

Abstract
Bacteria are surrounded by a complex cell envelope made up of one or two membranes supplemented with a layer of peptidoglycan (PG). The envelope is responsible for the protection of bacteria against lysis in their oft‐unpredictable environments and it contributes to cell integrity, morphology, signaling, nutrient/small‐molecule transport, and, in the case of pathogenic bacteria, host–pathogen interactions and virulence. The cell envelope requires considerable remodeling during cell division in order to produce genetically identical progeny. Several proteinaceous machines are responsible for the homeostasis of the cell envelope and their activities must be kept coordinated in order to ensure the remodeling of the envelope is temporally and spatially regulated correctly during multiple cycles of cell division and growth. This review aims to highlight the complexity of the components of the cell envelope, but focusses specifically on the molecular apparatuses involved in the synthesis of the PG wall, and the degree of cross talk necessary between the cell division and the cell wall remodeling machineries to coordinate PG remodeling during division. The current understanding of many of the proteins discussed here has relied on structural studies, and this review concentrates particularly on this structural work.
Keywords: cell division, cell wall, divisome, peptidoglycan
1. INTRODUCTION
Bacteria are classified into two groups, Gram‐negative and Gram‐positive, based on their response to the Gram stain. Gram‐negative cell envelopes consist of both an inner and an outer membrane (IM and OM, respectively), which encapsulate a relatively thin peptidoglycan (PG) layer a few nanometers (nm) thick.1 By contrast, Gram‐positive bacteria have only a single membrane surrounded by a much thicker PG layer, ranging from 30 to 100 nm in thickness.2 It is this thick, external layer of PG in Gram‐positive bacteria that allows for the retention of the Gram stain. It makes sense that the Gram nomenclature for bacterial cells has remained an important classification since its development by Hans Christian Gram in 1884, as it provides important information about the gross structure of bacterial cell walls. The PG layer is a defining feature of bacteria, distinguishing them from archaea or eukaryotes. The tensile strength of PG allows bacteria to thrive in a variety of environments, indeed, targeting the proteins responsible for PG renewal in bacteria has been central to mankind's fight against infectious diseases.3 While some chemical moieties in PG differ between species of bacteria, the general structure of PG comprises repeating disaccharides of N‐acetylglucosamine (GlcNAc) and N‐acetylmuramic acid (MurNAc), cross‐linked between MurNAcs via short (four or five residues) peptide stems (Figure 1), to form a lattice‐like arrangement.4 The fifth, terminal D‐alanine is normally lost during PG maturation. Some variations on this basic theme include additional short (one to five residue) peptide linkers between the peptide stems, which normally (but not exclusively) are cross‐linked between the amino group from the sidechain of residue 3 and the carboxyl group of D‐alanine at position 4 (a three to four cross‐link). The direct cross‐links typically also involve residues 3 and 4. Differences in the structure and regulation of PG and its synthesis are responsible for variations in cell integrity and morphology,4, 5 highlighting the importance of the PG layer to bacteria. The synthesis of PG is a complex multienzyme process initiated in the cytoplasm and subsequently linked to the inner (and outer) leaflet of the cell membrane. PG synthesis has been studied relatively extensively, particularly in the rod‐shaped model organisms Escherichia coli and Bacillus subtilis, which are representatives of the Gram‐negative and Gram‐positive groups, respectively. Consequently, the anabolism of PG is fairly well understood.6 The coupling of PG hydrolysis and resynthesis during cell division is essential for the cell to avoid an untimely death; however, the study of this synergy is a field that remains in its infancy.
Figure 1.
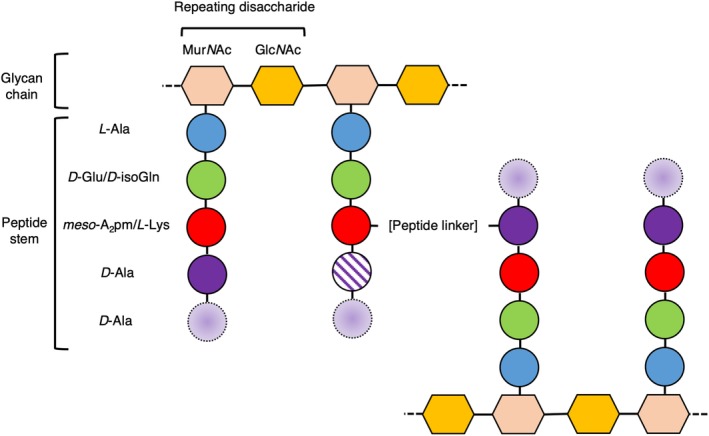
The general structure of peptidoglycan (PG). The general structure of the matrix of PG is shown as a cartoon in which sugar moieties are represented as hexagons and the amino acids that comprise the peptide stems are shown as circles. The identity of the residues present in the cartoon represent the majority of Gram‐negative/Gram‐positive peptide stems, respectively. Variability in the presence of the D‐Ala residues in mature PG at positions 4 (which is sometimes lost) and 5 (which is always lost) of the peptide stem is represented by a cross‐hatched fill and a dashed outline with a gradient fill, respectively. The most common peptide linker in PG is formed between positions 3 and 4 of the peptide stem (as shown here). The chemical structure of the peptide cross‐link varies, and may be composed of a direct link between residues in the peptide stem, or may be comprised of a peptide link, such as the penta‐glycine linker present typically in S. aureus
In the majority of cases, bacteria undertake a process of binary fission through which two identical daughter cells are produced from a parental cell, which involves the establishment of a site of division, elongation (in some cases), chromosome replication and segregation, generation and closure of a septum, and finally separation of the two cells at the septum. Cell division is thus an exceptionally complicated cycle of events requiring a multitude of spatial and temporal backstops for it to be undertaken successfully. There is, however, insufficient scope in this review to consider the spatiotemporal coordination of chromosome replication and segregation with the later steps of cell division. While the fundamental events in this process are conserved across the entire bacterial kingdom, species‐specific nuances are observed, often related to the structure of the cell envelope, variations in morphology, or the identity and nature of some of the regulators. Defining mid‐cell is a critical step of division and is undertaken differently depending on the bacterial species. In rod‐shaped bacteria, such as B. subtilis and E. coli, the mid‐cell is defined at the mid‐point of the longest edge of the cell, where the septum forms in a ring across the shortest width of the cell. In spherical bacteria (cocci), such as the Staphylococci, mid‐cell is defined at the point at which there is the longest diameter of cross section of the spherical cell, and the septum forms around the circumference of the cell in a ring,7, 8, 9 and subsequent division planes are placed orthogonally to the previous because the PG “pie‐crust” rings are important topological markers of past sites of division in Staphylococcus aureus.9 In ovococci, like Streptococcus pneumoniae, both cell length and diameter considerations are important for determining mid‐cell.10 The location of septum formation is a good example of the level of control involved throughout; in many rod‐shaped bacteria (such as E. coli) its position rarely varies in location beyond a few percent, resulting in progeny that rarely vary in volume outside of this margin (~4%).11 Placement of the septum is aided by the Min and nucleoid occlusion systems, which both act as inhibitors of the septal ring progenitor protein, FtsZ.12, 13, 14, 15 Some bacteria that lack the Min system, such as Mycobacterium tuberculosis, seem to utilize a different, albeit less‐efficient system for determining mid‐cell as cell division of this species results in daughter cells of different sizes.12 Septum formation and constriction of the cell into two daughter cells are common themes between both Gram‐negative and Gram‐positive bacteria; however, Gram‐positive bacteria form a complete septum across the mid‐cell before division occurs, whereas a Gram‐negative bacterium divides while developing its septum simultaneously.7 Whether cell elongation occurs during/prior to this process is also species specific and is not necessarily predictable by morphology and/or cell wall composition alone.
There are obviously a great number of fine details relevant to these processes suitable for review but, for the sake of brevity, we will discuss briefly the make‐up of the envelope of both Gram‐negative and Gram‐positive bacteria, before discussing the mechanisms through which bacteria build their wall, the proteins involved in regulating cell division in context of the life cycle of the cell, and the significance of understanding these processes with regards to their potential for exploitation in the pursuit of much needed new antibiotics.
2. THE GRAM‐NEGATIVE ENVELOPE
The OM of Gram‐negative bacteria is an asymmetrical bilayer (Figure 2) consisting of phospholipids and lipopolysaccharides, which assemble into the inner and outer leaflets of the membrane, respectively.1, 2 The structure, function, and synthesis of the Gram‐negative OM are all complex discussion points appropriate for review in their own right.1, 2, 16 Broadly speaking, the OM serves as a protective layer for Gram‐negative bacteria, functioning as a semi‐permeable barrier to the periplasm,17 and as an added layer of protection from environmental turgor.2 The OM accommodates a variety of OM proteins (OMPs), which consist of porins, proteases, lipases, transporters, and various receptors embedded into the OM.16 Unsurprisingly given their nomenclature, porins and transporters are responsible for the diffusion/transport of small molecules into the periplasm, respectively,18 and are responsible for the semi‐permeable nature of the OM. Several members of the porin family of OMPs have been amenable to structural study by crystallography19, 20, 21 and the resulting structural models have had essential roles in the elucidation of molecular mechanisms of transport, highlighting the complexity of the OM. The OM also requires remodeling during cell division and a more complete picture of the OM and its constituents is thus key to understanding how this is regulated on a molecular level,22, 23 and an integrative approach21 will lead to a far greater mechanistic understanding than with structure alone.
Figure 2.
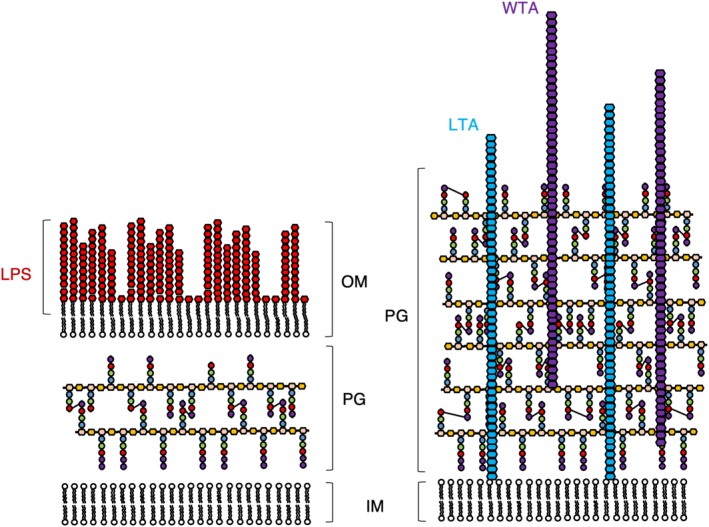
Comparison of the cell envelopes of Gram‐negative (left) and Gram‐positive (right) bacteria. The cell envelope of Gram‐negative bacteria comprises of an inner (IM) and outer membrane (OM) decorated with lipopolysaccharide (LPS), sandwiching a relatively thin layer of peptidoglycan (PG). By contrast, the cell envelope of Gram‐positive bacteria is comprised of a single cell membrane surrounded much thicker PG layer complemented with lipoteichoic acid (LTA) and wall teichoic acid (WTA). Cross‐links between peptide stems are shown as three to four cross‐links for the sake of simplicity. Not shown are the multitude of proteins that sit in the inner and OM of Gram‐negative bacteria, nor those that reside in the membrane or the PG of Gram‐positive bacteria. The components of PG are displayed using the same scheme as in Figure 1, with the nascent PG chain containing D‐Ala at positions 4 and 5. The mature PG is represented without the D‐Ala at position 5 and occasionally also without the D‐Ala at position 4 to represent the natural variability of the peptide stem in the mature PG mesh
3. THE GRAM‐POSITIVE CELL ENVELOPE
Gram‐positive bacteria compensate for the lack of the protective/stabilizing presence of the OM with a thicker layer of PG.2, 24 As well as the obvious difference in thickness, the PG layer of Gram‐positive bacteria is distinguished from Gram‐negative bacteria by its decoration with anionic glycopolymers called teichoic acids (TAs).25, 26 TAs constitute 30–60% of the Gram‐positive cell wall. There are two types of TA; wall TAs (WTAs), which are covalently attached to the PG, and lipoteichoic acids (LTAs), which are attached to the cytoplasmic membrane and extend deep into the wall (Figure 2).27, 28 TAs may act as a functional substitute for the OMPs present in Gram‐negative bacteria as they are also capable of affecting permeability and integrity, and are responsible for host–pathogen interactions.29 It follows that TAs are a virulence factor for Gram‐positive pathogens and for this reason are of interest for study, as their synthesis pathways are also a potential target for future antibiotics.30, 31, 32 The TA chemical structure, similarly to PG, also varies from species to species. The general structure is a β‐1 → 4 linked N‐acetylmannosamine/GlcNAc (ManNAc/GlcNAc) disaccharide, a glycerophosphate linker made of three phosphoglycerol molecules, followed by a long chain of glycerol‐ or ribitol‐phosphate repeats.33 In WTAs, the ManNAc/GlcNAc is linked to the MurNAc residues of the glycan chain of PG by a phosphodiester bond.34 In LTAs, a lipid anchors the TA to the cell membrane and the lipid anchor is another example of a species‐specific variable region in the molecule.34
The cell walls of Gram‐positive bacteria, not unlike the OMs of Gram‐negative bacteria, also contain surface proteins. The surface proteins present in the wall are transported out of the cell by secretory systems such as the Sec or TAT pathways.35, 36 Following secretion, surface proteins are attached either to the IM by way of lipid anchors/transmembrane domains, or to the PG/TAs that make up the cell wall.37
4. PG SYNTHESIS
4.1. Cytosolic pathway
The cytosolic reactions of PG synthesis are undertaken chiefly by a family of ligases designated “Mur” (MurA, MurB, etc.); these enzymes are involved in the production of the PG precursor, Lipid II, in the inner leaflet of the IM.38 The first step in the cytosolic pathway of PG synthesis is undertaken by MurA, which catalyzes the production of uridine diphosphate (UDP)‐GlcNAc‐enolpyruvate from phosphoenolpyruvate (PEP), and UDP‐GlcNAc.39 The production of UDP‐GlcNAc‐enolpyruvate is followed by its reduction into UDP‐MurNAc, catalyzed by MurB.40 Extension of UDP‐MurNAc by sequential addition of the residues present in the stem peptide is taken on by the enzymes MurC‐F to produce Park's nucleotide. In Gram‐negative bacteria Park's nucleotide comprises UDP‐MurNAc, L‐alanine (L‐Ala), D‐glutamine (D‐Gln), meso‐diaminopimelic acid (meso‐A2pm), D‐alanine (D‐Ala), D‐Ala. Park's nucleotide is attached to undecaprenyl (C 55) diphosphate by MraY, resulting in a lipid PG precursor referred to as Lipid I.41 The final cytoplasmic enzymatic step involves the formation of the β‐1 → 4 glycosidic bond of MurNAc with GlcNAc via a glycosyltransferase (MurG) to produce Lipid II,42 which is then flipped to the outside of the membrane of the cell by a flippase.43 The structure of each of the Mur enzymes from at least one species of bacteria have now been solved,38 a process that started with the structure of MurA in 1996.44 The structure of MurA was solved in complex with both its substrate UDP‐GlcNAc and the antibiotic fosfomycin,44 revealing both the mechanism of action45 and the mode of inhibition of MurA. Similar studies have been carried out on the remainder of the Mur enzymes in this pathway (Figure 3), lending the entire cytoplasmic pathway of the process to rational drug design.38, 46
Figure 3.
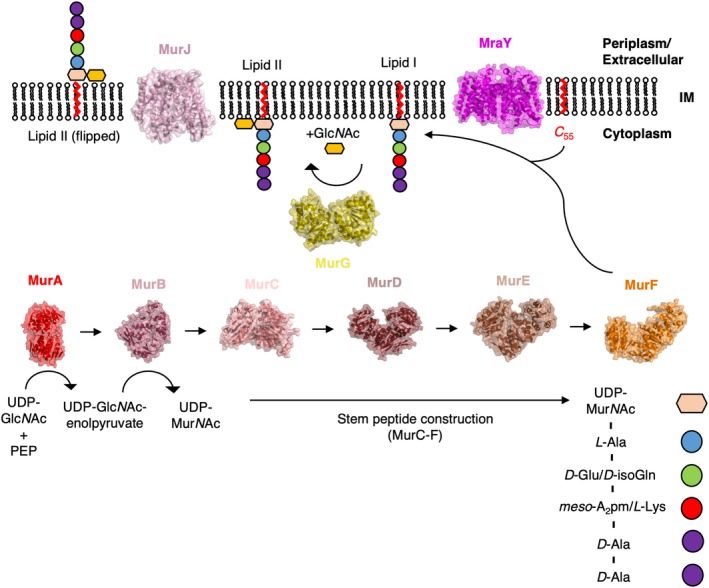
A structural overview of Lipid II construction. The structure of every enzyme involved in the cytoplasmic pathway of Lipid II synthesis has now been determined by X‐ray crystallographic techniques. Here, the structures of every enzyme involved in Lipid II synthesis are displayed alongside the reaction(s) they are responsible for catalyzing on either face of the cell membrane (IM). For the sake of consistency, the structures shown here are those derived from E. coli
4.2. The elusive flippase
The identity of the flippase responsible for the transfer of Lipid II across the cell membrane has recently been a subject of some debate. Three major candidates for the flippase were initially proposed, FtsW, RodA, and MurJ.47, 48, 49 All three proteins are integral membrane proteins, with 10 (FtsW/RodA) and 14 (MurJ) predicted transmembrane domains. All three proteins are highly conserved and essential50 for growth in both Gram‐positive and Gram‐negative bacteria, which would suggest that they do not play redundant roles in the cell. The first studies that appeared to observe FtsW as the Lipid II flippase were in vitro experiments in which the ability of E. coli FtsW to flip fluorescently Lipid II was measured using model membranes/liposomes.47 These initial experiments suggested that FtsW was indeed capable of flipping Lipid II whereas, under the same conditions, MurJ was not.47 Follow‐up experiments worked to identify the region of the protein responsible for the flippase activity by removing transmembrane domains of FtsW and performing the same fluorescence‐based flipping assays, with concomitant microscopy experiments on E. coli cells harboring the same FtsW mutants.51 The in vitro and in vivo experiments performed in this study suggested that FtsW likely acted through a pore‐like mechanism.51 MurJ was proposed as an alternative Lipid II flippase in E. coli through a combination of a bioinformatics approach coupled with in vivo genetic analyses.52 Mutational studies of E. coli harboring MurJ mutations revealed an accumulation of intracellular PG precursors,53 suggesting an inability to flip Lipid II. More recent work now suggests that both FtsW and RodA function as PG polymerases,32, 54, 55 swaying the flippase argument toward MurJ as the Lipid II flippase. Aided by recent advancements in membrane protein crystallographic techniques,56 the crystal structure of MurJ from the extremophile Thermosipho africanus was solved in an inward conformation, allowing for the generation of an alternative access model of Lipid II flipping based on a combination of in silico docking and in vivo experiments.57 Subsequently, the structure of MurJ in several different conformations was solved, allowing for modelling of the mechanism of Lipid II flipping.58 The structure of MurJ from E. coli has also since been solved, and high‐throughput mutagenesis performed, leading to the identification of sites for potential inhibitor development.59
4.3. Extracellular pathway
The extracellular members of the PG synthesis pathway are membrane‐associated penicillin‐binding proteins (PBPs), named for their affinity for penicillin. The PBPs are responsible for the polymerization of the growing glycan chain, and/or for linking the peptide chains extruding from the glycan backbone; these are glycosyltransferase and transpeptidase activities, respectively.60 Some PBPs also have carboxypeptidase activity; these are responsible for the trimming of the peptide stem as a mechanism of regulating the number of cross‐links formed in the cell wall and are responsible for cell shape maintenance in E. coli.7, 61 PBPs can be separated into two groups, those that solely have transpeptidase activity (Class B PBPs), or bifunctional PBPs that possess both activities (Class A PBPs). Some examples of bifunctional PBPs include PBP1a, b, and c in E. coli; PBP1, 2c, and 4 in B. subtilis; and PBP1a, 2a, and 1b in S. pneumonia.62, 63, 64 Glycosyltransferases act early in the extracellular pathway to catalyze the polymerization of the nonlipid region of Lipid II into the nascent glycan chain,60, 64 while downstream transpeptidases are responsible for the linking of the D‐Ala residues of the extruding peptide chain to form the strong and stable PG mesh.60, 64 The transpeptidase forms an acyl‐enzyme intermediate with the D‐Ala residue of the acceptor peptide, before being cross‐linked to the donor peptide via an amino group, which in Gram‐positive organisms is glycine or lysine.4, 65 Structural studies of PBPs have been performed extensively in several Gram‐negative and Gram‐positive species of bacteria, and their activities and inhibition by antibiotics have been reviewed excellently elsewhere.6, 60, 64, 66, 67
4.4. Differences between PG from Gram‐negative and Gram‐positive bacteria
As well as the obvious differences in thickness of the PG layer and associated macromolecules (TAs/surface proteins) described above, there are further subtle nuances to the precise chemical make‐up of the PG from Gram‐positive and Gram‐negative bacteria. Chemical differences of the glycan chain between Gram‐negative and Gram‐positive species include variations in modifications such as glycosylation, phosphorylation, and deacetylation.68 In contrast to the lack of chemical diversity in the glycan chain, differences in the peptide stems of PG between Gram‐negative and Gram‐positive bacteria are far more common.68 The majority of peptide stems in the PG of Gram‐negative bacteria follows the pattern L‐Ala, D‐glutamate (D‐Glu), meso‐A2pm, followed by two D‐Ala residues. In Gram‐positive bacteria, however, the residues present in the stem peptide vary at positions 2 and 3, at which D‐isoglutamine (D‐isoGln) and L‐lysine (L‐Lys) are prevalent, respectively.68 In both Gram‐positive and Gram‐negative bacteria, D‐Glu is initially added to the growing peptide stem by MurD at position 2 during construction of the peptide stem.4, 6 In those Gram‐positive species where D‐isoGln is found at Position 2, D‐Glu is enzymatically deamidated to D‐isoGln by an enzyme complex of MurT/GatD.69 The structure of the MurT/GatD complex has been solved recently, revealing the mechanism of PG amidation by MurT, and also the mechanism through which GatD produces and channels the ammonia required for PG amidation to the MurT active site.70
5. COORDINATION OF CELL WALL SYNTHESIS WITH CELL DIVISION
5.1. The divisome
The collection of ~20–30 proteins responsible for regulating cell division has come to be known as the “divisome” (Figure 4). Proteins in the divisome ensure that one round of division occurs at a time, with one copy of the chromosome present in each cell, that cell wall synthesis is undertaken appropriately to circumvent lysis during septum formation, and that cell separation occurs through the function of cell wall hydrolases (autolysins).7 Investigation of the divisome has chiefly been undertaken in rod‐shaped bacteria, yet there is still a lot to learn about divisome formation and organization in these bacteria as well as in other bacteria with different shapes, such as the spherical cocci. Some archaea with profound differences in morphology and cell division, such as the triangular Haloferax volcanii which divides by a process of ternary fission, also utilize some of the same proteins as in bacteria when regulating division.71 The divisome is an attractive target for the generation of novel antimicrobials, as disruption of the machinery responsible for organizing cell division would ultimately result in a reduction and cessation of propagation. A lack of structural and functional information about lesser studied components of the divisome, and the divisome itself as a dynamic molecular machine, presents a stumbling block in the antimicrobial pipeline and is thus a potentially lucrative area of research. The function of the divisome to coordinate chromosome replication and segregation with cytokinesis is common to both Gram‐negative and Gram‐positive organisms, but the precise constituents of the divisome vary across species.72
Figure 4.
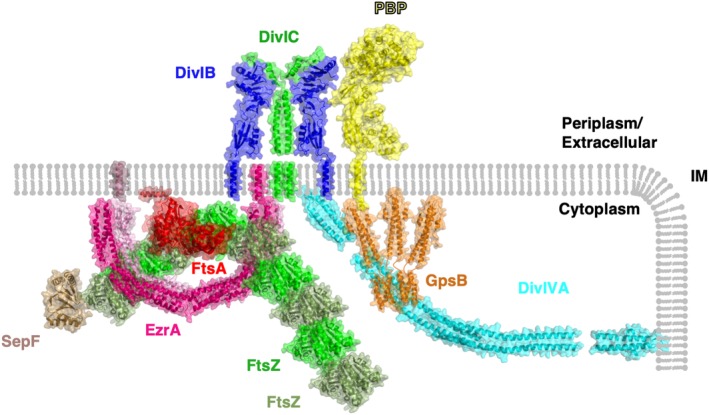
Structural model of the divisome. A selection of divisome proteins for which atomic structures have been determined are shown in context of the cell membrane (IM). Where structural data regarding protein–protein interactions are known, this figure reflects this information. Where available, structures have been taken from the model organism B. subtilis; however, in the cases where there is a lack of structural data from this organism, the structures with the greatest degree of sequence homology are shown. FtsZ is colored alternately in shades of green to denote filament formation, FtsA is shown in red, an antiparallel EzrA dimer is colored in shades of pink, SepF is shown in sand (note that the membrane‐anchoring region of SepF was not resolved in its crystal structure analysis), a 2:2 complex of DivIB and DivIC is shown in blue and green, respectively, a GpsB hexamer is shown in orange, a representative class B PBP is shown in yellow and DivIVA in cyan
5.2. The Z‐ring
The best studied of the divisome proteins is FtsZ, and it is arguably the central protein to the function of the divisome in cell division. FtsZ, named after its mutants' abilities to cause E. coli cells to divide in a filamentous fashion at nonpermissive temperatures (filamentous temperature sensitive),73 is a cytosolic protein known to accumulate at the site of division across all bacterial species.74, 75 The discovery of FtsZ set the groundwork for much of the research conducted on the divisome in the past ~25 years. The crystal structure of FtsZ provided the first definitive evidence of a bacterial cytoskeleton, as FtsZ is a clear structural homolog of tubulin despite sharing no significant amino acid sequence homology.76 FtsZ polymerizes into filaments in a GTP‐dependent manner rapidly “treadmilling”77 in an agile and nimble ring‐like structure, dubbed the “Z‐ring,” adjacent to the cytoplasmic face of the membrane. Prior to the observation of the treadmilling of FtsZ, the Z‐ring was thought to represent a semicontinuous ring at the mid‐cell which constricted during septum formation.74, 75 There are several molecular mechanisms that govern the placement of the Z‐ring, for instance, the Min system functions in an inhibitory manner, preventing formation of the Z‐ring away from midcell,12 while Zap proteins function in a stimulatory manner, encouraging the formation of lateral interactions between filaments of FtsZ.12, 78 FtsZ acts as a scaffold onto which protein–protein interactions with other members of the divisome come together to stimulate division (Figure 4). A well‐studied protein–protein interaction of FtsZ is with FtsA, a well‐conserved “early” divisome component, which self‐organizes with FtsZ and tethers it to the membrane.79 The FtsA‐mediated membrane tethering is essential to ensure constriction of the membrane at the septum, as FtsZ does not interact with the membrane of its own accord. ZipA works alongside FtsA in Gram‐negative bacteria to tether FtsZ to the membrane80; however, no such homolog is present in Gram‐positive bacteria, instead, SepF has been proposed to fulfil a similar function.81 The combination of the polymerized FtsZ, along with SepF/ZipA and FtsA forms a structure called the proto‐ring.82
5.3. Recruitment of downstream proteins to the divisome
Following the formation of the proto‐ring, the “late” division proteins start to assemble to promote downstream processes involved in the remodeling of the cell wall. Divisome assembly has perhaps been studied most extensively in E. coli; the proteins known to accumulate are FtsN, FtsI, FtsEX, FtsQ, FtsL, FtsB, FtsW, FtsK, and PBP2B.83 However, the conservation of many of these components is not necessarily maintained in Gram‐positive bacteria, and the essentiality of the components is also not strictly conserved. One example of a widely conserved and essential subcomponent of the divisome is a trimeric complex of DivIB, DivIC, and FtsL in Gram‐positives (FtsQ, FtsB, and FtsL in Gram‐negatives, respectively). DivIB/FtsQ, DivIC/FtsB, and FtsL are transmembrane proteins, each with a single transmembrane helix, and form a complex independent of other divisome proteins in E. coli and B. subtilis.84, 85 DivIB/FtsQ interacts with PG through its extracellular PASTA domain and is required as a cell division checkpoint based on studies in S. aureus.86 The extracellular portion of DivIB/FtsQ is also known to interact with PBPs, such as PBP2b in B. subtilis.87 It appears that the DivIB/DivIC/FtsL (FtsQ/FtsB/FtsL) complex links the early intracellular stages of cell division with PG remodeling, which is predominantly an extracellular process.88 The structure of E. coli FtsQ in complex with a fragment of FtsB has recently been solved.89 Based on the essentiality of the interaction between FtsQ/DivIB with FtsB/DivIC, and their location of interaction on the outside surface of the cell, the structure of the complex should allow for the considered design of antibiotics, without a need to consider cell membrane traversal.
6. REGULATORS OF CELL DIVISION AND PG REMODELLING
DivIVA is a member of the Gram‐positive divisome with divergent roles in those bacteria that encode DivIVA orthologs. One feature common to all DivIVA homologs, however, is an apparent innate ability to sense membrane curvature.90 The bulk of the published information about DivIVA comes from studies in B. subtilis where, among other things, it functions as a topological marker for the Min system,12 a cell division inhibitor. X‐ray crystallographic studies of the isolated N‐ and C‐terminal domains of DivIVA revealed that it formed tetramers made up of an antiparallel arrangement of parallel coiled‐coils.91 This model clashes somewhat with earlier EM studies suggesting that DivIVA forms higher order oligomers; however, these studies were performed on mutants of DivIVA purposefully designed to alter oligomeric state and may not accurately represent the wild‐type protein.92 The crystal structure of the N‐terminal domain of DivIVA also provided some insight into how membrane binding might be possible; a conserved Phe‐Arg motif oriented outwards from the protein is positioned to form hydrophobic and electrostatic interactions with the membrane, respectively.91 Since DivIVA interacts with several cytoplasmic proteins through its C‐terminal domain, it may act somewhat promiscuously as a topological marker for several intracellular systems.93 The situation is complicated as a result of DivIVA's divergent roles, for instance, S. aureus lacks MinC/D, and the interactions of DivIVA with divisome components in S. aureus have yet to be elucidated fully.
GpsB, a homolog of DivIVA present in Gram‐positive organisms, is also involved in the coordination of PG synthesis at the septum. GpsB and DivIVA share sequence and structural homology at their N‐termini; however, this homology between the two proteins decreases significantly in their C‐terminal regions. The difference in C‐terminal structure between the two proteins is responsible for the differences in oligomeric state between the two proteins.91, 94 Integrative structural and biochemical studies on GpsB from Listeria monocytogenes and B. subtilis revealed that GpsB binds the cytoplasmic microdomains of PBPs and forms a hexamer in solution, allowing for the generation of a model through which GpsB likely coordinates the activities of PBPs.94, 95 Interactions between GpsB and PBPs from a wider range of Gram‐positive bacteria including B. subtilis, L. monocytogenes, and S. pneumoniae have since been interrogated structurally and biochemically, leading to the identification of motifs required for binding.96 A greater mechanistic understanding of the interactions between GpsB and PBP homologs in various species has aided the identification of novel interactions between GpsB and the PG remodeling enzymes YpbE and YrrS, leading to the conclusion that GpsB acts as an adaptor protein.96 Recent work on GpsB from S. aureus suggests a novel action of GpsB in the stimulation of lateral interactions between FtsZ filaments.97 No interactions between FtsZ and GpsB have been detected in any other species of bacteria tested; therefore, this finding would suggest a divergent role for GpsB in S. aureus.97
Negative regulation is also necessary in order to prevent aberrant Z‐ring formation; EzrA is an example of such a negative regulator in Gram‐positive bacteria. EzrA was first identified in B. subtilis, and its colocalization with FtsZ first observed by fluorescence microscopy.98 EzrA has a predicted TM helix at its N‐terminus to link it, and potentially its interaction partners, to the membrane. B. subtilis EzrA inhibits the formation of FtsZ filaments in vitro and in vivo, and complete deletion of EzrA results in an increased frequency of FtsZ ring formation.98, 99, 100 Recent studies in S. aureus and B. subtilis have implicated EzrA in the control of PG synthesis through direct interactions with PG synthases.101, 102 EzrA has been found by bacterial two‐hybrid to interact with a multitude of divisome proteins in S. aureus, as well as the PG synthases PBP1, 2, and 3.102 The crystal structure of EzrA from B. subtilis revealed that EzrA forms antiparallel dimers forming an overall crescent shape, with each monomer made up of repeating three‐helical bundles that have structural homology to the spectrin repeat fold found in eukaryotic cytoskeletal proteins.103 The space within the arch of the crescent is sufficient to enclose an FtsZ filament, to sterically hinder the formation of lateral interactions between filaments.103, 104
As stated above, ZipA is involved in the regulation of divisome assembly and Z‐ring placement in Gram‐negative bacteria.105 ZipA has been studied most extensively in E. coli, where it is essential.106 ZipA is predominantly cytoplasmic, with a single transmembrane helix and a micro‐domain in the periplasm. ZipA binds to FtsZ and tethers it to the cell membrane as well as FtsA.80 The functional significance of ZipA was brought into question by a gain of function mutant of FtsA which allowed for the bypassing of ZipA107; however, more recent studies have suggested that ZipA functions by protecting FtsZ from degradation by cytoplasmic proteases, a role that cannot be substituted for by FtsA.108 The stimulatory or inhibitory effects of ZipA on Z‐ring formation and FtsZ bundling are currently unknown; while early studies on ZipA provided contradictory evidence for stimulatory109 and inhibitory79 roles of ZipA, more recent work suggests that ZipA has neither a stimulatory nor an inhibitory effect on lateral interactions between FtsZ filaments, and that it simply acts as a passive membrane tether for FtsZ.105 More work is clearly required to elucidate the functional significance of ZipA for divisome formation in Gram‐negative bacteria.
7. ANTIBIOTICS TARGETING THE CELL DIVISION/PG SYNTHESIS MACHINERY
Beta‐lactam antibiotics inhibit the transpeptidase action of PBPs by mimicking the structure of the terminal d‐Ala‐D‐Ala residues of the glycan‐attached peptide of PG, sequestering the active site serine in a covalent adduct and rendering the transpeptidase domain of the PBPs inactive.110 The inhibition of the cell wall synthesis machinery ultimately results in the lysis of the cell due to the inability of the bacteria to generate new cell wall material to replace the parts it has degraded; this is the major cause of cell death by beta‐lactam antibiotics.110 Secondary mechanisms of cell death have also been observed, however, in which nonlytic cells undergo cell death as a result of futile cycles of PG precursor synthesis, for instance.111 The introduction of beta‐lactams ushered in a golden age of antibiotics, during which many diseases previously fatal became treatable. The overuse of antibiotics, combined with the short life cycle and rapid evolution of bacteria, has resulted in the generation of antibiotic‐resistant strains of bacteria which, in certain clinical scenarios, often have fatal consequences.3 PG homeostasis and regulation thus remain an attractive target for antibiotic drug design.
Two major mechanisms of resistance to beta‐lactams have arisen: first, an accumulation of mutations in PBPs renders them insensitive to beta‐lactams and second, enzymes capable of degrading beta‐lactams, the beta‐lactamases, have evolved. For instance, methicillin resistance in methicillin‐resistant S. aureus (MRSA) is caused by the introduction of the mecA gene coding for PBP2a, where the active site serine is located in a pocket that is occluded from beta‐lactams.112 A multitude of new PBP2a‐targetting antibiotics have been developed, including the cephalosporin subgroup of beta‐lactams, ceftobiprole, and ceftaroline.113 These molecules appear to target both the PBP2a active site as well as a putative allosteric site,114, 115 but resistance to these new drugs has already emerged.112 Beta‐lactam resistance is also caused by the evolution of new beta‐lactamases, which form acyl‐enzyme intermediates with beta‐lactams as in PBPs; however, these enzymes can break open the beta‐lactam ring to form products that do not inhibit PBPs.116 Beta‐lactamases were present in bacteria prior to the use of antibiotics in clinical and agricultural scenarios, but they have evolved into a very efficient beta‐lactam resistance mechanism thanks to increased selection pressure from antibiotic overuse by mankind.117, 118 The somewhat unfortunately named New‐Delhi metallo‐beta‐lactamases (NDMs) are one such example of this type of evolution; NDMs were first isolated and identified from a patient with a Klebsiella pneumoniae infection in 2009119 and have since become a cause of global concern.120 Novel antibiotics targeting NDMs are now necessary to combat the rise of superbugs which harbor these enzymes, and excellent progress is being made toward this end.121
The gap between antibiotic drug discovery and the development of antimicrobial resistance is becoming shorter with each iteration. In fact, the first report of penicillin resistance in E. coli appeared in 1940 before penicillin was in general public use.122 Alternative means of disrupting cell division to fight infection are, therefore, an attractive method of sidestepping this problem and there is potential for divisome components to provide the necessary novel target. Inhibitors of divisome components have shown promising preliminary results against bacterial infections, specifically in the case of FtsZ. An inhibitor of FtsZ, PC190723, causes cell elongation in B. subtilis, and cell enlargement in S. aureus, and is effective against MRSA strains.123 Computational ligand docking of PC190723 has identified a potential site of action at an allosteric site away from the nucleotide binding region of FtsZ.124, 125 This is the first viable mechanism found for alternative antibiotics targeting the divisome, but with the influx of structures becoming available for several divisome components, there is much promise for antibiotics targeting cell division to be developed to take back control from the march of infectious disease.
8. CONCLUSION
Here, we have shown a brief overview of the complexities of the cell envelopes of Gram‐negative and Gram‐positive bacteria. Although much progress has been made in the last decade or so in some areas, such as the atomic detail of PG synthesis, there is still much work to be done. Focus now should be turned to the molecular nuances of envelope modification during division and how environmental factors influence cell division and PG synthesis. From a translational standpoint, a lack of structural and functional information about lesser studied members of the divisome presents a stumbling block in the antimicrobial pipeline and is thus a potentially lucrative area of research.
ACKNOWLEDGMENTS
Research in the Lewis laboratory has been funded by Newcastle University in a studentship to S.B., the UK BBSRC and MRC, and the European Commission.
Booth S, Lewis RJ. Structural basis for the coordination of cell division with the synthesis of the bacterial cell envelope. Protein Science. 2019;28:2042–2054. 10.1002/pro.3722
Funding information Newcastle University
REFERENCES
- 1. Nikaido H. Outer membrane, Gram‐negative bacteria Encyclopedia of microbiology. 3rd ed. Academic Press, Cambridge Mass. USA, 2009; p. 439–452. [Google Scholar]
- 2. Silhavy TJ, Kahne D, Walker S. The bacterial cell envelope. Cold Spring Harb Perspect Biol. 2010;2:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lobanovska M, Pilla G. Penicillin's discovery and antibiotic resistance: Lessons for the future? Yale J Biol Med. 2017;90:135–145. [PMC free article] [PubMed] [Google Scholar]
- 4. Vollmer W, Blanot D, De Pedro MA. Peptidoglycan structure and architecture. FEMS Microbiol Rev. 2008;32:149–167. [DOI] [PubMed] [Google Scholar]
- 5. Turner RD, Vollmer W, Foster SJ. Different walls for rods and balls: The diversity of peptidoglycan. Mol Microbiol. 2014;91:862–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lovering AL, Safadi SS, Strynadka NCJ. Structural perspective of peptidoglycan biosynthesis and assembly. Annu Rev Biochem. 2012;81:451–478. [DOI] [PubMed] [Google Scholar]
- 7. Egan AJF, Vollmer W. The physiology of bacterial cell division. Ann N Y Acad Sci. 2013;1277:8–28. [DOI] [PubMed] [Google Scholar]
- 8. Lutkenhaus J. The regulation of bacterial cell division: A time and place for it. Curr Opin Microbiol. 1998;1:210–215. [DOI] [PubMed] [Google Scholar]
- 9. Turner RD, Ratcliffe EC, Wheeler R, Golestanian R, Hobbs JK, Foster SJ. Peptidoglycan architecture can specify division planes in Staphylococcus aureus . Nat Commun. 2010;1:26. [DOI] [PubMed] [Google Scholar]
- 10. Garcia PS, Simorre JP, Brochier‐Armanet C, Grangeasse C. Cell division of Streptococcus pneumoniae: Think positive! Curr Opin Microbiol. 2016;34:18–23. [DOI] [PubMed] [Google Scholar]
- 11. Männik J, Wu F, Hol FJH, et al. Robustness and accuracy of cell division in Escherichia coli in diverse cell shapes. Proc Natl Acad Sci U S A. 2012;109:6957–6962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rowlett VW, Margolin W. The bacterial Min system. Curr Biol. 2013;23:R553–R556. [DOI] [PubMed] [Google Scholar]
- 13. Wu LJ, Errington J. Nucleoid occlusion and bacterial cell division. Nat Rev Microbiol. 2012;10:8–12. [DOI] [PubMed] [Google Scholar]
- 14. Rowlett VW, Margolin W. The Min system and other nucleoid‐independent regulators of Z ring positioning. Front Microbiol. 2015;6:478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Adams DW, Wu LJ, Errington J. Cell cycle regulation by the bacterial nucleoid. Curr Opin Microbiol. 2014;22:94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rollauer SE, Sooreshjani MA, Noinaj N, Buchanan SK. Outer membrane protein biogenesis in Gram‐negative bacteria. Philos Trans R Soc B Biol Sci. 2015;370:20150023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ruiz N, Kahne D, Silhavy TJ. Advances in understanding bacterial outer‐membrane biogenesis. Nat Rev Microbiol. 2006;4:57–66. [DOI] [PubMed] [Google Scholar]
- 18. Achouak W. Multiple facets of bacterial porins. FEMS Microbiol Lett. 2001;199:1–7. [DOI] [PubMed] [Google Scholar]
- 19. Cowan SW, Garavito RM, Jansonius JN, et al. The structure of OmpF porin in a tetragonal crystal form. Structure. 1995;3:1041–1050. [DOI] [PubMed] [Google Scholar]
- 20. Weiss MS, Schulz GE. Structure of porin refined at 1.8 Å resolution. J Mol Biol. 1992;227:493–509. [DOI] [PubMed] [Google Scholar]
- 21. Glenwright AJ, Pothula KR, Bhamidimarri SP, et al. Structural basis for nutrient acquisition by dominant members of the human gut microbiota. Nature. 2017;541:407–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Egan AJF. Bacterial outer membrane constriction. Mol Microbiol. 2018;107:676–687. [DOI] [PubMed] [Google Scholar]
- 23. Gray AN, Egan AJ, Van't Veer IL, et al. Coordination of peptidoglycan synthesis and outer membrane constriction during Escherichia coli cell division. Elife. 2015;4:e07118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vollmer W, Massidda O, Tomasz A. The cell wall of Streptococcus pneumoniae . Microbiol Spectr. 2019;7:1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brown S, Santa Maria JP, Walker S. Wall teichoic acids of Gram‐positive bacteria. Annu Rev Microbiol. 2013;67:313–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Percy MG, Gründling A. Lipoteichoic acid synthesis and function in Gram‐positive bacteria. Annu Rev Microbiol. 2014;68:81–100. [DOI] [PubMed] [Google Scholar]
- 27. Formstone A, Carballido‐Lopez R, Noirot P, Errington J, Scheffers DJ. Localization and interactions of teichoic acid synthetic enzymes in Bacillus subtilis . J Bacteriol. 2008;190:1812–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rausch M, Deisinger JP, Ulm H, et al. Coordination of capsule assembly and cell wall biosynthesis in Staphylococcus aureus . Nat Commun. 2019;10:1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wanner S, Schade J, Keinhörster D, et al. Wall teichoic acids mediate increased virulence in Staphylococcus aureus . Nat Microbiol. 2017;2:16257. [DOI] [PubMed] [Google Scholar]
- 30. Swoboda JG, Meredith TC, Campbell J, et al. Discovery of a small molecule that blocks wall teichoic acid biosynthesis in Staphylococcus aureus . ACS Chem Biol. 2010;4:875–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Farha MA, Leung A, Sewell EW, et al. Inhibition of WTA synthesis blocks the cooperative action of PBPs and sensitizes MRSA to β‐lactams. ACS Chem Biol. 2013;8:226–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Meeske AJ, Riley EP, Robins WP, et al. SEDS proteins are a widespread family of bacterial cell wall polymerases. Nature. 2016;537:634–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sanderson AR, Strominger JL, Nathenson SG, Sanderson R. Chemical structure of teichoic acid from chemical structure of teichoic acid from Staphylococcus aureus, strain Copenhagen. J Biol Chem. 1962;237:3603–3613. [PubMed] [Google Scholar]
- 34. Neuhaus FC, Baddiley JA. A continuum of anionic charge: Structures and functions of D‐alanyl‐teichoic acids in Gram‐positive bacteria. Microbiol Mol Biol Rev. 2003;67:686–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tsirigotaki A, De Geyter J, Šoštaric N, Economou A, Karamanou S. Protein export through the bacterial Sec pathway. Nat Rev Microbiol. 2017;15:21–36. [DOI] [PubMed] [Google Scholar]
- 36. Palmer T, Berks BC. The twin‐arginine translocation (Tat) protein export pathway. Nat Rev Microbiol. 2015;10:483–496. [DOI] [PubMed] [Google Scholar]
- 37. Scott JR, Barnett TC. Surface proteins of Gram‐positive bacteria and how they get there. Annu Rev Microbiol. 2006;60:397–423. [DOI] [PubMed] [Google Scholar]
- 38. El Zoeiby A, Sanschagrin F, Levesque RC. Structure and function of the Mur enzymes: Development of novel inhibitors. Mol Microbiol. 2002;47:1–12. [DOI] [PubMed] [Google Scholar]
- 39. Brown ED, Vivas EI, Walsh CT, Kolter R. MurA (MurZ), the enzyme that catalyzes the first committed step in peptidoglycan biosynthesis, is essential in Escherichia coli . J Bacteriol. 1995;177:4194–4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sylvester DR, Alvarez E, Patel A, Ratnam K, Kallender H, Wallis NG. Identification and characterization of UDP‐N‐acetylenolpyruvylglucosamine reductase (MurB) from the Gram‐positive pathogen Streptococcus pneumoniae . Biochem J. 2001;355:431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Van Nieuwenhze MS, Mauldin SC, Zia‐Ebrahimi M, Aikins JA, Blaszczak LC. The total synthesis of lipid I. J Am Chem Soc. 2001;123:6983–6988. [DOI] [PubMed] [Google Scholar]
- 42. de Kruijff B, van Dam V, Breukink E. Lipid II: A central component in bacterial cell wall synthesis and a target for antibiotics. Prostag Leukotr Ess Fat Acids. 2008;79:117–121. [DOI] [PubMed] [Google Scholar]
- 43. Ruiz N. Lipid flippases for bacterial peptidoglycan biosynthesis. Lipid Insights. 2016;8:21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Skarzynski T, Mistry A, Wonacott A, Hutchinson SE, Kelly VA, Duncan K. Structure of UDP‐N‐acetylglucosamine enolpyruvyl transferase, an enzyme essential for the synthesis of bacterial peptidoglycan, complexed with substrate UDP‐N‐acetylglucosamine and the drug fosfomycin. Structure. 1996;4:1465–1474. [DOI] [PubMed] [Google Scholar]
- 45. Eschenburg S, Kabsch W, Healy ML, Schönbrunn E. A new view of the mechanisms of UDP‐N‐acetylglucosamine enolpyruvyl transferase (MurA) and 5‐enolpyruvylshikimate‐3‐phosphate synthase (AroA) derived from X‐ray structures of their tetrahedral reaction intermediate states. J Biol Chem. 2003;278:49215–49222. [DOI] [PubMed] [Google Scholar]
- 46. Hrast M, Sosič I, Šink R, Gobec S. Inhibitors of the peptidoglycan biosynthesis enzymes MurA‐F. Bioorg Chem. 2014;55:2–15. [DOI] [PubMed] [Google Scholar]
- 47. Mohammadi T, van Dam V, Sijbrandi R, et al. Identification of FtsW as a transporter of lipid‐linked cell wall precursors across the membrane. EMBO J. 2011;30:1425–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sham LT, Butler EK, Lebar MD, Kahne D, Bernhardt TG, Ruiz N. MurJ is the flippase of lipid‐linked precursors for peptidoglycan biogenesis. Science. 2014;345:220–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sieger B, Schubert K, Donovan C, Bramkamp M. The lipid II flippase RodA determines morphology and growth in Corynebacterium glutamicum . Mol Microbiol. 2013;90:966–982. [DOI] [PubMed] [Google Scholar]
- 50. Boyle DS, Khattar MM, Addinall SG, Lutkenhaus J, Donachie WD. ftsW is an essential cell‐division gene in Escherichia coli . Mol Microbiol. 1997;24:1263–1273. [DOI] [PubMed] [Google Scholar]
- 51. Mohammadi T, Sijbrandi R, Lutters M, et al. Specificity of the transport of lipid II by FtsW in Escherichia coli . J Biol Chem. 2014;289:14707–14718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ruiz N. Bioinformatics identification of MurJ (MviN) as the peptidoglycan lipid II flippase in Escherichia coli . Proc Natl Acad Sci U S A. 2008;105:15553–15557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Inoue A, Murata Y, Takahashi H, Tsuji N, Fujisaki S, Kato J. Involvement of an essential gene, MviN, in murein synthesis in Escherichia coli . J Bacteriol. 2008;190:7298–7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Taguchi A, Welsh MA, Marmont LS, et al. FtsW is a peptidoglycan polymerase that is functional only in complex with its cognate penicillin‐binding protein. Nat Microbiol. 2019;4:587–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Emami K, Guyet A, Kawai Y, et al. RodA as the missing glycosyltransferase in Bacillus subtilis and antibiotic discovery for the peptidoglycan polymerase pathway. Nat Microbiol. 2017;2:16253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Caffrey M. Crystallizing membrane proteins for structure‐function studies using lipidic mesophases. Biochem Soc Trans. 2011;39:725–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kuk ACY, Mashalidis EH, Lee SY. Crystal structure of the MOP flippase MurJ in an inward‐facing conformation. Nat Struct Mol Biol. 2017;24:24171–24176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kuk ACY, Hao A, Guan Z, Lee SY. Visualizing conformation transitions of the lipid II flippase MurJ. Nat Commun. 2019;10:1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zheng S, Sham LT, Rubino FA, et al. Structure and mutagenic analysis of the lipid II flippase MurJ from Escherichia coli . Proc Natl Acad Sci U S A. 2018;115:6709–6714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Egan AJF, Biboy J, van't Veer I, Breukink E, Vollmer W. Activities and regulation of peptidoglycan synthases. Philos Trans R Soc B Biol Sci. 2015;370:20150031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Peters K, Kannan S, Rao VA, et al. The redundancy of peptidoglycan carboxypeptidases ensures robust cell shape maintenance in Escherichia coli . MBio. 2016;7:e00819–e00816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Marie A, Guilmi D, Dideberg O, Vernet T. Functional characterization of penicillin‐binding protein 1b from Streptococcus pneumoniae . J Bacteriol. 2003;185:1650–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Marie A, Guilmi D, Dessen A, Dideberg O, Vernet T. Bifunctional penicillin‐binding proteins: Focus on the glycosyltransferase domain and its specific inhibitor moenomycin. Curr Pharm Biotechnol. 2010;3:63–75. [DOI] [PubMed] [Google Scholar]
- 64. Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P. The penicillin‐binding proteins: Structure and role in peptidoglycan biosynthesis. FEMS Microbiol Rev. 2008;32:234–258. [DOI] [PubMed] [Google Scholar]
- 65. Kim SJ, Chang J, Singh M. Peptidoglycan architecture of Gram‐positive bacteria by solid‐state NMR. Biochim Biophys Acta. 2015;1848:350–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Schneider T, Sahl HG. An oldie but a goodie—Cell wall biosynthesis as antibiotic target pathway. Int J Med Microbiol. 2010;300:161–169. [DOI] [PubMed] [Google Scholar]
- 67. Moon TM, D'Andréa ÉD, Lee CW, et al. The structures of penicillin binding protein 4 (PBP4) and PBP5 from Enterococci provide structural insights into β–lactam resistance. J Biol Chem. 2018;293:18574–18584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Vollmer W. Structural variation in the glycan strands of bacterial peptidoglycan. FEMS Microbiol Rev. 2008;32:287–306. [DOI] [PubMed] [Google Scholar]
- 69. Zapun A, Philippe J, Abrahams KA, et al. In vitro reconstitution of peptidoglycan assembly from the Gram‐positive pathogen Streptococcus pneumoniae . ACS Chem Biol. 2013;8:2688–2696. [DOI] [PubMed] [Google Scholar]
- 70. Morlot C, Straume D, Peters K, et al. Structure of the essential peptidoglycan amidotransferase MurT/GatD complex from Streptococcus pneumoniae . Nat Commun. 2018;9:3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Walsh JC, Angstmann CN, Bisson‐Filho AW, Garner EC, Duggin IG, Curmi PMG. Division plane placement in pleomorphic archaea is dynamically coupled to cell shape. Mol Microbiol. 2019;112:785–799. 10.1111/mmi.14316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. den Blaauwen T, Hamoen LW, Levin PA. The divisome at 25: The road ahead. Curr Opin Microbiol. 2017;36:85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lutkenhaus JF, Wolf‐Watz H, Donachie WD. Organization of genes in the ftsA‐envA region of the Escherichia coli genetic map and identification of a new fts locus (ftsZ). J Bacteriol. 1990;142:615–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Erickson H. FtsZ, a tubulin homologue in prokaryote cell division. Trends Cell Biol. 1997;7:362–367. [DOI] [PubMed] [Google Scholar]
- 75. Lutkenhaus J, Addinall SG. Bacterial cell division and the Z ring. Annu Rev Biochem. 1997;66:93–116. [DOI] [PubMed] [Google Scholar]
- 76. Löwe J, Amos LA. Crystal structure of the bacterial cell‐division protein FtsZ. Nature. 1998;391:203–206. [DOI] [PubMed] [Google Scholar]
- 77. Bisson‐Filho AW, Hsu YP, Squyres GR, et al. Treadmilling by FtsZ filaments drives peptidoglycan synthesis and bacterial cell division. Science. 2017;355:739–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Buss JA, Peters NT, Xiao J, Bernhardt TG. ZapA and ZapB form an FtsZ‐independent structure at midcell. Mol Microbiol. 2017;104:652–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Loose M, Mitchison TJ. The bacterial cell division proteins FtsA and FtsZ self‐organize into dynamic cytoskeletal patterns. Nat Cell Biol. 2014;16:38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Pazos M, Peters K, Casanova M, et al. Z‐ring membrane anchors associate with cell wall synthases to initiate bacterial cell division. Nat Commun. 2018;9:5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hamoen LW, Meile JC, de Jong W, Noirot P, Errington J. SepF, a novel FtsZ‐interacting protein required for a late step in cell division. Mol Microbiol. 2006;59:989–999. [DOI] [PubMed] [Google Scholar]
- 82. Rico AI, Krupka M, Vicente M. In the beginning, Escherichia coli assembled the proto‐ring: An initial phase of division. J Biol Chem. 2013;288:20830–20836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Haeusser DP, Margolin W. Splitsville: Structural and functional insights into the dynamic bacterial Z ring. Nat Rev Microbiol. 2016;14:305–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Robson SA, King GF. Domain architecture and structure of the bacterial cell division protein DivIB. Proc Natl Acad Sci U S A. 2006;103:6700–6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Buddelmeijer N, Beckwith J. A complex of the Escherichia coli cell division proteins FtsL, FtsB and FtsQ forms independently of its localization to the septal region. Mol Microbiol. 2004;52:1315–1327. [DOI] [PubMed] [Google Scholar]
- 86. Bottomley AL, Kabli AF, Hurd AF, Turner RD, Garcia‐Lara J, Foster SJ. Staphylococcus aureus DivIB is a peptidoglycan‐binding protein that is required for a morphological checkpoint in cell division. Mol Microbiol. 2014;94:1041–1064. [DOI] [PubMed] [Google Scholar]
- 87. Angeles DM, Macia‐Valero A, Bororquez LC, Scheffers DJ. The PASTA domains of Bacillus subtilis PBP2B stabilize the interaction of PBP2B with DivIB. bioRxiv. 2019; 10.1101/713677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Boes A, Olatunji S, Breukink E, Terrak M. Regulation of the peptidoglycan polymerase activity of PBP1b by antagonist actions of the core divisome proteins FtsBLQ and FtsN. MBio. 2019;10:e01912–e01918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kureisaite‐Ciziene D, Kureisaite‐Ciziene D, Varadajan A, et al. Structural analysis of the interaction between the bacterial cell division proteins FtsQ and FtsB. MBio. 2018;9:e01346–e01318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Lenarcic R, Halbedel S, Visser L, et al. Localisation of DivIVA by targeting to negatively curved membranes. EMBO J. 2009;28:2272–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Oliva MA, Halbedel S, Freund SM, et al. Features critical for membrane binding revealed by DivIVA crystal structure. EMBO J. 2010;29:1988–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Stahlberg H, Kutejová E, Muchová K, et al. Oligomeric structure of the Bacillus subtilis cell division protien DivIVA determined by transmission microscopy. Mol Microbiol. 2004;52:1281–1290. [DOI] [PubMed] [Google Scholar]
- 93. Halbedel S, Lewis RJ. Structural basis for interaction of DivIVA/GpsB proteins with their ligands. Mol Microbiol. 2019;111:1404–1415. [DOI] [PubMed] [Google Scholar]
- 94. Cleverley RM, Rismondo J, Lockhart‐Cairns MP, et al. Subunit arrangement in GpsB, a regulator of cell wall biosynthesis. Microb Drug Resist. 2016;22:446–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Rismondo J, Cleverley RM, Lane HV, et al. Structure of the bacterial cell division determinant GpsB and its interaction with penicillin‐binding proteins. Mol Microbiol. 2016;99:978–998. [DOI] [PubMed] [Google Scholar]
- 96. Cleverley RM, Rutter ZJ, Rismondo J, et al. The cell cycle regulator GpsB functions as cytosolic adaptor for multiple cell wall enzymes. Nat Commun. 2019;10:261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Eswara PJ, Brzozowski RS, Viola MG, et al. An essential Staphylococcus aureus cell division protein directly regulates FtsZ dynamics. Elife. 2018;7:e38856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Levin PA, Kurtser IG, Grossman AD. Identification and characterization of a negative regulator of FtsZ ring formation in Bacillus subtilis . Proc Natl Acad Sci U S A. 1999;96:9642–9647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Land AD, Luo Q, Levin PA. Functional domain analysis of the cell division inhibitor EzrA. PLoS One. 2014;9:e102616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Singh JK, Makde RD, Kumar V, Panda D. A membrane protein, EzrA, regulates assembly dynamics of FtsZ by interacting with the C‐terminal tail of FtsZ. Biochemistry. 2007;46:11013–11022. [DOI] [PubMed] [Google Scholar]
- 101. Claessen D, Emmins R, Hamoen LW, Daniel RA, Errington J, Edwards DH. Control of the cell elongation–division cycle by shuttling of PBP1 protein in Bacillus subtilis . Mol Microbiol. 2008;68:1029–1046. [DOI] [PubMed] [Google Scholar]
- 102. Steele VR, Bottomley AL, Garcia‐Lara J, Kasturiarachchi J, Foster SJ. Multiple essential roles for EzrA in cell division of Staphylococcus aureus . Mol Microbiol. 2011;80:542–555. [DOI] [PubMed] [Google Scholar]
- 103. Cleverley RM, Barrett JR, Baslé A, et al. Structure and function of a spectrin‐like regulator of bacterial cytokinesis. Nat Commun. 2014;5:5421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Cleverley R, Lewis RJ. EzrA: A spectrin‐like scaffold in the bacterial cell division machinery. Microb Cell. 2015;2:59–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Krupka M, Sobrinos‐Sanguino M, Jiménez M, Rivas G, Margolin W. Escherichia coli ZipA organizes FtsZ polymers into dynamic ring‐like protofilament structures. MBio. 2018;9:e01008–e01018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Hale CA, de Boer PA. Direct binding of FtsZ to ZipA, an essential component of the septal ring structure that mediates cell division in E. coli . Cell. 1997;88:175–185. [DOI] [PubMed] [Google Scholar]
- 107. Geissler B, Elraheb D, Margolin W. A gain‐of‐function mutation in ftsA bypasses the requirement for the essential cell division gene zipA in Escherichia coli . Proc Natl Acad Sci U S A. 2003;100:4197–4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Pazos M, Natale P, Vicente M. A specific role for the ZipA protein in cell division. J Biol Chem. 2013;288:3219–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Chen Y, Huang H, Osawa M, Erickson HP. ZipA and FtsA* stabilize FtsZ‐GDP miniring structures. Sci Rep. 2017;7:3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Waxman DJ, Strominger JL. Penicillin‐binding proteins and the mechanism of action of beta‐lactam antibiotics. Annu Rev Biochem. 1983;52:825–869. [DOI] [PubMed] [Google Scholar]
- 111. Cho H, Uehara T, Bernhardt TG. Beta‐lactam antibiotics induce a lethal malfunctioning of the bacterial cell wall synthesis machinery. Cell. 2014;159:1300–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Peacock SJ, Paterson GK. Mechanisms of methicillin resistance in Staphylococcus aureus . Annu Rev Biochem. 2015;84:577–601. [DOI] [PubMed] [Google Scholar]
- 113. Foster TJ. Antibiotic resistance in Staphylococcus aureus. Current status and future prospects. FEMS Microbiol Rev. 2017;41:430–449. [DOI] [PubMed] [Google Scholar]
- 114. Otero LH, Rojas‐Altuve A, Llarrull LI, et al. How allosteric control of Staphylococcus aureus penicillin binding protein 2a enables methicillin resistance and physiological function. Proc Natl Acad Sci U S A. 2013;110:16808–16813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Mahasenan KV, Molina R, Bouley R, et al. Conformational dynamics in penicillin‐binding protein 2a of methicillin‐resistant Staphylococcus aureus, allosteric communication network and enablement of catalysis. J Am Chem Soc. 2017;139:2102–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Fernandes R, Amador P, Prudêncio C. β‐Lactams: Chemical structure, mode of action and mechanisms of resistance. Rev Med Microbiol. 2013;24:7–17. [Google Scholar]
- 117. Bonomo RA. β‐Lactamases: A focus on current challenges. Cold Spring Harb Perspect Med. 2017;7:a025239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Hall BG, Barlow M. Evolution of the serine β‐lactamases: Past, present and future. Drug Resist Updat. 2004;7:111–123. [DOI] [PubMed] [Google Scholar]
- 119. Yong D, Toleman MA, Giske CG, et al. Characterization of a new metallo‐lactamase gene, blaNDM‐1, and a novel erythromycin esterase gene carried on a unique genetic structure in Klebsiella pneumoniae sequence type 14 from India. Antimicrob Agents Chemother. 2009;53:5046–5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Zmarlicka MT, Nailor MD, Nicolau DP. Impact of the New Delhi metallo‐beta‐lactamase on beta‐lactam antibiotics. Infect Drug Resist. 2015;8:297–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Ma B, Fang C, Lu L, et al. The antimicrobial peptide thanatin disrupts the bacterial outer membrane and inactivates the NDM‐1 metallo‐β‐lactamase. Nat Commun. 2019;10:3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Abraham EP, Chain E. An enzyme from bacteria able to destroy penicillin. Nature. 1940;146:837–837. [PubMed] [Google Scholar]
- 123. Andreu JM, Schaffner‐Barbero C, Huecas S, et al. The antibacterial cell division inhibitor PC190723 is an FtsZ polymer‐stabilizing agent that induces filament assembly and condensation. J Biol Chem. 2010;285:14239–14246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Elsen NL, Lu J, Parthasarathy G, et al. Mechanism of action of the cell‐division inhibitor PC190723: Modulation of FtsZ assembly cooperativity. J Am Chem Soc. 2012;134:12342–12345. [DOI] [PubMed] [Google Scholar]
- 125. Haydon DJ, Stokes NR, Ure R, et al. An inhibitor of FtsZ with potent and selective anti‐Staphylococcal activity. Science. 2008;321:1673–1675. [DOI] [PubMed] [Google Scholar]