

Abstract
Background
Sickle cell disease (SCD) is a group of inherited disorders of haemoglobin (Hb) structure in a person who has inherited two mutant globin genes (one from each parent), at least one of which is always the sickle mutation. It is estimated that between 5% and 7% of the world's population are carriers of the mutant Hb gene, and SCD is the most commonly inherited blood disorder.
SCD is characterized by distorted sickle‐shaped red blood cells. Manifestations of the disease are attributed to either haemolysis (premature red cell destruction) or vaso‐occlusion (obstruction of blood flow, the most common manifestation). Shortened lifespans are attributable to serious comorbidities associated with the disease, including renal failure, acute cholecystitis, pulmonary hypertension, aplastic crisis, pulmonary embolus, stroke, acute chest syndrome, and sepsis.
Vaso‐occlusion can lead to an acute, painful crisis (sickle cell crisis, vaso‐occlusive crisis (VOC) or vaso‐occlusive episode). Pain is most often reported in the joints, extremities, back or chest, but it can occur anywhere and can last for several days or weeks. The bone and muscle pain experienced during a sickle cell crisis is both acute and recurrent.
Key pharmacological treatments for VOC include opioid analgesics, non‐opioid analgesics, and combinations of drugs. Non‐pharmacological approaches, such as relaxation, hypnosis, heat, ice and acupuncture, have been used in conjunction to rehydrating the patient and reduce the sickling process.
Objectives
To assess the analgesic efficacy and adverse events of pharmacological interventions to treat acute painful sickle cell vaso‐occlusive crises in adults, in any setting.
Search methods
We searched the Cochrane Central Register of Controlled Trials (CENTRAL) via the Cochrane Register of Studies Online, MEDLINE via Ovid, Embase via Ovid and LILACS, from inception to September 2019. We also searched the reference lists of retrieved studies and reviews, and searched online clinical trial registries.
Selection criteria
Randomized, controlled, double‐blind trials of pharmacological interventions, of any dose and by any route, compared to placebo or any active comparator, for the treatment (not prevention) of painful sickle cell VOC in adults.
Data collection and analysis
Three review authors independently assessed studies for eligibility. We planned to use dichotomous data to calculate risk ratio (RR) and number needed to treat for one additional event, using standard methods. Our primary outcomes were participant‐reported pain relief of 50%, or 30%, or greater; Patient Global Impression of Change (PGIC) very much improved, or much or very much improved. Our secondary outcomes included adverse events, serious adverse events, and withdrawals due to adverse events. We assessed GRADE and created three 'Summary of findings' tables.
Main results
We included nine studies with data for 638 VOC events and 594 participants aged 17 to 42 years with SCD presenting to a hospital emergency department in a painful VOC. Three studies investigated a non‐steroidal anti‐inflammatory drug (NSAID) compared to placebo. One study compared an opioid with a placebo, two studies compared an opioid with an active comparator, two studies compared an anticoagulant with a placebo, and one study compared a combination of three drugs with a combination of four drugs.
Risk of bias across the nine studies varied. Studies were primarily at an unclear risk of selection, performance, and detection bias. Studies were primarily at a high risk of bias for size with fewer than 50 participants per treatment arm; two studies had 50 to 199 participants per treatment arm (unclear risk).
Non‐steroidal anti‐inflammatory drugs (NSAID) compared with placebo
No data were reported regarding participant‐reported pain relief of 50% or 30% or greater.
The efficacy was uncertain regarding PGIC very much improved, and PGIC much or very much improved (no difference; 1 study, 21 participants; very low‐quality evidence).
Very low‐quality, uncertain results suggested similar rates of adverse events across both the NSAIDs group (16/45 adverse events, 1/56 serious adverse events, and 1/56 withdrawal due to adverse events) and the placebo group (19/45 adverse events, 2/56 serious adverse events, and 1/56 withdrawal due to adverse events).
Opioids compared with placebo
No data were reported regarding participant‐reported pain relief of 50% or 30%, PGIC, or adverse events (any adverse event, serious adverse events, and withdrawals due to adverse events).
Opioids compared with active comparator
No data were reported regarding participant‐reported pain relief of 50% or 30% or greater.
The results were uncertain regarding PGIC very much improved (33% of the opioids group versus 19% of the placebo group). No data were reported regarding PGIC much or very much improved.
Very low‐quality, uncertain results suggested similar rates of adverse events across both the opioids group (9/66 adverse events, and 0/66 serious adverse events) and the placebo group (7/64 adverse events, 0/66 serious adverse events). No data were reported regarding withdrawal due to adverse events.
Quality of the evidence
We downgraded the quality of the evidence by three levels to very low‐quality because there are too few data to have confidence in results (e.g. too few participants per treatment arm). Where no data were reported for an outcome, we had no evidence to support or refute (quality of the evidence is unknown).
Authors' conclusions
This review identified only nine studies, with insufficient data for all pharmacological interventions for analysis.
The available evidence is very uncertain regarding the efficacy or harm from pharmacological interventions used to treat pain related to sickle cell VOC in adults. This area could benefit most from more high quality, certain evidence, as well as the establishment of suitable registries which record interventions and outcomes for this group of people.
Plain language summary
Medicines for treating painful sickle cell crises in adults
Bottom line
We are uncertain which medicines provide the best pain relief for adults experiencing a painful sickle cell crisis.
Background
People with sickle cell disease have abnormally shaped red cells in their blood. Sickle cell disease is the most common inherited blood disorder around the world. It is estimated globally that 367 million to 500 million people are carriers. People with sickle cell disease have a higher chance of life‐threatening complications, such as infection, severe chest pain and stroke in early life, and kidney or liver damage in adulthood.
A pain crisis is the most common problem of sickle cell disease and can require several treatments at once, usually in an emergency situation. The first priority is to control the pain, using medicines (such as opioids, non‐steroidal anti‐inflammatories, paracetamol, and blood thinners) or relaxation, hypnosis, heat, ice, or acupuncture.
Study characteristics
In September 2019, we searched for clinical trials that used medicines in any setting to treat painful sickle cell crises. We found nine trials, with 594 adults (aged 17 to 42 years) who had sickle cell disease, experiencing a combined total of 638 painful episodes.
Key results
The studies looked at different comparisons of the medicines butorphanol, cetiedil, fentanyl, ketoprofen, ketorolac, metoclopramide, morphine, paracetamol, placebo, tinzaparin, and tramadol. Only three studies compared the same two drugs (non‐steroidal anti‐inflammatory drugs such as ibuprofen, aspirin, or naproxen, with a placebo (pretend treatment)) and we had very limited data to be able to investigate the effects on pain scores from these medicines.
Side effects were rare and were generally mild.
Quality of the evidence
We rated the quality of the evidence from studies using four levels: very low, low, moderate, or high. Very low‐quality evidence means that we are very uncertain about the results. High‐quality evidence means that we are very confident in the results. For pain relief and side effects, we rated the quality of evidence as very low.
We downgraded the quality of the evidence to very low because there were not enough data (e.g. too few participants). For some outcomes the quality of the evidence is unknown because there was no evidence available.
Summary of findings
Background
A previous review, entitled 'Pain management for sickle cell disease in children and adults' was withdrawn as it was out of date (Dunlop 2014). This is a completely new review focusing only on the treatment of painful vaso‐occlusive crises (VOC; or episodes) of sickle cell disease and it excludes the paediatric population.
Description of the condition
Sickle cell disease is a generic term for a group of inherited disorders of haemoglobin (Hb) structure in which the affected person inherits two mutant globin genes (one from each parent), at least one of which is always the sickle mutation (WHO 2010). The latter results from a single nucleotide change (GAT→GTT) in the sixth codon of exon 1 of the β‐globin gene responsible for the synthesis of the β‐globin chain. The resulting replacement of the normal glutamic acid by valine at position 6 in the β chain leads to the formation of sickle Hb (Hb S). Sickle cell anaemia is the homozygous state, in which the sickle gene is inherited from both parents. Other sickle cell syndromes result from the coinheritance of the sickle gene and a non‐sickle gene, such as in Hb C, Hb OArab, Hb D, β+‐thalassaemia or β0‐thalassemia (NIH 2014).
JB Herrick first described the abnormally shaped red blood cell in 1910 (Herrick 1910). Sickle cell originated in Sub‐Saharan Africa and the Indian subcontinent (Martí‐Carvajal 2009; Stuart 2004; Weatherall 2006), and has persisted as a recessive trait due to the survival advantage against malaria experienced by people with the heterozygote form (Weatherall 2001). In recent years, the United Nations and the World Health Organization have recognized sickle cell disease as a global public health problem due to population mobility, with increased migration from high‐ to low‐frequency sickle Hb areas (WHO 2010). Combined with the global increase in population size, reduction in child mortality and improved adult survival, the worldwide burden of sickle cell disease is predicted to increase (Al Hajeri 2007; Martí‐Carvajal 2009; Piel 2013).
Sickle cell disease is the most common inherited blood disorder globally. Epidemiologically, it is estimated that between 5% (approximately 367 million people) (WHO 2011) and 7% (approximately 500 million people) (Weatherall 2001) of the world's population are carriers of the mutant Hb gene.
Worldwide, the African region has the highest prevalence with an estimated 200,000 babies born with sickle cell disease every year (Diallo 2002). In 2010, approximately 75% of the globally estimated homozygous sickle cell disease newborns were from Sub‐Saharan Africa (Piel 2013). In the US, the number of people living with sickle cell disease is approximately 100,000 and it predominantly affects Americans of African descent (NIH 2015). In addition to this, 3.5 million African‐Americans have the sickle cell trait HbAS genotype (i.e. are heterozygote carriers of sickle Hb) (Yawn 2014). The UK reports an estimated prevalence of 12,500 people with the disease (National Screening Committee 2006).
Shortened lifespans are attributable to serious comorbidities associated with the disease. People with sickle cell disease have a higher chance of life‐threatening complications, such as sepsis, acute chest syndrome and stroke in early life, and end organ damage in adulthood.(Tanabe 2012). For example, in the 1990s, African‐Americans with sickle cell disease had a life expectancy of less than 50 years, compared with the life expectancy of African‐Americans without sickle cell disease of more than 70 years (Platt 1994).
Sickle cell disease is characterized by the presence of distorted sickle‐shaped red blood cells in the bloodstream. These sickle‐shaped cells arise as a result of the "polymerization (gelling of the molecules) of the abnormal Hb in the red blood cells when they release their combined oxygen" (Al Hajeri 2007). Many manifestations of the disease are attributed to either haemolysis (premature red cell destruction) or vaso‐occlusion (obstruction of blood flow), which is the most common manifestation (Al Hajeri 2007). Red cell dehydration contributes to vaso‐occlusion: dehydrated red cells adhere to vascular endothelium, which results in blockage of blood flow (Lutz 2015). Other severe outcomes of the manifestations of sickle cell disease can include acute chest syndrome (chest crisis), priapism, acute cholecystitis, acute stroke and aplastic crisis (NIH 2014).
Vaso‐occlusion can lead to acute, painful crisis. This is also known as a sickle cell crisis, VOC or vaso‐occlusive episode. Pain is most often reported in the joints, extremities, back or chest, but it can occur anywhere and can last for several days or weeks. The frequency and severity of the painful episodes vary widely within and between individuals. The peak incidence is in late adolescence and early adulthood (Ballas 2007). The bone and muscle pain experienced during a sickle cell crisis is both acute (sudden onset of severe intensity) and recurrent (reoccurring unpredictably and intermittently over long periods while fluctuating in frequency and quality) (Serjeant 1994; Thienhaus 2002).
This review focused on the vaso‐occlusive manifestation of the disease, which causes the acute and painful crises (episodes), VOC. It excluded the other possible outcomes in people with sickle cell disease.
Description of the intervention
A VOC can require several interventions, depending on the person and situation. Treatment is primarily symptomatic and aims to stabilize pain. As well as pharmacological agents, non‐pharmacological approaches, such as relaxation, hypnosis, heat, ice and acupuncture, have been used to rehydrate the person and reduce the sickling process (Ballas 2005; Ballas 2007; De Ceulaer 1982; Gaston 1986; Lane 2001; Okomo 2015).
In this review, we assessed pharmacological interventions used to treat VOC in adults living with sickle cell disease. These included (but were not limited to) opioid and non‐opioid analgesics, as well as partial agonists, mixed agonists–antagonists, antagonists and adjuvants (Ballas 2007).
In context, a VOC is the hallmark of sickle cell disease and is the most common cause of hospitalization (up to 90%) (Ballas 2005; Dampier 2013). Therefore, treatment for adults with VOC is usually on presentation to an emergency healthcare facility, requiring immediate treatment for acute and debilitating pain. In addition, some people with sickle cell disease often self medicate at home with pharmacological agents. In both settings, primary management should focus on rapid pain control, whereby fluids and analgesics should be administered immediately (Gillis 2012).
VOC can be treated with various drugs (described below). As with all pharmacotherapies, the risk of adverse effects can be minimized by prescribing the lowest dose for the shortest duration possible to control the symptoms (NICE 2015).
For all analgesia, the person's respiratory rate, pain and sedation are assessed at 20‐minute intervals until pain control is reached, then monitored with analgesia readministered every four hours (NICE 2015; Rees 2003). Patient‐controlled administration (PCA) is an option used in adolescents transitioning into adult care (Telfer 2014).
Opioid analgesics
Opioid analgesics are recommended as the primary choice of pain relief in VOC management (NICE 2015). They include (but are not limited to): codeine, hydrocodone/paracetamol (hydrocodone/acetaminophen), hydrocodone/ibuprofen, oxycodone (and with codeine), morphine, hydromorphone, oxymorphone, methadone, diamorphine and fentanyl (Ballas 2007). Opioids are generally available in healthcare settings in most Western countries and are often delivered as intravenous (IV) morphine every four to six hours. In addition, oral opioids are prescribed as PCA after a VOC episode (NICE 2015).
Most analgesic opioids have a half‐life of two to four hours, with the exception of methadone, propoxyphene and meperidine, for which this can be one to several days (Trescot 2008). Although the standard dosing is generally every four to six hours, some people develop tolerance to opioids, thus requiring doses every two hours (Okpala 2002). Combination with diclofenac or paracetamol (which have different mechanisms of action) helps keep opioid use to a minimum (Okpala 2002).
The adverse effects of analgesic opioids include: (short‐term) respiratory depression, constipation, vomiting, nausea, pruritus and hives (Ballas 2007), addiction, and withdrawals (Rosenblum 2008).
Non‐opioid analgesics
Non‐opioid analgesics are the other drugs of choice for managing a VOC (NICE 2015). These include: paracetamol, non‐selective cyclo‐oxygenase (COX) inhibitors (aspirin (acetylsalicylic acid), ibuprofen, naproxen and ketorolac) and selective COX‐2 inhibitors (celecoxib) (Ballas 2007).
The most commonly used drugs from this family are non‐steroidal anti‐inflammatory drugs (NSAIDs), paracetamol, diclofenac and ketorolac. They are commonly used for mild‐to‐moderate pain either alone or in combination with opioid analgesia (Rees 2003).
NSAIDs are available in most countries. The adverse effects of NSAIDs include gastrointestinal complications and they should be administered with caution in people with a history of renal failure, bleeding tendencies, asthma or peptic ulcers (Rees 2003).
Paracetamol is also widely available in Western countries and is administered orally at a dosage of 200 mg to 500 mg every four to six hours until satisfactory pain relief has been achieved. The adverse effects of paracetamol are influenced by the dosage and duration of use and can include liver failure (Rees 2003).
Other pharmacotherapeutic drugs
Other pharmacotherapeutic drugs to be considered for analysis in this review include partial agonists (e.g. buprenorphine and buprenorphine/naloxone), mixed agonist‐antagonists (e.g. pentazocine, nalbuphine and butorphanol) and hydroxyurea (Ballas 2007). Finally, we will also consider any other pharmacotherapeutic drugs that we find, which are used to treat VOC.
How the intervention might work
Analgesics work in several ways to treat VOC, thus the key factor is whether the drugs provide relief of participant‐reported pain. Therefore, we were interested in gathering a comprehensive list of all pharmacotherapeutic treatments that have been trialled in a clinical setting to treat a new or recurrent painful crisis in people with sickle cell disease. We were also interested in combination drug therapies.
Why it is important to do this review
Globally, sickle cell disease is one of the four most common autosomal‐recessive disorders, along with thalassaemia, Tay‐Sachs disease and cystic fibrosis (Hussein 2015). In many countries, there has been an improvement in the survival rate of children with sickle cell disease who now live on into adulthood, thus increasing the adult population living with this condition (Quinn 2010; Sasongko 2013).
We are unaware of any recent or up‐to‐date systematic reviews that draw on comparisons of a full range of pharmacotherapies used specifically to treat VOC in adults. Therefore, this review aimed to address and analyse all available pharmacotherapies, including drug combinations.
This systematic review also addressed the issue of tolerance or habituation to opioids where this was reported in people with sickle cell disease (Tanabe 2012; Waldrop 1995). Regular analgesia should be given for acute pain when presenting in the emergency setting. Due to people developing a tolerance and requiring more frequent doses of opioids, it has been suggested that efforts should be made to prevent such tolerance developing in new patients because there is a limited choice of injectable opioids that can be used in acute painful episodes (Okpala 2002).
A systematic review of the current evidence on the reported benefits and harms is required to clarify the most effective pharmacotherapeutic options (and combinations) for effective pain relief interventions in adults with acute painful sickle cell VOC.
Objectives
To assess the analgesic efficacy and adverse events of pharmacological interventions to treat acute painful sickle cell vaso‐occlusive crises in adults, in any setting.
Methods
Criteria for considering studies for this review
Types of studies
We included randomized controlled trials (RCTs) with double‐blind assessment of participant outcomes following immediate treatment for acute painful sickle cell vaso‐occlusive episodes. We considered studies of parallel and crossover trial designs. We required full journal publication, with the exception of online clinical trial results, summaries of otherwise unpublished clinical trials and extended abstracts of otherwise unpublished clinical trials if sufficient data could be analysed. We excluded studies that were non‐randomized, case reports and clinical observations.
Types of participants
Studies included adults, aged 18 years and above, diagnosed with sickle cell disease and who had an acute painful sickle cell VOC (new or recurring).
We anticipated that some studies would contain some participants below 18 years of age (such as adolescents mixed with adults over 18 years of age). In the protocol, we planned to extract the data on participants aged 18 years and above and to contact the study authors for the separate adult data if necessary. At review stage, we changed this approach to include the study and data on all its participants if less than 20% of the participants were aged 17 or under. If greater than 20% of the participants were aged 17 years or under, we planned to extract the data on the participants aged 18 years and above, and contact the authors of the studies for separate adult data if necessary, However, this did not occur.
Types of interventions
Any pharmacological intervention at any dose, by any route, administered for the relief of acute pain associated with a sickle cell vaso‐occlusive episode or event (new or recurring) and compared to placebo or any active comparator.
We included studies treating the crisis in any healthcare setting, as well as combination drug regimens. IN fluid replacement therapy is a primary step in both the National Institute of Health and Care Excellence (NICE) and National Institutes of Health (NIH) guidelines for managing a sickle cell crisis (NICE 2015; NIH 2014), and was, therefore, considered standard practice and not an active comparator for the purpose of this review. We excluded studies using agents to treat pain resulting from other causes and studies using agents to attempt to treat the cause of sickle cell disease, chronic pain or non‐painful symptoms of the disease.
Types of outcome measures
Studies had to report pain assessment as either a primary or secondary outcome to be eligible for this review.
We anticipated that studies would use a variety of outcome measures, with the majority of studies using standard subjective scales (numerical rating scale (NRS) or visual analogue scale (VAS)) for pain intensity or pain relief, or both (Dworkin 2008).
We used the following dichotomous measures of pain: at least 50% pain relief over baseline (substantial), at least 30% pain relief over baseline (moderate), very much improved on the Patient Global Impression of Change (PGIC) (substantial), and much or very much improved on the PGIC (moderate). These outcomes are different from those used in most earlier reviews (Wiffen 2005), and we recognize that continuous responses to chronic pain generally do not follow a normal (Gaussian) distribution. People with chronic pain desire high levels of pain relief, ideally more than 50% and ideally leading to no more than mild pain (Moore 2013; O'Brien 2010). For the purpose of this review, we considered it appropriate to adapt these measurements of chronic pain to acute painful episodes, as the outcomes used to measure the end points of chronic pain are the same outcomes used to measure end points of acute pain.
We included three 'Summary of findings' tables. The 'Summary of findings' tables included outcomes of at least 50% and at least 30% pain intensity reduction, PGIC, serious adverse events (which included death) and withdrawals due to adverse events. We used the GRADE approach to assess the quality of the evidence related to each of the key outcomes listed in here (Higgins 2011a), as appropriate (see Data synthesis).
Primary outcomes
Participant‐reported pain relief of 50% or greater at 6, 12, 24, 48 hours and at the end of treatment.
Participant‐reported pain relief of 30% or greater at 6, 12, 24, 48 hours and at the end of treatment.
Patient global impression of change (PGIC) very much improved.
Patient global impression of change (PGIC) much or very much improved.
Secondary outcomes
Opioid consumption.
Time to pain resolution.
Length of hospitalization.
Participants experiencing any adverse or serious adverse event. Serious adverse events typically include any untoward medical occurrence or effect that at any dose resulted in death, was life‐threatening, required hospitalization or prolongation of existing hospitalization, resulted in persistent or significant disability or incapacity, was a congenital anomaly or birth defect, was an 'important medical event' that may have jeopardized the person, or may have required an intervention to prevent one of the above characteristics or consequences. Acute chest syndrome is well documented as a reported adverse event of VOC and we planned to assess the issues around tolerability of the drugs.
Any pain‐related outcome indicating some improvement, such as sleep quality or activities of daily living.
Withdrawals due to lack of efficacy, adverse events and for any cause.
Reports of tolerance or habituation to opioids.
Search methods for identification of studies
The Information Specialist carried out the searches.
Electronic searches
We searched the following databases without language restrictions:
Cochrane Central Register of Controlled Trials (CENTRAL; via the Cochrane Library) searched 16 September 2019;
MEDLINE and MEDLINE in Process (via Ovid) searched 1947 to 16 September 2019;
Embase (via Ovid) searched 1974 to 2019 week 38;
LILACS (via BIRME) searched 1982 to September 2019.
We used MeSH or equivalent and text‐word terms. We tailored the searches to the individual databases.
The search strategies for CENTRAL, MEDLINE, Embase and LILACS are in Appendix 1.
Searching other resources
We searched the metaRegister of controlled trials (mRCT) (www.isrctn.com/), ClinicalTrials.gov (www.clinicaltrials.gov), and the World Health Organization (WHO) International Clinical Trials Registry Platform (ICTRP) (apps.who.int/trialsearch/) for ongoing trials. We also checked the reference lists of reviews and retrieved articles for additional studies and performed citation searches on key articles.
Data collection and analysis
Selection of studies
Three review authors (TC SB, and BJ) independently determined the eligibility of each study identified by the search by reading the title and abstract. The review authors then independently eliminated studies that clearly did not satisfy the inclusion criteria and obtained full copies of the remaining studies.
Three review authors (TC SB, and BJ) independently read these reports to select relevant studies. In the event of disagreement, a fourth review author (PW) adjudicated. We did not anonymize the studies before assessment. We included a PRISMA flow chart, which shows the status of identified studies (Moher 2009), as recommended in Part 2 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a). We included studies in the review irrespective of whether measured outcome data were reported in a 'usable' way.
Data extraction and management
Two review authors (TC and IH) independently extracted data using a standard piloted form, and checked for agreement before entry into Review Manager 5 (Review Manager 2014).
We included information about the pain condition, number of participants treated, drug and dosing regimen, study design (placebo or active control), study duration and follow‐up, analgesic outcome measures and results, withdrawals and adverse events (participants experiencing any adverse event or serious adverse event).
We collated multiple reports of the same study, so that each study, rather than each report, was the unit of interest in the review.
We collected characteristics of the studies in sufficient detail to complete the Characteristics of included studies table.
Assessment of risk of bias in included studies
Two review authors (TC and IH) independently assessed the risk of bias for each study, using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011b), adapted from those used by the Cochrane Pregnancy and Childbirth Group, with any disagreements resolved by discussion. We completed a 'Risk of bias' table for each included study using the 'Risk of bias' tool in Review Manager 5 (Review Manager 2014).
We assessed the following for each study.
Random sequence generation (checking for possible selection bias). We assessed the method used to generate the allocation sequence as: low risk of bias (any truly random process, e.g. random number table; computer random number generator); unclear risk of bias (method used to generate sequence not clearly stated). We excluded studies using a non‐random process (e.g. odd or even date of birth; hospital or clinic record number).
Allocation concealment (checking for possible selection bias). The method used to conceal allocation to interventions prior to assignment determines whether intervention allocation could have been foreseen in advance of, or during, recruitment or changed after assignment. We assessed the methods as: low risk of bias (e.g. telephone or central randomization; consecutively numbered, sealed, opaque envelopes); unclear risk of bias (method not clearly stated). We excluded studies that did not conceal allocation (e.g. open list).
Blinding of participants and personnel (checking for possible performance bias). We assessed the methods used to blind the participants and personnel from knowledge of which intervention a participant received. We assessed the methods as: low risk of bias (study stated that the participants and personnel involved were blinded to treatment groups); unclear risk of bias (study did not state whether or not participants and personnel were blinded to treatment groups); or high risk of bias (participants or personnel were not blinded) (as stated in Types of studies, we included trials with or without blinding, and participant‐ or observer‐reported outcomes).
Blinding of outcome assessment (checking for possible detection bias). We assessed the methods used to blind outcome assessors from knowledge of which intervention a participant received. We assessed the methods as: low risk of bias (study stated that it was blinded and described the method used to achieve blinding, e.g. identical tablets; matched in appearance and smell); unclear risk of bias (study stated that it was blinded but did not provide an adequate description of how it was achieved). We excluded studies that were not double‐blind.
Incomplete outcome data (checking for possible attrition bias due to the amount, nature and handling of incomplete outcome data). We assessed the methods used to deal with incomplete data as: low risk of bias (less than 10% of participants did not complete the study or used 'baseline observation carried forward' analysis, or both); unclear risk of bias (used 'last observation carried forward' analysis); high risk of bias (used 'completer' analysis).
Selective reporting (checking for possible reporting bias). We assessed the methods used to report the outcomes of the study as: low risk of bias (if all planned outcomes in the protocol or methods were reported in the results); unclear risk of bias (if there was no clear distinction between planned outcomes and reported outcomes); or high risk of bias (if some planned outcomes from the protocol or methods were clearly not reported in the results).
Size of study (checking for possible biases confounded by small‐study size). We assessed studies as being at low risk of bias (200 participants or more per treatment arm); unclear risk of bias (50 to 199 participants per treatment arm); high risk of bias (fewer than 50 participants per treatment arm).
Other bias, such as multiple publications, financial declarations, participants with conflicts of interest. We assessed studies for any additional sources of bias as low, unclear or high risk of bias, and provided rationales.
Measures of treatment effect
We used dichotomous data to calculate risk ratios (RR) with 95% confidence intervals (CI) using a fixed‐effect model unless there was significant statistical heterogeneity (see Assessment of heterogeneity).
We calculated numbers needed to treat for an additional beneficial outcome (NNTB) as the reciprocal of the absolute risk reduction (ARR; McQuay 1998). For unwanted effects, the NNTB becomes the number needed to treat for an additional harmful outcome (NNTH) and is calculated in the same manner. We planned to use the term number needed to treat to prevent an additional outcome (NNTp) where the unwanted effect is less common with treatment than control.
For our primary outcomes measuring pain, we planned not to use continuous data in analyses because it is inappropriate where there is underlying skewed distribution, as is usually the case with analgesic response. For our secondary outcomes, we used continuous data for the meta‐analysis where appropriate.
Unit of analysis issues
We expected that the unit of analysis would be at the participant level, with each participant providing one pain episode per trial. It was possible that a trial may have included multiple pain events per participant, introducing a statistical clustering effect with two pain events in the same participant likely to be more similar than two independent pain events from two different participants; however, this did not occur in any of our included studies. We had planned to use the number of trial clusters and an estimate of the intraclass correlation coefficient to inflate the standard errors associated with each clustered trial in this scenario.
We had planned for the unit of analysis to be each participant from the first phase of treatment before crossing over (who, where randomised, took at least one dose of study, and provided at least one outcome score) if any included studies were crossover studies. We would include the second phase of treatment if there was a sufficient washout period and results were reported clearly and separately. If not, and the results appeared unclear from which phase they were taken, we would contact the authors and attempt to gain access to the original data. Otherwise, we would not use the data.
Dealing with missing data
We used intention‐to‐treat analysis where the intention‐to‐treat population consisted of participants who were randomized, received at least one dose of the assigned study treatment and provided at least one post‐baseline assessment. We assigned missing participants zero improvement wherever possible.
Assessment of heterogeneity
We dealt with clinical heterogeneity (variation in participants, interventions or outcomes) by combining studies that examined similar conditions when possible. We assessed statistical heterogeneity visually (L'Abbé 1987), and with the I² statistic. We considered the possible reasons when the I² value was greater than 50%.
Assessment of reporting biases
The aim of this review was to use dichotomous outcomes of known utility and of value to patients (Hoffman 2010; Moore 2010a; Moore 2010b; Moore 2010c; Moore 2013). The review did not depend on what the authors of the original studies chose to report or not, though difficulties arose in studies failing to report any dichotomous results. We extracted and used continuous data where possible, which poorly reflected efficacy and utility but was useful for illustrative purposes only.
We assessed publication bias using a method designed to detect the amount of unpublished data with a null effect required to make any result clinically irrelevant (usually taken to mean an NNTB of 10 or higher; Moore 2008).
Data synthesis
We planned to meta‐analyse data from similar comparisons using a fixed‐effect model in Review Manager 5 (Review Manager 2014). Due to the lack of available data, we used Review Manager 5 to undertake a summary of effects from individual studies, fixed‐effects and without group totals (Analysis 1.1; Analysis 1.2; Analysis 1.3; Analysis 1.4).
1.1. Analysis.
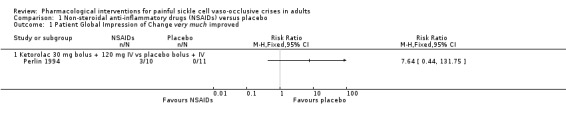
Comparison 1 Non‐steroidal anti‐inflammatory drugs (NSAIDs) versus placebo, Outcome 1 Patient Global Impression of Change very much improved.
1.2. Analysis.
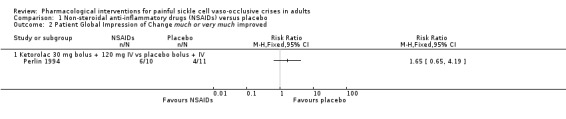
Comparison 1 Non‐steroidal anti‐inflammatory drugs (NSAIDs) versus placebo, Outcome 2 Patient Global Impression of Change much or very much improved.
1.3. Analysis.
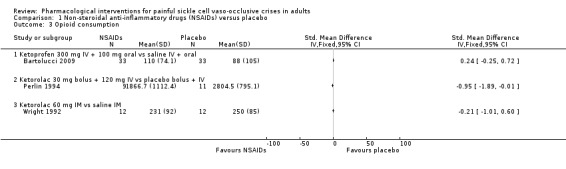
Comparison 1 Non‐steroidal anti‐inflammatory drugs (NSAIDs) versus placebo, Outcome 3 Opioid consumption.
1.4. Analysis.
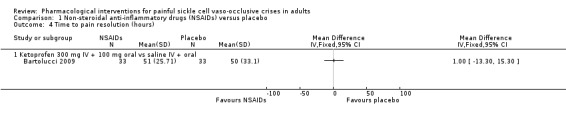
Comparison 1 Non‐steroidal anti‐inflammatory drugs (NSAIDs) versus placebo, Outcome 4 Time to pain resolution (hours).
We planned to have used a random‐effects model for meta‐analysis if there was significant clinical heterogeneity and it was considered appropriate to combine studies. This did not occur.
Quality of the evidence
Two review authors (TC, PW) independently rated the quality of the evidence for each outcome. We used the GRADE system to rank the quality of the evidence using the GRADEprofiler Guideline Development Tool software (GRADEpro GDT), and the guidelines provided in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a).
The GRADE approach uses five considerations (study limitations, consistency of effect, imprecision, indirectness and publication bias) to assess the quality of the body of evidence for each outcome. The GRADE system uses the following criteria for assigning the grade of evidence.
High: further research is very unlikely to change our confidence in the estimate of effect.
Moderate: further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate.
Low: further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate.
Very low: any estimate of effect is very uncertain.
Reasons for decreasing grade are due to:
serious (–1) or very serious (–2) limitation to study quality;
important inconsistency (–1);
some (–1) or major (–2) uncertainty about directness;
imprecise or sparse data (–1);
high probability of reporting bias (–1).
In addition, we anticipated there may have been circumstances where the overall rating for a particular outcome would need to be adjusted per GRADE guidelines (Guyatt 2013a; Guyatt 2013b). For example, if there were so few data that the results are highly susceptible to the random play of chance, or if studies used last observation carried forward imputation in circumstances where there were substantial differences in adverse event withdrawals, or where there were a small number of participants per treatment arm, one would have no confidence in the result, and would need to downgrade the quality of the evidence by three levels to very low quality.
In circumstances where no data were reported for an outcome, the quality of the evidence is unknown and we reported this as 'no evidence to support or refute'.
Subgroup analysis and investigation of heterogeneity
We planned no subgroup analyses since experience from previous reviews suggests a limited chance of sufficient data. We would have considered subgroup classification between genotypes should the data be available; however, the available data proved to be insufficient for this purpose.
Sensitivity analysis
We planned no sensitivity analysis because the evidence base is known to be too small to allow reliable analysis. We did not plan to pool results from sickle cell pain of different origins in the primary analyses.
Results
Description of studies
Results of the search
A PRISMA flow diagram of the search results is shown in Figure 1.
1.
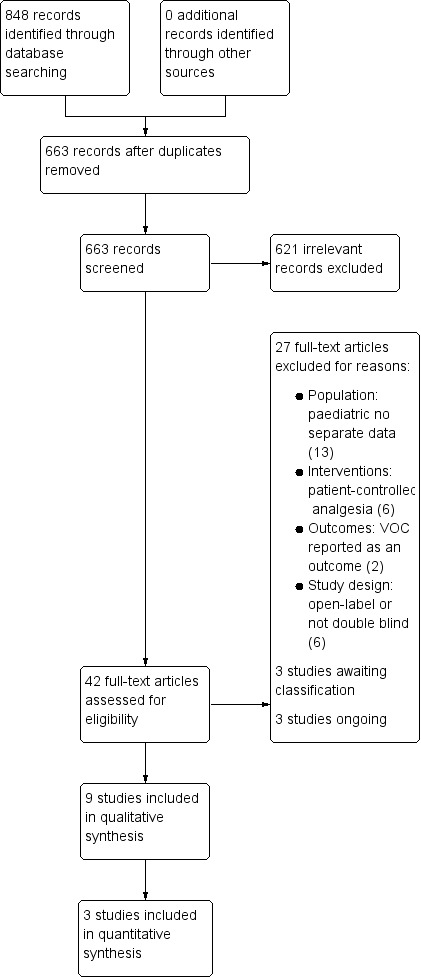
Study flow diagram.
The three main database searches found 848 records, of which we removed 185 duplicates. We also searched clinicaltrials.gov and app.who.int/trialsearch/ and found no additional eligible studies.
We screened the remaining 663 titles and abstracts for eligibility, of which we removed 621 as clearly irrelevant studies.
We retrieved the full‐text reports of the 42 remaining studies. Twenty‐seven were ineligible and excluded (Characteristics of excluded studies table). Three studies are awaiting classification (Characteristics of studies awaiting classification table), and three studies are ongoing (Characteristics of ongoing studies table).
Nine studies fulfilled the eligibility criteria, which we included in the qualitative synthesis. We entered three of these studies into a quantitative analysis.
Included studies
We included nine studies in this review. See Characteristics of included studies table.
Design
All nine studies were randomized double‐blind placebo‐controlled trials with two arms. Eight studies were parallel groups and one study had crossover arms.
Sample sizes
The sample sizes of the studies ranged from 18 participants (Gonzalez 1988; Wright 1992) to 253 participants (Qari 2007).
Setting
Two studies were multicentre studies and seven were single‐centre studies. All studies were based in emergency departments of hospitals from France, Italy, Saudi Arabia, and the USA. No included studies were based in home or other settings.
Participants
The age ranges were adults from 17 to 42 years old. Gender ratios were roughly equal between women and men across all studies. All nine studies had inclusion criteria of pre‐existing diagnosis of sickle cell disease and presenting to the emergency department in a VOC.
Interventions
Analgesic interventions varied including butorphanol, cetiedil, fentanyl, hydromorphone, ketamine, ketoprofen, metoclopramide, morphine, paracetamol, tinzaparin and tramadol.
Six studies administered IV bolus injections (Arambasik 2013; Benjamin 1986; Gonzalez 1988; Qari 2007; Rehmani 2013; Wright 1992), three administered IV infusions (Bartolucci 2009; De Franceschi 2016) and one administered an IV bolus followed by an IV infusion (Perlin 1994). Two studies were a single dose of the drug only and seven administered multiple doses of the treatments (Rehmani 2013; Wright 1992).
Outcomes
All nine studies reported pain outcomes using a validated pain scale or global assessment scores. Four studies reported opioid consumption (Bartolucci 2009; Perlin 1994; Rehmani 2013; Wright 1992). Two studies reported time to pain resolution (Bartolucci 2009; Qari 2007). Two studies reported the duration of time spent in hospital (Perlin 1994; Qari 2007). All but two studies reported adverse events and withdrawals (Arambasik 2013; Qari 2007).
Excluded studies
We excluded 27 studies in this review. See Characteristics of excluded studies table.
Thriteen studies were in paediatric or combined paediatric‐adult populations, with no separate data available for adults. Six studies implemented patient‐controlled analgesia (PCA). In two studies, pain was not measured as an outcome but VOC was measured as the outcome. Six studies were either not double‐blind or were an open‐label trial.
Studies awaiting classification
See Studies awaiting classification table.
De Castro 2013 and Perlin 1988 are conference abstracts which currently meet eligibility criteria but require outcome data. We contacted the study authors for further information so these remain in studies awaiting classification. In Teuscher 1989, it was unclear whether pain was reported as a primary or secondary outcome. We contacted the study author for further information but this has not been received.
Ongoing studies
We identified three ongoing studies (IRCT2016072511956N6; NCT03431285; NCT03978156). See Characteristics of ongoing studies table.
Risk of bias in included studies
A summary of the risk of bias assessment is available in Figure 2.
2.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Full details of risk of bias assessments are available in the Characteristics of included studies table.
Allocation
Random sequence generation
Four studies adequately described the methods used to randomize participants (computer‐generated randomization) and were at low risk of selection bias for random sequence generation (Benjamin 1986; De Franceschi 2016; Perlin 1994; Wright 1992). Five studies did not adequately describe the methods used to randomize participants and were at unclear risk of selection bias for random sequence generation (Arambasik 2013; Bartolucci 2009; Gonzalez 1988; Qari 2007; Rehmani 2013). No studies displayed a high risk of selection bias for random sequence generation.
Allocation concealment
Three studies adequately described the methods used to conceal allocation of treatment to each group and were at low risk of selection bias for allocation concealment (Benjamin 1986; Perlin 1994; Wright 1992). Six studies did not adequately describe the methods used to conceal allocation of treatment to each group and were at unclear risk of selection bias for allocation concealment (Arambasik 2013; Bartolucci 2009; De Franceschi 2016; Gonzalez 1988; Qari 2007; Rehmani 2013). No studies displayed a high risk of selection bias for allocation concealment.
Blinding
Performance bias
Four studies adequately described the methods to blind both participants and study personnel from knowledge of the treatment groups and were at low risk of performance bias (Benjamin 1986; Gonzalez 1988; Perlin 1994; Wright 1992). Five studies did not adequately describe the methods to blind both participants and study personnel from knowledge of the treatment groups and were at unclear risk of performance bias (Arambasik 2013; Bartolucci 2009; De Franceschi 2016; Qari 2007; Rehmani 2013). No studies displayed a high risk of performance bias.
Detection bias
Three studies adequately described the methods used to conceal and blind the outcome assessors from knowledge of the treatment groups and were at low risk of detection bias (De Franceschi 2016; Gonzalez 1988; Perlin 1994). Six studies did not adequately describe the methods used to conceal and blind the outcome assessors from knowledge of the treatment groups and were at unclear risk of detection bias (Arambasik 2013; Bartolucci 2009; Benjamin 1986; Qari 2007; Rehmani 2013; Wright 1992). No studies displayed a high risk of detection bias.
Incomplete outcome data
Two studies adequately accounted for all participants from the recruitment stage, through randomization until follow‐up, including counting all withdrawals and were at low risk of attrition bias (De Franceschi 2016; Wright 1992). Six studies did not adequately account for all participants from the recruitment stage, through randomization until follow‐up, including counting all withdrawals and were at unclear risk of attrition bias (Arambasik 2013; Benjamin 1986; Gonzalez 1988; Perlin 1994; Qari 2007; Rehmani 2013). One study reported 20% attrition, the outcome reporting ignored these randomized participants and analyses did not appear to be by intention to treat (Bartolucci 2009). This was at high risk of attrition bias.
Selective reporting
Six studies adequately reported in their results all the outcomes that were planned in the methods sections and were at low risk of reporting bias (Bartolucci 2009; Benjamin 1986; De Franceschi 2016; Gonzalez 1988; Perlin 1994; Wright 1992). One study displayed an unclear risk of reporting bias and was at unclear risk of reporting bias (Arambasik 2013). Two studies, planned to measure outcomes in their methods, but did not mention them when reporting results and were at high risk of reporting bias (Qari 2007; Rehmani 2013). Qari 2007 did not report on adverse events or opioid consumption, as planned. Rehmani 2013 did not report on the VRS, as planned.
Other potential sources of bias
Size
Two studies investigated between 50 and 200 participants per treatment arm and were at unclear risk of bias in relation to size (Qari 2007; Rehmani 2013).
Seven studies investigated fewer than 50 participants per treatment arm and were at high risk of bias in relation to size (Arambasik 2013; Bartolucci 2009; Benjamin 1986; De Franceschi 2016; Gonzalez 1988; Perlin 1994; Wright 1992). No studies investigated more than 200 participants per treatment arm.
Other
Gonzalez 1988 displayed a high risk of bias in relation to a unit of analysis issue, 12 events per treatment arm. However, the unit of randomization was the painful crisis (45 randomized crises events among 18 participants), random‐effects analysis took this unit of analysis issue appropriately into account. This was judged at high risk of 'other' bias.
Bartolucci 2009 displayed some unclear risk of bias in relation to a unit of analysis error. The unit of randomization was the painful crisis (66 randomized crises among 54 participants), but the unit of analysis ignored the clustered nature of the data which were analysed as if all randomized events were independent. This was judged at unclear risk of 'other' bias.
Arambasik 2013 displayed some unclear risk of bias as we could not conclude there was low risk of bias due to only the abstract being available. This was judged at unclear risk of 'other' bias.
For the remaining six studies, we found no other potential sources of bias and they were at low risk of 'other' bias.
Effects of interventions
See: Table 1; Table 2; Table 3
Summary of findings for the main comparison. Non‐steroidal anti‐inflammatory drugs (NSAID) compared with placebo for painful sickle cell vaso‐occlusive crises.
| NSAIDs compared with placebo for painful sickle cell vaso‐occlusive crises | ||||||
|
Patient or population: adults with sickle cell disease in a vaso‐occlusive crises Settings: emergency departments Intervention: NSAIDs (ketorolac or ketoprofen) Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Placebo | NSAIDs | |||||
| Participant‐reported pain relief of 50% or greater | No data | No data | No data | No data | No dataa | Quality of the evidence is unknown |
| Participant‐reported pain relief of 30% or greater | No data | No data | No data | No data | No dataa | Quality of the evidence is unknown |
|
PGIC very much improved (Follow‐up: up to 5 days) |
0/11 | 3/10 | RR 7.64 (0.44 to 131.75) | 21 participants, 21 VOC events (1 study) | ⊕⊝⊝⊝ Very lowb | — |
|
PGIC much or very much improved (Follow‐up: up to 5 days) |
4/11 | 6/10 | RR 1.65 (0.65 to 4.19) | 21 participants, 21 VOC events (1 study) | ⊕⊝⊝⊝ Very lowb | — |
|
Any adverse event (Follow‐up: up to 5 days) |
19/45 | 16/45 | N/A | 72 participants, 90 VOC events (2 studies) | ⊕⊝⊝⊝ Very lowb | — |
|
Serious adverse events (Follow‐up: up to 5 days) |
2/56 | 1/55 | N/A | 93 participants, 111 VOC events (3 studies) | ⊕⊝⊝⊝ Very lowb | — |
|
Withdrawals due to adverse events (Follow‐up: up to 5 days) |
1/56 | 1/55 | N/A | 93 participants, 111 VOC events (3 studies) | ⊕⊝⊝⊝ Very lowb | — |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; N/A: not applicable; NSAID: non‐steroidal anti‐inflammatory drug; PGIC: Patient Global Impression of Change; RR: risk ratio; VOC: vaso‐occlusive crisis. | ||||||
| GRADE Working Group grades of evidence High quality: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate quality: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of effect, but there is a possibility that it is substantially different. Low quality: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low quality: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
a No data available, therefore no GRADE rating has been performed for this outcome and the quality of the evidence is unknown.
b Downgraded three levels; too few data (limited number of participants per treatment arm) to have confidence in results.
Summary of findings 2. Opioids compared with placebo for painful sickle cell vaso‐occlusive crises.
| Opioids compared with placebo for painful sickle cell vaso‐occlusive crises | ||||||
|
Patient or population: adults with sickle cell disease in a vaso‐occlusive crisis Settings: emergency departments Intervention: opioids Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Placebo | Opioids | |||||
| Participant‐reported pain relief of 50% or greater | No data | No data | N/A | N/A | No dataa | Quality of the evidence is unknown |
| Participant‐reported pain relief of 30% or greater | No data | No data | N/A | N/A | No dataa | Quality of the evidence is unknown |
| PGIC very much improved | No data | No data | N/A | N/A | No dataa | Quality of the evidence is unknown |
| PGIC much or very much improved | No data | No data | N/A | N/A | No dataa | Quality of the evidence is unknown |
| Any adverse event | No data | No data | N/A | N/A | No dataa | Quality of the evidence is unknown |
| Serious adverse event | No data | No data | N/A | N/A | No dataa | Quality of the evidence is unknown |
| Withdrawals due to adverse events | No data | No data | N/A | N/A | No dataa | Quality of the evidence is unknown |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; N/A: not applicable; PGIC: Patient Global Impression of Change; RR: risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: we are very confident that the true effect lies close to that of the estimate of the effect; Moderate quality: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of effect, but there is a possibility that it is substantially different; Low quality: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect; Very low quality: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
a No data available, therefore no GRADE rating has been performed for this outcome and the quality of the evidence is unknown.
Summary of findings 3. Opioids compared with active comparator for painful sickle cell vaso‐occlusive crises.
| Opioids compared with active comparator for painful sickle cell vaso‐occlusive crises | ||||||
|
Patient or population: adults with sickle cell disease in a vaso‐occlusive crisis Settings: emergency departments Intervention: opioids (morphine) Comparison: active comparator (butorphanol or paracetamol) | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Active comparator | Opioids | |||||
| Participant‐reported pain relief of 50% or greater | No data | No data | N/A | N/A | No dataa | Quality of the evidence is unknown |
| Participant‐reported pain relief of 30% or greater | No data | No data | N/A | N/A | No dataa | Quality of the evidence is unknown |
|
PGIC very much improved (Follow‐up: within 1 day) |
19.1 per 100 | 33.3 per 100 | N/A | 18 participants, 24 VOC events (1 study) | ⊕⊝⊝⊝ Very lowb | — |
| PGIC much or very much improved | No data | No data | N/A | N/A | No dataa | Quality of the evidence is unknown |
|
Any adverse event (Follow‐up: within 1 day) |
7/64 | 9/66 | N/A | 124 participants, 130 VOC events (2 studies) | ⊕⊝⊝⊝ Very lowb | — |
|
Serious adverse event (Follow‐up: within 1 day) |
0/64 | 0/66 | N/A | 124 participants, 130 VOC events (2 studies) | ⊕⊝⊝⊝ Very lowb | — |
| Withdrawals due to adverse events | No data | No data | N/A | N/A | No dataa | Quality of the evidence is unknown |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; N/A: not applicable; PGIC: Patient Global Impression of Change; RR: risk ratio; VOC: vaso‐occlusive crisis. | ||||||
| GRADE Working Group grades of evidence High quality: we are very confident that the true effect lies close to that of the estimate of the effect; Moderate quality: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of effect, but there is a possibility that it is substantially different; Low quality: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect; Very low quality: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
a No data available, therefore no GRADE rating has been performed for this outcome and the quality of the evidence is unknown.
b Downgraded three levels; too few data (limited number of participants per treatment arm) to have confidence in results.
For results and adverse events of individual studies, see Appendix 2 (primary outcome data: pain outcomes); Appendix 3 (secondary outcome data); and Appendix 4 (adverse events and withdrawals).
Table 1 displays the types of pharmacological intervention comparisons by study.
Table 1: types of drug comparisons
| Study | Interventions | Comparison pair |
| Arambasik 2013 | Ketamine + hydromorphone vs placebo + hydromorphone | Opioid vs placebo |
| Bartolucci 2009 | Ketoprofen vs placebo | NSAID vs placebo |
| Benjamin 1986 | Cetiedil vs placebo | Vasodilator (anti‐sickling) vs placebo |
| De Franceschi 2016 | Ketorolac + tramadol + metoclopramide + fentanyl vs ketorolac + tramadol + metoclopramide | Combination vs combination |
| Gonzalez 1988 | Butorphanol vs morphine | Opioid vs opioid |
| Perlin 1994 | Ketorolac vs placebo | NSAID vs placebo |
| Qari 2007 | Tinzaparin vs placebo | Anticoagulant vs placebo |
| Rehmani 2013 | Morphine vs paracetamol | Opioid vs paracetamol |
| Wright 1992 | Ketorolac vs placebo | NSAID vs placebo |
Comparison 1: non‐steroidal anti‐inflammatory drugs versus placebo
Three studies compared an NSAID with placebo (Bartolucci 2009; Perlin 1994; Wright 1992). See Table 1.
Primary outcomes
Participant‐reported pain relief of 50% or greater
None of the studies reported participants‐reported pain relief of 50% or greater.
As there were no data for this outcome, the quality of evidence is unknown, and there is no evidence to support or refute.
Participant‐reported pain relief of 30% or greater
None of the studies reported participant‐reported pain relief of 30% or greater.
As there were no data for this outcome, the quality of evidence is unknown, and there is no evidence to support or refute.
Patient Global Impression of Change very much improved
One study reported PGIC very much improved (Perlin 1994). As there was only one study reporting this comparison, we did not undertake a meta‐analysis but provided a summary of effects from the individual study (RR: risk ratio); Analysis 1.1; Figure 3.
3.

Forest plot of comparison: 1 Non‐steroidal anti‐inflammatory drugs (NSAIDs) versus placebo, outcome: 1.1 Patient Global Impression of Change very much improved.
Perlin 1994 reported very much improved for 3/10 participants in the ketorolac group, and for 0/11 participants in the placebo group (RR 7.64, 95% CI 0.44 to 131.75; P = 0.16; Analysis 1.1; Figure 3). As there were too few data for this outcome (limited number of participants per treatment arm), we had no confidence in the results and we downgraded the quality of evidence by three levels to very low quality.
Patient Global Impression of Change much or very much improved
One study reported PGIC much or very much improved (Perlin 1994). As there was only one study reporting this comparison, we did not undertake a meta‐analysis but provided a summary of effects from the individual study (RR: risk ratio); Analysis 1.2; Figure 4.
4.

Forest plot of comparison: 1 Non‐steroidal anti‐inflammatory drugs (NSAIDs) versus placebo, outcome: 1.2 Patient Global Impression of Change much or very much improved.
Perlin 1994 reported the combination of much or very much improved for 6/10 participants in the ketorolac group, and 4/11 participants in the placebo group (RR 1.65, 95% CI 0.65 to 4.19; P = 0.29; Analysis 1.2; Figure 4).
As there were too few data for this outcome (limited number of participants per treatment arm), we had no confidence in the results and we downgraded the quality of evidence by three levels to very low quality.
Secondary outcomes
Opioid consumption
Three studies reported opioid consumption (Bartolucci 2009; Perlin 1994; Wright 1992).
Bartolucci 2009 reported the use of additional rescue morphine. The overall median (interquartile range (IQR)) consumption of morphine was 110 mg (IQR 46 to 195) in the ketoprofen group and 88 mg (IQR 52.5 to 262.5) in the placebo group (33 participants per group).
Perlin 1994 reported the use of additional rescue meperidine. The mean daily dose of meperidine was 523.6 mg (standard deviation (SD) 222.1) in the ketorolac group (10 participants) and 662.4 mg (SD 68.6) in the placebo group (11 participants). The overall mean consumption of meperidine was 1866.7 mg (SD 1112.4) in the ketorolac group and 2804.5 mg (SD 795.1) in the placebo group.
Wright 1992 reported the use of additional rescue meperidine. The overall mean consumption of meperidine was 231 mg (SD 92) in the ketorolac group and 250 mg (SD 85) in the placebo group (12 participants per group).
As there were too few data for this outcome (limited number of participants per treatment arm), we had no confidence in the results and we downgraded the quality of evidence by three levels to very low quality.
Time to pain resolution
One study reported time to pain resolution (Bartolucci 2009).
Bartolucci 2009 reported median time to participants' resolution of pain as 51 hours (IQR 35.5 to 87) in the ketoprofen group and 50 hours (IQR 36 to 103) in the placebo group (33 participants per group). As there were too few data for this outcome (limited number of participants per treatment arm), we had no confidence in the results and we downgraded the quality of evidence by three levels to very low quality.
Length of hospitalization
One study reported length of hospitalization (Perlin 1994).
Perlin 1994 reported mean length of hospitalization as 3.3 days in the ketorolac group (10 participants) and 7.2 days in the placebo group (11 participants) (P < 0.05). As there were too few data for this outcome (limited number of participants per treatment arm), we had no confidence in the results and we downgraded the quality of evidence by three levels to very low quality.
Participants experiencing any adverse or serious adverse event
Two studies reported adverse events (Bartolucci 2009; Wright 1992).
Bartolucci 2009 reported any mild adverse events in 16/33 participants in the ketoprofen group and 19/33 participants in the placebo group.
Wright 1992 reported adverse events in 0/12 participants in the ketorolac group and 0/12 participants in the placebo group.
Three studies reported serious adverse events (Bartolucci 2009; Perlin 1994; Wright 1992).
Bartolucci 2009 reported serious adverse events in 1/33 participants in the ketoprofen group, and 2/33 participants in the placebo group.
Perlin 1994 reported serious adverse events in 0/10 participants in the ketorolac group and 0/11 participants in the placebo group.
Wright 1992 reported serious adverse events in 0/12 participants in the ketorolac group and 0/ 12 participants in the placebo group.
As there were too few data for this outcome (limited number of participants per treatment arm), we had no confidence in the results and we downgraded the quality of evidence by three levels to very low quality.
Any pain‐related outcome indicating some improvement
Three studies reported additional pain related outcome data (Bartolucci 2009; Perlin 1994; Wright 1992).
Bartolucci 2009 reported the mean daily scores on a categorical pain scale (0 to 3) as median 0.4 (IQR 0.2 to 0.7) in the ketoprofen group and 0.4 (IQR 0.2 to 0.7) in the placebo group. The authors also reported mean daily scores on a VAS (0 mm to 100 mm, where 0 = no pain) as median 12.6 (IQR 4.8 to 23.2) in the ketoprofen group and 9.6 (IQR 5.8 to 33.2) in the placebo group.
Perlin 1994 reported the pain scores at baseline on a VAS (0 mm to 100 mm), as mean 77.7 (95% CI 69.1, 86.2) in the ketorolac group and 79.1 (95% CI 72.1 to 86.0) in the placebo group. Pain scores at 24 hours on a VAS (0 mm to 100 mm) were mean 58.6 (95% CI 48.6 to 68.5) in the ketorolac group and 72.6 (95% CI 62.4 to 82.8) in the placebo group (P < 0.05). Mean pain scores at baseline on a visual categorical scale (0 to 3) were 2.5 in the ketorolac group and 2.6 in the placebo group. Mean pain scores at 24 hours on a visual categorical scale (0 to 3) were 2.0 in the ketorolac group and 2.4 in the placebo group. Mean pain scores at 24 hours on a VRS (0 to 4) were 1.8 in the ketorolac group and 1.9 in the placebo group.
Wright 1992 reported mean pain scores at baseline on a VAS (0 mm to 100 mm) were 70.3 in the ketorolac group and 79.3 in the placebo group (P = 0.26). Mean pain scores at four hours on a VAS (0 mm to 100 mm) were 44 (SD 34) in the ketorolac group and 37 (SD 31) in the placebo group (P = 0.49).
As there were too few data for this outcome (limited number of participants per treatment arm), we had no confidence in the results and we downgraded the quality of evidence by three levels to very low quality.
Withdrawals: due to lack of efficacy, adverse events and for any cause
Two studies reported all‐cause withdrawals (Perlin 1994; Wright 1992).
Perlin 1994 reported total all‐cause withdrawals in 1/10 participants in the ketorolac group and 0/11 participants in the placebo group.
Wright 1992 reported total all‐cause withdrawals in 0/12 participants in the ketorolac group and 0/12 participants in the placebo group.
Three studies reported withdrawals due to adverse events (Bartolucci 2009; Perlin 1994; Wright 1992).
Bartolucci 2009 reported withdrawals due to adverse events in 1/33 participants in the ketoprofen group and 1/33 participants in the placebo group.
Perlin 1994 reported withdrawals due to adverse events in 0/10 participants in the ketorolac group and 0/11 participants in the placebo group.
Wright 1992 reported withdrawals due to adverse events in 0/12 participants in the ketorolac group and 0/12 participants in the placebo group.
As there were too few data for this outcome (limited number of participants per treatment arm), we had no confidence in the results and we downgraded the quality of evidence by three levels to very low quality.
Reports of tolerance or habituation to opioids
None of the studies reported tolerance or habituation to opioids.
As there were no data for this outcome, the quality of evidence is unknown, and there is no evidence to support or refute.
Comparison 2: opioids versus placebo
One study compared ketamine plus hydromorphone with placebo plus hydromorphone (Arambasik 2013). See Table 2.
Primary outcomes
Participant‐reported pain relief of 50% or greater
The study did not report participant‐reported pain relief of 50% or greater.
As there were no data for this outcome, the quality of evidence is unknown, and there is no evidence to support or refute.
Participant‐reported pain relief of 30% or greater
The study did not report participant‐reported pain relief of 30% or greater.
As there were no data for this outcome, the quality of evidence is unknown, and there is no evidence to support or refute.
Patient Global Impression of Change very much improved
The study did not report PGIC very much improved.
As there were no data for this outcome, the quality of evidence is unknown, and there is no evidence to support or refute.
Patient Global Impression of Change much or very much improved
The study did not report PGIC much or very much improved.
As there were no data for this outcome, the quality of evidence is unknown, and there is no evidence to support or refute.
Secondary outcomes
Opioid consumption
The study did not report opioid consumption.
As there were no data for this outcome, the quality of evidence is unknown, and there is no evidence to support or refute.
Time to pain resolution
The study did not report time to pain resolution.
As there were no data for this outcome, the quality of evidence is unknown, and there is no evidence to support or refute.
Length of hospitalization
The study did not report length of hospitalization.
As there were no data for this outcome, the quality of evidence is unknown, and there is no evidence to support or refute.
Participants experiencing any adverse or serious adverse event
The study did not report any adverse event or serious adverse event.
As there were no data for this outcome, the quality of evidence is unknown, and there is no evidence to support or refute.
Any pain‐related outcome indicating some improvement
Arambasik 2013 reported mean pain scores on a VAS (1 to 10, where 1 implies none or minimal pain), on arrival, of 8.7 (95% CI 8.07 to 9.29) in the ketamine plus hydromorphone group and 8.5 (95% CI 7.90 to 9.05) in the placebo plus hydromorphone group. At administration of the study drugs, mean pain scores were 6.0 (95% CI 4.71 to 7.29; a 31% decrease from arrival) in the ketamine plus hydromorphone group and 7.0 (95% CI 6.20 to 7.85; a 17.6% decrease from arrival) in the placebo plus hydromorphone group. Once doses were administered, the mean pain scores were 5.2 (95% CI 4.01 to 6.46; a 40.2% decrease from arrival) in the ketamine plus hydromorphone group and 5.6 (95% CI 4.27 to 6.93; a 34.1% decrease from arrival) in the placebo plus hydromorphone group.
As there were too few data for this outcome (limited number of participants per treatment arm), we had no confidence in the results and we downgraded the quality of evidence by three levels to very low quality.
Withdrawals: due to lack of efficacy, adverse events and for any cause
The study did not report withdrawals.
As there were no data for this outcome, the quality of evidence is unknown, and there is no evidence to support or refute.
Reports of tolerance or habituation to opioids
The study did not report tolerance or habituation to opioids.
As there were no data for this outcome, the quality of evidence is unknown, and there is no evidence to support or refute.
Comparison 3: opioids versus active comparator
Two studies compared an opioid with an active comparator: Gonzalez 1988: morphine versus butorphanol; Rehmani 2013: morphine versus paracetamol. See Table 3.
Primary outcomes
Participant‐reported pain relief of 50% or greater
Neither study reported participant‐reported pain relief of 50% or greater.
As there were no data for this outcome, the quality of evidence is unknown, and there is no evidence to support or refute.
Participant‐reported pain relief of 30% or greater
Neither study reported participant‐reported pain relief of 30% or greater.
As there were no data for this outcome, the quality of evidence is unknown, and there is no evidence to support or refute.
Patient Global Impression of Change very much improved
One study reported PGIC very much improved (Gonzalez 1988).
Gonzalez 1988 reported an equivalent scale of very much improved as 'excellent' for 19.1% of events in the butorphanol group and 33.3% of events in the morphine group.
As there were too few data for this outcome (limited number of participants per treatment arm), we had no confidence in the results and we downgraded the quality of evidence by three levels to very low quality.
Patient Global Impression of Change much or very much improved
One study reported PGIC much or very much improved (Gonzalez 1988).
Gonzalez 1988 reported equivalent scales of 'good' and 'excellent'. The study reported 'good' improvement from 47.6% in the butorphanol group and 38.1% in the morphine group. The study reported 'excellent' improvement from 19.1% in the butorphanol group and 33.3% in the morphine group. The study did not report a combination of 'good and excellent' (equivalent scale of much and very much improved) and the available percentages did not seem to match the raw data, so it was not possible to calculate a combined percentage for this outcome.
As there were no data for this outcome, the quality of evidence is unknown, and there is no evidence to support or refute.
Secondary outcomes
Opioid consumption
One study reported opioid consumption.
Rehmani 2013 reported the use of additional rescue morphine 0.1 mg/kg at 30 minutes for 27/54 participants in the morphine group, and 24/52 participants in the paracetamol group.
As there were too few data for this outcome (limited number of participants per treatment arm), we had no confidence in the results and we downgraded the quality of evidence by three levels to very low quality.
Time to pain resolution
Neither study reported time to pain resolution.
As there were no data for this outcome, the quality of evidence is unknown, and there is no evidence to support or refute.
Length of hospitalization
Neither study reported length of hospitalization.
As there were no data for this outcome, the quality of evidence is unknown, and there is no evidence to support or refute.
Participants experiencing any adverse or serious adverse event
Both studies reported adverse events and serious adverse events.
Gonzalez 1988 reported nausea and vomiting in 4/12 participants in the butorphanol group and 4/12 participants in the morphine group.
Rehmani 2013 reported mild adverse events in 5/54 participants in the morphine group and 3/52 participants in the paracetamol group.
Gonzalez 1988 reported serious adverse events in 0/12 participants in the morphine group and 0/12 participants in the butorphanol group.
Rehmani 2013 reported serious adverse events 0/54 participants in the morphine group and 0/52 participants in the paracetamol group.
As there were too few data for this outcome (limited number of participants per treatment arm), we had no confidence in the results and we downgraded the quality of evidence by three levels to very low quality.
Any pain‐related outcome indicating some improvement
Both studies reported mean pain scores, at different time points.
Gonzalez 1988 reported adjusted mean overall pain scores, on a VAS (0 mm to 100 mm, where 0 = no pain) of 46.08 in the morphine group and 44.42 in the butorphanol group. Adjusted mean overall pain scores, on a Pain Relief Scale (0 mm to 100 mm, where 0 = no pain) were 55.50 in the morphine group and 43.79 in the butorphanol group.
Rehmani 2013 reported mean pain scores, on a VAS (0 mm to 100 mm, where 0 = no pain), at 30 minutes, of 44 (95% CI 33 to 56) in the morphine group and 41 (95% CI 32 to 49) in the butorphanol group. This was not statistically significant (P = 0.72).
As there were too few data for this outcome (limited number of participants per treatment arm), we had no confidence in the results and we downgraded the quality of evidence by three levels to very low quality.
Withdrawals: due to lack of efficacy, adverse events and for any cause
Neither study reported withdrawals.
As there were no data for this outcome, the quality of evidence is unknown, and there is no evidence to support or refute.
Reports of tolerance or habituation to opioids
Neither study reported tolerance or habituation to opioids.
As there were no data for this outcome, the quality of evidence is unknown, and there is no evidence to support or refute.
Comparison 4: 'other' active comparators versus placebo
Two studies investigated an active comparator compared with a placebo: Benjamin 1986: vasodilator (cetiedil) versus placebo; Qari 2007: anticoagulant (tinzaparin) versus placebo.
Primary outcomes
Participant‐reported pain relief of 50% or greater
Neither study reported participant‐reported pain relief of 50% or greater.
As there were no data for this outcome, the quality of evidence is unknown, and there is no evidence to support or refute.
Participant‐reported pain relief of 30% or greater
Neither study reported participant‐reported pain relief of 30% or greater.
As there were no data for this outcome, the quality of evidence is unknown, and there is no evidence to support or refute.
Patient Global Impression of Change very much improved
Neither study reported PGIC very much improved.
See Appendix 2 for further details reported by Benjamin 1986.
As there were no data for this outcome, the quality of evidence is unknown, and there is no evidence to support or refute.
Patient Global Impression of Change much or very much improved
One study reported PGIC much or very much improved (Benjamin 1986).
Benjamin 1986 reported 'good + excellent' (an equivalent scale to much or very much improvement) for 9/16 participants in the cetiedil 0.2 mg/kg group, 10/18 participants in the cetiedil 0.3 mg/kg group, 11/13 participants in the cetiedil 0.4 mg/kg group and 4/16 participants in the placebo group.
As there were too few data for this outcome (limited number of participants per treatment arm), we had no confidence in the results and we downgraded the quality of evidence by three levels to very low quality.
Secondary outcomes
Opioid consumption
Neither study reported opioid consumption.
As there were no data for this outcome, the quality of evidence is unknown, and there is no evidence to support or refute.
Time to pain resolution
One study reported time to pain resolution.
Qari 2007 reported mean time to participants' resolution of pain as 61.68 hours in the tinzaparin group (127 participants) and 104.4 hours in the placebo group (126 participants).
We considered the available data for this outcome to be low‐quality evidence, downgraded once for sparse data, and once for high probability of reporting bias.
Length of hospitalization
One study reported length of hospitalization.
Qari 2007 reported mean length of hospitalization as 7.08 days hours in the tinzaparin group (127 participants) and 12.06 in the placebo group (126 participants).
We considered the available data for this outcome to be low‐quality evidence, downgraded once for sparse data, and once for high probability of reporting bias.
Participants experiencing any adverse or serious adverse event
One study reported adverse events and serious adverse events.
Benjamin 1986 reported headache, nausea, vomiting and dry mouth in 2/17 participants in the cetiedil 0.2 mg/kg group, 9/18 participants in the cetiedil 0.3 mg/kg group, 8/14 participants in the cetiedil 0.4 mg/kg group and 8/18 participants in the placebo group.
Benjamin 1986 reported no serious adverse events in the cetiedil 0.2 mg/kg group (0/16 participants), cetiedil 0.3 mg/kg group (0/18 participants), cetiedil 0.4 mg/kg group (0/13 participants) and the placebo group (0/16 participants).
As there were too few data for this outcome (limited number of participants per treatment arm), we had no confidence in the results and we downgraded the quality of evidence by three levels to very low quality.
Any pain‐related outcome indicating some improvement
Neither study reported any pain‐related outcome indicating some improvement.
As there were no data for this outcome, the quality of evidence is unknown, and there is no evidence to support or refute.
Withdrawals: due to lack of efficacy, adverse events and for any cause
Neither study reported withdrawals.
As there were no data for this outcome, the quality of evidence is unknown, and there is no evidence to support or refute.
Reports of tolerance or habituation to opioids
Neither study reported tolerance or habituation to opioids.
As there were no data for this outcome, the quality of evidence is unknown, and there is no evidence to support or refute.
Comparison 5: combination pharmacotherapy versus combination pharmacotherapy
One study compared ketorolac plus tramadol plus metoclopramide plus fentanyl (KTMF) with ketorolac plus tramadol plus metoclopramide (KTM) (De Franceschi 2016).
Primary outcomes
Participant‐reported pain relief of 50% or greater
De Franceschi 2016 reported participant‐reported pain relief of 50% or greater at six hours for 12/20 participants in the KTMF group and 0/20 participants in the KTM group.
De Franceschi 2016 reported participant‐reported pain relief of 50% or greater at 12 hours for 18/20 participants in the KTMF group and 4/20 participants in the KTM group.
As there were too few data for this outcome (limited number of participants per treatment arm), we had no confidence in the results and we downgraded the quality of evidence by three levels to very low quality.
Participant‐reported pain relief of 30% or greater
De Franceschi 2016 reported participant‐reported pain relief of 30% or greater at six hours for 20/20 participants in the KTMF group and 1/20 participants in the KTM group.
De Franceschi 2016 reported participant‐reported pain relief of 30% or greater at 12 hours for 20/20 participants in the KTMF group and 4/20 participants in the KTM group.
As there were too few data for this outcome (limited number of participants per treatment arm), we had no confidence in the results and we downgraded the quality of evidence by three levels to very low quality.
Patient Global Impression of Change very much improved
The study did not report PGIC very much improved.
As there were no data for this outcome, the quality of evidence is unknown, and there is no evidence to support or refute.
Patient Global Impression of Change much or very much improved
The study did not report PGIC much or very much improved.
As there were no data for this outcome, the quality of evidence is unknown, and there is no evidence to support or refute.
Secondary outcomes
Opioid consumption
The study did not report opioid consumption.
As there were no data for this outcome, the quality of evidence is unknown, and there is no evidence to support or refute.
Time to pain resolution
The study did not report time to pain resolution.
As there were no data for this outcome, the quality of evidence is unknown, and there is no evidence to support or refute.
Length of hospitalization
The study did not report length of hospitalization.
As there were no data for this outcome, the quality of evidence is unknown, and there is no evidence to support or refute.
Participants experiencing any adverse or serious adverse event
De Franceschi 2016 reported adverse events as 0/20 in the KTMF group and 0/20 in the KTM group.
De Franceschi 2016 reported serious adverse events as 0/20 in the KTMF group and 0/20 in the KTM group.
As there were too few data for this outcome (limited number of participants per treatment arm), we had no confidence in the results and we downgraded the quality of evidence by three levels to very low quality.
Any pain‐related outcome indicating some improvement
The study did not report any pain‐related outcome indicating some improvement.
As there were no data for this outcome, the quality of evidence is unknown, and there is no evidence to support or refute.
Withdrawals: due to lack of efficacy, adverse events and for any cause
De Franceschi 2016 reported total all‐cause withdrawals as 0/20 in the KTMF group and 0/20 in the KTM group.
De Franceschi 2016 reported withdrawals due to adverse events as 0/20 in the KTMF group and 0/20 in the KTM group.
As there were too few data for this outcome (limited number of participants per treatment arm), we had no confidence in the results and we downgraded the quality of evidence by three levels to very low quality.
Reports of tolerance or habituation to opioids
The study did not report tolerance or habituation to opioids.
As there were no data for this outcome, the quality of evidence is unknown, and there is no evidence to support or refute.
Discussion
Summary of main results
We included nine studies in this review with data for 638 VOC events and 594 participants aged 17 to 42 years with sickle cell disease presenting to a hospital emergency department in a painful VOC.
Three studies compared NSAID with placebo for which we were unable to undertake a meta‐analysis. One study compared an opioid with a placebo, two studies compared an opioid with an active comparator, two studies compared an anticoagulant with a placebo, and one study compared a combination of three drugs with a combination of four drugs.
Risk of bias across the nine studies varied. For randomization and allocation concealment, studies were mostly at an unclear risk of bias, with only three studies adequately describing their randomization methods (low risk). For blinding, studies primarily displayed an unclear risk of bias, with only two studies clearly describing blinding methods of participants, personnel and outcome assessors methods (low risk). For attrition, studies primarily displayed an unclear risk of bias, with only two studies clearly accounting for all participants (low risk), and one study not at all (high risk). For reporting, studies primarily displayed a low risk of bias, with two studies poorly reporting planned outcomes (high risk) and one study unclearly reporting planned outcomes (low risk). For size, studies primarily displayed a high risk of bias with fewer than 50 participants per treatment arm, and two studies with 50 to 199 participants per treatment arm (unclear risk). For other risks of bias, studies primarily displayed a low risk of bias, with two studies showing uncertainties in their procedures (unclear risk) and one study displaying a high risk of bias.
Pain outcomes: only one study (under comparison 5: Combination pharmacotherapy versus combination pharmacotherapy) reported participant‐reported pain relief of 50% or greater and 30% or greater; so due to a lack of available data we were unable to undertake a meta‐analysis (De Franceschi 2016). Three studies reported Patient Global Impression of Change very much improved, and much or very much improved, however, due to lack of data, we were unable to undertake a meta‐analysis.
1: Non‐steroidal anti‐inflammatory drugs (NSAID) compared with placebo
For PGIC very much improved, we found no differences between ketorolac and placebo (RR 7.64, 95% CI 0.44 to 131.75; P = 0.16; Analysis 1.1; Figure 3), or for PGIC much or very much improved (RR 1.65, 95% CI 0.65 to 4.19; P = 0.29; Analysis 1.2; Figure 4) (one study, 21 participants, very low‐quality evidence).
For opioid consumption (rescue morphine), we found no differences between ketoprofen (overall median consumption 110 mg, IQR 46 to 195) and placebo (88 mg, IQR 52.5 to 262.5) (one study, very low‐quality evidence). The same for rescue meperidine, no differences were reported between ketorolac (1866.7 mg, SD 1112.4) compared with placebo (2804.5 mg, SD 795.1), and ketorolac (231 mg, SD 92) compared with placebo (250 mg, SD 85) (two non‐comparable RCTs, very low‐quality evidence).
For median time to participants' resolution of pain, we found no differences between ketoprofen (51 hours, IQR 35.5 to 87) and placebo (50 hours, IQR 36 to 103) (33 participants per group, one study, very low‐quality evidence).
For mean length of hospitalization, we found a difference between ketorolac (3.3 days) and placebo (7.2 days) (P < 0.05) (21 participants, one study, very low‐quality evidence).
Mild adverse events were reported in the ketoprofen (16/33), placebo (19/45), and ketorolac (0/12) study arms (two studies, very low‐quality evidence). Serious adverse events (not specified) were reported in the ketoprofen (1/33), placebo (2/56), and ketorolac (0/22) study arms (three studies, very low‐quality evidence).
For any pain‐related outcome indication some improvement, we found a difference in mean change in pain scores between ketorolac and placebo, from baseline to 24 hours, on a VAS 0 to 100 mm: ketorolac baseline (77.7, 95% CI 69.1, 86.2) to 24 hours (58.6, 95% CI 48.6 to 68.5), and placebo baseline (79.1, 95% CI 72.1 to 86.0) to 24 hours (72.6, 95% CI 62.4 to 82.8) (P < 0.05) (one study, very low‐quality evidence). However, other scales used to measure pain (VCS 0 to 3, VRS 0 to 4, VAS at 4 hours) reported no differences between ketorolac and placebo, the same for ketoprofen compared with placebo (three studies, very low‐quality evidence).
Withdrawals due to adverse events were reported in ketoprofen (1/33), ketorolac (0/22), and placebo (1/56) (three studies, very low‐quality evidence).
2: Opioids compared with placebo
For any pain‐related outcome indication some improvement, we found no difference in mean pain scores between ketamine/hydromorphone and placebo/hydromorphone on a VAS (1 to 10 cm): ketamine/hydromorphone (5.2 cm; 95% CI 4.01 to 6.46; 40.2% decrease from baseline), placebo/hydromorphone (5.6 cm; 95% CI 4.27 to 6.93; 34.1% decrease from baseline) (one study, very low‐quality evidence).
3: Opioids compared with active comparator
For PGIC very much improved, the outcome was reported as 'excellent' in 19.1% of butorphanol and 33% of morphine events (one study, very low‐quality evidence).
For opioid consumption (rescue morphine 0.1 mg/kg taken at 30 minutes), we found no difference between morphine (27/54) and paracetamol participants (24/52) (one study, very low‐quality evidence).
Mild adverse events (nausea and vomiting) were reported in butorphanol (4/12), morphine (9/66), and paracetamol (3/52). No serious adverse events were reported in any treatment group (0/132 participants) (two studies, very low‐quality evidence).
For any pain‐related outcome indication some improvement, we found no difference between the adjusted mean pain scores using: VAS (0 to 100 mm) morphine (46.08 mm) and butorphanol (44.42 mm); or PRS (0 to 100 mm) morphine (55.50 mm) and butorphanol (43.79 mm) (one study, very low‐quality evidence).
4: Other active comparator compared with placebo
For PGIC much or very much improved, we found no differences between cetiedil 0.2 mg/kg (9/16 participants), cetiedil 0.3 mg/kg (10/18), cetiedil 0.4 mg/kg (11/13), and placebo (4/16) (one study, very low‐quality evidence).
For mean time to participants' resolution of pain, we found no differences between tinzaparin (61.68 hours, 127 participants) and placebo (104.4 hours, 126 participants) (one study, low quality evidence).
For mean length of hospitalization, we found no differences between tinzaparin (7.08 days, 127 participants) and placebo (12.06 days, 126 participants) (one study, low quality evidence).
Any adverse events (headache, nausea, vomiting and dry mouth) were reported in cetiedil 0.2 mg/kg (2/17), cetiedil 0.3 mg/kg (9/18), cetiedil 0.4 mg/kg (8/14), and placebo (8/18) (one study, very low‐quality evidence). No serious adverse events were reported in any groups (one study, very low‐quality evidence).
5: Combination pharmacotherapy compared with combination pharmacotherapy
For participant‐reported pain relief of 50% or greater, we found no differences between ketorolac plus tramadol plus metoclopramide plus fentanyl (KTMF) (12/20) and ketorolac plus tramadol plus metoclopramide (KTM) (0/20) at six hours, and no differences between KTMF (18/20) and KTM (4/20) at 12 hours (one study, very low‐quality evidence).
For participant‐reported pain relief of 30% or greater, we found this was reported in 20/20 participants for KTMF and in 1/20 participants for KTM (at six hours), and in 20/20 participants for KTMF and in 4/20 participants for KTM (at 12 hours) (one study, very low‐quality evidence).
Adverse events, serious adverse events, and total all‐cause withdrawals were reported in KTMF (0/20) and KTM (0/20) (one study, very low‐quality evidence).
Overall completeness and applicability of evidence
This review identified only nine studies, with insufficient data for all pharmacological interventions for analysis.
As we were unable to undertake a meta‐analysis for each drug comparison (only a summary of effects from individual studies), we were unable to comment about efficacy or harm from the use of pharmacological interventions to treat painful sickle cell VOC in adults. Similarly we could not comment on our remaining secondary outcomes: opioid consumption, time to pain resolution, length of hospitalization and opioid tolerance.
Quality of the evidence
Of the nine included studies, only four adequately described randomization methods, and only three adequately described single‐ or double‐blinding methods. One study displayed a high risk of attrition bias and two studies displayed a high risk of selective reporting bias. Seven studies displayed a high risk of size bias with study samples fewer than 50 participants per treatment arm, and the remaining two were unclear risk of size bias. Only seven studies provided sufficient information about adverse events, and four studies about withdrawals.
The studies recruited participants with adequate baseline pain in a VOC, but not all reported clinically useful outcome measures.
The studies themselves were of moderate quality; however, the number of studies and sample sizes for some comparisons were limited, given what is known about study size and estimates of effect for outcomes derived from studies with few participants and events (Dechartres 2013; Dechartres 2014; McQuay 1998; Nüesch 2010; Thorlund 2011).
We downgraded the quality of the evidence for our primary outcomes to very low quality for the reason:
Due to too few data for these outcomes (limited number of participants per treatment arm) as we had no confidence in the results.
In some comparisons, and for some outcomes, there were no data available, so we reported this as:
There were no data for this outcome, the quality of evidence is unknown, and there is no evidence to support or refute.
We applied the same approach to our secondary outcomes.
Potential biases in the review process
We used a comprehensive and highly sensitive search strategy in the major databases, in addition to two large clinical trial registries. We consider it unlikely that we missed relevant studies.
We followed standard Cochrane methods and there were no contestable decisions relating to the inclusion or exclusion of studies, data analyses, or assessing risk of bias. There were no contestable decisions relating to the synthesis of studies.
Agreements and disagreements with other studies or reviews
This is the first Cochrane Review, and the first systematic review that we are aware of that encompasses the search of all drugs implemented during a VOC.
Authors' conclusions
Implications for practice.
For people with sickle cell disease
The amount and quality of evidence around the use of any pharmacological treatment for painful sickle cell vaso‐occlusive crises (VOC) is very low. This means treatment is based on clinical experience and advice from respected authorities. We could make no judgement about which pharmacological treatment is more effective than any other to reduce the painful crisis. We could make no judgement about adverse events or withdrawals.
Sickle cell pain should be treated by specialists in this area.
For clinicians
The amount and quality of evidence around the use of any pharmacological treatment for painful sickle cell VOC is very low. This means treatment is based on clinical experience and advice from respected authorities. We could make no judgement about which pharmacological treatment is more effective than any other to reduce the painful crisis. We could make no judgement about adverse events or withdrawals.
Clinicians who do not have expertise in this field should defer to clinicians with specialist knowledge.
For policy makers and funders of the intervention
The amount and quality of evidence around the use of any pharmacological treatment for painful sickle cell VOC is very low. This means treatment is based on clinical experience and advice from respected authorities. We could make no judgement about which pharmacological treatment is more effective than any other to reduce the painful crisis. We could make no judgement about adverse events or withdrawals.
Specialist services treating people with sickle cell disease will need to be supported (by clinical expertise and multi‐factorial service models) to provide adequate treatment.
Implications for research.
The results of this review are disappointing with fewer than 600 participants included in relevant randomized controlled trials. While it would be easy to suggest more trials are needed, and they probably are, it may be that a different approach is necessary. One possible route is the establishment of suitable registries for this group of people which records interventions and outcomes. To achieve any progress, thought needs to be given to relevant outcomes for this patient group.
With the advent of precision medicine, the use of analgesics in general and opioids in particular should be based on recent advances in the pharmacodynamics and pharmacokinetics of opioids. Pharmacodynamically, opioids are ligands that bind and activate specific receptors that modulate the transmission of painful stimuli in the central nervous system. Accordingly, the analgesic potential of a specific opioid depends on the number of receptors and the ability of the opioid to bind to these receptors. Since these factors vary among people, so does the analgesic potential. In addition, pharmacokinetically, opioids are metabolized by specific enzymes into metabolites that could be analgesically active or inactive depending on the opioid being used. These enzymes could be duplicated, triplicated, mutated or deleted in different people thus causing significant variation in pain relief among patients. Hopefully, these pharmacological markers could be determined for each person in the future which allows the choice of the best opioid compatible with the person in question.
What's new
| Date | Event | Description |
|---|---|---|
| 18 February 2020 | Amended | Clarification added to Declarations of interest. |
| 22 November 2019 | Review declared as stable | See Published notes. |
History
Protocol first published: Issue 5, 2016 Review first published: Issue 11, 2019
| Date | Event | Description |
|---|---|---|
| 18 July 2017 | Amended | Contact details updated. |
Notes
The research area is not active and we do not expect new RCTs to be published. Therefore, this review has now been stabilised following discussion with the authors and editors. The review will be assessed for updating in five years. If appropriate, we will update the review before this date if new evidence likely to change the conclusions is published, or if standards change substantially which necessitate major revisions.
Acknowledgements
Institutional support is provided by the Oxford Pain Relief Trust.
Cochrane Review Group funding acknowledgement: this project was supported by the National Institute for Health Research (NIHR), via Cochrane Infrastructure funding to the Cochrane Pain, Palliative and Supportive Care Review Group (PaPaS). The views and opinions expressed herein are those of the review authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, National Health Service or the Department of Health.
The review was carried out in conjunction with the Cochrane Cystic Fibrosis and Genetic Disorders Group.
The authors would like to thank the following peer reviewers of the review: Ndi Euphrasia Ebai‐Atuh, and the two peer reviewers who did not wish to be named.
Appendices
Appendix 1. Database search strategies
CENTRAL search strategy (via CRSO)
#1 MeSH DESCRIPTOR Anemia, Sickle Cell EXPLODE ALL TREES
#2 (sickle cell):TI,AB,KY
#3 "SCD":TI,AB,KY
#4 ("sickling disorder"):TI,AB,KY
#5 "HBS":TI,AB,KY
#6 ((haemoglobin near1 disease)):TI,AB,KY
#7 ((haemoglobin near1 disease)):TI,AB,KY
#8 #1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 30
#9 MeSH DESCRIPTOR Pain EXPLODE ALL TREES
#10 pain*:TI,AB,KY
#11 #9 OR #10
#12 ((acetaminophen or "acetylsalicylic acid" or "alendronic acid" or alfentanil or amitriptyline or aspirin or baclofen or benzocaine or bupivacaine or buprenorphine or butorphanol or carbamazepine or chloroprocaine or "choline magnesium trisalicylate" or clonazepam or clonidine or codeine or dexamethasone or dexmetetomidine or dextroamphetamine or dextropropoxyphene or diamorphine or diazepam or diclofenac or dihydrocodeine or domperidone or fentanyl or fluoxetine or gabapentin or hydrocodone or hydromorphone or "hyoscine hydrobromide" or ibuprofen or ketamine or ketoprofen or ketorolac or "levo bupivacaine" or lidocaine or loperamide or lorazepam or mefenamic acid or meperidine or methadone or methylphenidate or midazolam or morphine or naproxen or nitrous oxide or nortriptyline or oxycodone or pamidronate or paracetamol or paroxetine or pentazocine or pethidine or phenobarbital or "phenytoin" or piroxicam or pregabalin or propoxyphene or "risedronate sodium" or "sodium clodronate" or tetracaine or tramadol or "valproic acid")):TI,AB,KY
#13 MeSH DESCRIPTOR Analgesics EXPLODE ALL TREES
#14 MeSH DESCRIPTOR Anesthesia, Local
#15 MeSH DESCRIPTOR Anti‐Inflammatory Agents, Non‐Steroidal EXPLODE ALL TREES
#16 #12 OR #13 OR #14 OR #15
#17 #8 AND #11 AND #16
MEDLINE search strategy (via Ovid)
1 exp Anemia, Sickle Cell/
2 sickle cell.tw.
3 "SCD".tw.
4 "sickling disorder".tw.
5 "HBS".tw.
6 (hemoglobin adj1 disease).tw.
7 (haemoglobin adj1 disease).tw.
8 or/1‐7
9 exp Pain/
10 pain*.tw.
11 or/9‐10
12 (acetaminophen or "acetylsalicylic acid" or "alendronic acid" or alfentanil or amitriptyline or aspirin or baclofen or benzocaine or bupivacaine or buprenorphine or butorphanol or carbamazepine or chloroprocaine or "choline magnesium trisalicylate" or clonazepam or clonidine or codeine or dexamethasone or dexmetetomidine or dextroamphetamine or dextropropoxyphene or diamorphine or diazepam or diclofenac or dihydrocodeine or domperidone or fentanyl or fluoxetine or gabapentin or hydrocodone or hydromorphone or "hyoscine hydrobromide" or ibuprofen or ketamine or ketoprofen or ketorolac or "levo bupivacaine" or lidocaine or loperamide or lorazepam or mefenamic acid or meperidine or methadone or methylphenidate or midazolam or morphine or naproxen or nitrous oxide or nortriptyline or oxycodone or pamidronate or paracetamol or paroxetine or pentazocine or pethidine or phenobarbital or "phenytoin" or piroxicam or pregabalin or propoxyphene or "risedronate sodium" or "sodium clodronate" or tetracaine or tramadol or "valproic acid").tw.
13 exp Analgesics/
14 Anesthesia, Local/
15 exp Anti‐Inflammatory Agents, Non‐Steroidal/
16 or/12‐15
17 8 and 11 and 16
18 randomized controlled trial.pt.
19 controlled clinical trial.pt.
20 randomized.ab.
21 placebo.ab.
22 drug therapy.fs.
23 randomly.ab.
24 trial.ab.
25 groups.ab.
26 18 or 19 or 20 or 21 or 22 or 23 or 24 or 25
27 exp animals/ not humans.sh.
28 26 not 27
29 17 and 28
Embase search strategy (via Ovid)
1 exp Anemia, Sickle Cell/
2 sickle cell.tw.
3 "SCD".tw.
4 "sickling disorder".tw.
5 "HBS".tw.
6 (hemoglobin adj1 disease).tw.
7 (haemoglobin adj1 disease).tw.
8 or/1‐7
9 exp Pain/
10 pain*.tw.
11 or/9‐10
12 (acetaminophen or "acetylsalicylic acid" or "alendronic acid" or alfentanil or amitriptyline or aspirin or baclofen or benzocaine or bupivacaine or buprenorphine or butorphanol or carbamazepine or chloroprocaine or "choline magnesium trisalicylate" or clonazepam or clonidine or codeine or dexamethasone or dexmetetomidine or dextroamphetamine or dextropropoxyphene or diamorphine or diazepam or diclofenac or dihydrocodeine or domperidone or fentanyl or fluoxetine or gabapentin or hydrocodone or hydromorphone or "hyoscine hydrobromide" or ibuprofen or ketamine or ketoprofen or ketorolac or "levo bupivacaine" or lidocaine or loperamide or lorazepam or mefenamic acid or meperidine or methadone or methylphenidate or midazolam or morphine or naproxen or nitrous oxide or nortriptyline or oxycodone or pamidronate or paracetamol or paroxetine or pentazocine or pethidine or phenobarbital or "phenytoin" or piroxicam or pregabalin or propoxyphene or "risedronate sodium" or "sodium clodronate" or tetracaine or tramadol or "valproic acid").tw.
13 exp Analgesics/
14 Anesthesia, Local/
15 exp Anti‐Inflammatory Agents, Non‐Steroidal/
16 or/12‐15
17 8 and 11 and 16
18 Randomized controlled trial/
19 Controlled clinical study/
20 18 or 19
21 Random$.ti,ab.
22 randomization/
23 intermethod comparison/
24 placebo.ti,ab.
25 (compare or compared or comparison).ti.
26 ((evaluated or evaluate or evaluating or assessed or assess) and (compare or compared or comparing or comparison)).ab.
27 (open adj label).ti,ab.
28 ((double or single or doubly or singly) adj (blind or blinded or blindly)).ti,ab.
29 double blind procedure/
30 parallel group$1.ti,ab.
31 (crossover or cross over).ti,ab.
32 ((assign$ or match or matched or allocation) adj5 (alternate or group$1 or intervention$1 or patient$1 or subject$1 or participant$1)).ti,ab.
33 (assigned or allocated).ti,ab.
34 (controlled adj7 (study or design or trial)).ti,ab.
35 (volunteer or volunteers).ti,ab.
36 human experiment/
37 trial.ti.
38 or/20‐37
39 17 and 38
LILACS search strategy (via BIRME)
(sickle cell) or SCD or (sickling disorder) or HBS or (haemoglobin disease) or (haemoglobin disease) [Words] and pain$ [Words] and (acetaminophen or (acetylsalicylic acid) or (alendronic acid) or alfentanil or amitriptyline or aspirin or baclofen or benzocaine or bupivacaine or buprenorphine or butorphanol or carbamazepine or chloroprocaine or (choline magnesium trisalicylate) or clonazepam or clonidine or codeine or dexamethasone or dexmetetomidine or dextroamphetamine or dextropropoxyphene or diamorphine or diazepam or diclofenac or dihydrocodeine or domperidone or fentanyl or fluoxetine or gabapentin or hydrocodone or hydromorphone or (hyoscine hydrobromide) or ibuprofen or ketamine or ketoprofen or ketorolac or (levo bupivacaine) or lidocaine or loperamide or lorazepam or mefenamic acid or meperidine or methadone or methylphenidate or midazolam or morphine or naproxen or nitrous oxide or nortriptyline or oxycodone or pamidronate or paracetamol or paroxetine or pentazocine or pethidine or phenobarbital or (phenytoin) or piroxicam or pregabalin or propoxyphene or (risedronate sodium) or (sodium clodronate) or tetracaine or tramadol or (valproic acid)) [Words]
Appendix 2. Primary outcome data
| Study | Treatment | Participant‐reported pain relief of 50% or greater | Participant‐reported pain relief of 30% or greater | Patient Global Impression of Change: both 'much improved' & 'much or very much improved' | Additional pain data reported by study |
| Arambasik 2013 |
Treatment group (17 participants; 17 VOC episodes):
Control group (20 participants; 20 VOC episodes):
|
Ketamine: no data Placebo: no data |
Ketamine: no data Placebo: no data |
Ketamine: no data Placebo: no data |
VAS (1–10); mean (% decrease from arrival) At arrival Ketamine: 8.7 (95% CI 8.07 to 9.29) Placebo: 8.5 (95% CI 7.90 to 9.05) |
|
At administration Ketamine: 6.0 (95% CI 4.71, to 7.29); 31% Placebo: 7.0 (95% CI 6.20 to 7.85); 17.6% | |||||
|
At disposition Ketamine: 5.2 (95% CI 4.01 to 6.46); 40.2% Placebo: 5.6 (95% CI 4.27 to 6.93); 34.1% | |||||
| Bartolucci 2009 |
Ketoprofen (33 VOC episodes): Days 1 and 2: continuous IV ketoprofen infusion, 300 mg/day, 2 days Days 3–5: oral ketoprofen 100 mg, every 8 hr, 3 days Placebo (33 VOC episodes): Days 1 and 2: physiological saline continuous IV infusion, 2 days. Days 3–5: oral placebo tablet, every 8 hr, 3 days |
Ketoprofen: no data Placebo: no data |
Ketoprofen: no data Placebo: no data |
Ketoprofen: no data Placebo: no data |
CPS (0–3) daily mean scores; median Ketoprofen: 0.4 (IQR 0.2–0.7) Placebo: 0.4 (IQR 0.2–0.7) |
| VAS (0–100) daily mean scores;median Ketoprofen: 12.6 (IQR 4.8–23.2) Placebo: 9.6 (IQR 5.8–33.2) | |||||
| Benjamin 1986 |
Cetiedil 0.2 (16 participants; 16 VOC episodes): 0.2 mg/kg IV infusion, every 8 hr, 4 days, total 12 doses Cetiedil 0.3 (18 participants; 18 VOC episodes): 0.3 mg/kg IV infusion, every 8 hr, 4 days, total 12 doses Cetiedil 0.4 (13 participants; 13 VOC episodes): 0.4 mg/kg IV infusion, every 8 hr, 4 days, total 12 doses Placebo (16 participants; 16 VOC episodes): saline IV infusion, every 8 hr, 4 days, total 12 doses |
Cetiedil 0.2 mg: no data Cetiedil 0.3 mg: no data Cetiedil 0.4 mg: no data Placebo: no data |
Cetiedil 0.2 mg: no data Cetiedil 0.3 mg: no data Cetiedil 0.4 mg: no data Placebo: no data |
Overall evaluation of treatment (good) Cetiedil 0.2 mg: 5/16 Cetiedil 0.3 mg: 4/18 Cetiedil 0.4 mg: 4/13 Placebo: 2/16 |
Cetiedil 0.2 mg: no data Cetiedil 0.3 mg: no data Cetiedil 0.4 mg: no data Placebo: no data |
| Overall evaluation of treatment (good + excellent) Cetiedil 0.2 mg: 9/16 Cetiedil 0.3 mg: 10/18 Cetiedil 0.4 mg: 11/13 Placebo: 4/16 | |||||
| De Franceschi 2016 |
Treatment group VOC1 (KTM) (10 participants; 20 VOC episodes): continuous IV infusion ketorolac 0.86 mg/kg/day + tramadol 7.2 mg/kg/day + metoclopramide 0.57 mg/kg/day, for maximum of 72 hr Treatment group VOC2 (KT MF) (10 participants; 20 VOC episodes): continuous IV infusion ketorolac 0.86 mg/kg/day + tramadol 7.2 mg/kg/day + metoclopramide 0.57 mg/kg/day + fentanyl buccal single tablet 100 μg after 3 hr from beginning of multimodal analgesia, for maximum of 72 hr |
At 6 hr KTM: 0/20 KTMF: 12/20 |
At 6 hr KTM: 1/20 KTMF: 20/20 |
KTM: no data KTMF: no data |
KTM: no data KTMF: no data |
| At 12 hr KTM: 4/20 KTMF: 18/20 |
At 12 hr KTM: 4/20 KTMF: 20/20 |
||||
| Gonzalez 1988 |
Butorphanol (9 participants; 12 VOC episodes): IM butorphanol 2 mg, repeated within 30–60 min until pain relief obtained, repeat every 2–4 hr, until discharge. Morphine (9 participants; 12 VOC episodes): IM morphine 6 mg, repeat within 30–60 min until pain relief obtained, repeated every 2–4 hr, until discharge |
Butorphanol: no data Morphine: no data |
Butorphanol: no data Morphine: no data |
Good Butorphanol: 47.6% Morphine: 38.1% |
VAS (0–100 mm); adjusted means overall Butorphanol: 44.42 Morphine: 46.08 |
| Excellent Butorphanol: 19.1% Morphine: 33.3% |
Pain relief (0–100 mm); adjusted means overall Butorphanol: 43.79 Morphine: 55.50 |
||||
| Perlin 1994 |
Ketorolac (10 participants; 10 VOC episodes): ketorolac 30 mg bolus; continuous IV infusion ketorolac 120 mg, 5 mg/hr, total dose 150 mg (on first day), maximum 120 mg/day, up to 5 days Placebo (11 participants; 11 VOC episodes): bolus saline; continuous IV infusion saline, up to 5 days |
Ketorolac: no data Placebo: no data |
Ketorolac: no data Placebo: no data |
Better Ketorolac: 3/10 Placebo: 4/11 |
VAS (0–100 mm); mean At baseline Ketorolac: 77.7 (95% CI 69.1 to 86.2) Placebo: 79.1 (95% CI 72.1 to 86.0) At 24 hr Ketorolac: 58.6 (95% CI 48.6 to 68.5) Placebo: 72.6 (95% CI 62.4 to 82.8) P < 0.05 |
| Much better Ketorolac: 3/10 Placebo: 0/11 |
VCS (0–3); mean At baseline Ketorolac: 2.5 Placebo: 2.6 At 24 hr Ketorolac: 2.0 Placebo: 2.4 |
||||
| Better + much better Ketorolac: 6/10 Placebo: 4/11 |
VRS (0–4) at 24 hr; mean Ketorolac: 1.8 Placebo: 1.9 |
||||
| Qari 2007 |
Tinzaparin (127 participants; 127 VOC episodes): subcutaneous tinzaparin 175 IU/kg, once daily, 7 days Placebo (126 participants; 126 VOC episodes): subcutaneous placebo, once daily, 7 days |
Tinzaparin: no data Placebo: no data |
Tinzaparin: no data Placebo: no data |
Tinzaparin: no data Placebo: no data |
Tinzaparin: no data Placebo: no data |
| Rehmani 2013 |
Morphine (54 participants; 54 VOC episodes): IV morphine 0.1 mg/kg, single dose Paracetamol (52 participants; 52 VOC episodes): IV paracetamol 1 g, single dose |
Morphine: no data Paracetamol: no data |
Morphine: no data Paracetamol: no data |
Morphine: no data Paracetamol: no data |
VAS (0–100 mm) at 30 min; mean Morphine: 44 (95% CI 33 to 56) Paracetamol: 41 (95% CI 32 to 49) P = 0.72 |
| Wright 1992 |
Ketorolac (12 VOC episodes): IM ketorolac 60 mg, single dose Placebo (12 VOC episodes): IM saline, single dose |
Ketorolac: no data Placebo: no data |
Ketorolac: no data Placebo: no data |
Ketorolac: no data Placebo: no data |
VAS (0–100 mm) at baseline; mean Ketorolac: 70.3 Placebo: 79.3 P = 0.26 |
| VAS (0–100 mm) at 4 hr; mean Ketorolac: 44 (SD 34) Placebo: 37 (SD 31) P = 0.49 | |||||
| CI: confidence interval; CPS: categorical pain scale; hr: hour; IM: intramuscular; IQR: interquartile range; IV: intravenous; KTM: ketorolac + tramadol + metoclopramide; KTMF: ketorolac + tramadol + metoclopramide + fentanyl;min: minute; SD: standard deviation; VAS: visual analogue scale; VCS: visual categorical score; VOC: vaso‐occlusive crisis; VRS: verbal rating scale. | |||||
Appendix 3. Secondary outcome data
| Study | Treatment | Opioid consumption | Time to pain resolution (hours) | Length of hospitalization (days) | Tolerance to opioids |
| Arambasik 2013 |
Treatment group (17 participants; 17 VOC episodes):
Control group (20 participants; 20 VOC episodes):
|
Ketamine: no data Placebo: no data |
Ketamine: no data Placebo: no data |
Ketamine: no data Placebo: no data |
Ketamine: no data Placebo: no data |
| Bartolucci 2009 |
Ketoprofen (33 VOC episodes): Day 1 and 2: continuous IV infusion ketoprofen, 300 mg/day, 2 days Days 3–5: 100 mg oral ketoprofen, every 8 hr, 3 days Placebo (33 VOC episodes): Day 1 and 2: physiological saline continuous IV infusion, 2 days Days 3–5: oral placebo tablet, every 8 hr, 3 days |
Median, mg Ketoprofen: 110 (IQR 46–195) Placebo: 88 (IQR 52.5–262.5) |
Median, hours Ketoprofen: 51 (IQR 35.5–87) Placebo: 50 (IQR 36–103) |
Ketoprofen: no data Placebo: no data |
Ketoprofen: no data Placebo: no data |
| Benjamin 1986 |
Cetiedil 0.2 (16 participants; 16 VOC episodes): 0.2 mg/kg IV infusion, every 8 hr, 4 days, total 12 doses Cetiedil 0.3 (18 participants; 18 VOC episodes): 0.3 mg/kg IV infusion, every 8 hr, 4 days, total 12 doses Cetiedil 0.4 (13 participants; 13 VOC episodes): 0.4 mg/kg IV infusion, every 8 hr, 4 days, total 12 doses Placebo (16 participants; 16 VOC episodes): saline IV infusion, every 8 hr, 4 days, total 12 doses |
Cetiedil 0.2 mg: no data Cetiedil 0.3 mg: no data Cetiedil 0.4 mg: no data Placebo: no data P > 0.05 |
Cetiedil 0.2 mg: no data Cetiedil 0.3 mg: no data Cetiedil 0.4 mg: no data Placebo: no data P > 0.05 |
Cetiedil 0.2 mg: no data Cetiedil 0.3 mg: no data Cetiedil 0.4 mg: no data Placebo: no data |
Cetiedil 0.2 mg: no data Cetiedil 0.3 mg: no data Cetiedil 0.4 mg: no data Placebo: no data |
| De Franceschi 2016 |
Treatment group VOC1 (KTM) (10 participants; 20 VOC episodes): continuous IV infusion ketorolac 0.86 mg/kg/day + tramadol 7.2 mg/kg/day + metoclopramide 0.57 mg/kg/day, for maximum of 72 hr Treatment group VOC2 (KTMF) (10 participants; 20 VOC episodes): continuous IV infusion ketorolac 0.86 mg/kg/day + tramadol 7.2 mg/kg/day + metoclopramide 0.57 mg/kg/day + fentanyl buccal single tablet 100 μg after 3 hr from beginning of multimodal analgesia, for maximum of 72 hr |
KTM: no data KTMF: no data |
KTM: no data KTMF: no data |
KTM: no data KTMF: no data |
KTM: no data KTMF: no data |
| Gonzalez 1988 |
Butorphanol (9 participants; 12 VOC episodes): IM butorphanol 2 mg, repeated within 30–60 min until pain relief obtained, repeat every 2–4 hr, until discharge Morphine (9 participants; 12 VOC episodes): IM morphine 6 mg, repeat within 30–60 min until pain relief obtained, repeated every 2–4 hr, until discharge |
Morphine: no data butorphanol: no data |
Butorphanol: no data Morphine: no data |
Butorphanol: no data Morphine: no data |
Butorphanol: no data Morphine: no data |
| Perlin 1994 |
Ketorolac (10 participants; 10 VOC episodes): bolus ketorolac 30 mg; continuous IV infusion ketorolac 120 mg, 5 mg/hr, total dose 150 mg (on first day), maximum 120 mg/day, up to 5 days Placebo (11 participants; 11 VOC episodes): bolus saline; continuous IV infusion saline, up to 5 days |
Meperidine mg mean total dose Ketorolac: 1866.7 (SD 1112.4) Placebo: 2804.5 (SD 795.1) |
Ketorolac: no data Placebo: no data |
Ketorolac: 3.3 Placebo: 7.2 P < 0.05 |
Ketorolac: no data Placebo: no data |
|
Mean daily dose Ketorolac: 523.6 (SD 222.1) Placebo: 662.4 (SD 68.6) | |||||
| Qari 2007 |
Tinzaparin (127 participants; 127 VOC episodes): subcutaneous tinzaparin 175 IU/kg, once daily, 7 days Placebo (126 participants; 126 VOC episodes): subcutaneous placebo, once daily, 7 days |
Morphine Tinzaparin: unclear Placebo: unclear |
Mean hours Tinzaparin: 61.68 Placebo: 104.4 |
Mean days Tinzaparin: 7.08 Placebo: 12.06 |
Tinzaparin: no data Placebo: no data |
| Rehmani 2013 |
Morphine (54 participants; 54 VOC episodes): IV morphine 0.1 mg/kg, single dose Paracetamol (52 participants; 52 VOC episodes): IV paracetamol 1 g, single dose |
Rescue morphine (0.1 mg/kg) at 30 min: Morphine: 27/54 Paracetamol: 24/52 |
Morphine: no data Paracetamol: no data |
Morphine: no data Paracetamol: no data |
Morphine: no data Paracetamol: no data |
| Wright 1992 |
Ketorolac (12 VOC episodes): IM ketorolac 60 mg, single dose Placebo (12 VOC episodes): IM saline, single dose |
Mean (SD) meperidine mg Ketorolac: 231(92) Placebo: 250(85) P = 0.61 |
Ketorolac: no data Placebo: no data |
Ketorolac: no data Placebo: no data |
Ketorolac: no data Placebo: no data |
| CPS: categorical pain scale; hr: hour; IM: intramuscular; IQR: interquartile range; IV: intravenous; KTM: ketorolac + tramadol + metoclopramide; KTMF: ketorolac + tramadol + metoclopramide + fentanyl;min: minutes; SD: standard deviation; VAS: visual analogue scale; VCS: visual categorical score; VOC: vaso‐occlusive crisis; VRS: verbal rating scale. | |||||
Appendix 4. Adverse events and withdrawals
| Study | Treatment | Total number of participants reporting ≥ 1adverse event | Serious adverse events | Total all‐cause withdrawals | Withdrawals due to adverse events |
| Arambasik 2013 |
Treatment group (17 participants; 17 VOC episodes):
Control group (20 participants; 20 VOC episodes):
|
Ketamine: no data Placebo: no data |
Ketamine: no data Placebo: no data |
Ketamine: no data Placebo: no data |
Ketamine: no data Placebo: no data |
| Bartolucci 2009 |
Ketoprofen (33 VOC episodes): Days 1 and 2: continuous IV infusion ketoprofen 300 mg/day, 2 days Days 3–5: 100 mg oral ketoprofen, every 8 hr, 3 days Placebo (33 VOC episodes): Days 1 and 2: physiological saline continuous IV infusion, 2 days Days 3–5: oral placebo tablet, every 8 hr, 3 days |
Ketoprofen: 16/33 Placebo: 19/33 |
Ketoprofen: 1/33 Placebo: 2/33 |
Ketoprofen: unclear Placebo: unclear |
Ketoprofen: 1/33 Placebo: 1/33 |
| Benjamin 1986 |
Cetiedil 0.2 (16 participants; 16 VOC episodes): 0.2 mg/kg IV infusion, every 8 hr, 4 days, total 12 doses Cetiedil 0.3 (18 participants; 18 VOC episodes): 0.3 mg/kg IV infusion, every 8 hr, 4 days, total 12 doses Cetiedil 0.4 (13 participants; 13 VOC episodes): 0.4 mg/kg IV infusion, every 8 hr, 4 days, total 12 doses Placebo (16 participants; 16 VOC episodes): saline IV infusion, every 8 hr, 4 days, total 12 doses |
Cetiedil 0.2 mg: 2/17 Cetiedil 0.3 mg: 9/18 Cetiedil 0.4 mg: 8/14 Placebo: 8/18 "The incidence of adverse effects was similar for the cetiedil 0.3, 0.4 and placebo groups, and significantly lower for the cetiedil 0.2 group (p = 0.04). The most commonly reported AE were headache and nausea and committing, and dry mouth." |
Cetiedil 0.2 mg: 0/16 Cetiedil 0.3 mg: 0/18 Cetiedil 0.4 mg: 0/13 Placebo: 0/16 |
Cetiedil 0.2 mg: unclear Cetiedil 0.3 mg: unclear Cetiedil 0.4 mg: unclear Placebo: unclear |
Cetiedil 0.2 mg: unclear Cetiedil 0.3 mg: unclear Cetiedil 0.4 mg: unclear Placebo: unclear |
| De Franceschi 2016 |
Treatment group VOC1 (KTM) (10 participants; 20 VOC episodes): continuous IV infusion ketorolac 0.86 mg/kg/day + tramadol 7.2 mg/kg/day + metoclopramide 0.57 mg/kg/day, for maximum of 72 hr Treatment group VOC2 (KT MF) (10 participants; 20 VOC episodes): continuous IV infusion ketorolac 0.86 mg/kg/day + tramadol 7.2 mg/kg/day + metoclopramide 0.57 mg/kg/day + fentanyl buccal single tablet 100 μg after 3 hr from beginning of multimodal analgesia, for maximum of 72 hr |
KTM: 0/20 KTMF: 0/20 |
KTM: 0/20 KTMF: 0/20 |
KTM: 0/20 KTMF: 0/20 |
KTM: 0/20 KTMF: 0/20 |
| Gonzalez 1988 |
Butorphanol (9 participants; 12 VOC episodes): IM butorphanol 2 mg, repeated within 30–60 min until pain relief obtained, repeat every 2–4 hr, until discharge. Morphine (9 participants; 12 VOC episodes): IM morphine 6 mg, repeat within 30–60 min until pain relief obtained, repeated every 2–4 hr, until discharge |
Butorphanol: 4/12 (nausea and vomiting) Morphine: 4/12 (nausea and vomiting) |
Butorphanol: 0/12 Morphine: 0/12 |
Butorphanol: no data Morphine: no data |
Butorphanol: no data Morphine: no data |
| Perlin 1994 |
Ketorolac (10 participants; 10 VOC episodes): bolus ketorolac 30 mg; continuous IV infusion ketorolac 120 mg, 5 mg/hr, total dose 150 mg (on first day), maximum 120 mg/day, up to 5 days Placebo (11 participants; 11 VOC episodes): bolus saline; continuous IV infusion saline, up to 5 days |
Ketorolac: unclear Placebo: unclear |
Ketorolac: 0/10 Placebo: 0/11 |
Ketorolac: 1/10 Placebo: 0/11 |
Ketorolac: 0/10 Placebo: 0/11 |
| Qari 2007 |
Tinzaparin (127 participants; 127 VOC episodes): subcutaneous tinzaparin 175 IU/kg, once daily, 7 days Placebo (126 participants; 126 VOC episodes): subcutaneous placebo, once daily, 7 days |
Tinzaparin: no data Placebo: no data |
Tinzaparin: no data Placebo: no data |
Tinzaparin: no data Placebo: no data |
Tinzaparin: no data Placebo: no data |
| Rehmani 2013 |
Morphine (54 participants; 54 VOC episodes): IV morphine 0.1 mg/kg, single dose Paracetamol (52 participants; 52 VOC episodes): IV paracetamol 1 g, single dose |
Morphine: 5/54 Paracetamol: 3/52 |
Morphine: 0/54 Paracetamol: 0/52 |
Morphine: no data Paracetamol: no data |
Morphine: no data Paracetamol: no data |
| Wright 1992 |
Ketorolac (12 VOC episodes): IM ketorolac 60 mg, single dose Placebo (12 VOC episodes): IM saline, single dose |
Ketorolac: 0/12 Placebo: 0/12 |
Ketorolac: 0/12 Placebo: 0/12 |
Ketorolac: 0/12 Placebo: 0/12 |
Ketorolac: 0/12 Placebo: 0/12 |
| CPS: categorical pain scale; hr: hour; IM: intramuscular; IQR: interquartile range; IV: intravenous; KTM: ketorolac + tramadol + metoclopramide; KTMF: ketorolac + tramadol + metoclopramide + fentanyl;VAS: visual analogue scale; VCS: visual categorical score; VOC: vaso‐occlusive crisis; VRS: verbal rating scale. | |||||
Data and analyses
Comparison 1. Non‐steroidal anti‐inflammatory drugs (NSAIDs) versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Patient Global Impression of Change very much improved | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.1 Ketorolac 30 mg bolus + 120 mg IV vs placebo bolus + IV | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Patient Global Impression of Change much or very much improved | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2.1 Ketorolac 30 mg bolus + 120 mg IV vs placebo bolus + IV | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Opioid consumption | 3 | Std. Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 3.1 Ketoprofen 300 mg IV + 100 mg oral vs saline IV + oral | 1 | Std. Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Ketorolac 30 mg bolus + 120 mg IV vs placebo bolus + IV | 1 | Std. Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.3 Ketorolac 60 mg IM vs saline IM | 1 | Std. Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Time to pain resolution (hours) | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 4.1 Ketoprofen 300 mg IV + 100 mg oral vs saline IV + oral | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Arambasik 2013.
| Methods |
Allocation: randomized Controlled: placebo and active comparator Blinding: double‐blind Arm: 2‐arm, parallel treatment group, pilot study Centre: single centre Study dates and duration: 16‐month period trial (June 2011 to October 2012) |
|
| Participants |
Inclusion criteria: not reported Exclusion criteria: not reported Baseline characteristics N: 37 participants; 37 VOC episodes Age (mean): 29.9 years Gender: F 13, M 24 Number randomized: 17 intervention, 20 control Number completed: number of participants at each follow‐up time not explicitly stated Setting of recruitment and treatment: not reported Country and sites: Akron General Medical Centre, Akron, OH, USA |
|
| Interventions |
Duration of treatment: individual time to resolution of crisis Follow‐up period: not reported Treatment group (17 participants; 17 VOC episodes):
Control group (20 participants; 20 VOC episodes):
Cointerventions/additional analgesia: not reported |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes | Abstract only, sufficient data on pain scores Sources of funding: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk |
Quote: "this pilot study was randomised, prospective, double‐blinded trial." Comment: unclear details of randomization. |
| Allocation concealment (selection bias) | Unclear risk |
Quote: "this pilot study was randomised, prospective, double‐blinded trial." Comment: unclear details of concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk |
Quote: "this pilot study was... double‐blind..." Comment: insufficient information, unclear method of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: insufficient information from abstract. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: insufficient information from abstract. |
| Selective reporting (reporting bias) | Unclear risk | Comment: primary outcome was reported in methods and results. However, unclear what other outcomes were from this abstract alone. |
| Size | High risk | Comment: total 37 participants, fewer than 50 participants per treatment arm. |
| Other bias | Unclear risk | Comment: unclear due to only abstract available. |
Bartolucci 2009.
| Methods |
Allocation: randomized Controlled: placebo and active comparator Blinding: double‐blind Arm: 2‐arm, parallel treatment groups Centre: single centre Study duration: 39‐month period trial (August 2000 to March 2003) |
|
| Participants |
Inclusion criteria: homozygous SCD participants' who had a severe VOC requiring hospitalization in the internal medicine department. Exclusion criteria: VOC lasting > 72 hr or < 24 hr; parenteral hydration > 24 hr; blood transfusion during the previous month; any NSAID intake during the previous 7 days; pregnancy; history of drug abuse; hypertension; leukocyte count > 30 × 109/L or < 4 × 109/L; presence at inclusion of an acute chest syndrome, defined as the association of 2 criteria among chest pain, radiological infiltrate and auscultatory abnormality; severe anaemia requiring a blood transfusion at inclusion; psychiatric disorder or progressive visceral disease; and ketoprofen allergy or NSAID contraindication; peptic ulcer or treated with 1 of the following drugs the week before enrolment: aspirin, valproic acid, macrolides, anti‐H2, imidazole, rifampicin, phenobarbital, carbamazepine, phenytoin, heparin, vitamin K antagonist, ticlopidine, lithium, methotrexate, interferon‐α, diuretics or antihypertensive agents Baseline characteristics N: 54 participants; 66 VOC episodes Gender: F 20, M 34 Age (mean): ≥ 15 years overall: 27 (SD 7) years intervention; 26 (SD 7) years control Number randomized: 33 intervention; 33 control (VOC episodes) Number completed: 26 intervention; 26 control (VOC episodes) Setting and location: Adult Sickle Cell Referral Centre, Paris University Hospital, France |
|
| Interventions |
Treatment group (33 VOC episodes): Days 1 and 2:
Days 3–5:
Control group (33 VOC episodes): Days 1 and 2:
Days 3–5:
Duration of treatment: 5 days Follow‐up period: up to 14 days postdischarge Cointerventions/additional analgesia: adjunctive standardized treatment: bed rest, < 3 L of 5% glucose infusion, 1 L/day oral alkaline water, 20 mg/day folic acid, morphine dose not reported and IV paracetamol 2 g every 8 hr for 48 hr, then 1 g every 8 hr) |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes | Sources of funding: Inserm was the trial promoter. Publication costs of the article were defrayed in part by page charge payments. The article was marked as advertising. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk |
Quote: "patients were randomly assigned in a 1:1 ratio to either the intervention or control group." Comment: no details of randomization method. |
| Allocation concealment (selection bias) | Unclear risk | Comment: insufficient information of concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: no information of details of the blinding process for either IV saline or oral placebo. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: no statement of outcome assessment of blinding. |
| Incomplete outcome data (attrition bias) All outcomes | High risk |
Quote: "7 VOCs each group were excluded from the analysis because of treatment failures." Comment: 20% attrition, and similar in each group. But outcome reporting ignored these randomized participants, and analyses did not seem to be ITT. |
| Selective reporting (reporting bias) | Low risk | Comment: all end points listed in the methods were reported in the results. |
| Size | High risk | Comment: total participants 54 (66 events), < 50 participants per treatment arm. |
| Other bias | Unclear risk | Comment: unit of analysis error. Unit of randomization was the painful crisis (66 randomized crises among 54 participants), but unit of analysis ignored the clustered nature of the data; analysed as if all randomized events were independent. |
Benjamin 1986.
| Methods |
Allocation: randomized Controlled: placebo Blinding: double‐blind Arm: 4 arms, parallel treatment groups Centre: multicentre (5 centres) Study dates and duration: not reported |
|
| Participants |
Inclusion criteria: known history of SCD (SS, SB/Thal), in a crisis lasting 4–24 hr. 1 crisis per participant Exclusion criteria: pregnant, women of childbearing potential, incarceration, drug abuse history, transfusion 90 days prior to trial, acute cerebrovascular accident, overt infection, renal failure with a serum creatinine concentration > 2 mg percent (0.18 mmol/L), clinical or roentgenographic evidence of pulmonary oedema, glaucoma, urinary retention, high sensitivity to anticholinergic or atropine‐like drugs Baseline characteristics N: 67 participants; 67 VOC episodes Age: ≥ 19 years (67% aged 20–29) years Gender: F 27, M 36 Setting and location: 5 state and university hospitals, and clinical centres, USA Number randomized: 16 cetiedil 0.2 mg/kg group; 18 cetiedil 0.3 mg/kg group; 13 cetiedil 0.4 mg/kg group; 16 control group Number completed: 16 cetiedil 0.2 mg/kg group; 18 cetiedil 0.3 mg/kg group; 13 cetiedil 0.4 mg/kg group; 16 control group (63 included in efficacy analysis, 4 high haemoglobin A, all 67 included in safety analysis |
|
| Interventions |
Treatment group A (16 participants; 16 VOC episodes):
Treatment group B (18 participants; 18 VOC episodes):
Treatment group C (13 participants; 13 VOC episodes):
Control group (16 participants; 16 VOC episodes):
Duration of treatment period: 4 days Follow‐up period: not reported Cointerventions/additional analgesia: additional analgesics if required – limited to parenteral meperidine hydrochloride 1.2 mg/kg, or oral paracetamol 300 mg with codeine phosphate 30 mg. Standard fluid replacement therapy |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes | Sources of funding: supported by Johnson and Johnson Baby Products, McNeil Pharmaceutical, Inc and by General Clinical Research Center Grants (RR 00102 and RR 00046) from the National Institutes of Health and by the Veterans Administration Medical Service Research Fund. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were randomly assigned at a central data centre." "The randomization plan specified a coded ampule and a volume dosage for each patient." |
| Allocation concealment (selection bias) | Low risk | Quote: "Drug, active or placebo, was presented in identically appearing ampules. The investigators were aware of the volume of medication but were not aware of the ampule's contents." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The study was double‐blind." "The investigators were aware of the volume of medication by were not aware of the ampule's contents." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk |
Quote: "The investigators were aware of the volume of medication but were not aware of the ampule's contents." Comment: not entirely clear whether this was extended to the pain evaluators or research nurses being blind to the treatment groups when assessing pain. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk |
Quote: "Sixty‐seven patients were entered in the trial. Sixty‐three patients were included in the analysis of efficacy. Four patients were found retrospectively to have high hemoglobin." Comment: reasonable retroactive application of exclusion criteria. Likely to be fairly small attrition bias effect, but small numbers per treatment group, so this randomization 'error' was unfortunate. |
| Selective reporting (reporting bias) | Low risk | Comment: no protocol available, but all planned end points were reported in results. |
| Size | High risk | Comment: total participants 67, < 50 participants per treatment arm. |
| Other bias | Low risk | Comment: no other possible sources of bias. |
De Franceschi 2016.
| Methods |
Allocation: randomized Controlled: active comparators ‐ combination pharmacotherapy Blinding: double‐blind Arm: 2‐arm crossover treatment Centre: single centre Study duration: January 2010 to July 2013 |
|
| Participants |
Inclusion criteria: adults (aged 18–45 years) prior diagnosis of SCD, presenting to emergency department with severe VOC (VAS 7+). Exclusion criteria: opioid consumption in prior 2 weeks; history of opioid consumption (VOC or non‐SCD related); altered conscious state; pregnancy or lactating; hepatic or renal failures (or both); gastritis; peptic ulcer; allergies to study drugs; cardiopulmonary disease, psychiatric conditions which may compromise study responses. Baseline characteristics N: 20 participants; 40 VOC episodes Gender: F 11, M 9 Age (median): 22 years (range 17–28 years) Number randomized: 10 intervention; 10 control (VOC episodes) Number completed: 10 intervention; 10 control (VOC episodes) Setting and location: Department of Medicine, University of Verona, Italy |
|
| Interventions |
Treatment group VOC1 (10 participants; 20 VOC episodes):
Treatment group VOC2 (10 participants; 20 VOC episodes):
|
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes | Sources of funding: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Comment: not applicable, crossover trial. |
| Allocation concealment (selection bias) | Unclear risk | Comment: insufficient information. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk |
Quote: "data were collected by blinded physicians and nurses." Comment: insufficient information. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "data were collected by blinded physicians and nurses." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: all participants were accounted for from randomization through allocation, results and follow‐up. |
| Selective reporting (reporting bias) | Low risk | Comment: study authors reported all of the outcomes planned in their methods. |
| Size | High risk | Comment: total participants 20, < 50 participants per treatment arm. |
| Other bias | Low risk | Comment: we found no other potential sources of bias. |
Gonzalez 1988.
| Methods |
Allocation: randomized Controlled: active comparator Blinding: double‐blind Arm: 2‐arm, parallel treatment groups Centre: single centre Study duration: not reported |
|
| Participants |
Inclusion criteria: adults aged 18–65 years with SCD treated in the emergency department Exclusion criteria: history of drug or alcohol abuse; opioid tolerance; hypersensitivity; allergy to morphine or butorphanol; pregnancy; breastfeeding; acute myocardial infarction within 6 months; daily use of narcotic analgesic during the last week Baseline characteristics N: 18 participants Gender: F 6, M 12 Age (mean): 29.3 (SD 7.7) years Number randomized: 9 butorphanol; 9 morphine Number completed: 9 butorphanol; 9 morphine Setting and location: primary emergency department, Medical College of Virginia Hospital, USA |
|
| Interventions |
Duration of treatment: not explicitly stated Follow‐up period: not explicitly stated Treatment group (9 participants; 12 VOC episodes):
Control group (9 participants; 12 VOC episodes):
Standard treatments to all groups: dextrose 5% and 0.45% saline IV infusion 150 mL/hr Cointerventions: IM prochlorperazine 5–10 mg as needed for nausea or vomiting Cointerventions/additional analgesia: standard IV hydration |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes | Sources of funding: supported by a grant from Bristol‐Meyers United States Pharmaceutical Group, Evansville, IN, USA. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "patients were randomly assigned on each visit to receive either 2 mg IM butorphanol or 6 mg IM morphine." |
| Allocation concealment (selection bias) | Unclear risk | Comment: no technical details of allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "the patient and the medical and nursing staff were blinded to the identity of the assigned drug." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk |
Quote: "the patient and the medical and nursing staff were blinded to the identity of the assigned drug" Comment: nursing staff made patient assessments, so likely to be adequate. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: all participants were accounted for from randomization, through treatment, to follow‐up. However, see 'Size' and 'Other Bias' domains below. |
| Selective reporting (reporting bias) | Low risk | Comment: no protocol available, but all planed outcomes were reported in results. |
| Size | High risk | Comment: total 18 participants (45 events), some randomized twice, < 50 participants per treatment arm. |
| Other bias | High risk | Comment: unit of analysis issue, 12 events per treatment arm. However, unit of randomization was the painful crisis (45 randomized crises events among 18 participants), random‐effects analysis took this unit of analysis issue appropriately into account. |
Perlin 1994.
| Methods |
Allocation: randomized Controlled: placebo Blinding: double‐blind Arm: 2‐arm, parallel‐group design Centre: single centre Study Duration: unclear |
|
| Participants |
Inclusion criteria: aged ≥ 15 years, people with SCD admitted to emergency department with pain of VOC Exclusion criteria: active peptic ulcer disease; systemic bleeding disorders; impaired renal function (urea > 20 mg/dL /or serum creatinine > 1 mg/dL, or both); other medical condition; history of hypersensitivity to NSAIDs; pregnant women Baseline characteristics N: 21 participants; 21 VOC episodes Gender: F 10, M 11 Age (range): 19–41 years Number randomized: 10 intervention; 11 control Number completed: 9 intervention; 9 control Setting and location: Howard University Hospital, Washington, DC, USA |
|
| Interventions |
Treatment group (10 participants; 10 VOC episodes):
Control group (11 participants; 11 VOC episodes):
Duration of treatment: duration of crisis, 5‐day infusion period Follow‐up period: not stated Cointerventions/additional analgesia: supplemental IM meperidine 100 mg as needed, frequency no more than 3 hr |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes | Sources of funding: supported in part by a grant from Syntex Research, a division of Syntex (USA), Inc, who manufacture ketorolac as Toradol | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "the study was randomized," "The study drugs were prepared by a designated hospital pharmacist (J.P.) and allocated according to a predetermined, computer generated random code, balanced in blocks of four." |
| Allocation concealment (selection bias) | Low risk | Comment: preparations by a hospital pharmacist, not study investigators. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk |
Quote: "the study drugs were prepared by a designated hospital pharmacist (J.P.) and allocated according to a predetermined, computer generated random code, balanced in blocks of four." Comment: not explicitly stated, but likely that investigators remained blinded to treatment allocation. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: not explicitly stated, but likely that investigators remained blinded to treatment allocation. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk |
Quote: "3 participants withdrawn prematurely (1‐active, 2‐control)...ITT analysis to account for withdrawals." Comment: effect not likely to be small, but appropriate ITT analysis to cope with withdrawals. |
| Selective reporting (reporting bias) | Low risk | Comment: no protocol available, but all planned outcomes were reported in results |
| Size | High risk | Comment: total participants 21, fewer than 50 participants per treatment arm. |
| Other bias | Low risk | Comment: no other potential sources of bias. General comment: ITT analysis performed. |
Qari 2007.
| Methods |
Allocation: randomized Controlled: placebo Blinding: double‐blind Arm: 2‐arm, parallel‐group design Centre: multicentre (3 centres) Study duration: 48 months |
|
| Participants |
Inclusion criteria: participants aged ≥ 12 years with acute SCD/SCA crisis and no other complications; pain severe enough to require more than paracetamol; SCA participants with homozygous sickle cell (SS) disease; participants admitted through the emergency department with painful VOC severe enough to require narcotic analgesia Exclusion criteria: presence of medical or surgical contraindication to LMWH; pregnancy; low platelet counts (< 100,000/dL) or impaired haemostasis on admission in the form of INR > 1.4 or prolonged aPTT > 5 seconds of the hospital normal range; complicated SCA; history of cerebrovascular accident; current aplasia; acute chest syndrome; exchange transfusion; sequestration; anticoagulants therapy for other aetiology; participants with painful crises within the month before this admission; women receiving hormonal contraception Baseline characteristics N: 253 participants; 253 VOC episodes Gender: F 132, M 121 Age (mean): 22.8 (SD 4.5) years intervention; 21.6 (SD 3.8) years control Number randomized: 127 intervention; 126 control Number completed: 127 intervention; 126 control Setting and location: King Abdulaziz University Hospital, King Abdulaziz Oncology Centre, Jeddah, Saudi Arabia |
|
| Interventions |
Treatment group (127 participants; 127 VOC episodes):
Control group (126 participants; 126 VOC episodes):
Duration of treatment: 7 days Follow‐up period: not reported Cointerventions/additional analgesia: (preintervention) supportive analgesia: IV morphine 1 mg/hr + hydration IV fluids |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes | Sources of funding: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk |
Quote: "patients were randomized consecutively into either study group." Comment: no method of randomization provided. |
| Allocation concealment (selection bias) | Unclear risk | Comment: no technical details of allocation concealment provided. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk |
Quote: "Tinzaparin and placebo were provided by the manufacturer. All drug supplies were appropriately packaged, labelled, and kept in a locked, safe area under appropriate storage conditions with access limited to persons authorized by the investigator and those who directly involved in the study." Comment: no technical details. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: no technical details. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: insufficient information, not reported on withdrawals or treatment failures. |
| Selective reporting (reporting bias) | High risk | Comment: no available protocol, some outcomes listed in the methods were not reported in the results: adverse effects, opioid consumption. |
| Size | Unclear risk | Comment: total participants 253, 50 to 199 participants per treatment arm. |
| Other bias | Low risk | Comment: no other possible sources of bias. |
Rehmani 2013.
| Methods |
Allocation: randomized Controlled: active comparator Blinding: double‐blind Arm: 2‐arm, parallel‐group design Centre: single centre Study duration: not reported |
|
| Participants |
Inclusion criteria: participants presenting to emergency department with acute painful crisis of SCD Exclusion criteria: not reported Baseline characteristics N: 106 participants; 106 VOC episodes Gender: not reported in abstract Age: not reported Number randomized: 54 intervention; 52 control Number completed: not reported Setting and location: King Abdulaziz Hospital, Al Ahsa, Saudi Arabia |
|
| Interventions |
Treatment group (54 participants; 54 VOC episodes):
Control group (52 participants; 52 VOC episodes):
Duration of treatment: duration of painful crisis Follow‐up period: not reported Cointerventions/additional analgesia: rescue analgesics |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes | Sources of funding: not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk |
Quote: "106 patients were randomised to treatment, morphine or paracetamol." Comment: no clear method of randomization. |
| Allocation concealment (selection bias) | Unclear risk | Comment: insufficient information. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Comment: insufficient information. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Comment: insufficient information. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: insufficient information. |
| Selective reporting (reporting bias) | High risk | Comment: not all outcomes listed in methods were reported in the results, e.g. VRS; however, unclear due to abstract only. |
| Size | Unclear risk | Comment: total participants 106, 50–199 participants per treatment arm. |
| Other bias | Low risk | Comment: no other possible sources of bias. |
Wright 1992.
| Methods |
Allocation: randomized Controlled: placebo Blinding: double‐blind Arm: 2‐arm, parallel‐group trial (single dose) Centre: multicentre Study duration: 10 months |
|
| Participants |
Inclusion criteria: men and women presenting to emergency department with chief complaint of crisis pain, must have self‐rated moderate or severe pain intensity on a categorical scale Exclusion criteria: allergy to study drugs; active peptic ulcer; bleeding disorders; analgesics or central nervous system‐active drugs during the 3‐hr period before administration of study medication Baseline characteristics N: 18 participants; 24 events Gender: F 15, M 9 Age (mean): 29.8 years intervention; 31.9 years control Number randomized: 12 events intervention; 12 events control Number completed: unclear Setting and location: Vanderbilt University Hospital or Metropolitan Nashville General Hospital, USA |
|
| Interventions |
Treatment group (12 VOC episodes):
Control group (12 VOC episodes):
Duration of treatment: 4 hr Follow‐up period: not reported Cointerventions/additional analgesia: both groups received IV meperidine 50 mg and IV promethazine 12.5 mg on presentation and a standardized dose of meperidine every 30 minutes during 4‐hr observation. |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
|
| Notes | Sources of funding: study was supported by Biomedical Research Support Grant RR‐05424. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "the study medication was assigned in accordance with a computer‐generated randomization schedule, and the drugs were administered in identical syringes." |
| Allocation concealment (selection bias) | Low risk | Quote: "patients were enrolled in a prospective double‐blind fashion. They were randomly assigned to receive either ketorolac 60 mg IM or saline placebo IM." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "the drugs were administered in identical syringes." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk |
Quote: "a single nurse observer was responsible for all data collection and drug administration for all enrolled patients." Comment: unclear role and knowledge of the study drugs. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: all participants were accounted for from randomization, through intervention, to follow‐up. |
| Selective reporting (reporting bias) | Low risk | Comment: all planned outcomes in the methods were reported in the results. |
| Size | High risk | Comment: total participants 18, 12 events per treatment arm, < 50 participants per treatment arm. |
| Other bias | Low risk | Comment: no other possible source of bias. |
aPTT: partial thromboplastin time bleeding test; CPS: categorical pain score; F: female; hr: hour; IM: intramuscular; INR: international normalized ratio; ITT: intention to treat; IV: intravenous; LAS: lung allocation score; LMWH: low‐molecular‐weight heparin; M: male; NSAID: non‐steroidal anti‐inflammatory drugs; N: number of sample size; NPS: numerical pain score; NRS: numerical rating scale; SB/Thal: sickling beta‐thalassaemia; SCA: sickle cell anaemia; SCD: sickle cell disease; SD: standard deviation; SPID24: sum of the pain intensity difference in 24 hours; VAS: visual analogue scale; VOC: vaso‐occlusive crisis; VRS: verbal rating scale.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Al‐Jam'a 1999 | Participants: children and adults aged ≥ 2.5 years. Age data not reported separately. |
| Ataga 2018 | Participants: VOC (crisis event) as an outcome, not the condition/population. |
| Ballas 2010 | Outcomes: did not report on pain as an outcome as per eligibility criteria. |
| Cho 2016 | Intervention: patient‐controlled analgesia. |
| Dampier 2010 | Participants: aged ≥ 10 years, no separate data reported for ≥ 18 years (no full‐text publication available). |
| Dampier 2013 |
Study design: not double‐blind (single blind with participants knowing the treatment and assessors blinded). Notes: terminated early due to low accrual. |
| Desai 2013 | Outcomes: did not report on pain as an outcome as per eligibility criteria. |
| Euctr 2008 | Intervention: patient‐controlled analgesia. |
| Euctr 2011 | Participants: paediatric population aged < 18 years. |
| Gonzalez 1991 | Intervention: patient‐controlled analgesia. |
| Isrctn 2006 | Intervention: patient‐controlled analgesia. |
| Isrctn 2009 | Participants: paediatric population aged < 18 years. |
| Morris 2009 | Participants: paediatric population aged < 18 years (age range 3–19 years). |
| Nct 2019a | Participants: paediatric population aged < 18 years. |
| Nct 2019b | Study design: open‐label study design, not blinded as per eligibility criteria. |
| Niihara 2018 | Participants: VOC (crisis event) as an outcome, not the condition/population. |
| Orringer 2001 | Participants: children and adults (age range 8–60 years). Pain data available for < 15 years but not adults. |
| Pactr 2018 | Study design: open‐label trial, not blinded as per eligibility criteria. |
| Puri 2019 | Participants: paediatric population aged 1‐21 years. No separate data available for 18‐21 years in the full‐text publication. |
| Rousseau 2015 | Participants: paediatric population (age range 6–21 years). |
| Sandoval 2013 | Study design: open‐label case series, no evidence of controls or randomization. Not a randomized controlled trial. |
| Shah 2019 | Participants: VOC (crisis event) as an outcome, not the condition/population. |
| Smith 2011 | Intervention: patient‐controlled analgesia. |
| Tanabe 2018 | Study design: open‐label trial, not blinded as per inclusion criteria. |
| Telen 2015 | Participants: 54 adults, 20 children. Data not reported separately. |
| Uzun 2010 | Study design: not double‐blind as per eligibility criteria. |
| van Beers 2007 | Intervention: patient‐controlled analgesia. |
VOC: vaso‐occlusive crisis.
Characteristics of studies awaiting assessment [ordered by study ID]
De Castro 2013.
| Methods |
Allocation: randomized Controlled: placebo Blinding: double‐blind Arm: 2‐arm parallel group Centre: multicentre Study dates and duration: not reported |
| Participants |
Diagnostic criteria/inclusion criteria: people hospitalized for VOC Exclusion criteria: not reported in abstract Number: not reported in abstract Age: 12–60 years Gender: not reported Setting of recruitment and treatment: not reported in abstract Country and sites: not reported in abstract |
| Interventions |
Treatment group (number not reported): multiple IV doses of GMI 1070 Loading dose to achieve steady state drug level was followed by maintenance doses every 12 hr. Study dose was doubled per protocol after interim pharmacokinetic analysis. Control group (number not reported): not reported in abstract |
| Outcomes |
Primary
Secondary
|
| Notes | Conference abstract only. Insufficient data to analyse. Attempted to contact authors. |
Perlin 1988.
| Methods |
Allocation: randomized by unclear method Controlled: placebo Blinding: double‐blind |
| Participants |
Number: not reported Age: not reported Gender: not reported Inclusion criteria: not reported Exclusion criteria: not reported |
| Interventions |
Study period: 5 days Both groups: IM meperidine or hydroxyzine (or both), dose not reported, every 3–4 hr as needed. Intervention group (number not reported): oral diflunisal 1000 mg loading dose + oral diflunisal 500 mg every 12 hr Control group (number not reported): oral placebo, loading dose + oral placebo, every 12 hrs |
| Outcomes |
Primary
|
| Notes | Conference abstract only. Insufficient data to analyse. Attempted to contact authors |
Teuscher 1989.
| Methods |
Allocation: randomized by unclear method Controlled: placebo Blinding: not reported Arm: 2‐arm, parallel group Centre: single Study duration: not reported |
| Participants |
Inclusion criteria: not reported Exclusion criteria: not reported Baseline characteristics 36 participants Gender: not reported in abstract Ages: not reported in abstract Number randomized: not reported Number completed: not reported Setting and location: rural hospital, Togo, West Africa |
| Interventions |
Duration of treatment: not reported Follow‐up period: not reported Treatment group (number not reported): not reported Control group (number not reported): placebo not reported Cointerventions/additional analgesia: not reported |
| Outcomes |
Primary: not reported Secondary: not reported |
| Notes | Unclear from abstract whether the study reported on pain as an outcome as per eligibility criteria. |
hr: hour; IM: intramuscular; IV: intravenous; VAS: visual analogue scale; VOC; vaso‐occlusive crisis.
Characteristics of ongoing studies [ordered by study ID]
IRCT2016072511956N6.
| Trial name or title | To relieve crisis pain in sickle cell anemia patients |
| Methods |
Allocation: randomized Controlled: active comparator Blinding: double‐blind Arm: 2 parallel Centre: single Study duration: February 2017 (recruited, ongoing, data not published) |
| Participants |
Inclusion criteria: adults with sickle cell anemia; no prior history of cardiovascular or respiratory disease; no hypertension; not using other analgesic drugs Exclusion criteria: known major side effects of trial medications; addiction, especially to morphine Baseline characteristics N: target sample size 84, recruitment complete Gender: female and male Ages: no age limit (if included, need separate adult data only) Setting and location: Emam Reza Hospital, Mashhad University of Medical Sciences, Iran |
| Interventions |
Treatment group: 30 mg injection ketorolac Control group: 4‐10 mg injection morphine at 4 to 5 ml injection water solution |
| Outcomes |
Primary outcomes
Secondary outcomes
|
| Starting date | 19 February 2017 |
| Contact information | Somaye Sadat Hosseiny, Emam Reza Hospital, Emam Reza Square, Ebne Sina Avenue Mshhad Iran Email: somaye_2004a@yahoo.com |
| Notes | If study is included in future updates, need to separate adult data only |
NCT03431285.
| Trial name or title | Ketamine for acute painful crisis in sickle cell disease patients |
| Methods |
Allocation: randomized Controlled: active comparator Blinding: triple Arm: 3 arm, parallel groups Centre: single Study duration: 1 January 2018 (recruiting, ongoing) |
| Participants |
Inclusion criteria: known diagnosis of sickle cell disease based on sickle cell tests and haemoglobin electrophoresis; acute onset of painful crises, defined as having an onset within 7 days Exclusion criteria: healthy volunteers; other comorbidities; allergies to study medication; pregnancy or breastfeeding Baseline characteristics N: 264 participants, still recruiting Gender: men and women Ages: 18–60 years Setting and location: Dammam University, Saudi Arabia |
| Interventions |
Treatment group: morphine Control group: ketamine Other group: standard intravenous hydration |
| Outcomes |
Primary outcomes
Secondary outcomes
|
| Starting date | 1 January 2018 |
| Contact information | Mohammed SS Alshahrani, MD, Dammam, Eastern, Saudi Arabia, 31952 |
| Notes |
NCT03978156.
| Trial name or title | Dronabinol for pain and inflammation in adults living with sickle cell disease |
| Methods |
Allocation: randomized Controlled: placebo Blinding: double‐blind Arm: 2 crossover Centre: single Study duration: June 2019 (recruiting). Estimated completion June 2020. |
| Participants |
Inclusion criteria: adults over 18 clinical diagnosis of SCD; baseline score <60 ACQ‐Me 7‐day pain domain Exclusion criteria: intolerance to dronabinol, sesame oil, or marijuana; psychiatric disorder; concomitant medical condition; pregnant Baseline characteristics N: 30 Gender: female and male Ages: 18 years or older Setting and location: Yale New Haven Hospital, USA |
| Interventions |
Treatment group: 2.5 mg dronabinol oral tablet (titrated to 10 mg twice daily) Control group: placebo oral tablet |
| Outcomes |
Primary outcomes
Secondary outcomes
|
| Starting date | 6 June 2019 |
| Contact information | Susanna Curtis, Yale New Haven Hospital Smilow Cancer Centre, New Haven, Connecticut, 06510, USA. Yale University School of Medicine Oncology Section. Email: susanna.curtis@yale.edu |
| Notes |
mg: milligrams; ml: millilitres; N: number of participants; SCD: sickle cell disease; VAS: visual analogue scale
Differences between protocol and review
Types of participants: in the protocol, for any studies we identified that included participants aged 17 years and under, we planned to extract the data on participants aged 18 years and above and to contact the study authors for the separate adult data if necessary. We changed this approach to include the study and data on all its participants if less than 20% of the participants were aged 17 years or under. The reason for this change was because we felt that less than 20% would not make a significant impact on the results.
If greater than 20% of the participants were aged 17 years or under, we planned to extract the data on the participants aged 18 years and above, and contact the authors of the studies for separate adult data if necessary.
Outcomes planned: in the protocol, we expressed that death would be sought as a secondary outcome. This was not the intention and we have not investigated it as an outcome. The implication is that the outcome, 'serious adverse events' includes death. This has been amended in the 'Methods' section.
Risk of bias domains: in the protocol, we unintentionally omitted three domains (blinding of participants and personnel, selective reporting bias, other). These are now outlined in the 'Methods' section.
GRADE quality of the evidence: we unintentionally omitted a final reason for downgrading to very low quality in one step (by three levels). This is now outlined in the 'Methods' section.
Measures of treatment effect: we clarified that we planned not to use continuous data for our primary outcomes measuring pain. For our secondary outcomes, we used continuous data for the meta‐analysis where appropriate.
Contributions of authors
TC registered the title.
TC, IH, SB and PW wrote the 2016 protocol.
TC, IH, SB, PW and BJ screened, extracted, analysed and graded the data, and wrote the full review.
TC will be responsible for updates.
Sources of support
Internal sources
Oxford Pain Relief Trust, UK.
External sources
No sources of support supplied
Declarations of interest
TC: none known.
IH: none known.
SB is an internist specializing in basic and clinical haematology with an emphasis on the management of adult and children with sickle cell disease and pain. SB has been in the Speakers Bureau of Novartis with an emphasis on Deferasirox (both Exjade and Jadenu formulations) in September 2016.
BJ: none known.
PW undertakes work for GSK under the auspices of his company Oxford Systematic Review Services. The work is related to over‐the‐counter analgesics.
The protocol for this review was identified in a 2019 audit as not meeting the current definition of the Cochrane Commercial Sponsorship policy. At the time of its publication it was compliant with the interpretation of the existing policy. A new author team fully compliant with the 2014 policy completed the review. As with all reviews, new and updated, at update this review will be revised according to 2020 policy update.
Stable (no update expected for reasons given in 'What's new')
References
References to studies included in this review
Arambasik 2013 {published data only}
- Arambasik J, Griffin P, Zimmerman J, Beeson M, McMullen MJ, Griffin G, et al. The use of low dose ketamine and hydromorphone for patients in sickle cell crisis. Academic Emergency Medicine 2013;1:S149. [Google Scholar]
Bartolucci 2009 {published data only}
- Bartolucci P, Murr T, Roudot‐Thoraval F, Habibi A, Santin A, Renaud B, et al. A randomized, controlled clinical trial of ketoprofen for sickle‐cell disease vaso‐occlusive crises in adults. Blood 2009;114(18):3742‐7. [DOI: 10.1182/blood-2009-06-227330] [DOI] [PubMed] [Google Scholar]
Benjamin 1986 {published data only}
- Benjamin LJ, Berkowitz LR, Orringer E. A collaborative, double‐blind randomized study of cetiedil citrate in sickle cell crisis. Blood 1986;67(5):1442‐7. [online ISSN 1528‐0020] [PubMed] [Google Scholar]
De Franceschi 2016 {published data only}
- Franceschi L, Mura P, Schweiger V, Venacto E, Quaglia FM, Delmonte L, et al. Fentanyl buccal tablet: a new breakthrough pain medication in early management of severe vaso‐occlusive crisis in sickle cell disease. Pain Practice 2016;16(3):680‐7. [DOI] [PubMed] [Google Scholar]
Gonzalez 1988 {published data only}
- Gonzalez ER, Ornato JP, Ware D, Bull D, Evens RP. Comparison of intramuscular analgesic activity of butorphanol and morphine in patients with sickle cell disease. Annals of Emergency Medicine 1988;17(8):788‐91. [DOI: 10.1016/S0196-0644(88)80554-7] [DOI] [PubMed] [Google Scholar]
Perlin 1994 {published data only}
- Perlin E. Intravenous ketorolac tromethamine enhances pain control in sickle cell vaso‐occlusive crisis [abstract]. 18th Annual Meeting of the National Sickle Cell Disease Program; Philadelphia, PA, USA. (May 24‐26) 1993; Vol. 65a.
- Perlin E, Finke H, Castro O, Rana S, Pittman J, Burt R, et al. Enhancement of pain control with ketorolac tromethamine in patients with sickle cell vaso‐occlusive crisis. American Journal of Hematology 1994;46(1):43‐7. [DOI] [PubMed] [Google Scholar]
Qari 2007 {published data only}
- Qari MH, Aljaouni SK, Alardawi MS, Fatani H, Alsayes FM, Zografos P, et al. Reduction of painful vaso‐occlusive crisis of sickle cell anaemia by tinzaparin in a double‐blind randomized trial. Thrombosis and Haemostasis 2007;98(2):392‐6. [PubMed] [Google Scholar]
Rehmani 2013 {published data only}
- Rehmani R. A randomized controlled trial of paracetamol versus morphine for the treatment of acute painful crisis of sickle cell disease. Academic Emergency Medicine 2013;20(5 Suppl 1):S149. [DOI: 10.1111/acem.12115] [DOI] [Google Scholar]
Wright 1992 {published data only}
- Wright SW, Norris RL, Mitchell TR. Ketorolac for sickle cell vaso‐occlusive crisis pain in the emergency department: lack of a narcotic‐sparing effect. Annals of Emergency Medicine 1992;21(8):925‐8. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Al‐Jam'a 1999 {published data only}
- Al‐Jam'a AH, Al‐Dabbous IA, Rafiq MS, Al‐Khatti A, Al‐Salem AH, Al‐Bahrna A, et al. Isoxsuprine in the treatment of sickle cell painful crises: a double‐blind comparative study with narcotic analgesic. Annals of Saudi Medicine 1999;19(2):97‐100. [DOI] [PubMed] [Google Scholar]
Ataga 2018 {published data only}
- Ataga KI, Hoppe CC, Ware RE, Howard J, Tonda M, Ganju J, et al. Novel trial design to evaluate oral voxelotor for the treatment of sick cell disease: The phase 3 hemoglobin oxygen affinity modulation to inhibit sickle hemoglobin polymerization (HOPE) trial. HemaSphere 2018;2(Suppl 2):670. [Google Scholar]
Ballas 2010 {published data only}
- Ballas SK, Bauserman RL, McCarthy WF, Castro OL, Smith WR, Waclawiw MA. Hydroxyurea and acute painful crises in sickle cell anemia: effects of hospital length of stay and opioid utilization during hospitalisation, outpatient acute care contacts, and at home. Journal of Pain and Symptom Management 2010;40(6):870‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cho 2016 {published data only}
- Cho G, Anie KA, Buckton K, Kiilu P, Layton M, Alexander L, et al. SWIM (Sickle With Ibuprofen and Morphine) randomized controlled trial fails to recruit: lessons learnt. BMJ Open 2016;6:e011276. [DOI: 10.1136/bmjopen-2016-011276] [DOI] [PMC free article] [PubMed] [Google Scholar]
Dampier 2010 {published data only}
- Dampier C, Smith WR, Wager C, Bell M, Eckman JR, Hsu LL, et al. Initial experience with the IMPROVE trial – a phase III analgesic trial for hospitalized sickle cell painful episodes. Blood 2010;116(21):2667. [Google Scholar]
Dampier 2013 {published data only}
- Dampier CD, Smith WR, Wager CG, Kim HY, Bell MC, Miller ST, et al. IMPROVE trial: a randomized controlled trial of patient‐controlled analgesia for sickle cell painful episodes: rationale, design challenges, initial experience, and recommendations for future studies. Clinical Trials 2013;10(2):319‐31. [DOI: 10.1177/1740774513475850] [DOI] [PMC free article] [PubMed] [Google Scholar]
Desai 2013 {published data only}
- Desai PC, Brittain JE, Jones SK, McDonald A, Wilson DR, Dominik R. A pilot study of eptifibatide for treatment of acute pain episodes in sickle cell disease. Thrombosis Research 2013;132(3):341‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Euctr 2008 {published data only}
- Euctr GB. An evaluation of the effectiveness of ibuprofen and morphine for acute pain in sickle cell disease ‐ ibuprofen and morphine for acute sickle cell pain. http://www.who.int/trialsearch/Trial2.aspx?TriallD=EUCTR2008‐0068‐24‐GB 2008.
Euctr 2011 {published data only}
- Euctr IE. Sickle cell anaemia is an inherited blood disorder which results in abnormal sickle shaped red blood cells which do not fit well through small blood vessels. These blockages prevention oxygen (in blood) from reaching different parts of the body resulting in painful crisis. This study will compared the effectiveness of two types of pain medication, one given through a vein and one squirted up the nose. http://www.who.int/trialsearch/Trial2.aspx?TriallD=EUCTR2011‐005161‐20‐IE 2011.
Gonzalez 1991 {published data only}
- Gonzalez ER, Bahal N, Hansen LA, Ware D, Bull DS, Ornato JP, et al. Intermittent injection vs patient‐controlled analgesia for sickle cell crisis pain. Comparison in patients in the emergency department. Archives of Internal Medicine 1991;151(7):1373‐8. [PubMed] [Google Scholar]
Isrctn 2006 {published data only}
Isrctn 2009 {published data only}
- Isrctn. Use of intravenous paracetamol in combination with morphine in sickle cell disease children with vaso‐occlusive crisis. http://www.who.int/trialsearch/Trial2.aspx?TriallD=ISRCTN34491330 2009.
Morris 2009 {published data only}
- Morris CR, Ansari M, Lavrisha L, Sweeters N, Kuypers FA, Vichinsky EP. Arginine therapy for vaso‐occlusive pain episodes in sickle cell disease. Blood 2009;144(22):238. [DOI] [PMC free article] [PubMed] [Google Scholar]
Nct 2019a {published data only}
- Nct. Nebulized sub‐dissociative dose ketamine at three different dosing regimens for treating acute pain in the pediatric ED. https://clinicaltrials.gov/show/NCT03950817 2019.
Nct 2019b {published data only}
- Nct. Ketamine infusion for sickle cell pain crisis. https://clinicaltrials.gov/show/NCT04005209 2019.
Niihara 2018 {published data only}
- Niihara Y, Stark CW, Razon R, Tran LT, Becerra JM, Panosyan EH, Lasky JL. Consistent benefit of L‐glutamine observed across patients with low, medium, and high number of crises reported in the year prior to screening‐analysis from the phase 3 study of L‐glutamine in sickle cell anemia. Blood 2018;132(Suppl 1):1065. [Google Scholar]
Orringer 2001 {published data only}
- Orringer EP, Casella JF, Ataga KI, Koshy M, Adams‐Graves P, Luchtman‐Jones L, et al. Purified poloxamer 188 for treatment of acute vaso‐occlusive crisis of sickle cell disease: a randomized controlled trial. Journal of the American Medical Association 2001;286(17):2099‐106. [DOI] [PubMed] [Google Scholar]
Pactr 2018 {published data only}
- Pactr. A comparative analysis of parenteral opioids in the management of acute sickle cell crisis in an urban primary care centre (Korle bu Polyclinic) in Accra. http://www/who.int/trialsearch/Trial2.aspx?TriallD=PACTR201812917283565 2018.
Puri 2019 {published data only}
- Puri L, Anghelescu D, Hankins J, Nottage K, Wang W, Kang G, et al. Gabapentin for pain in sickle cell disease: Results of a randomized phase II clinical trial. Pediatric Blood and Cancer 2019;66:S7. [Google Scholar]
Rousseau 2015 {published data only}
- Rousseau V, Arriuberge C, Chabaud S, Launay V, Thouvenin S, Tourniaire B, et al. Efficacy and tolerance of lidocaine 5% patches in neuropathic pain and vaso‐occlusive sickle‐cell crises pain in children: a prospective multi‐centre clinical study. Pediatric Blood and Cancer 2015;62:S384. [DOI] [PubMed] [Google Scholar]
Sandoval 2013 {published data only}
- Sandoval M, Coleman P, Govani R, Siddiqui S, Todd KH. Pilot study of human recombinant hyaluronidase‐enhance subcutaneous hydration and opioid administration for sickle cell disease acute pain episodes. Journal of Pain & Palliative Care Pharmacotherapy 2013;27(1):10‐8. [DOI] [PubMed] [Google Scholar]
Shah 2019 {published data only}
- Shah N, Boccia R, Kraft WK, Hardesty BM, Paulose J, Lain D, et al. Pro3 successor study: treatment and health care resource utilization by sickle cell patients who participated in the sustain study in the United States. Value in Health 2019;22(Suppl 2):S335. [Google Scholar]
Smith 2011 {published data only}
- Smith W, Dampier C, Kim HY, Wager C, Bell M, Eckman J, et al. Pain intensity during the initial experience with the IMPROVE trial – a phase III analgesic trial for hospitalised sickle cell painful episodes. American Journal of Hematology 2011;86(10):E7‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tanabe 2018 {published data only}
- Tanabe P, Silva S, Bosworth HB, Crawford R, Paice JA, Richardson LD, et al. A randomized controlled trial comparing two vaso‐occlusive episdoe (VOE) protocols in sickle cell disease (SCD). American Journal of Hematology 2018;93(2):159‐168. [DOI] [PMC free article] [PubMed] [Google Scholar]
Telen 2015 {published data only}
- Telen MJ, Wun T, McCavit TL, Castro LM, Krishnamurti L, Lanzkron S. GMI 1070: reduction in time to resolution of vaso‐occlusive crisis and decreased opioid use in a prospective, randomized, multi‐centre double blind, adaptive phase 2 study in sickle cell disease. Blood 2013; Vol. 122:21.
- Telen MJ, Wun T, McCavit TL, Castro LM, Krishnamurti L, Lanzkron S, et al. Randomized phase 2 study of GMI‐1070 in SCD: reduction in time to resolution of vaso‐occlusive events and decreased opioid use. Blood 2015;125(17):2656‐64. [DOI: 10.1182/blood-2014-583351] [DOI] [PMC free article] [PubMed] [Google Scholar]
Uzun 2010 {published data only}
- Uzun B, Kekec Z, Gurkan E. Efficacy of tramadol vs meperidine in vasoocclusive sickle cell crisis. American Journal of Emergency Medicine 2010;28(4):445‐9. [DOI] [PubMed] [Google Scholar]
van Beers 2007 {published data only}
- Beers EJ, Tuijn CF, Nieuwkerk PT, Friederich PW, Vranken JH, Biemond BJ. Patient‐controlled analgesia versus continuous infusion of morphine during vaso‐occlusive crisis in sickle cell disease, a randomized controlled trial. American Journal of Hematology 2007;82(11):955‐60. [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
De Castro 2013 {published data only}
- Castro LM, Wun T, Lanzkron S, Smith WR, Hassell KL, Kutlar A, et al. Effects of GMI‐1070, a pan‐selection inhibitor, on pain intensity and opioid utilization in sickle cell disease. Blood 2013;122(21):775. [Google Scholar]
Perlin 1988 {published data only}
- Perlin E, Finke H, Castro O, Bang KM, Rana S, Taylor R, et al. Treatment of sickle cell pain crisis. A clinical trial of diflunisal. Clinical Trials Journal 1988;25(4):254‐64. [Google Scholar]
Teuscher 1989 {published data only}
- Teuscher T, Ahe CW, Balillod P, Holzer B. Double‐blind randomised clinical trial of pentoxiphyllin in vaso‐occlusive sickle cell crisis. Tropical & Geographical Medicine 1989;41(4):320‐5. [PubMed] [Google Scholar]
References to ongoing studies
IRCT2016072511956N6 {published data only}
- IRCT2016072511956N6. To relieve crisis pain in sickle cell anemia patients. http://apps.who.int/trialsearch/Trial2.aspx?TrialID=IRCT2016072511956N6 (accessed September 2019).
NCT03431285 {published data only}
- NCT03431285. Ketamine for acute painful crisis in sickle cell disease patients. clinicaltrials.gov/ct2/show/NCT03431285 (first received 13 February 2018).
NCT03978156 {published data only}
- NCT03978156. Dronabinol for pain and inflammation in adults living with sickle cell disease. https://clinicaltrials.gov/show/NCT03978156 (accessed September 2019) 2019.
Additional references
Al Hajeri 2007
- Al Hajeri A, Fedorowicz Z, Omran A, Tadmouri GO. Piracetam for reducing the incidence of painful sickle cell disease crises. Cochrane Database of Systematic Reviews 2007, Issue 2. [DOI: 10.1002/14651858.CD006111.pub2] [DOI] [PubMed] [Google Scholar]
Ballas 2005
- Ballas SK. Pain management of sickle cell disease. Hematology/Oncology Clinics of North America 2005;19(5):785‐802. [DOI: 10.1016/j.hoc.2005.07.008] [DOI] [PubMed] [Google Scholar]
Ballas 2007
- Ballas SK. Current issues in sickle cell pain and its management. Hematology 2007;2007(1):97‐105. [DOI: 10.1182/asheducation-2007.1.97] [DOI] [PubMed] [Google Scholar]
De Ceulaer 1982
- Ceulaer K, Hayes R, Gruber C, Serjeant GR. Medroxyprogesterone acetate and homozygous sickle‐cell disease. Lancet 1982;2(8292):229‐31. [DOI: 10.1016/S0140-6736(82)90320-8] [DOI] [PubMed] [Google Scholar]
Dechartres 2013
- Dechartres A, Trinquart L, Boutron I, Ravaud P. Influence of trial sample size on treatment effect estimates: meta‐epidemiological study. BMJ 2013;346:f2304. [DOI: 10.1136/bmj.f2304] [DOI] [PMC free article] [PubMed] [Google Scholar]
Dechartres 2014
- Dechartres A, Altman DG, Trinquart L, Boutron I, Ravaud P. Association between analytic strategy and estimates of treatment outcomes in meta‐analyses. JAMA 2014;312:623‐30. [DOI: 10.1001/jama.2014.8166] [DOI] [PubMed] [Google Scholar]
Diallo 2002
- Diallo D, Tchernia G. Sickle cell disease in Africa. Current Opinion in Hematology 2002;9(2):111‐6. [ISSN: 1065‐6251] [DOI] [PubMed] [Google Scholar]
Dunlop 2014
- Dunlop R, Bennet KC. Pain management for sickle cell disease in children and adults. Cochrane Database of Systematic Reviews 2014, Issue 4. [DOI: 10.1002/14651858.CD003350.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Dworkin 2008
- Dworkin RH, Turk DC, Wyrwich KW, Beaton D, Cleeland CS, Farrar JT, et al. Interpreting the clinical importance of treatment outcomes in chronic pain clinical trials: IMMPACT recommendations. Journal of Pain 2008;9(2):105‐21. [DOI: 10.1016/j.jpain.2007.09.005] [DOI] [PubMed] [Google Scholar]
Gaston 1986
- Gaston MH, Verter JI, Woods G, Pegelow C, Kelleher J, Presbury G, et al. Prophylaxis with oral penicillin in children with sickle cell anemia. A randomized trial. New England Journal of Medicine 1986;314(25):1593‐9. [DOI: 10.1056/NEJM198606193142501] [DOI] [PubMed] [Google Scholar]
Gillis 2012
- Gillis VL, Senthinathan A, Dzingina M, Chamberlain K, Banks E, Baker MR, Longson D. Management of an acute painful sickle cell episode in hospital: summary of NICE guidance. BMJ 2012;344:e4063. [DOI] [PubMed] [Google Scholar]
GRADEpro GDT [Computer program]
- McMaster University (developed by Evidence Prime). GRADEpro GDT. Hamilton (ON): McMaster University (developed by Evidence Prime), 2015.
Guyatt 2013a
- Guyatt G, Oxman AD, Sultan S, Brozek J, Glasziou P, Alonso‐Coelle P, et al. Making an overall rating of the confidence in effect estimates for a single outcome and for all outcomes. Journal of Clinical Epidemiology 2013;66(2):151‐7. [DOI: 10.1016/j.jclinepi.2012.01.006] [DOI] [PubMed] [Google Scholar]
Guyatt 2013b
- Guyatt GH, Oxman AD, Santesso N, Helfand M, Vist G, Kunz R, et al. GRADE guidelines: 12. Preparing summary of findings tables – binary outcomes. Journal of Clinical Epidemiology 2013;66(2):158‐72. [DOI: 10.1016/j.jclinepi.2012.01.012] [DOI] [PubMed] [Google Scholar]
Herrick 1910
- Herrick J. Peculiar elongated and sickle‐shaped red blood corpuscles in a case of severe anemia. Archives of Internal Medicine 1910;5(5):517‐21. [DOI: ] [Google Scholar]
Higgins 2011a
- Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2011b
- Higgins JP, Altman DG, Sterne JA. Chapter 8: Assessing risk of bias in included studies. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Hoffman 2010
- Hoffman DL, Sadosky A, Dukes EM, Alvir J. How do changes in pain severity levels correspond to changes in health status and function in patients with painful diabetic peripheral neuropathy?. Pain 2010;149(2):194‐201. [DOI: 10.1016/j.pain.2009.09.017] [DOI] [PubMed] [Google Scholar]
Hussein 2015
- Hussein N, Weng SF, Kai J, Kleijnen J, Quershi N. Preconception risk assessment for thalassaemia, sickle cell disease, cystic fibrosis and Tay‐Sachs disease. Cochrane Database of Systematic Reviews 2015, Issue 8. [DOI: 10.1002/14651858.CD010849.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
L'Abbé 1987
- L'Abbé KA, Detsky AS, O'Rourke K. Meta‐analysis in clinical research. Annals of Internal Medicine 1987;107(2):224‐33. [DOI: 10.7326/0003-4819-107-2-224] [DOI] [PubMed] [Google Scholar]
Lane 2001
- Lane PA, Nuss R, Ambruso D. Hematologic disorders. In: Hay WW, Hayward AR, Levin MJ, Sondheimer JM editor(s). Current Pediatric Diagnosis and Treatment. 15th Edition. New York (NY): McGraw‐Hill Companies Inc, 2001:756‐9. [Google Scholar]
Lutz 2015
- Lutz B, Meiler SE, Bekker A, Tao YX. Updated mechanisms of sickle cell disease‐associated chronic pain. Translational Perioperative and Pain Medicine 2015;2(2):8‐17. [PUBMED: PMC4542088] [PMC free article] [PubMed] [Google Scholar]
Martí‐Carvajal 2009
- Martí‐Carvajal AJ, Peña‐Martí GE, Comunián‐Carrasco G, Martí‐Peña AJ. Interventions for treating painful sickle cell crisis during pregnancy. Cochrane Database of Systematic Reviews 2009, Issue 1. [DOI: 10.1002/14651858.CD006786.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
McQuay 1998
- McQuay H, Moore R. An Evidence‐based Resource for Pain Relief. Oxford: Oxford University Press, 1998. [ISBN: 0‐19‐263048‐2] [Google Scholar]
Moher 2009
- Moher D, Liberati A, Tetzlaff J, Altman DG, the PRISMA Group. Preferred Reporting Items for Systematic Reviews and Meta‐Analyses: the PRISMA statement. PLoS Medicine 2009;6(7):e1000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2008
- Moore RA, Barden J, Derry S, McQuay HJ. Managing potential publication bias. In: McQuay HJ, Kalso E, Moore RA editor(s). Systematic Reviews in Pain Research: Methodology Refined. Seattle (WA): IASP Press, 2008:15‐24. [ISBN: 978–0–931092–69–5] [Google Scholar]
Moore 2010a
- Moore RA, Straube S, Paine J, Phillips CJ, Derry S, McQuay HJ. Fibromyalgia: moderate and substantial pain intensity reduction predicts improvement in other outcomes and substantial quality of life gain. Pain 2010;149(2):360‐4. [DOI: 10.1016/j.pain.2010.02.039] [DOI] [PubMed] [Google Scholar]
Moore 2010b
- Moore RA, Moore OA, Derry S, Peloso PM, Gammaitoni AR, Wang H. Responder analysis for pain relief and numbers needed to treat in a meta‐analysis of etoricoxib osteoarthritis trials: bridging a gap between clinical trials and clinical practice. Annals of the Rheumatic Diseases 2010;69(2):374‐9. [DOI: 10.1136/ard.2009.107805] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2010c
- Moore RA, Smugar SS, Wang H, Peloso PM, Gammaitoni A. Numbers‐needed‐to‐treat analyses – do timing, dropouts, and outcome matter? Pooled analysis of two randomized, placebo‐controlled chronic low back pain trials. Pain 2010;151(3):592‐7. [DOI: 10.1016/j.pain.2010.07.013] [DOI] [PubMed] [Google Scholar]
Moore 2013
- Moore RA, Straube S, Aldington D. Pain measures and cut‐offs – 'no worse than mild pain' as a simple, universal outcome. Anaesthesia 2013;68(4):400‐12. [DOI: 10.1111/anae.12148] [DOI] [PubMed] [Google Scholar]
National Screening Committee 2006
- National Screening Committee for Sickle Cell Disease and Thalassaemia. NHS Sickle Cell and Thalassaemia Screening Programme, 2006. www.gov.uk/topic/population‐screening‐programmes/sickle‐cell‐thalassaemia (accessed 7 September 2015).
NICE 2015
- National Institute of Health and Care Excellence (NICE). Guidance – sickle cell acute painful episode, 2015. (accessed 7 September 2015).
NIH 2014
- National Institutes of Health. Evidence‐based management of sickle cell disease: expert panel report, 2014. www.nhlbi.nih.gov/guidelines (accessed 15 August 2017).
NIH 2015
- National Heart, Lung and Blood Institute. Sickle cell disease, 2015. www.nhlbi.nih.gov/health/health‐topics/topics/sca/atrisk (accessed 12 October 2015).
Nüesch 2010
- Nüesch E, Trelle S, Reichenbach S, Rutjes AW, Tschannen B, Altman DG, et al. Small study effects in meta‐analyses of osteoarthritis trials: meta‐epidemiological study. BMJ 2010;341:c3515. [DOI: 10.1136/bmj.c3515] [DOI] [PMC free article] [PubMed] [Google Scholar]
O'Brien 2010
- O'Brien EM, Staud RM, Hassinger AD, McCulloch RC, Craggs JG, Atchison JW, et al. Patient‐centered perspective on treatment outcomes in chronic pain. Pain Medicine 2010;11(1):6‐15. [DOI: 10.1111/j.1526-4637.2009.00685.x] [DOI] [PubMed] [Google Scholar]
Okomo 2015
- Okomo U, Meremikwu MM. Fluid replacement therapy for acute episodes of pain in people with sickle cell disease. Cochrane Database of Systematic Reviews 2015, Issue 3. [DOI: 10.1002/14651858.CD005406.pub4] [DOI] [PubMed] [Google Scholar]
Okpala 2002
- Okpala I, Tawil A. Management of pain in sickle‐cell disease. Journal of the Royal Society of Medicine 2002;95(9):456‐8. [PUBMED: PMC1279994] [DOI] [PMC free article] [PubMed] [Google Scholar]
Piel 2013
- Piel FB, Patil AP, Howes RE, Nyangiri OA, Gething PW, Dewi M, et al. Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model‐based map and population estimates. Lancet 2013;381(9861):142‐51. [DOI: 10.1016/S0140-6736(12)61229-X] [DOI] [PMC free article] [PubMed] [Google Scholar]
Platt 1994
- Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, et al. Mortality in sickle cell disease: life expectancy and risk factors for early death. New England Journal of Medicine 1994;330(23):1639‐44. [DOI: 10.1056/NEJM199406093302303] [DOI] [PubMed] [Google Scholar]
Quinn 2010
- Quinn CT, Rogers ZR, McCavit TL, Buchanan GR. Improved survival of children and adolescents with sickle cell disease. Blood 2010;115(17):3447‐52. [DOI: 10.1182/blood-2009-07-233700] [DOI] [PMC free article] [PubMed] [Google Scholar]
Rees 2003
- Rees DC, Olujohungbe AD, Parker NE, Stephens AD, Telfer P, Wright J. Guidelines for the management of the acute painful crisis in sickle cell disease. British Journal of Haematology 2003;120(5):744. [DOI: 10.1046/j.1365-2141.2003.04193] [DOI] [PubMed] [Google Scholar]
Review Manager 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Rosenblum 2008
- Rosenblum A, Marsch LA, Joseph H, Portenoy RK. Opioids and the treatment of chronic pain: controversies, current status, and future directions. Experimental and Clinical Psychopharmacology 2008;16(5):405‐16. [DOI: 10.1037/a0013628] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sasongko 2013
- Sasongko TH, Nagalla S, Ballas SK. Angiotensin‐converting enzyme (ACE) inhibitors for proteinuria and microalbuminuria in people with sickle cell disease. Cochrane Database of Systematic Reviews 2013, Issue 3. [DOI: 10.1002/14651858.CD009191.pub2] [DOI] [PubMed] [Google Scholar]
Serjeant 1994
- Serjeant GR. The geography of sickle‐cell disease: opportunity for understanding its diversity. Annals of Saudi Medicine 1994;14(3):237‐46. [PUBMED: 17586900] [DOI] [PubMed] [Google Scholar]
Stuart 2004
- Stuart MJ, Nagel RL. Sickle‐cell disease. Lancet 2004;364(9442):1343‐60. [DOI: 10.1016/S0140-6736(04)17192-4] [DOI] [PubMed] [Google Scholar]
Tanabe 2012
- Tanabe P, Hafner J, Martinovich Z, Artz N. Adult emergency department patients with sickle cell pain crisis: results from a quality improvement learning collaborative model to improve analgesic management. Academic Emergency Medicine 2012;9(4):430‐8. [DOI: 10.1111/j.1553-2712.2012.01330.x] [DOI] [PMC free article] [PubMed] [Google Scholar]
Telfer 2014
- Telfer P, Bahal N, Lo A, Challands J. Management of the acute painful crisis in sickle cell disease – a re‐evaluation of the use of opioids in adult patients. British Journal of Haematology 2014;166:157‐64. [DOI: 10.1111/bjh.12879] [DOI] [PubMed] [Google Scholar]
Thienhaus 2002
- Thienhaus O, Cole BE. Classification of pain. In: Weiner RS editor(s). Pain management: a practical guide for clinicians. 6th Edition. New York (NY): CRC Press, 2002. [Google Scholar]
Thorlund 2011
- Thorlund K, Imberger G, Walsh M, Chu R, Gluud C, Wetterslev J, et al. The number of patients and events required to limit the risk of overestimation of intervention effects in meta‐analysis – a simulation study. PloS One 2011;6(10):e25491. [DOI: 10.1371/journal.pone.0025491] [DOI] [PMC free article] [PubMed] [Google Scholar]
Trescot 2008
- Trescot M, Datta S, Lee M, Hansen H. Opioid pharmacology. Pain Physician Journal 2008;11(2S):S133‐53. [ISSN: 1533‐3159; PUBMED: 18443637] [PubMed] [Google Scholar]
Waldrop 1995
- Waldrop RD, Mandry C. Health professional perceptions of opioid depended among patients with pain. American Journal of Emergency Medicine 1995;13(5):529‐31. [DOI: 10.1016/0735-6757(95)90163-9] [DOI] [PubMed] [Google Scholar]
Weatherall 2001
- Weatherall DJ, Clegg JB. Inherited haemoglobin disorders: an increasing global health problem. Bulletin of the World Health Organization 2001;79(8):704‐12. [DOI: 10.1590/S0042-96862001000800005] [DOI] [PMC free article] [PubMed] [Google Scholar]
Weatherall 2006
- Weatherall D, Akinyanju O, Fucharoen S, Olivieri N, Musgrove P. Inherited disorders of hemoglobin. Disease Control Priorities in Developing Countries. 2nd Edition. Washington (DC): World Bank and Oxford University Press, 2006:663‐80. [Google Scholar]
WHO 2010
- World Health Organization. Sickle‐cell disease: a strategy for the WHO African region 2010. www.afro.who.int/en/sixtieth‐session.html (accessed 19 October 2015).
WHO 2011
- World Health Organization. Sickle‐cell disease and other haemoglobin disorders: fact sheet N°308. Geneva: World Health Organization, 2011. [Google Scholar]
Wiffen 2005
- Wiffen PJ, McQuay HJ, Edwards JE, Moore RA. Gabapentin for acute and chronic pain. Cochrane Database of Systematic Reviews 2005, Issue 3. [DOI: 10.1002/14651858.CD005452] [DOI] [PubMed] [Google Scholar]
Yawn 2014
- Yawn BP, Buchanan GR, Afenyi‐Annan AN, Ballas SK, Hassell KL, James AH, et al. Management of sickle cell disease: summary of the 2014 evidence‐based report by expert panel members. JAMA 2014;312(10):1033‐48. [DOI: 10.1001/jama.2014.10517] [DOI] [PubMed] [Google Scholar]