

SUMMARY
Mammalian erythropoiesis yields a highly specialized cell type, the mature erythrocyte, evolved to meet the organismal needs of increased oxygen-carrying capacity. To better understand the regulation of erythropoiesis, we performed genome-wide studies of chromatin accessibility, DNA methylation, and transcriptomics using a recently developed strategy to obtain highly purified populations of primary human erythroid cells. The integration of gene expression, DNA methylation, and chromatin state dynamics reveals that stage-specific gene regulation during erythropoiesis is a stepwise and hierarchical process involving many cis-regulatory elements. Erythroid-specific, nonpromoter sites of chromatin accessibility are linked to erythroid cell phenotypic variation and inherited disease. Comparative analyses of stage-specific chromatin accessibility indicate that there is limited early chromatin priming of erythroid genes during hematopoiesis. The epigenome of terminally differentiating erythroid cells defines a distinct subset of highly specialized cells that are vastly dissimilar from other hematopoietic and nonhematopoietic cell types. These epigenomic and transcriptome data are powerful tools to study human erythropoiesis.
Graphical Abstract
In Brief
Schulz et al. use genome-wide studies of chromatin accessibility, DNA methylation, and transcriptomes in primary human erythroid cells to reveal important characteristics of erythropoiesis. Chromatin accessibility of terminal erythroid differentiation is markedly dissimilar from other hematopoietic cell types. Epigenomic changes are linked to erythroid cell traits and disease genes.
INTRODUCTION
Erythropoiesis occurs in a series of stages, with early developmental stages marked by the commitment of multilineage progenitor cells into erythroid progenitors with subsequent cellular proliferation and differentiation, followed by terminal erythroid differentiation, enucleation, and reticulocyte maturation into mature erythrocytes (Dzierzak and Philipsen, 2013). Mature mammalian erythrocytes are unique among the vertebrates, lacking nuclei and cellular organelles. An enucleate erythrocyte is a trait attributed to the organismal need to develop strategies to increase oxygen-carrying capacity (Scott, 2015; Scott and Milsom, 2006; Lenfant and Johansen, 1972) to adapt to increased oxygen demands (Glomski and Tamburlin, 1990; Glomski et al., 1992,1997; Nikinmaa, 1997). Thus, mature erythrocytes cannot undergo cell division or perform RNA synthesis, and they have a limited ability for self-repair.
Throughout the process of erythropoiesis, erythroid cells exhibit patterns of gene expression that direct a precise ensemble of proteins required for cellular structure and function. During terminal erythroid differentiation, erythroblasts decrease in size, nuclear chromatin condenses, cells produce large amounts of hemoglobin, membranes undergo reorganization, cell volume and surface area decrease, and enucleation occurs (Hattangadi et al., 2011; Wong et al., 2011). To meet the requirements for these precise cellular functions, hundreds of genes change their patterns of expression between each stage of terminal erythroid differentiation (An et al., 2014). In nonerythroid cells, epigenetic regulation has been shown to establish patterns of gene expression for developmental and stage-specific cell types via the differential utilization of gene regulatory elements (Song et al., 2011; Heintzman et al., 2009; West et al., 2014).
Identification and characterization of the epigenomic landscape and its contribution to the control of programs of gene expression in highly specialized human erythroid cells will allow a better understanding of erythropoiesis by revealing the temporal dynamics of regulatory element usage (Park, 2009; Farnham, 2009; Barski and Zhao, 2009; Kadauke and Blobel, 2009). In addition, linking regions with appropriate chromatin accessibility and/or DNA methylation to relevant SNPs will provide mechanistic insights into erythroid cell phenotypic variation in normal individuals (Visel et al., 2009; Pilon et al., 2011; Tallack et al., 2010; Xu et al., 2012; Wilson et al., 2009; Yu et al., 2009; Fujiwara et al., 2009; Kadauke et al., 2012; Chlon et al., 2012; Doré et al., 2012; Palii et al., 2011; Kassouf et al., 2010; Cheng et al., 2009; Su et al., 2013) and into pathways dysregulated in inherited and acquired diseases of the erythrocyte (Bauer et al., 2012, 2013; Sankaran et al., 2011) by capturing the regulatory landscapes central to human health and disease (Nord et al., 2013).
In this report, we define and correlate genome-wide chromatin accessibility, DNA methylation, and genomic organization with transcriptome analyses in highly purified populations of primary human erythroid cells at distinct stages of erythroid development and differentiation. These epigenomic and expression data are integrated with cis-genetic variants that are associated with phenotypic variation in erythroid cell traits and with acquired and inherited diseases of the erythrocyte.
Widespread changes in stage-specific patterns of chromatin accessibility and DNA methylation occurred, marking dynamic regulatory elements associated with patterns of gene expression influencing erythroid cell development and differentiation. Stage-specific patterns of chromatin accessibility were linked to genetic loci associated with erythroid cell traits and inherited erythroid diseases. Comparative analyses revealed a new conceptual framework for understanding terminal erythroid differentiation. Indistinguishable from other cells via morphologic characteristics, the epigenome of differentiating human erythroid cells defines a unique subset of highly specialized cells that are vastly dissimilar from all other hematopoietic and nonhematopoietic cell types. These epigenomes direct a cellular program that ultimately produces the highly specialized morphologically and functionally distinct mature erythrocyte. These epigenomic and transcriptome data are important tools to study human erythropoiesis and will be of benefit to a wide range of investigators studying inherited and acquired diseases of the erythrocyte.
RESULTS
Gene Expression during Erythropoiesis
A strategy using fluorescence-activated cell sorting (FACS)-based methods to purify morphologically and functionally discrete populations of cells at differing stages of erythroid development and differentiation was used to obtain pure populations of erythroid cells at distinct developmental and differentiation stages (An et al., 2014; Li et al., 2014; Chen et al., 2009).
RNA was prepared from umbilical cord-derived CD34+ hematopoietic stem and progenitor cells (HSPCs) and from BFU-E (burst-forming unit erythroid) and CFU-E (colony-forming unit erythroid) cells obtained after FACS sorting of cells derived from the erythroid cell culture of umbilical cord-derived HSPCs (Figure S1) (An et al., 2014; Li et al., 2014). RNA sequencing (RNA-seq) was performed and a series of unbiased, stage-specific transcriptomes was prepared.
Analysis of these progenitor cell transcriptomes and previously published transcriptomes from umbilical cord-derived proerythroblast through polychromatic erythroblast stage cells from terminal erythroid differentiation (An et al., 2014; Li et al., 2014) revealed tight clustering of transcriptomes from differing stages, even between biologically different replicates (Figure S2). There are marked differences between stages, with both shared and dissimilar gene expression profiles defining each stage (data not shown). There are numerous significant (false discovery rate [FDR] < 0.05; Table S1) temporal changes in gene expression, with each developmental (BFU-E, CFU-E) and differentiation (proerythroblast to orthochromatic erythroblast) stage exhibiting unique transcriptomes. Gene Ontology analyses reveal varying patterns of expression enriched for genes of differing function (Figure S3). These data indicate that differing programs of regulation likely control these varying stage-specific expression patterns.
Changes in Chromatin Accessibility throughout Erythropoiesis
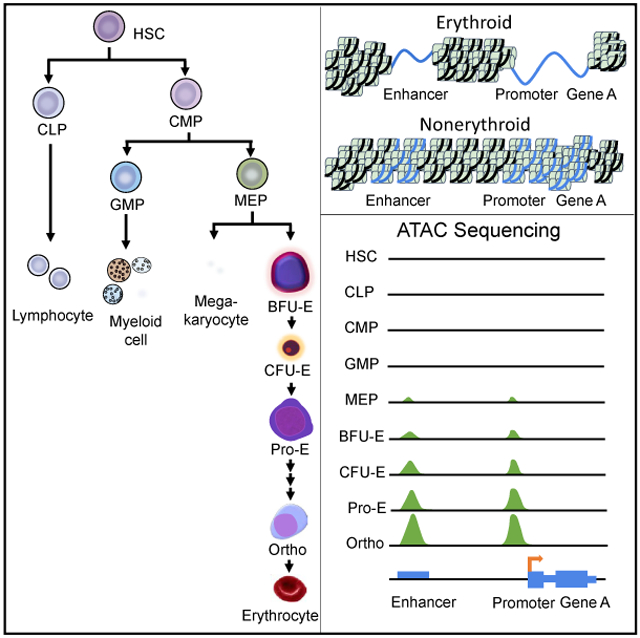
Nucleosome-depleted regions of open, accessible chromatin are found around the transcription start sites (TSSs) of active genes (Teif et al., 2012) and mark important regulatory elements, including promoters, enhancers, and insulators (Teif et al., 2012; Heintzman et al., 2007; Kellis et al., 2014; Thurman et al., 2012; Xi et al., 2007). The study of chromatin accessibility has yielded many insights into erythroid gene regulation. For instance, the identification of DNaseI hypersensitive sites (DHSs), regions of DNA sensitive to digestion because of an “open” chromatin state, led to the identification of the globin gene locus control region (LCR) and an erythroid enhancer in the BCL11A gene linked to hemoglobin F levels, now a target of treatment strategies to treat β-thalassemia and sickle cell disease (Li et al., 2002; Liang et al., 2008; Bauer et al., 2013). We used the assay for transposase-accessible chromatin using sequencing (ATAC-seq), a rapid, sensitive technique for identifying sites of open chromatin on a genome-wide scale (Corces et al., 2016). We applied this technique to human erythroid cells cultured from umbilical cord-derived HSPCs at differing stages of erythroid development and differentiation using FACS-based methods to purify morphologically and functionally discrete populations of cells (Figure S1 and S4) (An et al., 2014; Li et al., 2014; Chen et al., 2009). Sequence and read mapping data are provided in Figure S2). Principal-component analyses revealed stage-dependent patterns that ordered as expected during erythropoiesis (Figure S2).
We detected changes in chromatin accessibility across erythroid development and differentiation, with sites of chromatin accessibility gained or lost during erythropoiesis. Examples of changes in the patterns of chromatin accessibility are shown at the β-globin-like gene cluster and BCL11A gene locus, where ATAC peaks localize at gene promoters and known erythroid cell enhancers and regulatory elements (Figures 1A and 1B). Large numbers of ATAC peaks were acquired during the developmental stages of erythropoiesis, between HSPC to BFU-E, BFU-E to CFU-E, and notably from CFU-E to proerythroblast stage (ProE), while large numbers of ATAC peaks were lost between HSPC and BFU-E and late basophilic erythroblast (LBaso) to polychromatic erythroblast stages (Figure 1C). For unknown reasons, there were no large changes in ATAC peaks between three of the four transitions during terminal erythroid differentiation. Analysis of ATAC peak localization revealed the proportion of nonpromoter peaks (intergenic + 5′ and 3′ distal peaks) decreased by stage, from BFU-E throughout erythroid development and differentiation, while the proportion of promoter (≤1,000 bp from the TSS) peaks increased (Figure 1D). To assess whether these changes affected putative enhancers or other regulatory elements, we compared nonpromoter (>1,000 and <50,000 bp from the TSS) ATAC peaks lost during erythroid development and differentiation with genomic data from human HSPC and mixed populations of cultured human erythroblasts (Xu et al., 2012, 2015; Steiner et al., 2016). Of the lost nonpromoter ATAC peaks, 28% were putative active enhancers (defined as the presence of monomethyl histone 3 lysine 4 [H3K4me1] and acetyl histone 3 lysine 27 [H3K27Ac] and the absence of trimethyl histone 3 lysine 27 [H3K27me3] in HSPCs [p ≤ 0.0001]), suggesting enhancer decommissioning during erythropoiesis. In parallel, 10% of the lost nonpromoter ATAC peaks exhibited H3K27 trimethylation in erythroblasts (p ≤ 0.0001). Eleven and nine percent of the lost nonpromoter ATAC peaks had co-localizing CTCF or cohesinSA-1 respectively (both p ≤ 0.0001).
Figure 1. Regions of Open Chromatin Identified during Erythropoiesis.

(A) ATAC peaks at the β-globin-like gene locus.
(B)ATAC peaks at the BCL11A gene locus.
(C)Bar chart representation of differential changes in open chromatin as identified by ATAC peaks by stages of erythropoiesis, with sites of acquired ATAC peaks shown in red and sites of lost ATAC peaks shown in blue.
(D)Distribution of ATAC peaks in human erythroid cells at differing stages of erythroid development and differentiation. The human genome was portioned into seven bins relative to RefSeq genes. The percentage of the human genome represented by each bin was color coded, and the distribution of ATAC peaks placed in each bin was graphed on the color-coded bar. The Genome Bar is a graphical representation of the relative proportions of the different location categories in the entire human genome.
TES, transcriptional end site; TSS, transcriptional start site.
Analyses of associated functional terms with differential ATAC peaks revealed enrichment for erythroid-related terms, particularly for regions showing increased chromatin accessibility in the transitions from BFU-E to CFU-E, CFU-E to proerythroblast, and late basophilic erythroblast to polychromatic erythroblast stages (Table S2).
Erythroid Cells Exhibit a Distinct Pattern of Chromatin Accessibility
One goal of our study was to identify regions of open chromatin unique to erythroid cells, as markers of critical erythroid-specific regulatory elements. To do this, we examined patterns of chromatin accessibility in erythroid cells and compared them to patterns in other hematopoietic and nonhematopoietic cell types. To separate direct promoter-associated sites and nonpromoter-associated sites, we analyzed chromatin accessibility at both promoter and nonpromoter locations, respectively, by erythroid-specific stage (Figure 2). Many (4,386) erythroid-specific nonpromoter sites exhibiting distinct patterns of chromatin accessibility compared to other hematopoietic cell types were identified. An example of erythroid-specific ATAC peaks at the hexokinase-1 (HK1) gene locus is shown in Figure 2A. Regions of chromatin accessibility representing stages of terminal erythroid differentiation clustered together tightly and were the most different from all of the other hematopoietic cell types (Figures 2B and 2C). Functional analyses using the genomic regions enrichment of annotations tool (GREAT) algorithm (McLean et al., 2010) of nonpromoter differential ATAC peaks revealed enrichment for erythroid-related terms, particularly terms related to red blood cell phenotypic traits (Figure 2D). Erythroid cell promoter sites exhibited similar distinctive patterns of chromatin accessibility with functional correlates linked to erythroid cell phenotypes (Table S2; data not shown). The stages of terminal erythroid differentiation clustered together tightly and were the most different from all of the other hematopoietic cell types.
Figure 2. Erythroid Cells Exhibit Unique Stage- and Cell Type-Specific Patterns of Nonpromoter Chromatin Accessibility.

(A)Chromatin accessibility at the HK1 locus demonstrating erythroid-specific (red) ATAC peaks.
(B)Read count correlation of ATAC peaks in erythroid and nonerythroid hematopoietic cells.
(C)Heatmap of differential ATAC regions between erythroid cells and other hematopoietic cell types. The regions at the top far right, representing terminal erythroid differentiation, show high accessibility in erythroid cells, and the regions at bottom have low accessibility in erythroid cells compared to other hematopoietic cell types.
(D)GREAT (genomic regions enrichment of annotations tool) analysis of erythroid-specific regions of chromatin accessibility revealed significantly enriched terms for erythroid-related traits. The log10 binomial p value of each category is shown at right.
(E)Principal-component analysis of patterns of chromatin accessibility in erythroid, nonerythroid hematopoietic, and nonhematopoietic cells.
(F)Hierarchical cluster analysis shows terminally differentiating erythroid cells clustering separately from all of the other hematopoietic and nonhematopoietic cells (orange box, lower left corner).
The distinct pattern of chromatin accessibility of cells from terminal differentiation was even more apparent when nonhematopoietic cells were included in the comparison, in which they widely separate in both principal-component analysis and hierarchical clustering analysis (Figures 2E and 2F). These observations support the hypothesis that erythroid cells possess a specialized pattern of regulatory elements to direct patterns of gene expression that define stage-specific cellular phenotype.
Correlation of ATAC Peaks and Gene Expression
Regions of chromatin accessibility found at gene promoters and at linked regulatory elements are expected to be associated with expressed, actively transcribing genes. To assess the relation between chromatin accessibility and gene expression, we correlated promoter-associated and nonpromoter-associated ATAC peaks with the expression of nearby genes. While the expression of linked genes with an ATAC peak at only the promoter would be expected to be lower than the expression of genes with an ATAC peak in both the promoter and at a nearby nonpromoter site, this was not observed in erythroid progenitor cells (HSPC p = 0.38, BFU-E p = 1.0, and CFU-E p = 0.14; Figure 3A). However, the expression of linked genes with an ATAC peak at both the promoter and at a nearby nonpromoter site was higher in cells undergoing terminal erythroid differentiation (proerythroblasts, early and late basophilic erythroblasts, orthochromatic and polychromatic erythroblasts, all p < 2.2e–16). The association of highly expressed erythroid genes with increasing numbers of nonpromoter-associated ATAC peaks during terminal erythroid differentiation suggests the acquisition of erythroid enhancers (Figure S5). When correlating the amount of gene expression assayed by RNA-seq transcripts per million reads (TPM) and the amount of open chromatin determined by ATAC-seq reads per kilobase per million reads (RPKM), the correlation was stronger in terminal erythropoiesis than in progenitors and was stronger in promoter than in nonpromoter regions (Figure S5).
Figure 3. Chromatin Accessibility and Gene Expression.

Levels of gene expression determined by RNA-seq were correlated with the location of ATAC peaks in linked gene promoters, linked nonpromoter regions, both, or neither.
(A)Patterns of gene expression associated with ATAC peak presence or absence as described are shown in CFU-E, early basophilic erythroblasts, and orthochromatic erythroblasts.
(B)Levels of gene expression and gain or loss of ATAC peaks. In general, as gene loci acquired ATAC peaks, levels of gene expression increased. Shown here in the transitions from HSPC to BFU-E, BFU-E to CFU-E, and CFU-E to proerythroblast (ProE), the gene expression of linked genes that lost ATAC peaks (blue) is significantly lower than the expression of linked genes that gained ATAC peaks (red).
(C)SLC2A1 gene locus. Patterns of chromatin accessibility denoted by ATAC peaks (blue) are apparent in erythroid differentiation before the onset of detectable gene expression identified by RNA-seq (red).
A pattern of chromatin opening in one stage followed by the acquisition of linked gene expression in subsequent stages, while not uniform, was common throughout erythroid development and differentiation. In the transitions from HSPC to BFU-E, BFU-E to CFU-E, and CFU-E to proerythroblast, the gene expression of linked genes that lost ATAC peaks was significantly lower than the expression of linked genes that gained ATAC peaks (p= 1.8e–5, 2.2e–16, and 2.2e–16, respectively; Figure 3B). For example, at the single-gene level, promoter and nonpromoter-associated ATAC peaks that are not present in erythroid progenitor cells are acquired during erythroid differentiation, paralleling the onset of detectable gene expression at the GLUT1 (SLC2A1) Ggene locus (Figure 3C).
Open chromatin regions identified by ATAC-seq or DNaseI hypersensitive mapping are enriched for known and novel cell-type biologically-relevant transcription factor binding motifs (Neph et al., 2012; Sheffield et al., 2013). We performed motif finding using the hypergeometric optimization of motif enrichment (HOMER) algorithm to determine whether specific motifs are enriched during erythropoiesis (Heinz et al., 2010). In HSPCs and at all erythroid stages, the top enriched motif was CTCF, similar to other reports in which the CTCF is the most common motif found at sites of open chromatin (Table S3) (Xi et al., 2007).
When analyzing differentially acquired ATAC peaks between stages, CEBPα, ETS, GATA, and RUNX were the top motifs acquired between HSPC and BFU-E cells; GATA, AP1/NFE2, KLF, and RUNX were the top motifs acquired between BFU-E and CFU-E cells; and GATA, KLF, CCAAT, and CTCF were the top motifs acquired between CFU-E and proerythroblast cells (Figure 4A; Table S4). When examining terminal erythroid differentiation (proerythroblast to orthochromatic erythroblast), the acquired ATAC peaks had high amounts of NFE2 and KLF1 consensus motifs, paralleling the increases in NFE2 and KLF1 gene and protein expression (Figures 4B and 4C; Table S1) (Gautier et al., 2016). In contrast, ATAC peaks that decrease from proerythroblast to orthochromatic erythroblast stages have high amounts of GATA consensus motifs (46%; Table S4). This change parallels the decrease in GATA1 expression between these stages of differentiation (Figures 4B and 4C; Table S1). The decrease in GATA1 expression led us to examine whether GATA motifs were changing in other ATAC peaks, including those that do not change during terminal erythroid differentiation.
Figure 4. Transcription Factor Activity Changes across Erythropoiesis.

(A)The top regulatory protein-binding sites identified by the HOMER algorithm searching ±50 bp from summits of differential increasing stage to stage ATAC peaks. The top four motifs ranked by p value are shown for erythroid cell type transitions.
(B)Gene expression of critical erythroid transcription factors are shown for each stage of erythropoiesis. Transcriptome data are from An et al. (2014) and the present article.
(C)Protein expression of critical erythroid transcription factors are shown for each stage of erythropoiesis. Protein data are from Gautier et al. (2016).
Analysis of differential digital footprints obtained with the HINT (Hmm-based identification of transcription factor footprints) algorithm in the complete set of ATAC peaks revealed that GATA footprints decreased globally (p = 0.033), not just in the subset of decreasing ATAC peaks, as terminal erythroid differentiation progressed (Figure S6A) (Li et al., 2019). These data suggest that GATA1 activity may lose importance in the late stages of terminal erythroid differentiation.
A smaller number of erythroid cell type-specific ATAC peaks that decrease from proerythroblast to orthochromatic erythroblast stages have CTCF consensus motifs (6%; Table S4). Like GATA1, CTCF protein decreased in terminal erythroid differentiation (Table S1). CTCF binding sites are classified as those conserved in multiple cell types and those that are cell type specific (Steiner et al., 2016). Differential footprint analysis in the complete set of ATAC peaks showed no difference in the CTCF motifs as terminal erythroid differentiation progressed (p = 0.85), suggesting that there was no global rearrangement in CTCF occupancy in conserved, non-cell type-specific regions (Figure S6B). The CTCF-associated ATAC peaks lost during terminal erythroid differentiation were not subject to differential changes in methylation (Wilcox p = 0.23).
Genomic ChIP-seq (chromatin immunoprecipitation with massively parallel DNA sequencing) data of RUNX1 and PU.1 from HSPC and GATA1, TAL1, and NFE2 mixed populations of cultured human erythroblasts (Xu et al., 2012, 2015; Steiner et al., 2016) were compared to ATAC peaks in HSPCs and cells from all stages of erythropoiesis (Figure S6). GATA1 and TAL1 demonstrated higher co-localization with ATAC peaks from earlier stages of erythropoiesis compared with the later polychromatic and orthochromatic stages of erythropoiesis, which is consistent with the data above. These findings demonstrate that key erythroid transcription factors have dynamic roles in the regulation of programs of gene expression throughout erythropoiesis.
Epigenetic Bookmarking
Bookmarking is an epigenetic mechanism for transmitting the cellular memory of the pattern of gene expression in a cell throughout mitosis to its daughter cells. This is vital for maintaining the phenotype in a lineage of cells (e.g., erythroid cells divide into erythroid cells and not some other cell type). Thus, the term bookmarking has been extended to apply to changes in the Epigenomics landscape as indicators of cell fate transitions, lineage relations, and cellular dysfunction (Stergachis et al., 2013). We categorized open chromatin regions that exhibited characteristics of bookmarking (i.e., present in a stem or progenitor cell and all subsequent stages). Sites bookmarked in HSPC (375), BFU-E (847), CFU-E (1,460), and proerythroblast (1,647) cells and not present in B and T lymphocytes were studied (Figures 5A and 5B; Table S5). Bookmarked sites were associated with the increased expression of linked genes and erythroid-linked functional terms, including phenotype traits and associated disorders (Table S5). Examples of the levels of linked gene expression with sites bookmarked in BFU-E and CFU-E are shown in Figures 5C and 5D.
Figure 5. Chromatin Accessibility Bookmarking and Gene Expression.

Sites of chromatin accessibility identified in HSPC, BFU-E, CFU-E, or ProE cells, respectively, and all of the other subsequent erythroid stages were defined as exhibiting bookmarking.
(A and B) Examples of bookmarked regions of open chromatin in HSPCs in the HBS1L/MYB intergenic region (A) and the SOX6 locus (B) are shown. Both of these bookmarked sites contain SNPs (associated dbSNP identifiers) linked to genome-wide association study (GWAS)-associated erythroid cell phenotypic traits.
(C and D) Comparison of gene expression of bookmarked-linked genes (red) compared to control, non-bookmarked genes (blue) is shown in BFU-E (C) and in CFU-E (D) cells.
Characterization of the transcription factor-binding sites under bookmarked ATAC peaks revealed that the overwhelming majority were GATA sites. Remarkably, 55% of bookmarked sites in HSPCs (p = 1e–225) had underlying GATA motifs. In BFU-E, GATA motifs were present in 71% of bookmarked sites (p = 1e–117), in 66% of bookmarked sites in CFU-E (p = 1e–416), and in 52% of bookmarked sites in proerythroblasts (p = 1e–335).
Conservation Analyses of Chromatin Accessibility
Evolutionary constraint in regions of noncoding DNA has served as a proxy for functional constraint in the identification of candidate regulatory elements that are also frequently marked by ATAC peaks. Many regulatory elements are rapidly evolving, and in some species, many elements are evolutionarily young and species specific (Blow et al., 2010; Schmidt et al., 2010). PhastCons analyses use a hidden Markov model method on aligned genomic sequences to estimate a probability that any nucleotide is conserved and predicts whether a region of DNA contains a functional regulatory element (Cheng et al., 2008; Kim et al., 2007; King et al., 2005; Siepel and Haussler, 2004). Maximum 46-way placental mammal PhastCons conservation scores were calculated for erythroid-specific ATAC peaks, hematopoietic ATAC peaks that were not erythroid specific, and a randomized group of ATAC peaks. Moderate conservation was seen for erythroid-specific ATAC peaks (median value 0.63), and strong conservation was seen for shared peaks (0.84) compared to low conservation for random regions (0.42), suggesting that these regions are not only conserved but likely contain functional regulatory elements.
Changes in DNA Methylation throughout Erythropoiesis
We performed genome-wide DNA methylation analyses using enhanced reduced representation bisulfite sequencing (ERRBS) across human erythroid development and differentiation. A pattern of gradual, global demethylation during erythropoiesis was observed (Figures 6A and 6B), similar to the data reported in erythropoiesis in mouse fetal liver erythroblasts and human umbilical cord-derived and mobilized erythroid cells (Shearstone et al., 2011; Yu et al., 2013; Bartholdy et al., 2018). These dynamic changes in demethylation were most prominent along CpG shores (<2 kb flanking CpG islands, proerythroblast to orthochromatic erythroblast p < 2.2e–16) and along CpG shelves (<2 kb flanking outward from a CpG shore, proerythroblast to orthochromatic erythroblast, p < 2.2e–16) (Figure S7), with lower CpG density regions often associated with dynamic changes in methylation. As expected, global methylation at CpG islands including CpG promoters was low, with minimal changes throughout erythropoiesis (Figure S7) (Gentleman et al., 2004; Ziller et al., 2013). While global methylation at CpG promoters was low without significant changes (p = 0.61), there was a decrease in the methylation of non-CpG island promoters throughout erythropoiesis (p < 2.16e–9; Figure S7).
Figure 6. Dynamic Changes in Methylation across Erythropoiesis.

(A)Boxplots of the percentage of methylation of 5-kb genomic windows show a gradual decrease in methylation.
(B)Dynamic changes in methylation in the regions of the RBM38 gene locus. Changes from DNA methylation (red, height indicates number of methylated reads) to demethylation (blue, height indicates number of unmethylated reads) are observed.
(C)Numbers of differentially methylated regions are shown in 5-kb tiled regions across the genome. Red indicates regions of gained methylation and blue indicates regions of lost methylation.
(D)Numbers of differentially methylated regions located in open chromatin.
(E)Heatmap display of methylation levels (%) in open chromatin regions with dynamic methylation.
(F)Dynamic changes in DNA methylation, alterations in chromatin configuration, and pattern of gene expression at the ZFPM1 gene locus.
Comparing differential methylation across the stages of erythropoiesis, many regions became demethylated in the transition between BFU-E and CFU-E stages, and a large number of regions gained methylation between CFU-E and proerythroblast stages (Figure 6C).
Correlation of Chromatin Accessibility and DNA Methylation during Erythropoiesis
Sites of dynamic patterns of methylation located in regions of open chromatin were sought, as these sites often occur near enhancers and other regulatory elements (Lessard et al., 2015; Varley et al., 2013). The most dramatic changes were observed between the BFU-E and CFU-E stages, with 504 regions (of 24,688 methylated regions) undergoing demethylation (Figures 6D and 6E). These regions show significant increases in chromatin opening (p < 2.2e–16). An example of the dynamic changes in DNA methylation, alterations in chromatin accessibility, and changes in gene expression is shown at the ZFPM1 gene locus (Figure 6F). Overall integration of gene expression, DNA methylation, and chromatin state dynamics reveal that stage-specific gene regulation during erythropoiesis is a stepwise and hierarchical process involving many cis-regulatory elements.
ATAC Peaks Localize to Sites Linked to Erythroid Cell Traits and Erythroid Disease Genes
Genetic variants that modify chromatin accessibility and transcription factor binding are major factors in which genetic variation leads to differences in gene expression (Corces et al., 2016; Degner et al., 2012; Kellis et al., 2014; Ulirsch et al., 2014; Birney et al., 2010; Qu et al., 2015; Armstrong et al., 2014; Kumasaka et al., 2016). We explored whether SNPs associated with erythroid cell traits were enriched in biologically relevant epigenomic changes. We used the set of noncoding SNPs from the genome-wide association study (GWAS) catalog of the National Human Genome Research Institute (NHGRI) associated with erythroid cell terms (https://www.genome.gov/gwastudies) and mapped ATAC peaks and methylation sites by erythroid cell stages. A total of 497 SNPs associated with erythroid cell traits were found in regions of open chromatin (Table S6).
One hundred seventy-three SNPs found in ATAC peaks associated with erythroid cell traits were within 1 kb of a gene promoter, while 324 were not. Examples of erythroid trait-associated SNPs bookmarked in CFU-E cells that were also erythroid specific are shown in Figures 7A and 7B. Many of these SNP-associated ATAC peaks were bookmarked, as noted above, especially those found in HSPCs, CFU-E, and proerythroblasts (Figure 7C). Of the SNP-associated ATAC peaks, 103 were erythroid specific (Table S4); 19 differentially methylated regions were associated with SNP-associated ATAC peaks.
Figure 7. Sites of Chromatin Accessibility Containing Genome-wide Association Study-Linked SNPs Linked to Erythroid Cell Traits.

ATAC peaks were mapped onto the GWAS catalog of the National Human Genome Research Institute associated with erythroid cell terms.
(A and B) Examples of erythroid-specific, erythroid trait-associated SNPs in ATAC peaks bookmarked in CFU-E cells are shown at the RCL1 gene locus (A, two SNPs) and the PIEZO1 gene locus (B, one SNP).
(C) The x axis represents the 497 SNPs associated with erythroid cell traits ordered from right to left. The top of the heatmap (red) shows each ATAC peak that contains a GWAS erythroid-associated SNP by erythroid stage. The bottom half of the heatmap (blue) shows the trait associated with each ATAC peak-containing GWAS SNP. Many SNPs were bookmarked, and most were associated with more than one erythroid cell trait.
Several bookmarked regions of open chromatin contained SNPs linked to GWAS-associated erythroid cell phenotypic traits, 11 in HSPCs, 6 in BFU-E cells, 38 in CFU-E cells, and 29 in proerythroblast cells (Table S4). Two of the GWAS-associated sites in HSPCs were at or near sites encoding critical regulatory genes (Figures 5A and 5B). One was located in the HBS1L-MYB intergenic region of chromosome 6 and the other in an intronic site in the SOX6 gene. The HBS1L-MYB intergenic SNP, rs9494142, and other SNPs in the region have been associated with hemoglobin, hematocrit, hemoglobin F, hemoglobin A2, mean cell volume, and mean corpuscular hemoglobin concentration (MCHC). This bookmarked ATAC peak SNP is associated with a GATA-containing enhancer linked to the MYB gene via chromosome conformation capture, influencing its expression (Stadhouders et al., 2014). The intronic SOX6 SNP, rs17462448, has been associated with MCHC and red blood cell distribution width. Notable GWAS-associated sites bookmarked in BFU-Es include rs35152987 at the β-globin gene locus, rs737092 at the RBM38 gene locus, and rs2703485 at the KIT locus (not shown).
We compared regions of chromatin accessibility in a well-characterized, comprehensive panel of genes linked to inherited anemia (Mayo Clinic ID: NGHHA, with STOM and UGT1A1 genes removed and HBA1, HBA2, and KCNN4 genes added). All but 4 of the 40 genes had regions of chromatin accessibility in ≥1 stages of erythroid development or differentiation (data not shown). Of note, 76 erythroid-specific regions of chromatin accessibility were observed in 24 of these genes (p < 0.0001; c.f., randomized genomic intervals).
DISCUSSION
Human erythropoiesis is one of the best-characterized models of cell development and differentiation serving as a paradigm for understanding mechanisms of development and differentiation in other cell types (Lloyd, 2018; Tsiftsoglou et al., 2009; Xu et al., 2012, 2015). Epigenetic studies have shown that critical regulatory elements appear to be gained and lost during hematopoiesis (Xu et al., 2012; Lara-Astiaso et al., 2014; Heuston et al., 2018). Our erythroid cell data confirm and expand these observations, with sites of chromatin accessibility gained and lost throughout erythropoiesis. Our data also reveal that chromatin states differ by specific erythroid stages, with marked transitions between some stages, refining results compared to previous studies of mixed populations of erythroid cells (Corces et al., 2016), indicating that cell stage-specific activation of gene loci during erythropoiesis is a stepwise and hierarchical process that involves many cis-regulatory elements (Bonifer and Cockerill, 2017).
Tissues adopt varying strategies to establish downstream gene regulatory networks. For instance, chromatin architecture in intestinal stem cells exhibited features of active enhancers, including regions of chromatin accessibility, that were then broadly conserved in differentiating intestinal progenitor cells at or near genes exclusively expressed in intestinal cells (Kim et al., 2014). In contrast, in a murine hematopoiesis model, active regulatory elements were dynamically regulated, with stem and progenitor cell populations showing very few regions of chromatin marking active elements, which were then acquired de novo during lineage commitment and differentiation (Luyten et al., 2014). A limited number of sites of chromatin accessibility identified in HSPCs were identified in cells committed to the erythroid lineage. This is similar to B lymphocytes, in which the majority of the active regulatory elements in differentiated B lymphocytes were not marked in HSPCs (Miyai et al., 2018; Choukrallah et al., 2015). These observations demonstrate that a dynamic program of chromatin accessibility is shaped during lineage commitment and differentiation in hematopoiesis, including erythropoiesis, and that this program is independent of early chromatin priming.
DHS mapping and ATAC-seq across many cell types have allowed the construction of transcription factor regulatory circuits and demonstrated various degrees of similarity between cell types (Neph et al., 2012; Sheffield et al., 2013). Our data demonstrate that genome-wide patterns of chromatin accessibility are unique to cells in the terminal stages of erythroid differentiation, indicating that a highly specialized regulatory DNA landscape is present, likely evolved to direct a program conferring specialized functions to the mature erythrocyte. In parallel, they also reveal that patterns of chromatin accessibility in erythropoiesis are the most divergent among hematopoietic cell types, again likely attributable to erythroid cell function.
It will be interesting to compare conservation of the human erythroid epigenetic landscape to other species, allowing us to highlight shared pathways of erythroid cell function (Kellis et al., 2014). Comparison of human and murine regulatory elements in proerythroblasts suggest that evolutionary divergence drives the changes in gene expression (Ulirsch et al., 2014). Human erythroid regulatory elements appear to be under weak evolutionary constraint, especially when compared to placental mammals and nonplacental vertebrates, and many are species specific and evolutionarily young (Su et al., 2013). This is likely because mammalian erythrocytes are one of the most highly specialized cell types known. The mature erythrocyte has evolved to a cell that lacks a nucleus with unique membrane function and excess membrane surface area relative to cell volume that enable efficient oxygen delivery to meet the increased oxygen demand of evolving homeotherms (Scott and Milsom, 2006).
A limitation of our study is the use of cell culture to obtain adequate numbers of primary erythroid cells, with the results likely suffering from artifacts of culture conditions per se. A goal is to perform epigenomic and transcriptomic studies on uncultured primary human erythroid cells isolated directly from the bone marrow of normal individuals at steady state.
Regions of chromatin accessibility, identified by DHS sites and/or ATAC peaks, often map to enhancers and other regulatory elements. The DNA sequence under DHS sites and ATAC peaks yields a collection of highly enriched transcription factor-binding motifs, as DHS sites and ATAC peaks define a much more limited DNA region compared to the broad regions of histone modifications (Song et al., 2011; Thurman et al., 2012). DHS mapping and ATAC-seq have also revealed that chromatin accessibility patterns may exhibit individual- and allele-specific chromatin signatures (Birney et al., 2010; Qu et al., 2015; Xu et al., 2017). The mapping of DHS sites and ATAC peaks to genetic loci associated with various genetic traits by genome-wide association studies is a powerful technique that has yielded novel insights into quantitative trait variation and genetic disease (Kumasaka et al., 2016; Abraham et al.; 2013; Bao et al., 2015; Degner et al., 2012; Frank et al., 2015; Koh et al., 2016; Stergachis et al., 2013). We found large numbers of epigenomic changes at SNPs linked to GWAS-associated erythroid cell traits near genes encoding proteins that are critical for erythroid cell development and function (Table S2).
These previously undescribed stage-specific patterns of chromatin accessibility linked to genetic loci associated with erythroid traits and inherited erythrocyte disorders will allow the interrogation of changes in the epigenome and associated gene expression changes and target them to specific erythroid stages. For instance, in the early stages of terminal erythroid differentiation, iron accumulates in ferritin (Philpott, 2018). As differentiation proceeds into the basophilic and orthochromatic erythroblast stages, large amounts of iron are imported through ferritin in parallel with high rates of iron transfer to the mitochondria. The programs of gene and protein expression required for these processes can now be better understood in a stage-specific manner.
Defining these regulatory regions in erythroid cells will also be useful in the genetic diagnosis of patients with hematologic disease. In some recessively inherited diseases, deleterious coding region mutations are identified in causative genes only on one allele, with the causative mutation in trans not identified. A subset of patients with recessively inherited pyruvate kinase deficiency has disease-associated mutations on one or neither PKLR gene alleles after mutation screening of the promoter and coding exons (Bianchi et al., 2019). In recessively inherited congenital dyserythropoietic anemia type II (CDAII), a subset of patients who exhibit all of the phenotypic and laboratory characteristics of CDAII, a disease-associated SEC23B mutation has been identified on only one allele (Iolascon et al., 2010; Russo et al., 2010). Both genes have candidate ATAC peaks in the genomic vicinity, making them candidate regions for disease-associated mutations. The data and tools provided here should serve as helpful resources for the study of normal and perturbed human erythropoiesis.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
For further information and requests for resources should be directed to and will be fulfilled by Patrick G. Gallagher (patrick.gallagher@yale.edu).
Materials Availability Statement: This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Subjects
Human cord blood was obtained from the National Cord Blood Program, New York Blood Center. Studies were conducted with Institutional Review Board approval of the New York Blood Center. All umbilical cord samples were completely de-identified; thus sex, race, and ethnic background of the donors were not provided.
Model
Erythroid cell culture of human umbilical cord-derived CD34+ hematopoietic stem and progenitor cells (HSPCs) and fluorescence-activated cell sorting to isolate discrete, highly purified populations of cells at differing stages of erythroid development and differentiation were performed as described below (An et al., 2014; Li et al., 2014; Chen et al., 2009).
Isolation and culture
CD34+ cells were purified from umbilical cord blood by positive selection using the CD34 MicroBead Kit UltraPure (Miltenyi Biotec), according to the manufacturer’s instructions. Purity of isolated CD34+ cells was 90%–98%. To enrich for erythroid progenitor cells, CD34+ cells were cultured in expansion medium with StemSpan SFEM (StemCell technologies) as base medium for 4 days supplemented with 10% FBS (GE Healthcare), 10 ng/mL IL-3 (StemCell technologies), 50ng/mL stem cell factor (StemCell Technologies), 1IU/mL erythropoietin (ThermoFisher Scientific) and 0.06mM α-thioglycerol (Sigma) starting at a cell concentration of 105/mL (Hu et al., 2013). To promote erythroid differentiation, cells were then cultured in a series of 3 phases.2 Composition of the base culture medium was Iscove’s Modified Dulbecco’s Medium, 2% human peripheral blood plasma (StemCell Technologies), 3% human AB serum (Atlanta Biologicals), 3 IU/mL heparin (StemCell Technologies), and 10 μg/mL insulin (Sigma). In the first phase (day 0-6), CD34+ cells at a concentration of 105/mL were cultured in the presence of 1ng/mL IL-3, 10ng/mL stem cell factor, 3IU/mL erythropoietin and 200μg/mL Holo-human transferrin (Sigma). In the second phase (day 7-11), IL-3 was omitted and erythropoietin was reduced to 1IU/mL. In the third phase, stem cell factor was removed, holo-human transferrin was increased to 1mg/mL and cell concentration was adjusted to 106/mL on day 11 and to 5 × 106/mL on day 15, respectively. All cell cultures were incubated at 37°C in the presence of 5% CO2.
Fluorescence-activated cell sorting
Cells cultured in expansion medium for 4 days were used for sorting of BFU-E and CFU-E cells, separately. Harvested cells were washed with cold PBS/0.5% BSA and suspended in PBS/0.5% BSA at a concentration of 20 million/mL, blocked with 4% human AB serum for 10 min, then incubated with PE-conjugated mouse anti-human CD34 (BD Biosciences), FITC-conjugated mouse anti-human CD36 (BD Biosciences), APC-conjugated mouse anti-human glycophorin A (BD Biosciences) and PE-Cy7-conjugated mouse anti-human IL3-R (ThermoFisher Scientific) at 4°C for 30 min in the dark. Cells were washed twice with PBS/0.5% BSA, re-suspended in PBS/0.5% BSA at a concentration of 15 million/mL and stained with cell viability dye 7-AAD on ice for 10 min in the dark. Cell debris and dead cells were excluded from analysis based on scatter signal and 7-AAD fluorescence. BFU-E colony forming IL3R-GPA-CD34+CD36- cells and CFU-E colony forming IL3R-GPA-CD34-CD36+ cells, respectively, were collected (Figure S1).
To isolate erythroblasts representing stages of terminal erythroid differentiation, in the third phase of culture, proerythroblast, early basophilic erythroblasts and late basophilic erythroblasts were isolated after 7 days of culture and polychromatic and orthochromatic erythroblasts were isolated after 13-14 days of culture. Cells were harvested and prepared as described above, then stained with PE-conjugated mouse anti-human GPA (BD Biosciences), FITC-conjugated mouse anti-human band 3 (New York Blood Center) and APC-conjugated mouse anti-human a4-integrin (Miltenyi Biotec). Cell debris and dead cells were excluded from analysis based on scatter signal and 7-AAD fluorescence. From GPA+ cells, proerythroblasts, early basophilic erythroblasts, late basophilic erythroblasts, polychromatic erythroblasts, and orthochromatic erythroblasts were gated and collected based on their expression level of Band 3 and μ4-integrin (Figure S5). Sorting was performed on a MOFLO high-speed cell sorter (Beckman-Coulter).
Validation: Sorting, colony assay and morphology
Purities of all sorted populations were examined by analytic flow cytometry analysis using the antibodies and sorting schemes described above. Colony assays were performed to validate the sorted progenitor populations (BFU-E and CFU-E) functionally. Two semi-solid media, MethoCult H4434 classic media and MethoCult H4330 EPO-alone media (Stem Cell Technologies), were used for colony assay to validate the colony forming potential of sorted erythroid progenitor cells, according the manufacturer’s instruction. Collected IL3R-GPA-CD34+CD36- cells and IL3R-GPA-CD34-CD36+ cells were diluted to a density of 200 cells/ml of semi-solid medium, cultured at 37°C in the presence of 5% CO2. CFU-E and BFU-E colonies were defined according to the criteria described by Dover et al. (1983). CFU-E colonies in EPO-alone media were counted on day 7 of culture and BFU-E colonies in classic media were counted on day14 of culture.
Cytospin preparations were prepared to validate the sorted erythroblasts morphologically. 105 sorted erythroblasts from each stage were resuspended in 200μl PBS for cytospin preparation on coated slides using a ThermoScientific Shandon4 Cytospin. Slides were stained with May-Grunwald (Sigma) solution for 5 min, rinsed in 40mM Tris buffer (pH 7.2) for 90 s, and subsequently stained with Giemsa solution (Sigma) for 15 min. After staining, slides were rinsed in double distilled water and dried at room temperature. Slides were imaged using a Leica DM2000 inverted microscope with images obtained at 100 × magnification.
METHOD DETAILS
RNA Sequencing
BFU-E and CFU-E. RNA was prepared from HSPCs and BFU-E and CFU-E primary human erythroid progenitor cells for RNA-seq analyses using an RNeasy mini kit (QIAGEN) per manufacturer instructions on three biologic replicates from unrelated donors (Steiner et al., 2009). RNA samples were treated with RNase-free DNase I (Takara, Otsu, Japan), quantified using a NanoDrop1000 (Thermo-Fisher, Waltham, MA) and assessed with an Agilent2100 Bioanalyzer (Agilent, Santa Clara, CA). The RNA integrity number of each sample was >9. DNA libraries were prepared according to manufacturer’s instructions (Illumina, San Diego, CA). Each library was sequenced on the Illumina HiSeq2000 platform using a 50-bp single-end, non-strand specific sequencing strategy (An et al., 2014). Deep sequencing was performed on polyA+ mRNA from the 3 biological replicates of HSPCs and each erythroid stage. RNA-seq data for human primary erythroblasts representing erythroblast through orthochromatic erythroblast stages were obtained from NCBI GEO: GSE53983. Primary and downloaded data were analyzed and subjected to quality control analyses using the FastQC and QoRTS packages (Hartley and Mullikin, 2015). Sequence reads were mapped to the human genome version hg19 using TopHat version 2.0.13 (Kim et al., 2013). Reads were counted for each gene using featureCounts (Liao et al., 2014).
Gene expression was analyzed using the EdgeR package (Robinson et al., 2010). Only genes with 3 or more samples in the dataset with >1 count per million reads were analyzed. The Trimmed Mean of M values (TMM) method was used for normalization (Robinson and Oshlack, 2010). Principal component analysis (PCA) was performed on log transformed counts per million values for expressed genes. Differentially expressed genes in adjacent stages, HSPC to ProE, and ProE to Ortho were identified using the EdgeR exact-Test. The set of 341 highly expressed erythroid genes was defined by having orthochromatic erythroblast RPKM > 64 and >32-fold increase compared to HSPC. Clustering of differentially expressed genes with similar expression patterns was performed using k-means with 25 clusters, and clusters with visually similar patterns were merged together. Gene ontology analysis was performed using the goseq package (Young et al., 2010).
ATAC sequencing
ATAC sequencing was performed using 50,000 primary erythroid cells per reaction (Buenrostro et al., 2013; Corces et al., 2016) from two biological replicates of HSPCs and each erythroid stage from unrelated donors. After washing once with cold PBS, cells were lysed using cold lysis buffer (10mM TrisHCl, pH 7.4, 10mM NaCl, 3mM MgCl2 and 0.1% IGEPAL CA-630). Nuclei were collected by centrifugation, with the pellet resuspended in 50ul transposase reaction mix (25 μL 2 × TD buffer, 2.5 mL transposase (Illumina Nextera FC121-1030 TDE1 and TD Buffer) and 22.5 μL nuclease-free water). The transposition reaction was carried out for 30 min at 37°C in a thermocycler. Following transposition, samples were purified using a MinElute kit (QIAGEN) following manufacturer’s instructions and eluted in 24 μL.
Library assembly was done by PCR in a reaction mix containing 25uL HiFi 2x mix (HiFi HotStart ReadyMix PCR Kit (Kapa), 24uL of tagmented DNA and 1uL primer mix (12.5uM each of AD1 and AD2 primer mix) (Buenrostro et al., 2015). PCR conditions were 72°C for 5 min (adaptor extension), 98°C for 45 s (denaturation) and 5 cycles (98°C for 15 s, 63°C for 30 s, 72°C for 30 s), ending with 72°C for 1 min. The reaction was cleaned with the MinElute kit (QIAGEN) and eluted in 50 μL. Size selection was done with an EZNA size select kit (Omega) for isolation of 150-700bp fragments following manufacturer’s instructions. Q-PCR library amplification test was done to determine the additional number of cycles needed for the sequencing library in a mixture containing 5uL size-selected DNA, 1uL SYBR, 1uL G3306/3307 primers, 3uL H2O, and 10uL 2x KAPA HiFi mix for 20 cycles (95°C for 45 s, 20 × (95°C for 15 s, 63°C for 30 s and 72°C for 30 s). Primer sequences were: G3306 5′-AATGATACGGCGACCACCGA-3′ and G3307 5′-CAAGCAGAAGACGGCATACGA-3′. PCR library amplification was done in a reaction of 5uL size-selected DNA, 1uL G3308/3309 primers, 19uL H2O and 25uL 2x KAPA HiFi mix with PCR amplification for Y number of cycles determined by Q-PCR: 98°C for 45 s, Y x (98°C for 15 s, 63°C for 30 s, and 72°C for 30 s), ending with 72°C for 1 min. Primer sequences were: G3308 5′- AATGATACGGCGACCACCGAGATCTACA*C-3′ and G3309 5′- CAAGCAGAAGACGGCATACGAGA*T-3′ with *phosphorothioate bond. PCR amplified sequencing libraries were cleaned with 0.9 volumes of AMPure Beads (Beckman Coulter). Quality of the library was determined by analysis on an Agilent Bioanalyzer.
For each sample, 47-85 million 76 base, single end sequence reads were obtained on an Illumina HiSeq 2500 sequencer (Table S2). Sequenced reads were trimmed using Trimmomatic version 0.32 and mapped to the human genome (hg19 GRCh37) using the Burrows-Wheeler Aligner version 0.7.9a mem alignment program (Bolger et al., 2014; Li and Durbin, 2010). Reads with poor mapping quality or that mapped to the ENCODE project blacklist of repetitive regions and mitochondrial regions were removed (Carroll et al., 2014). Coverage files for genomic visualization in IGV were generated using bedtools after extending reads by 100 base and normalized per million reads (Quinlan and Hall, 2010). MACS2 program version 2.1.1.20160309 was used to identify peaks using parameters nomodel shift −100 extsize 200 with a q value of < 0.05 (Zhang et al., 2008). To pass quality control, sequencing datasets were required to have PHRED score >35 assessed using FASTQC, >10 million reads, >10,000 called peaks, and good peak quality in visual inspection of read coverage profiles. Other ATAC datasets downloaded from GEO were processed and assessed in the same way, using only the first read even if paired end reads were available. Read counts for open chromatin regions in all samples were obtained using the DiffBind version 2.4.8 package with DESEQ2 normalization using all aligned reads (parameter bFullLibrarySize = TRUE) (Ross-Innes et al., 2012). Statistically significant differentially accessible regions were identified by DiffBind using the DESEQ2 method with default parameters (Love et al., 2014). ATAC peaks/regions were linked to genes using the single nearest transcription start site (TSS) for RefSeq transcripts < 50000bp away utilizing the GenomicRanges package (Lawrence et al., 2013). ATAC peaks/regions < 1000bp from the nearest TSS were designated as promoters.
Erythroid specific peaks with 16-fold change were identified using DESeq2 version_1.16.1 comparing the erythroid read counts to hematopoietic progenitor, dendritic, B and T lymphocyte cell counts, in the merged set of regions in all datasets (Love et al., 2014).
Overrepresented motifs in ATAC peak sets were identified using the HOMER (Hypergeometric Optimization of Motif EnRichment) suite of tools for Motif Discovery (Heinz et al., 2010). Peak regions were intersected with the MACS2 summit positions, and overrepresented motifs in the 200 base pair region flanking the summits within the repeat masked Hg19 genome. Differential footprints between proerythroblasts and orthochromatic erythroblasts were identified using the HINT algorithm utilizing the mapped reads and peak calls from one replicate of each cell type (Li et al., 2019).
Functional analysis of ATAC peak sets was performed using the GREAT algorithm (Genomic Regions Enrichment of Annotations Tool) (McLean et al., 2010). Default parameters were used (Basal plus extension, 5kb upstream 1kb downstream plus distal up to 1MB, utilizing curated regulatory domains), and mouse phenotype category enriched terms are reported.
ATAC peak localization was compared to downloaded ChIP-seq datasets. ChIP-seq datasets were mapped as above, and peaks were called using MACS2 using default parameters for transcription factors, using-broad-broad-cutoff 0.01-qvalue 0.01-nomodel-extsize 147 for H3K27Ac, and using-broad-broad-cutoff 0.001-qvalue0.001-nomodel-extsize 147 for H3K4me1. Sicer was used to identify enriched regions in H3K27me3 that were further filtered to keep regions with ≥ 3-fold enrichment. Overlap of regions was assessed using bedtools Jaccard and with testing of overlap compared to multiple shuffles of randomized regions.
Conservation analysis of ATAC regions used 46 way placental mammal PhastCons scores downloaded from the UCSC database (http://hgdownload.cse.ucsc.edu/goldenpath/hg19/phastCons46way/placentalMammals/) that gives a conservation score of each base in the human genome compared to the alignment of 45 vertebrate genomes (six fish, one amphibian, one reptile, two avian, three non-placental mammal and thirty two placental mammals) (Siepel et al., 2005; Siepel and Haussler, 2004). The maximum phast-Cons score for each erythroid specific genomic interval to be tested was obtained, and the set of maximum scores was compared to the maximum scores of randomized intervals or non-erythroid specific ATAC regions. The randomized intervals were obtained using the Bedtools shuffle command on erythroid-specific regions. Promoter and exonic regions were removed from all three region sets prior to conservation analysis, since changes in promoter and exon composition could skew the conservation analysis.
Methylation assays
Genome-wide DNA methylation profiles were created using an enhanced reduced representation bisulfite-based sequencing (ERRBS) technique (Akalin et al., 2012a; Garrett-Bakelman et al., 2015). Three biological replicates of HSPCs and each erythroid stage were utilized from unrelated donors. Genomic DNA was extracted from stage-specific erythroid progenitor or erythroblast cells using the PureGene Kit (QIAGEN) per manufacturer’s instructions. 100ng DNA was digested with 200U Mspl (New England Biolabs) in a 100 mL reaction at 37°C for 18 h, then extracted with phenol-chloroform followed by ethanol precipitation and resuspended in 30 μL 10mM TrisCl pH 8.0. End repair of extracted DNA was performed in 100 μL of T4 DNA buffer, 15U of T4 DNA polymerase, 5U Klenow DNA polymerase, 50U T4 Polynucleotide Kinase and 4 μL premixed nucleotide triphosphates (All reagents from New England Biolabs) with incubation at 20°C for 30 min. DNA was extracted using QIAquick PCR columns per manufacturer’s instructions (QIAGEN). Adenylation was performed in 32 μL of DNA solution in elution buffer, 15U Klenow fragment and 10 μL of dATP at 1mM in Klenow buffer added (All reagents, New England Biolabs) for total reaction volume of 50 μl, with incubation at 37°C for 30 min. DNA was purified using MinElute PCR columns per manufacturer’s instructions (QIAGEN) into 10 μL of elution buffer. Adaptor ligation was performed in a total volume of 50 μL, 2000U T4 DNA ligase (New England Biolabs) and the Methylated Illumina adapters at a final concentration of 1.2mM with incubation for 16 h at 16°C. Products were isolated using PCR MinElute as previously described into 10 mL of elution buffer. For size selection, library fragment lengths of 150-250bp and 250-400bp were gel selected from a 1.5% agarose gel then isolated using QIAquick gel extraction kit (QIAGEN) per manufacturer’s instructions into 40 μL of elution buffer. Bisulfite conversion was performed using an EZ DNA methylation kit (Zymo Research) as per manufacturer’s instructions with the following modifications; after addition of CT conversion agent, the incubation was done on a thermocycler for 55 cycles of 95°C for 30 s, 50°C for 15 min, followed by elution of products into 40 μL of nuclease free water. PCR amplification was performed in a reaction volume of 200 mμL containing 2 mμL FastStart High Fidelity DNA polymerase (Roche), 0.5 mM each of the PE1.0 and PE2.0 Illumina PCR primers and 0.25mM of each nucleotide triphosphate using buffer 2. The reaction was split into four 50 mμL tubes and with thermal cycling as follows: 94°C for 5 min, 18 cycles of 20 s at 94°C, 30 s at 65°C, 1 min at 72°C, followed by 3 min at 72°C. PCR products from the combined 4 tubes were isolated using AMPure XP beads (Beckman Coulter) per manufacturer’s instructions. Libraries underwent quality control using Quant-IT dsDNA High Sensitivity Assay Kits on a Qubit 1.0 fluorometer.
Amplified libraries were sequenced using 50bp single-end read runs on an Illumina HiSeq2000 sequencer. Image capture, analysis, and base calling were performed using CASAVA 1.8 (Illumina). Reads were trimmed using Trim Galore v0.4.0 in rrbs mode (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/), and were then mapped to the hg19 genome using Bismark_v0.14.3 (Krueger and Andrews, 2011). Differential methylation was identified using methylKit v1.2.4 (Akalin et al., 2012b). Regions with less than 10 read coverage or greater than 99.9th percentile of coverage were discarded. Coverage values between samples were normalized using a scaling factor derived from differences between median of coverage distributions. Only CpG’s with 2 or more covered samples per group were analyzed. Differential methylation in CpG or different classes of genomic regions was identified by comparing two cell types using the MethylKit calculateDiffMeth function with a q-value cutoff of 0.01 requiring 25% change in methylation.
Genome-Wide Association Study Information
The NHGRI catalog of GWAS SNPs was downloaded from https://www.ebi.ac.uk/gwas/docs/file-downloads (MacArthur et al., 2017). Erythroid terms related to anemia, corpuscular, erythrocyte, F-cell, ferritin, HBA2 levels, hematocrit, hematological phenotypes, hemochromatosis, hemoglobin, iron, red blood, red cell, transferrin saturation were used to select 3645 Erythroid trait SNP’s. PLINK 1.9 was used to identify SNPs within one megabase in high linkage, R2 > 0.8 in the European samples of the 1000 genomes database (Chang et al., 2015). The combined set of 6844 GWAS plus linked SNPs was used for further analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was conducted using R version 3.4.1 (http://www.R-project.org/). The Wilcox test was used except as otherwise noted. Significance of overlap of genomic intervals was assessed by comparing the number of overlaps with the number of overlaps from multiple shuffles of randomized genomic regions. HOMER motif enrichment uses a hypergeometric distribution to calculate p values.
DATA AND CODE AVAILABILITY
The raw data files have been submitted to the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) and the VISION databases (http://www.bx.psu.edu/~giardine/vision/). The accession number for the RNA-seq, ATAC-seq and methylation analyses reported in this paper is GEO: GSE128269.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PE-Cy7-conjugated mouse anti-human IL3-R | Thermofisher Scientific | Cat# 25-1239-42; RRID: AB_1257136 |
| APC-conjugated mouse anti-human Glycophorin A | BD Biosciences | Cat# 551336; RRID: AB_398499 |
| PE-conjugated mouse anti-human CD34 | BD Biosciences | Cat# 555822; RRID: AB_396151 |
| FITC-conjugated mouse anti-human CD36 | BD Biosciences | Cat# 555454; RRID: AB_2291112 |
| PE-conjugated mouse anti-human GPA | BD Biosciences | Cat# 555570; RRID: AB_395949 |
| APC-conjugated mouse anti-human α4-integrin | Miltenyi Biotec | Cat# 130-093-281; RRID: AB_1036218 |
| FITC-conjugated mouse anti-human band 3 | NY Blood Center | mnarla@nybc.org |
| 7-AAD | BD Biosciences | Cat# 559925 |
| Biological Samples | ||
| Human cord blood | New York Blood Center | https://nybloodcenter.org/products-and-services/blood-products/research-products/ |
| Chemicals, Peptides, and Recombinant Proteins | ||
| SFEM | StemCell Technologies | Cat# 09650 |
| FBS | GE Healthcare | Cat# S40110H |
| IL-3 | StemCell Technologies | Cat# 78040 |
| SCF | StemCell Technologies | Cat# 78062 |
| EPO | ThermoFisher Scientific | Cat# PHC2054 |
| α-thioglycerol | Sigma-Aldrich | Cat# M6145-25ML |
| human peripheral blood plasma | StemCell Technologies | Cat# 70039.5 |
| human type AB serum | Atlanta Biologicals | Cat# S40110H |
| heparin | StemCell technologies | Cat# 07980 |
| insulin | Sigma-Aldrich | Cat# I9278 |
| Holo-human transferrin | Sigma-Aldrich | Cat# T4132-1G |
| BSA | Sigma-Aldrich | Cat# A7030-100G |
| May-Grunwald stain | Sigma-Aldrich | Cat# MG128-4L |
| Giemsa stain | Sigma-Aldrich | Cat# GS1L-1L |
| MethoCult H4434 Classic | StemCell Technologies | Cat# 04434 |
| MethoCult H4330 | StemCell Technologies | Cat# 04330 |
| MspI restriction enzyme | New England Biolabs | Cat# R0106T |
| T4 DNA polymerase | New England Biolabs | Cat# M0203S |
| T4 DNA ligase reaction buffer | New England Biolabs | Cat# B0202S |
| DNA polymerase I, Large (Klenow) fragment | New England Biolabs | Cat# M0210S |
| T4 polynucleotide kinase | New England Biolabs | Cat# M0201S |
| Deoxynucleotide (dNTP) solution set | New England Biolabs | Cat# N0446S |
| dATP solution | New England Biolabs | Cat# N0440S |
| NE Buffer 2 | New England Biolabs | Cat# B7002S |
| T4 DNA ligase | New England Biolabs | Cat# M0202T |
| Critical Commercial Assays | ||
| Gentra Puregene Blood Kit | QIAGEN | Cat# 158445 |
| QIAquick PCR Purification Kit | QIAGEN | Cat# 28104 |
| MinElute PCR Purficiation Kit | QIAGEN | Cat# 28004 |
| QIAQuick gel extraction set | QIAGEN | Cat# 28115 |
| EZ DNA Methylation Kit | Zymo Research | Cat# D5001 |
| FastStart High Fidelity PCR System | Roche | Cat# 3553400001 |
| AMPure XP for PCR Purification | Beckman Coulter | Cat# A63881 |
| Quant-It dsDNA Assay Kit, High Sensitivity | Invitrogen | Cat# Q33120 |
| Transposase (TDE1) Nextera DNA Sample Prep Kit | Illumina | Cat# FC-131-1024 |
| KAPA HiFi HotStart ReadyMix (2x) | Kapa Biosystems | Cat#KK2602 |
| AMPureXP | Beckman Coulter | Cat#A63880 |
| Omega EZNA size select kit | Omega | Cat#D6488-01 |
| Deposited Data | ||
| Raw and analyzed ATAC data | This paper | GEO: GSE128266 |
| Raw and analyzed methylation data | This paper | GEO: GSE128267 |
| Raw and analyzed RNA-seq data | This paper | GEO: GSE128268 |
| Human dendritic cell: ATAC-seq | NCBI GEO | GEO: GSM1565735 |
| Human corneal epithelial cell: ATAC-seq | NCBI GEO | GEO: GSM1662255 |
| Human fibroblast cell: ATAC-seq | NCBI GEO | GEO: GSM1662258 |
| Human cardiac smooth muscle cell: ATAC-seq | NCBI GEO | GEO: GSM1876021 |
| Human LMPP cell: ATAC-seq | NCBI GEO | GEO: GSM1937378 |
| Human bone marrow CD34+ cell: ATAC-seq | NCBI GEO | GEO: GSM1937399 |
| Human umbilical cord CD34+ cell” ATAC-seq | NCBI GEO | GEO: GSM1937401 |
| Human CD4 T lymphocyte: ATAC-seq | NCBI GEO | GEO: GSM1937406 |
| Human CD8 T lymphocyte: ATAC-seq | NCBI GEO | GEO: GSM1937407 |
| Human CLP: ATAC-seq | NCBI GEO | GEO: GSM1937408 |
| Human CMP: ATAC-seq | NCBI GEO | GEO: GSM1937410 |
| Human GMP: ATAC-seq | NCBI GEO | GEO: GSM1937415 |
| Human HSC: ATAC-seq | NCBI GEO | GEO: GSM1937416 |
| Human MEP: ATAC-seq | NCBI GEO | GEO: GSM1937418 |
| Human MPP: ATAC-seq | NCBI GEO | GEO: GSM19374119 |
| Human MPP: ATAC-seq | NCBI GEO | GEO: GSM1937419 |
| Human NK cell: ATAC-seq | NCBI GEO | GEO: GSM1937421 |
| Human B lymphocyte: ATAC-seq | NCBI GEO | GEO: GSM1937423 |
| Human pancreatic alpha cell: ATAC-seq | NCBI GEO | GEO: GSM1978243 |
| Human pancreatic beta cell: ATAC-seq | NCBI GEO | GEO: GSM1978246 |
| Human pancreatic Ac: ATAC-seq | NCBI GEO | GEO: GSM1978249 |
| Human human keratinocyte: ATAC-seq | NCBI GEO | GEO: GSM2035768 |
| Human neuron: ATAC-seq | NCBI GEO | GEO: GSM2199916 |
| Human non-neuron: ATAC-seq | NCBI GEO | GEO: GSM2199924 |
| Human HUVEC: ATAC-seq | NCBI GEO | GEO: GSM2202931 |
| Human cardiac mesoderm: ATAC-seq | NCBI GEO | GEO: GSM2257299 |
| Human CD8 naive lymphocyte: ATAC-seq | NCBI GEO | GEO: GSM2365846 |
| Human CD8 effector lymphocyte: ATAC-seq | NCBI GEO | GEO: GSM2365849 |
| Human CD8 memory lymphocyte: ATAC-seq | NCBI GEO | GEO: GSM2365852 |
| Human: aortic endothelial cell: ATAC-seq | NCBI GEO | GEO: GSM2394391 |
| Human HSPC H3K27me3: ChIP-seq | NCBI GEO | GEO: GSM1816077 |
| Human mixed proerythroblast H3K27me3: ChIP-seq | NCBI GEO | GEO: GSM1816078 |
| Human HSPC H3K4me1: ChIP-seq | NCBI GEO | GEO: GSM1816066 |
| Human mixed proerythroblast H3K4me1: ChIP-seq | NCBI GEO | GEO: GSM1816067 |
| Human HSPC H3K27Ac: ChIP-seq | NCBI GEO | GEO: GSM1816073 |
| Human mixed proerythroblast H3K27Ac: ChIP-seq | NCBI GEO | GEO: GSM1816074 |
| Human mixed proerythroblast TAL1: ChIP-seq | NCBI GEO | GEO: GSM1816083 |
| Human mixed proerythroblast GATA1: ChIP-seq | NCBI GEO | GEO: GSM970257 |
| Human mixed proerythroblast NFE2: ChIP-seq | NCBI GEO | GEO: GSM1816086 |
| Human HSPC PU1: ChIP-seq | NCBI GEO | GEO: GSM1816089 |
| Human HSPC RUNX1: ChIP-seq | NCBI GEO | GEO: GSM1816091 |
| Human HSPC CTFC 1: ChIP-seq | NCBI GEO | GEO: GSM1655735 |
| Human HSPC SA1 1: ChIP-seq | NCBI GEO | GEO: GSM1655736 |
| Human Erythroid CTFC 1: ChIP-seq | NCBI GEO | GEO: GSM1655739 |
| Human Erythroid SA1 1: ChIP-seq | NCBI GEO | GEO: GSM1655740 |
| Human Proerythroblast 1: RNA-seq | NCBI GEO | GEO: GSM1304777 |
| Human Proerythroblast 2: RNA-seq | NCBI GEO | GEO: GSM1304778 |
| Human Proerythroblast 3: RNA-seq | NCBI GEO | GEO: GSM1304779 |
| Human Early Basophilic 1: RNA-seq | NCBI GEO | GEO: GSM1304780 |
| Human Early Basophilic 2: RNA-seq | NCBI GEO | GEO: GSM1304781 |
| Human Early Basophilic 3: RNA-seq | NCBI GEO | GEO: GSM1304782 |
| Human Late Basophilic 1: RNA-seq | NCBI GEO | GEO: GSM1304783 |
| Human Late Basophilic 2: RNA-seq | NCBI GEO | GEO: GSM1304784 |
| Human Late Basophilic 3: RNA-seq | NCBI GEO | GEO: GSM1304785 |
| Human Polychromatic 1: RNA-seq | NCBI GEO | GEO: GSM1304786 |
| Human Polychromatic 2: RNA-seq | NCBI GEO | GEO: GSM1304787 |
| Human Polychromatic 3: RNA-seq | NCBI GEO | GEO: GSM1304788 |
| Human Orthochromatic 1: RNA-seq | NCBI GEO | GEO: GSM1304789 |
| Human Orthochromatic 2: RNA-seq | NCBI GEO | GEO: GSM1304790 |
| Human Orthochromatic 3: RNA-seq | NCBI GEO | GEO: GSM1304791 |
| Oligonucleotides | ||
| pGATcGGAAGAGcGGTTcAGcAGGAATGccGAG | Gu et al., 2011 | PE1 |
| AcAcTcTTTcccTAcAcGAcGcTcTTccGATcxT | Gu et al., 2011 | PE2 |
| AATGATACGGCGACCACCGAGATCTACACTTTCCCTACACGACGCTCTTCCGATCxT | Gu et al., 2011 | PE1.0 |
| CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCxT | Gu et al., 2011 | PE 2.0 |
| Software and Algorithms | ||
| EdgeR | Robinson et al., 2010 | https://bioconductor.org/packages/release/bioc/html/edgeR.html, RRID:SCR_012802 |
| Trimmomatic version 0.32 | Bolger et al., 2014 | http://www.usadellab.org/cms/?page=trimmomatic, RRID:SCR_011848 |
| Goseq | Young et al., 2010 | https://bioconductor.org/packages/release/bioc/html/goseq.html, RRID:SCR_017052 |
| Burrows-Wheeler Aligner version 0.7.9a | Li and Durbin, 2010 | http://bio-bwa.sourceforge.net/, RRID:SCR_010910 |
| MACS2 program version 2.1.1.20160309 | Zhang et al., 2008 | http://liulab.dfci.harvard.edu/MACS/ |
| DiffBind version 2.4.8 | Ross-Innes et al., 2012 | http://bioconductor.org/packages/release/bioc/vignettes/DiffBind/inst/doc/DiffBind.pdf: SCR_012918 |
| GenomicRanges | Lawrence et al., 2013 | https://bioconductor.org/packages/release/bioc/html/GenomicRanges.html, RRID:SCR_000025 |
| Chipseeker version 1.12.1 | Yu et al., 2015 | https://bioconductor.org/packages/release/bioc/html/ChIPseeker.html |
| GREAT | McLean et al., 2010 | http://great.stanford.edu/public/html/, RRID:SCR_005807 |
| HOMER | Heinz et al., 2010 | http://homer.ucsd.edu/homer/index.html, RRID:SCR_010881 |
| PhastCons | King et al., 2005 | https://www.rdocumentation.org/packages/rphast/versions/1.6.9/topics/phastCons |
| Trim Galore v0.4.0 | Babraham Bioinformatics | http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/, RRID:SCR_011847 |
| Bismark | Krueger and Andrews, 2011 | www.bioinformatics.bbsrc.ac.uk/projects/bismark/, RRID:SCR_005604 |
| MethylKit | Akalin et al., 2012b | https://bioconductor.riken.jp/packages/3.5/bioc/vignettes/methylKit/inst/doc/methylKit.html |
| PLINK | Chang et al., 2015 | https://www.cog-genomics.org/plink2/, RRID:SCR_005177 |
Highlights.
Epigenomic landscape of erythropoiesis reveals stage-specific patterns of regulation
Epigenomic changes in erythropoiesis are linked to erythroid traits and disease genes
Erythroid cells exhibit chromatin accessibility patterns distinct from other cell types
ACKNOWLEDGMENTS
This study was supported in part by grants R01AR068994, UM1HG009409, R01DK104046, and P01DK032094 from the NIH. We would like to thank the Yale Center for Genome Analysis and the Epigenomics Core Facility at Weill Cornell Medicine, as well as Jackie Knobelsdorf, Carson McCann, and Siri Palreddy for manuscript assistance.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.08.020.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abraham BJ, Cui K, Tang Q, and Zhao K (2013). Dynamic regulation of epigenomic landscapes during hematopoiesis. BMC Genomics 14, 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akalin A, Garrett-Bakelman FE, Kormaksson M, Busuttil J, Zhang L, Khrebtukova I, Milne TA, Huang Y, Biswas D, Hess JL, et al. (2012a). Base-pair resolution DNA methylation sequencing reveals profoundly divergent epigenetic landscapes in acute myeloid leukemia. PLoS Genet. 8, e1002781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akalin A, Kormaksson M, Li S, Garrett-Bakelman FE, Figueroa ME, Melnick A, and Mason CE (2012b). methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 13, R87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An X, Schulz VP, Li J, Wu K, Liu J, Xue F, Hu J, Mohandas N, and Gallagher PG (2014). Global transcriptome analyses of human and murine terminal erythroid differentiation. Blood 123, 3466–3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong DL, Zidovetzki R, Alarcón-Riquelme ME, Tsao BP, Criswell LA, Kimberly RP, Harley JB, Sivils KL, Vyse TJ, Gaffney PM, et al. (2014). GWAS identifies novel SLE susceptibility genes and explains the association of the HLA region. Genes Immun. 15, 347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao X, Rubin AJ, Qu K, Zhang J, Giresi PG, Chang HY, and Khavari PA (2015). A novel ATAC-seq approach reveals lineage-specific reinforcement of the open chromatin landscape via cooperation between BAF and p63. Genome Biol. 16, 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, and Zhao K (2009). Genomic location analysis by ChIP-Seq. J. Cell. Biochem. 107, 11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartholdy B, Lajugie J, Yan Z, Zhang S, Mukhopadhyay R, Greally JM, Suzuki M, and Bouhassira EE (2018). Mechanisms of establishment and functional significance of DNA demethylation during erythroid differentiation. Blood Adv. 2, 1833–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer DE, Kamran SC, and Orkin SH (2012). Reawakening fetal hemoglobin: prospects for new therapies for the β-globin disorders. Blood 120, 2945–2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer DE, Kamran SC, Lessard S, Xu J, Fujiwara Y, Lin C, Shao Z, Canver MC, Smith EC, Pinello L, et al. (2013). An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science 342, 253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi P, Fermo E, Glader B, Kanno H, Agarwal A, Barcellini W, Eber S, Hoyer JD, Kuter DJ, Maia TM, et al. (2019). Addressing the diagnostic gaps in pyruvate kinase deficiency: consensus recommendations on the diagnosis of pyruvate kinase deficiency. Am. J. Hematol. 94, 149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birney E, Lieb JD, Furey TS, Crawford GE, and Iyer VR (2010). Allele-specific and heritable chromatin signatures in humans. Hum. Mol. Genet. 19 (R2), R204–R209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blow MJ, McCulley DJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. (2010). ChIP-Seq identification of weakly conserved heart enhancers. Nat. Genet. 42, 806–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifer C, and Cockerill PN (2017). Chromatin priming of genes in development: concepts, mechanisms and consequences. Exp. Hematol. 49, 1–8. [DOI] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Wu B, Chang HY, and Greenleaf WJ (2015). ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol 109,21.29.1–21.29.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll TS, Liang Z, Salama R, Stark R, and de Santiago I (2014). Impact of artifact removal on ChIP quality metrics in ChIP-seq and ChIP-exo data. Front. Genet 5, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, and Lee JJ (2015). Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Liu J, Heck S, Chasis JA, An X, and Mohandas N (2009). Resolving the distinct stages in erythroid differentiation based on dynamic changes in membrane protein expression during erythropoiesis. Proc. Natl. Acad. Sci. USA 106, 17413–17418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, King DC, Doré LC, Zhang X, Zhou Y, Zhang Y, Dorman C, Abebe D, Kumar SA, Chiaromonte F, et al. (2008). Transcriptional enhancement by GATA1-occupied DNA segments is strongly associated with evolutionary constraint on the binding site motif. Genome Res. 18, 1896–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Wu W, Kumar SA, Yu D, Deng W, Tripic T, King DC, Chen KB, Zhang Y, Drautz D, et al. (2009). Erythroid GATA1 function revealed by genome-wide analysis of transcription factor occupancy, histone modifications, and mRNA expression. Genome Res. 19, 2172–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chlon TM, Dore LC, and Crispino JD (2012). Cofactor-mediated restriction of GATA-1 chromatin occupancy coordinates lineage-specific gene expression. Mol. Cell 47, 608–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choukrallah MA, Song S, Rolink AG, Burger L, and Matthias P (2015). Enhancer repertoires are reshaped independently of early priming and heterochromatin dynamics during B cell differentiation. Nat. Commun. 6, 8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Buenrostro JD, Wu B, Greenside PG, Chan SM, Koenig JL, Snyder MP, Pritchard JK, Kundaje A, Greenleaf WJ, et al. (2016). Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet. 48, 1193–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degner JF, Pai AA, Pique-Regi R, Veyrieras JB, Gaffney DJ, Pickrell JK, De Leon S, Michelini K, Lewellen N, Crawford GE, et al. (2012). DNase I sensitivity QTLs are a major determinant of human expression variation. Nature 482, 390–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doré LC, Chlon TM, Brown CD, White KP, and Crispino JD (2012). Chromatin occupancy analysis reveals genome-wide GATA factor switching during hematopoiesis. Blood 119, 3724–3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dover GJ, Chan T, and Sieber F (1983). Fetal hemoglobin production in cultures of primitive and mature human erythroid progenitors: differentiation affects the quantity of fetal hemoglobin produced per fetal-hemoglobin-containing cell. Blood 61, 1242–1246. [PubMed] [Google Scholar]
- Dzierzak E, and Philipsen S (2013). Erythropoiesis: development and differentiation. Cold Spring Harb. Perspect. Med 3, a011601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnham PJ (2009). Insights from genomic profiling of transcription factors. Nat. Rev. Genet 10, 605–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank CL, Liu F, Wijayatunge R, Song L, Biegler MT, Yang MG, Vockley CM, Safi A, Gersbach CA, Crawford GE, and West AE (2015). Regulation of chromatin accessibility and Zic binding at enhancers in the developing cerebellum. Nat. Neurosci 18, 647–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara T, O’Geen H, Keles S, Blahnik K, Linnemann AK, Kang YA, Choi K, Farnham PJ, and Bresnick EH (2009). Discovering hematopoietic mechanisms through genome-wide analysis of GATA factor chromatin occupancy. Mol. Cell 36, 667–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett-Bakelman FE, Sheridan CK, Kacmarczyk TJ, Ishii J, Betel D, Alonso A, Mason CE, Figueroa ME, and Melnick AM (2015). Enhanced reduced representation bisulfite sequencing for assessment of DNA methylation at base pair resolution. J. Vis. Exp (96), e52246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier EF, Ducamp S, Leduc M, Salnot V, Guillonneau F, Dussiot M, Hale J, Giarratana MC, Raimbault A, Douay L, et al. (2016). Comprehensive Proteomic Analysis of Human Erythropoiesis. Cell Rep. 16, 1470–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. (2004). Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5, R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glomski CA, and Tamburlin J (1990). The phylogenetic odyssey of the erythrocyte. II. The early or invertebrate prototypes. Histol. Histopathol. 5, 513–525. [PubMed] [Google Scholar]
- Glomski CA, Tamburlin J, and Chainani M (1992). The phylogenetic odyssey of the erythrocyte. III. Fish, the lower vertebrate experience. Histol. Histopathol 7, 501–528. [PubMed] [Google Scholar]
- Glomski CA, Tamburlin J, Hard R, and Chainani M (1997). The phylogenetic odyssey of the erythrocyte. IV. The amphibians. Histol. Histopathol. 12, 147–170. [PubMed] [Google Scholar]
- Gu H, Smith ZD, Bock C, Boyle P, Gnirke A, and Meissner A (2011). Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat. Protoc. 6, 468–481. [DOI] [PubMed] [Google Scholar]
- Hartley SW, and Mullikin JC (2015). QoRTs: a comprehensive toolset for quality control and data processing of RNA-Seq experiments. BMC Bioinformatics 16, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattangadi SM, Wong P, Zhang L, Flygare J, and Lodish HF (2011). From stem cell to red cell: regulation of erythropoiesis at multiple levels by multiple proteins, RNAs, and chromatin modifications. Blood 118, 6258–6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, et al. (2007). Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 39, 311–318. [DOI] [PubMed] [Google Scholar]
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. (2009). Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineagedetermining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuston EF, Keller CA, Lichtenberg J, Giardine B, Anderson SM, Hardison RC, and Bodine DM; NIH Intramural Sequencing Center (2018). Establishment of regulatory elements during erythro-megakaryopoiesis identifies hematopoietic lineage-commitment points. Epigenetics Chromatin 11, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Liu J, Xue F, Halverson G, Reid M, Guo A, Chen L, Raza A, Galili N, Jaffray J, et al. (2013). Isolation and functional characterization of human erythroblasts at distinct stages: implications for understanding of normal and disordered erythropoiesis in vivo. Blood 121, 3246–3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iolascon A, Russo R, Esposito MR, Asci R, Piscopo C, Perrotta S, Fénéant-Thibault M, Garcon L, and Delaunay J (2010). Molecular analysis of 42 patients with congenital dyserythropoietic anemia type II: new mutations in the SEC23B gene and a search for a genotype-phenotype relationship. Haematologica 95, 708–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadauke S, and Blobel GA (2009). Chromatin loops in gene regulation. Biochim. Biophys. Acta 1789, 17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadauke S, Udugama MI, Pawlicki JM, Achtman JC, Jain DP, Cheng Y, Hardison RC, and Blobel GA (2012). Tissue-specific mitotic bookmarking by hematopoietic transcription factor GATA1. Cell 150, 725–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassouf MT, Hughes JR, Taylor S, McGowan SJ, Soneji S, Green AL, Vyas P, and Porcher C (2010). Genome-wide identification of TAL1’s functional targets: insights into its mechanisms of action in primary erythroid cells. Genome Res. 20, 1064–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellis M, Wold B, Snyder MP, Bernstein BE, Kundaje A, Marinov GK, Ward LD, Birney E, Crawford GE, Dekker J, et al. (2014). Defining functional DNA elements in the human genome. Proc. Natl. Acad. Sci. USA 111, 6131–6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TH, Abdullaev ZK, Smith AD, Ching KA, Loukinov DI, Green RD, Zhang MQ, Lobanenkov VV, and Ren B (2007). Analysis of the verte-brate insulator protein CTCF-binding sites in the human genome. Cell 128, 1231–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TH, Li F, Ferreiro-Neira I, Ho LL, Luyten A, Nalapareddy K, Long H, Verzi M, and Shivdasani RA (2014). Broadly permissive intestinal chromatin underlies lateral inhibition and cell plasticity. Nature 506, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King DC, Taylor J, Elnitski L, Chiaromonte F, Miller W, and Hardison RC (2005). Evaluation of regulatory potential and conservation scores for detecting cis-regulatory modules in aligned mammalian genome sequences. Genome Res. 15, 1051–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh PW, Sinha R, Barkal AA, Morganti RM, Chen A, Weissman IL, Ang LT, Kundaje A, and Loh KM (2016). An atlas of transcriptional, chromatin accessibility, and surface marker changes in human mesoderm development. Sci. Data 3, 160109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F, and Andrews SR (2011). Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27, 1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumasaka N, Knights AJ, and Gaffney DJ (2016). Fine-mapping cellular QTLs with RASQUAL and ATAC-seq. Nat. Genet. 48, 206–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara-Astiaso D, Weiner A, Lorenzo-Vivas E, Zaretsky I, Jaitin DA, David E, Keren-Shaul H, Mildner A, Winter D, Jung S, et al. (2014). Immunogenetics. Chromatin state dynamics during blood formation. Science 345, 943–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, and Carey VJ (2013). Software for computing and annotating genomic ranges. PLoS Comput. Biol. 9, e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenfant C, and Johansen K (1972). Gas exchange in gill, skin, and lung breathing. Respir. Physiol. 14, 211–218. [DOI] [PubMed] [Google Scholar]
- Lessard S, Beaudoin M, Benkirane K, and Lettre G (2015). Comparison of DNA methylation profiles in human fetal and adult red blood cell progenitors. Genome Med. 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Peterson KR, Fang X, and Stamatoyannopoulos G (2002). Locus control regions. Blood 100, 3077–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Hale J, Bhagia P, Xue F, Chen L, Jaffray J, Yan H, Lane J, Gallagher PG, Mohandas N, et al. (2014). Isolation and transcriptome analyses of human erythroid progenitors: BFU-E and CFU-E. Blood 124, 3636–3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Schulz MH, Look T, Begemann M, Zenke M, and Costa IG (2019). Identification of transcription factor binding sites using ATAC-seq. Genome Biol. 20, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang S, Moghimi B, Yang TP, Strouboulis J, and Bungert J (2008). Locus control region mediated regulation of adult beta-globin gene expression. J. Cell. Biochem. 105, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30,923–930. [DOI] [PubMed] [Google Scholar]
- Lloyd JA (2018). An Introduction to Erythropoiesis Approaches. Methods Mol. Biol. 1698, 1–10. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luyten A, Zang C, Liu XS, and Shivdasani RA (2014). Active enhancers are delineated de novo during hematopoiesis, with limited lineage fidelity among specified primary blood cells. Genes Dev. 28, 1827–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, Hastings E, Junkins H, McMahon A, Milano A, Morales J, et al. (2017). The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 45 (D1), D896–D901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, and Bejerano G (2010). GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyai T, Takano J, Endo TA, Kawakami E, Agata Y, Motomura Y, Kubo M, Kashima Y, Suzuki Y, Kawamoto H, and Ikawa T (2018). Three-step transcriptional priming that drives the commitment of multipotent progenitors toward B cells. Genes Dev. 32, 112–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neph S, Stergachis AB, Reynolds A, Sandstrom R, Borenstein E, and Stamatoyannopoulos JA (2012). Circuitry and dynamics of human transcription factor regulatory networks. Cell 150, 1274–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikinmaa M (1997). Oxygen and carbon dioxide transport in vertebrate erythrocytes: an evolutionary change in the role of membrane transport. J. Exp. Biol. 200, 369–380. [DOI] [PubMed] [Google Scholar]
- Nord AS, Blow MJ, Attanasio C, Akiyama JA, Holt A, Hosseini R, Phouanenavong S, Plajzer-Frick I, Shoukry M, Afzal V, et al. (2013). Rapid and pervasive changes in genome-wide enhancer usage during mammalian development. Cell 155, 1521–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palii CG, Perez-Iratxeta C, Yao Z, Cao Y, Dai F, Davison J, Atkins H, Allan D, Dilworth FJ, Gentleman R, et al. (2011). Differential genomic targeting of the transcription factor TAL1 in alternate haematopoietic lineages. EMBO J. 30, 494–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park PJ (2009). ChIP-seq: advantages and challenges of a maturing technology. Nat. Rev. Genet. 10, 669–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philpott CC (2018). The flux of iron through ferritin in erythrocyte development. Curr. Opin. Hematol. 25, 183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilon AM, Ajay SS, Kumar SA, Steiner LA, Cherukuri PF, Wincovitch S, Anderson SM, Mullikin JC, Gallagher PG, Hardison RC, et al. ; NISC Comparative Sequencing Center (2011). Genome-wide ChIP-Seq reveals a dramatic shift in the binding of the transcription factor erythroid Kruppel-like factor during erythrocyte differentiation. Blood 118, e139–e148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu K, Zaba LC, Giresi PG, Li R, Longmire M, Kim YH, Greenleaf WJ, and Chang HY (2015). Individuality and variation of personal regulomes in primary human T cells. Cell Syst. 1, 51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, and Oshlack A (2010). A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, et al. (2012). Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo R, Esposito MR, Asci R, Gambale A, Perrotta S, Ramenghi U, Forni GL, Uygun V, Delaunay J, and Iolascon A (2010). Mutational spectrum in congenital dyserythropoietic anemia type II: identification of 19 novel variants in SEC23B gene. Am. J. Hematol. 85, 915–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaran VG, Xu J, Byron R, Greisman HA, Fisher C, Weatherall DJ, Sabath DE, Groudine M, Orkin SH, Premawardhena A, and Bender MA (2011). A functional element necessary for fetal hemoglobin silencing. N.Engl. J. Med. 365, 807–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt D, Wilson MD, Ballester B, Schwalie PC, Brown GD, Marshall A, Kutter C, Watt S, Martinez-Jimenez CP, Mackay S, et al. (2010). Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science 328, 1036–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott GR (2015). Early insights into the evolution of respiratory and cardiovascular physiology in vertebrates. J. Exp. Biol. 218, 2818–2820. [DOI] [PubMed] [Google Scholar]
- Scott GR, and Milsom WK (2006). Flying high: a theoretical analysis of the factors limiting exercise performance in birds at altitude. Respir. Physiol. Neurobiol. 154, 284–301. [DOI] [PubMed] [Google Scholar]
- Shearstone JR, Pop R, Bock C, Boyle P, Meissner A, and Socolovsky M (2011). Global DNA demethylation during mouse erythropoiesis in vivo. Science 334, 799–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield NC, Thurman RE, Song L, Safi A, Stamatoyannopoulos JA, Lenhard B, Crawford GE, and Furey TS (2013). Patterns of regulatory activity across diverse human cell types predict tissue identity, transcription factor binding, and long-range interactions. Genome Res. 23, 777–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, and Haussler D (2004). Combining phylogenetic and hidden Markov models in biosequence analysis. J. Comput. Biol. 11, 413–428. [DOI] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. (2005). Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15, 1034–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Zhang Z, Grasfeder LL, Boyle AP, Giresi PG, Lee BK, Sheffield NC, Graf S, Huss M, Keefe D, et al. (2011). Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res. 21, 1757–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadhouders R, Aktuna S, Thongjuea S, Aghajanirefah A, Pourfarzad F, van Ijcken W, Lenhard B, Rooks H, Best S, Menzel S, et al. (2014). HBS1L-MYB intergenic variants modulate fetal hemoglobin via long-range MYB enhancers. J. Clin. Invest. 124, 1699–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner LA, Maksimova Y, Schulz V, Wong C, Raha D, Mahajan MC, Weissman SM, and Gallagher PG (2009). Chromatin architecture and transcription factor binding regulate expression of erythrocyte membrane protein genes. Mol. Cell. Biol. 29, 5399–5412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner LA, Schulz V, Makismova Y, Lezon-Geyda K, and Gallagher PG (2016). CTCF and CohesinSA-1 Mark Active Promoters and Boundaries of Repressive Chromatin Domains in Primary Human Erythroid Cells. PLoS One 11, e0155378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stergachis AB, Neph S, Reynolds A, Humbert R, Miller B, Paige SL, Vernot B, Cheng JB, Thurman RE, Sandstrom R, et al. (2013). Developmental fate and cellular maturity encoded in human regulatory DNA landscapes. Cell 154, 888–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su MY, Steiner LA, Bogardus H, Mishra T, Schulz VP, Hardison RC, and Gallagher PG (2013). Identification of biologically relevant enhancers in human erythroid cells. J. Biol. Chem. 288, 8433–8444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallack MR, Whitington T, Yuen WS, Wainwright EN, Keys JR, Gardiner BB, Nourbakhsh E, Cloonan N, Grimmond SM, Bailey TL, and Perkins AC (2010). A global role for KLF1 in erythropoiesis revealed by ChIP-seq in primary erythroid cells. Genome Res. 20, 1052–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teif VB, Vainshtein Y, Caudron-Herger M, Mallm JP, Marth C, Höfer T, and Rippe K (2012). Genome-wide nucleosome positioning during embryonic stem cell development. Nat. Struct. Mol. Biol. 19, 1185–1192. [DOI] [PubMed] [Google Scholar]
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. (2012). The accessible chromatin landscape of the human genome. Nature 489, 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsiftsoglou AS, Vizirianakis IS, and Strouboulis J (2009). Erythropoiesis: model systems, molecular regulators, and developmental programs. IUBMB Life 61, 800–830. [DOI] [PubMed] [Google Scholar]
- Ulirsch JC, Lacy JN, An X, Mohandas N, Mikkelsen TS, and Sankaran VG (2014). Altered chromatin occupancy of master regulators underlies evolutionary divergence in the transcriptional landscape of erythroid differentiation. PLoS Genet. 10, e1004890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varley KE, Gertz J, Bowling KM, Parker SL, Reddy TE, Pauli-Behn F, Cross MK, Williams BA, Stamatoyannopoulos JA, Crawford GE, et al. (2013). Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 23, 555–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Rubin EM, and Pennacchio LA (2009). Genomic views of distantacting enhancers. Nature 461, 199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JA, Cook A, Alver BH, Stadtfeld M, Deaton AM, Hochedlinger K, Park PJ, Tolstorukov MY, and Kingston RE (2014). Nucleosomal occupancy changes locally over key regulatory regions during cell differentiation and reprogramming. Nat. Commun. 5, 4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson NK, Miranda-Saavedra D, Kinston S, Bonadies N, Foster SD, Calero-Nieto F, Dawson MA, Donaldson IJ, Dumon S, Frampton J, et al. (2009). The transcriptional program controlled by the stem cell leukemia gene Scl/Tal1 during early embryonic hematopoietic development. Blood 113, 5456–5465. [DOI] [PubMed] [Google Scholar]
- Wong P, Hattangadi SM, Cheng AW, Frampton GM, Young RA, and Lodish HF (2011). Gene induction and repression during terminal erythropoiesis are mediated by distinct epigenetic changes. Blood 118, e128–e138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi H, Shulha HP, Lin JM, Vales TR, Fu Y, Bodine DM, McKay RD, Chenoweth JG, Tesar PJ, Furey TS, et al. (2007). Identification and characterization of cell type-specific and ubiquitous chromatin regulatory structures in the human genome. PLoS Genet. 3, e136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Shao Z, Glass K, Bauer DE, Pinello L, Van Handel B, Hou S, Stamatoyannopoulos JA, Mikkola HK, Yuan GC, and Orkin SH (2012). Combinatorial assembly of developmental stage-specific enhancers controls gene expression programs during human erythropoiesis. Dev. Cell 23, 796–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Shao Z, Li D, Xie H, Kim W, Huang J, Taylor JE, Pinello L, Glass K, Jaffe JD, et al. (2015). Developmental control of polycomb subunit composition by GATA factors mediates a switch to non-canonical functions. Mol. Cell 57, 304–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Carter AC, Gendrel AV, Attia M, Loftus J, Greenleaf WJ, Tibshirani R, Heard E, and Chang HY (2017). Landscape of monoallelic DNA accessibility in mouse embryonic stem cells and neural progenitor cells. Nat. Genet. 49, 377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young MD, Wakefield MJ, Smyth GK, and Oshlack A (2010). Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 11, R14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, and He QY (2015). ChIPseeker: an R/Bioconductor pack-age for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383. [DOI] [PubMed] [Google Scholar]
- Yu M, Riva L, Xie H, Schindler Y, Moran TB, Cheng Y, Yu D, Hardison R, Weiss MJ, Orkin SH, et al. (2009). Insights into GATA-1-mediated gene activation versus repression via genome-wide chromatin occupancy analysis. Mol. Cell 36, 682–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Mo Y, Ebenezer D, Bhattacharyya S, Liu H, Sundaravel S, Giricz O, Wontakal S, Cartier J, Caces B, et al. (2013). High resolution methylome analysis reveals widespread functional hypomethylation during adult human erythropoiesis. J. Biol. Chem. 288, 8805–8814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziller MJ, Gu H, Müller F, Donaghey J, Tsai LT, Kohlbacher O, De Jager PL, Rosen ED, Bennett DA, Bernstein BE, et al. (2013). Charting a dynamic DNA methylation landscape of the human genome. Nature 500, 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data files have been submitted to the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) and the VISION databases (http://www.bx.psu.edu/~giardine/vision/). The accession number for the RNA-seq, ATAC-seq and methylation analyses reported in this paper is GEO: GSE128269.