

Abstract
Prion diseases in mammals are caused by a conformational transition of the cellular prion protein from its native conformation (PrPC) to a pathological isoform called “prion protein scrapie” (PrPSc). A molecular level of understanding of this conformational transition will be helpful in unveiling the disease etiology. Experimental structural biological techniques (NMR and X‐ray crystallography) have been used to unravel the atomic level structural information for the prion and its binding partners. More than one hundred three‐dimensional structures of the mammalian prions have been deposited in the protein databank. Structural studies on the prion protein and its structural transitions will deepen our understanding of the molecular basis of prion pathogenesis and will provide valuable guidance for future structure‐based drug discovery endeavors.
Keywords: prion, mis‐folding, neurodegenerative diseases, prion protein
Abbreviations
- NMR
nuclear magnetic resonance spectroscopy
- PDB
protein data bank
- TSEs
stransmissible spongiform encephalopathies
1. INTRODUCTION
Prion diseases or transmissible spongiform encephalopathies (TSEs) include Creutzfeldt–Jakob disease, Gerstmann–Sträussler–Scheinker syndrome, and fatal familial insomnia in humans, bovine spongiform encephalopathy in cattle, scrapie in goats and sheep, and chronic wasting disease in cervids.1 The “protein‐only” hypothesis posits that the key event in the prion disease pathogenesis occurs when the cellular prion protein (PrPC) undergoes a conformational transition from the mainly α‐helix‐rich folded structure into an infectious and pathogenic β‐sheet‐rich conformer (PrPSc). Importantly, PrPSc possesses abnormal physiological properties such as resistance to proteolytic degradation, relative insolubility, and the propensity to polymerize into scrapie agents.2, 3 In the last three decades, prion diseases have received tremendous attention from the scientific community because of their unique properties in disease transmission, pathophysiology as well as the ambiguous roles that they have in several physiological processes.
Prion diseases fall into three main categories; sporadic, acquired, and genetic forms.2 The genetic forms of the prion diseases are caused by point mutations in the prion gene.4 The sporadic or spontaneous forms of prion diseases have no cause ascertained as yet; at least some cases may represent unrecognized transmission or may be caused by somatic mutations in the PRNP gene that encodes PrP. Healthy organisms carrying the cellular PrPC may develop infectious TSE following inoculation with transmissible biological material containing PrPSc.5 Prions transmit efficiently between organisms of the same species, whereas there are “species barriers” that limit interspecies transfer. Species barriers may abolish completely or may suppress prion transmission by having a longer incubation time.6 There have been numerous prion strains identified having unique structural, biochemical, and physiological properties.7
The cellular prion protein is expressed almost ubiquitously throughout the human bodily tissues but larger quantities are present in the central nervous system. The only function of PrPC that is well‐understood at the molecular level is its role as the physiological ligand of the G protein‐coupled receptor, Gpr126.8 Studies involving PrPC‐knockout mouse models have suggested an involvement of this protein in several behavioral and physiological processes involving regulation of circadian rhythm, synaptic function conservation, and peripheral myelin maintenance.9, 10, 11 The cellular PrPC physiology is associated with maintaining a balance of the divalent metal ions Cu2+ and Zn2+ in the central nervous system.12, 13 Four octapeptide‐repeat amino acid sequences present in the N‐terminal region of PrPC binds the metal ions through molecular coordination.14 PrPC is increasingly being recognized as a promiscuous regulator of neuronal signaling; PrPC has been shown to interact with, and to modulate, a surprisingly large number of membrane receptors, including ionotropic and metabotropic glutamate receptors, ion channels, and amino acid transporters.15 Functional interactions with PrPC have recently been reported for the α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid receptor and the N‐methyl‐d‐aspartate glutamate receptor, as well as the Type 1 metabotropic glutamate receptors mGluR1 and mGluR5.16, 17, 18 In addition, PrPC has been shown to regulate glutamate uptake by the excitatory amino acid transporter EAAT3 that further supports it's active role in neurotransmission and protection from excitotoxicity.19
In this review, we provide an overview of the structure–function relationship of the mammalian prion proteins, its fragments, and the scrapie prions. Structural data have been obtained through various experimental techniques: X‐ray crystallography, nuclear magnetic resonance spectroscopy, cryo‐electron microscopy, and CD spectroscopy to name a few. There has been a consensus on the overall structural fold as experimentally determined for the cellular prion proteins; the N‐terminal region consists of a long unstructured, flexible part whereas the C‐terminus contains the folded, structured domain. Several unique structural features have been discovered over past years by the use of complementary structural biology tools and together those provide plausible answers for the crucial misfolding process.
1.1. Solution state structural studies by nuclear magnetic resonance spectroscopy
In vivo prion proteins exist as extracellular glycosylphosphatidylinositol (GPI)‐anchored glycoproteins composed of 209 residues after the enzymatic removal of the N‐terminal 22 amino acid long signal peptide sequence.20 The majority of the structural studies have been carried out with either the recombinant full‐length protein or a truncated PrPC that lack both the glycans at the two glycosylation sites and the GPI‐anchor.
Prior to the structural resolution of PrPC by nuclear magnetic resonance (NMR), it was thought that PrPC would consist of four alpha helices.21 According that model, these helices would give rise to stacked beta sheets during the conversion into PrPSc. However, the first NMR spectroscopic “in solution” structure of the moPrPC (mouse prion protein) revealed a completely different structure. Surprisingly, PrPC was found to contain two parts, each having different structural and dynamical features (Figure 1a).22, 23 It consisted of a highly flexible N‐terminal segment encompassing residues 23–124 and a folded C‐terminal structured domain comprising residues ~125–231. The globular, folded domain contains the structural motifs of three α‐helices and two short antiparallel β‐strands; residues 144–154 (helix α1), 173–194 (helix α2), 200–228 (helix α3), and residues 128–131 (strand β1) and 161–164 (strand β2), respectively. A disulfide bond between Cys179 and Cys214 covalently links two of the helical structures, α2 and α3. Due to the discrepancies in the amino acid numbering scheme between the human and the mouse prion protein, the human prion protein amino acid numbering has been adopted in this review. NMR spectroscopic structure determinations of other mammalian species show similar overall folding.
Figure 1.
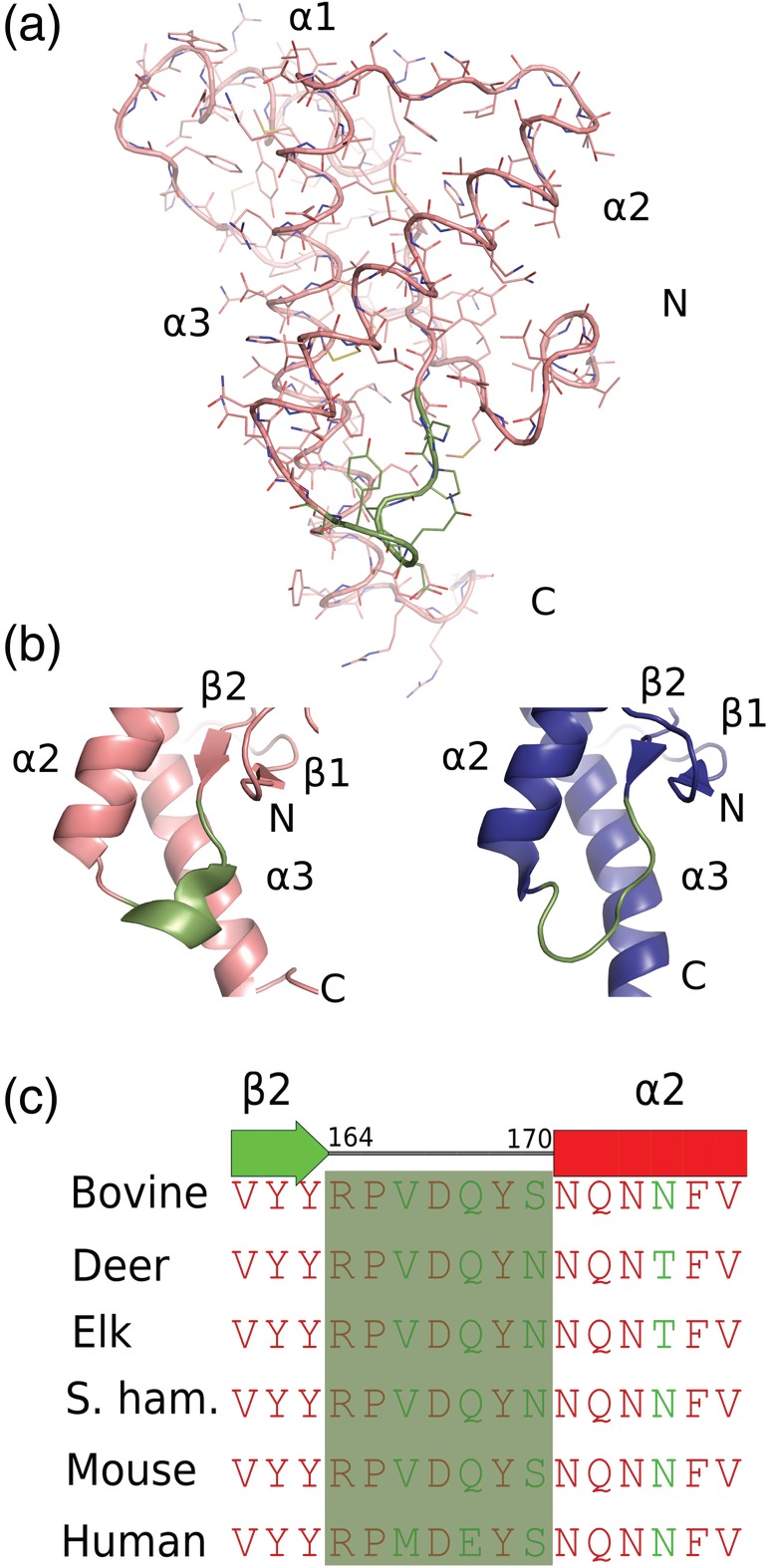
(a) The nuclear magnetic resonance (NMR) structure of moPrP(121–231; PDB ID 2L39) represented as a ribbon model with side chains as sticks (b) the β2–α2 loops are shown as cartoon from mouse NMR structures (PDB codes 2L39 and 2L40). A 310‐helical turn conformation is shown in teal color (PDB:2L39), whereas (PDB:2L40) shown in deep blue adopts a Type I β‐turn conformation. (c) Amino acid sequence alignment for the polypeptide segment of the β2–α2 loops of bovine, deer, elk, S. hamster, mouse, and human. The red color indicates identical residues whereas the green color shows the amino acid substitutions. The β2–α2 loop segments are shown in a transparent green color
Highly conserved amino acid sequences among the mammalian prion proteins of different species adopt similar tertiary structures; however, some structural variations have been observed at the interface of the β2–α2 loop and in the C‐terminal part of helix α3.24 Structural mapping of these sequence variations amongst mammalian prion proteins has revealed that the loop between strand β2 and helix α2 contains a greater number of differing residues compared to rest of the protein.25 Species‐specific variations of the conformational ensembles sampled by the β2–α2 loop have also been observed.26 In the mouse prion protein, the β2–α2 loop adopts a 310‐helical conformation, with only a minor population forming a Type I β‐turn.27 The exchange between the 310‐helical conformation and a Type I β‐turn occurs on the millisecond to microsecond timescale at physiological temperatures. NMR spectroscopic analysis of PrPC (121–231) from elk, deer, bank vole, rabbit, wallaby, and horse revealed a well‐defined 310‐helix for these species at 20°C; whereas all other mammalian PrPC species show a disordered Type I β‐turn at this temperature by NMR.25, 28 Due to the inherent flexible nature of the prion proteins, NMR parameters collected during the structure determination undergo different spatial and temporal averaging. Therefore, some polypeptide segments show reduced precision of the structure determination such as the C‐terminal parts of the helices α2 and α3, and the loop linking β‐strand β2 and the helix α2.29
The β2–α2 loop adopts only a 310‐helical conformation in the prion protein structures determined by X‐ray crystallography. Human prion protein crystal structures that have been determined with the help of different antibody Fab fragments such as POM1 Fab30 and ICSM18 Fab31 as well as with nanobody Nb484,32 show similar β2–α2 loop conformations (Figure 2a–d). These crystal structures have been determined under different crystallization conditions and the protein crystals adopted different space groups. However, in the human prion protein that has been determined without a binding partner, a unique 310‐helical conformation was observed.33 In this conformation, the side chain of Tyr169 is flipped outwards into the aqueous solution and does not make any contacts with other residues from the loop region. This residue has been considered one of the key residues that is responsible for imparting structural stability to the β2–α2 loop region by making hydrogen bonding interactions with Asp178 on helix α2 and through hydrophobic contacts.27, 34 This region of the human prion protein in the unliganded conformation makes extensive crystal contacts with a neighboring prion molecule. Whether the unique 310‐helical conformation observed in this human prion protein is the result of the crystal‐lattice contacts or whether it is the result of the flexible nature of this part of the prion protein remains a point of discussion. Nevertheless, none of the prion crystal structures, adopt the Type‐I β‐turn conformation reported by the NMR. In several of the NMR structures of the mouse prion protein, this loop is reported to have different conformations (Figure 2e–h). In these several loop conformations, Tyr169 is reported to adopt multiple conformations, from a completely buried state to a fully exposed state and in many states in between.24, 27
Figure 2.
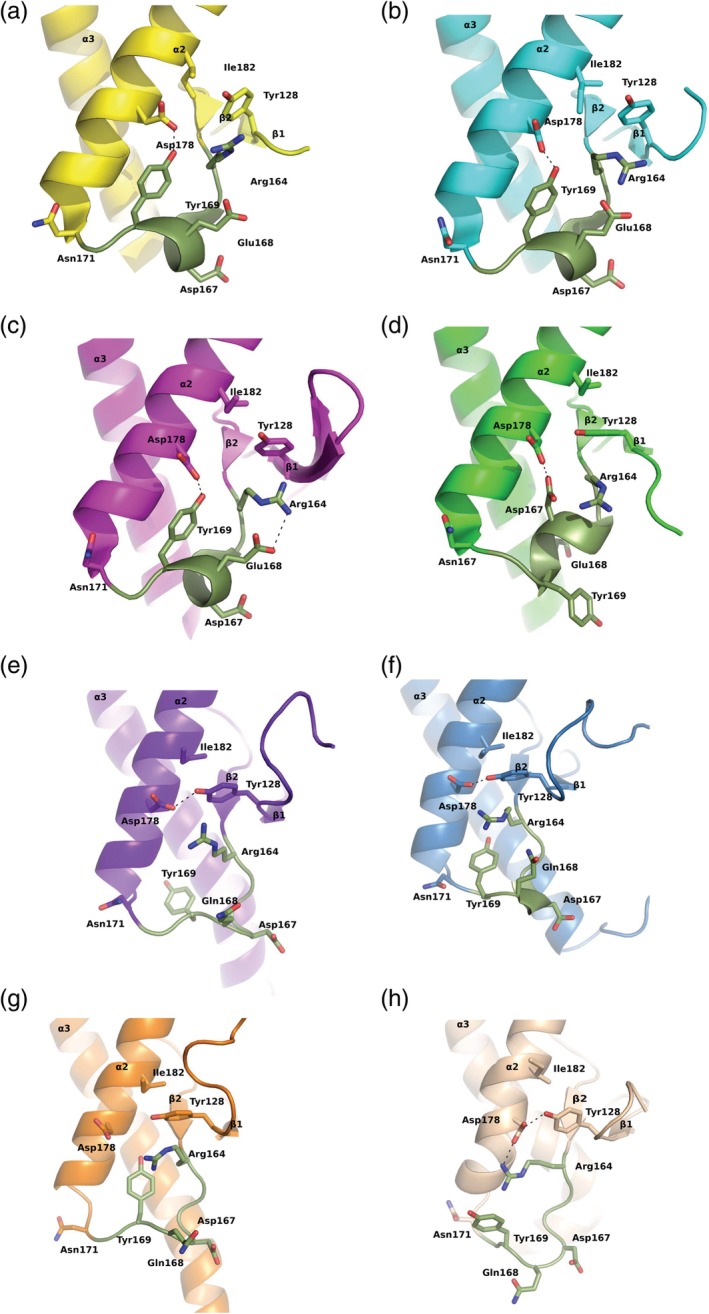
Different conformations of the β2–α2 loop observed by X‐ray crystallography and by nuclear magnetic resonance (NMR) spectroscopic techniques. Several of the key side chains in that region are shown as stick models and hydrogen bonding interactions in black dashed lines. (a) The human prion protein in complex with POM1 Fab crystal structure (yellow); Tyr128, Asp178 form a hydrogen bond (PDB ID:4DGI); (b) human prion protein in complex with ICSM18 Fab crystal structure (cyan), two hydrogen bonding interactions between Tyr169:Asp178 and Arg164:Glu168 (PDB ID:2W9E); (c) human prion protein in complex with Nb484 nanobody (magenta) two hydrogen bonding interactions between Tyr169:Asp178 and Arg164:Glu168 (PDB ID:4KML); (d) human prion protein crystal structure without any binding partner (green), hydrogen bonding interaction between Asp178 and Asp167; (e) mouse prion NMR structure (purple; PDB ID:1XYX), Tyr128 is hydrogen bonded to Asp178, and Tyr169 is fully buried; (f) mouse prion nuclear magnetic resonance (NMR) structure (blue; PDB ID:2L39) Tyr128 is hydrogen bonded with Asp178, Tyr 169 is partially buried, with an altered loop conformation; (g) mouse prion NMR structure (orange; PDB ID:2L1H) no hydrogen bonding interactions among Tyr128, Tyr169, and Asp178; (h) mouse prion NMR structure (wheat; PDB ID:1AG2), hydrogen bonds are present between Tyr128:Asp178 and Asp178:Arg164, and Tyr169 is completely surface exposed. The β2–α2 loop segments are shown in deep green color
Prion transmissibility between different species is dependent on the β2–α2 loop conformations.26 It was shown that inoculation with PrPSc from a species with a similar β2–α2 loop structure results in efficient transmission of prion disease in a transgenic mouse expressing mammalian prion proteins.35 On the other hand, those species having dissimilar β2–α2 loop structures exhibited large prion transmission barriers. Few amino acid changes in the β2–α2 loop at residues 170 and 174 could lead to a rigid loop in the mouse and exhibit resistance to transmission when challenged with normal brain homogenate (Figure 1c).26 It has also been suggested that the β2–α2 loop is part of a recognition site for an unknown binding partner “Protein X” that would regulate the conversion from PrPC to PrPSc.36
1.2. Three‐dimensional structure determination by X‐ray crystallography
The first three‐dimensional (3D) atomic structure of the wild‐type (WT) recombinant huPrPC determined by X‐ray crystallography was reported in 2001 (Figure 3a).37 This structure revealed an unexpected domain‐swapped dimer; helix α3 that packs against helix α2 through an intrasubunit Cys179‐Cys214 disulfide bond is shared with the accompanying monomer of the dimer in this structure. Domain swapping possibilities of human prion protein suggested that the dimeric oligomerization event may be the initial event underlying the conversion of PrPC into the pathological PrPSc. Interestingly, similar phenomena have also been implicated in other amyloidogenic protein oligomerization events.38 Apparently for the swapping of helix 3 to occur,it would require reduction of the disulfide bond then re‐oxidation of thedisulfide to form the helix‐swapped dimer.
Figure 3.
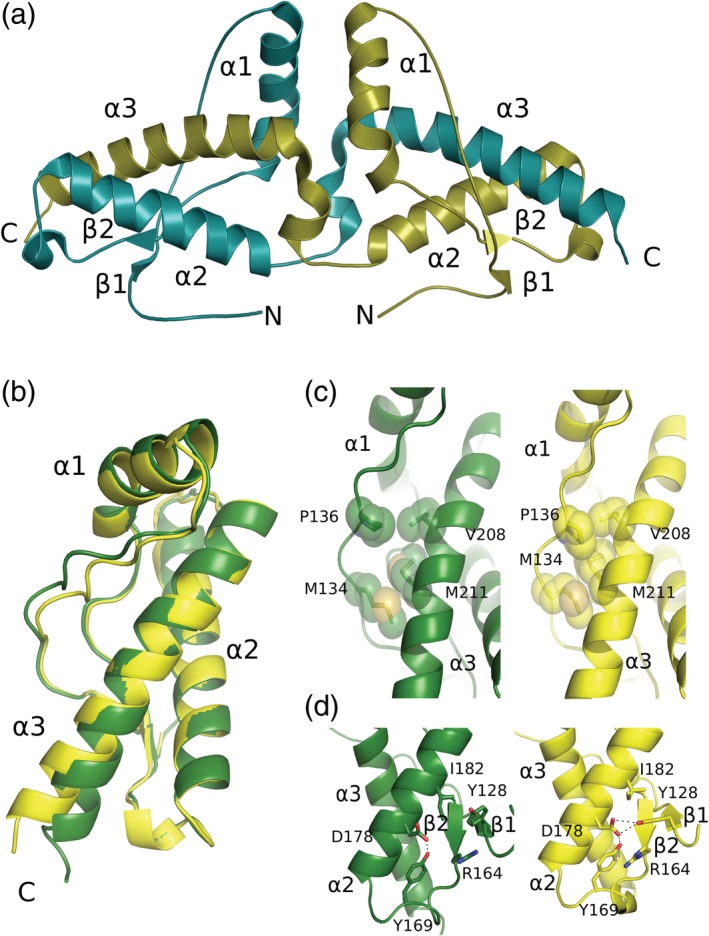
(a) Cartoon representation of the human prion protein dimer crystal structure (PDB ID: 1I4M). The two peptide chains are shown in olive and deep teal, with their N‐ and C‐termini labeled. (b) Structural superposition of the moPrP from the protein complexes of moPrP:POM6 Fab in yellow and moPrP:POM1 Fab in green (PDB ID: 4H88 and 6AQ7). (c) Hydrophobic contacts between residues of the β1‐α1 loop regions and helix α3. The residues involved in the contact are represented as sticks and spheres. The right panel shows the contacts in moPrP from moPrP: POM6 Fab whereas the left panel shows the contacts from moPrP from moPrP: POM1 Fab. (d) In the right panel of moPrP: POM6 Fab, hydrogen bonds are shown as dashed lines; Tyr128, Asp178, Arg164, and Tyr169 form a hydrogen bond network whereas in the moPrP: POM1 Fab crystal structure (green) this network is disrupted, in particular Tyr128 lacks a hydrogen bond to Asp178
Several crystal structures of the folded domain of the prion protein have been determined as the monomeric domain. WT huPrPC containing V129, and four of the familial variants D178N and F198S, containing either M129 or V129 were determined by the same research group.33 Structural comparisons of these variants with one another and with the previously published WT huPrPC structures containing M129 revealed that only WT huPrPCs were found to crystallize as domain‐swapped dimers or closed monomers. The four mutant huPrPCs crystallized as nonswapped dimers. Three of the four mutant huPrPC species aligned through the crystal contacts to form intermolecular beta‐sheets involving the M/V129. The common methionine/valine polymorphism at residue 129 in the human prion protein has been known to influence the disease susceptibility and phenotype.39, 40 In the crystallographic structure of rabbit PrPC, a key helix‐capping motif (residues 170–174) has been identified as leading to higher resistance to prion disease transmission in rabbits.41 One of the important residues, Asn171 makes hydrogen‐bonding interactions to the main chain amide functional group of Asn174 through the carbonyl group of its side chain. In the several human prion protein crystal structures they all have similar orientations having the side chain of Asn171 orientated toward the N‐terminal part of helix α2 (Figure 2). Whereas this residue seems to adopt very different conformations in the mouse prion proteins as observed through NMR spectroscopic techniques. Experimental evidence suggests that mammals vary in their prion disease susceptibility; hamsters and mice show relatively high susceptibility, whereas rabbits, horses, and dogs show a greater resistance to prion transmission.42 This susceptibility of some prion proteins has been associated with the intrinsic propensity of mammalian PrPC to convert from the native, α‐helical state to a cytotoxic β‐structured state that exists in a monomer‐octamer equilibrium. This capping motif increases the β‐structure folding propensity of mouse, a species more susceptible to prion disease.
Crystallization of the unbound prion proteins presents a tremendous challenge due to the intrinsic flexibility of the structured domain.43 The long flexible N‐terminal part of the molecule could also affect the crystal contacts between the monomers. So, many of the mammalian prion protein crystal structures have been determined by making complexes of the prion proteins with the Fab fragments of anti‐prion antibodies such as VRQ14, ICSM18, POM1, and POM6.30, 44, 45, 46 These anti‐prion monoclonal antibodies were being developed to act as therapeutic agents against the prion diseases by stabilizing the structured domain of the prion protein. Surprisingly, many of the C‐terminal specific antibodies exhibited highly toxic behavior in biological experiments.47 POM1 was found to be a highly toxic antibody that recognizes noncontiguous prion epitopes; the N‐terminal part of helix α1, the loop structure joining strand β1 and helix α1, and few residues from helix α3.30 The binding epitopes of an innocuous antibody POM6 and the purportedly therapeutic antibody ICSM18 are nonoverlapping but adjacent to the prion epitope recognized by POM1 Fab.45, 46 Structural comparisons of POM1 Fab: moPrP and POM6 Fab: moPrP complexes reveal subtle structural changes in the folded domain that could lead to the toxic misfolding process.46 Several differences in hydrogen bonding interactions among residues from the β2‐α2 loop and Y128 from strand‐β1 have been observed along with the differences in the hydrophobic residue packing between helix α3 and the loop joining strand β1 and helix α1. The N‐terminal region‐specific antibodies have been found to be therapeutic and prevent the scrapie challenge in POSCA (prion organotypic slice culture assay).47 A high‐resolution crystal structure of POM2 Fab has been determined bound to one octa‐peptide amino acid segment from the four octa‐peptide repeats present in the N‐terminal segment of the prion protein.48
Several small molecules have been found to act as anti‐prion therapeutics; they belong to a wide range of chemical groups.49 These compounds are known to reduce the scrapie burden in cell‐based assays, but their effectiveness has never been confirmed in clinical trials.50 Our group has managed to produce 3D crystal structures of promazine and chlorpromazine bound to the mouse prion protein.51 The phenothiazine tricyclic small molecule complexes have been determined with help of the POM1 Fab bound to the prion protein. These tricyclic anti‐prion compounds bind to a hydrophobic pocket at the interface of the β1‐β2 region and helix α3 while inducing the formation of another antiparallel β strand preceding strand β1 (Figure 4a). These anti‐prion small molecules seem to provide stability to several suspected misfolding initiator motifs in the prion protein. The hydrophobic amino acid side chain substitution of a protective G127V variant prion protein is also found in the same binding region as the phenothiazine compounds and perhaps exerts a similar stabilization effect on the folded domain. Surprisingly, in the crystal structure of huPrPC determined with a therapeutic nanobody (Nb484), the N‐terminally conserved hydrophobic motif AGAAAAGA (residues 113–120) makes a three‐stranded antiparallel β‐sheet with the β1 and β2 strands comparable to the one that the phenothiazine bound mouse prion structure exhibits.32 These observations indicate a unique structural arrangement of β0‐β1‐α1‐β2‐α2‐α3 as compared to the canonical β1‐α1‐β2‐α2‐α3 prion‐like fold.
Figure 4.
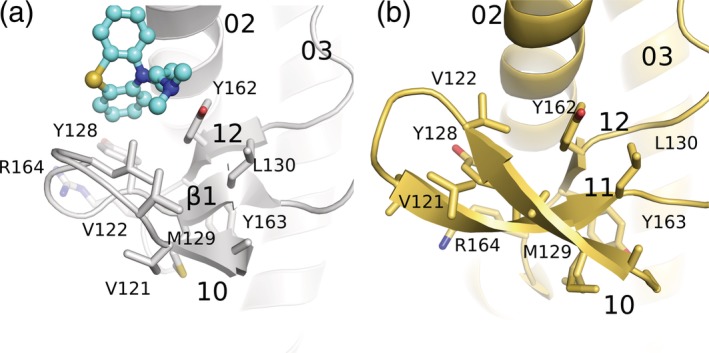
Formation of an additional β‐strand antiparallel to β1‐β2 at the N‐terminal region of the folded domain of the prion protein. (a) Cartoon representation of the N‐terminal region of moPrP in the promazine‐bound structure (PDB ID: 4MA7) shown in a gray color with side chains represented as stick models. The bound promazine is represented as a ball and stick model and in cyan color. (b) Cartoon representation of the N‐terminal part of a nanobody bound to huPrP (PDB ID:4KML) shown in a yellow color. Promazine and the nanobody Nb484 cause similar structural rearrangements of the N‐terminal region forming an additional β‐strand anti‐parallel to strand β1
1.3. Structural models for the scrapie agents
Aggregated, infectious PrPSc poses challenges for X‐ray crystallization and NMR spectroscopy that are attributable to its unique properties. Due to the lack of high‐resolution three‐dimensional structural data for PrPSc not much information is available about the detailed architecture of the scrapie agents. Although PrPC and PrPSc have the same amino acid sequences, they exhibit huge differences in their structural properties. Various low‐resolution structural techniques such as Fourier transform infrared and circular dichroism spectroscopy have revealed a significant content of β‐sheet for the PrPSc contrary to the α‐helical nature of the PrPC.52 A number of structural models have been proposed from different groups based on their experimental observations; a β‐helix model, the β‐spiral model, and the extended in‐register β‐sheet model are the most popular ones.53, 54, 55 The β‐helix model has been proposed based on electron microscopic studies, in which the N‐terminal region of PrPC undergoes major refolding to form left‐handed β‐helices stacked in a trimeric arrangement, whereas the α‐helical structure of the C‐terminal part is unaffected.53 The β‐spiral model has been proposed, based on molecular dynamics simulation studies that indicated extended sheets comprised of β‐strands. The C‐terminal helical structure is still preserved in the β‐spiral model.54 The extended in‐register β‐sheet model has been suggested based on the observations from experimental techniques such as site‐directed spin labeling in combination with electron paramagnetic resonance and hydrogen/deuterium exchange coupled with mass spectrometry.56 This model exhibits striking differences compared to the β‐helix and β‐spiral models. The folded domain undergoes complete transformation into a β‐strand structure. The extended in‐register β‐sheet model proposes that the C‐terminal residues from a single molecule align on top of one another forming parallel β‐strands. For this in‐register intermolecular arrangement, the helical nature of the structured domain is completely lost. A four‐rung β‐solenoid architecture has recently been proposed for PrPSc based on cryo‐electron microscopy and 3D reconstruction experiments (Figure 5).57 The average molecular height of each PrPSc molecule along the fibril axis was taken to be ~19.2 Å; in this proposal two monomers lie in a potential head‐to‐head or tail‐to‐tail configuration.58 In a recent paper on the atomistic model of the scrapie prion structure, the four‐rung β‐solenoid model has been generated based on different experimental data and computational techniques.59 In this model, the inner core of the model is constituted by mainly hydrophobic or mildly polar side‐chains whereas charged residues are exposed to the outside. Molecular dynamic simulation analysis on this model also confirmed a stability similar to the HET‐S prion from yeast. However, high‐resolution experimental structural data on the molecular arrangement of the scrapie are still lacking.
Figure 5.
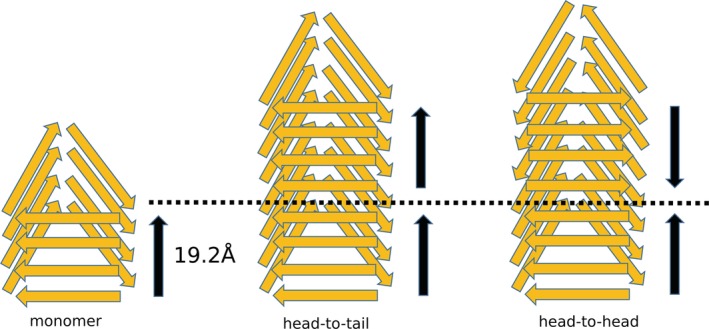
Proposed four‐rung β‐solenoid representations of PrPSc amyloid fibrils possible head‐to‐tail and head‐to‐head (or tail‐to‐tail) architectures. The measured length of the monomer parallel to the fibril axis is 19.2 Å. A head‐to‐tail stacking architecture would lead to a polar fibril while a head‐to‐head (or tail‐to‐tail) architecture would give rise to a nonpolar fibril
A combination of high‐resolution and low‐resolution experimental techniques has been very useful in gathering structural information on prion proteins and the scrapie agents. Such knowledge, along with the results from numerous biological experiments provides vital information regarding the structure–function relationship of a highly flexible protein. Despite the tremendous progress made in our understanding of prion biology, a great deal of structural detail is still lacking. Structural analysis on the prion misfolded intermediates and on the oligomeric structures will prove hugely valuable in piecing together the PrPc to PrPSc conversion map.
ACKNOWLEDGMENTS
This work has been funded by PrioNet Canada and APRI/ALMA in grants to M.N.G.J. AA is the recipient of an Advanced Grant of the European Research Council (Prion2020), the Swiss National Foundation, the Clinical Research Priority Programs “Small RNAs” and “Human Hemato‐Lymphatic Diseases”, and SystemsX.ch.
Baral PK, Yin J, Aguzzi A, James MNG. Transition of the prion protein from a structured cellular form (PrPC) to the infectious scrapie agent (PrPSc). Protein Science. 2019;28:2055–2063. 10.1002/pro.3735
Funding information Alberta Innovates, Grant/Award Number: 201600017; Clinical Research Priority Programs; Swiss National Foundation; European Research Council (Prion2020); APRI/ALMA; PrioNet Canada
REFERENCES
- 1. Aguzzi A, Polymenidou M. Mammalian prion biology: One century of evolving concepts. Cell. 2004;116:313–327. [DOI] [PubMed] [Google Scholar]
- 2. Collinge J. Prion diseases of humans and animals: Their causes and molecular basis. Annu Rev Neurosci. 2001;24:519–550. [DOI] [PubMed] [Google Scholar]
- 3. Prusiner SB. Molecular biology of prion diseases. Science. 1991;252:1515–1522. [DOI] [PubMed] [Google Scholar]
- 4. Lloyd SE, Mead S, Collinge J. Genetics of prion diseases. Curr Opin Genet Dev. 2013;23:345–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Weissmann C, Enari M, Klohn PC, Rossi D, Flechsig E. Transmission of prions. Proc Natl Acad Sci U S A. 2002;99:16378–16383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Raymond GJ, Bossers A, Raymond LD, et al. Evidence of a molecular barrier limiting susceptibility of humans, cattle and sheep to chronic wasting disease. EMBO J. 2000;19:4425–4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science. 2007;318:930–936. [DOI] [PubMed] [Google Scholar]
- 8. Küffer A, Lakkaraju AKK, Mogha A, et al. The prion protein is an agonistic ligand of the G protein‐coupled receptor Adgrg6. Nature. 2016;536:464–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Steele AD, Lindquist S, Aguzzi A. The prion protein knockout mouse: A phenotype under challenge. Prion. 2007;1:83–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tobler I, Gaus SE, Deboer T, et al. Altered circadian activity rhythms and sleep in mice devoid of prion protein. Nature. 1996;380:639–642. [DOI] [PubMed] [Google Scholar]
- 11. Collinge J, Whittington MA, Sidle KCL, et al. Prion protein is necessary for normal synaptic function. Nature. 1994;370:295–297. [DOI] [PubMed] [Google Scholar]
- 12. Pauly PC, Harris DA. Copper stimulates endocytosis of the prion protein. J Biol Chem. 1998;273:33107–33110. [DOI] [PubMed] [Google Scholar]
- 13. Perera WS, Hooper NM. Ablation of the metal ion‐induced endocytosis of the prion protein by disease‐associated mutation of the octarepeat region. Curr Biol. 2001;11:519–523. [DOI] [PubMed] [Google Scholar]
- 14. Hooper NM, Taylor DR, Watt NT. Mechanism of the metal‐mediated endocytosis of the prion protein. Biochem Soc Trans. 2008;36:1272–1276. [DOI] [PubMed] [Google Scholar]
- 15. Wulf MA, Senatore A, Aguzzi A. The biological function of the cellular prion protein: An update. BMC Biol. 2017;15:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. You H, Tsutsui S, Hameed S, et al. Aβ neurotoxicity depends on interactions between copper ions, prion protein, and N‐methyl‐D‐aspartate receptors. Proc Natl Acad Sci USA. 2012;109:1737–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Haas LT, Salazar SV, Kostylev MA, Um JW, Kaufman AC, Strittmatter SM. Metabotropic glutamate receptor 5 couples cellular prion protein to intracellular signalling in Alzheimer's disease. Brain. 2016;139:526–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Goniotaki D, Lakkaraju AKK, Shrivastava AN, et al. Inhibition of group‐I metabotropic glutamate receptors protects against prion toxicity. PLoS Pathog. 2017;13:e1006733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Guitart K, Loers G, Schachner M, Kleene R. Prion protein regulates glutathione metabolism and neural glutamate and cysteine uptake via excitatory amino acid transporter 3. J Neurochem. 2015;133:558–571. [DOI] [PubMed] [Google Scholar]
- 20. Aguzzi A, Baumann F, Bremer J. The prion's elusive reason for being. Annu Rev Neurosci. 2008;31:439–477. [DOI] [PubMed] [Google Scholar]
- 21. Gasset M, Baldwin MA, Lloyd DH, et al. Predicted alpha‐helical regions of the prion protein when synthesized as peptides form amyloid. Proc Natl Acad Sci U S A. 1992;89:10940–10944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Riek R, Hornemann S, Wider G, Billeter M, Glockshuber R, Wuthrich K. NMR structure of the mouse prion protein domain PrP(121‐231). Nature. 1996;382:180–182. [DOI] [PubMed] [Google Scholar]
- 23. Riek R, Hornemann S, Wider G, Glockshuber R, Wüthrich K. NMR characterization of the full‐length recombinant murine prion protein, mPrP(23‐231). FEBS Lett. 1997;413:282–288. [DOI] [PubMed] [Google Scholar]
- 24. Gossert AD, Bonjour S, Lysek DA, Fiorito F, Wuthrich K. Prion protein NMR structures of elk and of mouse/elk hybrids. Proc Natl Acad Sci U S A. 2005;102:646–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lysek DA, Schorn C, Nivon LG, et al. Prion protein NMR structures of cats, dogs, pigs, and sheep. Proc Natl Acad Sci U S A. 2005;102:640–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sigurdson CJ, Nilsson KPR, Hornemann S, et al. A molecular switch controls interspecies prion disease transmission in mice. J Clin Invest. 2010;120:2590–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Damberger FF, Christen B, Perez DR, Hornemann S, Wuthrich K. Cellular prion protein conformation and function. Proc Natl Acad Sci U S A. 2011;108:17308–17313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Perez DR, Damberger FF, Wuthrich K. Horse prion protein NMR structure and comparisons with related variants of the mouse prion protein. J Mol Biol. 2010;400:121–128. [DOI] [PubMed] [Google Scholar]
- 29. Zahn R, Liu A, Lührs T, et al. NMR solution structure of the human prion protein. Proc Natl Acad Sci U S A. 2000;97:145–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Baral PK, Wieland B, Swayampakula M, et al. Structural studies on the folded domain of the human prion protein bound to the fab fragment of the antibody POM1. Acta Cryst. 2012;D68:1501–1512. [DOI] [PubMed] [Google Scholar]
- 31. Nicoll AJ, Trevitt CR, Tattum MH, et al. Pharmacological chaperone for the structured domain of human prion protein. Proc Natl Acad Sci U S A. 2010;107:17610–17615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Abskharon RN, Giachin G, Wohlkonig A, et al. Probing the N‐terminal beta‐sheet conversion in the crystal structure of the human prion protein bound to a nanobody. J Am Chem Soc. 2014;136:937–944. [DOI] [PubMed] [Google Scholar]
- 33. Lee S, Antony L, Hartmann R, et al. Conformational diversity in prion protein variants influences intermolecular beta‐sheet formation. EMBO J. 2010;29:251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huang D, Caflisch A. Evolutionary conserved Tyr169 stabilizes the beta2‐alpha2 loop of the prion protein. J Am Chem Soc. 2015;137:2948–2957. [DOI] [PubMed] [Google Scholar]
- 35. Christen B, Damberger FF, Perez DR, Hornemann S, Wuthrich K. Structural plasticity of the cellular prion protein and implications in health and disease. Proc Natl Acad Sci U S A. 2013;110:8549–8554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kaneko K, Zulianello L, Scott M, et al. Evidence for protein X binding to a discontinuous epitope on the cellular prion protein during scrapie prion propagation. Proc Natl Acad Sci U S A. 1997;94:10069–10074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Knaus KJ, Morillas M, Swietnicki W, Malone M, Surewicz WK, Yee VC. Crystal structure of the human prion protein reveals a mechanism for oligomerization. Nat Struct Biol. 2001;8:770–774. [DOI] [PubMed] [Google Scholar]
- 38. Rousseau F, Schymkowitz J, Itzhaki LS. Implications of 3D domain swapping for protein folding, misfolding and function. Adv Exp Med Biol. 2012;747:137–152. [DOI] [PubMed] [Google Scholar]
- 39. Wadsworth JD, Asante EA, Desbruslais M, et al. Human prion protein with valine 129 prevents expression of variant CJD phenotype. Science. 2004;306:1793–1796. [DOI] [PubMed] [Google Scholar]
- 40. Hosszu LL, Jackson GS, Trevitt CR, et al. The residue 129 polymorphism in human prion protein does not confer susceptibility to Creutzfeldt‐Jakob disease by altering the structure or global stability of PrPC. J Biol Chem. 2004;279:28515–28521. [DOI] [PubMed] [Google Scholar]
- 41. Khan MQ, Sweeting B, Mulligan VK, et al. Prion disease susceptibility is affected by beta‐structure folding propensity and local side‐chain interactions in PrP. Proc Natl Acad Sci U S A. 2010;107:19808–19813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Beringue V, Vilotte JL, Laude H. Prion agent diversity and species barrier. Vet Res. 2008;39:47. [DOI] [PubMed] [Google Scholar]
- 43. Baral PK, Wieland B, Swayampakula M, et al. Crystallization and preliminary X‐ray diffraction analysis of prion protein bound to the Fab fragment of the POM1 antibody. Acta Cryst. 2011;F67:1211–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Eghiaian F, Grosclaude J, Lesceu S, et al. Insight into the PrPC→PrPSc conversion from the structures of antibody‐bound ovine prion scrapie‐susceptibility variants. Proc Natl Acad Sci U S A. 2004;101:10254–10259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Antonyuk SV, Trevitt CR, Strange RW, et al. Crystal structure of human prion protein bound to a therapeutic antibody. Proc Natl Acad Sci U S A. 2009;106:2554–2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Baral PK, Swayampakula M, Aguzzi A, James MNG. Structural characterization of POM6 Fab and mouse prion protein complex identifies key regions for prions conformational conversion. FEBS J. 2018;285:1701–1714. [DOI] [PubMed] [Google Scholar]
- 47. Sonati T, Reimann RR, Falsig J, et al. The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein. Nature. 2013;501:102–106. [DOI] [PubMed] [Google Scholar]
- 48. Swayampakula M, Baral PK, Aguzzi A, Kav NN, James MN. The crystal structure of an octapeptide repeat of the prion protein in complex with a Fab fragment of the POM2 antibody. Protein Sci. 2013;22:893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kocisko DA, Baron GS, Rubenstein R, Chen J, Kuizon S, Caughey B. New inhibitors of scrapie‐associated prion protein formation in a library of 2000 drugs and natural products. J Virol. 2003;77:10288–10294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Geschwind MD, Kuo AL, Wong KS, et al. Quinacrine treatment trial for sporadic Creutzfeldt‐Jakob disease. Neurology. 2013;81:2015–2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Baral PK, Swayampakula M, Rout MK, et al. Structural basis of prion inhibition by phenothiazine compounds. Structure. 2014;22:291–303. [DOI] [PubMed] [Google Scholar]
- 52. Caughey BW, Dong A, Bhat KS, Ernst D, Hayes SF, Caughey WS. Secondary structure analysis of the scrapie‐associated protein PrP 27‐30 in water by infrared spectroscopy. Biochemistry. 1991;30:7672–7680. [DOI] [PubMed] [Google Scholar]
- 53. Govaerts C, Wille H, Prusiner SB, Cohen FE. Evidence for assembly of prions with left‐handed beta‐helices into trimers. Proc Natl Acad Sci U S A. 2004;101:8342–8347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. DeMarco ML, Daggett V. From conversion to aggregation: Protofibril formation of the prion protein. Proc Natl Acad Sci U S A. 2004;101:2293–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cobb NJ, Sonnichsen FD, McHaourab H, Surewicz WK. Molecular architecture of human prion protein amyloid: A parallel, in‐register beta‐structure. Proc Natl Acad Sci U S A. 2007;104:18946–18951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Smirnovas V, Baron GS, Offerdahl DK, Raymond GJ, Caughey B, Surewicz WK. Structural organization of brain‐derived mammalian prions examined by hydrogen‐deuterium exchange. Nat Struct Mol Biol. 2011;18:504–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Vazquez‐Fernandez E, Vos MR, Afanasyev P, et al. The structural architecture of an infectious mammalian prion using electron cryomicroscopy. PLoS Pathog. 2016;12:e1005835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wille H, Requena JR. The structure of PrP(Sc) prions. Pathogens. 2018;7:E20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Spagnolli G, Rigoli M, Orioli S, et al. Full atomistic model of prion structure and conversion. PLoS Pathog. 2019;15:e1007864. [DOI] [PMC free article] [PubMed] [Google Scholar]