

Abstract
Nicotine can produce antinociception in preclinical pain models; however, the ability of nicotine to augment the antinociceptive effects of opioid agonists has not been investigated. The present experiments were conducted to determine how nicotine modifies the effects of opioid agonists differing in efficacy. Male squirrel monkeys responded for the delivery of milk under a fixed ratio 10 schedule of reinforcement. During the 30-second timeout period following each milk delivery, the subject’s tail was immersed in 35, 50, 52, or 55°C water, and the latency to remove the tail was recorded. Dose-response functions for tail-withdrawal latency and operant performance were determined for fentanyl, oxycodone, buprenorphine, and nalbuphine alone and after treatment with nicotine. Excepting nalbuphine, all opioids produced dose-related disruptions in food-maintained responding and increases in tail-withdrawal latency at each water temperature. Nicotine did not exacerbate the behaviorally disruptive effects of the μ-opioids on operant performance but produced a significant mecamylamine-sensitive enhancement of the antinociceptive potency of each opioid. Failure of arecoline to augment the antinociceptive effects of oxycodone and antagonism by mecamylamine suggests this nicotine-induced augmentation of prescription opioid antinociception was nicotinic acetylcholine receptor (nAChR) mediated. This was reflected in leftward shifts in the antinociceptive dose–response curve of each opioid, ranging from 2- to 7-fold increases in the potency of oxycodone across all water temperatures to an approximately 70-fold leftward shift in the antinociceptive dose-response curve of nalbuphine at the lower and intermediate water temperatures. These results suggest that nicotine may enhance μ-opioid antinociceptive effects without concomitantly exacerbating their behaviorally disruptive effects.
SIGNIFICANCE STATEMENT
Prescription opioids remain the most effective pain-management pharmacotherapeutics but are limited by their adverse effects. The present results indicate that nicotine enhances antinociceptive effects of various opioid agonists in nonhuman primates without increasing their disruptive effects on operant performance. These results suggest that nicotine might function as an opioid adjuvant for pain management by enabling decreased clinically effective analgesic doses of prescription opioids without exacerbating their adverse behavioral effects.
Introduction
Pain is one of the leading causes of hospital visits and presents a significant health and financial burden to the United States (Institutes of Medicine, 2011). Opioid agonists are among the most commonly prescribed and effective analgesics (Paulozzie and Ryan, 2006; Clark and Schumacher, 2017). However, the use of opioid agonists is constrained by their adverse effects (e.g., respiratory depression, addiction, behavioral inhibition, etc.), spurring the search for novel and improved pain-management strategies (Schug et al., 1992). One strategy involves the combination of opioid agonists and nonopioid ligands. The basis of this approach is the idea that if a nonopioid can augment the clinically beneficial effects of opioid agonists without exacerbating their adverse effects, then effective analgesia may be provided by lower doses of opioids with less side-effect liability (for review, see Li, 2019). In this regard, previous reports indicate that various drug classes can augment the antinociceptive effects of opioids, encouraging the view that this approach may yield a clinically useful pain-management strategy. Drug classes that have been examined include: imidazoline I2 receptor agonists (Siemian et al., 2016; Li, 2017), cannabinoids (Maguire and France, 2014, 2018), antiepileptics (Tomic et al., 2018), κ and δ agonists (Negus et al., 2008, 2009, 2012), and adrenergic α2 receptor agonists (Blaudszun et al., 2012; Engelman and Marsala, 2013).
Although studies have illustrated the ability of nonopioid ligands to augment opioid antinociception, the role of opioid efficacy in such interactions is presently unclear. For instance, Maguire and France (2014) reported that cannabinoid agonists such as Δ9-THC and CP55,940 enhanced the antinociceptive effects of higher efficacy opioid agonists (e.g., fentanyl) more effectively than those of lower efficacy agonists (e.g., buprenorphine and nalbuphine) in rhesus monkeys. On the other hand, studies in rats have found that cannabinoid agonists enhanced the effects of morphine to a greater extent than those of the high efficacy agonist etorphine (Maguire and France, 2018). In general agreement with the latter results, the imidazoline I2 receptor agonist 2-(2-benzofuranyl)-2-imidazoline hydrochloride (2-BFI) also appeared to enhance the antinociceptive effects of lower-efficacy opioids to a greater extent than higher-efficacy opioids (Siemian et al., 2016). It is unclear whether the dissimilar relationships between opioid efficacy and the enhancement of opioid antinociception by nonopioid drugs in the above studies reflect differences in opioid actions in rats and monkeys or unique features of the opioid ligands themselves.
The cholinergic agonist nicotine has been shown to produce antinociception in preclinical studies (Wewers et al., 1999; Zarrindast et al., 1999; Berrendero et al., 2002; Kyte et al., 2018). It has been reported that nicotine can induce the release of endogenous opioids, and that crosstolerance between nicotine and μ-opioid agonists develops, coupled with changes in μ-opioid receptor (MOR) expression levels (Kishioka et al., 2000). Some studies suggest that tobacco users are especially sensitive to chronic pain or require higher doses of opioids to manage chronic pain, possibly reflecting nicotine-induced crosstolerance to MOR agonists (Yoon et al., 2015; however, see Ackerman, 2012; Plesner et al., 2016; Oh et al., 2018; De Vita et al., 2019). However, as of 2017, only approximately 14% of the Americans smoked cigarettes (Centers for Disease Control and Prevention, 2018), leaving the possibility that a large nonsmoking population may benefit from nicotine’s analgesic effects. Although nicotine itself is most probably not an effective analgesic from a clinical perspective (Mishriky and Habib, 2014), some evidence suggests that nicotine also can enhance opioid-induced analgesia (McMillan and Tyndale, 2015). Yet, the ability of nicotine to augment opioid-induced analgesia has not yet been systematically investigated. The present studies were conducted to address this issue, using a nociception assay in which tail-withdrawal latency from warm water and operant performance were concurrently measured in squirrel monkeys (Withey et al., 2018a). The ratio of 50% effective dose (ED50) values on both measures has been used previously to construct a preclinical therapeutic index, i.e., a quantitative indicator of the potential behavioral side-effect liability of prescription opioid analgesics (Withey et al., 2018a). Opioid agonists that differ in MOR efficacy (fentanyl > oxycodone > buprenorphine > nalbuphine) were studied to also evaluate the possible relationship between efficacy and nicotine-induced improvement in the preclinical therapeutic index (Picker and Yarbrough, 1991; Walker et al., 1993; McPherson et al., 2010). A selective enhancement of opioid antinociception, reflected in an improved index, would suggest that nicotine could be a clinically valuable adjuvant that allows reduction in the effective doses of opioids needed to produce analgesia without altering their behaviorally disruptive effects.
Methods
Subjects.
Adult male squirrel monkeys (n = 4; Saimiri sciureus) were housed in stainless steel cages in a climate-controlled vivarium under a 12-hour light/dark cycle at the McLean Hospital Animal Care Facility (licensed by the United States Department of Agriculture). Subjects had unrestricted access to water and were fed a daily allotment of high-protein primate chow (Purina Monkey Chow, St. Louis, MO), supplemented with multivitamins and fruit, per veterinary recommendation. All subjects previously served in behavioral studies with other drug classes and had been drug-free for at least 3 months prior to the present study. All procedures and protocols were approved by the Institutional Animal Care and Use Committee at McLean Hospital, and housing was compliant with guidelines provided by the Committee on Care and Use of Laboratory Animals of the Institute of Laboratory Animals Resources, Commission on Life Sciences, National Research Council (2011).
Apparatus.
Experimental sessions were conducted 5 days a week, 9 AM to 1 PM. During experimental sessions, subjects were placed in customized Plexiglas chairs as previously described (Withey et al., 2018a). Briefly, each chair was designed so that the subject faced two levers, each 8 cm below a stimulus light that could be illuminated during the session. The subjects also faced and had easy access to a customized Plexiglas receptacle between the levers into which fluid could be delivered via a syringe pump (Model PHM-100-10; Med Associates, Inc., Georgia, VT) outside the chamber. This pump could be operated to deliver sweetened condensed milk via Tygon tubing into the receptacle’s reservoir (0.15 ml in 0.84 seconds). The rear portion of the chair was designed so that the tail of the subject could hang freely, permitting the distal portion to be immersed in a water-filled container; only the distal 3-4 cm of the tail were immersed in water during experiments, allowing the subject to remove its tail from the water at any time without assistance. All experimental events were controlled and recorded through a commercially available interface and program (MED-PC; Med Associates Inc., St. Albans, VT).
Behavioral Procedures.
The procedures in this study follow methods described in Withey et al. (2018a). When the red stimulus light above the active lever was illuminated, the completion of 10 lever-presses (FR10) within 20 seconds [limited hold (LH) 20-second] triggered the delivery of 30% milk in water (v/v) into the reservoir. A 30-second short timeout (STO) followed the completion of the FR requirement or the expiration of the LH time. During the 30-second STO, the distal portion of the subject’s tail was submerged in either 35, 50, 52, or 55°C water, and the latency to withdraw the tail from the water was recorded. The trial was terminated by the withdrawal of the subject’s tail from the water or by the elapse of 10 seconds at which time the experimenter removed the tail from the water to avoid potential tissue damage (10-second cutoff). Sessions consisted of four 15-minute components, each divided into a 10-minute long timeout (LTO) followed by a 5-minute period during which the FR10/STO30-second schedule was in effect. These schedule parameters permitted at least six tail-withdrawal measurements within each component. The order of water temperatures within and across components was randomized, with the proviso that determinations in each component occurred no more than once at 50, 52, or 55°C and at least three times at 35°C.
Drug Testing Procedures.
Dose-response functions for the rate-decreasing and antinociceptive effects of nicotine and of the opioid agonists fentanyl, oxycodone, buprenorphine, and nalbuphine were determined in all subjects using cumulative-dosing procedures, i.e., by administering cumulative doses at the beginning of each component (10 minutes before the FR30/STO30-second schedule was in effect). In other experiments, the effects of each opioid also were determined after administration of saline or nicotine (0.1 or 0.18 mg/kg) at the beginning of the first component. In these tests, the first dose of each opioid was administered in the second component. The role of selective nicotinic actions in the ability of nicotine to augment the behavioral effects of opioids were explored by determining whether: 1) The muscarinic agonist arecoline, administered at the beginning of the first component, similarly modified the effects of cumulative doses of oxycodone and 2) the nicotinic receptor blocker mecamylamine, administered 5 minutes prior to nicotine, could antagonize nicotine’s modulation of the effects of oxycodone or buprenorphine. The doses of arecoline and mecamylamine were selected on the basis of previous studies in squirrel monkeys (Withey et al., 2018b). Finally, changes in tail-withdrawal latency produced by 0.1 mg/kg of oxycodone were examined alone or in the presence of 0.1 mg/kg of nicotine, administered either concurrently or 30 minutes prior to oxycodone administration. Drug testing occurred no more than once per week.
Data Analyses.
The effects of successive injections of saline on both tail-withdrawal latency and response rates across components were determined at least monthly (control sessions). Tail-withdrawal latencies and response rates were averaged across components of each control session to obtain single control values for each subject. Overall baseline values for tail-withdrawal latency and response rate in the present study are expressed as the mean of control values across individual subject (±S.E.M.). For each subject, tail-withdrawal latency during a test session was expressed as the number of seconds over which the subject’s tail remained in warm water after immersion; response rate was expressed as a percentage of the subject’s overall rate of responding during control sessions. The effects of each drug test are represented as the mean of data obtained in all subjects (±S.E.M.).
Linear regression was used to fit lines to the linear portion of dose-response functions in individual subjects (GraphPad Prism version 5.0 for Windows; GraphPad Software, San Diego, CA). For response rate, the linear portion of the curve was defined to include not more than one dose that produced <20% of control response rate, and not more than one dose that produced >80% of control response rate. For tail-withdrawal latency, the linear portion of the curve was defined to include not more than one dose that produced <2-second latency, and not more than one dose that produced >8-second latency.
The effects of an opioid alone or following treatment with either nicotine or arecoline alone or nicotine in the presence of mecamylamine or saline were fitted to straight lines and were considered significant if the slope differed significantly from 0. The slopes and intercepts of the dose-response functions for an opioid alone or after each type of treatment were compared using an F-ratio test (GraphPad Prism), with statistical significance set at P < 0.05. ED50 values were determined using linear interpolation for opioids alone and in combination with nicotine, arecoline, or nicotine/mecamylamine. Linear regression was used to determine a common slope to calculate potency ratios with corresponding 95% confidence limits (95% CL) per Tallarida (2000). If the 95% confidence limits of a potency ratio did not include 1, the ED50 values were considered significantly different. ED50 ratios previously defined in Withey et al. (2018a) were used to quantify the preclinical therapeutic index for opioid agonists alone and in the presence of nicotine according to the following formula:
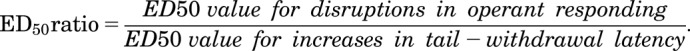 |
Finally, the effects of 0.1 mg/kg of oxycodone in the presence of 0.1 mg/kg of nicotine administered either concurrently or 30 minutes prior to oxycodone administration were compared using a repeated-measures one-way analysis of variance with a Newman-Keuls multiple comparisons test.
Drugs.
All doses of a drug were calculated in terms of its base weight and were administered intramuscularly. Drugs used in this study were fentanyl citrate (Sigma-Aldrich, St. Louis, MO), oxycodone hydrochloride (Sigma-Aldrich), buprenorphine hydrochloride (National Institute on Drug Abuse, Rockville, MD), nalbuphine hydrochloride hydrate (Sigma-Aldrich), nicotine (Wako Chemicals USA, Inc., Richmond, VA), arecoline hydrobromide (Sigma-Aldrich), and mecamylamine hydrochloride (Sigma-Aldrich).
Results
Control Response Rate and Tail-Withdrawal Latencies.
Control rates of responding (i.e., after saline administration) did not vary greatly throughout the session in individual subjects. Response rates also did not vary greatly from one control session to the next: Mean values for the group of four subjects ranged from 2.27 (±0.29) responses/s to 2.95 (±0.39) responses/s over the course of the present experiments. Tail-withdrawal latencies (mean ± S.E.M.) following the administration of saline averaged 1.5 (±0.23), 1.4 (±0.21), and 1.2 (±0.13) seconds at 50, 52, and 55°C, respectively (Fig. 1).
Fig. 1.

Rate-decreasing effects and tail-withdrawal latencies at 50, 52, and 55°C for fentanyl (open circle), buprenorphine (triangle), nicotine (filled circle), oxycodone (square), and nalbuphine (inverted triangle) administered i.m. (n = 4). Top panel: ordinate, response rate normalized to baseline rate of responding. Bottom panels: ordinate, tail-withdrawal latency in seconds. Error bars depict ±S.E.M.
Effects of Nicotine and Opioids Alone.
Nicotine dose-dependently reduced response rate under the FR schedule, with an ED50 value of 0.15 mg/kg (95% CL: 0.11–0.20 mg/kg; Fig. 1, upper panel). Nicotine also dose-dependently increased tail-withdrawal latencies at all three temperatures, though it was most effective with the subject’s tail in water warmed only to 50°C. Tail-withdrawal latencies following the highest dose (3.2 mg/kg) of nicotine reached 10 (±0), 9.3 (±1.2), and 4.6 (±1.2) seconds at 50, 52, and 55°C, respectively (Fig. 1, bottom panels).
Fentanyl, oxycodone, and buprenorphine also dose-dependently decreased rates of responding, with ED50 (95% CL) values of, respectively, 0.0034 (0.0023–0.0050), 0.45 (0.22–0.91), and 0.031 (0.014–0.069) mg/kg (Fig. 1, top panel). In contrast, doses of the low-efficacy opioid nalbuphine failed to reduce responding under the FR schedule at any dose (up to 3.2 mg/kg). All opioids dose-dependently increased tail-withdrawal latencies at each water temperature, excepting nalbuphine at 55°C (Fig. 1, bottom panels). The antinociceptive potency of fentanyl did not change in relation to increases in warm water temperature; however, the potency of oxycodone and buprenorphine was 5.3- and 2.4-fold, respectively, less at 55°C than at 50°C. Furthermore, the maximum tail-withdrawal latencies engendered by oxycodone, buprenorphine, and nalbuphine dropped from 10 (±0), 9.4 (±0.65), and 6.4 (±1.3) seconds at 50°C to 8.8 (±1.2), 7.2 (±2.1), and 1.3 (±0.10) seconds at 55°C. ED50 values for fentanyl, oxycodone, buprenorphine, and nalbuphine to increase tail-withdrawal latencies are summarized in Table 1.
TABLE 1.
ED50 values and potency ratios of fentanyl, oxycodone, buprenorphine, and nalbuphine alone and in combination with various doses of nicotine
ED50 values in milligrams per kilogram with corresponding 95% confidence limits for fentanyl, oxycodone, buprenorphine, and nalbuphine alone and in the presence of various doses of nicotine in 50, 52, and 55°C warm water. Potency ratios are calculated when available with corresponding 95% confidence limits. Increases in antinociceptive potency are considered statistically significant if the 95% confidence limits of the potency ratios of drug alone or in the presence of nicotine does not include 1.
| Drug (in mg/kg) | 50°C | 52°C | 55°C | |||
|---|---|---|---|---|---|---|
| ED50 (95% CL) | PR (95% CL) | ED50 (95% CL) | PR (95% CL) | ED50 (95% CL) | PR (95% CL) | |
| Fentanyl | 0.0033 (0.0013–0.0086) | 0.0031 (0.0020–0.0050) | 0.0036 (0.0018–0.0070) | |||
| +0.1 Nic | 0.0061 (0.0036–0.01) | 0.6 (0.3–1.4) | 0.0051 (0.0042–0.0061) | 0.6 (0.3–1.1) | 0.0061 (0.0036–0.01) | 0.5 (0.3–1.0) |
| +0.18 Nic | 0.00049 (0.00021–0.0011) | 7.9 (2.2–28) | 0.0015 (0.00091–0.0024) | 2.1 (1.2–3.8) | NA | NA |
| Oxycodone | 0.11 (0.053–0.22) | 0.16 (0.067–0.36) | 0.58 (0.29–1.2) | |||
| +0.032 Nic | 0.039 (0.015–0.10) | 2.0 (0.8–5.3) | 0.11 (0.054–0.21) | 1.3 (0.4–3.8) | 0.36 (0.086–1.5) | 1.6 (0.6–3.8) |
| +0.1 Nic | 0.018 (0.027–0.29) | 5.1 (2.2–12) | 0.031 (0.008–0.12) | 4.0 (1.2–14) | 0.11 (0.053–0.23) | 5.2 (2.2–12) |
| Buprenorphine | 0.026 (0.016–0.041) | 0.031 (0.014–0.066) | 0.063 (0.035–0.11) | |||
| +0.01 Nic | 0.014 (0.010–0.019) | 1.9 (1.1–3.2) | 0.031 (0.019–0.052) | 1.0 (0.4–2.3) | 0.038 (0.023–0.061) | 1.7 (0.8–3.7) |
| +0.032 Nic | 0.0054 (0.0028–0.010) | 4.6 (2.3–9.3) | 0.0084 (0.0052–0.014) | 3.6 (1.6–8.1) | 0.012 (0.0065–0.021) | 5.6 (2.4–13.1) |
| +0.1 Nic | 0.0036 (0.0027–0.0049) | 7.1 (4.2–12) | 0.0050 (0.0042–0.0060) | 6.1 (3.1–12) | 0.0057 (0.0035–0.0091) | 12 (5.4–25) |
| Nalbuphine | 1.6 (0.82–3.1) | 1.9* (0.54–7.0) | NA | |||
| +0.032 Nic | 0.12 (0.038–0.37) | 13 (4.3–39) | 0.26* (0.13–0.49) | 19 (6.1–61) | NA | NA |
| +0.1 Nic | 0.069 (0.035–0.14) | 68 (21–220) | 0.025* (0.0071–0.088) | 76 (20–281) | NA | NA |
ED25 value (in milligrams per kilogram).
Indicate the potency ratio was determined calculating the ED25 value as tail-withdrawal latency did not exceed 5 seconds.
Nicotine Modulation of Opioid Behavioral Effects.
Pretreatment with nicotine did not significantly alter the effects of any of the opioids on rates of responding (P > 0.05; Figs. 2 and 3). However, nicotine did modify the antinociceptive effects of all opioid agonists, as evident by increases in the potency of each opioid in delaying tail withdrawal from at least one temperature of warmed water. The magnitude of these effects differed among opioids. Fentanyl was the least sensitive to nicotine: A dose of nicotine (0.18 mg/kg) larger than those effective with other opioids shifted the tail-withdrawal latency dose-response function for fentanyl only 2- to 8-fold leftward in, respectively, 50 and 52°C water, and was ineffective in altering the antinociceptive potency of fentanyl in 55°C water (Fig. 2, left). In contrast, nicotine augmented the antinociceptive potency of nalbuphine, oxycodone, and buprenorphine at all water temperatures examined (Figs. 2 and 3). Nalbuphine was the most sensitive to the effects of nicotine inasmuch as 0.1 mg/kg of nicotine produced a 76-fold increase in the antinociceptive potency of nalbuphine in water warmed to 50°C (Fig. 2, right panels). Although nalbuphine alone did not increase tail-withdrawal latency at 55°C, nicotine increased the maximum antinociceptive effect of nalbuphine from a 1-second to a 5.1-second tail-withdrawal latency at this temperature of warmed water. As such, nicotine not only increased the antinociceptive potency of nalbuphine but also its effectiveness. Nicotine also significantly increased the antinociceptive potency of oxycodone and buprenorphine at all water temperatures, as evident in the 4- to 12-fold leftward shifts in their dose-response functions following 0.1 mg/kg of nicotine (Fig. 3). It should be noted that 0.032 mg/kg of nicotine enhanced the antinociceptive effects of oxycodone, buprenorphine, and nalbuphine, albeit more modestly. The impact of increasing water temperatures on the capacity of nicotine to augment opioid-induced antinociception was not uniform. For example, 0.18 mg/kg of nicotine shifted the dose-response function leftward for fentanyl 7.9-fold at 50°C, but only 2.1-fold at 52°C. Conversely, the antinociceptive effects of buprenorphine were enhanced by nicotine 12-fold at 55°C, but only 6.1-fold at 52°C. All increases in antinociceptive potency of the opioids in the presence of various nicotine doses are summarized in Table 1.
Fig. 2.

Rate-decreasing effects and tail-withdrawal latencies at 50, 52, and 55°C for (a) fentanyl alone and in combination with 0.1 and 0.18 mg/kg of nicotine, and (b) nalbuphine alone and in combination with 0.032 and 0.1 mg/kg of nicotine administered i.m. (n = 4). Top panels: ordinate, response rate normalized to baseline rate of responding; abscissa, drug dose in milligrams per kilogram. Bottom panels: ordinate, tail-withdrawal latency in seconds; abscissa, drug dose in milligrams per kilogram. Error bars depict ±S.E.M.
Fig. 3.

Rate-decreasing effects and tail-withdrawal latencies at 50, 52, and 55°C for (a) oxycodone alone and in combination with 0.032 and 0.1 mg/kg of nicotine, and/or 0.1 mg/kg of mecamylamine; and (b) buprenorphine alone and in combination with 0.01, 0.032, and 0.1 mg/kg of nicotine, and/or 0.1 mg/kg of mecamylamine administered i.m. (n = 4). Top panels: ordinate, response rate normalized to baseline rate of responding; abscissa, drug dose in milligrams per kilogram. Bottom panels: ordinate, tail-withdrawal latency in seconds; abscissa, drug dose in milligrams per kilogram. Error bars depict ±S.E.M.
Mechanism of Nicotine Action.
The nicotinic receptor blocker mecamylamine (0.1 mg/kg) did not alter the antinociceptive effects of oxycodone or buprenorphine (P > 0.05; Fig. 3) but attenuated the nicotine-induced enhancement of the antinociceptive potency of both opioids. Mecamylamine, like nicotine, also did not alter the potency with which either opioid disrupted operant responding (P > 0.05; Fig. 3, right panels). However, the muscarinic agonist arecoline exacerbated the effects of oxycodone on operant responding at doses that did not influence its antinociceptive effects. As shown in Fig. 4 (top panel), 0.1 mg/kg of arecoline produced a significant increase in the potency with which oxycodone decreased response rates (F2,22 = 8.4, P = 0.002) but did not significantly alter its capacity to produce increases in tail-withdrawal latency at 50°C (0.1 mg/kg: F2,22 = 0.24, P = 0.79), 52°C (0.1 mg/kg: F2,22 = 0.97, P = 0.40), or 55°C (0.1 mg/kg: F2,18 = 1.2, P = 0.32) (Fig. 4, bottom panels).
Fig. 4.

Rate-decreasing effects and tail-withdrawal latencies at 50, 52, and 55°C for oxycodone alone and in combination with 0.032 and 0.1 mg/kg of arecoline administered i.m. (n = 4). Top panels: ordinate, response rate normalized to baseline rate of responding; abscissa- drug dose in milligrams per kilogram. Bottom panels: ordinate- tail-withdrawal latency in seconds; abscissa, drug dose in milligrams per kilogram. Error bars depict ±S.E.M.
Nicotine/Oxycodone Time-Course Studies.
Administration of 0.1 mg/kg of nicotine and 0.1 mg/kg of oxycodone significantly increased tail-withdrawal latencies at 50°C (F6,26 = 3.7, P = 0.012), 52°C (F6,26 = 7.1, P = 0.0004), and 55°C (F6,26 = 3.2, P = 0.022) (Fig. 5). Post-hoc analysis of data at 50 and 52°C revealed that both concurrent administration of nicotine and oxycodone and the administration of oxycodone 30 minutes following nicotine administration generated tail-withdrawal latencies significantly greater than following oxycodone alone (Fig. 5). At a warm water temperature of 55°C, post-hoc analysis revealed that concurrent administration of 0.1 mg/kg of nicotine and 0.1 mg/kg of oxycodone produced a significantly greater tail-withdrawal latency than 0.1 mg/kg of oxycodone alone.
Fig. 5.

Tail-withdrawal latencies at 50, 52, and 55°C for 0.1 mg/kg of oxycodone alone, or in combination with 0.1 mg/kg of nicotine administered concurrently or 30 minutes prior to oxycodone administration (i.m.; n = 4). Ordinate, tail-withdrawal latency in seconds; abscissa, condition (drug in milligrams per kilogram). Error bars depict ±S.E.M. *P < 0.05.
Nicotine and the Preclinical Therapeutic Index for μ-Opioid Agonists.
The ED50 ratio of disruptions in operant responding and tail-withdrawal latency can be a useful preclinical estimate of the therapeutic window of analgesic drugs (Withey et al., 2018a). As summarized in Table 2, pretreatment with nicotine increased the preclinical therapeutic index (PTI) for fentanyl, oxycodone, and buprenorphine up to 25-fold at 50°C, 15-fold at 52°C, and 5.4-fold at 55°C. These increases in PTI were nicotine dose–dependent. Changes in the PTIs of oxycodone and buprenorphine were greater than those observed with fentanyl when examined in the presence of 0.1 mg/kg of nicotine. For example, at 52°C, the PTIs of oxycodone and buprenorphine were 15 and 6.2, respectively, compared with 0.67 for fentanyl. However, even in combination with a larger nicotine dose (0.18 mg/kg), the PTI of fentanyl at 52°C (2.3) was still less than that seen with oxycodone and buprenorphine. This differential modulation of opioid PTI by nicotine most probably reflects differences in opioid agonist efficacy. As expected, however, PTI values were smaller when the subjects were presented with greater noxious stimuli (i.e., 55 vs. 50 or 52°C warm water). This was most apparent with oxycodone in combination with 0.1 mg/kg of nicotine, as PTIs for nicotine were 25, 15, and 4.1 at 50, 52, and 55°C, respectively. The absence of effects of nalbuphine on operant responding precluded estimation of nicotine-induced changes in PTI values.
TABLE 2.
Preclinical therapeutic index of fentanyl, oxycodone, and buprenorphine alone or in combination with nicotine
ED50 ratios comparing rate of responding over tail-withdrawal latencies at 50, 52, and 55°C warm water. These ratios are an index of the therapeutic window of the drug alone or in the presence of nicotine. This ratio could not be calculated for nalbuphine as response rate did not decrease as a function of nalbuphine dose.
| Drug | 50°C | 52°C | 55°C |
|---|---|---|---|
| Fentanyl | 1.0 | 1.1 | 0.94 |
| +0.1 mg/kg of nicotine | 0.56 | 0.67 | 0.56 |
| +0.18 mg/kg of nicotine | 6.9 | 2.3 | NA |
| Oxycodone | 4.1 | 2.8 | 0.78 |
| +0.032 mg/kg of nicotine | 12 | 4.1 | 1.3 |
| +0.1 mg/kg of nicotine | 25 | 15 | 4.1 |
| Buprenorphine | 1.2 | 1.0 | 0.50 |
| +0.01 mg/kg of nicotine | 2.2 | 1.0 | 0.82 |
| +0.032 mg/kg of nicotine | 5.7 | 3.7 | 2.6 |
| +0.1 mg/kg of nicotine | 8.6 | 6.2 | 5.4 |
Discussion
The treatment of pain with opioid-based analgesics is complicated by the presence of significant unwanted effects (Schug et al., 1992), indicating the need for novel pain-management strategies with fewer deleterious effects. The present experiments investigated an opioid-sparing approach by determining whether nicotine could significantly augment the antinociceptive effects of several prescription opioid analgesics without concomitant increases in their behaviorally disruptive effects. Except nalbuphine at the highest temperature of water, all opioid agonists dose-dependently increased tail-withdrawal latencies. The potency of opioids to increase the latency with which subjects removed their tails from warm water was comparable to those previously observed in other studies using squirrel monkeys (Withey et al., 2018a). Presession administration of nicotine dose-dependently increased the antinociceptive potency of all opioid agonists without exacerbating their behaviorally disruptive effects.
As in previous studies, the ability of opioids to increase tail-withdrawal latency was inversely related to opioid efficacy and nociceptive stimulus intensity, i.e., water temperature (Walker et al., 1993). The present results indicate that the ability of nicotine to augment the antinociceptive effects of opioids varied likewise along both dimensions. For example, nicotine was much more effective in enhancing the effects of the low-efficacy opioid nalbuphine than the high-efficacy opioid fentanyl. Additionally, nicotine was most effective in enhancing opioid antinociception at the lowest temperature (50°C) and least effective at the highest temperature (55°C) of warmed water—even though the control latency to tail withdrawal did not differ greatly across temperatures (1.2 vs. 1.5 seconds). These findings suggest that, notwithstanding some enhancement of the effects of the high-efficacy opioid fentanyl with the highest dose of nicotine, the potential utility of nicotine as an adjunct medication in an opioid-sparing approach may be greatest when it is used for the production of antinociception in combination with lower to intermediate-efficacy opioids, e.g., with nalbuphine. This conclusion is in general agreement with previous studies showing that the opioid-sparing effects of imidazoline I2 and cannabinoid ligands also are more pronounced when combined with opioid partial agonists than with opioid full agonists (Siemian et al., 2016; Maguire and France, 2018).
Nicotine did not have consistent effects on response rate throughout the present study (i.e., in the first component of test sessions prior to opioid administration). Variability in nicotinic acetylcholine receptor (nAChR) sensitivity probably cannot explain the irregularity in the effects of nicotine on response rates as nicotine reliably enhanced the antinociceptive effects of all the opioids tested. Another possible explanation for the variability in nicotine’s effects is that tolerance selectively developed to the rate-decreasing effects of nicotine over the course of the study. Previous reports have demonstrated that various nAChR subtypes differentially mediate the behavioral effects of nicotine. For example, the α7 nAChR antagonist MLA has been shown to antagonize the antinociceptive effects of nicotine (Kyte et al., 2018) but not its discriminative stimulus or rate-decreasing effects (de Moura and McMahon, 2017). That different nicotinic receptor mechanisms may be responsible for the opioid antinociception enhancing effects of nicotine, tolerance may have selectively developed only to the operant suppressant effects of nicotine. Future studies should attempt to identify, if possible, activation of which specific nAChR subtype augments the antinociceptive effects of nicotine.
The nonselective, noncompetitive nAChR antagonist mecamylamine has been shown previously to antagonize the rate-decreasing and antinociceptive effects of nicotine (Martin et al., 1990; de Moura and McMahon, 2016; Withey et al., 2018b). In this regard, mecamylamine was a useful pharmacological tool to elucidate the involvement of nicotinic receptors in the current study. The ability of mecamylamine to block nicotine’s effects suggests that the enhancement of opioid antinociception can be attributed to activation of nAChRs following nicotine administration.
Previous preclinical reports have characterized the antinociceptive effects of the muscarinic receptor agonist arecoline (Sheardown et al., 1997; for review, see Naser and Kuner. 2018). In the present study, arecoline (0.1 mg/kg) increased tail-withdrawal latencies at 50 and 52°C water, suggestive of antinociception. However, unlike nicotine, arecoline did not enhance the antinociceptive effects of oxycodone. The inability of arecoline to accentuate opioid antinociception even at doses that greatly disrupted operant responding provides further evidence for nAChR-selective (i.e., not resulting from activation of muscarinic acetylcholine receptors) actions of nicotine in the present studies (Liu et al., 2016).
The last dose of each opioid during cumulative dose–response determinations for MOR agonists was tested 45 minutes after nicotine pretreatment. Nicotine’s duration of action is relatively short (Rodriguez et al., 2014), and it is possible that nicotine may not have been effective throughout the course of the experimental sessions. However, the half-life of nicotine in nonhuman primates is 2 and 3 hours (O’Leary et al., 2008; Moerke et al., 2017), suggesting that nicotine should retain the ability to augment opioid-induced antinociception throughout cumulative-dosing sessions. This suggestion was supported by results showing that 0.1 mg/kg of nicotine administered 30 minutes prior to 0.1 mg/kg of oxycodone, i.e., 40–45 minutes prior to testing, retained the ability to enhance oxycodone antinociception.
Despite previous information on nicotine antinociception (Wewers et al., 1999; Zarrindast et al., 1999; Berrendero et al., 2002; Kyte et al., 2018) and nicotine-opioid interactions (Patterson et al., 2012; Kishioka et al., 2014; Plesner et al., 2016; De Vita et al., 2019), the use of nicotine to promote opioid antinociception has not yet been systematically evaluated clinically, perhaps owing to previous reports of increased opioid use in tobacco users (Skurtveit et al., 2010; Yoon et al., 2015; but see Ackerman, 2012; Oh et al., 2018). However, the present results indicate that nonsmokers might benefit from the use of nicotine as an adjuvant for opioid analgesia, an idea that is supported by clinical data obtained in nonsmokers. For example, treatment with nicotine prior to emergence from anesthesia following uterine surgery in women decreased pain scores and morphine exposure (Flood and Daniel, 2004). Likewise, Habib et al. (2008) found that a transdermal nicotine patch applied to patients 30–60 minutes prior to induction of anesthesia decreased postoperative opioid usage. These results highlight the benefits of such an opioid-sparing approach, i.e., lower opioid dosages providing effective analgesia (Habib et al., 2008).
Warm water tail-withdrawal assays in nonhuman primates have been used to identify and characterize the analgesic potential of various drugs (Walker et al., 1993; Maguire and France, 2018). However, the interpretation of tail-withdrawal latency data can be complicated by evidence of dose-related behavioral disruption that also may be produced by effective doses of a drug. For example, in the present studies, nicotine increased tail-withdrawal latencies at all warm water temperatures at a dose (0.32 mg/kg) that significantly decreased rates of operant responding. Thus, it is unclear whether increases in tail-withdrawal latencies produced by nicotine itself are a function of its antinociceptive properties or are a byproduct of nonspecific disruptions in behavior. On the other hand, nicotine was able to accentuate opioid antinociception but not exacerbate the effects of opioids on operant behavior. Indeed, with nicotine on board, comparable levels of antinociception could be achieved with lower doses of the opioid and, consequently, fewer (or in some cases, no) disruptions in operant responding. To the extent that disruptions in operant behavior serve as a predictor of a drug’s behavioral side-effects in a clinical setting, the present findings suggest that incorporating nicotinic ligands as adjuvants for pain management with opioids may be therapeutically beneficial.
Acknowledgments
The authors would like to thank Dr. Roger Spealman for his comments on a previous version of this manuscript.
Abbreviations
- CP55,940
2-[(1R,2R,5R)-5-hydroxy-2-(3-hydroxypropyl)cyclohexyl]-5-(2-methyloctan-2-yl)phenol
- Δ9-THC
(6aR,10aR)-6,6,9-trimethyl-3-pentyl-6a,7,8,10a-tetrahydro-6H-benzo[c]chromen-1-ol
- ED50
50% effective dose
- MLA
methyllycaconitine
- MOR
μ-opioid receptor
- nAChR
nicotinic acetylcholine receptor
- PTI
preclinical therapeutic index
- STO
short timeout
Authorship Contributions
Participated in research design: Moura, Withey, Bergman.
Conducted experiments: Moura, Withey.
Performed data analysis: Moura.
Wrote or contributed to the writing of the manuscript: Moura, Withey, Bergman.
Footnotes
This work was supported by the National Institutes of Health National Institutes on Drug Abuse [Grant DA035857]; and Harvard Medical School Livingston Fellowship.
Parts of this work were previously presented as de Moura FB (2018) Nicotine as an adjuvant to μ-opioids: tail-withdrawal latency and disruptions in operant responding. Behavioral Pharmacology Society Annual Meeting; San Diego, CA; April 20–21, 2018.
References
- Ackerman WE., III (2012) The effect of cigarette smoking on hydrocodone efficacy in chronic pain patients. J Ark Med Soc 109:90–93. [PubMed] [Google Scholar]
- Berrendero F, Kieffer BL, Maldonado R. (2002) Attenuation of nicotine-induced antinociception, rewarding effects, and dependence in mu-opioid receptor knock-out mice. J Neurosci 22:10935–10940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaudszun G, Lysakowski C, Elia N, Tramèr MR. (2012) Effect of perioperative systemic α2 agonists on postoperative morphine consumption and pain intensity: systematic review and meta-analysis of randomized controlled trials. Anesthesiology 116:1312–1322. [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention (2018) Current cigarette smoking among adults- United States, 2007. MMWR Morb Mortal Wkly Rep 67:1225–1232.30408019 [Google Scholar]
- Clark DJ, Schumacher MA. (2017) America’s Opioid Epidemic: Supply and Demand Considerations. Anesth Analg 125 (5):1667–1674. [DOI] [PubMed] [Google Scholar]
- de Moura FB, McMahon LR. (2016) Differential antagonism and tolerance/cross-tolerance among nicotinic acetylcholine receptor agonists: scheduled-controlled responding and hypothermia in C57BL/6J mice. Behav Pharmacol 27:240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Moura FB, McMahon LR. (2017) The contribution of α4β2 and non-α4β2 nicotinic acetylcholine receptors to the discriminative stimulus effects of nicotine and varenicline in mice. Psychopharmacology (Berl) 234:781–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vita MJ, Maisto SA, Ansell EB, Zale EL, Ditre JW. (2019) Pack-years of tobacco cigarette smoking as a predictor of spontaneous pain reporting and experimental pain reactivity. Exp Clin Psychopharmacol DOI: 10.1037/pha0000258 [published ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman E, Marsala C. (2013) Efficacy of adding clonidine to intrathecal morphine in acute postoperative pain: meta-analysis. Br J Anaesth 110:21–27. [DOI] [PubMed] [Google Scholar]
- Flood P, Daniel D. (2004) Intranasal nicotine for postoperative pain treatment. Anesthesiology 101:1417–1421. [DOI] [PubMed] [Google Scholar]
- Institute of Medicine (2011) Committee on Advancing Pain Research, Care, and Education. Washington (DC), National Academies Press, US. [Google Scholar]
- Kishioka S, Kiguchi N, Kobayashi Y, Saika F. (2014) Nicotine effects and the endogenous opioid system. J Pharmacol Sci 125:117–124. [DOI] [PubMed] [Google Scholar]
- Kishioka S, Paronis CA, Lewis JW, Woods JH. (2000) Buprenorphine and methoclocinnamox: agonist and antagonist effects on respiratory function in rhesus monkeys. Eur J Pharmacol 391:289–297. [DOI] [PubMed] [Google Scholar]
- Kyte SL, Toma W, Bagdas D, Meade JA, Schurman LD, Lichtman AH, Chen ZJ, Del Fabbro E, Fang X, Bigbee JW, et al. (2018) Nicotine prevents and reverses paclitaxel-induced mechanical allodynia in a mouse model of CIPN. J Pharmacol Exp Ther 364:110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JX. (2017) Imidazoline I2 receptors: an update. Pharmacol Ther 178:48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JX. (2019) Combining opioids and non-opioids for pain management: current status. Neuropharmacology 158:107619. [DOI] [PubMed] [Google Scholar]
- Liu YJ, Peng W, Hu MB, Xu M, Wu CJ. (2016) The pharmacology, toxicology and potential applications of arecoline: a review. Pharm Biol 54:2753–2760. [DOI] [PubMed] [Google Scholar]
- Maguire DR, France CP. (2014) Impact of efficacy at the μ-opioid receptor on antinociceptive effects of combinations of μ-opioid receptor agonists and cannabinoid receptor agonists. J Pharmacol Exp Ther 351:383–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire DR, France CP. (2018) Antinociceptive effects of mixtures of mu opioid receptor agonists and cannabinoid receptor agonists in rats: impact of drug and fixed-dose ratio. Eur J Pharmacol 819:217–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin TJ, Suchocki J, May EL, Martin BR. (1990) Pharmacological evaluation of the antagonism of nicotine’s central effects by mecamylamine and pempidine. J Pharmacol Exp Ther 254:45–51. [PubMed] [Google Scholar]
- McMillan DM, Tyndale RF. (2015) Nicotine increases codeine analgesia through the induction of brain CYP2D and central activation of codeine to morphine. Neuropsychopharmacology 40:1804–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherson J, Rivero G, Baptist M, Llorente J, Al-Sabah S, Krasel C, Dewey WL, Bailey CP, Rosethorne EM, Charlton SJ, et al. (2010) μ-opioid receptors: correlation of agonist efficacy for signalling with ability to activate internalization. Mol Pharmacol 78:756–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishriky BM, Habib AS. (2014) Nicotine for postoperative analgesia: a systematic review and meta-analysis. Anesth Analg 119:268–275. [DOI] [PubMed] [Google Scholar]
- Moerke MJ, Zhu AZX, Tyndale RF, Javors MA, McMahon LR. (2017) The discriminative stimulus effects of i.v. nicotine in rhesus monkeys: pharmacokinetics and apparent pA2 analysis with dihydro-β-erythroidine. Neuropharmacology 116:9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naser PV, Kuner R. (2018) Molecular, cellular and circuit basis of cholinergic modulation of pain. Neuroscience 387:135–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negus SS, Bear AE, Folk JE, Rice KC. (2009) Role of delta opioid efficacy as a determinant of mu/delta opioid interactions in rhesus monkeys. Eur J Pharmacol 602:92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negus SS, Morrissey EM, Folk JE, Rice KC. (2012) Interaction between mu and delta opioid receptor agonists in an assay of capsaicin-induced thermal allodynia in rhesus monkeys. Pain Res Treat 2012:867067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negus SS, Schrode K, Stevenson GW. (2008) Micro/kappa opioid interactions in rhesus monkeys: implications for analgesia and abuse liability. Exp Clin Psychopharmacol 16:386–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh TK, Jeon JH, Lee JM, Kim MS, Kim JH, Lee SJ, Eom W. (2018) Chronic smoking is not associated with increased postoperative opioid use in patients with lung cancer or esophageal cancer. Pain Physician 21:E49–E55. [PubMed] [Google Scholar]
- O’Leary K, Parameswaran N, McIntosh JM, Quik M. (2008) Cotinine selectively activates a subpopulation of α3/α6β2 nicotinic receptors in monkey striatum. J Pharmacol Exp Ther 325:646–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson AL, Gritzner S, Resnick MP, Dobscha SK, Turk DC, Morasco BJ. (2012) Smoking cigarettes as a coping strategy for chronic pain is associated with greater pain intensity and poorer pain-related function. J Pain 13:285–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulozzi LJ, Ryan GW. (2006) Opioid analgesics and rates of fatal drug poisoning in the United States. Am J Prev Med 31 (6):506–511. [DOI] [PubMed] [Google Scholar]
- Picker MJ, Yarbrough J. (1991) Cross-tolerance and enhanced sensitivity to the response rate-decreasing effects of opioids with varying degrees of efficacy at the mu receptor. Psychopharmacology (Berl) 105:459–466. [DOI] [PubMed] [Google Scholar]
- Plesner K, Jensen HI, Højsted J. (2016) Smoking history, nicotine dependence and opioid use in patients with chronic non-malignant pain. Acta Anaesthesiol Scand 60:988–994. [DOI] [PubMed] [Google Scholar]
- Rodriguez JS, Cunningham CS, Moura FB, Ondachi P, Carroll FI, McMahon LR. (2014) Discriminative stimulus and hypothermic effects of some derivatives of the nAChR agonist epibatidine in mice. Psychopharmacology (Berl) 231:4455–4466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheardown MJ, Shannon HE, Swedberg MD, Suzdak PD, Bymaster FP, Olesen PH, Mitch CH, Ward JS, Sauerberg P. (1997) M1 receptor agonist activity is not a requirement for muscarinic antinociception. J Pharmacol Exp Ther 281:868–875. [PubMed] [Google Scholar]
- Siemian JN, Obeng S, Zhang Y, Zhang Y, Li JX. (2016) Antinociceptive interactions between the imidazoline I2 receptor agonist 2-BFI and opioids in rats: role of efficacy at the µ-opioid receptor. J Pharmacol Exp Ther 357:509–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skurtveit S, Furu K, Selmer R, Handal M, Tverdal A. (2010) Nicotine dependence predicts repeated use of prescribed opioids. Prospective population-based cohort study. Ann Epidemiol 20:890–897. [DOI] [PubMed] [Google Scholar]
- Tallarida RJ. (2000) Drug Synergism and Dose-Effect Data Analysis, Chapman and Hall/CRC, Boca Raton. [Google Scholar]
- Tomić M, Pecikoza U, Micov A, Vučković S, Stepanović-Petrović R. (2018) Antiepileptic drugs as analgesics/adjuvants in inflammatory pain: current preclinical evidence. Pharmacol Ther 192:42–64. [DOI] [PubMed] [Google Scholar]
- Walker EA, Butelman ER, DeCosta BR, Woods JH. (1993) Opioid thermal antinociception in rhesus monkeys: receptor mechanisms and temperature dependency. J Pharmacol Exp Ther 267:280–286. [PubMed] [Google Scholar]
- Wewers ME, Dhatt RK, Snively TA, Tejwani GA. (1999) The effect of chronic administration of nicotine on antinociception, opioid receptor binding and met-enkelphalin levels in rats. Brain Res 822:107–113. [DOI] [PubMed] [Google Scholar]
- Withey SL, Doyle MR, Bergman J, Desai RI. (2018b) Involvement of nicotinic receptor subtypes in the behavioral effects of nicotinic drugs in squirrel monkeys. J Pharmacol Exp Ther 366:397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Withey SL, Paronis CA, Bergman J. (2018a) Concurrent assessment of the antinociceptive and behaviorally disruptive effects of opioids in squirrel monkeys. J Pain 19:728–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JH, Lane SD, Weaver MF. (2015) Opioid analgesics and nicotine: more than blowing smoke. J Pain Palliat Care Pharmacother 29:281–289. [DOI] [PubMed] [Google Scholar]
- Zarrindast MR, Khoshayand MR, Shafaghi B. (1999) The development of cross-tolerance between morphine and nicotine in mice. Eur Neuropsychopharmacol 9:227–233. [DOI] [PubMed] [Google Scholar]