

Abstract
Describing the bovine vaginal microbiota is essential to better understand its physiology and its impact on health maintenance. Despite the economic importance of reproduction of these animals, bovine vaginal microbial community is still poorly described in comparison with rumen microbiome. Previous studies of our group described the vaginal microbiota of Nellore, an important Bos taurus indicus breed, using metagenomics. In order to better understand this microbiota, the present work aims to investigate another important breed, Gyr. Results have shown bacterial dominance over Archaea and Fungi was observed, with the most abundant bacterial phylum (Firmicutes) representing 40–50% of bacterial population, followed by Bacteroidetes, Proteobacteria, and Actinobacteria. The Fungi kingdom had the Mycosphaerella genus as its main representative, followed by Cladosporium. Archaea were observed at a very low abundance in all animals, with a high relative abundance of Methanobrevibacter genus. These results demonstrate a high microbial diversity on vaginal tract of Gyr, as demonstrated for Nellore and different from the previously described for other species. Our results indicate a great similarity between vaginal microbiota of Nellore and Gyr despite the differences in animal handling and genetic improvement. As observed for both breeds, individual variation is the largest source of microbial diversity between animals.
Keywords: Microbiota, Metagenomics, Bovine, Gyr, Vaginal tract
Introduction
The study of the microbiota has provided important insights into health and pathology of animals of economic importance [1]. To date, most of the research has focused on the ruminal microbiota due its biotechnological interest [2] and in order to understand and reduce the pressing environmental problems associated with greenhouse gas emissions from ruminant livestock production [3, 4]. The economic importance of cattle, as well as the need for a better understanding of resident microbial communities, has led to the development of many studies to elucidate the relationship of the indigenous microbiota with the physiology of these animals. However, now, other ecological niches as vaginal tract are beginning to be explored in cattle [5–8]; once the knowledge is still incipient despite its importance for further understanding reproductive biology and diseases, especially those of microbial etiology.
The vaginal microbiota of women has been widely studied and was shown to be dominated by the Lactobacillus genus, an environment of low microbial diversity [9–11]. Although the healthy human vaginal microbiota comprises around 70% of lactobacilli, other mammals have < 1% of these bacteria [12, 13]. A study conducted in nine primate species have shown a great difference between humans and non-human primates (NHPs), with an abundance of Lactobacillus lower than 5% in all the NHPs [14]. A recent work evaluating the species composition of vaginal microbiota showed that lactobacilli is a common finding in ewes and cows (80% and 90%, respectively), although in low abundance [15].
According to the common function hypothesis, bacteria other than Lactobacillus spp. may protect hosts via mechanisms as production of bacteriocins and other antimicrobial compounds. Different microbiota taxonomic composition was also revealed in studies conducted in monkeys [13, 14, 16] and rats [17].
Despite its economic importance, much needs to be learned about bovine reproductive biology and microbiota components in the different breeds. We have just started to know the microbiota components in different breeds. Studies using culture-dependent techniques have shown a low microbial abundance in cows [18–21]. A deeper understanding has been gained with metagenomics studies that began to elucidate the microbiota composition in the Nellore breed, the most economically relevant in Brazil [5]. However, additional studies are needed to establish a microbiota core and understanding this microenvironment, what could lead to an improvement of animal health.
For this purpose, the present study aims to evaluate the vaginal microbiota of Gyr cattle, the Zebuine breed most used in dairy production in Brazil, composed by animals that are capable of maintaining good production standards under different management systems and climates, in comparison to the vaginal microbiota of Nellore cattle.
Materials and methods
Farm, animals, and samples
In order to investigate the unbiased indigenous vaginal microbiota, an elite herd was selected, composed of pure-by-origin Gyr cattle, from Agricultural Research Company of Minas Gerais State (EPAMIG). This livestock system has a medium milk production of 3700 kg/lactation/cow. The reproduction is realized by fixed time artificial insemination or embryo transfer, without the presence of bulls or teasers. Within the herd, four non-pregnant animals that did not present any reproductive pathology clinical signs for the past 12 months were selected and divided in two groups: two heifers (H) and two cows (C). Heifers were not older than 18 months and cows were older than 2 years. Sample collection took place in May 2013 and this study was approved by the Ethics Committee in Animal Experimentation of the Universidade Federal de Minas Gerais, Brazil (CETEA/UFMG - 95/2012) with all experiments performed in accordance with relevant guidelines and regulations. The samples collection was accompanied by the herd manager. For sampling, the vulva was washed with distilled water and 70% ethanol, and then 50 ml of sterile saline solution was introduced into the vaginal cavity of the animal, through a syringe coupled to a sterile probe. The vaginal wash then was aspirated and kept at 4 °C until processing, which occurred after 5 h.
The material was lyophilized to reduce sample volume and the high and the large quantity of mucus, after that total DNA was extracted using DNeasy Blood & Cell Culture DNA Midi Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The extracted DNA was used as template to the PCR reactions using universal primers for Bacteria, Archaea, and Fungi (Table 1). The reactions were conducted as follows: for Bacteria—a mix containing 10 pmol of each primer, 4 μl of 5× buffer (Promega, Fitchburg, Wisconsin, USA), 1 U of GoTaq (Promega), 200 μM of dNTP, 1.5 mM of MgCl2, 20 ng of template DNA, and nuclease-free water to a final volume of 20 μl was subjected to the following cycling: 95 °C for 5 min, 30 cycles of 95 °C for 40 s, 55 °C for 40 s, 72 °C for 1 min, and finally 72 °C for 7 min; for Archaea—a mix containing 25 pmol of each primer, 45 μl of Taq Platinum Supermix (Thermo Fischer Scientific, Waltham, USA), 80 ng of template DNA and nuclease-free water to a final volume of 50 μl was subjected to the following cycling: 94 °C for 4 min, 35 cycles of 94 °C for 30 s, 60 °C for 30 s, 72 °C for 40 s, and finally 72 °C for 10 min; for Fungi—a mix containing 10 pmol of each primer, 12.5 μl of 2× GoTaq Green Master Mix (Promega), 40 ng of template DNA and nuclease-free water to a final volume of 25 μl was subjected to the following cycling: 95 °C for 2 min, 35 cycles of 95 °C for 15 s, 54 °C for 25 s, 72 °C for 20 s, and finally 72 °C for 10 min.
Table 1.
PCR reactions using universal primers
| Organism | Bacteria | Archaea | Fungi |
|---|---|---|---|
| Target region | 16S rRNA | 16S rRNA | D1/D2 |
| Primer (Forward) | 784F: AGG ATT AGA TAC CCT GGT A | 300fEyAr: AGC RRG AGC CCG GAG ATG G | NL1: GCA TAT CAA TAA GCG GAG GAA AAG |
| Primer (Reverse) | 1061R: CRR CAC GAG CTG ACG AC | 954rEyAr: CGG CGT TGA RTC CAA TTA AAC | NL4: GGT CCG TGT TTC AAG ACG G |
| Expected amplicon size | ~ 280 bp | ~ 500–600 bp | ~ 400–1000 bp |
| Reference | [22] | [23] | [24] |
Universal primers used in the PCR reaction for Bacteria, Archaea, and Fungi; R, degenerated nucleotides (A/G)
Construction of libraries and sample sequencing
Paired-end libraries were constructed using 50 ng of each PCR product (amplicon from each animal and target—bacteria, archaea and fungi). For Bacteria, with a 280 bp amplicon, DNA fragments were coupled directly to specific adapters using the TruSeq Nano DNA Library Preparation Kit (Illumina, San Diego, USA) according to the manufacturer’s instructions. Next, the DNA-adapter molecules were purified and submitted to amplification reactions using specific primers targeting the adapters. A quantification of amplified products was conducted using SYBR Fast qPCR Kit (Kapa Biosystems, Wilmington, USA). Next, the libraries were diluted in a Tris-HCl and 0.1% Tween solution, deposited on a flow cell and subjected to 600 sequencing cycles (2 × 300 bp) using the MiSeq Reagent Kit v3 (Illumina). For Archaea and Fungi, DNA amplicons of each animal were submitted to a random fragmentation, once some of the amplicons were as big as 1000 bp. DNA samples were fragmented to smaller segments and coupled to specific adapters, using the Nextera XT DNA Library Preparation Kit (Illumina), according to the manufacturer’s instructions. Amplification, quantification, and sequencing protocols were performed as described above for bacterial libraries. Then, two libraries were constructed and sequenced comprising all four animals, one for Bacteria and one for Archaea/Fungi. Samples of the same animal were marked with a distinct index, using the Nextera XT Index Kit (24 indices, 96 samples) (Illumina). Thus, it was possible to sequence all animals in the same flow cell and to separate the results according to the index. The sequences have been deposited into NCBI SRA database. The datasets supporting the conclusions of this article are available in the NCBI SRA repository, with the BioProject accession number PRJNA435944.
The paired-end libraries sequenced were then submitted to the MG-RAST server [25] where the library pairs where merged based on the homology of the different ends, eliminating those which lacked their respective pair-ends. The merged pairs where then submitted to the server’s pipeline. Sample dereplication was performed in order to remove artificial replicates produced by amplification and sequencing, and sequences were screened for removal of Bos taurus sequences. The dynamic trimming parameter was selected, aiming to eliminate low-quality sequences, requiring a minimal Phred score of 15 for each base and a limit of five low-quality bases for each fragment. The sequences of high quality-generated were used to further analysis, which required 98% minimal identity, 100 bp minimal alignment and maximum E-value of 10-8 to determine an Operational Taxonomic Unit (OTU). M5RNA database—which comprises Greengenes, RDP, and Silva databases—was selected for annotation comparison, being a suitable base for various rRNA sequence analysis.
In order to reduce the influence of extreme values and to obtain a high-fidelity result, a maximum of one value of each group was excluded in data analysis, when it had a significant difference from the group average, allowing a more realistic overview of the group’s vaginal microbiota. In order to show the importance of the individual variation among animals, the sequences were submitted to principal component analysis (PCA). Nellore data was also submitted to this statistical procedure thus enabling to correlate the vaginal microbiota of these two breeds. All the results were analyzed on STAMP v2.1.3. by ANOVA with the Tukey-Kramer post-test and p < 0.05 [26].
Results
In continuation to a recent work published by our group on the vaginal microbiota of Nellore cattle using the same methodological approach of the present work [5], this study presents the analysis of this microbiota in pure-by-origin dairy Gyr cattle (2 heifers and 2 cows non-pregnant). This Gyr herd has high sanitary indices and the reproduction is realized by artificial insemination or embryo transfer, without the presence of bulls or teasers.
Bacterial phyla
Major bacterial phyla present in this study were basically the same as those found in Nellore [5]. The most abundant phylum was Firmicutes, representing 40–50% of the bacterial population, followed by Bacteroidetes, Proteobacteria, and Actinobacteria. Unclassified bacterial sequences, the second most abundant OTU at this taxonomic level, represented 18–30% (Fig. 1).
Fig. 1.
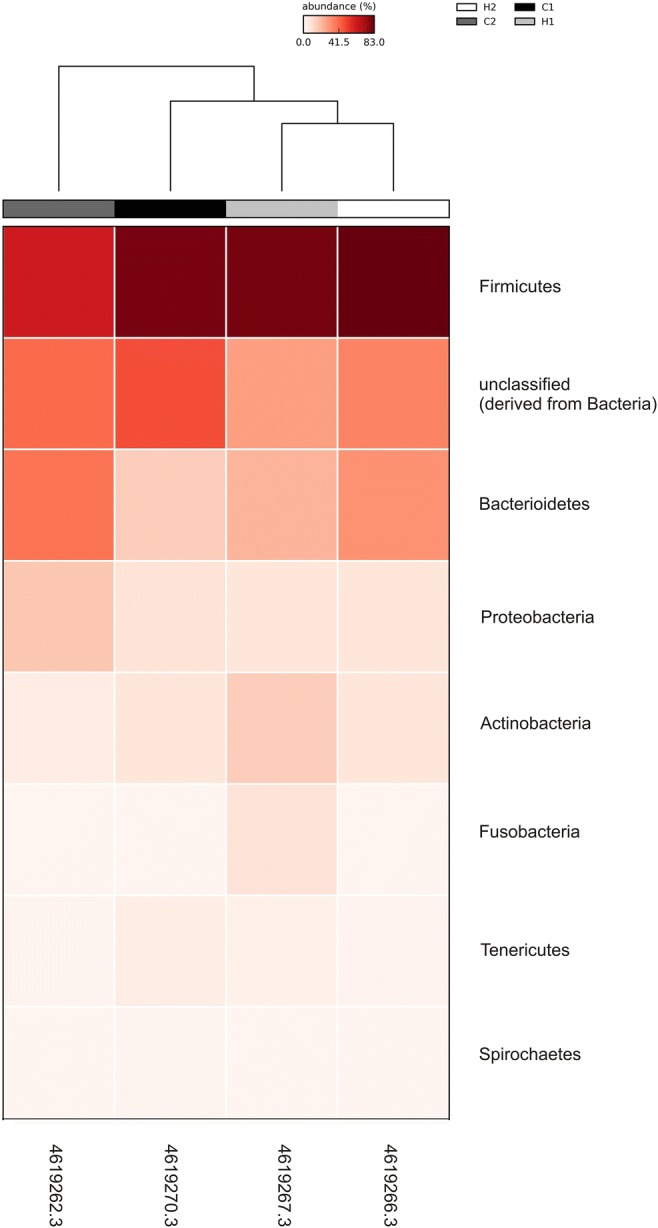
Heat map of the relative abundance of each bacterial OTU at phylum level in all four animals. H, heifers; C, cows
The analysis of microbiota from Gyr vaginal tract using a culture-independent approach showed, similarly observed in Nellore breed, a bacterial dominance over Archaea and Fungi, with the most abundant bacterial phylum being Firmicutes, which represented 40–50% of the bacterial population, followed by Bacteroidetes, Proteobacteria, and Actinobacteria phyla. The most predominant known taxa were Aeribacillus, Bacillus, Clostridium, Ruminococcus, Bacteroides, Rikenella, and Alistipes, composing seven OTUs which were to those observed in Nellore and it was not possible to associate a specific vaginal microbiota composition with cows and heifers. A principal component analysis (PCA) of the four Gyr and 20 Nellore samples clearly showed that the samples did not group, independently of breed or age (cow and heifers) supporting our data, even from a small sample size (Fig. 2) and making it possible to infer that, although there is a similar vaginal bacterial composition in the two analyzed breeds, the marked individual difference between the animals is quite important to establish the composition of microbiota.
Fig. 2.

PCA analysis of Gyr (gray markers) and Nellore (black markers) cows and heifers
These results indicate a dominance of these genera in bovine species, since the animals investigated in these two studies are of different breeds (Nellore vs Gyr), with different management systems (beef herd and dairy herd) located in different farms and geographical regions. None of the animal evaluated presented a microbiota dominated by obligate anaerobes, as what was observed in Nellore in our previous report, suggesting that the microbiota of a healthy bovine reproductive tract consists in a combination of aerobic, facultative anaerobic, and obligate anaerobic microorganisms [19, 27].
Bacterial genera
Genus level analysis revealed (Fig. 3) the most frequent OTUs in Gyr vaginal tract. The unclassified sequences group was generally the most abundant, although the aerotolerant genera (Aeribacillus and Bacillus) together reaches the values around those presented by unclassified. It is important to highlight that among the OTUs genus, the difference observed in groups was the presence of Porphyromonas only in heifers, although this difference should be observed with attention, due to the small number of animals analyzed.
Fig. 3.

Relative abundance of the ten major bacterial OTUs in Gyr vaginal tract at genus level. Black bars represent aerotolerant genera, and gray bars represent obligate anaerobes. H, heifers; C, cows
The sampled animals presented a predominance of aerotolerant organisms, but all had a mix that also included anaerobic genera (Fig. 4). Interestingly, individual variation is evident and no animal presented a microbiota dominated by obligate anaerobes, as observed in Nellore [5].
Fig. 4.
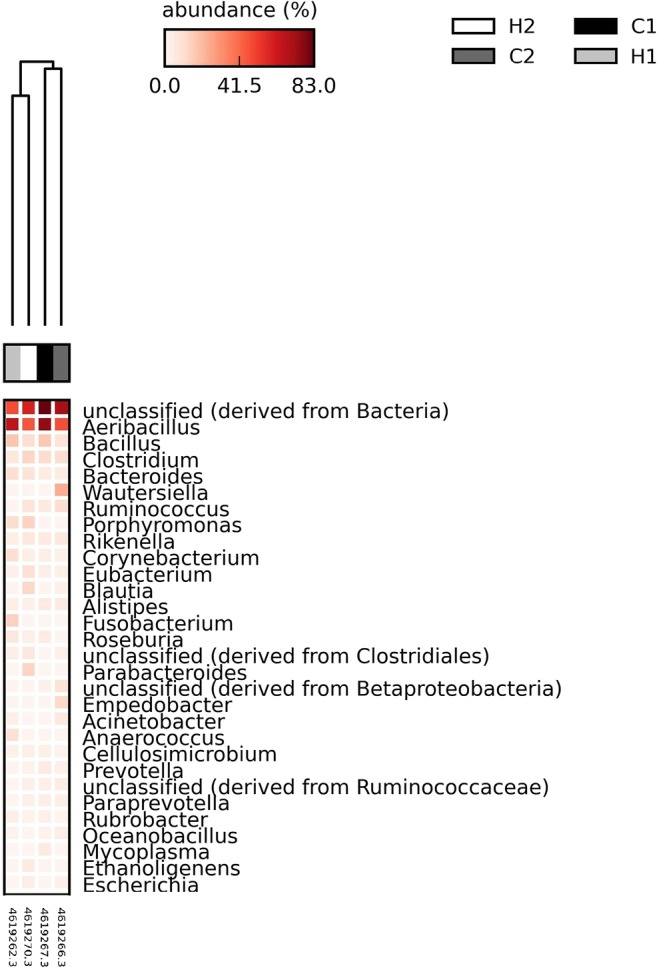
Heat map of the relative abundance of the 30 major bacterial OTUs in Gyr vaginal tract at genus level. H, heifers; C, cows
Fungal genera
Among the fungal OTU, Ascomycota dominates with the Mycosphaerella genus displaying an abundance ranging from 30 to 82% in heifers and cows. There was a predominance of the Mycosphaerella genus in three animals (H1, H2, and C2), with C1 presenting a similar abundance of Cladosporium and Mycosphaerella genera (Fig. 5).
Fig. 5.
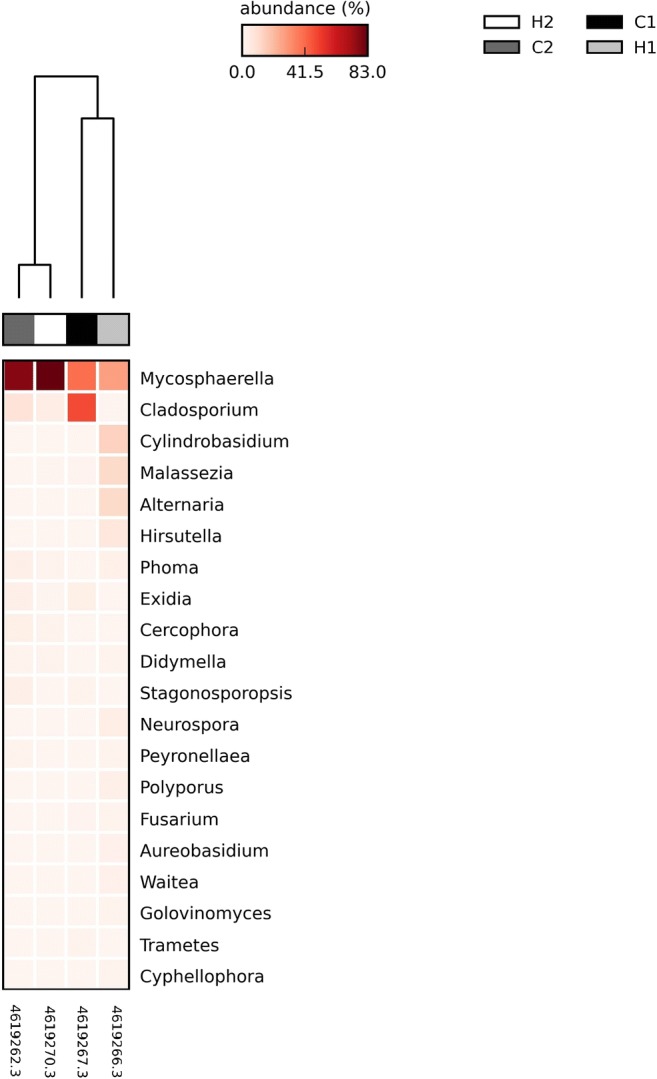
Heat map of the relative abundance of each fungal OTU at genus level. H, heifers; C, cows
With the second largest abundance, the Fungi kingdom had the Mycosphaerella genus as its main representative, followed by Cladosporium genus. The genus Mycosphaerella is one of the largest genera of ascomycetes, comprising many plant pathogens of economically important crops, but also saprophytic species [28].
Archaeal genera
Archaea were observed at low abundance in all four animals with a dominance of the Methanobrevibacter genus, showing no difference between heifers and cows (Fig. 6). The same profile was observed in Nellore animals, reinforcing a microbial composition similarity between these two breeds. This result is in accordance with what was observed in animals in Nellore cattle [5].
Fig. 6.

Heat map of the relative abundance of each archaeal OTU at genus level. H, heifers; C, cows
In the present work, Archaea was observed in low abundance and with dominance of Methanobrevibacter genus in the four animals analyzed, all of the non-pregnant (cows and heifers). In our previous work with Nellore cattle, Methanobrevibacter was also the dominant genus in all analyzed groups (pregnant and non-pregnant, cows and heifers), highlighting that Archaea showed great abundance in pregnant animals comparing with non-pregnant animals [5].
Discussion
Bacterial phyla
The literature reports a similar study conducted in vaginal washes from 20 crossbred beef cows (artificially inseminated or embryo transfer recipient, pregnant or non-pregnant) where authors reported a great bacterial diversity in all the studied animals, and no difference was seen in the microbiota composition in relation to age and method of fertilization. The most abundant phyla were Proteobacteria, Fusobacteria, Bacteroidetes, and Firmicutes, differing from our findings in order of abundance, however, being coincident in 3 of 4 phyla found. These differences could be related to the breed studied (crossbred beef and pure Gyr cattle), the gene region 16S rRNA amplified (V3-V4 and V5-V6 in the present work) and also differences in handling or diet, once that gastrointestinal tract (GIT) may influence in vaginal microbiota [5, 15].
In accordance with our data and supporting the hypothesis that the vaginal microbiota is influenced by GIT, a recent study with rumen microbiota of 45 crossbreed dairy calves found a similar microbial composition to the findings of this present work. The authors reported a great abundance of Firmicutes, Bacteroidetes, Actinobacteria, and Proteobacteria, in a similar distribution to that observed for vaginal microbiota in Gyr and Nellore cattle [29].
Due to the importance of the reproductive hormone therapies, a study has been recently conducted to compare the resident microbial population of the cranial vaginal mucosa exposed to a progesterone-releasing intravaginal device (PRID) in animals with reproductive disorders in crossbred Bradford heifers and cows. Using the PCR-DGGE approach, the authors reported results very similar to the microbiota found in Gyr and Nellore, where Bacteroidetes, Proteobacteria, and Firmicutes were the dominant phyla associated to healthy status and Actinobacteria being less frequently observed and only in healthy heifers under reproductive hormone therapies. Firmicutes and Bacteroidetes were also present in samples from cows presenting metritis and heifers presenting vaginitis. The phylum Proteobacteria was found to be almost exclusively associated with metritis in cows [7].
Investigating the vaginal microbiota of Holstein dairy cows during the transition period using next-generation DNA sequencing of the bacterial 16S rRNA gene, other authors found that phyla associated with uterine disease and related risk factors were Proteobacteria, Bacteroidetes, and Fusobacteria, two of them being the same phyla found in healthy Gyr as reported in the present study. However, analyzing vaginal samples in different days pre and post-parturition and correlating with total bacteria load (TBL, accessed by kinetics PCR) found that at the day of parturition healthy cows as well as cows presenting reproductive failures had similar TBL but at day 7 postpartum, cows with retained placenta and metritis showed a significantly higher TBL, mainly driven by higher estimated loads of Fusobacteria/Bacteroidetes and Proteobacteria, respectively [6]. The similarity of phyla in healthy animals with those observed in animals presenting reproductive failure suggest that there are specific intrinsic factors in microorganisms present in each ecological niche that may be more biologically relevant than only the abundance.
Bacterial genera
The aerotolerant genus Aeribacillus (Firmicutes) dominated both when bacteria of cows and heifers were compared and when the most abundant OTU’s were evaluated in animals individually. This genus which houses thermophilic bacteria was firstly described in 2010 [30, 31] in the skin microbiota of six species of fish evaluated, representing 19% of all clones detected by ribosomal internal spacer analysis and 16S rRNA gene sequencing [32]. Interestingly, a study evaluating mastitis in two large dairy farms by comparison of the microbial diversity in healthy, sub-clinically and clinically diseased quarters as well as in milk samples (using pyrosequencing of the 16S rRNA genes), showed that the Aeribacillus genus with three other genera (Faecalibacterium, unclassified Lachnospiraceae, and Propionibacterium) were present in all the samples healthy quarters, regardless of their somatic cell counts or culture status [33]. In the work investigating vaginal tract of crossbred Bradford cited above, Aeribacillus was observed in healthy heifers or presenting vaginitis and in cows with metritis, pointing out that in this study healthy cows were not analyzed [7]. Therefore, the great abundance of Aeribacillus found in bovine vaginal tract from healthy Gyr and Nellore (Brazil), as well as in crossed Bradford animals (Argentine-healthy or presenting reproductive diseases) and in milk from healthy quarters in dairy cattle (USA) point out the probable importance of this bacterium in the bovine microbiota.
In the present work, the vaginal microbiota was accessed in Gyr breed and compared with results described in our previous work with Nellore cattle [5]. Although the Gyr and Nellore breeds present differences in the production and environmental systems, the main difference observed was among individuals. Since its domestication, different bovine breeds have been exposed to intense selective forces for commercially important traits related to production and adaptation. According to Randhawa et al. (2016), the bovine genome has undergone changes at the underlying regions of functional genetic variants. Studies conducted on gut microbial communities of vertebrates have shown that the dominant microbial groups are exceptionally diverse between individuals [34]. Working with quantitative pyrosequencing assay in a large number of inbred mice it was possible to define a core measurable microbiota (CMM) consisting of 64 conserved taxonomic groups [35]. These authors found that CMM was highly variable in most animal populations. Single-nucleotide polymorphism markers evaluated in relation to the CMM abundancies identified 18 host quantitative trait loci. These data provide the importance of host genetics in establishing the individual microbiota diversity in mammals. These gut data can be extended to another tract and reinforces the individual microbiota diversity described in bovine vaginal environment.
Fungal genera
The most abundant fungal genus, Mycosphaerella, is an endophytic fungus genus commonly found in soil, and for few species associates with grass and ferns [36]. Some authors have reported the efficiency of these fungi in producing compounds against other pathogenic fungi [37]. As colonization of the cattle vagina is influenced by proximity of the gastrointestinal tract, the presence of Mycosphaerella could be related to the ingestion of this fungus and its antagonist activity could explain the dominance of its OTU over other fungal genera.
Many sources of food supplement of protein and fiber for animal production have been used such as agroindustrial residues. In a study aiming to determine the presence and levels of microbiota in brewers grains (beer industry residue) used for bovine intensive rearing showed that Cladosporium spp. and Aspergillus spp. were the most prevalent genera isolated from pre- and post-stored samples [38]. These results reinforce the hypothesis that the presence of Cladosporium genus in the present work could be related to its ingestion. The air of animal dwellings can contain great amounts of bioaerosol composed of dust, bacteria, fungi, and toxins. Then, another source of fungus contamination can be from the air of dwellings whose composition may depend on animal species, building construction, animal accommodation, and microclimate parameters. Analyzing airborne fungi in dwellings for dairy cows and laying hens in Croatia, Cladosporium spp. was among the most common fungi found in both environment [39]. The presence of Mycosphaerella and Cladosporium in these animals increases the suspicion of an environmental colonization of the vaginal tract by these fungi or accidental presence of these organisms originally from the gastrointestinal tract.
A study conducted with crossbred beef cows and Rambouillet ewes either reported low abundance of Archaea in both ruminant species (around 1.3% of all analyzed reads), although Archaea was detected in 95% of all collected vaginal samples and the order assigned was Desulfurococcales, instead of the Methanobacteriales found in the present work [15].
Archaeal genera
It is interesting to point out that the first description of Methanobrevibacter in vaginal microbiota was done in 1990, in samples collected from 12 women healthy or presenting vaginosis. Methanogenic activity was detected only in two samples (16.6%), both of which were from patients with bacterial vaginosis (and none of the samples from healthy patients). Both isolates were identified as Methanobrevibacter smithii [40].
More recently, an improved protocol to detect Archaea was described and tested in stool specimens collected from 16 children younger than 2 years of age. DNA of M. smithii was detected in 95.7% and M. stadtmaniae in 29.4% of the specimens. According to authors, there is a high prevalence of the methanogens M. smithii and M. stadtmaniae in human gut, with the former being an almost ubiquitous inhabitant of the intestinal microbiota and that the acquisition of M. smithii is an early event in newborns, resulting from exposure to maternal or environmental source [41]. Other reports show a relationship in the presence of Archaea in disease, as in the case of periodontal disease where the archaeal community at infected sites was dominated by a Methanobrevibacter oralis-like phylotype and a distinct Methanobrevibacter subpopulation related to archaea that inhabit the gut of numerous animals [42]. Methanobrevibacter genus has also a central role in rumen on the metabolism of plant compounds [43], being present in gastrointestinal tract of bovines and other animals, including humans. The presence of this Archaea in the vaginal tract seen in the present work can be due to the anatomical characteristics of bovine species that has the vaginal lumen exposed to the intestinal content. However, the presence of Archaea and its correlation with healthy or vaginosis in humans [40] point out that the direct and indirect role of this microorganism in maintenance of the healthy and disease status needs to be elucidate, including in the vaginal microbiome.
Conclusions
Despite the fitness, geographical distance, and differences in animal handling, Gyr and Nellore present a similar vaginal microbiota. Aeribacillus, Bacillus, Clostridium, Bacteroides, and Ruminococcus are among the major bacterial genera found, and Mycosphaerella is the dominant fungus in this microbiota. Archaea is poorly represented, with Methanobrevibacter being the most abundant genus. These data start to unravel the bovine vaginal microbiota, and a wide picture is being defined with the major microbial agents from this tract. It is important to highlight that this work permits studies of probiotics, prebiotics, and the understanding of the vaginal microbial communities.
Acknowledgments
The study was supported by grants from the Brazilian Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).
Author contributions
Conceived and designed the experiments: SGF, MLN, JRN, GCO and EFBS. Performed the experiments: SGF, MLN, MRG, FMGA, ACMS and APO. Analyzed the data: SGF, MLN and LRL. Contributed reagents/materials/analysis tools: JRN, GCO, FGF and EFBS. Wrote the paper: SGF, MLN, JRN, GCO, FGF and EFBS. All authors reviewed the manuscript.
Funding
This study was supported by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (Grant ID 473879/2013-1).
Data availability
The datasets generated and analyzed during the current study are available in the NCBI SRA repository (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA435944).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval
All procedures performed in studies involving animals were in accordance with the ethical standards of Ethics Committee in Animal Experimentation of the Universidade Federal de Minas Gerais, Brazil (CETEA/UFMG - 95/2012) at which the studies were conducted.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Silvia Giannattasio-Ferraz and Mateus Laguardia-Nascimento contributed equally to this work.
References
- 1.Garoussi MT, Eidi S, Mehravaran M. Isolation and comparative investigation of vaginal mycoflora in feline population of urban and dairy cattle herds. J M Mycol. 2016;26:22–27. doi: 10.1016/j.mycmed.2015.10.013. [DOI] [PubMed] [Google Scholar]
- 2.Loaces I, Amarelle V, Muñoz-Gutierrez I, Fabiano E, Martinez A, Noya F. Improved ethanol production from biomass by a rumen metagenomic DNA fragment expressed in Escherichia coli MS04 during fermentation. Appl Microbiol Biotechnol. 2015;99:9049–9060. doi: 10.1007/s00253-015-6801-0. [DOI] [PubMed] [Google Scholar]
- 3.Roehe R, Dewhurst RJ, Duthie CA, Rooke JA, McKain N, Ross DW, Hyslop JJ, Waterhouse A, Freeman TC, Watson M, Wallace RJ. Bovine host genetic variation influences rumen microbial methane production with best selection criterion for low methane emitting and efficiently feed converting hosts based on metagenomic gene abundance. PLoS Genet. 2016;12:e1005846. doi: 10.1371/journal.pgen.1005846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wallace RJ, Snelling TJ, McCartney CA, Tapio I, Strozzi F. Application of meta-omics techniques to understand greenhouse gas emissions originating from ruminal metabolism. Genet Sel Evol. 2017;49:9. doi: 10.1186/s12711-017-0285-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Laguardia-Nascimento M, Branco KMGR, Gasparini MR, Giannattasio-Ferraz S, Leite LR, Araujo FMG, Salim ACM, Nicoli JR, de Oliveira GC, Barbosa-Stancioli EF. Vaginal microbiome characterization of Nellore cattle using metagenomic analysis. PLoS One. 2015;10:e0143294. doi: 10.1371/journal.pone.0143294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bicalho ML, Santin T, Rodrigues MX, Marques CE, Lima SF, Bicalho RC. Dynamics of the microbiota found in the vaginas of dairy cows during the transition period: associations with uterine diseases and reproductive outcome. J Dairy Sci. 2017;100:3043–3058. doi: 10.3168/jds.2016-11623. [DOI] [PubMed] [Google Scholar]
- 7.Gonzalez-Moreno C, Fontana C, Cocconcelli PS, Callegari ML, Otero MC. Vaginal microbial communities from synchronized heifers and cows with reproductive disorders. J Appl Microbiol. 2016;121:1232–1241. doi: 10.1111/jam.13239. [DOI] [PubMed] [Google Scholar]
- 8.Zinicola M, Lima F, Lima S, Machado V, Gomez M, Döpfer D, Guard C, Bicalho R. Altered microbiomes in bovine digital dermatitis lesions, and the gut as a pathogen reservoir. PLoS One. 2015;10:e0120504. doi: 10.1371/journal.pone.0120504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Antonio MA, Hawes SE, Hillier SL. The identification of vaginal Lactobacillus species and the demographic and microbiologic characteristics of women colonized by these species. J Infect Dis. 1999;180:1950–1956. doi: 10.1086/315109. [DOI] [PubMed] [Google Scholar]
- 10.Dumonceaux TJ, Schellenberg J, Goleski V, Hill JE, Jaoko W, Kimani J, Money D, Ball TB, Plummer FA, Severini A. Multiplex detection of bacteria associated with normal microbiota and with bacterial vaginosis in vaginal swabs by use of oligonucleotide-coupled fluorescent microspheres. J Clin Microbiol. 2009;47:4067–4077. doi: 10.1128/JCM.00112-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Branco KMGR, Nardi RMD, Moreira JLS, Nunes AC, Farias LM, Nicoli JR, Carvalho MAR. Identification and in vitro production of Lactobacillus antagonists from women with or without bacterial vaginosis. Braz J Med Biol Res. 2010;43:338–344. doi: 10.1590/S0100-879X2010007500013. [DOI] [PubMed] [Google Scholar]
- 12.Miller EA, Beasley DE, Dunn RR, Archie EA (2016) Lactobacilli dominance and vaginal pH: why is the human vaginal microbiome unique? Front Microbiol 7. 10.3389/fmicb.2016.01936 [DOI] [PMC free article] [PubMed]
- 13.Spear GT, Kersh E, Guenthner P, Vishwanathan SA, Gilbert D, Zariffard MR, Mirmonsef P, Landay A, Zheng L, Gillevet P. Longitudinal assessment of pigtailed macaque lower genital tract microbiota by pyrosequencing reveals dissimilarity to the genital microbiota of healthy humans. AIDS Res Hum Retrovir. 2012;28:1244–1249. doi: 10.1089/aid.2011.0382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yildirim S, Yeoman CJ, Janga SC, Thomas SM, Ho M, Leigh SR, Consortium PM, White BA, Wilson BA, Stumpf RM. Primate vaginal microbiomes exhibit species specificity without universal Lactobacillus dominance. ISME J. 2014;8:2431–2444. doi: 10.1038/ismej.2014.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Swartz JD, Lachman M, Westveer K, O’Neill T, Geary T, Kott R et al (2014) Characterization of the vaginal microbiota of ewes and cows reveals a unique microbiota with low levels of lactobacilli and near-neutral pH. Front Vet Sci 1. 10.3389/fvets.2014.00019 [DOI] [PMC free article] [PubMed]
- 16.Herthelius M, Gorbach SL, Mollby R, Nord CE, Pettersson L, Winberg J. Elimination of vaginal colonization with Escherichia coli by administration of indigenous flora. Infect Immun. 1989;57:2447–2451. doi: 10.1128/IAI.57.8.2447-2451.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reid G, Chan RCY, Bruce AW, Costerton JW. Prevention of urinary tract infection in rats with an indigenous Lactobacillus casei strain. Infect Immun. 1985;49:320–324. doi: 10.1128/IAI.49.2.320-324.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amin JD, Zaria LT, Malgwi RM. Vaginal aerobic bacterial flora of apparently healthy cattle in various stages of the reproductive cycle in the Sahel region of Nigeria. Bull Anim Heal Prod Africa. 1996;44:15–18. [Google Scholar]
- 19.Otero C, Ruiz CS, Iba R, Wilde OR, Holgado PDR. Lactobacilli and enterococci isolated from the bovine vagina during the estrous cycle. Anaerobe. 1999;5:305–307. doi: 10.1006/anae.1999.0245. [DOI] [Google Scholar]
- 20.Otero C, Saavedra L, Silva-De-Ruiz C, Wilde O, Holgado AR, Nader-Macías ME. Vaginal bacterial microflora modifications during the growth of healthy cows. Lett Appl Microbiol. 2000;31:251–254. doi: 10.1046/j.1365-2672.2000.00809.x. [DOI] [PubMed] [Google Scholar]
- 21.Sharda R, Monghe MN, Tanwani SK. Antibiotic sensitivity pattern of bacteria isolated from repeat breeding animals. Indian Vet J. 1991;68:197–200. [Google Scholar]
- 22.Andersson AF, Lindberg M, Jakobsson H, Bäckhed F, Nyrén P, Engstrand L. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS One. 2008;3:e2836. doi: 10.1371/journal.pone.0002836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Faveri M, Gonçalves LFH, Feres M, Figueiredo LC, Gouveia LA, Shibli JA, Mayer MP. Prevalence and microbiological diversity of archaea in peri-implantitis subjects by 16S ribosomal RNA clonal analysis. J Periodontal Res. 2011;46:338–344. doi: 10.1111/j.1600-0765.2011.01347.x. [DOI] [PubMed] [Google Scholar]
- 24.Kwiatkowski NP, Babiker WM, Merz WG, Carroll KC, Zhang SX. Evaluation of nucleic acid sequencing of the D1/D2 region of the large subunit of the 28S rDNA and the internal transcribed spacer region using smartgene idn software for identification of filamentous fungi in a clinical laboratory. J Mol Diagnostics. 2012;14:393–401. doi: 10.1016/j.jmoldx.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 25.Meyer F, Paarmann D, D’Souza M, Oslon R, Kubal M et al (2008) The metagenomics RAST server—a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinf 9. 10.1186/1471-2105-9-386 [DOI] [PMC free article] [PubMed]
- 26.Parks DH, Tyson GW, Hugenholtz P, Beiko RG. STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics. 2014;30:3123–3124. doi: 10.1093/bioinformatics/btu494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Ametaj BN, Ambrose DJ, Gänzle MG. Characterization of the bacterial microbiota of the vagina of dairy cows and isolation of pediocin-producing Pediococcus acidilactici. BMC Microbiol. 2013;13:19. doi: 10.1186/1471-2180-13-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Verkley JMG, Crous PW, Groenewald JZ, Braun U, Aptroot A. Mycosphaerella punctiformis revisited: morphology, phylogeny, and epitypification of the type species of the genus Mycosphaerella (Dothideales, Ascomycota) Mycol Res. 2004;108:1271–1282. doi: 10.1017/S0953756204001054. [DOI] [PubMed] [Google Scholar]
- 29.Dias J, Marcondes MI, Noronha MF, Resende RT, Machado FS, Mantovani HC, Dill-McFarland KA, Suen G (2017) Effect of pre-weaning diet on the ruminal archaeal, bacterial, and fungal communities of dairy calves. Front Microbiol 8. 10.3389/fmicb.2017.01553 [DOI] [PMC free article] [PubMed]
- 30.Miñana-Galbis D, Pinzón DL, Lorén JG, Manresa A, Oliart-Ros RM. Reclassification of Geobacillus pallidus (Scholz et al. 1988) Banat et al. 2004 as Aeribacillus pallidus gen. Nov., comb. nov. Int J Syst Evol Microbiol. 2010;60:1600–1604. doi: 10.1099/ijs.0.003699-0. [DOI] [PubMed] [Google Scholar]
- 31.Sevim E, Gaballa A, Beldüz AO, Helmann JD. DNA-binding properties of the Bacillus subtilis and Aeribacillus pallidus AC6 σD proteins. J Bacteriol. 2011;193:575–579. doi: 10.1128/JB.01193-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Larsen A, Tao Z, Bullard SA, Arias CR. Diversity of the skin microbiota of fishes: evidence for host species specificity. FEMS Microbiol Ecol. 2013;85:483–494. doi: 10.1111/1574-6941.12136. [DOI] [PubMed] [Google Scholar]
- 33.Oikonomou Georgios, Bicalho Marcela Lucas, Meira Enoch, Rossi Rodolfo Elke, Foditsch Carla, Machado Vinicius Silva, Teixeira Andre Gustavo Vieira, Santisteban Carlos, Schukken Ynte Hein, Bicalho Rodrigo Carvalho. Microbiota of Cow’s Milk; Distinguishing Healthy, Sub-Clinically and Clinically Diseased Quarters. PLoS ONE. 2014;9(1):e85904. doi: 10.1371/journal.pone.0085904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Randhawa IAS, Khatkar MS, Thomson PC, Raadsma HW. A meta-assembly of delection signatures in cattle. PLoS One. 2016;11:e0153013. doi: 10.1371/journal.pone.0153013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benson AK, Kelly SA, Legge R, Ma F, Low SJ, Kim J, Zhang M, Oh PL, Nehrenberg D, Hua K, Kachman SD, Moriyama EN, Walter J, Peterson DA, Pomp D. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. PNAS. 2010;107:18933–18938. doi: 10.1073/pnas.1007028107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guatimosim E, Schwartsburd PB, Barreto RW, Crous PW. Novel fungi from an ancient niche: cercosporoid and related sexual morphs on ferns. Persoonia. 2017;37:106–141. doi: 10.3767/003158516X690934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pereira CB, Pereira-de-Sá N, Borelli BM, Rosa CA, Barbeira PJ, Cota BB, Johann S. Antifungal activity of eicosanoic acids isolated from the endophytic fungus Mycosphaerella sp. against Cryptococcus neoformans and C. gattii. Microb Pathog. 2016;100:205–212. doi: 10.1016/j.micpath.2016.09.022. [DOI] [PubMed] [Google Scholar]
- 38.Keller LAM, Pereyra CM, Cavaglieri LR, Dalcero AM, Rosa CAR. Fungi and mycotoxins from pre- and poststorage Brewer’s grain intended for bovine intensive rearing. ISRN Vet Sci. 2012;2012:1–6. doi: 10.5402/2012/396590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matković K, Vucemilo M, Vinković B. Airborne fungi in dwellings for dairy cows and laying hens. Arh Hig Rada Toksikol. 2009;60(4):395–399. doi: 10.2478/10004-1254-60-2009-1970. [DOI] [PubMed] [Google Scholar]
- 40.Belay N, Mukhopadhyay B, Conway-de-Macario E, Galask R, Daniels L. Methanogenic bacteria in human vaginal samples. J Clin Microbiol. 1990;28(7):1666–1668. doi: 10.1128/JCM.28.7.1666-1668.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dridi B, Henry M, El Khéchine A, Raoult D, Drancourt M. High prevalence of Methanobrevibacter smithii and Methanosphaera stadtmanae detected in the human gut using an improved DNA detection protocol. PLoS One. 2009;4:e7063. doi: 10.1371/journal.pone.0007063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lepp PW, Brinig MM, Ouverney CC, Palm K, Armitage GC, Relman DA. Methanogenic Archaea and human periodontal disease. PNAS. 2004;101:6176–6181. doi: 10.1073/pnas.0308766101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carberry CA, Kenny DA, Kelly AK, Waters SM. Quantitative analysis of ruminal methanogenic microbial populations in beef cattle divergent in phenotypic residual feed intake (RFI) offered contrasting diets. J Anim Sci Biotechnol. 2014;5:41. doi: 10.1186/2049-1891-5-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated and analyzed during the current study are available in the NCBI SRA repository (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA435944).