

Abstract

The wild type protein, transthyretin (TTR), and over 120 genetic TTR variants are amyloidogenic and cause, respectively, sporadic and hereditary systemic TTR amyloidosis. The homotetrameric TTR contains two identical thyroxine binding pockets, occupation of which by specific ligands can inhibit TTR amyloidogenesis in vitro. Ligand binding stabilizes the tetramer, inhibiting its proteolytic cleavage and its dissociation. Here, we show with solution-state NMR that ligand binding induces long-distance conformational changes in the TTR that have not previously been detected by X-ray crystallography, consistently with the inhibition of the cleavage of the DE loop. The NMR findings, coupled with surface plasmon resonance measurements, have identified dynamic exchange processes underlying the negative cooperativity of binding of “monovalent” ligand tafamidis. In contrast, mds84, our prototypic “bivalent” ligand, which is a more potent stabilizer of TTR in vitro that occupies both thyroxine pockets and the intramolecular channel between them, has greater structural effects.
Introduction
The normal wild type plasma protein, transthyretin (TTR) is inherently amyloidogenic, forming pathogenic extracellular amyloid deposits, most notably in the heart and predominantly in elderly men. In addition, there are over 120 different TTR gene mutations1 that cause autosomal dominant hereditary systemic TTR amyloidosis, affecting about 10 000 individuals worldwide.2 Crystallographic studies, pioneered by Blake & Oatley in 1977,3 showed the noncovalent, homotetrameric structure of TTR (a dimer of dimers) and the presence of two identical binding sites located in a channel formed by the dimer–dimer interface and crossing the protein molecule. The first drug licensed for treatment of TTR amyloidosis, tafamidis,4 was devised as a specific ligand for TTR and developed on the basis of its capacity to inhibit aggregation of TTR at low pH in vitro.5 Other small molecules with this property have been extensively investigated6 but all share the characteristic of being “monovalent”, that is, each ligand molecule only occupies a single thyroxine binding pocket in a single native tetrameric TTR molecule. In contrast, we devised “bivalent” TTR ligand compounds that are avidly, indeed pseudo-irreversibly, bound by native TTR under physiological conditions, occupying both binding pockets simultaneously and also the intramolecular channel that connects them.7 These compounds are much more potent inhibitors of acid-mediated TTR denaturation and aggregation in vitro.7 Crucially, the bivalent ligands are also more effective inhibitors of the physiologically relevant mechanism of TTR amyloid fibrillogenesis that involves mechanical forces and the specific, catalytic, proteolytic cleavage of TTR at K48,8−10 a process that is apparently mediated by plasmin in vivo.11
More recent high-resolution X-ray studies have shed light on the interaction between specific ligands and TTR and have informed chemical modifications to improve the binding properties of new ligands.12 However, a comparative analysis of crystallographic structures in the presence of ligands (holo-TTR) or in their absence (apo-TTR) failed to identify significant differences in the protein structure.13 In contrast, our recent work on limited proteolysis of TTR suggests that, upon binding, changes on TTR conformation and/or dynamics might occur, even far from the site of interaction, particularly at loop CD, where the proteolytic cleavage of the K48–T49 peptide bond occurs.9 While maximum resistance to proteolytic cleavage of the holo-state of TTR, and thus protection against amyloidogenesis, is obtained when both sites are occupied by ligands,10 the known negative cooperativity of binding of the majority of TTR ligands14 militates strongly against this. Monovalent ligands such as tafamidis are bound by TTR, as is thyroxine, with negative cooperativity, with dissociation constants Kd1 and Kd2 of ∼2 and ∼200 nM, respectively.4
A recent study demonstrated that, in contrast to X-ray crystallography, NMR spectroscopy is able to detect changes in the solution structure of TTR caused by pathogenic amino acid mutations.15 Furthermore, it was found that conformational changes associated with different mutations were not restricted to the immediate proximity of the mutation site but included remote allosteric effects. Here, we used NMR to probe two prototypic TTR ligands: the monovalent ligand, tafamidis (Figure 1A)4 and the symmetrical bivalent compound, mds84 (Figure 1B).7 We demonstrate that the unique simultaneous occupancy of both binding sites and the inner TTR channel by the bivalent ligand, in addition to short range effects, has major allosteric structural effects.
Figure 1.

Effects of tafamidis and mds84 binding by TTR shown by X-ray crystal structures and NMR spectra. (A,B) Chemical structures of tafamidis and mds84. (C,D) X-ray crystallographic structures of TTR–tafamidis (C, pdb 3TCT) and TTR–mds84 complexes (D, pdb 3IPE) in which the heavy atom rmsd difference from the apo-TTR form (pdb 5CN3) is highlighted using a color gradient from minimal (blue) to maximal (red) differences. The average heavy atom rmsd values are 0.844 and 0.771 Å in the presence of tafamidis and mds84, respectively. (E,F) Overlay of the 2D [1H, 15N] TROSY spectra, acquired at 800 MHz, of apo-TTR (black) and holo-TTR in the presence of tafamidis at 2:1 ratio (E, red) and mds84 at 1:1 ratio (F, purple).
Results
NMR Titrations Illuminate the Mechanism of Negative Cooperativity of TTR Ligand Binding
More than 25 very high-resolution X-ray structures have been reported of free wild type TTR as well as the complex of TTR bound to tafamidis and mds84, respectively. Figure S1 shows the TTR dimer with the labeling for each of the eight strands. The X-ray crystallographic structures of the apo- and holo-forms, with each of these different ligands, are almost identical. The heavy atom root mean square deviation (rmsd) is <1 Å (Figure 1C,D), although the holo-forms with both tafamidis and mds84 have a marginally higher rmsd for the C-terminal end of helix 1 and for loops BC and FG than for the rest of the protein. The capacity of these ligands to inhibit TTR amyloid formation in vitro,9 by reducing the extent of proteolytic cleavage of the K48–T49 peptide bond, therefore suggests that ligand binding may produce allosteric modifications in TTR that may be revealed by solution studies despite not being detected in the crystal structures. The NMR backbone chemical shift assignment of wild type TTR has previously been reported under non-physiological pH and temperature conditions16 and a new assignment at neutral pH has recently been deposited in the bmrb databank.17
Here, we used TROSY multidimensional experiments18,19 to determine a de novo assignment for our TTR preparation, at 310 K and pH values of 7.0 and 7.4. At pH 7.4, the resonances of residues 4–6 and 9–10 in the N-terminal unstructured region, residues 56–57 in the long DE loop, 91 and 95 at the beginning and end of strand F, and 116 and 119–121 in strand H were not observed in the apo-TTR spectrum because of fast amide hydrogen exchange with the solvent. At pH 7.0, only residues 56–57 were still not detected. The assignment of TTR resonances when bound to each of the two prototypic ligands, respectively, the monovalent tafamidis4 and the bivalent mds84,7 were confirmed by 3D [1H, 13C, 15N] TROSY HNCA. Based on these assignments, we have compared the respective holo-TTR forms with apo-TTR.
According to the Kd values of 2 and 200 nM reported for the first and second sites occupied in the TTR–tafamidis complex,10 at a protein concentration of 50 μM and 1 equiv of tafamidis, 91% of TTR molecules have a single ligand bound and 4% have two ligands bound. The NMR resonances will thus be dominated by the singly bound form (but note this gives rise to two different signals, corresponding to the different chemical environments in the two-halves of the TTR tetramer). In the presence of 2 equiv of tafamidis, 8% have a single ligand bound and 92% have two ligands bound, producing spectra dominated by the fully bound form. The avid pseudo-irreversible binding of mds84 and the simultaneous occupancy of the two binding sites7 allows the complete saturation of TTR by a single equivalent of the bivalent ligand. Based on these known stoichiometric states, NMR can highlight the structural modifications caused by the occupancy of one or both sites by the monovalent ligand and both sites by the bivalent ligand.
The appearance of NMR resonances during titration with a ligand is determined by the exchange rate, kex, relative to the frequency difference, Δω, between the free and bound state. When chemical shift differences are large (Δω ≫ kex), the exchange is slow so that free and bound peaks are observed simultaneously. Alternatively, if chemical shift differences are small (Δω ≪ kex), then the resonance will be in the fast exchange regime and the observed resonance position is the population-weighted average of the free and bound chemical shift.20 Moreover, changes in NMR line shapes across titrations can also be highly sensitive to microscopic reaction mechanisms.21,22
The mechanism of the peculiar negative cooperativity of binding of TTR to thyroxine, tafamidis and most other monovalent ligands, can be explored by NMR. At first glance, inspection of 2D [1H, 15N]-TROSY spectra acquired at varying ligand concentrations showed the simultaneous presence of peaks of the free and bound form (Figure S2), consistent with slow exchange on the chemical shift timescale both for tafamidis and mds84, but more thorough analysis revealed more complex exchange behavior. The numerous peaks that shift in the holo-form are clearly observable for tafamidis and mds84 in Figure 1E,F.
Figure 2 shows magnified views of the behavior of two representative TTR residues, E72 and H88, across the titration with mds84. For E72 (Figure 2A), which is representative of the majority of TTR resonances, mds84 induced a progressive decrease of the peak intensity of the apo-form and an increase of the same peak intensity in the holo-form, that is, slow exchange. For a group of 14 residues (Figure 2B), exemplified by H88, the corresponding peaks were not observed in the holo-TTR, indicating the presence of either intermediate exchange or micro second–millisecond dynamics within the bound state. The chemical shift perturbation (CSP) values between the apo and holo form are reported in Figure 2C and Movie M1.
Figure 2.

TTR binds the symmetrical bivalent ligand, mds84, in a slow exchange regime. The histograms represent the abundance of the TTR populations with 0, 1, and 2 binding sites occupied at molar mds84/TTR ratios of 0, 0.5, and 1, based on the binding data of the literature.7 (A) E72 illustrating the behavior of most of the shifting peaks, 29 of 43, upon binding of mds84, according to a two-state model. The initial and final positions of the peaks are designated by i and f, respectively, in the absence of ligand and with full occupancy of the two binding sites. (B) H88, a representative of the 14 peaks whose bound form is not observed, namely, L17, A19, R21, G22, L58, H88, E92, T96, A108, A109, L110, Y114, S117, and T118. (C) Combined chemical shift variations illustrated in a green-light blue-blue gradient color according to the scale shown, on the structure of the 1:1 TTR complex with mds84 (PDB: 3IPE). (D) Peaks observed with intermediate exchange regime shown as purple spheres.
A titration of TTR with tafamidis disclosed a more complex NMR chemical exchange behavior. This binding can be represented by three different classes of peaks exemplified by D99, S117, and L55 (Figure 3). According to the observed chemical shift changes, two phases can be distinguished: the first, during the occupancy of the first binding site is followed by a second phase when both sites are saturated. In the first phase, two of the four TTR subunits are directly involved in the tafamidis binding (bound subunits) while the remaining two (unbound subunits) are not.
Figure 3.

TTR binding of the monovalent ligand, tafamidis, and negative cooperativity. The histograms illustrate the populations with 0, 1, and 2 binding sites occupied by tafamidis at molar tafamidis/TTR ratios 0, 0.5, 1, 1.5, and 2. Behavior of three prototypic peaks during the titration with tafamidis is illustrated. (A) D99, that exemplifies 10 residues, following a two-state model; (B) S117, that exemplifies 6 residues, showing an intermediate exchange, and (C) L55 exemplifying the majority of shifting peaks, 35 of 51, whose bound form not only decreases in intensity upon tafamidis binding, but also shifts, as indicated by the red arrow. The initial and final positions of the peaks, designated by i and f, are, respectively, in the absence of ligand and with full occupancy of the two binding sites. (D) Only one of the two binding sites occupied at a 1:1 tafamidis/TTR ratio. The two opposite chains of TTR not bound to tafamidis are colored with a yellow-light blue-blue gradient, and the chains involved in the binding with a green-light blue-blue gradient, each according to their CSPs variations with the respective scale shown. (E) Variations of the combined chemical shift for holo-TTR saturated with two-fold molar excess of tafamidis are illustrated by a green-light blue-blue gradient according to the scale shown (PDB: 3TCT).
During titration from a tafamidis/TTR ratio of 0:1 to 1:1, shown in Figure 3A, the initial D99 peak corresponding to unbound TTR subunits progressively decreased, while the peak corresponding to bound subunits increased, similar to the slow exchange behavior of E72 during addition of mds84. In contrast, the initial unbound S117 resonance decreased and broadened prior to the observations of resonances from bound subunits (Figure 3B). Additionally, for a subset of residues including L55, progressive chemical shift changes and a decrease in intensity were observed from the initial unbound resonance up to a 1:1 ratio (Figure 3C, red arrows). The resonance at this point corresponds to unoccupied subunits within singly-bound TTR tetramers, and the observed changes indicate both that a component of the binding process occurs within the fast exchange regime, and that the conformation of these subunits is altered relative to the apo state. Simultaneously, a second peak was observed, increasing in intensity and corresponding to bound subunits within occupied TTR tetramers. A small change in its chemical shift was observed between 1 and 2 equiv, also indicating a change in the conformation of subunits between singly and fully occupied TTR tetramers. For all the three classes of peaks, in the second phase of the reaction, the apo-form vanished and the holo-form reached its maximum intensity. Figure 3D,E and Movies M2 and M3 summarize the conformational changes occurring in the first and second phase, respectively. This complex behavior cannot be obtained from a simple two-state binding equilibrium. Instead, the simplest model consistent with the observed NMR lineshapes21 is that the first equivalent of tafamidis binds through a scheme where the initial binding occurs in fast exchange, followed by a slower intramolecular rearrangement. A second molecule of tafamidis may then bind to this singly-bound state, although it is not clear from our data whether this occurs in a simple two-state manner or again via a more complex multistep mechanism.
At a 1:1 ratio, the residues involved in the fast formation of the complex and those involved in the final rearrangement of the complex are shown in Figure 3D. The residues within the binding site showing a chemical shift change were K15, L17 within strand A and R103 and R104, located just before and at the N-terminus of strand G. Other residues involved, but located outside of the binding site, were A25 and I26 in the A′B loop, S52 in the CD loop, and H90 and V94 in strand F. The chemical shift variations of the peaks in the rearranged complex outnumbered those of the initially formed complex and were mostly located in the dimer interface and in particular throughout strand A, A′, at A120 and V122 of strand H, strand C, and the C-term of strand G, localizing the site of interaction with tafamidis. However, variations also propagated to the outer strand F (H90 and V94) to the helix with the NH and the indole side chain of W79 where conformational changes may be hypothesized.
Surface Plasmon Resonance Analysis Reveals Two Different Binding Kinetics of Tafamidis by TTR
The NMR-based evidence describes, for the first time, that a structural rearrangement occurs in TTR upon occupancy of the first site. We used surface plasmon resonance (SPR) to analyze the TTR tafamidis interaction in order to measure the contribution of kon and koff to the binding dynamics that produce the observed negative cooperativity. Global fitting of the data from the association and dissociation phases (Figure 4) provided estimates of the on-rate (k1 and k3) and off-rate (k2 and k4) (Table 1) for Scheme 1 described in the Experimental Section.
Figure 4.

SPR analysis of ligand binding by TTR. (A,B) Association and dissociation curves derived by titration of immobilized TTR with tafamidis. The black symbols show experimental measurements and red lines are global fitting of the results, as described in the Experimental Section.
Table 1. Association and Dissociation Parameters for the Binding of Tafamidis to TTRa.
| k1 | (4.5 ± 2.2) × 106 M–1 s–1 |
| k2 | (0.008 ± 0.001) s–1 |
| k3 | (3.5 ± 1.0) × 106 M–1 s–1 |
| k4 | (0.060 ± 0.006) s–1 |
| Kd1 | (3 ± 1) × 10–9 M |
| Kd2 | (22 ± 7) × 10–9 M |
Kinetics and thermodynamics constants are reported with their standard deviations. The Kd1 and Kd2 are reported as the averages of the ratios of k2/k1 and k4/k3, respectively, of three independent experiments.
Scheme 1.
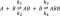
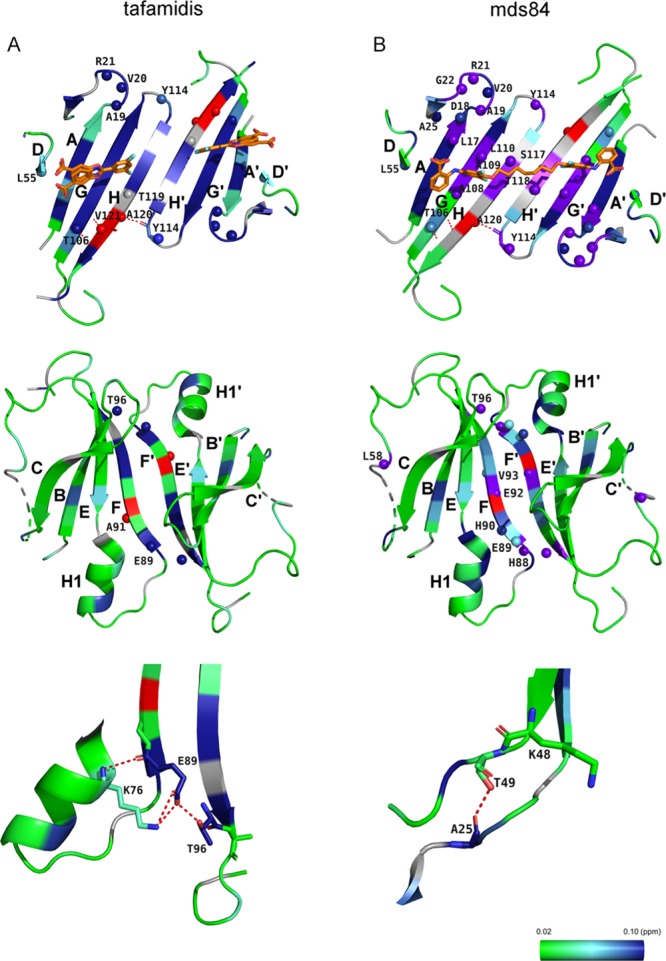
The calculated association rate constants were comparable for both sites in three independent experiments. However, the dissociation rate constants were distinctly different. The rate at the second site was an order of magnitude faster than at the first site, indicating that the primary driver of the observed negative cooperativity may be a faster dissociation from the second binding site than from the first. The NMR slow exchange phase is in full agreement with the off-rates determined by SPR and represents the SPR rate-determining step, as, in contrast to NMR, SPR cannot detect structural variations. The following comparison considers that while NMR exchange regimes are determined by the exchange rate, kex = kon[L] + koff, relative to the frequency difference between states, and are therefore technically dependent on the free ligand concentration, at 50% occupancy, where the appearance of the spectrum is most sensitive to chemical exchange, kex is simply equal to twice the off rate. The off-rate, k2 = 0.008 s–1, for occupation of a single binding site, can be compared to the chemical shift difference calculated between the apo-form and the TTR–tafamidis complex at 1:1 stoichiometry. It resulted in an average value Δω1H (rad/s) = 511.5 (min = 90.4, max = 2281.0) and Δω15N (rad/s) = 242.0 (min = 62.7, max = 891.6). Similarly, the off-rate, k4 = 0.06 s–1, for the occupation of the second binding site compares to the chemical shift difference between holo-TTR and TTR with a single binding site occupied, where the average values were Δω1H (rad/s) = 465.6 (min = 175.9, max = 1286.8) and Δω15N (rad/s) = 203.8 (min = 63.2, max = 874.8). Both off-rates were much smaller than the Δω for 1H and 15N nuclei, confirming a slow exchange on the chemical shift timescale.
Comparative Analysis of Saturation Binding of Different Ligands Reveals Global Protein Structural Effects on TTR
An in-depth analysis of the NMR spectra at complete saturation with both ligands was carried out. The numerous CSPs observed in holo-TTR with tafamidis and mds84, respectively, are shown in Figure 5, and highlighted in blue in Figure 6.
Figure 5.

Observed CSPs of TTR in the presence of tafamidis and mds84. (A) Combined amide 1H and 15N chemical shift for holo-TTR with tafamidis at 2:1 and (B) in the presence of mds84 at a 1:1 ratio. The dashed line shows the mean chemical shift variation after the removal of outliers.
Figure 6.
Structural effects of bound tafamidis and mds84 at 2:1 and 1:1 molar ratios to TTR, respectively. (A) Effects of tafamidis on residues within the protein central β-sheet (upper panel) and in its external surface (middle panel). The lower panel illustrates a network of H-bonds between K76, E89 of chain A and T96 of chain B. (B) Effects of mds84 on residues within the protein central β-sheet (upper panel) and in its external surface (middle panel). The lower panel illustrates the H-bond between A25 and T49. A green-light blue-blue gradient depicts the extent of chemical shift change and those that become visible only once the ligand is bound are shown in red. Purple spheres represent amides that disappear only upon binding. When no information is available, the residues are shown in gray. The PDB codes are 3TCT for the TTR complex with tafamidis and 3IPE with mds84.
Both ligands produced a marked perturbation at the dimer–dimer interface (strands DAGH–H′G′A′D′; Figure 6A,B upper panel) in regions corresponding to the three halogen binding pockets (HBPs) originally described by Blake & Oatley3 and then refined by Palaninathan.13 HBP1 is the wide outer pocket formed by M13, K15, L17, T106, A108, V121, and HBP2 comprises L17, A108, A109, L110, and the inner pocket, HBP3, is composed of A108, L110, S117, and T119. Other effects specific for tafamidis or mds84 were also detected. In particular, the full occupancy of the two sites affected all the observed residues of the HBPs HBP3–HBP1. Moreover, V121, not observable in the apo-form, became visible with tafamidis only. CSPs were also detected in strands B, E, and F of the outer β-sheet (Figure 6A,B middle panel), as well as loops AB, BC, only with tafamidis, and CD, DE, EH1, only with mds84, and H1 helix. These surprisingly significant remote perturbations in the BEF strands and in the helix, the outer part of the molecule distant by about 12–36 Å from the ligand binding pocket (Figure 6A,B middle panel), were not predicted, according to the HBP model, to change their conformation upon binding, in contrast to the chemical shift deviations expected for the actual binding residues.
The residues not belonging to HBPs and shifting more than 1 standard deviation from the mean were all in loop AB; 19–21 with bound tafamidis and 18, 20, and 25 with mds84. The binding of mds84 caused the greatest CSP in residue A25, which makes a H-bond with T49Oγ1 (Figure 6B lower panel), suggesting that strengthening of this H-bond may be responsible for inhibition of the amyloidogenic cleavage of the residue 48–49 peptide bond by serine proteases. We note that loop AB is central to the weak dimer–dimer association, as it interacts with the opposite loop GH of a different chain that was also perturbed upon binding. The bivalent ligand, mds84, was particularly effective in perturbing loop CD, strands D and E and helix H1. Binding of either ligand had major effects on strand F, with residues E89 and T96 shifted more than 3 standard deviations from the mean in the presence of tafamidis. Their side chains participate in an interchain hydrogen bond, also involving residue K76, to form a network connecting the apices of two monomers. In the presence of mds84, the major shifts were in strand F residues H90 and V93 (Figure 6A lower panel).
Interestingly, a few peaks (in red in Figure 6) became visible only when the ligands occupied the HBPs: K9 and A120 appeared with both tafamidis and mds8, whereas A91 and V121 were observed only in the presence of the monovalent ligand (Figure S3). The appearance of A120 and V121, located in strand H, could be due to a slower exchange rate of the amide with water than in the apo-form. This may reflect the stabilization of the interchain hydrogen bonds between A120 (chain A, C) and Y114 of strand H′ (chain B, D) and the intraprotomer hydrogen bond between strand G residues V121 and T106, confirmed by a temperature coefficient for the amide hydrogen of A120 (see Table S1) higher than −4.5 ppb/K.23 Interestingly, the amide of L55, exhibiting a small chemical shift change both with tafamidis and mds84, became H-bonded in the holo-form (Table S1). Its structural location, in front of K48, is consistent with the ligand-mediated reduced susceptibility to proteolysis of the CD loop.
Discussion and Conclusions
We report here two key molecular structural aspects of ligand binding by TTR, elucidated by NMR analysis:
-
1
We have shown that an intermediate conformational state forms when TTR binds a monovalent ligand in only one of its two identical binding sites;
-
2
We have discovered long distance (12–36 Å) structural effects on the protein when the binding sites are occupied by ligands and we have shown that the structural effects are more extensive with a bivalent than a monovalent ligand.
Negative cooperativity of binding of thyroxine, the natural TTR ligand, first described in 1975,24 has been attributed to putative subtle differences between the two binding sites.25−28 This poorly understood aspect of negative cooperativity is nevertheless important because it affects the possibility to fully saturate plasma TTR with stabilizing ligand compounds to most effectively protect against TTR amyloid fibril formation.10 Although the 2D [1H, 15N] TROSY spectra do not reveal any asymmetry of apo-TTR, we have shown here that occupancy of the first site by tafamidis is followed by a rearrangement of the binding site, involving not only residues located at the dimer interface but also residues in the outer TTR surface and not in contact with the ligand. The scheme in Figure 7A summarizes what emerges from our current study of an unusually complex equilibrium pattern. Our results clearly show an induced fit mechanism in which the initial fast occupancy of the first binding site is followed by a slower rearrangement of the site enabling a tighter embrace of the monovalent ligand. Moreover, the geometry of the binding pocket is altered when two ligands, rather than one, are bound. The simpler equilibrium pattern in the case of the binding of mds84 is described in Figure 7B.
Figure 7.
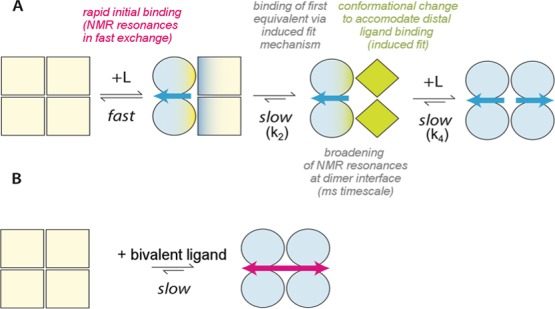
Scheme of the binding of tafamidis and mds84 by TTR. (A) Binding of tafamidis. Apo-TTR, which is a symmetrical homotetramer is shown as pale-yellow squares. Binding of a single molecule of tafamidis initially induces changes in the occupied half molecule dimer, shown here as light-blue green circles, and is then followed by conformational changes in the unoccupied dimer, shown as green diamonds. When the second tafamidis molecule is bound, the TTR tetramer becomes symmetrical again, with the protomers illustrated as light blue circles. The off-rates measured by SPR, k2 = 0.008 Hz and k4 = 0.06 Hz, are consistent with global slow exchange in which the slower k4 is responsible for the negative cooperative effect of binding of this monovalent ligand. (B) Binding of mds84 induces symmetrical changes into the holo-TTR shown as light blue circles.
This NMR derived model is supported by direct SPR measurements of the binding dynamics, in which the 7-fold difference in the observed affinities at the two sites is driven by different off-rates (k2 and k4, Table 1). The calculated affinity of the second site based on the SPR data is higher than previously reported4 (22 nM vs 200 nM) and the reasons of this are not clear. One possibility is that immobilization of the tetramer to the SPR surface impacts on the dynamics of the conformational changes observed in solution, and limiting the change in off rate induced by binding at the first site.
Our NMR findings with full occupancy of TTR by tafamidis or mds84 show that substantial perturbations are not confined to the proximity of ligands but are also propagated into the outer region, comprising the BCEF β-sheet and the helix. Aromatic rings, present in tafamidis and mds84, are well known to affect the chemical shift of neighboring amides, but this effect decreases with the third power of the distance from the center of the ring20 and cannot cause the distant CSPs we report here. A radius 12 Å sphere centered on the ligand rings defines a region, external to the spheres, where any perturbation can only be attributed to allosteric structural changes induced by ligand binding. Furthermore, significant stabilization by additional hydrogen bonding, as witnessed by temperature coefficients measurements, can be envisaged for residues L55, A120, and V121. L55 is located opposite to K48; A120 is involved in the intermolecular pairing between strands H and H′; and V121 forms an intramolecular H-bond with T106 of the facing strand G. Evidence from 19F NMR also suggests that A120, together with F87, may additionally be critical for TTR stabilization as shown by the A120L TTR variant favoring a mis-packed state.17
Our data show that the binding of the monovalent and the bivalent ligands affects protein dynamics differentially. The disappearance of amides within the dimer interface from 2D TROSY maps when mds84 is bound is attributable to protein dynamics in the micro second–millisecond time scale, unfavorable for NMR observation. Residues in loops AB, DE, H1F, and strand F are similarly affected, probably reflecting a change in mobility due to the occupancy of the central part of the TTR channel by the linker chain between the two head groups of mds84. Neutron scattering studies of H/D exchange in the most highly amyloidogenic TTR variant, S52P, show, counterintuitively, greater exchange when tafamidis is bound than in the TTR protein alone.29 Ligand binding by TTR evidently can increase accessibility/mobility and normal mode analysis simulations have previously shown low frequency subunit motion in the apo-protein that can be modified in the holo-form.27 Remarkably, residue A25, which is not within the HBPs, shows the largest NMR chemical shift change in the mds84 complex and just above the mean with bound tafamidis. The carbonyl of A25 is hydrogen bonded to the side chain of T49 and the stronger observed intensity of the A25 peak shift is consistent with our experimental observation that mds84 confers the highest protection against the proteolytic cleavage of the peptide bond 48–49.10 As previously highlighted by Mangione et al.,8 conformational changes favoring stability of the cluster of H-bonds involving residues S50, S52, and E54, that are partially disrupted in the highly amyloidogenic variant S52P, should protect against this amyloidogenic cleavage. The observed CSPs of S50, G53, E54, and L55, together with the H-bond of the latter when the HBPs are occupied, are therefore interpretable as decreased accessibility to proteolysis of the K48–T49 bond.
The changes reported here in solution have not previously been detected in a plethora of X-ray structures of TTR. As discussed extensively by Palaninathan,13 the large number of reported X-ray crystal structures, including wild type TTR, numerous TTR variants and TTR complexed with a variety of different ligands, do not show any structural alterations that could account for their different propensity to unfold and then aggregate into amyloid fibrils. In particular, a detailed analysis of the pairwise superimposition of the X-ray structure of TTR complexed with tafamidis (3TCT)4 or mds84 (3IPE)7 with the structure of apo-TTR (5CN3) gives Cα rmsd values of 0.618 and 0.315 Å, respectively, accounted for by a small deviation in the FG loop (residues 100–103).
In conclusion, our present NMR observations of free TTR alone compared to TTR complexes with different ligands, have revealed conformational changes never previously seen with other approaches including high-resolution X-ray crystallography. The direct NMR structural evidence of long distance conformational changes induced by ligands is consistent with the indirect evidence of long distance effects provided by experimental observations of the inhibitory effects of ligand binding on limited proteolysis of TTR and its amyloidogenesis. In the future, NMR signatures of the effects of ligand binding could make a valuable contribution to design and development of novel ligands for use as inhibitors of TTR amyloid fibril formation.
Experimental Section
Ligand Preparation
Tafamidis was synthesized by Selcia Ltd, Ongar, Essex UK. Analysis was performed in accordance with Internal quality program procedures by the Analytical Support Group, Selcia. Identity of the test item is confirmed by NMR and LCMS analysis. Chemical purity is determined by 1H NMR and HPLC–UV analysis. Chemical purity was assessed as 94% (relative area by NMR); of the 6% impurities, 2.3% is attributable to methanol. Chemical purity is 98.4% (% AUC).
Mds84 was synthesized by WuXi AppTec, Shanghai, China. The sample was >99% pure by HPLC–UV analysis. 1H NMR analysis is consistent with the purity.
NMR Spectroscopy
NMR spectra were acquired at 310 K with a Bruker AVANCE 700 and 800 MHz NMR spectrometer on a U-2H, 13C, 15N-labeled recombinant TTR samples at concentrations of 50–90 μM in PBS buffer at pH 7.0 and 7.4 in a mixture H2O/D2O 90/10%.
The addition of 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) for reference purposes led to the observation that DSS binds to the TTR HBP (see Figure S4).
Ligand stock solutions at 10 mM were prepared in DMSO-d6 and additions up to 6 μL into 600 μL of 50 μM protein solution were made. The effect of DMSO-d6 at up to 1% v/v on chemical shift was negligible (see Figure S5). 2D [1H, 15N] TROSY spectra were acquired at ligand/protein ratios of 0, 0.5, 1.0, 1.5, and 2.0 with tafamidis, the monovalent ligand, and at 0, 0.5, and 1.0 with mds84, the bivalent ligand.
3D HNCA spectra were also recorded in the presence of the ligands to confirm the assignment.
To express changes in the chemical shifts of the individual amide pairs, a combined chemical shift change (in ppm) was defined30 as
The involvement of amide hydrogens in H-bonds was estimated by a series of 11 2D TROSY [1H, 15N] spectra acquired at temperature varying from 289 to 313 K for apo-TTR, for holo-TTR with tafamidis and mds84 in a 2:1 and 1:1 ligand/protein ratio, respectively.
Surface Plasmon Resonance
The binding of tafamidis to TTR was monitored using SPR performed using a Biacore T200 instrument (GE Healthcare) with streptavidin coated sensor chips.
Data were fitted to the model described by Scheme 1. The measured fractional response is then given by {[AB + (2ABB)]/Ai} where Ai is the notional concentration of TTR on the surface. Rate parameters were determined from the global fitting of the association data to the binding equilibria as defined by Scheme 1. The dissociation rates (k2 and k4) were separately derived from the fitting of the dissociation data to a double exponential equation with k2 and k4 being allocated to the first and second binding steps, respectively, based on the reduced contribution of a second exponential at the lower concentrations of tafamidis, and the global fit repeated to determine the on rate constants with the dissociation constants fixed. The affinity of tafamidis at each of the two binding sites was determined from the ratio of rate constants, Kd1 = k2/k1 and Kd2 = k4/k3, respectively.
Other Methods
Recombinant TTR production, details on NMR spectra acquisition, and on SPR data acquisition are described in Supporting Information Methods.
Acknowledgments
This work was supported by investment from the University College London Technology Fund and grants from the U.K. Medical Research Council (MR/K000187/1 and MR/R016984/1), the Rosetrees Trust/Royal Free Charity PhD programme (M427), the Cariplo Foundation (projects 2014-0700), the Italian Ministry of Health (Ricerca Finalizzata RF 2013 02355259) and the Istituto Nazionale di Biostrutture e Biosistemi. Core support for the Wolfson Drug Discovery Unit is provided by the UK National Institute for Health Research Biomedical Research Centre and Unit Funding Scheme via the UCLH/UCL Biomedical Research Centre and by the UCL Amyloidosis Research Fund. We acknowledge the use of the UCL NMR Centre and the MRC Biomedical NMR Centre at the Francis Crick Institute, London, and thank the staff for their support. We acknowledge MIUR Dipartimento di Eccellenza UniPV. We also thank Professor Trevor Forsyth for kindly providing the plasmid used for recombinant TTR production.
Glossary
Abbreviations
- TTR
transthyretin
- rmsd
root mean square deviation
- TROSY
transverse relaxation-optimized spectroscopy
- CSP
chemical shift perturbation
- SPR
surface plasmon resonance
- HBP
halogen binding pocket
- DSS
4,4-dimethyl-4-silapentane-1-sulfonic acid
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.9b01037.
Molecular formula strings (CSV)
Methods of TTR expression and purification, NMR spectra acquisition and on SPR data acquisition; additional table of NMR temperature coefficients of apo and holo TTR with tafamidis and mds84 (Table S1); and figures reporting TTR dimer structure, 2D TROSY spectra at sub-stoichiometric ligand/TTR ratios showing the presence of free and bound peaks, NMR peak intensities, DSS, and DMSO effects (PDF)
Structural effects of mds84 and tafamidis (MPG)
Structural effects of mds84 and tafamidis (MPG)
Structural effects of mds84 and tafamidis (MPG)
Author Contributions
The study was conceived, designed and supervised by V.B. and A.C. A.C., G.V., C.A.W., P.P.M., D.C., P.N. and G.W.T. performed research. A.C., G.V., C.A.W., J.C. analysed and interpreted NMR data. R.B. and I.U. performed and analysed Biacore data. J.C. and M.B.P. contributed to discussion. All the authors analysed and interpreted the data. The paper was written by V.B., A.C. and M.B.P. and reviewed and approved by all co-authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Rowczenio D. M.; Noor I.; Gillmore J. D.; Lachmann H. J.; Whelan C.; Hawkins P. N.; Obici L.; Westermark P.; Grateau G.; Wechalekar A. D.. Online registry for mutations in hereditary amyloidosis including nomenclature recommendations. Hum. Mutat.. 2014, 35–E2403-E2412. 10.1002/humu.22619 [DOI] [PubMed] [Google Scholar]
- Hawkins P. N.; Ando Y.; Dispenzeri A.; Gonzalez-Duarte A.; Adams D.; Suhr O. B. Evolving landscape in the management of transthyretin amyloidosis. Ann. Med. 2015, 47, 625–638. 10.3109/07853890.2015.1068949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake C. C. F.; Oatley S. J. Protein-DNA and protein-hormone interactions in prealbumin: a model of the thyroid hormone nuclear receptor?. Nature 1977, 268, 115–120. 10.1038/268115a0. [DOI] [PubMed] [Google Scholar]
- Bulawa C. E.; Connelly S.; Devit M.; Wang L.; Weigel C.; Fleming J. A.; Packman J.; Powers E. T.; Wiseman R. L.; Foss T. R.; Wilson I. A.; Kelly J. W.; Labaudiniere R. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 9629–9634. 10.1073/pnas.1121005109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly J.; Colon W.; Lai Z.; Lashuel H.; McCulloch J.; McCutchen S.; Miroy G.; Peterson S. Transthyretin quaternary and tertiary structural changes facilitate misassembly into amyloid. Adv. Protein Chem. 1997, 50, 161–181. 10.1016/s0065-3233(08)60321-6. [DOI] [PubMed] [Google Scholar]
- Connelly S.; Choi S.; Johnson S. M.; Kelly J. W.; Wilson I. A. Structure-based design of kinetic stabilizers that ameliorate the transthyretin amyloidoses. Curr. Opin. Struct. Biol. 2010, 20, 54–62. 10.1016/j.sbi.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolstoe S. E.; Mangione P. P.; Bellotti V.; Taylor G. W.; Tennent G. A.; Deroo S.; Morrison A. J.; Cobb A. J. A.; Coyne A.; McCammon M. G.; Warner T. D.; Mitchell J.; Gill R.; Smith M. D.; Ley S. V.; Robinson C. V.; Wood S. P.; Pepys M. B. Trapping of palindromic ligands within native transthyretin prevents amyloid formation. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 20483–20488. 10.1073/pnas.1008255107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangione P. P.; Porcari R.; Gillmore J. D.; Pucci P.; Monti M.; Porcari M.; Giorgetti S.; Marchese L.; Raimondi S.; Serpell L. C.; Chen W.; Relini A.; Marcoux J.; Clatworthy I. R.; Taylor G. W.; Tennent G. A.; Robinson C. V.; Hawkins P. N.; Stoppini M.; Wood S. P.; Pepys M. B.; Bellotti V. Proteolytic cleavage of Ser52Pro variant transthyretin triggers its amyloid fibrillogenesis. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 1539–1544. 10.1073/pnas.1317488111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcoux J.; Mangione P. P.; Porcari R.; Degiacomi M. T.; Verona G.; Taylor G. W.; Giorgetti S.; Raimondi S.; Sanglier-Cianférani S.; Benesch J. L.; Cecconi C.; Naqvi M. M.; Gillmore J. D.; Hawkins P. N.; Stoppini M.; Robinson C. V.; Pepys M. B.; Bellotti V. A novel mechano-enzymatic cleavage mechanism underlies transthyretin amyloidogenesis. EMBO Mol. Med. 2015, 7, 1337–1349. 10.15252/emmm.201505357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verona G.; Mangione P. P.; Raimondi S.; Giorgetti S.; Faravelli G.; Porcari R.; Corazza A.; Gillmore J. D.; Hawkins P. N.; Pepys M. B.; Taylor G. W.; Bellotti V. Inhibition of the mechano-enzymatic amyloidogenesis of transthyretin: role of ligand affinity, binding cooperativity and occupancy of the inner channel. Sci. Rep. 2017, 7, 182. 10.1038/s41598-017-00338-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangione P. P.; Verona G.; Corazza A.; Marcoux J.; Canetti D.; Giorgetti S.; Raimondi S.; Stoppini M.; Esposito M.; Relini A.; Canale C.; Valli M.; Marchese L.; Faravelli G.; Obici L.; Hawkins P. N.; Taylor G. W.; Gillmore J. D.; Pepys M. B.; Bellotti V. Plasminogen activation triggers transthyretin amyloidogenesis in vitro. J. Biol. Chem. 2018, 293, 14192–14199. 10.1074/jbc.ra118.003990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson S. M.; Connelly S.; Wilson I. A.; Kelly J. W. Toward optimization of the second aryl substructure common to transthyretin amyloidogenesis inhibitors using biochemical and structural studies. J. Med. Chem. 2009, 52, 1115–1125. 10.1021/jm801347s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palaninathan S. K. Nearly 200 X-ray crystal structures of transthyretin: what do they tell us about this protein and the design of drugs for TTR amyloidoses?. Curr. Med. Chem. 2012, 19, 2324–2342. 10.2174/092986712800269335. [DOI] [PubMed] [Google Scholar]
- Cho Y.; Baranczak A.; Helmke S.; Teruya S.; Horn E. M.; Maurer M. S.; Kelly J. W. Personalized medicine approach for optimizing the dose of tafamidis to potentially ameliorate wild-type transthyretin amyloidosis (cardiomyopathy). Amyloid 2015, 22, 175–180. 10.3109/13506129.2015.1063485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X.; Jaeger M.; Kelly J. W.; Dyson H. J.; Wright P. E. Mispacking of the Phe87 Side Chain Reduces the Kinetic Stability of Human Transthyretin. Biochemistry 2018, 57, 6919–6922. 10.1021/acs.biochem.8b01046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K.; Cho H. S.; Hoyt D. W.; Nguyen T. N.; Olds P.; Kelly J. W.; Wemmer D. E. Deuterium-proton exchange on the native wild-type transthyretin tetramer identifies the stable core of the individual subunits and indicates mobility at the subunit interface. J. Mol. Biol. 2000, 303, 555–565. 10.1006/jmbi.2000.4164. [DOI] [PubMed] [Google Scholar]
- Leach B. I.; Zhang X.; Kelly J. W.; Dyson H. J.; Wright P. E. NMR Measurements Reveal the Structural Basis of Transthyretin Destabilization by Pathogenic Mutations. Biochemistry 2018, 57, 4421–4430. 10.1021/acs.biochem.8b00642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikura M.; Kay L. E.; Bax A. A novel approach for sequential assignment of 1H, 13C, and 15N spectra of proteins: heteronuclear triple-resonance three-dimensional NMR spectroscopy. Application to calmodulin. Biochemistry 1990, 29, 4659–4667. 10.1021/bi00471a022. [DOI] [PubMed] [Google Scholar]
- Pervushin K.; Riek R.; Wider G.; Wuthrich K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc Natl Acad Sci U S A 1997, 94, 12366–12371. 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson M. P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 73, 1–16. 10.1016/j.pnmrs.2013.02.001. [DOI] [PubMed] [Google Scholar]
- Kovrigin E. L. NMR line shapes and multi-state binding equilibria. J. Biomol. NMR 2012, 53, 257–270. 10.1007/s10858-012-9636-3. [DOI] [PubMed] [Google Scholar]
- Waudby C. A.; Ramos A.; Cabrita L. D.; Christodoulou J. Two-Dimensional NMR Lineshape Analysis. Sci. Rep. 2016, 6, 24826. 10.1038/srep24826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter N. J.; Williamson M. P. Temperature dependence of 1H chemical shifts in proteins. J. Biomol. NMR 1997, 9, 359–369. 10.1023/a:1018334207887. [DOI] [PubMed] [Google Scholar]
- Ferguson R. N.; Edelhoch H.; Saroff H. A.; Robbins J.; Cahnmann H. J. Negative cooperativity in the binding of thyroxine to human serum prealbumin. Biochemistry 1975, 14, 282–289. 10.1021/bi00673a014. [DOI] [PubMed] [Google Scholar]
- Neumann P.; Cody V.; Wojtczak A. Structural basis of negative cooperativity in transthyretin. Acta Biochim Pol 2001, 48, 867–875. [PubMed] [Google Scholar]
- Tomar D.; Khan T.; Singh R. R.; Mishra S.; Gupta S.; Surolia A.; Salunke D. M. Crystallographic study of novel transthyretin ligands exhibiting negative-cooperativity between two thyroxine binding sites. PLoS One 2012, 7, e43522 10.1371/journal.pone.0043522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cianci M.; Folli C.; Zonta F.; Florio P.; Berni R.; Zanotti G. Structural evidence for asymmetric ligand binding to transthyretin. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2015, 71, 1582–1592. 10.1107/s1399004715010585. [DOI] [PubMed] [Google Scholar]
- Zanotti G.; Vallese F.; Ferrari A.; Menozzi I.; Saldaño T. E.; Berto P.; Fernandez-Alberti S.; Berni R. Structural and dynamics evidence for scaffold asymmetric flexibility of the human transthyretin tetramer. PLoS One 2017, 12, e0187716 10.1371/journal.pone.0187716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee A. W.; Aldeghi M.; Blakeley M. P.; Ostermann A.; Mas P. J.; Moulin M.; de Sanctis D.; Bowler M. W.; Mueller-Dieckmann C.; Mitchell E. P.; Haertlein M.; de Groot B. L.; Boeri Erba E.; Forsyth V. T. A molecular mechanism for transthyretin amyloidogenesis. Nat. Commun. 2019, 10, 925. 10.1038/s41467-019-08609-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder F. A. A.; Schipper D.; Bott R.; Boelens R. Altered flexibility in the substrate-binding site of related native and engineered high-alkaline Bacillus subtilisins. J. Mol. Biol. 1999, 292, 111–123. 10.1006/jmbi.1999.3034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.