

Abstract
The Central European Cooperative Oncology Group (CECOG) and ‘ESMO Open—Cancer Horizons’ roundtable discussion brought together stakeholders from several European Union (EU) countries involved in drug development, drug authorisation and reimbursement or otherwise affected by delayed and unequal access to innovative anticancer drugs. The approval process of drugs is well established and access delays can be caused directly or indirectly by national or regional decision-making processes on reimbursement. The two key aspects for those involved in reimbursement decisions are first the level of evidence required to decide and second pricing, which can be challenging for some innovative oncology compounds, especially in Eastern and South-Eastern European countries. Other important factors include: available healthcare budget; the structure and sophistication of healthcare authorities and health technology assessment processes; societal context and political will. From the point of view of the pharmaceutical industry, better alignment between stakeholders in the process and adaptive pathway initiatives is desirable. Key aspects for patients are improved access to clinical trials, preapproval availability and reports on real-world evidence. Restricted access limits oncologists’ daily work in Eastern and South-Eastern EU countries. The roundtable discussion suggested considering the sequencing of regulatory approval and reimbursement decisions together with more flexible contracting as a possible way forward. The panel concluded that early and regular dialogue between all stakeholders including regulators, payers, patient stakeholders and industry is required to improve the situation.
Keywords: cancer drugs, drug access, regulatory approval, early access to medicines, study design, benefit-risk assessment, cancer care
Video Abstract.
Introduction
The incidence of cancer has increased by more than 30% over the past decade in Europe.1 Cancer mortality has also increased, but at a lower rate (11%),1 showing that survival is improving. However, there are major differences within the European Union (EU) regarding cancer cure rates and survival.1
Access to innovative treatments, sustainable health systems and maintaining incentives for innovation are key objectives health policymakers seek to reconcile. While this holds true for the future, the advances of targeted medicine and immune-oncology have raised the hope of significantly improving cancer survival across a number of tumour types and have changed scientific development dramatically. From 2011 to 2016, 68 novel therapies have been launched globally for the treatment of cancer, with over 600 molecules in late-stage development, of which 90% is targeted therapy.2
This rapid innovation has many consequences. One is that, clinicians struggle to recruit patients for clinical trials given the volume of trials being initiated and consequently the size of clinical trials are reduced. This in turn reduces the opportunity to collect data on important endpoints of overall survival and quality of life (QoL). Another consequence is that in a fatal disease with often short survival times, there is a strong desire among all stakeholders to give patients the fastest possible access to novel treatments expected to be effective. Intermediate outcomes (eg, disease-free survival, event-free survival, progression-free survival and objective response rate) are being used; however, these can be controversial when the evidence is appraised, especially for reimbursement decisions.3 There will always be a trade-off between early access and uncertainties, mainly safety and efficacy aspects.
In an ideal world, all patients with cancer should have rapid access to all newly available anticancer drugs when approved. However, this is far from the reality. With the variations in economic standard across the EU, access to new cancer medicines differs significantly1: for instance, according to the European Federation of Pharmaceutical Industries and Associations (EFPIA) commissioned W.A.I.T. indicator (based on EFPIA member companies’ information), only 14% of recently approved medicines are available to patients in Latvia. In contrast, nearly 95% of all medicines newly authorised by the European Medicines Agency (EMA) are available to patients in Germany.4
There are also large differences in time to access: a patient in Serbia may wait nine times longer than a patient in Germany until a newly authorised drug becomes available on average; in Bulgaria, patients wait five times longer on average.4
Figure 1 illustrates the rate of availability measured by the number of oncology medicines available to patients in European countries as of 2018: for most countries, this is the point at which the product gains access to the reimbursement list.
Figure 1.
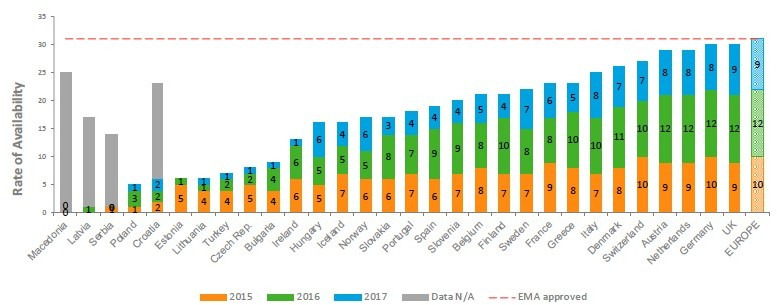
Rate of availability of oncology medicines available in the European countries (based on European Federation of Pharmaceutical Industries and Associations (EFPIA) member companies’ information). EMA, European Medicines Agency. Source: EFPIA Patient W.A.I.T. Indicator 2018 survey (4), Permission granted to reprint. Document publicly available.
The European Society for Medical Oncology (ESMO) has declared achieving equal access to cancer care as one of its major goals.
Physicians from Eastern and South-Eastern EU countries with delayed access to innovative medicines can be limited in their everyday work by the inability to provide the best standard-of-care for their patients. It is hard for them to explain to their patients and their families the lack of access to the most effective treatments despite living in societies with declared universal healthcare access.5
This article summarises a multidisciplinary roundtable discussion organised by the Central European Cooperative Oncology Group (CECOG, www.cecog.org), headquartered in Vienna, Austria, and ‘ESMO Open—Cancer Horizons’ in Vienna in December 2018. The roundtable panel with stakeholders from several EU countries identified several challenges to equal and timely access and potential solutions that could accelerate access to innovative anticancer drugs, particularly in Eastern and South-Eastern EU countries with known access delays. The participants and authors of this review are stakeholders with different perspectives on the topic and give the point of view of a regulator, a health economist, a patient representative, a clinician and the pharmaceutical industry, as well as the payer perspective.
The path of a new drug to the patient
New drugs pass various steps until they reach patients, as shown in figure 2.6 Barriers to rapid access can occur at different stages:
Figure 2.
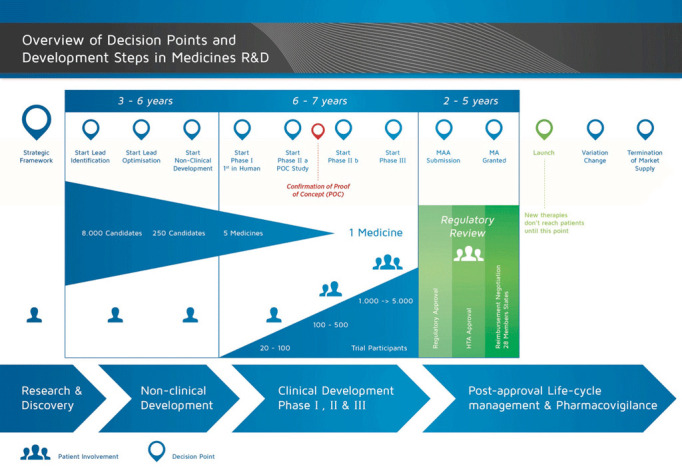
Overview of decision points and development steps in medicines’ research and development.6 HTA, health technology assessment. Source: European Patients Academy (EUPATI) (2015), Development of medicines.5 Permission granted to reprint.
Research and clinical trials.
Regulatory approval.
National and regional access decisions, including health technology assessment (HTA) and pricing and reimbursement decisions.
Uptake into local practice based on national/regional or local financing.
The roundtable started with discussing the EMA approval process, which has become more flexible in recent years in response to the growth of personalised medicine and immune-oncology.
Regulatory approval—required evidence and accelerated approval initiatives
New medicines for cancer treatment have to undergo regulatory assessment by EMA to be approved via the centralised marketing authorisation.7 This requirement intends to provide the basis for availability of new cancer treatments throughout all member states of the EU. The prerequisite for any type of marketing authorisation in the EU is a positive benefit-risk ratio, and confirmatory randomised controlled trials (RCTs) remain the gold standard for evaluation of anticancer drugs.
In addition to the classical setting of RCT, other study designs have been introduced in the development of anticancer drugs:
Especially in late line oncology situations, single-arm studies have been accepted to provide evidence of efficacy and safety for approval.8 9
Another innovative way to investigate products in precision medicine oncology is to study them in a histology-independent approach, that is, regardless of the location or tissue type of the tumour but dependent on a specific molecular alteration. This is also referred to as site-agnostic indication.10
Immunotherapy and targeted therapies are examples.
Here one can distinguish between
umbrella trials, where multiple targeted therapy products are studied in the context of a single type of cancer, and
basket trials, where a single targeted therapy medicine is investigated for its effect on multiple types of cancer.11
Finally, so-called platform trials allow to add or stop substudies like umbrella or basket trials in a dynamic approach. While these designs may deliver advantages in terms of efficiency of discovering new treatment paradigms, some caveats should be respected, especially methodological problems of multiplicity and possible inadequate pooling of data.12
The US Food and Drug Administration (FDA) has recently approved two drugs in tissue/site agnostic indications, that is, pembrolizumab for the treatment of unresectable or metastatic, microsatellite instability-high or mismatch-repair-deficient solid tumours, regardless of tumour site or histology13 and larotrectinib for treating solid tumours with a neurotrophic receptor tyrosine kinase gene fusion.14 An analogous application for larotrectinib was filed also to the EMA for evaluation and recently received a positive opinion by the Committee for Medicinal Products for Human Use (CHMP).
Accelerated approval initiatives
In order to foster early access to medicines, EMA has introduced several initiatives, which all focus on unmet medical need.15
With accelerated assessment over 150 days, medicines of major interest for public health can be authorised faster, especially those representing a therapeutic innovation.
Conditional approval allows the acceptance of less comprehensive clinical results than normally required, if the immediate value of the medicine outweighs the uncertainties that must be supplemented postauthorisation via legally binding specific obligations to collect further data.
An approval under exceptional circumstances may be granted when comprehensive efficacy and safety data can never be expected and specific obligations mostly for safety data are then imposed on the marketing authorisation holder.
Medicines still undergoing centralised marketing authorisation review or clinical trials may be supplied to patients via compassionate-use programmes.
To optimise the use of the above regulatory tools, EMA has launched the PRIME (PRIority MEdicines) scheme16 to provide support and interaction with frequent scientific advice and early involvement of HTA bodies (responsible for later reimbursement decisions) during the development of medicines that are anticipated as game-changing therapies.
Another concept for medicine development and data generation with the aim to facilitate patient access to promising medicines addressing high unmet need is ‘Medicines Adaptive pathways to Patients (MAPPs)’, which is a prospectively planned approach17 involving stakeholders from research through to treatment outcomes. The ADAPT-SMART platform18 funded by the EU’s Innovative Medicines Initiative is a multistakeholder platform for the coordination of adaptive pathways-related activities. The initiative is based on three principles, that is,
an iterative development with approval in stages, or on early surrogate data, or data in patient subpopulations,
further evidence generation through real-world data, and
early involvement of patients and HTA institutions.
The introduction of accelerated approval processes is however not without criticism.
A review of accelerated approvals of anticancer treatments by the FDA between 2009 and 2014 showed that all were based on intermediate endpoints.19 20 Intermediate endpoints have long been in use in a number of diseases and are acceptable provided they are a strong substitute for the primary outcome measure based on validation studies. However, the above review showed that in 84% of the accelerated approvals validation was absent, negative or weak.19
The uncertainty surrounding the effectiveness of these new treatments is high.21–24 In such situations of limited evidence on the extent of efficacy, specific considerations become the prime driver for regulators to approve a drug, such as the observation of durable overall response rates of a large magnitude, a strong scientific rationale and preclinical data supporting the mechanism of action, as well as the use of objective endpoints and a predictable course of the disease in well-defined patient populations. While all of these criteria may aid in strengthening of the evidence base and facilitate the interpretation of the results, considerable uncertainties in the outcomes remain and these are then passed on to HTA agencies and payers.
Nevertheless, intermediate endpoints are accepted in Europe on an exceptional basis only. Surrogacy is not accepted in anticancer EU guidelines and intermediate endpoints can only support harder endpoints except when they are related to a direct benefit (without prediction) and as such become clinically relevant endpoints in itself or if very large effects make it unlikely that no relevant benefit is observed in the end on hard endpoints.
The EMA assesses quality, safety and efficacy only, and economic and other considerations are exempted from the authorisation.7 Since nearly all new medicines in the EU undergo central regulatory approval by the EMA, access delays are caused by later steps in the process especially during national and regional reimbursement decisions.
Reimbursement decisions, payers’ concerns and potential solutions to early access
Reimbursement decisions are made at a national and regional level based on some form of HTA. The decision makers can be government bodies, HTA agencies, reimbursement agencies or the payers themselves.
The three most important questions in the evaluation for reimbursement are as follows:
What are the available alternatives?
Is the new drug better with respect to efficacy, safety and/or QoL?
Is the price worth the difference?
The key concern of payers around the world about cancer medicines is that they are too expensive. This is true for the USA, where high list prices have led to the introduction of the concept of ‘financial toxicity’25 and for payers and healthcare providers in Europe. Payers and the HTA agencies advising them perceive a disconnection between the certainty and the extent of the demonstrated value of a new medicine at the time of marketing authorisation and its price.26
Despite—or because of—possible reasons (such as small patient groups and lack of clinical equipoise for a randomised trial) for a company seeking marketing authorisation not being able to provide all the data that payers would like to have, research costs may not seem to justify the high asking prices for cancer products.27
Potentials solutions from the payers’ point of view to accelerating access to innovative anticancer drugs are given below:
Parallel consultation
The rationale behind this approach28 is to establish—alongside the scientific evidence needed for marketing authorisation by EMA—the evidence requirements by payers for reimbursement. The convergence between EMA parallel scientific advice and the European network for Health Technology Assessment (EUnetHTA) early dialogue is ongoing, and it involves participation by both regulators and HTA institutions in the process of advising pharmaceutical companies about their clinical development programmes. In the European context, the advantages for companies are obvious—they learn what payers need to know for making a positive reimbursement decision at a point in time where this information can be used in product development. However, one of payers’ concerns with this approach is prematurely making an implicit or explicit commitment about the acceptability of generated evidence. Another concern is the limited opportunity for involvement of small member states—given the interest of companies in large markets and the limited resources of small member states.
Use of real-world evidence and adaptive pathways
One approach to fill the lack of evidence regarding patient-relevant endpoints is to gather evidence after marketing authorisation. This can allow early, although conditional, access to new medicines for patients with dire, unmet needs—if all stakeholders agree to the conditions involved. This was the concept underlying the ‘adaptive pathways’ paradigm of EMA29 and the ADAPT-SMART project.18 Payers were sceptical of this idea,30 pointing out their difficulties, among others, in delisting approved products. Concerns regarding managed entry agreements were highlighted in a recent review,17 indicating the need for further refinement on agreements and procedures.
Adaptive/flexible pricing
This means paying a lower initial price for a new medicine with limited evidence, and adapting the price, based on subsequent evidence. These models are particularly attractive for therapies, which are based on the premise of being potentially curative.31 Legal agreements, based on robust parameters, are a prerequisite, as well as trust between parties. Both are difficult to achieve.
The local health-economic situation in Eastern and South-Eastern Europe
Eastern and South-Eastern EU countries have a fivefold to sixfold lower direct spending on cancer care than wealthy Western European countries32 as shown in figure 3. These differences are to some degree compensated by lower salaries for healthcare workers and lower direct costs, but costs for diagnostic equipment and pharmaceuticals all lie within a common price corridor. There may be undisclosed discounts that to some extent reduce costs, but most likely, these discounts will not make access to new anticancer drugs equal across Europe.1
Figure 3.

Large variations in spending on cancer per capita in Europe.1 Permission granted to reprint. PPP, purchasing power parity.
It is however not only the lower direct spending on cancer that explains the inequality in Eastern and South-Eastern EU countries that have faced transition and economic difficulties in recent decades. Healthcare systems also often have less capacity and specialisation and there can be a lack of public knowledge, societal dialogue and political will. The pace of innovation in oncology treatments is much faster than the ability of healthcare systems to adapt to this fast-changing environment.5 33 This is true even in some high income countries.34
A clinical example: access to innovative treatments for metastatic melanoma
One of the examples of major achievements in oncology is the treatment of metastatic melanoma, a chemotherapy-resistant cancer with a median survival of only 6 to 9 months prior to 2010.35
After 2011, a number of clinical breakthroughs have been achieved with targeted therapy and immunotherapy. This has led to significantly prolonged survival with nearly 50% of metastatic melanoma patients in good prognostic groups surviving up to 5 years based on recent trials.36 Even in patients with brain metastases, these treatments can have intracranial response rates of 44% to 58% with some patients achieving a long-term benefit.37 However, despite their high efficacy, there is restricted access to these treatments in parts of Europe34; in 2016, over 5000 patients did not have access.38
The EADO and MWS survey
The magnitude of the problem and major determinants of delayed access to innovative medicines were recently explored for metastatic melanoma in Europe and worldwide. The European Association of Dermato-Oncology (EADO) and Melanoma World Society (MWS) conducted a survey in 34 countries: USA, China, Australia, Argentina, Brazil, Chile, Mexico and 27 European countries, from September 2017 to December 2018.39 The aim of the study was to analyse the access to first-line recommended treatments for metastatic melanoma by current guidelines (National Comprehensive Cancer Network (NCCN), European Society for Medical Oncology (ESMO) and European Organisation for Research and Treatment of Cancer (EORTC)/EADO/ European Dermatology Forum (EDF)).
It was found that BRAF and MEK inhibitors combination and anti-programmed cell death protein 1 (PD-1) immunotherapy after centralised European authorisation were fully reimbursed in 20 out of 27 EU countries, combination anti-cytotoxic T-lymphocyte-associated protein 4 and anti-PD-1 therapies in 8 out of 27 and talimogene laherparepvec (T-Vec) in 3 of 27 EU countries. Delays in access ranged in Western Europe from 0 days in Switzerland to 1057 days in Portugal and in Central, Eastern and South-Eastern Europe from 501 days in Poland and Slovenia to 1486 days in Ukraine.
Delay in reimbursement was in correlation with scores of clinical benefits developed by major oncological organisations such as the American Society of Clinical Oncology Net Health Benefit Score (ASCO-NHB score) (rho=0.819; p=0.004) and the ESMO-Magnitude of Clinical Benefit Scale (ESMO-MCBS) (rho=0.933, p<0.01), as well as with median market price (rho=0.694, p=0.026). The medicines with the highest (ie, best) scores of clinical benefits were the ones with the longest delay in access. Also, it was in correlation with health expenditure per capita, health policy performance scores and HTA implementation (p<0.05). Countries without implemented HTA assessment process were the ones with the greatest reimbursement delays (median 743 vs 1088 days, p=0.057). In most countries (64%), governmental price control mechanisms including managed entry agreements with national authorities were necessary for reimbursement.40 Based on this survey, it was, however, also obvious that in some of the countries with medium to low healthcare expenditure per capita, the reimbursement of most medicines is given (table 1).39
Table 1.
Access to innovative medicines for metastatic melanoma and economic and healthcare system parameters39
| Number of reimbursed medicines | Delay in reimbursement | Restrictions in reimbursement | GDP* | Human development index | HEPC | Mackenbach score | HTA | Type of reimbursement | |
| Europe | |||||||||
| Austria | 8 | 185.5 | No | 50 078 | 0.885 | 5138 | 48 | Yes | Discount rebates |
| Belgium | 8 | 355 | Yes | 46 383 | 0.89 | 4782 | 17 | Yes | Price cap freezes |
| Denmark | 8 | 357 | Yes | 49 496 | 0.923 | 5083 | 43 | Yes | Price volume agreements |
| France | 7 | 543.5 | No | 41 466 | 0.888 | 4542 | 52 | Yes | Price volume agreements |
| Germany | 9 | 30 | No | 48 730 | 0.916 | 5357 | 35 | Yes | Discount rebates |
| Greece | 8 | 451.5 | No | 26 783 | 0.865 | 2204 | 16 | Yes | Discount rebates |
| Ireland | 6 | 636 | No | 68 883 | 0.923 | 5335 | 38 | Yes | Discount rebates |
| Italy | 6 | 724 | No | 38 161 | 0.873 | 3351 | 31 | Yes | Discount rebates |
| Portugal | 2 | 1057.5 | Yes | 30 624 | 0.83 | 2661 | 19 | Yes | Not known |
| Spain | 7 | 977.5 | No | 36 310 | 0.876 | 3183 | 35 | No | Discount rebates |
| Switzerland | 8 | 0 | No | 62 882 | 0.93 | 7583 | 46 | No | Not known |
| UK | 7 | 395.5 | No | 42 609 | 0.747 | 4145 | 37 | Yes | Discount rebates |
| Albania | 2 | 1314 | No | 11 929 | 0.733 | 774 | −13 | No | Not known |
| Belarus | 1 | 1314 | Yes | 18 060 | 0.798 | 1085 | −25 | Yes | Not known |
| Bosnia and Herzegovina | 0 | 1458 | No | 12 075 | 0.733 | 1102 | −60 | Yes | Not known |
| Bulgaria | 3 | 996 | No | 19 199 | 0.782 | 1492 | −33 | No | MEA |
| Croatia | 5 | 938 | Yes | 23 596 | 0.818 | 1656 | −17 | No | MEA |
| Czech Republic | 5 | 801.5 | Yes | 34 711 | 0.87 | 2470 | 12 | Yes | MEA |
| Estonia | 3 | 1149.5 | No | 29 365 | 0.861 | 1887 | −32 | Yes | MEA |
| Lithuania | 2 | 1367.5 | No | 29 966 | 0.839 | 1875 | −28 | Yes | MEA |
| FYR Macedonia | 2 | 933.5 | 15 121 | 0.747 | 857 | 0 | Yes | Price cap freezes | |
| Montenegro | 3 | 1303 | Yes | 16 854 | 0.802 | 957 | −18 | No | Not known |
| Poland | 7 | 501.5 | No | 27 811 | 0.843 | 1704 | -4 | Yes | Price volume agreements |
| Romania | 4 | 905.5 | No | 23 626 | 0.793 | 1090 | −42 | Yes | Price volume agreements |
| Serbia | 3 | 1130.5 | Yes | 14 512 | 0.771 | 1324 | −17 | Yes | MEA |
| Slovenia | 7 | 631 | No | 32 885 | 0.88 | 2734 | 15 | No | Discount rebates |
| Ukraine | 1 | 1274.5 | No | 8272 | 0.907 | 469 | −73 | No | Not known |
*GDP, gross domestic product, World Bank Data.
HEPC, health expenditure per capita; HTA, health technology assessment; MEA, managed entry agreements.
Possible solutions from the clinician’s point of view
Based on this and other studies, possible solutions for improved access could include further development of centralised HTA in the EU41 and innovative payment models.42 It would also be good to invest in education of national authorities on HTA assessment and governmental price control mechanisms.41 Legislation on cross-border participation in clinical trials that could enable early access to effective treatments for patients should be created. Early dialogue with decision makers should be developed.40 It is crucial to raise the public awareness and start a societal dialogue about rising incidence of cancer and recent revolutionary innovations in cancer treatment. In the short term, they are coming with rising costs of treatment, but on the long-run these innovations can change the survival and QoL of patients and their families, which will have an impact on society.40
The patient advocate’s point of view
From the patient perspective, there are many challenges in ensuring access to quality cancer care. These challenges begin when the quality, efficacy and safety of new treatments are being assessed and do not end once the patient has finished their initial treatment. A key aspect for patients is the access to information on clinical trials.
Clinical trials access
Patients see clinical trials as complicated, and the current search engines on clinicaltrials.gov and clinicaltrialsregister.eu are difficult for a lay person to use. Furthermore, the inclusion and exclusion criteria are complex, especially now with the use of biomarker testing in many studies. In order to support awareness and correct understanding of clinical trials, there is a need to promote patient-friendly clinical trial search engines, provide training on the terms used (such as ‘Phase 3’ and ‘randomisation’) and ensure that more information is available in languages other than English.
QoL and other patient-relevant outcomes
In all phase 2 and phase 3 clinical trials, it is vital that QoL and other patient-relevant outcomes are measured. When appraising the risks and benefits of a new cancer drug, we must capture the ways in which a drug changes how a patient functions and feels. Without this information, we do not have a complete understanding of the characteristics of that drug.43
Patient follow-up
Clinical trials and cancer registries should follow-up patients after their acute phase of treatment has ended, in order to quantify the QoL and late treatment effects experienced by cancer survivors.44
Compassionate use and named-patient use
If a patient is not eligible for any clinical trial, they may still seek access to new investigational therapies through a process known in Europe as compassionate use (preapproval access in the USA45). Compassionate access is not regulated by the EU but at national level.
Compassionate use gives patients the opportunity to take investigational therapies that are still in the process of being assessed by regulatory agencies for safety and efficacy. This type of access should be streamlined, to enable more patients to understand if they can access pipeline treatments if they are not eligible for clinical trials. There also needs to be clear differentiation between compassionate use programmes that are designed for groups of patients and are formally approved by regulatory authorities vs access to (yet) unapproved drugs provided for single patients on the basis of named-patient use, which is offered under the responsibility of the treating physician.
The use of real-world evidence
Real-world evidence (RWE) measures the effectiveness in a more diverse group of patients than those in clinical trials, under more realistic conditions, usually after a treatment is authorised.
By adding to data obtained in a narrow patient population in the clinical trial setting, RWE can also provide useful information about the safety of a drug in a larger and more heterogeneous population.
Furthermore, RWE generated from everyday patients in everyday settings can also improve the overall comprehension of the disease and help identify new points in the patient journey for treatment.46 RWE is important as one third of all cancer cases now occur in individuals over the age of 75 years.1 The efficacy and toxicity of new anticancer therapies may not be evaluated in this age group in clinical trials. If implemented correctly, RWE studies can also form the basis for better political decisions and adequate allocation of healthcare resources.
The view of the pharmaceutical industry
For drug manufacturers, delayed access or so-called launch delays are challenges that need to be resolved. Pricing policies such as External Reference Pricing can be one of the various causes. A key missing link from the pharmaceutical industry point of view revolves around evidence requirements and the willingness to pay for the evidence.47
For effective drug development, better alignment between stakeholders, especially with payers, is needed. This is most of all regarding clinical endpoints and clinical trial design, with ideally the acceptance of single-arm trials by payers.20 47 One can argue that while surrogate endpoints contain a level of uncertainty, granting access only based on overall survival delays access by several years.
Also more flexibility through access agreements can be a way to reduce inequalities as it allows to reflect individual affordability levels. Initiatives like ADAPT-SMART help and manage entry agreements including schemes like ‘coverage with evidence development’ can provide a more flexible and tailor-made framework to allow for earlier access while managing uncertainty.48
As timely and equal patient access needs an early dialogue of all stakeholders, including regulators, payers, governments, patients and industry, EFPIA has established a multistakeholder platform to facilitate exchange and alignment on access solutions.49
The health economist’s point of view
Looking at the bigger picture during the access process, the key challenge happens when it comes to payment decisions. Payers bear most of the risk, as new cancer products are often launched with limited information, at high prices, with a large impact on budgets. The effect of follow-up studies is small, as products are usually not taken off the market, even if validation studies are inconclusive or negative, and prices are not changed.
This equates to drug manufacturers taking an up-front payment for hope and incites some payers to delay access.
The pricing approach for targeted drugs must therefore change, and the question is whether there is a political will to do so. Much more flexible pricing is needed. If manufacturers are expected to start with low prices until confirmation studies are available, price increases need to be allowed if these studies are positive. Alternatively, prices need to be lowered or drugs de-reimbursed if the studies are negative.34
A suggested way forward with a changing order of events in the process could look as follows:
Pre-study joint consultation with EMA and HTA on the validity and validation of a surrogate endpoint.
Accelerated approval based on the surrogate endpoint.
A mandatory joint European clinical efficacy assessment (HTA).
A hypothetical cost-effectiveness model to assess the range of possible incremental cost-effectiveness ratios.
The introduction at ‘risk-limited’ price, variable depending on countries’ wealth (or one price for the whole EU, recovered at different levels from different countries).
Real-world and validation studies, jointly designed with the EMA and HTAs, could be supported by manufacturers and healthcare systems.
Evaluation of the follow-up studies and changes to price and/or reimbursements and/or market authorisation.
Recommendations from the roundtable
The members of the panel agreed on and all endorsed the following recommendations to accelerate access to innovative cancer drugs, especially in countries where delays are common:
Political and societal awareness
Promote more political and public awareness and discussion about the rising incidence of cancer and recent revolutionary innovations in cancer treatment.
Politicians and institutions should acknowledge the large differences in access to cancer care in the EU and should address these, ideally through a multistakeholder process (see next paragraph).
Early stakeholder communication and patient involvement
Timely and affordable patient access requires closer collaboration and dialogue between regulators, payers, governments, patient representatives and industry from early stages of drug development.
By working together with patient organisations, stakeholders can ensure that the patient perspective is taken into account. One example of such multistakeholder cooperation is the Mechanism of Coordinated Access to Orphan Medicinal Products (MoCA) process to improve access to orphan drugs (http://www.eurordis.org/content/moca).
Health technology assessment
Further support collaboration in HTA in the EU, that is, the clinical assessment, as outlined in the Proposal of the European Commission to reduce duplication and improve the situation in countries with less developed HTA processes.
Adaptive licensing and pricing approaches
Further evaluate approaches such as new access agreements (managed entry agreements, adaptive licensing, coverage with evidence development, etc) to secure timely patient access while addressing potential evidence uncertainties.
Clinical trials
Develop legislation on cross-border participation in clinical trials that could enable early access to effective treatments for patients and early dialogue with decision makers.
Provide patient-friendly clinical trial search engines in more languages than English and training on the terms used (such as ‘phase 3’ and ‘randomisation’).
Conclusion
There is no single simple solution according to patient groups, healthcare providers, pharmaceutical companies, regulators and HTA institutions/payers about the best way to provide seamless access, that is, efficient and effective access to cancer care. There is consensus that to ensure timely patient access, current access processes have to be adapted to keep pace with scientific developments and affordability needs. This will help to ensure that people living in EU countries with access delays and lower levels of economic development are also able to receive quality cost-effective care.
Closer collaboration and dialogue between regulators, payers, governments, patient stakeholders and industry are a first step.
Acknowledgments
We would like to thank Dr Christiane Rehwagen for her work organising the roundtable discussion and her writing and editorial support on the manuscript.
Footnotes
Twitter: @lydiamakaroff
Contributors: All authors have contributed in the roundtable discussion and to the manuscript and have reviewed and agreed on the final version.
Funding: This initiative is sponsored by the Central European Cooperative Group (CECOG) based upon educational grants by Merck Sharp & Dohme and Boehringer Ingelheim.
Competing interests: The current work of AB (for MoCA and as unpaid expert for the Austrian Federal Administrative Court) informs about payers’ attitudes/concerns regarding the reimbursement of expensive medicines. However, the opinions expressed here cannot be construed as an official position of any payer organisation. LKS received relevant financial funding for activities outside the submitted work, which are speaker’s fees from Roche, Novartis, BMS and MSD. GK received relevant financial funding outside the submitted work from MSD. AL: the views expressed in this article are the personal views of the the co-author and may not be understood or quoted as being made on behalf of or reflecting the position of the European Medicines Agency or one of its committees or working parties. LM is a paid employee of Fight Bladder Cancer UK, and a volunteer board member of the World Bladder Cancer Patient Coalition. Fight Bladder Cancer UK has received financial support from BMS, Janssen, MSD and F. Hoffman-La Roche AG. The World Bladder Cancer Patient Coalition has received financial support from AstraZeneca, Bayer, F. Hoffman-La Roche AG, Janssen, Ipsen, MSD and Photocure. AR is an employee of MSD International and owns shares of MSD. MSD has supported this meeting together with Boehringer Ingelheim. NW received project grants and consulting and speaking fees from a large number of pharmaceutical companies, including MSD. CZ received honoraria from Roche, Novartis, BMS, MSD, Imugene, Ariad, Pfizer, Merrimack/Shire, Merck KGaA, Fibrogen, AstraZeneca, Tesaro, Gilead, Servier, Eli Lilly and Amgen.
Patient consent for publication: Not required.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Joensson B, Hofmarcher T, Lindgren P, et al. Comparator report on patient access to cancer medicines in Europe revisited. IHE report, 2016. Available: https://ihe.se/wp-content/uploads/2016/08/IHE-Report_2016_4.pdf
- 2.Global Oncology Trends Advances, complexity and cost. Institute report, 2017. Available: https://www.iqvia.com/institute/reports/global-oncology-trends-2017-advances-complexity-and-cost
- 3.Booth CM, Eisenhauer EA. Progression-Free survival: meaningful or simply measurable? JCO 2012;30:1030–3. 10.1200/JCO.2011.38.7571 [DOI] [PubMed] [Google Scholar]
- 4.EFPIA European Federation of pharmaceutical industries and associations (EFPIA), 2018. Available: https://www.efpia.eu/media/412416/market-access-delays-2017-final-140318.pdf
- 5.Aapro M, Astier A, Audisio R, et al. Identifying critical steps towards improved access to innovation in cancer care: a European cancer organisation position paper. Eur J Cancer 2017;82:193–202. 10.1016/j.ejca.2017.04.014 [DOI] [PubMed] [Google Scholar]
- 6.EUPATI European patients' Academy (EUPATI), 2015. Available: https://www.eupati.eu/non-clinical-studies/discovery-development-medicines/
- 7.European Union Regulation (EC) NO 726/2004 of the European Parliament and of the Council of 31 March 2004, laying down community procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European medicines Agency. Available: https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/reg_2004_726/reg_2004_726_en.pdf
- 8.Pignatti F. Single-Arm trials in cancer drug evaluation. EMA-ESMO workshop on single-arm trials for cancer drug market access, 30thJune 2016. human medicines evaluation division, European medicines Agency. Available: https://www.ema.europa.eu/documents/presentation/presentation-single-arm-trials-cancer-drug-evaluation-problem-statement-francesco-pignatti_en.pdf
- 9.Blumenthal G. The role of single arm trials in oncology drug development. EMA-ESMO workshop on single-arm trials for cancer drug market access, 30thJune 2016. office of hematology and oncology products, U.S. food and drug administration. Available: https://www.ema.europa.eu/documents/presentation/presentation-role-single-arm-trials-oncology-drug-developmentgideon-blumenthal_en.pdf
- 10.Blumenthal GM, Pazdur R. Approvals in 2017: gene therapies and site-agnostic indications. Nat Rev Clin Oncol 2018;15:127–8. 10.1038/nrclinonc.2018.11 [DOI] [PubMed] [Google Scholar]
- 11.West H. Novel precision medicine trial designs – Umbrellas and baskets. JAMA Oncology 2017;3. [DOI] [PubMed] [Google Scholar]
- 12.Renfro LA, Sargent DJ. Statistical controversies in clinical research: basket trials, umbrella trials, and other master protocols: a review and examples. Ann Oncol 2017;28:34–43. 10.1093/annonc/mdw413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lemery S, Keegan P, Pazdur R. First FDA approval agnostic of cancer site — when a biomarker defines the indication. N Engl J Med 2017;377:1409–12. 10.1056/NEJMp1709968 [DOI] [PubMed] [Google Scholar]
- 14.FDA Fda approves an oncology drug that targets a key genetic driver of cancer, rather than a specific type of tumour, 2018. Available: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm626710.htm
- 15.European Medicines Agency Development support and regulatory tools for early access to medicines. EMA/531801/2015, 2016. Available: https://www.ema.europa.eu/documents/other/development-support-regulatory-tools-early-access-medicines_en.pdf
- 16.European Medicines Agency Prime priority medicines. Available: https://www.ema.europa.eu/en/human-regulatory/research-development/prime-priority-medicines
- 17.Bouvy JC, Sapede C, Garner S. Managed entry agreements for pharmaceuticals in the context of adaptive pathways in Europe. Front Pharmacol 2018;9:280 10.3389/fphar.2018.00280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.ADAPT-SMART Accelerated development of appropriate patient therapies – a sustainable, Multi-stakeholder approach from research to Treatment-outcomes. Available: http://adaptsmart.eu
- 19.Kemp R, Prasad V. Surrogate endpoints in oncology: when are they acceptable for regulatory and clinical decisions, and are they currently overused? BMC Med 2017;15:134 10.1186/s12916-017-0902-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.FDA Table of surrogate endpoints that were the basis of drug approval or licensure, 2018. Available: https://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/ucm613636.htm
- 21.Plourde G. Validation of surrogate endpoints for clinical trials. JPCR 2018;5 10.19080/JPCR.2018.05.555657 [DOI] [Google Scholar]
- 22.Ciani O, Buyse M, Garside R, et al. Comparison of treatment effect sizes associated with surrogate and final patient relevant outcomes in randomised controlled trials: meta-epidemiological study. BMJ 2013;346:f457 10.1136/bmj.f457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ciani O, Davis S, Tappenden P, et al. Validation of surrogate endpoints in advanced solid tumors: systematic review of statistical methods, results, and implications for policy makers. Int J Technol Assess Health Care 2014;30:312–24. 10.1017/S0266462314000300 [DOI] [PubMed] [Google Scholar]
- 24.Ciani O, Taylor RS. Surrogate, friend or foe? the need for case studies of the use of surrogate outcomes in cost-effectiveness analyses. Health Econ 2013;22:251–2. 10.1002/hec.2826 [DOI] [PubMed] [Google Scholar]
- 25.Zafar SY, Peppercorn JM, Schrag D, et al. The financial toxicity of cancer treatment: a pilot study assessing out-of-pocket expenses and the insured cancer patient's experience. Oncologist 2013;18:381–90. 10.1634/theoncologist.2012-0279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilking N, Lopes G, Meier K, et al. Can we continue to afford access to cancer treatment? European Oncology Haematology 2017;13:114–9. 10.17925/EOH.2017.13.02.114 [DOI] [Google Scholar]
- 27.Tay-Teo K, Ilbawi A, Hill SR. Comparison of sales income and research and development costs for FDA-approved cancer drugs sold by Originator drug companies. JAMA Network Open 2019;2:e186875 10.1001/jamanetworkopen.2018.6875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.European Medicines Agency Parallel consultation with regulators and health technology assessment bodies. Available: https://www.ema.europa.eu/en/human-regulatory/research-development/scientific-advice-protocol-assistance/parallel-consultation-regulators-health-technology-assessment-bodies
- 29.European Medicines Agency Adaptive pathways. Available: https://www.ema.europa.eu/en/human-regulatory/research-development/adaptive-pathways
- 30.Ermisch M, Bucsics A, Vella Bonanno P, et al. Payers' views of the changes arising through the possible adoption of adaptive pathways. Front Pharmacol 2016;7:305 10.3389/fphar.2016.00305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Institute for Clinical and Economic Review Chimeric antigen receptor T-cell therapy for B-cell cancers: effectiveness and value, 2018. Available: https://icer-review.org/wp-content/uploads/2017/07/ICER_CAR_T_Final_Evidence_Report_032318.pdf
- 32.Jönsson B, Hofmarcher T, Lindgren P, et al. The cost and burden of cancer in the European Union 1995–2014. Eur J Cancer 2016;66:162–70. 10.1016/j.ejca.2016.06.022 [DOI] [PubMed] [Google Scholar]
- 33.OECD/European Union “Health expenditure per capita”, in Health at a Glance: Europe 2016: State of Health in the EU Cycle, OECD Publishing, Paris, 2016. Available: 10.1787/health_glance_eur-2016-33-en [DOI]
- 34.Hofmacher T, Brådvik G, Lindgren P, et al. Comparator report on cancer in the Nordic countries – disease burden, costs and access to medicines. IHE Rapport. IHE: Lund, 2019. [Google Scholar]
- 35.Eigentler TK, Caroli UM, Radny P, et al. Palliative therapy of disseminated malignant melanoma: a systematic review of 41 randomised clinical trials. Lancet Oncol 2003;4:748–59. 10.1016/S1470-2045(03)01280-4 [DOI] [PubMed] [Google Scholar]
- 36.Ugurel S, Röhmel J, Ascierto PA, et al. Survival of patients with advanced metastatic melanoma: the impact of novel therapies–update 2017. Eur J Cancer 2017;83:247–57. 10.1016/j.ejca.2017.06.028 [DOI] [PubMed] [Google Scholar]
- 37.Long GV, Atkinson V, Lo S, et al. Combination nivolumab and ipilimumab or nivolumab alone in melanoma brain metastases: a multicentre randomised phase 2 study. Lancet Oncol 2018;19:672–81. 10.1016/S1470-2045(18)30139-6 [DOI] [PubMed] [Google Scholar]
- 38.Kandolf Sekulovic L, Peris K, Hauschild A, et al. More than 5000 patients with metastatic melanoma in Europe per year do not have access to recommended first-line innovative treatments. Eur J Cancer 2017;75:313–22. 10.1016/j.ejca.2017.01.012 [DOI] [PubMed] [Google Scholar]
- 39.Kandolf Sekulovic L, Guo J, Agarwala S, et al. Access to innovative medicines for metastatic melanoma worldwide: melanoma world Society and European association of Dermato-oncology survey in 34 countries. Eur J Cancer 2018;104:201–9. 10.1016/j.ejca.2018.09.013 [DOI] [PubMed] [Google Scholar]
- 40.European Cancer Patient Coalition Value of lnnovation in oncology. Brussels, Belgium: European cancer patient coalition, 2017. Available: www.ecpc.org/innovation
- 41.European Network for Health Technology Assessment Joint action 3, work package 7: national implementation and impact. Available: https://www.eunethta.eu/ja3-archive/work-package-7-national-implementation-and-impact/
- 42.European Union Innovative payment models for high-cost innovative medicines: report of the expert panel on effective ways of investing in health (EXPH). European Union, 2018. Available: https://ec.europa.eu/health/expert_panel/sites/expertpanel/files/docsdir/opinion_innovative_medicines_en.pdf
- 43.Basch E. Patient-Reported outcomes: an essential component of oncology drug development and regulatory review. Lancet Oncol 2018;19:595–7. 10.1016/S1470-2045(18)30141-4 [DOI] [PubMed] [Google Scholar]
- 44.De Lorenzo F, Apostolidis K, Florindi F, et al. Improving European policy to support cancer survivors. J Cancer Policy 2018;15:72–5. 10.1016/j.jcpo.2018.01.004 [DOI] [Google Scholar]
- 45.Kimberly LL, Beuttler MM, Shen M, et al. Pre-approval access terminology: a cause for confusion and a danger to patients. Ther Innov Regul Sci 2017;51:494–500. [DOI] [PubMed] [Google Scholar]
- 46.Wise J, Möller A, Christie D, et al. The positive impacts of real-world data on the challenges facing the evolution of biopharma. Drug Discov Today 2018;23:788–801. 10.1016/j.drudis.2018.01.034 [DOI] [PubMed] [Google Scholar]
- 47.Eichler H-G, Baird LG, Barker R, et al. From adaptive licensing to adaptive pathways: delivering a flexible life-span approach to bring new drugs to patients. Clin Pharmacol Ther 2015;97:234–46. 10.1002/cpt.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Towse A, Garrison LP. Canʼt get no satisfaction? will pay for performance help? Pharmacoeconomics 2010;28:93–102. 10.2165/11314080-000000000-00000 [DOI] [PubMed] [Google Scholar]
- 49.Fighting Cancer EFPIA oncology platform. Available: https://www.efpia.eu/about-medicines/use-of-medicines/disease-specific-groups/fighting-cancer/