

Introduction:
Sepsis is a life-threatening organ dysfunction which results from a dysregulated host response to infection and it remains a common, deadly, and expensive problem. 1 Treatment advances ranging from measured resuscitation strategies to broad spectrum antibiotics have improved the morbidity and mortality associated with sepsis. Despite these advances, nearly 50,000 cases occur annually in the United States and sepsis remains the leading cause of death in the non-coronary ICUs, costing more than $24 billion as of 2014. 2,3 In response to septic insults, a profound immune activation occurs as the body attempts to corral the infectious source. The result of such extreme immune activation is a multifactorial disharmony of immune cells. This disruption of immune homeostasis results in circulating immune cell influx into distal organs, leading to multi-organ failure. 4 This necessary balance between activation and suppression is mediated by immune checkpoint regulators. These regulators are implicated in multiple instances of immune imbalance, including in sepsis.
The immune system utilizes checkpoint regulators to balance immune activity throughout the body. Checkpoint regulators are membrane bound proteins which serve as a second signal to direct the immune response to a particular antigen. 5 An antigen bound by MHC class I or II receptors on an antigen presenting cell (APC) is presented to a T-cell Receptor (TCR), acting as the first signal. The second signal, from a checkpoint regulator, is necessary to instruct the T-cell on how to respond to this antigen. Without such signals the immune response is attenuated or absent. These proteins further allow for modification of the immune response over time, enabling different immune cells to respond to various environmental cues in unique ways. 6,7 Stimulatory signals from regulators can lead to activation, and subsequent cell and humoral mediated immunity, while inhibitory signals can lead to anergic T-cells unable to respond to further signals.8
In the setting of overwhelming infection checkpoint regulators serve as key mediators of the immune dysfunction that portends a risk of secondary infection. These regulators promote tolerance and can inhibit further immune reactivity to an infectious stimulus, traits that can be detrimental in the setting of overly profound infection.7 It has been demonstrated that alteration in signaling through a variety of such regulators can alter outcomes after sepsis in animal models, and this has introduced new therapeutic targets that are beginning to be tested in clinical settings. 9 These regulatory systems are made up of a variety of proteins with non-redundant spatial and temporal functions, yet, their ultimate signaling outcomes demonstrate substantial redundancy. The purpose of such repetition is incompletely understood. With a more complete understanding of these immune policing proteins it is thought that their power might be harnessed to better treat patients enduring septic insults. 10 We will elaborate on the roles of diverse checkpoint regulators, such as Programmed Cell Death-1, V-domain Ig suppressor of T-cell activation, Cytotoxic T-Lymphocyte Associated Protein 4, and Herpes Virus Entry Mediator in immune regulation after sepsis and their contribution to post-septic immunosuppression.
Programmed Cell Death −1 (PD-1, Pdcd1)
PD-1 is an immune checkpoint protein first identified as a classical programmed cell death induced gene, Pdcd1. PD-1 bears a V-immunoglobulin domain and belongs to the B7-CD28 superfamily of checkpoint proteins. Both PD-1 and it’s ligands are expressed on a variety of immune cell types, and the PD-1 signaling pathway uniquely allows for maintenance of both central and peripheral tolerance by limiting the activation of T-cells. 11
PD-1 and its Ligands
Non-redundant signaling pathway
PD-1 signals as a receptor tyrosine kinase. Its cytoplasmic domain contains an immunoreceptor tyrosine-based switch motif (ITSM) domain and an immunoreceptor tyrosine-based inhibitory motif (ITIM) domain. 12 Following ligation to the PD-1 ligands, the ITSM and ITIM domains are phosphorylated and Src Homology region 2 domain-containing Phosphatases (SHPs) 1 and 2 dock at the ITSM domain. Upon activation by PD-1, SHP-2 dephosphorylates kinases, phosphoinositide 3-kinase (PI3K) and ZAP70 to inhibit the Akt and Erk/MAPK pathways as shown in Figure 1. 13,14,15 Both TCR antigen stimulation and PD-1 ligation are requisite for PD-1 mediated suppression. 16
Figure 1. Checkpoint regulators serve as a necessary second signal for immune responses:
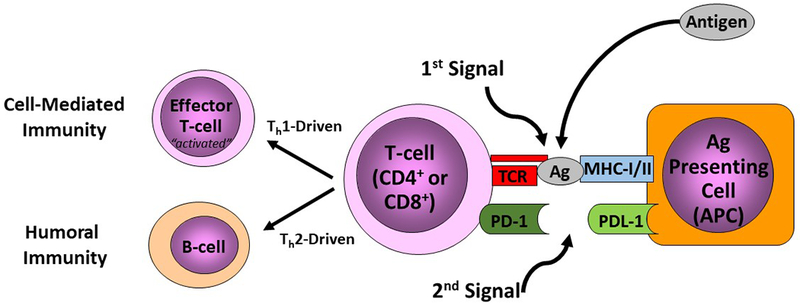
Checkpoint regulators are membrane bound proteins which serve as a second signal to direct the immune response to a particular antigen. When an antigen is present it is bound by MHC class I or II receptors on an antigen presenting cell (APC) and presented to a T-cell Receptor (TCR). Following this a second signal, from a checkpoint regulator, is necessary to instruct the T-cell on how to respond to this antigen, shown here as the PD-1/PDL-1 interaction. Stimulatory signals from regulators can lead to activation, and subsequent cell and humorally mediated immunity, while inhibitory signals can lead to anergic t-cells unable to respond to further signals.
Broad expression patterns promote tolerance
PD-1 is expressed on conventional T (Tconv) cells, regulatory T-cells (Tregs), Natural Killer T (NKT) cells, B-cells, dendritic cells (DCs), and macrophages. 17,18 Transcription factors such as NFAT2, STATs, Notch, and FoxO1 bind the Pdcd1 promoter to transiently upregulate PD-1 expression following T-cell receptor (TCR)-antigen stimulation. 19–21 The expression of PD-1 during antigen stimulation sets a threshold for reactivation of lymphocytes. 14 As lymphocyte stimulation subsides, PD-1 expression is downregulated by Blimp-1 and T-bet transcription factors to prevent overt T-cell exhaustion. 22,23
PD-L1 (B7-H1) and PD-L2 (B7-DC) are type 1 transmembrane proteins and CD28-B7 family members containing an extracellular region with IgV- and IgC- like domains. PD-L1 and PD-L2 compete for PD-1 binding with different affinities, and both bind additional ligand-specific partners B7-1 and repulsive guidance molecule b (RGMb), respectively. 24,25 The differential cell and tissue-specific expression patterns and competing molecular mechanisms of PD-L1 and PD-L2 promotes dynamic activity of PD-1 based on spatial and temporal context. 26
PD-L1 is broadly expressed on both lymphoid and non-lymphoid cells in peripheral tissues, mediating PD-1 activation at sites of infection. PD-L1 is constitutively expressed at high levels by naÏve CD4+T-cells, CD8+T-cells, B-cells, DCs, and macrophages. Upon activation of lymphoid cells PD-L1 surface expression is upregulated and induced on monocytes. 27 Non-lymphoid cells, including cardiac endothelium, lung, placenta, kidney, salivary gland, glial cells, muscle cells, epithelial cells, and liver non-parenchymal cells express PD-L1 constitutively as well. 28 These expression patterns ensure that activated T-cells are systemically regulated to maintain peripheral tolerance. 26 PD-L2 is more tightly regulated with weaker expression on a smaller subset of cells and high binding specificity to PD-1. PD-L2 expression is induced specifically on activated bone marrow-derived DCs and macrophages found in the liver, lung, and spleen. Despite the differences between these ligands, they do share some expression characteristics. Both PD-Ls are expressed on tumor cells, though PD-L1 expression extends to hematopoietic and non-hematopoietic tumors and PD-L2 is largely restricted to leukemias. In addition, PD-L1/2 expression on splenic DCs is upregulated by interferon-gamma (IFN-γ), granulocyte-macrophage colony-stimulating factor (GM-CSF), and interleukin-4 (IL-4). 27 PD-1 and its ligands mediate interactions between both immune and non-immune cell types to prevent autoreactivity in the periphery. However, the expansive influence of the PD-1 pathway has been exploited to curtail immune surveillance in cancer and chronic viral infections. This pathway has also demonstrated detrimental activity in other diseases such as sepsis and acute lung injury.
The Role of PD-1 in Immunopathology
Sepsis
As aforementioned, sepsis is characterized by systemic immune dysfunction. As with other immune-related diseases such as cancer and chronic viral infection, T-cell anergy is implicated in sepsis development. The large impact that lymphocyte regulation and activity have in immune dysfunction prompted Huang et al. to investigate the role of PD-1 in sepsis progression. This group found that adult PD-1−/− mice have a survival advantage following intra-abdominal septic insult via the cecal ligation and puncture (CLP) procedure. The PD-1 deficient mice maintained macrophage function, demonstrating improved bacterial clearance and reduced inflammatory cytokine production29. The survival results were recapitulated in a neonatal murine sepsis model using the neonatal cecal slurry technique. PD-1 deficiency promoted neonatal survival compared to Wild Type (WT) controls, but did not alter bactericidal efficacy. 30
Monaghan et al. expanded on the adult murine findings by analyzing the PD-1 expression patterns and cytokine profile of patients with septic shock. In septic patients, PD-1 was significantly upregulated upon circulating monocytes, granulocytes, and lymphocytes when compared to healthy controls. This upregulation positively correlated to IFN-γ, IL-4, and IL-2 levels which are associated with the T helper cell 1/2 (TH1/TH2) response and cytokine storm. The upregulation of PD-1 surface expression and cytokine production also correlated with the severity of illness as determined by the Acute Physiology And Chronic Health Evaluation II (APACHE II) score. 31,32 Thus, the increase in disease severity (APACHE II > 20) associated with PD-1 overexpression demonstrates the potential of PD-1 blockade as a therapeutic intervention for sepsis. 31
Acute Lung Injury and Respiratory Distress Syndrome
Acute lung injury (ALI) can develop following an infectious insult ranging from sepsis, trauma, shock or pneumonia, and can progress into the more severe acute respiratory distress syndrome (ARDS). ALI/ARDS development is largely induced by neutrophils, lung epithelial, and lung endothelial cells. As PD-1 and its ligands exert suppressive function in the periphery, it was postulated that this pathway may play a role in ALI/ARDS 33. In a murine model of indirect acute lung injury (iALI), PD-1 was upregulated on several immune populations, such as T-cells (CD4+), tissue resident DCs (CD11c+), and Gr1+ cells in the lung. This result was also mirrored in ARDS patients with a significant increase in PD-1 expressing CD3+ T-cells in the blood compared to healthy controls. In ARDS patients, the level of TNF-α found in the bronchoalveolar lavage fluid and immune cell apoptosis was significantly higher. The loss of lung barrier function due to apoptosis of epithelial and endothelial cells is another phenotype of ALI/ARDS patients, implicating TNF-α further in the development of this disease. In PD-1−/− mice, pathological indices of ARDS such as TNF-α levels, tissue congestion, neutrophil influx in the lungs, and immune cell apoptosis were significantly lower than WT mice. PD-1 deficient mice also demonstrated a survival advantage when compared to WT mice (70% v 31.25%). These murine and human results further support a role for PD-1 in the development of ALI/ARDS. 33
Viral control
Evading immune surveillance is a phenomenon that is typically associated with cancer. Antitumoral immunity can be dampened by exploiting negative checkpoint pathways, allowing tumors to expand and thrive in the host. The PD-1/ PD-L1 pathway has been targeted pharmacologically as an anticancer treatment with great efficacy, resulting in the development of multiple Food and Drug Administration (FDA) approved PD-1 inhibitors. 34 This method of immune evasion is not limited to cancer. Human Rhinovirus (HRV) is among the most common infections and has evolved a mechanism to escape the immune response through the PD-1/PD-L1 pathway. Kirchberger et al. demonstrated that HRV induces PD-L1 expression on DCs, blunting the ability to stimulate T-cells and promoting T-cell tolerance. In line with the known PD-1/PD-L1 function, T-cells were also reduced to a hypo-proliferative state. 35 These results support a clear role for the PD-1/PD-L1 pathway in sepsis, ARDS, and chronic infections.
V-domain Ig Suppressor of T-cell Activation (VISTA, Vsir, PD-1H)
VISTA is a negative checkpoint regulator that belongs to the B7-CD28 family and contains an IgV domain, a transmembrane domain, a cytoplasmic tail, and an extracellular domain homologous to PD-L1. Despite belonging to the B7-CD28 family, VISTA has several unique characteristics that are not shared with other family members.36 For instance, the cytoplasmic tail lacks both an ITIM and ITSM domain but contains proline residues, additional cysteines, and potential protein kinase c binding sites. It is theorized, based on these characteristics, that VISTA signals through a nonredundant pathway that remains undefined.37 Another unique characteristic of VISTA is its receptor and ligand-like structure, expression, and function.38 Acting as a receptor and ligand, VISTA inhibits T-cell proliferation, production of cytokines such as IL-2, and IFN-γ, and chemokine production following TCR activation.39 Through this inhibition, VISTA promotes peripheral tolerance.36
VISTA as a receptor and a ligand
VISTA-VISTA interaction promotes context dependent regulation
VISTA is almost exclusively expressed on hematopoietic lineages such as neutrophils, monocytes, DCs, and macrophages at high levels. VISTA is also constitutively expressed at lower levels on naÏve CD4+ T-cells, CD8+ T-cells, Tregs, and tumor infiltrating lymphocytes. 40 When expressed on Tconv cells, such as CD4+ T-cells, VISTA functions as a receptor and inhibits antigen-specific proliferation, cytokine, and chemokine production through cell intrinsic regulation 38. VISTA can also act as a ligand when expressed on APCs and Tregs. The VISTA ligand exerts cell extrinsic regulation of Tconv cells by interacting with the uncharacterized VISTA receptor on T-cells to suppress activation.41
The proposed VISTA-VISTA interaction between Tconv cells and Tregs could also promote the differentiation and suppressive function of Tregs.39 Interestingly, VISTA also appears to have a stimulatory role when expressed as a receptor on myeloid cells by enhancing inflammatory cytokine production and antigen presentation.37 Thus, the VISTA receptor inhibits Tconv cells whereas the VISTA ligand stimulates APC and Treg function. The ability to function as a ligand and a receptor depending on cellular context demonstrates the multi-faceted, dynamic regulatory role of VISTA.
VISTA-VSIG-3 interaction represents a novel checkpoint pathway
In addition to VISTA-VISTA interactions, a new VISTA ligand has been identified. V-set and Immunoglobulin domain containing 3 (VSIG-3, IGSF11, BT-IgSF) is a ligand in the immunoglobulin family and functionally interacts with VISTA expressed on T-cells.36 VSIG-3 is expressed in the brain, kidney, skeletal muscles, and germinal centers. It is also overexpressed in hepatic, colorectal, and gastric cancers.42 VSIG-3 is a homophilic adhesion molecule and shares structural homology with B7 family members. Association between the VSIG-3 and VISTA ectodomains suppress the T-cell response, cytokine, and chemokine production. 36 In parallel with other checkpoint proteins mentioned thus far, VISTA activity can also exacerbate immune related pathologies.
The Role of VISTA in Immunopathology
Sepsis
Though VISTA has mostly been discussed in the context of cancer and autoimmune disease, some recent work by Bharaj et al. demonstrated that monocytes upregulate VISTA following CLP in humanized BTL mice. BLT mice have severe immune-deficient non-obese diabetic phenotypes and produce inflammatory monocytes. Following CLP in BLT mice VISTA expression was significantly upregulated on monocytes43. These results support a role for VISTA in activating monocytes and contributing to the inflammatory response during sepsis progression.
Chronic Viral Infection (HIV)
As previously discussed, PD-1 plays an important role in chronic viral and bacterial infections. VISTA is no exception. In vitro treatment with Toll-like receptor (TLR) 3 and TLR5 correlated with significant VISTA upregulation in circulating CD14+ monocytes and macrophages. TLR3 and TLR5 are virus and bacteria-associated agonists respectively. Based on these preliminary data, Bharaj et al also investigated VISTA in the context of HIV-infected patients. This research group found significant upregulation of VISTA on monocytes associated with enhanced cytokine secretion in these individuals.37 It appears that the VISTA ligand plays a stimulatory role when expressed on monocytes during chronic viral infection. The differential role of VISTA as a ligand and receptor based on cell-type and disease context, further highlight the complex regulatory function of this checkpoint protein.
Cytotoxic T lymphocyte Ag-4 (CTLA-4)
CTLA-4 is a negative checkpoint regulator that was first identified as a homolog to costimulatory protein CD28 and belongs to the Ig superfamily. 44 CD28 and CTLA-4 are founding members of the CD28-B7 family and serve as the paradigm for immune checkpoint pathways. Both regulators bind B-cell activation antigens (B7-1/CD80 and B7-2/CD86) and are expressed on the surface of T-cells. B7-1 and B7-2 expression is restricted to lymphoid tissues. B7-1 expression is induced in T-cells, B-cells, monocytes, and DCs. B7-2 is constitutively expressed on B-cells, DCs, and monocytes. Following activation B7-2 is also upregulated on B-cells, DCs, and monocytes and induced on T-cells. 45 Despite structural similarity and shared ligands, CTLA-4 is a co-inhibitory protein whereas CD28 is co-stimulatory. CTLA-4 has higher affinity and avidity to B7s so it competes against CD28 to suppress activated T-cells. 46
Complex molecular mechanisms of CTLA-4-mediated suppression
CTLA-4 and CD28 target the same signaling pathway
Both CTLA-4 and CD28 associate with serine/threonine phosphatase PP2A to regulate T-cell activation. However, CTLA-4 is able to regulate T-cells through cell intrinsic and extrinsic inhibition as seen in Figure 2. When the TCR is stimulated, CTLA-4 recruits PP2A 47, inhibiting the Akt tyrosine kinase cascade responsible for potentiating T-cell activation. 48,47 This T-cell intrinsic mechanism targets Akt pathway kinases that are distal to the membrane whereas PD-1 targets this pathway proximal to the TCR (Fig 1 & 2a). In addition to AKT pathway inhibition, CTLA-4 competes for B7 binding to inhibit CD28 stimulatory activity and further suppress T-cell activation. 46
Figure 2. PD-1 exerts intrinsic suppression of T cell activity proximal to the TCR:
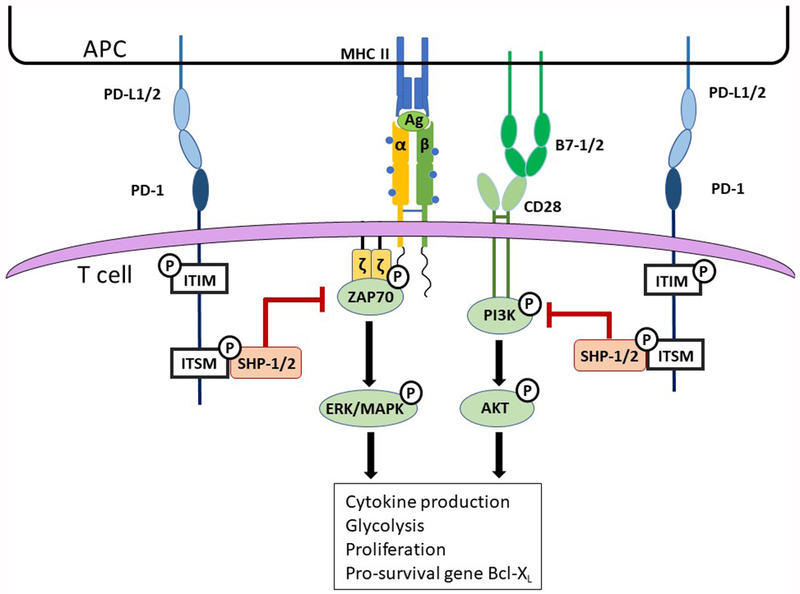
Ligation of PD-1 with its ligands results in recruitment of SHPs to the phosphorylated ITSM domain where they become functionally active. ZAP70 associates with the CD3-zeta chain and PI3K associates with the CD28 cytoplasmic tail upon TCR stimulation and CD28 ligation. ZAP70 is an adaptor protein that recruits and stabilizes a kinase complex to initiate the ERK/MAPK pathway. PI3K serves as the initial kinase in the AKT pathway. Active SHP-1/2 dephosphorylate ZAP70 and PI3K; thus, inhibiting these kinase pathways and preventing T cell activity, proper metabolic activity, T cell survival, and proliferation.
CTLA-4 also suppresses CD4+ and CD8+ T-cell activity through cell extrinsic mechanisms. Interaction of CTLA-4 with B7-1/ B7-2 promotes trans endocytosis to sequester these ligands from antigen presenting cells (APCs), preventing CD28 binding, thereby, inhibiting T-cell activation. Constitutively expressed CTLA-4 also enhances the suppressive function of Tregs to further inhibit Tconv activation. 49 Phosphorylated CTLA-4 also enhances the production of signaling molecules by T-cells. This downregulates B7 expression in the APC and reduces the ability of APCs to stimulate T-cells (Fig 2b). 50 The multiple molecular mechanisms by which CTLA-4 suppresses effector T-cells demonstrates the importance of this regulatory pathway in maintaining immune tolerance.
CTLA-4 expression is not restricted to lymphoid cells
Unlike CD28 which is constitutively expressed on T-cells, CTLA-4 expression is induced in naÏve T-cells upon TCR stimulation and constitutively expressed on Tregs. CTLA-4 is also expressed on B-cells, Natural Killer (NK) cells, NKT cells, and DCs. 51,52 This restricted expression can be attributed to strict regulation through multiple mechanisms including transcriptional control, post transcriptional modifications, and intracellular trafficking of CTLA-4. NFAT and Foxp3 are transcription factors that induce CTLA-4 transcription in Tconv cells and Tregs, respectively.53,54 MicroRNA activity and the post-transcriptional modifications of the CTLA-4 3′ UTR regulate CTLA-4 mRNA stability. These transcriptional and post-transcriptional mechanisms provide temporal control of CTLA-4 surface expression. 55,56
Intracellular trafficking regulates CTLA-4 surface expression via endocytic and secretory pathways, polarizing CTLA-4 surface expression to the immune synapse, and maintaining spatial control of CTLA-4 activity. 57,58 In CD4+ T-cells, CTLA-4 is localized within secretory lysosomes. Within these lysosomes, CTLA-4 is rapidly degraded when it is not being actively transcribed. When the TCR is stimulated, CTLA-4 gene expression is induced, intracellular CTLA-4 accumulates, and lysosomes secrete CTLA-4 to the cell surface.57 In CD8+ T-cells, intracellular trafficking of CTLA-4 is mediated through endocytic pathways. 58 In activated T-cells, the LRBA protein promotes migration of CTLA-4 containing endosomes to the plasma membrane.59 Once expressed proximal to the TCR, CTLA-4 effectively suppresses T-cell function. 47 To limit its activity and overall abundance in the absence of TCR stimulation, CTLA-4 is negatively regulated by clathrin-associated adaptor proteins AP-1 and AP-2. In resting T-cells, AP-1 and AP-2 interact with the unphosphorylated cytoplasmic tail of CTLA-4 to promote internalization of this receptor into the lysosomal network for degradation.59 This phosphorylation dependent trafficking allows for rapid and dynamic regulation of CTLA-4 at the cell surface.
CTLA-4 In Immunopathology:
CTLA-4 has been implicated in multiple roles in human disease processes varying from autoimmune phenomena to oncologic pathogenesis to infectious immunosuppression. Similar to other checkpoint regulators it often mediates overly robust tolerance of abnormal signals in the setting of profound disease, preventing necessary immune responsiveness to threat.
Sepsis
CTLA-4 has been implicated as a key mediator in septic immunosuppression. Early work by Inoue et al. demonstrated that CTLA-4 upregulation on CD4, CD8 and Tregs increased after experimental sepsis using a CLP model.60 Administration of intraperitoneal anti-CTLA-4 antibody generated a dose dependent survival benefit, with low doses producing profoundly improved survival associated decreased septic induced splenic apoptosis. Interestingly, high dose CTLA-4 was less protective inducing increased mortality.60
CTLA-4 has additionally demonstrated a significant role in the pathophysiology of both primary and secondary Candida albicans fungal sepsis 61. Survival was improved in mice treated with anti-CTLA-4 antibody both after primary fungal sepsis, induced through tail vein injection, or secondary fungal sepsis, modeled by tail vein injection 72 hours after CLP. This benefit was associated with an increase in splenocyte derived IFN-γ production suggesting that in vivo CTLA-4 expression inhibits this necessary protective IFN-γ phenotype.
CTLA-4’s role in septic immunosuppression extends to viral pathogens, with significant roles in the pathogenesis of two contemporary and highly morbid viruses, Human Immunodeficiency Virus (HIV) and Hepatitis C. While current anti-retroviral therapy has dramatically improved long-term outcomes in HIV by suppressing viral replication, latent stores of HIV have proven a major barrier to ultimate disease cure. CTLA-4 CD4+ T-cells have been identified as a major reservoir of latent viral particles, and therefore a target for future therapeutics.62 Further, specific genetic variants of CTLA-4 have been associated with chronic Hepatitis C infection, suggesting that some variants may predispose individuals to risk of chronic conversion upon viral exposure. 63
CTLA-4 in Critical Illness
Patients who are critically ill from non-septic sources are known to be similarly at risk of secondary infections. For example, patients with acute liver failure frequently overexpress CTLA-4 on circulating T-cells compared to healthy controls. 64 Further, T-cells isolated from patients with acute liver failure were hypo proliferative when challenged with CD3 or antigen stimulation. Serum from these patients possessed increased amounts of soluble B7 which was shown to induce CTLA-4 upregulation in healthy control T-cells.64 This implicates CTLA-4 as a prime mediator of decreased innate immune responses to infectious challenges in critical illness.
Herpes Virus Entry Mediator (HVEM)
Herpes Virus Entry Mediator (HVEM) is a type I transmembrane receptor member of the tumor necrosis factor receptor superfamily (TNFRSF) first discovered by Montgomery et al. as the necessary cell surface receptor for Herpes Simplex Virus-1 (HSV-1) entry. 65,66 It was separately identified in an expressed sequence tag survey seeking additional members of the TNFRSF. 67 The TNFRSF consists of 10 cell-surface proteins that regulate immune development and homeostasis. HVEM contains 4 cysteine rich domains (CRDs) in its extracellular region, a characteristic feature of TNFRSF utilized for ligand engagement. The cytoplasmic region of HVEM associates with TRAF1, TRAF2, TRAF3, TRAF5, and Stat3,and when transfected, cells demonstrate significant activation of NF-κB, Jun N-terminal kinase, and AP-1 67,68
Broad expression enables tissue specific behaviors:
Unlike many other checkpoint regulators whose expression is confined to immune cells, HVEM is expressed diffusely on multiple tissue types as well as many immune cell subsets. Northern blot survey of human tissue types demonstrated HVEM expression in most tissues, with the highest levels in adult spleen, peripheral blood leukocytes, fetal lung and kidney. 67 Immune cell characterization demonstrated monomeric HVEM expression on T-cells, B-cells, NK cells, DCs, and myeloid cells. 7 Specifically, human naÏve and memory B-cells express high levels of HVEM while germinal center B-cells lack HVEM expression, and T-cells constitutively express HVEM, unique from other TNFRs. 7
HVEM and its Ligands:
HVEM behaves as both a receptor and a ligand, with variable downstream effects:
HVEM has a total of 5 described ligands including members both within, and outside of, the TNFSF, a unique trait not possessed by other TNFRSF members.7 Immunoglobulin family ligands include B and T lymphocyte attenuator (BTLA), and CD160, while TNF superfamily ligands include Lymphotoxin Alpha (LTα), and LIGHT.69 Additionally, HVEM binds to Herpes Virus glycoprotein-D (HSV gD).65 The net function of HVEM, illustrated by HVEM−/− murine modeling, is inhibitory, with HVEM deficient T-cells exhibiting enhanced concanavalin (ConA) stimulation.70 HVEM deficient mice similarly demonstrate increased mortality in T-cell dependent autoimmune hepatitis models with increased T-cell proliferation and cytokine production. 70
HVEM signaling is variable, dependent on the ligand interacted with, the orientation of HVEM within the membrane, and the surrounding environment.69 HVEM binding TNFSF members generally results in immune stimulation via an NFκB dependent mechanism, while binding of immunoglobulin family members results in immune inhibition. 7,71 Interestingly, in addition to its role as a receptor, HVEM behaves as a ligand for BTLA and CD160, inducing inhibitory signaling within the immunoglobulin expressing cells. 72
LIGHT and Lymphotoxin Alpha (LTα):
LIGHT, which stands for “homologous to lymphotoxin, exhibits inducible expression and competes with HSV glycoprotein D for binding to herpesvirus entry mediator, a receptor expressed on T lymphocytes,” is also known as TNFSF14 or CD258. It is a 29kD, type II transmembrane protein, and member of the TNF family which formulates a homotrimer and exhibits highest expression in spleen, immature DC, granulocytes, and activated T-cells. 7,66,73 LIGHT acts as a ligand for both Lymphotoxin-β receptor (LTβR) and HVEM, which it binds in a 3:3 complex of trimeric LIGHT attached to 3 HVEM CRD2/3 regions.7,73 When binding LTβR, LIGHT activates a cell death pathway within T-lymphocytes resulting in chemokine production, TRAF2 degradation, and caspase 8 activation.71 However, when binding HVEM, LIGHT stimulates a robust stimulatory signal which, after TRAF2 recruitment results in NFκB activation, and promoting cell survival. 71
LTα is a compact trimer which is assembled from subunits expressed by B-cells, T-cells and NK cells. 7,66 Lacking a transmembrane domain, LTα is secreted in its homotrimeric form and binds to HVEM in a stimulatory manner similar to LIGHT resulting in NFκB recruitment and cell survival. 66 Like LIGHT, LTα binds to the CRD2/3 of the inner surface of HVEM forming a 3:3 complex on the membrane. 7
B and T Lymphocyte Attenuator (BTLA) and CD160:
A member of the immunoglobulin superfamily, BTLA possesses an intermediate type immunoglobulin fold in its ectodomain and two cytosolic ITIM inhibitory signaling domains.72,74 BTLA expression is highest in spleen, lymph nodes, activated T-cells, and resting B-cells. 7 BTLA engagement by HVEM results ITIM activation, inducing SHP-1/2 phosphatase recruitment, and subsequent attenuation of IL-2 within the BTLA expressing cell. 72,74 Within the HVEM expressing cell, BTLA engagement mirrors the stimulatory LIGHT and LTa pathway, resulting in NFκB activation. 69 This is supported by evidence that BTLA binding to HVEM promotes survival and memory generation in CD8+ T-cells.72 Given that the net function of HVEM signaling is inhibitory, the ultimate signal generated from BTLA and CD160 ligation remains unclear. Evidence points to inhibition, yet no mechanism connecting the known stimulatory activation of NFκB to an inhibitory end result has been discovered.7 It is this behavior, with inhibition when acting as a ligand, but stimulation while acting as receptor, that leads to the description of HVEM as a bidirectional switch.
CD160 was the last of HVEM’s ligands to be discovered, identified through an attempt to isolate NK cell-specific receptors. 7 It is a member of the immunoglobulin superfamily of receptors and contains a glycosylphosphatidylinositol (GPI)-anchor and single IgV-like domain.75 It is expressed highly in spleen, small intestine, as well as peripheral blood leukocytes, NK cells, and γδT-cells. 7 BTLA and CD160 both associate with the CRD1 domain of HVEM, demonstrating competitive binding. However, mutagenesis studies have demonstrated that the two possess overlapping but not identical binding domains within this CRD1 region. 76
Ligand binding can occur simultaneously:
HVEM’s binding complexity is increased by the discovery that its orientation within the membrane alters its binding site availability, affecting the manner in which it interacts with available ligands.77 Like other members of the TNF family, HVEM orients into a trimer within the membrane meaning it forms 3:3 complexes with many of its ligands.7 With its multiple CRDs located on alternate faces of the trimer complex this allows HVEM to bind both at its CRD1 and CRD2/3 regions simultaneously, forming complexes with 3 LIGHT or LTα molecules bound to one face and 3 BTLA or CD160 to the other when HVEM is expressed in its trans confirmation.7 In addition to these complexes, HVEM and BTLA can be co-expressed on single cells, forming a stable complex, with both proteins expressed in the cis confirmation on the membrane.77 This is seen almost exclusively in naÏve T-cells, where HVEM appears to associate with LIGHT, but no down-stream signaling, neither stimulatory nor inhibitory, is noted within the cells. 77 It is felt that this cis-complex competitively inhibits HVEM activation with the surrounding environmental ligands, maintaining T-cell naiveite.
HVEM’s Role in Septic Immunosuppression:
HVEM’s role in septic immunosuppression is best characterized by its behavior at mucosal barriers, but it has also been implicated in more systemic roles in indirect lung injury, viral illness and non-septic systemic critical illnesses, such as liver failure78–80. Unlike other checkpoint regulators, HVEM has been implicated both in septic immunosuppressive roles, mediating inappropriate levels of tolerance, and as mechanism of excessive immune activation, responsible for tissue injury. This represents a physical manifestation of its unique bidirectional behavior, making it an interesting therapeutic target allowing more context specificity.
HVEM is essential to mucosal immunity:
The mucosal surfaces serve as a primary entry site for many infectious threats, and the immune presence within these tissues is extensive. An investigation of innate lymphoid cell (ILC) checkpoint regulation demonstrated that HVEM signaling within the ILC3 subset was both necessary and sufficient to generate an appropriate IFN-γ response to protect against Yersinia enterocolitica infection.81 Improved survival, via IFN-γ production, was mediated through the HVEM-LIGHT axis, with no affect from BTLA or CD160.
Shui et al. echoed HVEM’s invaluable role in mucosal barrier signaling utilizing intestinal Citrobacter rodentium and Streptococcus pneumoniae pulmonary infection models. 68 HVEM stimulation, by either BTLA or CD160, in colonic epithelial cells induced STAT3 phosphorylation and innate inflammatory responses such as IL-6, CXCL1, and CCL20 production. HVEM−/− mice survived significantly worse than WT when subjected to Citrobacter rodentium infection, a surrogate for enteropathogenic Escherichia coli infection. These mice also had higher bacterial burden and lower STAT3 activation. They established this effect was mediated exclusively through CD160 interaction using BTLA−/−, LIGHT−/−and CD160 antibody administration.68 Their results were confirmed in mice subjected to a Streptococcus pneumoniae pulmonary infection model where again the HVEM-CD160 axis was essential to survival and bacterial clearance. In both examples the HVEM axis defends mucosal barriers against infection, with blockade of deletion resulting in decreased mucosal barrier defense, such as is common after sepsis.
HVEM in respiratory immunity:
The role of HVEM in immune dysfunction in respiratory tissues has been similarly well established. In a murine model of Chlamydia psittaci respiratory infection LIGHT−/−mice demonstrated a profound survival deficit with increased weight loss, higher bacterial burden, and heightened severity of lung tissue injury. 82 Lung tissue from these mice demonstrated decreased IFN-γ, TNF-α, and IL-12 mRNA levels, with elevated Treg abundance. Mice subjected to iALI using a double hit model of hemorrhage followed by CLP upregulate HVEM expression in lung tissue. 78 Administration of intratracheal HVEM siRNA attenuated this increased HVEM expression and conveyed a transient early survival benefit. 78 Similar to LIGHT−/− mice, HVEM siRNA treated mice had reduced cytokine and chemokine levels in respiratory mucosa, suggesting that HVEM signaling was necessary for this local inflammatory response, an example of inappropriate immune activation following sepsis. Together this demonstrates that the HVEM pathway, while necessary for response to respiratory infectious threats, can be inappropriately activated by distant infectious challenges resulting in inappropriate tissue injury.
HVEM and Herpes Simplex Virus-1:
In addition to allowing cell entry for Herpes Simplex Virus-1 (HSV-1), HVEM mediates HSV-1’s ability to chronically infect individuals by influencing the Treg population. 80 After HSV infection there is marked expansion of regulatory T-cell populations, HVEM is upregulated on CD4+FoxP3+ Tregs after HSV infection. HVEM−/− are more susceptible to HSV ocular disease and these mice had reduced T-cell expansion compared to wild type mice80. This suggests that HVEM regulation of Treg expansion initially aids in control of the infection and direct tissue injury but, may ultimately enable a definitive reservoir for chronic HSV infection.
HVEM expression in critically ill patients:
Expression of HVEM and its major ligand BTLA was explored in critically ill surgical patients by Shubin et al. demonstrating BTLA up regulation on CD4+ T-cells, monocytes and granulocytes and similar HVEM upregulation on granulocytes and monocytes in septic patients.83,84 Non-septic critically ill patients with >80% BTLA expression on CD4+ T-cells were at increased risk of developing secondary infection.84 Also, BTLA−/− mice demonstrate a survival benefit over WT controls with improved bacterial clearance after CLP. 83 Finally, critically ill patients with Hepatitis B-induced acute on chronic liver failure were shown to co-express HVEM, BTLA and fibrinogen-like protein (a virus induced molecule) on liver macrophages, implying absence of HVEM signaling by HVEM-BTLA complexing may play a role in Hepatitis pathogenesis and acute reactivation.79
Conclusion
Checkpoint regulators are crucial in producing an appropriate and controlled immune response to insults. Their expansive roles in immune modulation described above highlights their essential function. However, their powerful role is often mistakenly used to reduce immune reactions when true non-self-threats exist or to activate immune responses in the absence of infectious pathogens resulting in tissue injury. The immense role of checkpoint regulators in immune dysfunction, especially during sepsis progression, makes them an attractive target for therapeutic interventions.
Multiple clinical trials have been undertaken to investigate blockade of various checkpoint regulators during sepsis. Unfortunately, all reported clinical trials have demonstrated lackluster results thus far. The most recent trial of immunotherapy in sepsis, utilizing an anti-PD-L1 antibody in septic patients, demonstrated no change in mortality or cytokine levels. A modest increase in monocyte human leukocyte antigen-DR expression was obtained with anti-PD-L1, but only at higher doses. 85 Earlier immunotherapy trials demonstrated similarly disappointing results, as treatment with granulocyte-macrophage colony-stimulating factor (GM-CSF) provided no survival benefit and only a modest reduction in ventilatory days.86 However, treatment with IFN-γ correlates with decreased TNFα response to lipopolysaccharide stimulation and is FDA approved for treatment of fungal sepsis in patients with chronic granulomatous disease. Despite this small success no broadly applicable immunomodulatory agent is approved for use in sepsis treatment at this time.10
These underwhelming results may be due in part to the reliance on animal modeling to study these complex molecular mechanisms with limited confirmation from human sampling. Further, the lack of specific septic patient criteria make it difficult to select ideal patient cohorts for treatment as has successfully been done in cancer clinical trials. Finally, failures in sepsis clinical trials may stem from targeting single regulators in isolation, ignoring how molecules endogenously act in concert. The family of checkpoint regulators is extensive, diverse, and important. However, many of these regulators have overlapping roles without an obvious indication for such redundancy. Thus, a more thorough understanding of the behavior and hierarchy of checkpoint regulators in sepsis may afford a more effective combinatorial therapeutic approach.
Figure 3. CTLA-4 suppresses T cells through both T cell intrinsic and extrinsic mechanisms:
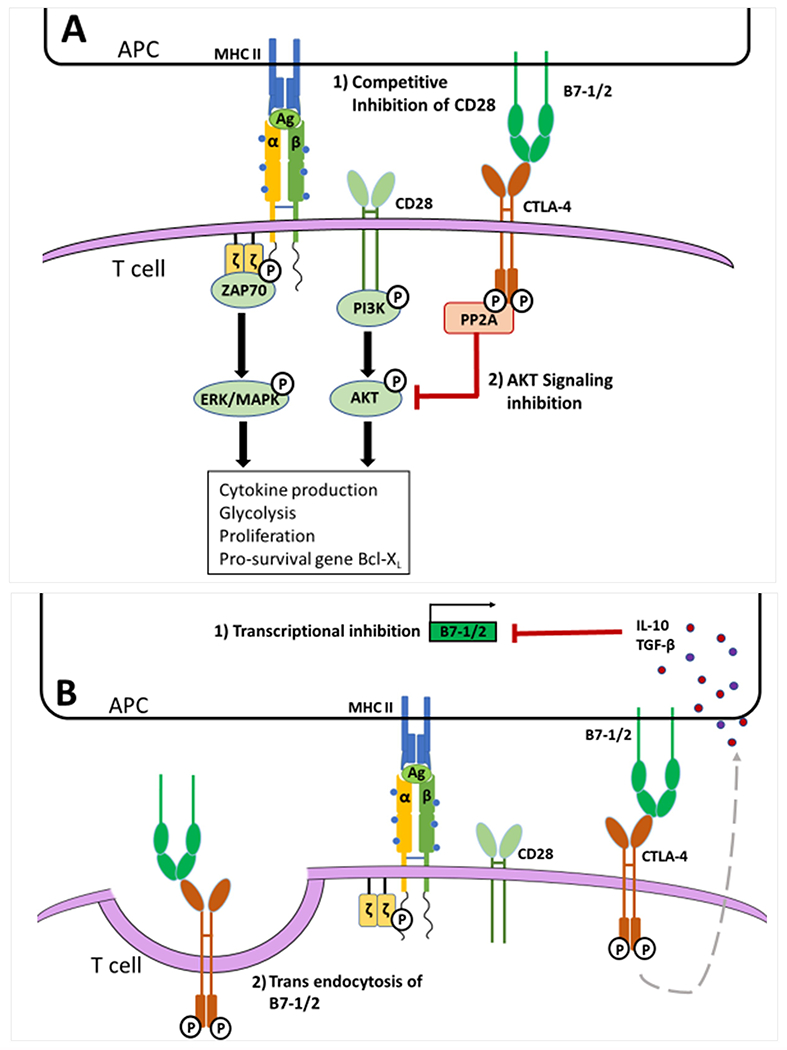
A. Upon TCR activation CTLA-4 is expressed on the T cell surface and exerts cell intrinsic inhibition. CTLA-4 outcompetes CD28 for the B7-1/2 ligands resulting in reduced CD28 stimulatory activity. CTLA-4 also recruits the phosphatase PP2A to its phosphorylated cytoplasmic tail. Active PP2A dephosphorylates AKT preventing downstream signaling; thus, inhibiting T cell activity, proliferation, and survival. B. Following CTLA-4 phosphorylation and ligation, a signal cascade is initiated by which IL-10, TGF-β, and soluble-CTLA-4 (not shown here) are upregulated. These signaling molecules are endocytosed by the APC and inhibit the transcription of B7-1 and B7-2, reducing the amount of B7-1/2 surface expression by APCs and preventing CD28 stimulation. When CTLA-4 binds to B7-1/2 it can also trigger trans endocytosis of these ligands. This reduces the surface expression of B7-1/2 on the APC and further hinders the ability of CD28 to be stimulatory.
Figure 4. HVEM behaves as bidirectional switch based on environmental signals:
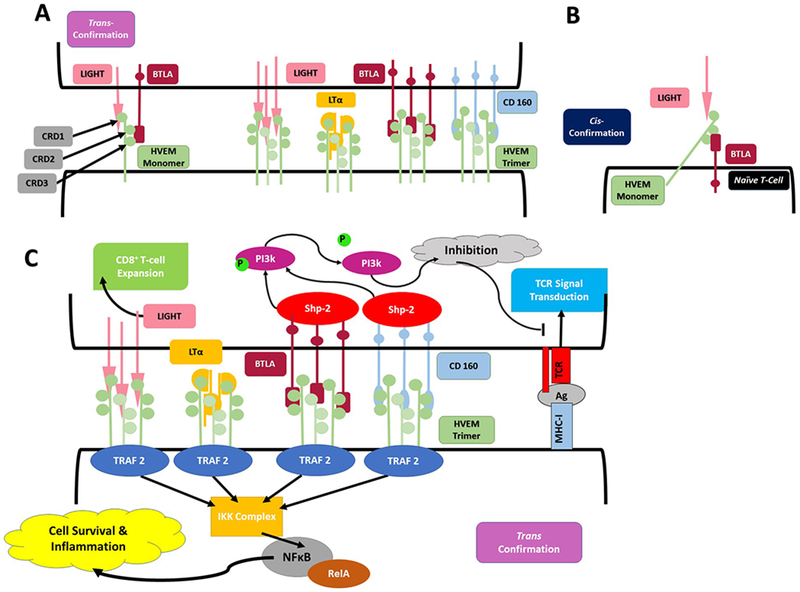
A. HVEM associates with LIGHT, LTα, BTLA, and CD160 in trimeric confirmations when expressed in trans confirmation. LIGHT and LTα associated with CRD1 binding domain, while BTLA and CD160 associate with CRD2/3. All ligands associate with HVEM in a trimeric confirmation, generating a 3:3:3 complex of 3 HVEM molecules with 3 BTLA or CD160 molecules and 3 LIGHT or LTα molecules. B. HVEM interacts with coexpressed BTLA to from an inert complex when both are expressed in their cis confirmation, most commonly on naÏve T-cells. In this formation HVEM can still associate with soluble LIGHT or LTα at its exposed CRD1 binding domain, yet no downstream signal is generated from the interaction. C. When HVEM is expressed in the trans confirmation on the membrane, ligation of LIGHT, LTα, BTLA or CD160 results in TRAF2 recruitment within the HVEM expressing cell. TRAF2 activates an IKK complex which in turn activates the RelA form of NFκB to promote cell survival. Within BTLA and CD160 expressing cells, ligation of HVEM results ITIM phosphorylation, recruiting SHP1 and SHP2, ultimately resulting in inhibitory signaling interrupting TCR signal transduction via dephosphorylation of PI3k -PKB pathway. Despite apparent absence of intracellular signaling motifs, LIGHT ligation of HVEM stimulates CD8+ T-cell expansion.
Key Points:
Checkpoint regulators are a diverse group of membrane bound proteins, with varied expression and notable redundancy, which dictate immune cell response to antigen presentation.
Septic Immunosuppression predisposing patients to secondary infection after a primary infectious insult is mediated, at least in part, by checkpoint regulators.
Checkpoint regulators have been manipulated in animal models improving outcomes after septic insult, but, have not been successfully harnessed as therapeutic targets in humans.
Synopsis:
Checkpoint regulators are a varied group of membrane bound receptors or ligands expressed on a variety of immune cells to regulate the immune cell response to antigen presentation and other immune stimuli such as cytokines, chemokines, and complement. In the context of profound immune activation such as sepsis, the immune system can be rendered anergic by these receptors to prevent excessive inflammation and tissue damage. However, if this septic immunosuppression is prolonged, the host is unable to mount the appropriate immune response to a secondary insult or infection. Here we describe the manner in which major regulators belonging to the B7-CD28 family, PD-1, VISTA, CTLA-4, HVEM, and their various ligands, mediate immunosuppression in sepsis.
Acknowledgments
Funding Sources:
This work was supported by the National Institutes of Health [grant numbers R35 GM118097 (AA), R25 GM083270 (CG), P20GM103652 (SFM), K08-GM110495 (DSH)] and Armand D. Versaci Research Scholar in Surgical Sciences Fellowship (MEW).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure Statement:
The authors report no proprietary or commercial interest in any product mentioned or concept discussed in this article.
References:
- 1.Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). Jama. 2016;315(8):801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murphy SL, Xu J, Kochanek KD. Deaths: Preliminary Data for 2010. National Vital Statistics Reports. 2012;60:1–52. [PubMed] [Google Scholar]
- 3.Rhee C, Murphy MV, Li LL, Platt R, Klompas M, Epictr CDCP. Comparison of Trends in Sepsis Incidence and Coding Using Administrative Claims Versus Objective Clinical Data. Clinical Infectious Diseases. 2015;60(1):88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boomer JS, To K, Chang KC, et al. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA. 2011;306(23):2594–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bretscher P, Cohn M. A theory of self-nonself discrimination. Science (New York, NY). 1970;169(3950):1042–1049. [DOI] [PubMed] [Google Scholar]
- 6.Lafferty KJ, Cunningham AJ. A new analysis of allogeneic interactions. The Australian journal of experimental biology and medical science. 1975;53(1):27–42. [DOI] [PubMed] [Google Scholar]
- 7.Cai G, Freeman GJ. The CD160, BTLA, LIGHT/HVEM pathway: a bidirectional switch regulating T-cell activation. Immunological reviews. 2009;229(1):244–258. [DOI] [PubMed] [Google Scholar]
- 8.Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol. 2003;77(8):4911–4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Biron BM, Ayala A, Lomas-Neira JL. Biomarkers for Sepsis: What Is and What Might Be? Biomarker Insights. 2015;10:7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hotchkiss RS, Monneret G, Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. The Lancet Infectious diseases. 2013;13(3):260–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Okazaki T, Honjo T. The PD-1-PD-L pathway in immunological tolerance. Trends Immunol. 2006;27(4):195–201. [DOI] [PubMed] [Google Scholar]
- 12.Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992;11(11):3887–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.So L, Fruman DA. PI3K signalling in B- and T-lymphocytes: new developments and therapeutic advances. Biochem J. 2012;442(3):465–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parry RV, Chemnitz JM, Frauwirth KA, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. 2005;25(21):9543–9553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol. 2004;173(2):945–954. [DOI] [PubMed] [Google Scholar]
- 17.Chang WS, Kim JY, Kim YJ, et al. Cutting edge: Programmed death-1/programmed death ligand 1 interaction regulates the induction and maintenance of invariant NKT cell anergy. J Immunol. 2008;181(10):6707–6710. [DOI] [PubMed] [Google Scholar]
- 18.Liu Y, Yu Y, Yang S, et al. Regulation of arginase I activity and expression by both PD-1 and CTLA-4 on the myeloid-derived suppressor cells. Cancer Immunol Immunother. 2009;58(5):687–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Austin JW, Lu P, Majumder P, Ahmed R, Boss JM. STAT3, STAT4, NFATc1, and CTCF regulate PD-1 through multiple novel regulatory regions in murine T cells. J Immunol. 2014;192(10):4876–4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cho HY, Lee SW, Seo SK, Choi IW, Choi I, Lee SW. Interferon-sensitive response element (ISRE) is mainly responsible for IFN-alpha-induced upregulation of programmed death-1 (PD-1) in macrophages. Biochim Biophys Acta. 2008;1779(12):811–819. [DOI] [PubMed] [Google Scholar]
- 21.Mathieu M, Cotta-Grand N, Daudelin JF, Thebault P, Labrecque N. Notch signaling regulates PD-1 expression during CD8(+) T-cell activation. Immunol Cell Biol. 2013;91(1):82–88. [DOI] [PubMed] [Google Scholar]
- 22.Lu P, Youngblood BA, Austin JW, et al. Blimp-1 represses CD8 T cell expression of PD-1 using a feed-forward transcriptional circuit during acute viral infection. J Exp Med. 2014;211(3):515–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kao C, Oestreich KJ, Paley MA, et al. Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8+ T cell responses during chronic infection. Nat Immunol. 2011;12(7):663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27(1):111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao Y, Yu S, Zhu B, et al. RGMb is a novel binding partner for PD-L2 and its engagement with PD-L2 promotes respiratory tolerance. J Exp Med. 2014;211(5):943–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghiotto M, Gauthier L, Serriari N, et al. PD-L1 and PD-L2 differ in their molecular mechanisms of interaction with PD-1. Int Immunol. 2010;22(8):651–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamazaki T, Akiba H, Iwai H, et al. Expression of programmed death 1 ligands by murine T cells and APC. J Immunol. 2002;169(10):5538–5545. [DOI] [PubMed] [Google Scholar]
- 28.Iwai Y, Terawaki S, Ikegawa M, Okazaki T, Honjo T. PD-1 inhibits antiviral immunity at the effector phase in the liver. J Exp Med. 2003;198(1):39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang X, Venet F, Wang YL, et al. PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc Natl Acad Sci U S A. 2009;106(15):6303–6308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Young WA, Fallon EA, Heffernan DS, Efron PA, Cioffi WG, Ayala A. Improved survival after induction of sepsis by cecal slurry in PD-1 knockout murine neonates. Surgery. 2017;161(5):1387–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Monaghan SF, Thakkar RK, Tran ML, et al. Programmed death 1 expression as a marker for immune and physiological dysfunction in the critically ill surgical patient. Shock. 2012;38(2):117–122. [DOI] [PubMed] [Google Scholar]
- 32.Knaus WA, Draper EA, Wagner DP, Zimmerman JE. APACHE II: a severity of disease classification system. Crit Care Med. 1985;13(10):818–829. [PubMed] [Google Scholar]
- 33.Monaghan SF, Thakkar RK, Heffernan DS, et al. Mechanisms of indirect acute lung injury: a novel role for the coinhibitory receptor, programmed death-1. Ann Surg. 2012;255(1):158–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gong J, Chehrazi-Raffle A, Reddi S, Salgia R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J Immunother Cancer. 2018;6(1):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kirchberger S, Majdic O, Steinberger P, et al. Human rhinoviruses inhibit the accessory function of dendritic cells by inducing sialoadhesin and B7-H1 expression. J Immunol. 2005;175(2):1145–1152. [DOI] [PubMed] [Google Scholar]
- 36.Wang J, Wu G, Manick B, et al. VSIG-3 as a ligand of VISTA inhibits human T-cell function. Immunology. 2019;156(1):74–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bharaj P, Chahar HS, Alozie OK, et al. Characterization of programmed death-1 homologue-1 (PD-1H) expression and function in normal and HIV infected individuals. PLoS One. 2014;9(10):e109103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang L, Rubinstein R, Lines JL, et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J Exp Med. 2011;208(3):577–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Le Mercier I, Chen W, Lines JL, et al. VISTA Regulates the Development of Protective Antitumor Immunity. Cancer Res. 2014;74(7):1933–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lines JL, Pantazi E, Mak J, et al. VISTA is an immune checkpoint molecule for human T cells. Cancer Res. 2014;74(7):1924–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Flies DB, Han X, Higuchi T, et al. Coinhibitory receptor PD-1H preferentially suppresses CD4(+) T cell-mediated immunity. J Clin Invest. 2014;124(5):1966–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Watanabe T, Suda T, Tsunoda T, et al. Identification of immunoglobulin superfamily 11 (IGSF11) as a novel target for cancer immunotherapy of gastrointestinal and hepatocellular carcinomas. Cancer Sci. 2005;96(8):498–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bharaj P, Ye C, Petersen S, et al. Gene array analysis of PD-1H overexpressing monocytes reveals a pro-inflammatory profile. Heliyon. 2018;4(2):e00545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Williams AF, Barclay AN. The immunoglobulin superfamily--domains for cell surface recognition. Annu Rev Immunol. 1988;6:381–405. [DOI] [PubMed] [Google Scholar]
- 45.Collins M, Ling V, Carreno BM. The B7 family of immune-regulatory ligands. Genome Biol. 2005;6(6):223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med. 1991;174(3):561–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chuang E, Fisher TS, Morgan RW, et al. The CD28 and CTLA-4 receptors associate with the serine/threonine phosphatase PP2A. Immunity. 2000;13(3):313–322. [DOI] [PubMed] [Google Scholar]
- 48.Kane LP, Weiss A. The PI-3 kinase/Akt pathway and T cell activation: pleiotropic pathways downstream of PIP3. Immunol Rev. 2003;192:7–20. [DOI] [PubMed] [Google Scholar]
- 49.Wang XB, Kakoulidou M, Giscombe R, et al. Abnormal expression of CTLA-4 by T cells from patients with myasthenia gravis: effect of an AT-rich gene sequence. Journal of neuroimmunology. 2002;130(1-2):224–232. [DOI] [PubMed] [Google Scholar]
- 50.Qureshi OS, Zheng Y, Nakamura K, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. 2011;332(6029):600–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kaufman KA, Bowen JA, Tsai AF, Bluestone JA, Hunt JS, Ober C. The CTLA-4 gene is expressed in placental fibroblasts. Mol Hum Reprod. 1999;5(1):84–87. [DOI] [PubMed] [Google Scholar]
- 52.Wang XB, Giscombe R, Yan Z, Heiden T, Xu D, Lefvert AK. Expression of CTLA-4 by human monocytes. Scand J Immunol. 2002;55(1):53–60. [DOI] [PubMed] [Google Scholar]
- 53.Gibson HM, Hedgcock CJ, Aufiero BM, et al. Induction of the CTLA-4 gene in human lymphocytes is dependent on NFAT binding the proximal promoter. J Immunol. 2007;179(6):3831–3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zheng Y, Josefowicz SZ, Kas A, Chu TT, Gavin MA, Rudensky AY. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature. 2007;445(7130):936–940. [DOI] [PubMed] [Google Scholar]
- 55.de Jong VM, Zaldumbide A, van der Slik AR, Persengiev SP, Roep BO, Koeleman BP. Post-transcriptional control of candidate risk genes for type 1 diabetes by rare genetic variants. Genes Immun. 2013;14(1):58–61. [DOI] [PubMed] [Google Scholar]
- 56.Sonkoly E, Janson P, Majuri ML, et al. MiR-155 is overexpressed in patients with atopic dermatitis and modulates T-cell proliferative responses by targeting cytotoxic T lymphocyte-associated antigen 4. J Allergy Clin Immunol. 2010;126(3):581–589 e581–520. [DOI] [PubMed] [Google Scholar]
- 57.Iida T, Ohno H, Nakaseko C, et al. Regulation of cell surface expression of CTLA-4 by secretion of CTLA-4-containing lysosomes upon activation of CD4+ T cells. J Immunol. 2000;165(9):5062–5068. [DOI] [PubMed] [Google Scholar]
- 58.Linsley PS, Bradshaw J, Greene J, Peach R, Bennett KL, Mittler RS. Intracellular trafficking of CTLA-4 and focal localization towards sites of TCR engagement. Immunity. 1996;4(6):535–543. [DOI] [PubMed] [Google Scholar]
- 59.Lo B, Zhang K, Lu W, et al. AUTOIMMUNE DISEASE. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science. 2015;349(6246):436–440. [DOI] [PubMed] [Google Scholar]
- 60.Inoue S, Bo L, Bian J, Unsinger J, Chang K, Hotchkiss RS. Dose-dependent effect of anti-CTLA-4 on survival in sepsis. Shock (Augusta, Ga). 2011;36(1):38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chang KC, Burnham CA, Compton SM, et al. Blockade of the negative co-stimulatory molecules PD-1 and CTLA-4 improves survival in primary and secondary fungal sepsis. Critical care (London, England). 2013;17(3):R85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McGary CS, Deleage C, Harper J, et al. CTLA-4(+)PD-1(−) Memory CD4(+) T Cells Critically Contribute to Viral Persistence in Antiretroviral Therapy-Suppressed, SIV-Infected Rhesus Macaques. Immunity. 2017;47(4):776–788. e775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sepahi S, Pasdar A, Gerayli S, Rostami S, Gholoobi A, Meshkat Z. CTLA-4 Gene Haplotypes and the Risk of Chronic Hepatitis C Infection; a Case Control Study. Reports of biochemistry & molecular biology. 2017;6(1):51–58. [PMC free article] [PubMed] [Google Scholar]
- 64.Khamri W, Abeles RD, Hou TZ, et al. Increased Expression of Cytotoxic T-Lymphocyte-Associated Protein 4 by T Cells, Induced by B7 in Sera, Reduces Adaptive Immunity in Patients With Acute Liver Failure. Gastroenterology. 2017;153(1):263–276. e268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Montgomery RI, Warner MS, Lum BJ, Spear PG. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell. 1996;87(3):427–436. [DOI] [PubMed] [Google Scholar]
- 66.Mauri DN, Ebner R, Montgomery RI, et al. LIGHT, a new member of the TNF superfamily, and lymphotoxin alpha are ligands for herpesvirus entry mediator. Immunity. 1998;8(1):21–30. [DOI] [PubMed] [Google Scholar]
- 67.Marsters SA, Ayres TM, Skubatch M, Gray CL, Rothe M, Ashkenazi A. Herpesvirus entry mediator, a member of the tumor necrosis factor receptor (TNFR) family, interacts with members of the TNFR-associated factor family and activates the transcription factors NF-kappaB and AP-1. The Journal of biological chemistry. 1997;272(22):14029–14032. [DOI] [PubMed] [Google Scholar]
- 68.Shui JW, Larange A, Kim G, et al. HVEM signalling at mucosal barriers provides host defence against pathogenic bacteria. Nature. 2012;488(7410):222–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cheung TC, Steinberg MW, Oborne LM, et al. Unconventional ligand activation of herpesvirus entry mediator signals cell survival. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(15):6244–6249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang Y, Subudhi SK, Anders RA, et al. The role of herpesvirus entry mediator as a negative regulator of T cell-mediated responses. The Journal of clinical investigation. 2005;115(3):711–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bechill J, Muller WJ. Herpesvirus entry mediator (HVEM) attenuates signals mediated by the lymphotoxin beta receptor (LTbetaR) in human cells stimulated by the shared ligand LIGHT. Molecular immunology. 2014;62(1):96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Steinberg MW, Huang Y, Wang-Zhu Y, Ware CF, Cheroutre H, Kronenberg M. BTLA interaction with HVEM expressed on CD8(+) T cells promotes survival and memory generation in response to a bacterial infection. PloS one. 2013;8(10):e77992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rooney IA, Butrovich KD, Glass AA, et al. The lymphotoxin-beta receptor is necessary and sufficient for LIGHT-mediated apoptosis of tumor cells. The Journal of biological chemistry. 2000;275(19):14307–14315. [DOI] [PubMed] [Google Scholar]
- 74.Watanabe N, Gavrieli M, Sedy JR, et al. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nature immunology. 2003;4(7):670–679. [DOI] [PubMed] [Google Scholar]
- 75.Giustiniani J, Bensussan A, Marie-Cardine A. Identification and characterization of a transmembrane isoform of CD160 (CD160-TM), a unique activating receptor selectively expressed upon human NK cell activation. Journal of immunology (Baltimore, Md: 1950). 2009;182(1):63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kojima R, Kajikawa M, Shiroishi M, Kuroki K, Maenaka K. Molecular basis for herpesvirus entry mediator recognition by the human immune inhibitory receptor CD160 and its relationship to the cosignaling molecules BTLA and LIGHT. Journal of molecular biology. 2011;413(4):762–772. [DOI] [PubMed] [Google Scholar]
- 77.Cheung TC, Oborne LM, Steinberg MW, et al. T cell intrinsic heterodimeric complexes between HVEM and BTLA determine receptivity to the surrounding microenvironment. Journal of immunology (Baltimore, Md: 1950). 2009;183(11):7286–7296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cheng T, Bai J, Chung CS, Chen Y, Fallon EA, Ayala A. Herpes Virus Entry Mediator (HVEM) Expression Promotes Inflammation/ Organ Injury in Response to Experimental Indirect-Acute Lung Injury. Shock (Augusta, Ga). 2019;51(4):487–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xu H, Cao D, Guo G, Ruan Z, Wu Y, Chen Y. The intrahepatic expression and distribution of BTLA and its ligand HVEM in patients with HBV-related acute-on-chronic liver failure. Diagn Pathol. 2012;7:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sharma S, Rajasagi NK, Veiga-Parga T, Rouse BT. Herpes virus entry mediator (HVEM) modulates proliferation and activation of regulatory T cells following HSV-1 infection. Microbes Infect. 2014;16(8):648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Seo GY, Shui JW, Takahashi D, et al. LIGHT-HVEM Signaling in Innate Lymphoid Cell Subsets Protects Against Enteric Bacterial Infection. Cell host & microbe. 2018;24(2):249–260. e244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cai H, Chen S, Xu S, et al. Deficiency of LIGHT signaling pathway exacerbates Chlamydia psittaci respiratory tract infection in mice. Microbial pathogenesis. 2016;100:250–256. [DOI] [PubMed] [Google Scholar]
- 83.Shubin NJ, Chung CS, Heffernan DS, Irwin LR, Monaghan SF, Ayala A. BTLA expression contributes to septic morbidity and mortality by inducing innate inflammatory cell dysfunction. J Leukoc Biol. 2012;92(3):593–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shubin NJ, Monaghan SF, Heffernan DS, Chung CS, Ayala A. B and T lymphocyte attenuator expression on CD4+ T-cells associates with sepsis and subsequent infections in ICU patients. Critical care (London, England). 2013;17(6):R276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hotchkiss RS, Colston E, Yende S, et al. Immune Checkpoint Inhibition in Sepsis: A Phase 1b Randomized, Placebo-Controlled, Single Ascending Dose Study of Antiprogrammed Cell Death-Ligand 1 (BMS-936559). Critical care medicine. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Meisel C, Schefold JC, Pschowski R, et al. Granulocyte-macrophage colony-stimulating factor to reverse sepsis-associated immunosuppression: a double-blind, randomized, placebo-controlled multicenter trial. Am J Respir Crit Care Med. 2009;180(7):640–648. [DOI] [PubMed] [Google Scholar]