

Abstract
Background:
CDK4/6-dependent cell-cycle regulation is disrupted in most glioblastomas. This study assesses the central nervous system (CNS) pharmacokinetics and tumor pharmacodynamics of ribociclib, a highly selective CDK4/6-inhibitor, in patients with recurrent glioblastoma.
Methods:
Recurrent glioblastoma patients with intact retinoblastoma protein (RB) expression and CDKN2A deletion or CDK4/6 amplification were treated with ribociclib daily (900 mg) for 5 days before tumor resection. Blood, tumor, and cerebrospinal fluid (CSF) samples were collected and total and unbound ribociclib concentrations were determined. Pharmacodynamic effects, assessed by RB and FOXM1 phosphorylation, were compared to matched archival tissue. Patients with positive pharmacokinetic and pharmacodynamic effects were enrolled into the expansion cohort for preliminary assessment of progression-free survival (PFS).
Results:
Twelve patients were enrolled. The mean unbound ribociclib concentrations in CSF, non-enhancing, and enhancing tumor regions were 0.374 μM, 0.560, and 2.152 μmol/kg, respectively, which were > 5-fold the in vitro IC50 for inhibition of CDK4/6 (0.04 μM). G1-to-S phase suppression was inferred by decreases in phosphorylation of RB (P < 0.01) and cellular proliferation (P < 0.05). Six of 12 patients were enrolled into the pharmacokinetic/pharmacodynamic-guided expansion cohort and demonstrated a median PFS of 9.7 weeks. Examination of recurrent tumors following monotherapy indicated upregulation of the PI3K/mTOR pathway.
Conclusion:
Ribociclib exhibited good CNS penetration and target modulation was indicated by inhibition of RB phosphorylation and tumor proliferation. Ribociclib monotherapy showed limited clinical efficacy in patients with recurrent glioblastoma. Combination therapy with CDK4/6 and PI3K/mTOR inhibitors may be explored for treating recurrent glioblastoma.
Keywords: Phase 0, glioblastoma, ribociclib, CDK4/6
Introduction
A hallmark of human glioblastoma is aberrant cell-cycle control, resulting in unlimited cell-cycle reentry and progression (1-3). The CDK4/6-Rb-E2F axis controls the cell-cycle and is tightly regulated by several factors such as cyclin D, INK4 family proteins, p21CIP1, and p27KIP1. According to The Cancer Genome Atlas database, homozygous deletion of the p16INK4a gene occurs in 50% of glioblastomas (1). Other common cell-cycle-related mutations in glioblastomas include amplification and overexpression of CDK4 and homozygous deletion/mutation of RB, followed in frequency by overexpression of cyclin D1 and amplification of CDK6 (2). Overall, the CDK4/6-Rb-E2F axis is deregulated in approximately 80% of glioblastomas, underscoring the clinical potential of cell-cycle targeting in this patient population. Preclinical studies demonstrated that tumor cells treated with CDK4/6 inhibitors exhibited reduction of phosphorylation of RB, G1 phase arrest, and decreased proliferation, and moreover, the inhibition of CDK4/6 activity slowed glioma progression (4).
Ribociclib (LEE011) is a highly specific, orally bioavailable, small-molecule inhibitor of CDK4 and CDK6 (5,6). In preclinical and clinical studies, ribociclib demonstrated a manageable tolerability and therapeutic potential for a variety of cancer types (5). Ribociclib was approved by the United States Food and Drug Administration (FDA) in 2017 for use in combination with an aromatase inhibitor (e.g., letrozole) for the treatment of postmenopausal women with advanced or metastatic breast cancer that is hormone receptor (HR) positive and human epidermal growth factor receptor 2 (HER2) negative (5,6). The efficacy and safety of single-agent ribociclib in patients with neuroblastoma was evaluated as part of the Phase I study in pediatric patients, where ribociclib demonstrated an acceptable toxicity profile and promising antitumor activity (7). Preclinical data suggest that ribociclib can cross the blood-brain barrier (BBB), supporting further clinical development for the treatment of central nervous system (CNS) tumors (8).
Phase 0 clinical trials are commonly defined as first-in-human studies with no therapeutic or diagnostic intent, a limited number of patients, and micro-dosing of the experimental agent (9,10). For brain tumor patients, however, the utility of these design elements is hampered by the BBB, a lack of predictive animal models, and the significant risks of tumor tissue acquisition. Here, we adapt the Phase 0 strategy (11,12) by employing “limited therapeutic” dosing (5 days of the maximally tolerated dose) instead of micro-dosing and uses matched archival controls (instead of pre- and post-treatment biopsies) to assess pharmacodynamic effects. To mitigate the ethical and accrual challenges of a nontherapeutic study, we incorporate a pharmacokinetic- and pharmacodynamic-guided “trigger” that graduates Phase 0 patients into a therapeutic expansion cohort.
Here, to explore the utility of ribociclib in treating recurrent glioblastoma patients, we completed a Phase 0 clinical trial with an expansion cohort for recurrent glioblastoma patients to examine pharmacokinetic and pharmacodynamic endpoints following a 5-day exposure to ribociclib (13). This study was designed to (i) provide in vivo pharmacokinetic data for non-enhancing and enhancing tumor tissue, (ii) identify the molecular effects of ribociclib in glioblastoma patients, and (iii) interrogate putative mechanisms of resistance in tumor recurrences. Positive results from the trial would accelerate our development of this combinatorial drug regimen and enable a larger-scale efficacy study, whereas negative results would enhance our understanding of tumor resistance and potentially identify an additional agent for combination drug therapy.
Methods
Patient Selection
Study patients were older than 18 years with a recurrent World Health Organization grade IV glioma. Prior to accrual, all patients had undergone resection of their primary tumor, followed by standard-of-care Stupp regimen with temozolomide and fractionated radiotherapy. The median time from initial diagnosis to recurrence was 271 days, whereas the median time from completion of radiation to recurrence was 200 days. Based on radiographic and/or clinical evidence of disease progression, all patients were previously scheduled for surgical re-resection, had an Eastern Cooperative Oncology Group performances status of <2, and exhibited adequate organ function (Table 1).
Table 1:
Patient demographics and clinical characteristics
| Characteristics | |
|---|---|
| Sex (male/female) | 8/4 |
| Age (years) | 49 (31-66) |
| Weight (kg) | 85 (63-109) |
| Height (cm) | 174 (154-193) |
| ECOG/Zubrod Performance Status, n (%) | |
| 0, n (%) | 3 (25%) |
| 1, n (%) | 7 (58%) |
| 2, n (%) | 2 (17%) |
| Extent of Resection | |
| GTR, n (%) | 8 (67%) |
| STR, n (%) | 4 (33%) |
| Unknown, n (%) | 0 (0%) |
| Prior bevacizumab, n (%) | 0 (0%) |
| Timing of ribociclib , n (%) | |
| First progression | 8 (67%) |
| Second progression | 1 (8%) |
| Third progression | 2 (17%) |
| Fourth progression | 1 (8%) |
| Alanine aminotransferase (IU/L) | 17.5 (10-39) |
| Aspartate aminotransferase (IU/L) | 16 (10-115) |
| Total bilirubin (mg/dl) | 0.6 (0.3-1) |
| Serum albumin | 4.2 (3.6-4.8) |
| Serum creatinine | 0.93 (0.67-3.8) |
Study Design and Drug Administration
This investigational, Phase 0, open-label, nonrandomized trial with a pharmacokinetic/pharmacodynamic-guided expansion cohort was conducted at the Ivy Brain Tumor Center at the Barrow Neurological Institute in Phoenix, Arizona in partnership with Karmanos Cancer Institute. The study () received approval from the institutional review board and was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice. Patients were accrued at the Barrow Neurological Institute and informed written consent was obtained from each patient in the study.
Enrolled Phase 0 patients were administered 900 mg/d of ribociclib for 5 days before planned cranial tumor resection. Patients were assigned to 3 time-escalation arms in which tumor resection was performed at 2, 8 or 24 hours, respectively, following the final dose of ribociclib. Intensive blood sampling was performed on the fourth day of drug administration. During tumor resection, blood, cerebrospinal fluid (CSF), and tumor samples from contrast-enhancing and non-enhancing regions (based on preoperative MRI and intraoperative neuronavigation) were collected for pharmacokinetic and pharmacodynamic analyses. We determined the maximum tolerated dose of ribociclib as 900 mg daily. To ensure tolerability of continuous treatment of ribociclib, patients enrolled into the expansion cohort were treated with a lower dose at 600 mg daily for 21 days on and 7 days off until disease progression was noted based on Response Assessment in Neuro-Oncology (RANO) criteria.
Statistical Methods
The primary objectives of this trial were to evaluate pharmacokinetic and pharmacodynamic endpoints. The secondary objectives were to explore progression-free survival (PFS) and overall survival (OS) among patients who exhibited positive pharmacokinetic and pharmacodynamic responses. Phase 0 patients were determined to be responders if both the unbound ribociclib concentration in the non-enhancing tumor exceeded 5-fold of the in vitro IC50 for CDK4/6 kinase activity (IC50, 0.04 μM) (6) and if we observed a > 20% decrease in pRB+ cells compared to matched archival tissue. Only Phase 0 responders were eligible to enroll into the pharmacokinetic/pharmacodynamic-guided expansion cohort of the trial. Pharmacokinetic/pharmacodynamic-guided expansion cohort patients were treated with ribociclib until disease progression. PFS was defined as the duration from the date of recurrent tumor resection to the date of disease progression or death from any cause, whichever occurred first. OS was defined as the duration from the date of recurrent tumor resection to the date of death. The pharmacokinetic/pharmacodynamic-guided expansion cohort study was exploratory for efficacy, and no formal statistical comparison was planned. Thus, the sample size was justified based on feasibility. For each of 3 time-escalation arms, 4 patients were allocated and, if no pharmacodynamic effects were observed in the setting of adequate drug penetration, time-escalation arms could be expanded to 8 patients to accommodate greater molecular diversity. Thus, the planned sample size for Phase 0 was 12 to 24 patients. Comparisons between 2 paired data and 2 unpaired data were carried out using a paired t-test and an unpaired t-test, respectively, after data transformation. The distributions of survival outcomes (PFS and OS) were graphically summarized using Kaplan-Meier curves, and their median times and associated 95% confidence intervals were estimated using Kaplan-Meier estimates.
Safety and Assessments
Demographic data and medical history were recorded for all study patients. Physical examination, vital signs, organ functions, and other safety assessments (Eastern Cooperative Oncology Group performance status, registration of concomitant medication, hematology, biochemistry, and urine analysis) were performed at baseline. Common Toxicity Criteria Adverse Event (CTC AE) 4.0 criteria were used to document adverse events.
Patient Enrollment Criteria with RB status and Genomic Screening
Specimen samples from prior tumor resections were examined with immunohistochemistry and array comparative genomic hybridization (aCGH). Immunohistochemistry staining for all samples was completed on the Leica Bond, a fully automated platform using optimized conditions that were standardized with archival glioblastoma tissue. Briefly, archival FFPE slides was stained with anti-RB (Cell Signaling, #9309, 1:100) for assessing the percentage of RB+ cells. The stained slides were imaged using Aperio Versa System (Leica) and analyzed using ImageScope software. In parallel, the slides were also analyzed by a board-certified neuropathologist. Array Comparative Genomic Hybridization (aCGH) was conducted within a CLIA-certified laboratory (MOgene LC, St. Louis, MO, USA). Genomic DNA from archival formalin-fixed paraffin-embedded (FFPE) sections was extracted with DNeasy Blood and Tissue kit (Qiagen, Valencia, CA, USA). Quality control analysis and DNA quantification were determined using Tapestation (Agilent Technologies, Santa Clara, CA) and a Qubit 2.0 fluorometer (Thermo Fisher Scientific, Waltham, MA, USA). Genomic DNA was then labeled and hybridized to a SurePrint G3 Human CGH microarray slide (Agilent Technologies). The array was then scanned using Agilent C Scanner and the image analyzed with Feature Extraction software. Patients who met all entry criteria, including positive RB status (> 20% RB+ cells) and loss of CDKN2A or amplification of CDK4 or CDK6 or CCND1/D2/D3 were eligible for enrollment.
Pharmacokinetic Evaluation
Blood pharmacokinetic samples were collected from each patient at pre-dosing, 0.5, 1, 2, 4, 6, 8 and 24 hours after the administration of the fourth presurgical dose of ribociclib. Plasma was separated from whole blood by centrifugation (at 4ºC, 1500 g for 10 minutes), and plasma samples were stored at −80ºC until analysis. Tumor resection was performed after the administration of the fifth presurgical dose of ribociclib. Blood, tumor (including contrast-enhancing and non-enhancing regions), and CSF samples were collected intraoperatively at predefined time points (i.e., 2, 8 or 24 hours after the fifth presurgical dose). Two tumor samples (0.5 cm3), corresponding to enhancing- and non-enhancing glioblastoma, were collected intraoperatively from each patient and divided into 2 equal portions. Specimen locations were recorded with intraoperative MRI neuronavigation system. Each tumor sample was immediately rinsed with ice-cold PBS to remove residual blood, then dried and snap frozen in liquid nitrogen. The total concentrations of ribociclib in plasma, tumor, and CSF samples were determined using a validated liquid chromatography with tandem mass spectrometry method (14). The fraction of unbound ribociclib in plasma and tumor tissues were determined by equilibrium dialysis, and unbound drug concentration was calculated as the product of total concentration and fraction unbound (14).
Pharmacokinetic Data Analysis
Plasma pharmacokinetic parameters of total and unbound ribociclib were estimated from individual plasma concentration-time profiles using the noncompartmental analysis with Phoenix WinNonlin software (Certara, Princeton, NJ, USA). Estimated plasma pharmacokinetic parameters, including the maximum plasma concentration (Cmax), time to reach Cmax (Tmax), area under the curve during one dosing interval (AUCτ), and apparent oral clearance (CL/F, estimated from dose/AUCτ) were summarized using descriptive statistics as the mean, median, range, and coefficient variation.
Because unbound drug concentrations are pharmacologically relevant (15), CNS pharmacokinetics were evaluated based on unbound ribociclib concentrations in CSF, non-enhancing, and enhancing tumor regions. The extents of drug penetration into the CNS and tumors were assessed by the CSF-to-plasma unbound drug concentration ratio (CSF/Pu), the tumor-to-plasma unbound drug concentration ratio (Tu/Pu), and the tumor-to-CSF unbound drug concentration ratio (Tu/CSF).
Pharmacodynamic Controls
To test the stability of proposed pharmacodynamic biomarkers (pRB, pFOXM1, MIB1, and cleaved caspase-3), we analyzed a historical cohort of 10 matched primary and recurrent glioblastoma patients who received standard-of-care Stupp regimen and were not enrolled in the study. FFPE tissues were stained with anti-pRB (Cell Signaling, #8516, 1:400), anti-pFOXM1 (Cell Signaling, #14655, 1:200), anti-MIB1 (DAKO, M724029, 1:100), and anti-cleaved caspase-3 (Cell Signaling, #9661, 1:300) using our standardized immunohistochemistry protocol with the BOND RX automated system (Leica Biosystems, Wetzlar, Germany). The stained slides were imaged and analyzed by a board-certified pathologist, and Aperio Image analysis software (Leica Biosystems) was used to assess differences in positivity for pRB, MIB1, and cleaved caspase-3 in primary vs. recurrent tumors. There were no significant differences between primary and recurrent tissues in the levels of the tested biomarkers.
Pharmacodynamic Assessment
FFPE tissue from the patient’s first tumor resection (at the time of initial diagnosis) was used as “archival” or treatment-naïve control tissue for comparison. Both archival FFPE tumor tissue and study specimens collected at the time of resection were assayed simultaneously using our standardized immunohistochemistry protocol with the Leica BOND RX automated system. For each run, we included positive (historical glioblastoma tissue) and negative controls (no primary antibody). The following antibodies were utilized in the study: anti-RB (Cell Signaling, #9309, 1:100), anti-pRB (Cell Signaling, #8516, 1:400), anti-pFOXM1 (Cell Signaling, #14655, 1:200), anti-FOXM1 (Sigma, HPA029974, 1:500), anti-MIB-1 (DAKO, M724029, 1:100), anti-activated caspase-3 (Cell Signaling, #9661, 1:300), anti-pS6 ribosomal protein (Cell Signaling, #4858, 1:100), and anti-4EBP1 (Cell Signaling, #2855, 1:500). Stained FFPE slides were imaged using a Leica DM55500 microscope and analyzed using Aperio Image analysis software. The function of “nucleus” in the software was used to systematically quantify the percentage of positive cells in a given area.
Once the percentage of positive cells for a given protein was quantified, a paired t-test was used to compare log-transformed percent positive cells between archival (predose) and Phase 0 surgical tissue (postdose) at a 2-sided 5% level. The comparisons were further carried out with the normalized percent positive cells to adjust the baseline (i.e., historical primary and recurrent controls) percent positive cells for both archival and surgical tissues. For normalization, all percent positive cells of the treated tissues were divided by the median of the percent positive cells of the untreated tissues (historical controls). The overall comparisons between pre- and postdose were performed regardless of specific time-escalation arms whereas the same comparisons were carried out for each of 3 time-escalation arms (at 2 hours, 8 hours, and 24 hours postdose).
Longitudinal Tissue Analysis
For patients in the expansion cohort, evidence of tumor recurrence led to routine clinical consideration of re-resection as part of the patient’s neurosurgical care. In cases where a re-resection was planned, enhancing tumor tissue, in addition to blood and CSF, were collected intraoperatively. Immunohistochemical analysis for phospho-S6, pFOXM1, pS6, and p4EBP1 (as listed above) was performed on archival-, surgical-, and post-treatment-derived tissue from each patient. Results were quantified using Aperio software, as described above. FFPE tumor tissue was analyzed using Vantage 3D solid tumor panel (28-plex protein expression; NanoString Technologies, Seattle, WA, USA). The quantification of each protein was normalized by internal Histone-3 expression levels. The ratio of phospho-form of protein to total protein was calculated for key components of the PI3K and MAPK pathways, including AKT, S6, 4EBP1, and ERK1/2. Lastly, RNA-sequencing was performed on FFPE tissue from all patients to characterize expression patterns following long-term ribociclib monotherapy.
Results
Patient population
Patient demographics and clinical characteristics are summarized in Table 1. All patients had completed the Stupp regimen prior to tumor recurrence, and no patients had received any other adjuvant chemotherapy prior to enrollment. Similarly, no patients had been treated with tumor-treating fields technology. Twelve adult patients with recurrent glioblastoma were enrolled in the Phase 0 component of the study and received 900 mg/d of ribociclib for 5 days prior to the scheduled brain tumor resection. Presurgical ribociclib was well-tolerated, and no patients were removed due to drug-related toxicities. All planned surgical resections occurred within the designated interval following the last presurgical dose of ribociclib (median error, ± 19.7 minutes).
Plasma and CNS Pharmacokinetics
Figure 1A illustrates the observed plasma concentration-time profiles of total and unbound ribociclib in individual glioblastoma patients following daily oral administration (900 mg) for 4 days. The plasma pharmacokinetics of total and unbound ribociclib are summarized (Figure 1B). Ribociclib achieved the peak plasma concentration at an average of 3.2 hours after oral administration. It exhibited a mean elimination half-life of 13.5 hours, based on which the steady-state drug exposure was considered achieved after 4 or 5 days of daily treatment. A large inter-individual pharmacokinetic variability was observed, as demonstrated by about 50% coefficient variation in the drug exposure (Cmax, Ctrough, and AUC0–24h) among 12 patients. Ribociclib was modestly bound to plasma proteins, with the mean fraction unbound of 0.12 in 12 glioblastoma patients. At the steady-state, the unbound drug concentrations fluctuated 3.4-fold, from 0.13 μM (mean trough level) to 0.44 μM (mean peak level; Figure 1B).
Figure 1.

Plasma pharmacokinetics of ribociclib in glioblastoma patients. (A) Observed plasma concentration-time profiles of total and unbound ribociclib in 12 glioblastoma patients following daily oral administration (900 mg) for 4 days. Symbols represent observed plasma concentrations, and dash lines represent the mean plasma concentration-time profiles fitted with the one-compartment model. (B) Plasma pharmacokinetic summary. Detailed summary of the steady-state plasma pharmacokinetic parameters of total and unbound ribociclib in glioblastoma patients are listed. Abbreviations: Cmax, maximum plasma concentration; Tmax, time to achieve the Cmax; Ctrough, trough plasma concentration (at ~ 24 h); AUC0-24h, area under the concentration-time curve during one dosing interval (24 h); CL/F, apparent oral clearance; T1/2, elimination half-life.
CNS pharmacokinetics, as assessed by unbound ribociclib concentrations in CSF, non-enhancing, and enhancing tumor regions (Figure 2A) after oral administration of the fifth presurgical dose are presented in Figure 2B and summarized (Figure 2C). Twelve patients were enrolled into 3 arms (with 4 patients per arm) for the assessment of CNS penetration and tumor pharmacodynamics at 2, 8, or 24 hours following the last dose of ribociclib. CSF drug concentrations are commonly used as a surrogate for unbound drug concentrations in the normal brain with intact BBB (16,17). Ribociclib reached maximum CNS exposure at 2 to 8 hours following the oral administration, which was in-line with the Tmax in plasma (Figure 2B). Drug elimination from the CNS (including CSF, non-enhancing, and enhancing tumor regions) appeared relatively slower than that from plasma, as indicated by the time-dependent increase of the CNS-to-plasma unbound drug concentration ratios (Figure 2C). At 2 to 24 hours after dosing, the unbound ribociclib concentrations (mean ± standard deviation) in CSF, non-enhancing, and enhancing tumor regions were 0.374 ± 0.272 μM, 0.560 ± 0.364 and 2.152 ± 1.559 μmol/kg, respectively. In 11 of 12 patients, the measured unbound drug concentrations in CSF, non-enhancing, and enhancing tumor regions at 2 to 24 h post dosing were all ≥ 5-fold of the in vitro IC50 for inhibiting CDK4/6 kinase activity (0.04 μM) (6).
Figure 2.
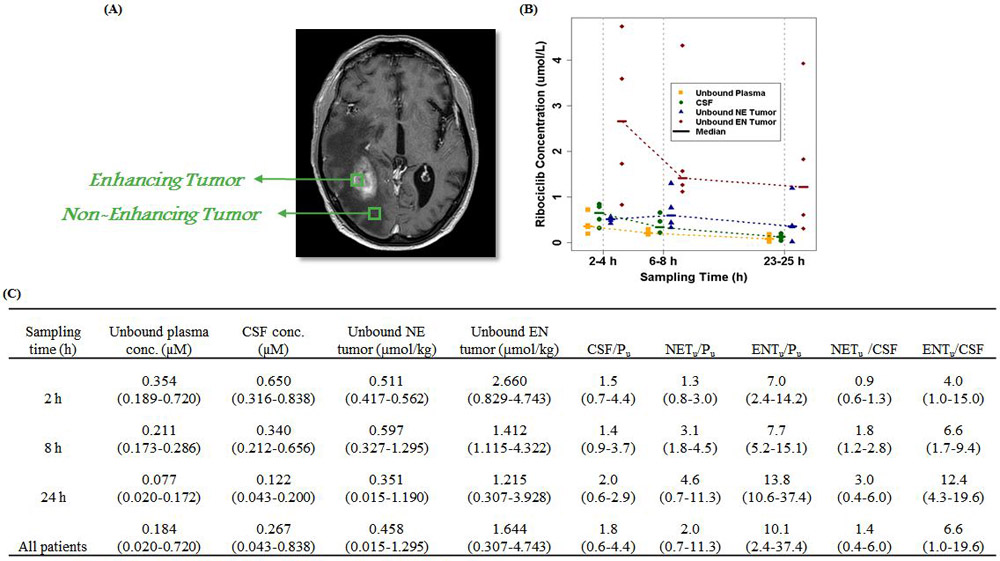
CNS pharmacokinetics of ribociclib in glioblastoma patients (A) Representative imaging indicating enhancing and non-enhancing region of the tumor. (B) Observed unbound ribociclib concentrations in plasma, CSF, non-enhancing, and enhancing glioblastoma tumor regions at 2 to 24 hours after oral administration of the fifth dose of ribociclib. Patients were treated with a daily oral dose of 900 mg for 5 days, and all samples were collected on day 5. Symbols represent observed concentrations, and dash lines represent the median concentration-time profiles. (C) Summary of pharmacokinetics characteristics of CNS ribociclib in glioblastoma patient. Data are presented as the median (range). Abbreviations: CNS, central nervous system; CSF, cerebrospinal fluid; NE, non-enhancing; EN, enhancing; CSF/Pu, CSF-to-plasma unbound drug concentration ratio; NETu/Pu, non-enhancing tumor-to-plasma unbound drug concentration ratio; ENTu/Pu, enhancing tumor-to-plasma unbound drug concentration ratio; NETu/CSF, non-enhancing tumor-to-CSF unbound drug concentration ratio; ENTu /CSF, enhancing tumor-to-CSF unbound drug concentration ratio.
Tumor Pharmacodynamics
To assess the pharmacodynamic effects of ribociclib in human glioblastoma, phosphorylation of RB was selected as the primary determinant. Other assayed biomarkers included phosphorylation of FOXM1, the proliferative marker MIB-1, and the apoptotic marker cleaved caspase-3. Among the 12 Phase 0 patients, 9 (75%) exhibited a > 20% of decrease in pRB levels (Figure 3A). On average, there was a 50% decrease in pRB levels among the 12 treated patients, which was significantly higher than paired control specimens (Figure 3B). With detailed quantification, both pRB and MIB-1 were significantly reduced following Phase 0 ribociclib exposure (P = 0.001 and 0.039, respectively), and cleaved caspase-3 also trended towards a concomitant increase (P = 0.19; Figure 3B). Observed declines in pRB and MIB-1 were most prominent in the 8-hour postdose cohort (P = 0.005 and 0.121, respectively). As a control, matched cohort analysis of 8 paired primary and recurrent glioblastoma samples without ribociclib exposure demonstrated no significant changes in any of the studied biomarkers (Figure 3). Interestingly, levels of pFOXM1 had a downward trend (P = 0.142) after normalization with historical control samples, primarily at the 8-hour postdose interval (Figure 3B). Both pRB and MIB-1 are also significantly decreased after normalization. Among the 12 phase 0 patients, 4 were diagnosed with histological pseudo-progression. After removing these 4 patients, the pharmacodynamic effects of ribociclib (n=8) remain evident with a significant decrease of pRB and MIB-1 (p=0.005 and 0.017, respectively; Supplemental Figure 1). Together, our pharmacodynamic analysis suggests that the targeted inhibition of CDK4/6 kinases is achieved in the recurrent tumor tissue from short-term ribociclib treatment.
Figure 3.

Pharmacodynamic analysis of surgical glioblastoma tissue after short-term ribociclib treatment. (A) Representative pRB immunohistochemistry staining for matched archival and surgical specimens from ribociclib treatment patients and historical control patients are shown. The scale bar is 100 μm. (B) Quantification of pRB, pFOXM1, MIB-1, and activated caspase-3 positive cells by Aperio imaging system from 8 matched archival and 12 surgical specimens are shown. Three time-escalating cohorts (2-, 8-, and 24-hour) after the last dose of ribociclib are indicated. Non-treatment control samples are in the left column whereas the treatment group results and the normalized results are shown in the middle and right columns. P-value of each marker is indicated.
Clinical Response and Longitudinal Tissue Analysis
Of the 12 Phase 0 study patients, 4 demonstrated histological evidence of pseudo-progression, rendering them ineligible for the expansion cohort study. Accordingly, acquired specimens from these 4 Phase 0 patients were employed for pharmacokinetics analysis only. Of the 8 Phase 0 patients with histologically evident glioblastoma recurrence, 6 (75%) were identified as responders based on their pharmacokinetic and pharmacodynamic results and were subsequently enrolled into the expansion cohort study to receive ribociclib continuously until recurrence is observed according to RANO criteria. No patients were removed from the expansion cohort due to drug-related toxicity. All patients ultimately progressed to tumor recurrence, with a PFS6 of 16.7% and a median OS of 7.8 months (Figure 4A). The clinical course of these 6 patients, as well as associated genomic characterizations, are summarized in Figure 3B. We observed no patterns of genomic alterations statistically associated with better clinical response.
Figure 4.

Progression-free survival (PFS) and overall survival (OS) of the expansion cohort. (A) Kaplan-Meier plot of the PFS and the OS. (B) Swimmer plot of the PFS for the 6 patients. Pharmacokinetic and pharmacodynamic characteristics of the patients are shown.
Three of 6 patients underwent planned re-resection based on evidence of tumor recurrence following ribociclib monotherapy (termed post-treatment in figures). In all 3 patients, a significant increase in pRB was observed, as compared to matched specimens collected during their Phase 0 operation (7.4% to 19.96%, P = 0.02; Figure 5A, B). For these patients, we also observed a trend towards increase in the MIB-1 index (2.3% to 7.55%, P = 0.23; Figure 5A, B). Interestingly, we detected a significant increase in the proportion of cells with mTOR/PI3K signaling pathway activity (Figure 5A, B). Specifically, as compared to matched Phase 0 samples, the post-treatment specimens exhibited significant increases in pS6 (21.3% to 36.7%, P = 0.009) and p4EBP1 levels (2.8% to 31.2%, P = 0.02; Figure 5A, B). To further examine mitogenic pathways in ribociclib-treated patients, we performed semi-quantitative protein analyses using the nCounter Vantage 3D protein solid tumor panel (NanoString Technologies, Seattle, WA, USA). The ratios of p4EBP-1/4EBP-1 in post-treatment samples were significantly higher compared to historical controls and accompanied by an upward trend in the ratio of pAKT/AKT (Figure 5C), suggesting that ribociclib therapy results in the upregulation of the mTOR/PI3K pathway. Furthermore, for the 3 post-treatment samples, we continued to observe an upward trend in p4EBP1 and pAKT levels (Figure 5D), indicating that the mTOR/PI3K pathway may serve as a resistance mechanism following CDK4/6 inhibition. No significant changes were observed in EGFR or MEK1/2 signaling pathways (Figure 5C, D).
Figure 5.

Longitudinal pharmacodynamic analysis of re-recurrent tumor specimens after continuous ribociclib treatment (post-treatment). (A) Representative immunohistochemistry images of pRB and pS6 are shown from matched tumor specimens (archival, surgical, and post-treatment). (B) Quantification and statistical analysis of percentage of pRB, MIB-1, pS6, and p4EBP1 are shown for 3 patients with re-recurrent surgery tumor specimens. (C, D) Protein array analysis was performed across study specimens and non-study control specimens. Quantification of the ratio of p4EBP-1/4EBP-1, pAKT/ATK, pERK/ERK, pS6/S6 and pEGFR/EGFR is shown with non-treatment historical control (n = 4) and ribociclib treatment cohort (n = 7) (C) and post-treatment re-recurrent cohort (n = 3) (D). P-value of each protein between samples was indicated.
Discussion
This study provides the first clinical evidence of CNS penetration of ribociclib and target modulation in patients with recurrent glioblastoma. Ribociclib achieves therapeutic concentrations not only in contrast-enhancing tumor regions with a disrupted BBB but also in non-enhancing tumor tissues with largely intact BBB. Because non-enhancing regions of glioblastoma are often unresectable, good penetration of ribociclib into these regions would represent a significant therapeutic advantage for treating glioblastoma. Following the administration of the last dose (900 mg), the median unbound concentrations of ribociclib in non-enhancing region were 0.51, 0.6 and 0.35 μmol/kg (or μM) at 2, 8 and 24 hours, respectively—all exceeding the pre-defined threshold of 0.2 μM (i.e., 5-fold of the in vitro IC50 for inhibition of CDK4/6). Given the linear pharmacokinetics of ribociclib, the median unbound drug concentrations in non-enhancing tumor regions following the therapeutic dose of 600 mg would be approximately 2/3 of those observed at the 900-mg dose, which could be adequate for target inhibition (> 0.2 μM). Despite this predicted correlation, the direct pharmacokinetic and pharmacodynamic effects of 600-mg dosing were not measured in this study, and our understanding of this dose-level will be enhanced by future tissue-based study. Nevertheless, evidence of CDK4/6 pathway inhibition after 5 days of drug exposure at 900 mg was suggested by significant reduction of RB phosphorylation and tumor proliferation. The reliability of this observation is tempered by the study’s need to employ archival tissues as the comparator for Phase 0-collected specimens.
Three orally bioavailable, highly-selective competitive inhibitors of CDK4/6, including palbociclib, abemaciclib, and ribociclib, have been FDA-approved for the treatment of breast cancer and are currently under clinical development for a variety of cancers including glioblastomas (18,19). In preclinical models, all 3 CDK4/6 inhibitors demonstrated brain penetration and antitumor activity against intracranial tumors (8,20). Palbociclib has been evaluated in 22 patients with recurrent RB-positive glioblastoma in a Phase 2 trial in which, despite evidence of tumor penetration (assessed by total drug tumor concentrations), it showed limited clinical efficacy in terms of prolonging PFS (21). Abemaciclib is currently under investigation for the treatment of patients with first recurrent glioblastoma [, ], and data are yet to be reported. Our present study on ribociclib complements previous knowledge on the therapeutic implications of modulating CDK4/6 signaling in glioblastomas.
Compared to historical controls for recurrent glioblastoma, the pharmacokinetic/pharmacodynamic-guided expansion cohort study suggest a limited clinical impact of ribociclib as monotherapy. Nevertheless, the accompanying longitudinal tissue analysis raises the possibility that ribociclib-treated glioblastomas utilize the PI3K/mTOR pathway for cellular resistance to CDK4/6 inhibition. Interestingly, previous work using glioma mouse models (19,22), as well as reports from non-CNS cancers (19,23), echo this observation. Taken together, these data identify ribociclib as a potential component of a combinatorial drug strategy for recurrent glioblastoma and suggests an mTOR inhibitor as a candidate pairing. While a regimen of ribociclib plus an mTOR inhibitor has yet to be tested in glioblastoma patients, a Phase 1 clinical trial for ribociclib plus everolimus plus exemestane in patients with endocrine-resistance advanced breast cancer was reported in 2017 (24). In that study, the maximally tolerated dose was not reached, and ribociclib 300 mg/d + everolimus 2.5 mg/d + exemestane 25 mg/d was declared the recommended Phase 2 dose. Importantly, the ribociclib Ctrough was similar to expected values, whereas the everolimus Ctrough was 2- to 3-fold greater than expected, resulting in everolimus blood exposure levels equivalent to 5 to 10 mg dosing of everolimus (24). Based on these findings, a combinatorial ribociclib plus everolimus Phase 0/2 study for recurrent glioblastoma is underway ().
The study design employed in this trial is an adaptation of conventional Phase 0 paradigms (10) to accommodate challenges associated with brain tumor patients, including the high degree of risk surrounding tumor acquisition, the unsuitability of microdosing when crossing the BBB, and the low accrual rates of nontherapeutic trials. Nevertheless, a number of important study design limitations exist. Conventional Phase 0 studies for non-CNS cancers often employ multiple biopsies before and after drug exposure. In contrast, Phase 0 trials such as this rely on archival tissue, instead, to serve as the baseline comparator. Additionally, inadequate penetration of therapeutic agents across the BBB remains a central obstacle for CNS drug development (25,26). For brain tumor patients, the utility of a microdosing strategy is limited by the BBB. Specifically, drug microdoses (typically < 1% of the therapeutic dose) are often undetectable within the CNS, even when using advanced bioanalytical methods. Consequently, brain tumor Phase 0 studies necessitate higher systemic drug concentrations and higher risks of associated side effects. Here, we used the maximally tolerated dose for a brief window of exposure prior to the planned resection. A final challenge for brain tumor Phase 0 studies is the dampening effect that non-therapeutic trials have on patient accrual. A Phase 0 trial that incorporates a pharmacokinetic- and pharmacodynamic-guided expansion cohort, graduating Phase 0 patients into a therapeutic continuous treatment cohort is more compelling for potential study candidates as it provides direct biological evidence supporting the decision for therapeutic dosing. Although the expansion cohort study is not a conventional phase 2 trial (27), it nevertheless provides a view of preliminary clinical responses and contextualizes these responses to therapy against the backdrop of detailed molecular and pharmacological analyses. Embracing and expanding this strategy for brain tumor patients may alleviate some of the challenges associated with conventional Phase 2 studies.
Conclusion
We conducted a Phase 0 clinical trial with a pharmacokinetic/pharmacodynamic-guided expansion cohort to assess plasma and tumor pharmacokinetic and pharmacodynamic endpoints in recurrent glioblastoma patients. Ribociclib penetrated well into not only human glioblastoma regions with disrupted BBB, but also tumor regions with largely intact BBB. Drug exposure was accompanied by a pharmacodynamic response, as indicated by significant reduction in RB phosphorylation and tumor proliferation. However, preliminary analysis from the expansion cohort suggests that ribociclib monotherapy showed limited clinical efficacy in recurrent glioblastoma. Our data support further investigation of combination therapy with CDK4/6 and PI3K/mTOR inhibitors for treating recurrent glioblastoma.
Supplementary Material
Statement of Translational Relevance.
The RB-CDK4/6-E2F signaling axis is frequently deregulated in glioblastoma, making it an ideal pathway to block tumor growth. In this phase 0 study, we assessed the CNS penetration and tumor pharmacodynamics of ribociclib, a CDK4/6 inhibitor, in patients with recurrent glioblastoma. Using an integrated approach involving quantitative analysis of drug penetration in the enhancing and non-enhancing tumor regions and downstream target inhibition, we provide the first clinical evidence for good CNS penetrance of ribociclib. The pharmacodynamic- and pharmacokinetic-guided expansion cohort study indicates limited clinical efficacy of ribociclib monotherapy. Longitudinal analysis suggests combination therapy with ribociclib and an mTOR inhibitor may provide therapeutic benefit in patients with recurrent glioblastoma.
Acknowledgment
The study was supported by a grant from the Ben and Catherine Ivy Foundation as well as the United States National Institute of Health Cancer Center Support Grant (P30 CA022453). We thank staff and research nurses who participated in this work at the Ivy Brain Tumor Center as well as Novartis for providing the study drug (ribociclib). We are indebted to the patients who enrolled in this study.
Funding: The Ben & Catherine Ivy Foundation and NIH Cancer Center Support Grant (P30 CA022453)
Footnotes
Conflicts: The authors declare no potential conflicts of interest
References
- 1.Cancer Genome Atlas Research N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008;455(7216):1061–8 doi 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008;321(5897):1807–12 doi 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144(5):646–74 doi 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 4.Michaud K, Solomon DA, Oermann E, Kim JS, Zhong WZ, Prados MD, et al. Pharmacologic inhibition of cyclin-dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res 2010;70(8):3228–38 doi 10.1158/0008-5472.CAN-09-4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Edessa D, Sisay M. Recent advances of cyclin-dependent kinases as potential therapeutic targets in HR+/HER2- metastatic breast cancer: a focus on ribociclib. Breast Cancer (Dove Med Press) 2017;9:567–79 doi 10.2147/BCTT.S150540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tripathy D, Bardia A, Sellers WR. Ribociclib (LEE011): Mechanism of Action and Clinical Impact of This Selective Cyclin-Dependent Kinase 4/6 Inhibitor in Various Solid Tumors. Clin Cancer Res 2017;23(13):3251–62 doi 10.1158/1078-0432.CCR-16-3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geoerger B, Bourdeaut F, DuBois SG, Fischer M, Geller JI, Gottardo NG, et al. A Phase I Study of the CDK4/6 Inhibitor Ribociclib (LEE011) in Pediatric Patients with Malignant Rhabdoid Tumors, Neuroblastoma, and Other Solid Tumors. Clin Cancer Res 2017;23(10):2433–41 doi 10.1158/1078-0432.CCR-16-2898. [DOI] [PubMed] [Google Scholar]
- 8.Raub TJ, Wishart GN, Kulanthaivel P, Staton BA, Ajamie RT, Sawada GA, et al. Brain Exposure of Two Selective Dual CDK4 and CDK6 Inhibitors and the Antitumor Activity of CDK4 and CDK6 Inhibition in Combination with Temozolomide in an Intracranial Glioblastoma Xenograft. Drug Metab Dispos 2015;43(9):1360–71 doi 10.1124/dmd.114.062745. [DOI] [PubMed] [Google Scholar]
- 9.Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov 2004;3(8):711–5 doi 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- 10.Kummar S, Kinders R, Rubinstein L, Parchment RE, Murgo AJ, Collins J, et al. Compressing drug development timelines in oncology using phase ‘0’ trials. Nat Rev Cancer 2007;7(2):131–9 doi 10.1038/nrc2066. [DOI] [PubMed] [Google Scholar]
- 11.Sanai N, Li J, Boerner J, Stark K, Wu J, Kim S, et al. Phase 0 Trial of AZD1775 in First-Recurrence Glioblastoma Patients. Clin Cancer Res 2018;24(16):3820–8 doi 10.1158/1078-0432.CCR-17-3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu R, Shimizu F, Hovinga K, Beal K, Karimi S, Droms L, et al. Molecular and Clinical Effects of Notch Inhibition in Glioma Patients: A Phase 0/I Trial. Clin Cancer Res 2016;22(19):4786–96 doi 10.1158/1078-0432.CCR-16-0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tien AC, Bao X, Derogatis A, Mehta S, Li J, Sanai N. Plasma and Tumor Pharmacokinetics of Ribociclib in Patients with Recurrent Glioblastoma. Clin Cancer Res 2018;(in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bao X, Wu J, Sanai N, Li J. Determination of total and unbound ribociclib in human plasma and brain tumor tissues using liquid chromatography coupled with tandem mass spectrometry. J Pharm Biomed Anal 2019;166:197–204 doi 10.1016/j.jpba.2019.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Waters NJ, Obach RS, Di L. Consideration of the unbound drug concentration in enzyme kinetics. Methods Mol Biol 2014;1113:119–45 doi 10.1007/978-1-62703-758-7_7. [DOI] [PubMed] [Google Scholar]
- 16.Mooberry SL, Hilinski MK, Clark EA, Wender PA. Function-oriented synthesis: biological evaluation of laulimalide analogues derived from a last step cross metathesis diversification strategy. Mol Pharm 2008;5(5):829–38 doi 10.1021/mp800043n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu X, Smith BJ, Chen C, Callegari E, Becker SL, Chen X, et al. Evaluation of cerebrospinal fluid concentration and plasma free concentration as a surrogate measurement for brain free concentration. Drug Metab Dispos 2006;34(9):1443–7 doi 10.1124/dmd.105.008201. [DOI] [PubMed] [Google Scholar]
- 18.Sherr CJ, Beach D, Shapiro GI. Targeting CDK4 and CDK6: From Discovery to Therapy. Cancer Discov 2016;6(4):353–67 doi 10.1158/2159-8290.CD-15-0894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Knudsen ES, Witkiewicz AK. The Strange Case of CDK4/6 Inhibitors: Mechanisms, Resistance, and Combination Strategies. Trends Cancer 2017;3(1):39–55 doi 10.1016/j.trecan.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patel Y PDTB-12. CNS PENETRATION OF THE CDK4/6 INHIBITOR RIBOCICLIB (LEE011) IN NON-TUMOR BEARING MICE AND MICE BEARING ORTHOTOPIC PEDIATRIC BRAIN TUMORS. Neuro-Oncology 2016;18(suppl_6):vi152–vi doi 10.1093/neuonc/now212.632. [DOI] [Google Scholar]
- 21.Taylor JW, Parikh M, Phillips JJ, James CD, Molinaro AM, Butowski NA, et al. Phase-2 trial of palbociclib in adult patients with recurrent RB1-positive glioblastoma. J Neurooncol 2018;140(2):477–83 doi 10.1007/s11060-018-2977-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Olmez I, Brenneman B, Xiao A, Serbulea V, Benamar M, Zhang Y, et al. Combined CDK4/6 and mTOR Inhibition Is Synergistic against Glioblastoma via Multiple Mechanisms. Clin Cancer Res 2017;23(22):6958–68 doi 10.1158/1078-0432.CCR-17-0803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.El-Naggar S, Liu Y, Dean DC. Mutation of the Rb1 pathway leads to overexpression of mTor, constitutive phosphorylation of Akt on serine 473, resistance to anoikis, and a block in c-Raf activation. Mol Cell Biol 2009;29(21):5710–7 doi 10.1128/MCB.00197-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sara Hurvitz DY, Amelia Zelnak, Angela DeMichele, Elizabeth Tan-Chiu, Cynthia Ma, Tania Small, Chris Tucci, Tanay Samant, Das Purkayastha, Stacy Moulder and Aditya Bardia. Ribociclib in combination with everolimus and exemestane in men and postmenopausal women with HR+/HER2- advanced breast cancer following progression on a CDK4/6 inhibitor: Safety, tolerability and pharmacokinetic results from Phase 1 of TRINITI-1 study (abstract). American Association for Cancer research 2017. [Google Scholar]
- 25.Bicker J, Alves G, Fortuna A, Falcao A. Blood-brain barrier models and their relevance for a successful development of CNS drug delivery systems: a review. Eur J Pharm Biopharm 2014;87(3):409–32 doi 10.1016/j.ejpb.2014.03.012. [DOI] [PubMed] [Google Scholar]
- 26.Parrish KE, Sarkaria JN, Elmquist WF. Improving drug delivery to primary and metastatic brain tumors: strategies to overcome the blood-brain barrier. Clin Pharmacol Ther 2015;97(4):336–46 doi 10.1002/cpt.71. [DOI] [PubMed] [Google Scholar]
- 27.Galanis E, Wu W, Cloughesy T, Lamborn K, Mann B, Wen PY, et al. Phase 2 trial design in neuro-oncology revisited: a report from the RANO group. Lancet Oncol 2012;13(5):e196–204 doi 10.1016/S1470-2045(11)70406-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.