiety (yellow sticks) and the residues defining the interaction with the inhibitor (cyan sticks) are shown. The electron density map (grey) around the bound inhibitor is contoured at 1σ. Hydrogen bonds are indicated by grey dotted lines. (B) The part of the protein surface (in the same colors as in A) showing the deep channel into the active site with Cys143 covalently modified by the fatty acid (in yellow sticks) is presented.
There are several hydrophobic patches on the surface of the AC enzyme around its active site (Figure 3). Based on biochemical and mutagenesis experiments, these hydrophobic surfaces were shown to be very important for substrate hydrolysis and for binding to membrane lipids in order to assess its substrates30. For instance, the mutagenesis experiments showed substantial reduction of ceramide hydrolysis for the AC variants where hydrophobic patches containing Leu80/Val165/Leu167 and Phe328/Phe329/Leu330 residues were mutated (>80% reduction). Our structural data also confirm these mutagenesis results: the loop (161–165) is involved in the inhibitor’s fatty acid moiety binding and simple modeling of a ceramide substrate also suggests that Val165, Phe166, Trp176, Phe227, Leu367, and Trp396 reside on the hydrophobic surface underneath the modeled d17 substrate moiety (Figure 3A). It should be noted that plausible interchain hydrophobic interactions could result in dimer formation as was observed by Al et al33. This dimer was disrupted with addition of Triton-100. In our work, we observed some interchain hydrophobic contacts between symmetry related molecules in the crystal packing, however, there is no biological or biochemical evidence that dimer or multimer formation have potential relevance for AC functions.
Figure 3. Hydrophobic surface of AC with a modeled ceramide substrate.

(A) The surface representation of the hydrophobic areas of the AC in ribbon diagram are colored yellow and the remaining residues are colored in magenta (α-subunit) and in cyan (β-subunit). The locations of the residues mutated in our previous work (black) and that of the residues forming plausible hydrophobic contacts with the substrate d17 moiety (red) are labeled30. (B) Close-up view of the modeled C8 ceramide substrate (d17:1/8:0, ball and stick in orange) interacting with enzyme residues in the substrate-binding area. The positions of several AC catalytic residues, which can form hypothetical interactions with the substrate in the tetrahedral complex, are indicated.
Optimal superposition of the fatty acid tail of ceramide substrate onto that of the carmofur molecule sitting in the enzyme active site can provide a reliable structural model for the acyl-enzyme intermediate. Our simple modeling based on this superposition followed by introduction of a covalent bond between the Cys143 Cγ and C8 ceramide substrate shows that the N-terminal amino group of Cys143, the backbone amide of Glu225 and the Nδ atom of Asn320 are at hydrogen-bonding distance (3.3 Å, 2.8 Å and 3.0 Å, respectively) from the carbonyl oxygen of the substrate scissile bond (Figure 3B). This suggests that these groups form the oxyanion hole that stabilizes the tetrahedral transition state. These three residues constituting the oxyanion hole are highly conserved among all acid ceramidase orthologs30. There are also other close contacts stabilizing the transition state around the catalytic Cys143 residue between the backbone amide of Asp162 and the amide of the scissile substrate bond (2.9 Å), meanwhile the N-terminal amino group of Cys143 and the guanidine group of Arg333 make a hydrogen bonds with the O19 atom of the substrate (2.7 Å and 3.4 Å, respectively, Figure 3B). In our previous work, we proposed a plausible mechanism of substrate hydrolysis by AC where all of the above mentioned residues were shown to be involved30.
CONCLUSION
In conclusion, a high quality structure of human acid ceramidase in complex with a potent covalent inhibitor, an enzyme which likely contributes to different pathological diseases via its biological functions, is presented. The new structural features of the inhibitor binding, the hydrophobic surfaces around the substrate binding site could be a foundation for an efficient structure-based drug design platform targeting the AC activities. Currently, there is no AC inhibitor clinically available to treat patients. This study is the first to crystallographically demonstrate that carmofur directly binds to AC, and prompts its inhibition, providing the detailed mechanism, which has eluded the scientific community for more than 30 years, of its mysterious 5-FU independent anti-tumor activity.…There is a strong clinical interest in developing AC inhibitors to treat GBM, prostate cancer, and melanoma13, 20–22, 34–37. This provides the impetus to now engage in clinical trials with carmofur for treatment of various types of cancers targeting AC as a new class of anticancer drugs.
Experimental Section
Protein Expression and Purification
Recombinant full-length AC (molecular weight ~ 50 kDa) was expressed as a secreted protein in Sf9 insect cells (Invitrogen) infected with baculovirus as previously described30. AC protein sample was isolated from culture media using nickel-nitrilotriacetic acid (Ni-NTA) resin (Thermo Fisher Scientific) and further purified by size exclusion chromatography on a Superdex 200 column (GE Healthcare) in 15 mM Tris-HCl buffer, pH 7.5, supplemented with 100 mM NaCl30. Protein for crystallization first underwent proteolytic autocleavage at 37 °C for 60 h. 100 mM sodium acetate solution, pH 5.0, was added for autocleavage and the solution was subsequently exchanged back into the neutral buffer followed by protein concentration up to 9.7 mg/ml30. Protein purity >95% was confirmed by gel electrophoresis.
Complex formation, crystallization and structure determination
A small aliquot of 1 M sodium-phosphate citrate buffer, pH 4.3, was added to the protein sample to adjust the pH value to 4.5. The complex between the protein and the inhibitor was formed by adding 30 mM carmofur (Sigma-Aldrich) in 100 % DMSO and 5 % Triton X-100 in water to the protein solution at a final concentrations of 1mM each. Initial crystals of the AC-carmofur complex were obtained at 16 °C from drops containing 1.5 μL of the protein sample and 1.5 μL of reservoir solution (0.2 M sodium-phosphate-citrate, pH 4.3, 0.275 M lithium sulfate, and 18% polyethylene glycol 1000). Protein crystals of the complex suitable for x-ray analysis were obtained after several cycles of microseeding under similar crystallization conditions.
For data collection, crystals were harvested with 20 % (v/v) glycerol in the reservoir solution. Diffraction data were collected from a single flash-frozen crystal on the SBC-CAT BM beamline (APS, Argonne National Laboratory) at 100 K. Data were indexed and processed with HKL-300038. The crystals belonged to the monoclinic space group C2 and contained two molecules of the complex per asymmetric unit. Cell parameters are listed in Table 1.
The structure of the complex was solved by molecular replacement using the PHASER program from the CCP4 software suite, with the structure of the activated apo-AC (PDB code 5U7Z) as a search model39, 40. The final model of the complex was obtained by carrying out several cycles consisting of manual model building using COOT, followed by structure isotropic refinement with Phenix.Refine from the PHENIX software suite41. Coordinates have been deposited in the Protein Data Bank (PDB ID 6MHM). Final refinement statistics are listed in Table 1.
The tetrahedral enzyme-substrate intermediate was modeled by superposition of the C8 fatty chain of ceramide substrate (d17:1/8:0) and free fatty acid tail of carmofur (5-fluoro-N-hexyl-2,4-dioxo-pyrimidine-1-carboxamide, C11H16FN3O3, molecular weight: 257.265 g/mol) from the covalent complex followed by introducing a covalent bond between Cys143 Sγ atom and the carbonyl carbon of the C8 ceramide scissile bond. The d17 moiety of the substrate was manually placed in the second hydrophobic cavity of the protein to avoid any steric clashes.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the staff at the SBC-CAT BM beamline of the Advanced Proton Source, Argonne National Laboratory, IL, where data were collected.
Funding: Musella Foundation Grant, Department of Neurosurgery, Mitchell Cancer Institute, University of South Alabama, NIH R01CA190688. The Structural Biology Center 19BM beamline is supported by the U. S. Department of Energy, Office of Biological and Environmental Research, under contract DE-AC02-06CH11357
ABBREVIATIONS USED
- AC
acid ceramidase
- S1P
sphingosine-1-phosphate
- 5-FU
5-fluorouracil
Footnotes
Supporting Information
Chemical structures of carmofur and its mechanism of inhibition is available free of charge at the http://pubs.acs.org.
Accession Codes
Structures and reflection data of AC-carmofur complexes reported here have been deposited to the Protein Data Bank under the accession code: 6MHM. Authors will release the atomic coordinates and experimental data upon article publication.
Conflict of interest:
The authors declare no competing financial interest
REFERENCES
- 1.Wang WB; Yang Y; Zhao YP; Zhang TP; Liao Q; Shu H Recent studies of 5-fluorouracil resistance in pancreatic cancer. World J Gastroenterol 2014, 20, 15682–15690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sara JD; Kaur J; Khodadadi R; Rehman M; Lobo R; Chakrabarti S; Herrmann J; Lerman A; Grothey A 5-fluorouracil and cardiotoxicity: a review. Ther Adv Med Oncol 2018, 10, 1758835918780140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sakamoto J; Ohashi Y; Hamada C; Buyse M; Burzykowski T; Piedbois P; Meta-Analysis Group of the Japanese Society for Cancer of the, C.; Rectum; Meta-Analysis Group in, C. Efficacy of oral adjuvant therapy after resection of colorectal cancer: 5-year results from three randomized trials. J Clin Oncol 2004, 22, 484–492. [DOI] [PubMed] [Google Scholar]
- 4.Kusumoto T; Maehara Y; Sakaguchi Y; Sugimachi K Hyperthermia enhances the in vitro activity of 1-hexylcarbamoyl-5-fluorouracil compared to that of 5-fluorouracil. Eur J Cancer Clin Oncol 1989, 25, 477–481. [DOI] [PubMed] [Google Scholar]
- 5.Ooi A; Ohkubo T; Higashigawa M; Kawasaki H; Kakito H; Kagawa Y; Kojima M; Sakurai M Plasma, intestine and tumor levels of 5-fluorouracil in mice bearing L1210 ascites tumor following oral administration of 5-fluorouracil, UFT (mixed compound of tegafur and uracil), carmofur and 5’-deoxy-5-fluorouridine. Biol Pharm Bull 2001, 24, 1329–1331. [DOI] [PubMed] [Google Scholar]
- 6.Sato S; Ueyama T; Fukui H; Miyazaki K; Kuwano M [Anti-tumor effects of carmofur on human 5-FU resistant cells]. Gan to kagaku ryoho. Cancer & chemotherapy 1999, 26, 1613–1616. [PubMed] [Google Scholar]
- 7.Liu P; Ma S; Liu H; Han H; Wang S HCFU inhibits cervical cancer cells growth and metastasis by inactivating wnt/beta-catenin pathway. J Cell Biochem [Online early access] DOI: doi: 10.1002/jcb.26570. Published Online: December 12, 2017. [DOI] [PubMed] [Google Scholar]
- 8.Yokoyama C; Sueyoshi Y; Ema M; Mori Y; Takaishi K; Hisatomi H Induction of oxidative stress by anticancer drugs in the presence and absence of cells. Oncol Lett 2017, 14, 6066–6070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sakamoto J; Hamada C; Rahman M; Kodaira S; Ito K; Nakazato H; Ohashi Y; Yasutomi M An individual patient data meta-analysis of adjuvant therapy with carmofur in patients with curatively resected colon cancer. Jpn J Clin Oncol 2005, 35, 536–544. [DOI] [PubMed] [Google Scholar]
- 10.Grohn P; Heinonen E; Kumpulainen E; Lansimies H; Lantto A; Salmi R; Pyrhonen S; Numminen S Oral carmofur in advanced gastrointestinal cancer. Am J Clin Oncol 1990, 13, 477–479. [DOI] [PubMed] [Google Scholar]
- 11.Maehara Y; Anai H; Kusumoto H; Kusumoto T; Sugimachi K Colorectal carcinoma in vitro is more sensitive to 1-hexylcarbamoyl-5-fluorouracil compared with six other antitumor drugs: carboquone, adriamycin, mitomycin C, aclacinomycin A, cisplatin, 5-fluorouracil. Dis Colon Rectum 1988, 31, 62–67. [DOI] [PubMed] [Google Scholar]
- 12.He X; Okino N; Dhami R; Dagan A; Gatt S; Schulze H; Sandhoff K; Schuchman EH Purification and characterization of recombinant, human acid ceramidase: catalytic reactions and interactions with acid sphingomyelinase. The Journal of biological chemistry 2003, 278, 32978–32986. [DOI] [PubMed] [Google Scholar]
- 13.Nguyen HS; Awad AJ; Shabani S; Doan N Molecular targeting of acid ceramidase in glioblastoma: a review of its role, potential treatment, and challenges. Pharmaceutics 2018, 10, 45–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park JH; Schuchman EH Acid ceramidase and human disease. Biochimica et biophysica acta 2006, 1758, 2133–2138. [DOI] [PubMed] [Google Scholar]
- 15.Mao C; Obeid LM Ceramidases: regulators of cellular responses mediated by ceramide, sphingosine, and sphingosine-1-phosphate. Biochimica et biophysica acta 2008, 1781, 424–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanger N; Ruckhaberle E; Gyorffy B; Engels K; Heinrich T; Fehm T; Graf A; Holtrich U; Becker S; Karn T Acid ceramidase is associated with an improved prognosis in both DCIS and invasive breast cancer. Mol Oncol 2015, 9, 58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seelan RS; Qian C; Yokomizo A; Bostwick DG; Smith DI; Liu W Human acid ceramidase is overexpressed but not mutated in prostate cancer. Genes, chromosomes & cancer 2000, 29, 137–146. [DOI] [PubMed] [Google Scholar]
- 18.Klobucar M; Grbcic P; Pavelic SK; Jonjic N; Visentin S; Sedic M Acid ceramidase inhibition sensitizes human colon cancer cells to oxaliplatin through downregulation of transglutaminase 2 and beta1 integrin/FAK-mediated signalling. Biochemical and biophysical research communications 2018, 503, 843–848. [DOI] [PubMed] [Google Scholar]
- 19.Realini N; Palese F; Pizzirani D; Pontis S; Basit A; Bach A; Ganesan A; Piomelli D Acid ceramidase in melanoma: expression, localization, and effects of pharmacological inhibition. The Journal of biological chemistry 2016, 291, 2422–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doan NB; Alhajala H; Al-Gizawiy MM; Mueller WM; Rand SD; Connelly JM; Cochran EJ; Chitambar CR; Clark P; Kuo J; Schmainda KM; Mirza SP Acid ceramidase and its inhibitors: a de novo drug target and a new class of drugs for killing glioblastoma cancer stem cells with high efficiency. Oncotarget 2017, 8, 112662–112674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doan NB; Nguyen HS; Al-Gizawiy MM; Mueller WM; Sabbadini RA; Rand SD; Connelly JM; Chitambar CR; Schmainda KM; Mirza SP Acid ceramidase confers radioresistance to glioblastoma cells. Oncol Rep 2017, 38, 1932–1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lai M; Realini N; La Ferla M; Passalacqua I; Matteoli G; Ganesan A; Pistello M; Mazzanti CM; Piomelli D Complete acid ceramidase ablation prevents cancer-initiating cell formation in melanoma cells. Scientific reports 2017, 7, 7411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doan NB; Nguyen HS; Montoure A; Al-Gizawiy MM; Mueller WM; Kurpad S; Rand SD; Connelly JM; Chitambar CR; Schmainda KM; Mirza SP Acid ceramidase is a novel drug target for pediatric brain tumors. Oncotarget 2017, 8, 24753–24761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nguyen HS; Shabani S; Awad AJ; Kaushal M; Doan N Molecular markers of therapy-resistant glioblastoma and potential strategy to combat resistance. Int J Mol Sci 2018, 19, 1765–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alhajala HS; Nguyen HS; Shabani S; Best B; Kaushal M; Al-Gizawiy MM; Erin Ahn EY; Knipstein JA; Mirza S; Schmainda KM; Chitambar CR; Doan NB Irradiation of pediatric glioblastoma cells promotes radioresistance and enhances glioma malignancy via genome-wide transcriptome changes. Oncotarget 2018, 9, 34122–34131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doan NB; Nguyen HS; Alhajala HS; Jaber B; Al-Gizawiy MM; Ahn EE; Mueller WM; Chitambar CR; Mirza SP; Schmainda KM Identification of radiation responsive genes and transcriptome profiling via complete RNA sequencing in a stable radioresistant U87 glioblastoma model. Oncotarget 2018, 9, 23532–23542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pizzirani D; Pagliuca C; Realini N; Branduardi D; Bottegoni G; Mor M; Bertozzi F; Scarpelli R; Piomelli D; Bandiera T Discovery of a new class of highly potent inhibitors of acid ceramidase: synthesis and structure-activity relationship (SAR). Journal of medicinal chemistry 2013, 56, 3518–3530. [DOI] [PubMed] [Google Scholar]
- 28.Realini N; Solorzano C; Pagliuca C; Pizzirani D; Armirotti A; Luciani R; Costi MP; Bandiera T; Piomelli D Discovery of highly potent acid ceramidase inhibitors with in vitro tumor chemosensitizing activity. Scientific reports 2013, 3, 1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pizzirani D; Bach A; Realini N; Armirotti A; Mengatto L; Bauer I; Girotto S; Pagliuca C; De Vivo M; Summa M; Ribeiro A; Piomelli D Benzoxazolone carboxamides: potent and systemically active inhibitors of intracellular acid ceramidase. Angew Chem Int Ed Engl 2015, 54, 485–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gebai A; Gorelik A; Li Z; Illes K; Nagar B Structural basis for the activation of acid ceramidase. Nat Commun 2018, 9, 1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shtraizent N; Eliyahu E; Park JH; He X; Shalgi R; Schuchman EH Autoproteolytic cleavage and activation of human acid ceramidase. The Journal of biological chemistry 2008, 283, 11253–11259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gorelik A; Gebai A; Illes K; Piomelli D; Nagar B Molecular mechanism of activation of the immunoregulatory amidase NAAA. Proc Natl Acad Sci U S A 2018, 115, E10032–E10040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Al BJ; Tiffany CW; Gomes de Mesquita DS; Moser HW; Tager JM; Schram AW Properties of acid ceramidase from human spleen. Biochimica et biophysica acta 1989, 1004, 245–251. [DOI] [PubMed] [Google Scholar]
- 34.Cheng JC; Bai A; Beckham TH; Marrison ST; Yount CL; Young K; Lu P; Bartlett AM; Wu BX; Keane BJ; Armeson KE; Marshall DT; Keane TE; Smith MT; Jones EE; Drake RR Jr.; Bielawska A; Norris JS; Liu X Radiation-induced acid ceramidase confers prostate cancer resistance and tumor relapse. The Journal of clinical investigation 2013, 123, 4344–4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doan NB; Nguyen HS; Montoure A; Al-Gizawiy MM; Mueller WM; Kurpad S; Rand SD; Connelly JM; Chitambar CR; Schmainda KM; Mirza SP Acid ceramidase is a novel drug target for pediatric brain tumors. Oncotarget 2017, 8, 24753–24761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu X; Elojeimy S; Turner LS; Mahdy AE; Zeidan YH; Bielawska A; Bielawski J; Dong JY; El-Zawahry AM; Guo GW; Hannun YA; Holman DH; Rubinchik S; Szulc Z; Keane TE; Tavassoli M; Norris JS Acid ceramidase inhibition: a novel target for cancer therapy. Frontiers in bioscience: a journal and virtual library 2008, 13, 2293–2298. [DOI] [PubMed] [Google Scholar]
- 37.Mahdy AE; Cheng JC; Li J; Elojeimy S; Meacham WD; Turner LS; Bai A; Gault CR; McPherson AS; Garcia N; Beckham TH; Saad A; Bielawska A; Bielawski J; Hannun YA; Keane TE; Taha MI; Hammouda HM; Norris JS; Liu X Acid ceramidase upregulation in prostate cancer cells confers resistance to radiation: AC inhibition, a potential radiosensitizer. Molecular therapy: the journal of the American Society of Gene Therapy 2009, 17, 430–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Otwinowski Z; Minor W Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol 1997, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- 39.McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Phaser crystallographic software. J Appl Crystallogr 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Winn MD; Ballard CC; Cowtan KD; Dodson EJ; Emsley P; Evans PR; Keegan RM; Krissinel EB; Leslie AG; McCoy A; McNicholas SJ; Murshudov GN; Pannu NS; Potterton EA; Powell HR; Read RJ; Vagin A; Wilson KS Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 2011, 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Adams PD; Afonine PV; Bunkoczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH PHENIX: a comprehensive python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.