

Abstract
Many picornaviruses hijack the Golgi resident Acyl‐coenzyme A binding domain containing 3 (ACBD3) protein in order to recruit the phosphatidylinositol 4‐kinase B (PI4KB) to viral replication organelles (ROs). PI4KB, once recruited and activated by ACBD3 protein, produces the lipid phosphatidylinositol 4‐phosphate (PI4P), which is a key step in the biogenesis of viral ROs. To do so, picornaviruses use their small nonstructural protein 3A that binds the Golgi dynamics domain of the ACBD3 protein. Here, we present the analysis of the highly flexible ACBD3 proteins and the viral 3A protein in solution using small‐angle X‐ray scattering and computer simulations. Our analysis revealed that both the ACBD3 protein and the 3A:ACBD3 protein complex have an extended and flexible conformation in solution.
Keywords: ACBD3, coarse‐grained simulations, host factor, intrinsically disordered regions, picornavirus, RNA virus, small‐angle X‐ray scattering (SAXS)
1. INTRODUCTION
Positive‐sense single‐stranded ribonucleic acid (+ RNA) viruses replicate at the surface of intracellular membranes where they establish their so‐called viral replication organelles (ROs), a membranous platform for viral replication with complex membrane topology.1 ROs provide shelter from innate immunity and also serve as a rendezvous point for viral and host proteins and for the assembly of new virions. A key step in the biogenesis of ROs is the recruitment of a phosphatidylinositol 4‐kinase (PI4K).2 Enteroviruses and kobuviruses, both members of the Picornaviridae family, use their small nonstructural 3A protein to recruit the phosphatidylinositol 4‐kinase B (PI4KB) kinase. 3A is a small (<100 AAs) protein with a predicted C‐terminal transmembrane helix.3 Its cytoplasmic part tightly interacts with the Acyl‐coenzyme A binding domain containing 3 (ACBD3) protein.4, 5
ACBD3 protein is a Golgi‐resident multifunctional, multidomain protein involved in maintenance of the Golgi apparatus.6 One of its key functions is the recruitment of the lipid kinase PI4KB,7 which together with the PI4K2A enzyme is responsible for the synthesis of the lipid hallmark of the Golgi—the phosphatidylinositol 4‐phosphate (PI4P) lipid.8, 9 ACBD3 consists of several domains connected by flexible linkers (Figure 1). Besides the N‐terminal ACBD domain, the protein also contains a Q (glutamine‐rich) domain that forms a helical bundle, which binds the most proximal N‐terminal helix of PI4KB with submicromolar affinity.7 The Golgi dynamics (GOLD) domain is localized at the C terminus and serves as a protein‐binding site. It is worth noting that most ACBD3 interacting proteins (giantin, TSPO, Rhes, PKARIα, and Golgin‐160) have been reported to bind to the GOLD domain.6, 10
Figure 1.
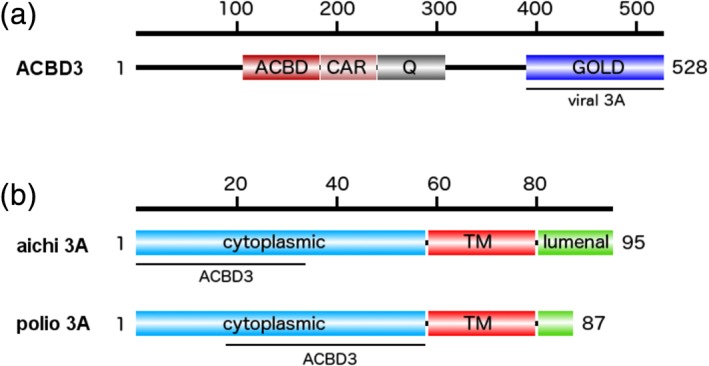
Schematic representation of the ACBD3 and Aichi 3A proteins with the binding sites for 3A and ACBD3 proteins highlighted
Recently, insights into interactions of the 3A protein from several picornaviruses, including the Aichi virus (genus Kobuvirus) and four representative enteroviruses (poliovirus, enterovirus A71, enterovirus D68, and rhinovirus B14), with the GOLD domain was gained crystallographically.11, 12 This structural analysis revealed that the viral 3A acts as a molecular harness—it wraps around the GOLD domain of ACBD3 and pins it down to the target membrane, which in turn leads to recruitment of the PI4KB lipid kinase and to the production of the PI4P. This lipid can be exchanged for other lipids such as cholesterol in order to build membranes of required properties for viral replication13 or to directly recruit viral RNA polymerase 3Dpol.14, 15
Crystallographic analysis of the full‐length ACBD3 and 3A was not possible because the ACBD3 protein contains large intrinsically disordered regions that connect the well‐defined domains. However, small‐angle X‐ray scattering (SAXS) combined with computer simulations is well suited for the analysis of large flexible proteins or protein complexes.16, 17 We used a combination of SAXS experiments and coarse‐grained molecular simulations to analyze the ACBD3 protein and its complex with the Aichi virus 3A protein.
2. MATERIALS AND METHODS
2.1. Protein expression and purification
Expression and purification of the full‐length ACBD3 and 3A protein from the human Aichi virus were described earlier.11 Briefly, the sequence encoding the full‐length ACBD3 protein was subcloned into the pRSFD plasmid with 6xHis tag fused to GB1 solubilization tag and TEV protease cleavage site.18 For the expression of the 3A protein, the sequence encoding for Aichi virus 3A (amino acids 2–58) was also inserted to the pRSFD plasmid with 6xHis tag fused to GB1 solubilization tag and TEV protease cleavage site and coexpressed with the GOLD domain from ACBD3 (ACBD3 amino acids 364–528) from the same plasmid because interaction of these partners stabilizes the 3A protein and protects it from degradation in bacteria. Proteins were expressed in E. coli BL21 Star in autoinduction ZY medium. Cells were lysed by sonication in lysis buffer (50 mM Tris, pH 8.0, 300 mM NaCl, 20 mM imidazole, 3 mM β‐mercaptoethanol, 10% glycerol), centrifuged, and the supernatant was used for the first step of purification by affinity chromatography (Ni‐NTA, Macherey‐Nagel). The 6xHis‐GB1 solubilization tag was cleaved by the TEV protease, and the proteins were further purified by size exclusion chromatography using Superdex 200 HiLoad 16/60 column (GE Healthcare) in 10 mM Tris, pH 8.0, 200 mM NaCl, and 3 mM β‐mercaptoethanol. For the preparation of samples for SAXS measurements, ACBD3 alone or incubated with 6× molar excess of the 3A protein was repurified on the Superdex 200 Increase 10/300 GL column (GE Healthcare) in 10 mM Tris at pH 7.4, 150 mM NaCl, and 2 mM tris(2‐carboxyethyl)phosphine. Purified proteins were concentrated, aliquoted, flash frozen in liquid nitrogen and stored in −80°C until needed.
2.2. SAXS data processing
The SAXS data were collected using BioSAXS Beamline BM29 (ESRF Grenoble) equipped with the Pilatus 1M detector (Dectris) at the ACBD3 protein concentrations of 5.8, 3.4, and 2.6 mg/mL. For each of the three concentrations, the SAXS data were frame averaged and buffer subtracted using PRIMUS.19 The resulting three data sets were then extrapolated to the dilute limit using the same software (Figure S1). The extrapolated SAXS data set was next used to model conformations of the ACBD3 protein, as detailed in the following section. In parallel, SAXS data were collected for the ACBD3:3A/AiV protein complex at concentrations of 4.7, 2.7, 1.8, and 0.9 mg/mL. These SAXS data were frame averaged, buffer subtracted, and subsequently extrapolated to the dilute limit in the same way that the ACBD3 SAXS data were processed (Figure S2). The extrapolated SAXS data set was used to model conformations of the ACBD3:3A/AiV protein complex in solution.
2.3. Molecular modeling and simulations
To efficiently sample conformations of the full‐length ACBD3 protein in solution, we used a coarse‐grained simulation model introduced by Kim and Hummer (KH).20 The KH model is equipped with a transferable energy function and devised to simulate conformational ensembles of large multidomain proteins and multiprotein complexes. It has been successfully applied to protein systems ranging from the endosomal sorting complexes required for transport membrane‐binding trafficking machinery21, 22, 23 to multidomain protein kinases24 and kinases in dynamic complexes with phosphatases.25, 26 Most recently, the KH model has been also used to study multidomain carbohydrate‐active enzymes,27, 28 flexible protein complexes containing lipid kinases,29, 30 and complexes of cell adhesion proteins involved in immunological responses.31 In the framework of the KM model, the solvent is implicit and amino acid residues are represented as single beads centered at α‐C atoms. The interactions between the pairs of residue beads are quantified by Lennard‐Jones (LJ) potentials and long‐range electrostatic potentials. The electrostatic interactions are described by the Debye‐Hückel equation at a screening length of 1 nm, which corresponds to physiological salt concentrations. The LJ interaction parameters are adapted from knowledge‐based statistical contact potentials and scaled relative to the electrostatic interaction energy. Importantly, folded‐protein domains are treated as rigid bodies, whereas disordered segments (e.g., flexible loops, linkers, and termini) are represented by bead polymers. In addition to the LJ potentials and the electrostatic potentials, the potential energy of the disordered segments contains contributions from bond‐length potentials, bond‐angle potentials, and torsion‐angle potentials.
As in recent coarse‐grained simulations of the ACBD3:PI4KB complex,30 here the structural model, that is, the full‐length ACBD3 protein comprises four rigid domains: (a) the crystallized portion of the GOLD domain with the PDB code 5LZ1 (residue numbers ranging 364–448 and 473–528); (b) the glutamine‐rich domain (Q domain, residue numbers ranging 241–308) given by the nuclear magnetic resonance structure with the PDB code 2N73; (c) the charged amino acid region (CAR, residue numbers ranging 180–240) represented by a single alpha‐helix, as proposed in Reference 32; and (d) a homology model of the ACBD domain (residue numbers ranging 82–179) based on the PDB entry 3FLV (29% sequence identity, 96% sequence coverage). All of the missing loops, disordered linkers, and terminal segments were built into the ACBD3 model using ModLoop33 and simulated as chains of amino acid beads with appropriate stretching, bending, and torsional potentials.20
Within the framework of the KH model, as described above, we performed replica exchange Monte Carlo (REMC) simulations with 16 replicas at temperatures ranging from 300 to 750 K. The basic MC moves were translations and rotations of the rigid domains. For flexible segments, in addition to local MC moves on each of the residue beads, crankshaft moves were employed to enhance sampling. After the initial 105 MC sweeps for equilibration, the REMC production run comprised 5 × 106 MC sweeps. The ACBD3 protein conformations were recorded every 103 MC sweeps. We thus obtained a pool of 8 × 104 conformations for further analysis.
The scattering intensity profile was computed for each of the simulated conformations individually using an algorithm codeveloped with the ensemble refinement of SAXS (EROS) method.23 The scattering intensity profile corresponding to a set S of conformations was then obtained from the scattering intensity profiles I k(q) of the individual conformations belonging to set S, that is,
The discrepancy between the experimental SAXS data, I exp(q), and the scattering intensity profile I S(q) of a given set of simulated conformations was quantified by
Here, N q is the number of SAXS data points, σ(q) is the statistical error of the extrapolated SAXS intensity I exp(q), whereas the scale factor a and offset b result from the conditions ∂χ 2/∂a = 0 and ∂χ 2/∂b = 0. The offset parameter b accounts for inaccuracies in the buffer subtraction and/or uncertainties in the electron density contrast. We applied a minimum‐ensemble method21, 34 to identify the smallest set of conformations consistent with the experimental SAXS data. We treated the electron density contrast of the protein hydration shell as a fitting parameter and varied it in the range between 0.01 and 0.05 e/Å3.
We performed analogous simulations and analyses for the ACBD3:3A/AiV protein complex. In the coarse‐grained simulations, the GOLD domain in complex with the N‐terminal part of the nonstructural 3A protein was treated as one rigid body based on the crystal structure with the PDB code 5LZ3. The C‐terminal tail of the 3A protein was simulated as a chain of amino acid beads. Other portions of the ACBD3 protein were represented in the manner as previously in the coarse‐grained simulations of the full‐length ACBD3 protein.
From the REMC simulations, we obtained a pool of 1.6 × 105 conformations of the ACBD3:3A/AiV protein complex. We postprocessed and analyzed these conformations in the same way as the simulated conformations of the full‐length ACBD3 protein in the absence of the 3A/AiV protein. Finally, we identified the smallest set of conformations consistent with the experimental SAXS data using the minimum‐ensemble method.21, 34
3. RESULTS AND DISCUSSION
We performed SAXS measurements at the ACBD3 concentrations of 5.8, 3.4, and 2.6 mg/mL. The resulting SAXS intensity profiles, I(q), were found to overlay to a great extent after rescaling (Figure S1a). However, a closer inspection revealed small systematic differences between the three I(q) data sets at the smallest scattering angles (Figure S1a–d). These systematic differences most likely reflect interactions between protein molecules in solution. Therefore, we extrapolated the three I(q) profiles to the dilute limit, that is, the formal limit of the protein concentration approaching zero (Figure S1e,f, and Figure 2a). The Guinier plot (Figure 2a, inset) of the extrapolated SAXS data yields a radius of gyration of about 69 Å, which suggest that the full‐length ACBD3 protein can attain relatively extended conformations in solution.
Figure 2.
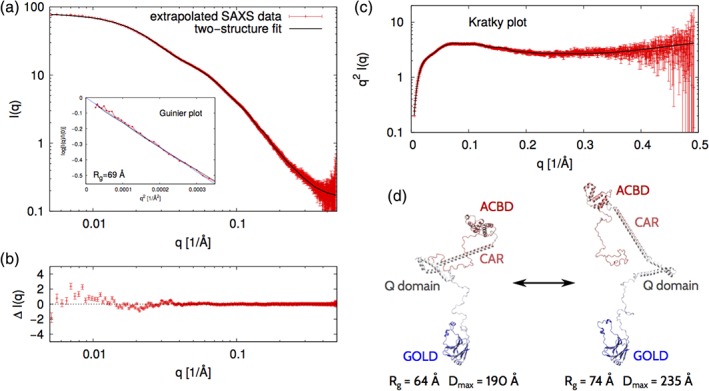
SAXS analysis and modeling of the full‐length ACBD3 protein. (a) The points in red constitute the SAXS intensity profile that results from extrapolating to the dilute limit three data sets collected at protein concentrations of 5.8, 3.4, and 2.6 mg/mL. The curve in black represents the theoretical SAXS intensity profile corresponding to an ACBD3 conformational ensemble that satisfactorily fits the experimental SAXS data (χ 2 = 1.7). The logarithmic scale is used on both the vertical and horizontal axes. The inset shows the Guinier plot, that is, log(I(q)/I(0)) versus q 2. The value range on the horizontal axis corresponds to the region qR g < 1.3 where the Guinier approximation is valid for globular proteins. The dashed line corresponds to the Guinier approximation with R g = 69 Å. (b) The difference between the experimental and simulated SAXS intensity profiles as computed from the data shown in panel A. (c) The Kratky plot, that is, q 2 I(q) versus q, corresponding to the data shown in panel A. (d) Two structural models that constitute the minimal representation of the ACBD3 conformational ensemble. These two models jointly yield the SAXS intensity profile shown in panel A in black and, thus, fit the experimental SAXS data with χ 2 = 1.7. The ACBD3 protein domains are indicated
We used the extrapolated SAXS intensity profile in combination with computer simulations to identify representative conformations of the full‐length ACBD3 protein in solution. To this end, we performed coarse‐grained simulations of the full‐length ACBD3 and quantitatively compared the SAXS intensity profiles of the simulated conformations to the experimental SAXS intensity profile. It turned out that none of the individual conformations that were obtained from the simulations could be fitted to the extrapolated SAXS data with χ 2 < 2.9. Since the full‐length ACBD3 protein contains disordered linkers, loops, and termini, we expected it to exhibit conformational flexibility and diversity. Therefore, we applied an EROS method,21, 34 and thus identified two conformations that jointly could account for the extrapolated SAXS intensity profile (Figure 2a). We found that the differences between the experimental and simulated SAXS intensity profiles were very small in the whole range of scattering angles (Figure 2b), yielding χ 2 = 1.7. Importantly, the Kratky plot (Figure 2c) shows the typical features of partially disordered proteins, which additionally supports our conclusion that the full‐length ACBD3 protein is flexible in solution. We found out that the minimum ensemble consistent with the experimental SAXS data was represented by two conformations, namely, one “extended” conformation with R g = 74 Å and D max = 235 Å, and one “compact” conformation with R g = 64 Å and D max = 190 Å (Figure 2d). These two conformations taken together with equal statistical weights fit the extrapolated SAXS intensity profile with χ 2 = 1.7 (Figure 2a,b). Interestingly, the individual domains of the ACBD3 protein do not make any extensive contacts in neither of these conformations.
Since the full‐length ACBD3 protein contains flexible linkers, loops and termini, it can attain multiple conformations, ranging from extended to compact ones. The two conformations identified in our SAXS analysis (Figure 2d) cannot fully represent the entire conformational ensemble of the ACBD3 protein in solution. However, the vast differences between the two conformations (Figure 2d) provide a glimpse of the conformational diversity of this highly flexible protein.
We also performed SAXS experiments on the ACBD3:3A/AiV complex at protein concentrations of 4.7, 2.7, 1.8, and 0.9 mg/mL. These protein concentrations are more than an order of magnitude larger than the dissociation constant of the ACBD3:3A/AiV protein complex (K d in the low micromolar range).11 We extrapolated the four I(q) profiles to the dilute limit (Figure S2 and Figure 3a). The Guinier plot (Figure 3a, inset) of the extrapolated SAXS data shows that the radius of gyration is about 70.5 Å, which is 1.5 Å more than the radius of gyration of the ACBD3 protein in the absence of the 3A protein, and which indicates that the ACBD3:3A/AiV protein complex attains extended conformations in solution.
Figure 3.
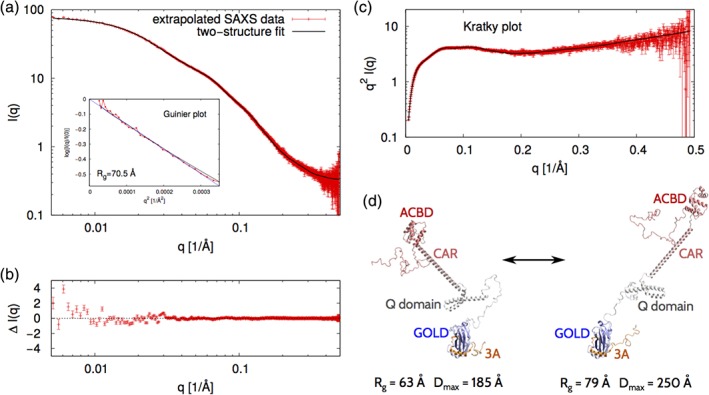
Analogous to Figure 2 but for the full‐length ACBD3:3A protein complex
The extrapolated SAXS intensity profile for ACBD3:3A/AiV (Figure 3a) looks very similar to the extrapolated SAXS intensity profile for ACBD3 (Figure 2a). Since each of the two SAXS intensity profiles contains the same number of data points (N q = 1,034), one way to quantify their similarity is to calculate χ 2. It turns out that χ 2 = 3.1 for these two SAXS curves, which indicates that the two SAXS profiles are clearly distinguishable within the experimental error despite their apparent similarity.
We next used coarse‐grained molecular simulations to structurally interpret the extrapolated SAXS intensity profile. The coarse‐grained simulations of the ACBD3:3A/AiV protein complex provided a large and diverse pool of conformations, but no single conformation from this pool was fully consistent with the experimental SAXS data (χ 2 > 3.4 for any of the simulated conformations). However, we found that a set of two conformations with equal statistical weights fitted the SAXS data with χ 2 = 1.7 (Figure 3a). We checked that the differences between the experimental and simulated SAXS intensity profiles were very small in the whole range of scattering angles (Figure 3b). The Kratky plot (Figure 3c) shows the typical features of partially disordered proteins, which supports our conclusion that the ACBD3:3A protein complex is flexible in solution. The two representative conformations of the ACBD3:3A protein complex are quite different, namely, the “extended” conformation has R g = 79 Å and D max = 250 Å, whereas the “compact” conformation has R g = 63 Å and D max = 185 Å (Figure 3d). However, neither of these conformations involves any extensive interdomain contacts. These observations indicate that the ACBD3:3A protein complex exhibits large conformational fluctuations in solution.
Taken together, our data clearly show that the ACBD3 protein is extended and flexible in solution—in both the presence and absence of the Aichi virus 3A protein. We did not observe any extensive contacts between the individual domains of the ACBD3 protein alone or when in complex with viral 3A protein, which implies that the binding of the Aichi virus 3A protein to the ACBD3 protein does not significantly affect the conformations of ACBD3 in solution. Our recent SAXS studies30 show that the ACBD3:PI4KB protein complex exhibits large conformational fluctuations and little interdomain contacts. Therefore, we also predict that the binding of ACBD3 to PI4KB should not significantly affect the conformational diversity of ACBD3 in solution.
ACBD3 is essential for the replication of all enteroviruses and kobuviruses, which have been tested in this respect.4, 35, 36, 37, 38 These results imply that viruses do not interfere with the conformation of ACBD3, but are consistent with the hypothesis that viruses use ACBD3 to recruit the lipid kinase PI4KB. This is also consistent with the fact that these viruses are sensitive to chemical inhibition of PI4KB.39, 40, 41, 42 However, because the 3A recruitment of ACBD3 also changes the cellular localization of ACBD3, it could also interfere with other, Golgi related, functions of ACBD3 such as intracellular transport for the benefit of the virus.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Appendix S1: Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the BioSAXS Beamline BM29 (ESRF Grenoble) and EMBL SAXS beamline P12 (Petra III DESY, Hamburg) for allocation of experimental beamtime. The project was supported by Czech Science Foundation grant number 17‐05200S (to EB), by the OP RDE; Project: “Chemical biology for drugging undruggable targets (ChemBioDrug)” (No. CZ.02.1.01/0.0/0.0/16_019/0000729) from the European Regional Development Fund, and by the National Science Centre, Poland, grant number 2016/21/B/NZ1/00006 (to BR). The Academy of Sciences of Czech Republic support (RVO:61388963) is also acknowledged.
Chalupska D, Różycki B, Klima M, Boura E. Structural insights into Acyl‐coenzyme A binding domain containing 3 (ACBD3) protein hijacking by picornaviruses. Protein Science. 2019;28:2073–2079. 10.1002/pro.3738
Funding information European Regional Development Fund, Grant/Award Number: CZ.02.1.01/0.0/0.0/16_019/0000729; National Science Centre, Poland; Czech Science Foundation, Grant/Award Number: 17‐05200S
REFERENCES
- 1. Harak C, Lohmann V. Ultrastructure of the replication sites of positive‐strand RNA viruses. Virology. 2015;479:418–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Altan‐Bonnet N, Balla T. Phosphatidylinositol 4‐kinases: Hostages harnessed to build panviral replication platforms. Trends Biochem Sci. 2012;37:293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fujita K, Krishnakumar SS, Franco D, Paul AV, London E, Wimmer E. Membrane topography of the hydrophobic anchor sequence of poliovirus 3A and 3AB proteins and the functional effect of 3A/3AB membrane association upon RNA replication. Biochemistry. 2007;46:5185–5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Greninger AL, Knudsen GM, Betegon M, Burlingame AL, Derisi JL. The 3A protein from multiple picornaviruses utilizes the golgi adaptor protein ACBD3 to recruit PI4KIIIbeta. J Virol. 2012;86:3605–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sasaki J, Ishikawa K, Arita M, Taniguchi K. ACBD3‐mediated recruitment of PI4KB to picornavirus RNA replication sites. EMBO J. 2012;31:754–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fan J, Liu J, Culty M, Papadopoulos V. Acyl‐coenzyme a binding domain containing 3 (ACBD3; PAP7; GCP60): An emerging signaling molecule. Prog Lipid Res. 2010;49:218–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Klima M, Toth DJ, Hexnerova R, et al. Structural insights and in vitro reconstitution of membrane targeting and activation of human PI4KB by the ACBD3 protein. Sci Rep. 2016;6:23641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boura E, Nencka R. Phosphatidylinositol 4‐kinases: Function, structure. and inhibition Exp Cell Res. 2015;337:136–145. [DOI] [PubMed] [Google Scholar]
- 9. Baumlova A, Chalupska D, Rozycki B, et al. The crystal structure of the phosphatidylinositol 4‐kinase IIalpha. EMBO Rep. 2014;15:1085–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sbodio JI, Paul BD, Machamer CE, Snyder SH. Golgi protein ACBD3 mediates neurotoxicity associated with Huntington's disease. Cell Rep. 2013;4:890–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Klima M, Chalupska D, Rozycki B, et al. Kobuviral non‐structural 3A proteins act as molecular harnesses to hijack the host ACBD3 protein. Structure. 2017;25:219–230. [DOI] [PubMed] [Google Scholar]
- 12. Horova V, Lyoo H, Rozycki B, et al. Convergent evolution in the mechanisms of ACBD3 recruitment to picornavirus replication sites. PLoS Pathog. 2019;15:e1007962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dorobantu CM, Albulescu L, Harak C, et al. Modulation of the host lipid landscape to promote RNA virus replication: The picornavirus encephalomyocarditis virus converges on the pathway used by hepatitis C virus. PLoS Pathog. 2015;11:e1005185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hsu NY, Ilnytska O, Belov G, et al. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell. 2010;141:799–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dubankova A, Humpolickova J, Klima M, Boura E. Negative charge and membrane‐tethered viral 3B cooperate to recruit viral RNA dependent RNA polymerase 3D (pol). Sci Rep. 2017;7:17309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rozycki B, Boura E. Large, dynamic, multi‐protein complexes: A challenge for structural biology. J Phys Condens Matter. 2014;26:463103. [DOI] [PubMed] [Google Scholar]
- 17. Peti W, Page R, Boura E, Rozycki B. Structures of dynamic protein complexes: Hybrid techniques to study MAP kinase complexes and the ESCRT system. Methods Mol Biol. 2018;1688:375–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Klima M, Baumlova A, Chalupska D, et al. The high‐resolution crystal structure of phosphatidylinositol 4‐kinase IIbeta and the crystal structure of phosphatidylinositol 4‐kinase IIalpha containing a nucleoside analogue provide a structural basis for isoform‐specific inhibitor design. Acta Crystallogr. 2015;D71:1555–1563. [DOI] [PubMed] [Google Scholar]
- 19. Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. PRIMUS: A windows PC‐based system for small‐angle scattering data analysis. J Appl Cryst. 2003;36:1277–1282. [Google Scholar]
- 20. Kim YC, Hummer G. Coarse‐grained models for simulations of multiprotein complexes: Application to ubiquitin binding. J Mol Biol. 2008;375:1416–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Boura E, Rozycki B, Herrick DZ, et al. Solution structure of the ESCRT‐I complex by small‐angle X‐ray scattering, EPR and FRET spectroscopy. Proc Natl Acad Sci U S A. 2011;108:9437–9442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boura E, Hurley JH. Structural basis for membrane targeting by the MVB12‐associated beta‐prism domain of the human ESCRT‐I MVB12 subunit. Proc Natl Acad Sci U S A. 2012;109:1901–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rozycki B, Kim YC, Hummer G. SAXS ensemble refinement of ESCRT‐III CHMP3 conformational transitions. Structure. 2011;19:109–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Leonard TA, Rozycki B, Saidi LF, Hummer G, Hurley JH. Crystal structure and allosteric activation of protein kinase C betaII. Cell. 2011;144:55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Francis DM, Rozycki B, Koveal D, Hummer G, Page R, Peti W. Structural basis of p38alpha regulation by hematopoietic tyrosine phosphatase. Nat Chem Biol. 2011;7:916–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Francis DM, Rozycki B, Tortajada A, Hummer G, Peti W, Page R. Resting and active states of the ERK2:HePTP complex. J Am Chem Soc. 2011;133:17138–17141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rozycki B, Cieplak M, Czjzek M. Large conformational fluctuations of the multi‐domain xylanase Z of clostridium thermocellum. J Struct Biol. 2015;191:68–75. [DOI] [PubMed] [Google Scholar]
- 28. Rozycki B, Cazade PA, O'Mahony S, Thompson D, Cieplak M. The length but not the sequence of peptide linker modules exerts the primary influence on the conformations of protein domains in cellulosome multi‐enzyme complexes. Phys Chem Chem Phys. 2017;19:21414–21425. [DOI] [PubMed] [Google Scholar]
- 29. Chalupska D, Eisenreichova A, Rozycki B, et al. Structural analysis of phosphatidylinositol 4‐kinase IIIbeta (PI4KB) ‐ 14‐3‐3 protein complex reveals internal flexibility and explains 14‐3‐3 mediated protection from degradation in vitro. J Struct Biol. 2017;200:36–44. [DOI] [PubMed] [Google Scholar]
- 30. Chalupska D, Rozycki B, Humpolickova J, Faltova L, Klima M, Boura E. Phosphatidylinositol 4‐kinase IIIbeta (PI4KB) forms highly flexible heterocomplexes that include ACBD3, 14‐3‐3, and Rab11 proteins. Sci Rep. 2019;9:567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Steinkuhler J, Rozycki B, Alvey C, et al. Membrane fluctuations and acidosis regulate cooperative binding of ‘marker of self’ protein CD47 with the macrophage checkpoint receptor SIRPα. J Cell Sci. 2018;132(4):jcs216770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Suveges D, Gaspari Z, Toth G, Nyitray L. Charged single alpha‐helix: A versatile protein structural motif. Proteins. 2009;74:905–916. [DOI] [PubMed] [Google Scholar]
- 33. Fiser A, Sali A. ModLoop: Automated modeling of loops in protein structures. Bioinformatics. 2003;19:2500–2501. [DOI] [PubMed] [Google Scholar]
- 34. Boura E, Rozycki B, Chung HS, et al. Solution structure of the ESCRT‐I and ‐II supercomplex: Implications for membrane budding and scission. Structure. 2012;20:874–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Greninger AL, Knudsen GM, Betegon M, Burlingame AL, DeRisi JL. ACBD3 interaction with TBC1 domain 22 protein is differentially affected by enteroviral and kobuviral 3A protein binding. MBio. 2013;4:e00098–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ishikawa‐Sasaki K, Sasaki J, Taniguchi K. A complex comprising phosphatidylinositol 4‐kinase IIIbeta, ACBD3, and Aichi virus proteins enhances phosphatidylinositol 4‐phosphate synthesis and is critical for formation of the viral replication complex. J Virol. 2014;88:6586–6598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lei X, Xiao X, Zhang Z, et al. The Golgi protein ACBD3 facilitates enterovirus 71 replication by interacting with 3A. Sci Rep. 2017;7:44592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lyoo H, van der Schaar HM, Dorobantu CM, Rabouw HH, Strating J, van Kuppeveld FJM. ACBD3 is an essential pan‐enterovirus host factor that mediates the interaction between viral 3A protein and cellular protein PI4KB. MBio. 2019;10:e02742–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mejdrova I, Chalupska D, Kogler M, et al. Highly selective phosphatidylinositol 4‐kinase IIIbeta inhibitors and structural insight into their mode of action. J Med Chem. 2015;58:3767–3793. [DOI] [PubMed] [Google Scholar]
- 40. Mejdrova I, Chalupska D, Plackova P, et al. Rational design of novel highly potent and selective phosphatidylinositol 4‐kinase IIIbeta (PI4KB) inhibitors as broad‐spectrum antiviral agents and tools for chemical biology. J Med Chem. 2017;60:100–118. [DOI] [PubMed] [Google Scholar]
- 41. Rutaganira FU, Fowler ML, McPhail JA, et al. Design and structural characterization of potent and selective inhibitors of phosphatidylinositol 4 kinase IIIbeta. J Med Chem. 2016;59:1830–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Humpolickova J, Mejdrova I, Matousova M, Nencka R, Boura E. Fluorescent inhibitors as tools to characterize enzymes: Case study of the lipid kinase phosphatidylinositol 4‐kinase IIIbeta (PI4KB). J Med Chem. 2017;60:119–127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supplementary Material