

Abstract
Current research on catalysts for proton exchange membrane fuel cells (PEMFC) is based on obtaining higher catalytic activity than platinum particle catalysts on porous carbon. In search of a more sustainable catalyst other than platinum for the catalytic conversion of water to hydrogen gas, a series of nanoparticles of transition metals viz., Rh, Co, Fe, Pt and their composites with functionalized graphene such as RhNPs@f-graphene, CoNPs@f-graphene, PtNPs@f-graphene were synthesized and characterized by SEM and TEM techniques. The SEM analysis indicates that the texture of RhNPs@f-graphene resemble the dispersion of water droplets on lotus leaf. TEM analysis indicates that RhNPs of <10 nm diameter are dispersed on the surface of f-graphene. The air-stable NPs and nanocomposites were used as electrocatalyts for conversion of acidic water to hydrogen gas. The composite RhNPs@f-graphene catalyses hydrogen gas evolution from water containing p-toluene sulphonic acid (p-TsOH) at an onset reduction potential, Ep, −0.117 V which is less than that of PtNPs@f-graphene (Ep, −0.380 V) under identical experimental conditions whereas the onset potential of CoNPs@f-graphene was at Ep, −0.97 V and the FeNPs@f-graphene displayed onset potential at Ep, −1.58 V. The pure rhodium nanoparticles, RhNPs also electrocatalyse at Ep, −0.186 V compared with that of PtNPs at Ep, −0.36 V and that of CoNPs at Ep, −0.98 V. The electrocatalytic experiments also indicate that the RhNPs and RhNPs@f-graphene are stable, durable and they can be recycled in several catalytic experiments after washing with water and drying. The results indicate that RhNPs and RhNPs@f-graphene are better nanoelectrocatalysts than PtNPs and the reduction potentials were much higher in other transition metal nanoparticles. The mechanism could involve a hydridic species, Rh-H− followed by interaction with protons to form hydrogen gas.
Subject terms: Chemistry, Nanoscience and technology
Introduction
The design of a complex that can catalyze electrochemical hydrogen activation at lower potentials than platinum electrode under ambient conditions is a challenge. The presence of the dimetallic iron and heterodimetallic iron-nickel centers at the active sites of the hydrogenases has invoked to search for a non-platinum catalyst material for the purpose1–8. Hydrogenases are enzymes that catalyze the interconversion of H2 and its constituents, two protons and two electrons as shown in Eq. (1).
| 1 |
The three known classes of hydrogenases, [NiFe]-, [FeFe]- and FeS-cluster free hydrogenases contain iron at their active sites which are coordinated by thiolates, CO, CN− or a light sensitive cofactor2. The [Fe4(μ3–S4)] cubane type sub-cluster obtains electrons from pyruvate oxidation and transfer those electrons to the [FeFe] site which utilizes them and form hydrogen gas in an unique reaction mechanism. The mechanism involves the replacement of the labile water ligand shown in Fig. 1 by a proton followed by reductive elimination supported by a free cysteine residue (Cys299) at the vicinity of the [FeFe] cluster1.
Figure 1.

H-cluster, the active site. Structure of the Hydrogen-cluster in the H2 evolving bacterium Clostridium pasteurianum1.
Although nature uses the potential redox active element “iron” in this active site, the hydrogen gas electrode is based on platinum metal. Electrocatalytic H2 gas generation is the conversion of electricity to H2 gas in the presence of a catalyst that can performance-wise replace the platinum electrode which functions at Ep, −0.413 V at a pH value of 7.08. Iron-containing electrochemical catalysts that are known to date include mononuclear iron(II) complexes, [FeII(S2C6H4)(CO)2(PMe3)2], [FeII(S2C6H2Cl2)(CO)2(PMe3)2], an FeS cluster with Fecubane(μ-SR)Fesubsite and [Fe2(CN)(CO)4H(PMe3)(S2C3H6)] which function at high reduction potentials, Ep ≈ −1.0 V9–16. However, several electrochemical catalysts that contain other transition metals are known, e.g. carbon nanotube supported [Ni(PPh2NPh2)2(CH3CN)][BF4]2, (CpMoμ-S)2S2CH2, cobalt macrocyclic glyoxime/tetraimine complexes and a nickel(II) dithiolene complex which function at considerably lower reduction potentials. Several other electrochemical hydrogen evolution catalysts have also been reported including metalloporphyrins, low-valent transition metal complexes forming hydrides upon reaction with acids, mononuclear iron(II) complexes, cobalt-dithiolene complexes and a nickel complex [Ni(PPh2NPh)2][BF4]2 (PPh2NPh = 1,3,6-triphenyl-1-aza-3,6-diphosphacycloheptane) which electrocatalyze H2 production with high turnover frequencies but at significantly high reduction potentials Ep > −1.1 V17–24. Various kinds of heterogeneous non-precious metal based electrocatalysts for hydrogen evolution reaction (HER) and oxygen reduction reaction (ORR) including metal sulfides, selenides, carbides, nitrides, phosphides, three dimensional porous carbon nanostructures and heteroatom doped nanocarbons have been reviewed25–27. Multilayer thin films of metal nanocrystals and graphene quantum dots have been prepared by layer-by-layer assembly approach and these composites demonstrate efficient and versatile electrocatalytic performances such as reduction of aromatic nitro compounds, methanol oxidation and water splitting28. Plasmonic TiO2 NRs@Ag@GQDs ternary heterostructures which have been prepared by a layer-by-layer assembly strategy combined with an in situ light irradiation display an enhanced photoelectrochemical water splitting performance29. Pd-CdS nanowire heterostructures display remarkable photocatalytic reduction of nitroarenes and photocatalytic hydrogen production under visible light irradiation30. Our research focused towards the electrochemical hydrogen evolution resulted in a mononuclear iron(III) dithiolene of severely distorted square pyramidal geometry and a nickel(II)-sulfur based radical ligand complex that catalyze electrochemical hydrogen gas evolution at lower potentials in CH3CN. In these cases, along with metals, the dithiolene ligands are also potentially redox active forming sulfur based radicals31–36.
It has been demonstrated that Pt3Ni(111) surface is 90-fold more active for ORR than the current platinum catalyst on porous carbon used in PEMFC37. Core-shelled TiC@TiO2 has been shown to be a promising catalyst support for proton exchange membrane fuel cells (PEMFCs). TiC is thermally stable with low solubility in sulfuric acid and high electronic conductivity. Both these materials are used as supports for platinum and platinum–palladium alloy catalysts (Pt/TiC, Pt3Pd/TiC and Pt3Pd/TiC@TiO2) and their catalytic activity toward ORR are much higher than those for Pt/TiC38. Earth abundant transition metal nanocatalysts that include Mn, Fe, Co, Ni and Cu and early transition metals such as Ti, V, Cr, Zr, Nb and their nanocomposites for reduction of various aromatic compounds have been reported39. Synthesis of different types of graphene-based composite photocatalysts and their applications in reduction of CO2, nitroarenes, methanol oxidation, elimination of pollutants and photochemical water splitting have been reviewed40. Au-Pd nanoalloys supported on graphene (Au-Pd/GR) have been reported which display higher photocatalytic performance than the monometallic, GR supported nanoparticles towards degradation of dye, rhodamine B (RhB)41. Basic principles of photocatalytic water splitting, engineering strategies for photocatalysts optimization, and promising photocatalytic materials for water splitting have been recently reviewed42. Various metal oxides, metal non-oxides and non-metal catalysts for oxygen evolution reaction (OER) have also been recently reviewed43. There are several reports on the catalytic activities of rhodium nanoparticles (RhNPs) and rhodium nanoparticles on polymer, graphene and carbon nanotube matrices for the reduction of aromatic compounds, amino boranes as a means of H2 storage. But there are no reports on the application of RhNPs and graphene supported RhNPs (RhNP@f-graphene) as electrocatalysts for the generation of hydrogen gas from water. Here in we report preparation, characterisation and electrocatalytic properties of rhodium nanoparticles (RhNPs) and graphene supported rhodium nanoparticles (RhNPs@f-graphene) which display better electrocatalytic performance than the platinum nanoparticles (PtNPs) and graphene supported platinum nanoparticles (PtNPs@f-graphene) under similar experimental conditions.
Results and Discussion
Preparation of nanoparticles of rhodium (RhNPs), platinum (PtNPs) and cobalt (CoNPs) (1–3)
The detailed synthetic procedures for the preparation of the three nanoparticles RhNPs, PtNPs and CoNPs, (1–3) are given in methods. The metal halides were reduced by NaBH4 in water at laboratory temperature (30 °C) under high dilution conditions. The black colored particles which precipitated out immediately were sonicated, centrifuged and isolated after 12 h. The transition metal nanoparticles (TMNP, 1–3) were insoluble in water, they were sonicated in water and deposited on to polymer coated carbon grids and aluminum stubs for transmission electron microscopic (TEM) and scanning electron microscopic (SEM) analysis respectively. The TEM analytical results of the rhodium nanoparticles are shown in Fig. 2A and a higher magnification image of RhNP is diaplayed in Fig. 2B. The RhNPs of <10 nm diameter are found to agglomerate to form nanostructures of 60 nm diameter as shown in Fig. 2B. The SEM images of RhNPs on aluminium stubs are displayed in Fig. 3A which further confirms the agglomeration of RhNPs to nanostructures of 60 nm diameter. The nanoparticles were confirmed to be pure rhodium nanoparticles by EDX analysis as shown in Fig. 3D. Powder XRD spectrum of RhNPs was measured in the range of 2θ from 5° to 90°. As shown in Fig. 3E, a sharp peak was observed at 2θ, 41.2° and broad, weak peaks were displayed at 2θ, 47.4°, 69.5° and 83.8°. The TEM images of the PtNPs and CoNPs are shown in Fig. 4. The PtNPs were polydispersed and are of 50 nm and 30 nm diameters as shown Fig. 4A and the CoNPs were found to group into spherical structures of 89 nm diameter as shown in Fig. 4B. The SEM image of the CoNPs also indicate the agglomeration of the particles as shown in Supplementary Information.
Figure 2.

TEM images of RhNPs and RhNPs@f-graphene. (A) RhNPs at low magnification; (B) RhNPs at high magnification; (C) RhNPs@f-graphene; (D) RhNPs@rGO.
Figure 3.

SEM analysis of RhNPs and RhNPs@f-graphene. (A) SEM image of RhNPs with inbox showing aggregation of nanoparticles; (B) SEM image of RhNPs@f-graphene at low magnification; (C) SEM image of RhNPs@f-graphene at higher magnification; (D) EDX analytical result of RhNPs; (E) Powder XRD spectrum of RhNPs; (F) EDX analytical result of RhNPs@f-graphene; (G) Powder XRD spectrum of RhNPs@f-graphene.
Figure 4.

TEM images of the composites. (A) TEM image of PtNPs@f-graphene; (B) TEM image of CoNPs@f-graphene; (C) Powder XRD spectrum of PtNPs@f-graphene.
Preparation of composites, TMNPs@f-graphene
The functionalized graphene was obtained from graphitic powder by following a previously reported procedure31. Graphitic powder was sonicated in tetrahydrofuran (thf) for 1 h followed by centrifugation. The supernatant thf layer was decanted and the residue was washed with acetone and dried thoroughly which was heated under reflux in a mixture of H2SO4 and HNO3. The composites of transition metal nanoparticles and functionalized graphene were obtained by sonication of both in acetonitrile. The solvent was evaporated and the residue was washed with acetone and then finally dried. The composites were characterized by TEM and SEM methods. The TEM images of the RhNP@f-graphene are given in Fig. 2C,D which clearly indicate the presence of graphene sheets and RhNPs. The TEM images further indicate that RhNPs of <10 nm diameter are dispersed on graphene sheets. The aggregated RhNPs are found to segregate upon formation of composite with f-graphene as shown in Fig. 2C. The SEM images of the RhNP@f-graphene composite shown in Fig. 3B,C indicate that the dispersion of RhNPs on graphene sheets resemble dispersion of water droplets on lotus leaves. The SEM images of the RhNP@f-graphene further confirm that the size of the rhodium nanoparticles is about 11–13 nm. The EDX analysis of the composite show that the percentage of rhodium is 66.2% and carbon content is 8.3% as shown in Fig. 3F. The powder XRD spectrum of RhNPs@-graphene in the range of 2θ from 5° to 90° is displayed in Fig. 3G which shows an additional weak and broad peak at 2θ, 24.6° along with the peaks at 2θ, 41.2°, 47.6°, 69.9°, 84.3°. The weak and broad signal at 2θ, 24.6° in RhNPs@f-graphene confirms the integration of RhNPs with f-graphene44. The TEM images of the platinum nanoparticles (PtNPs) and cobalt nanoparticles (CoNPs) are displayed in Supplementary Information whereas the TEM images of the f-graphene composites of PtNP and CoNP are displayed in Fig. 4A,B respectively. The PtNPs are polydispersed and are of 30–60 nm in diameter whereas the CoNPs agglomerated to form nanostructures of 80 nm in diameter. The TEM images of the f- graphene composites of PtNPs and CoNPs indicate that the metal nanoparticles are immobilized on the functionalized graphene and agglomerated as shown in Fig. 4A,B. The powder XRD spectrum of the composite, PtNPs@f-graphene is shown in Fig. 4C. The composite PtNPs@f-graphene displayed sharp peaks at 2θ, 39.7°, 46.2°, 67.5° and 81.1° which are the standard powder XRD signals for platinum nanoparticles along with a signal at 2θ, 26.3° indicating the integration of platinum nanoparticles with f-graphene45.
Electrocatalytic studies
The transition metal nanoparticles and their f-graphene composites were tested for electrocatalytic efficiency and hydrogen gas generation by cyclic voltammetry. The electrocatalysis was conducted in acidic water containing p-TsOH. Glassy carbon working, platinum wire auxillary and Ag/AgCl reference electrodes were used. In a typical experiment, nanoparticles (0.02 g) were taken in distilled water (8 ml) in an electrochemical cell, with potassium nitrate (0.1 mM) as a supporting electrolyte. Aqueous solutions of p-TsOH were added to the electrochemical cell and the cyclic voltammograms were recorded as a function of increasing concentrations of p-TsOH. The results of electrocatalytic experiments using RhNPs and RhNP@f-graphene as the catalysts are displayed in Figs 5 and 6 respectively. Pure RhNPs did not show any peak (Fig. 5, black). Upon adding aqueous solutions of p-TsOH (0.2 ml of 50 mM) a reduction signal was observed at Ep, −0.67 V (Fig. 5) whereas the RhNP@f-graphene displayed the reduction signal at Ep, −0.601 V (Fig. 6). The reduction onset potentials were observed at Ep, −0.186 V and −0.117 V for RhNPs and RhNP@f-graphene respectively. Compared with the pure RhNPs, the reduction potential of the composite RhNP@f-graphene was shifted by 60 mV towards Ep, 0.00 V. Additions of p-TsOH to the cyclic voltammetric cell resulted in increasing currents as shown in Figs 5 and 6. The RhNPs and RhNPs@f-graphene consumed 0.8 g of p-TsOH and the current exceeded 10 mA. Gas bubbles sticking to the glassy carbon working electrode were observed during electrolysis. The gas bubbles were analyzed and confirmed to be hydrogen gas by head space analysis32. A mixture of RhNPs@graphene (0.02 g) and p-TsOH (0.8 g, ~4 mmol) in water with KNO3 (0.2 M) as supporting electrolyte was purged with nitrogen gas for 15 min. A 2 ml syringe was inserted into the electrochemical cell and the argon gas supply inlet was closed. The reaction mixture was subjected to controlled potential electrolysis at −0.6 V. The gas bubbles formed at the glassy carbon working electrode surface were tapped off to the surface and the gas over the solution was taken into a syringe which was analysed by gas chromatography and confirmed to be H2 gas. Cyclic voltammograms of functionalized graphene without electrocatalysts were recorded as a function of addition of aqueous solutions of p-TsOH and the results are displayed in Fig. 7. As a function of increasing concentrations of added p-TsOH (black to blue) under similar experimental conditions, only graphene electrocatalyzes proton reduction at a potential of Ep, −1.7 V but the onset potentials were observed at Ep, −1.1 V. The reduction of pure p-TsOH in water in the absence of RhNPs or RhNPs@f-graphene was observed at a higher negative potential, Ep, −1.4 V vs. Ag/Ag+.
Figure 5.
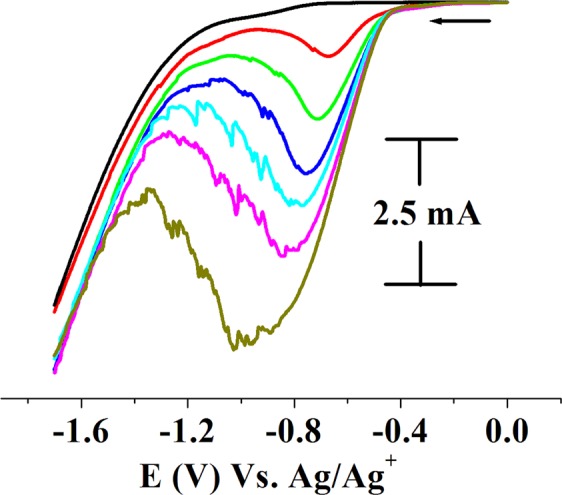
Electrocatalysis by RhNPs. Cyclic voltammograms of RhNPs as a function of increasing concentrations of added p-TsOH (red to olive green) in water. (0.5 M p-TsOH, 0.2 mL each in water). Displaying only forward reduction waves for clarity; scan rate of 100 mVs−1 (Supporting electrolyte, KNO3/H2O (0.2 M), GCE working, Pt wire auxillary and Ag/AgCl reference electrodes).
Figure 6.
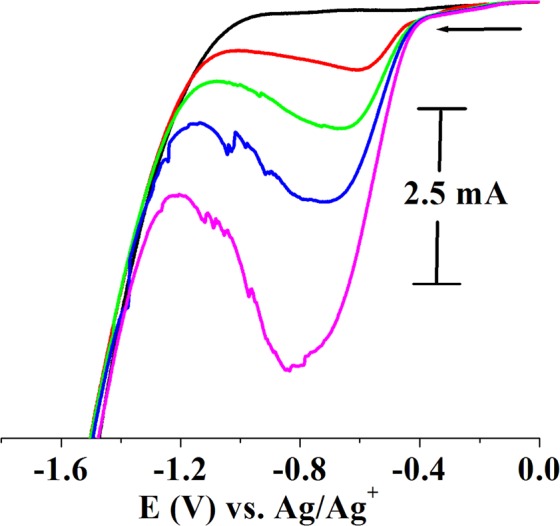
H2 evolution from acidic water by RhNPs@f-graphene. Cyclic voltammograms of RhNPs@f-graphene as a function of increasing concentrations of added p-TsOH (red to magenta) in water. (0.5 M p-TsOH, 0.2 mL each in water). Displaying only forward reduction waves for clarity; scan rate of 100 mVs−1 (Supporting electrolyte, KNO3/H2O (0.2 M), GCE working, Pt wire auxillary and Ag/AgCl reference electrodes).
Figure 7.
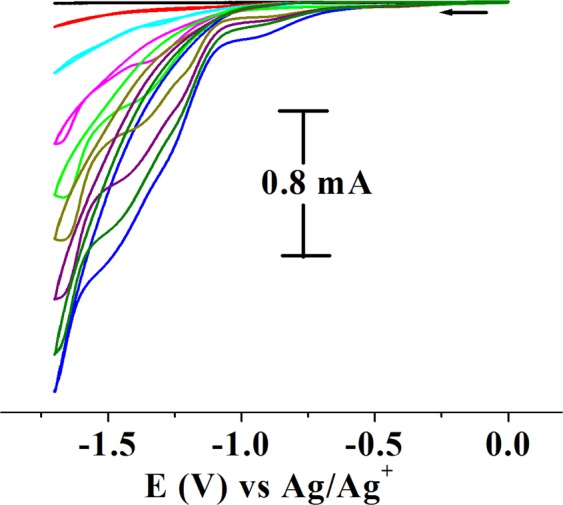
H2 evolution from acidic water by f-graphene in the absence of electrocatalysts. Cyclic voltammograms (black to blue) of f-graphene as a function of increasing concentrations of added p-TsOH in water. (0.5 M p-TsOH, 0.2 mL each in water). scan rate of 100 mVs−1 (Supporting electrolyte, KNO3/H2O (0.2 M), GCE working, Pt wire auxillary and Ag/AgCl reference electrodes).
Mechanism of proton activation
The SEM analytical results of the composite, RhNPs@f-graphene (Fig. 3C) indicate that the RhNPs are dispersed over f-graphene layers similar to the dispersion of water droplets on a lotus leaf. The interaction between the RhNPs and functionalized graphene could be π-bonding interaction or dihapto type (η2-type) of boding and protons could also bind to rhodium leading to the formation of Rh-H+ species. This could be reduced upon application of electrode potential to a hydridic species, Rh-H− as shown in Fig. 8. Structures of rhodium-hydride complexes coordinated to fullerenes (C60) in an η2-mode viz., (η2-C60)Rh(CO)H(PPh3)2 have been reviewed46. This Rh-H− species could interact with further protons and could form hydrogen gas. In the process, rhodium nanoparticles remain unaffected and provide site for fixing protons which in other words, an adsorption medium. The rhodium metal nanoparticles could play an important role in bringing down the reduction potential, Ep, because the reduction of protons and evolution of hydrogen gas takes place at higher reduction potentials when CoNPs (Ep, −0.98 V) and FeNPs (Ep, −1.59 V) were used as electrocatalysts. This might be due to the ease of formation and optimum stability of Rh-H− species.
Figure 8.

Mechanism of reduction of protons to hydrogen gas on RhNPs@f-graphene. Display of RhNPs on a functionalized graphene layer which take up protons and convert to hydrogen gas.
Recycling and durability of RhNPs and RhNPs@f-graphene
After the cyclic voltammetric experiments, the RhNPs and the RhNPs@f-graphene were filtered from the reaction mixture and were analysed by SEM and TEM techniques. The results obtained in the case of RhNPs are shown in Supplementary Information which indicate the presence of mixtures of large crystals along with the RhNPs. The large crystals could be KNO3 supporting electrolyte and p-toluene sulfonate used in the cyclic voltammetric experiment. The electrocatalysts, RhNPs and RhNPs@f-graphene were recycled as follows: After the cyclic voltammetric experiment, the RhNPs and RhNPs@f-graphene were filtered and were sonicated in water (50 mL) for 30 min. Then they were centrifuged and the water layer was decanted. The process was repeated for 6 times in order to get rid of KNO3 and p-toluene sulfonate and the recycled and purified RhNPs and RhNPs@f-graphene were reused as the electrocatalysts. The recycled RhNPs were found to electrocatalyze further generation of hydrogen gas by addition of p-TsOH (0.8 g) and the current again exceeded 10 mA. This proves that the RhNPs and RhNPs@f-graphene have excellent durability and electrocatalytic efficiency.
Comparison of electrocatalytic efficiency of RhNPs@f-graphene
In order to compare the catalytic efficiency of RhNPs and RhNP@f-graphene with that of Platinum electrodes under identical experimental conditions, electrocatalytic experiments were conducted using PtNPs and PtNPs@f-graphene. Electrocatalytic experiments were also conducted using CoNPs and CoNPs@f-graphene as electrocatalysts. The cyclic voltammetric results using PtNPs, PtNP@f-graphene, CoNPs, CoNP@f-graphene as electrocatalysts and as a function of increasing concentrations of three additions of p-TSOH are shown in Fig. 9. In these experiments the onset potential which is the lowest potential at which the reduction current starts to flow in the electrochemical system was monitored. The PtNPs displayed the onset potential at Ep, −0.367 V (Fig. 9B) where as CoNPs displayed the onset potential at Ep, −0.98 V (Fig. 9C). The composite PtNP@f-graphene displayed the onset potential at Ep, −0.38 V (Fig. 9E) and the composite, CoNP@f-graphene displayed the onset potential at Ep, −0.968 V (Fig. 9F). But the onset potentials for RhNPs and RhNPs@f-graphene are very low at Ep, −0.186 V (Fig. 9A) and Ep, −0.117 V (Fig. 9D) respectively. The onset potentials for the hydrogen evolution from acidic water by RhNPs@f-graphene, PtNPs@f-graphene and pure RhNPs are compared in Fig. 9G. The cyclic voltammetric profile obtained on using RhNPs@f-graphene as electrocatalyst is displayed in black, that for PtNPs@f-graphene in red and that for the pure RhNPs in blue. These onset potentials clearly indicate the higher catalytic performance of RhNPs and its composite, RhNP@f-graphene than even PtNPs and PtNP@f-graphene under identical experimental conditions. The RhNPs and RhNP@f-graphene were also found to be durable and extremely stable as explained above. Hence it can be concluded that the RhNPs immobilized on the functionalized graphene support is an ideal and better candidate that can substitute the platinum electrode (Platinum on porous carbon support) in fuel cells.
Figure 9.

Comparison of the onset potentials and electrocatalytic efficiency of RhNPs@f-graphene with other transition metal@f-graphene nanocomposites. Cyclic voltammograms of (A) RhNPs: (B) PtNPs; (C) CoNPs; (D) RhNPs@f-graphene; (E) PtNPs@f-graphene; (F) CoNPs@f-graphene as a function of increasing concentrations of added p-TsOH in water. (0.5 M p-TsOH, 0.2 mL each in water); (G) Comparison of reduction on-set potentials. black, onset potential for RhNPs@f-graphene, red, on-set potential for PtNPs@-graphene, Blue, onset potential for RhNPs. Displaying only forward reduction waves for clarity; scan rate of 100 mVs−1 (Supporting electrolyte, KNO3/H2O (0.2 M), GCE working, Pt wire auxillary and Ag/AgCl reference electrodes).
Conclusion
In summary, a series of transition metal nanoparticles (RhNPs, PtNPs, CoNPs, FeNPs) and their composites with f-graphene (RhNPs@f-graphene, PtNPs@f-graphene, CoNPs@f-graphene) have been prepared and characterized by SEM, TEM and p-XRD analytical methods. The nanoparticles were mostly aggregated and segregate upon composite formation with f-graphene. The RhNPs immobilized on f-graphene resemble the dispersion of water droplets on a lotus leaf. The RhNPs and the composite RhNPs@f-graphene electrocatalyze hydrogen evolution from acidic water at much lower potentials than that of PtNPs and PtNPs@f-graphene under identical experimental conditions. The reduction onset potentials for RhNPs and RhNPs@f-graphene were found to be Ep, −0.117 V and −0.186 V respectively whereas for the PtNPs and PtNPs@f-graphene were Ep, −0.367 V and −0.38 V respectively. The CoNPs, FeNPs and CoNPs@f-graphene electrocatalyze hydrogen evolution at higher reduction potentials Ep, −0.97 V, −1.58 V and −0.98 V respectively. The lower electropotentials and high current values observed in the case of RhNPs and RhNPs@-graphene reveal the crucial role played by rhodium in electrocatalytic hydrogen evolution. The plausible mechanism could be the formation of hydridic Rh-H− species under the influence of electrode potential which interact with further proton to form hydrogen gas.
Methods
Preparation of RhNPs, PtNPs, CoNPs and FeNPs
The metal halides, RhCl3.3H3O or anhydrous PtCl2 or CoCl2.6H2O or anhydrous FeCl3 (0.05 g) was stirred in distilled deionized water (30 mL) at 30 °C for 5 min. NaBH4 (0.02 g) was added to the reaction mixture in portions carefully. A bright red colored solution was obtained in the case of RhCl3.6H2O which became colorless after the precipitation of black particles. After stirring for 8 h, the black precipitate was collected by filtration. The precipitate was washed with water (10 mL), sonicated with acetone (25 mL), centrifuged and the acetone layer was decanted. The black precipitate thus obtained was dried and weighed (0.02 g; yield, >95%).
Preparation of functionalized grapheme
Graphite powder (0.5 g) was taken in THF (80 mL) and water (20 mL), stirred at 38 °C for 2 h and ultra-sonicated for 10 h. The solvents were decanted after centrifugation and the residue was dried thoroughly. This residue was treated carefully and dropwise with concentrated H2SO4 (30 mL) and fuming nitric acid (10 mL) at 0 °C. The reaction mixture was heated under reflux for 12 h and allowed to stand at 38 °C for 10 h. The supernatant acid layer was decanted and the residue was washed thoroughly with water by centrifugation and dried. FT-IR (v, cm−1): 3400 (br), 1714 (br.), 1600 (w).
Preparation of the Composite, TMNPs@graphene
RhNPs/PtNPs/CoNPs (0.02 g) was mixed with functionalized graphene (0.02 g) in degassed LC-MS grade CH3CN (12 mL) and ultra-sonicated in closed sample vials under argon atmosphere for 3 h. The reaction mixtures were then evaporated to dryness and the residues were used in catalytic experiments.
Supplementary information
Acknowledgements
A.B. thanks Department of Science and Technology (DST), New Delhi for the award of extramural research (EMR) project, EMR/2014/000246 and Alexander von Humboldt Foundation for the award of renewed research fellowship (2018). The authors thank the authorities at University of Paderborn, Germany and Sophisticated Analytical Instruments Facility (SAIF), All India Institute of Medical Sciences, New Delhi, India for SEM and TEM analysis. M.B. thanks University Grants Commission for D.S.K. Postdoctoral fellowship and G.M. thanks DST for National Postdoctoral fellowship.
Author contributions
A.B. designed and carried out the experiments. Some of the experiments were carried out by M.B. and G.M. with the supervision of A.B. The manuscript was written by A.B.
Competing interests
The authors have no competing interests as defined by Nature Publishing Group, or other interests that might be perceived to influence the results and/or discussion reported in this paper.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-019-53501-x.
References
- 1.Peters JW, Lawzilotta WN, Lemon BJ, Seefeldt LC. X-ray crystal structure of the Fe-only hydrogenase (CpI) from Clostridium pasteurianum to 1.8 angstrom resolution. Science. 1998;282:1853–1858. doi: 10.1126/science.282.5395.1853. [DOI] [PubMed] [Google Scholar]
- 2.Volbeda A, et al. Crystal structure of the nickel–iron hydrogenase from Desulfovibrio gigas. Nature. 1995;373:580–587. doi: 10.1038/373580a0. [DOI] [PubMed] [Google Scholar]
- 3.Shima S, et al. The cofactor of the iron–sulfur cluster free hydrogenase Hmd: structure of the light-inactivation product. Angew. Chem. Int. Ed. 2004;43:2547–2551. doi: 10.1002/anie.200353763. [DOI] [PubMed] [Google Scholar]
- 4.De Lacey AL, Fernandez VM, Rousset M, Cammack R. Activation and Inactivation of Hydrogenase Function and the Catalytic Cycle: Spectroelectrochemical Studies. Chem. Rev. 2007;107:4304–4330. doi: 10.1021/cr0501947. [DOI] [PubMed] [Google Scholar]
- 5.Lubitz W, Reijerse E, van Gastel M. [NiFe] and [FeFe] hydrogenases studied by advanced magnetic resonance techniques. Chem. Rev. 2007;107:4331–4365. doi: 10.1021/cr050186q. [DOI] [PubMed] [Google Scholar]
- 6.DuBois MR, DuBois DL. The roles of the first and second coordination spheres in the design of molecular catalysts for H2 production and oxidation. Chem. Soc. Rev. 2009;38:62–72. doi: 10.1039/B801197B. [DOI] [PubMed] [Google Scholar]
- 7.Fontecilla-Camps JC, Volbeda A, Cavazza C, Nicolet Y. Structure/Function relationships of [NiFe]- and [FeFe]-hydrogenases. Chem. Rev. 2007;107:4273–4303. doi: 10.1021/cr050195z. [DOI] [PubMed] [Google Scholar]
- 8.Kubas GJ. Fundamentals of H2 binding and reactivity on transition metals underlying hydrogenase function and H2 production and storage. Chem. Rev. 2007;107:4152–4205. doi: 10.1021/cr050197j. [DOI] [PubMed] [Google Scholar]
- 9.Le Goff A, et al. From hydrogenases to noble metal-free catalytic nanomaterials for H2 production and uptake. Science. 2009;326:1384–1387. doi: 10.1126/science.1179773. [DOI] [PubMed] [Google Scholar]
- 10.Wilson AD, et al. Agostic interaction and intramolecular proton transfer from the protonation of dihydrogen ortho metalated ruthenium complexes. PNAS. 2007;104:6945–6950. doi: 10.1073/pnas.0608979104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu X, Brunschwig BS, Peters JC. Electrocatalytic Hydrogen Evolution at Low Overpotentials by Cobalt Macrocyclic Glyoxime and Tetraimine Complexes. J. Am. Chem. Soc. 2007;129:8988–8998. doi: 10.1021/ja067876b. [DOI] [PubMed] [Google Scholar]
- 12.Wilson AD, et al. Hydrogen oxidation and production using nickel-based molecular catalysts with positioned proton relays. J. Am. Chem. Soc. 2006;128:358–366. doi: 10.1021/ja056442y. [DOI] [PubMed] [Google Scholar]
- 13.Appel AM, DuBois DL, DuBois MR. Molybdenum-sulfur dimers as electrocatalysts for the production of hydrogen at low overpotentials. J. Am. Chem. Soc. 2005;127:12717–12726. doi: 10.1021/ja054034o. [DOI] [PubMed] [Google Scholar]
- 14.Helm ML, Stewart MP, Bullock RM, Dubois MR, Dubois DL. A synthetic nickel electrocatalyst with a turnover frequency above 100,000 s−1 for H2 production. Science. 2011;333:863–866. doi: 10.1126/science.1205864. [DOI] [PubMed] [Google Scholar]
- 15.McNamara WR, et al. Cobalt-dithiolene complexes for the photocatalytic and electrocatalytic reduction of protons in aqueous solutions. PNAS. 2012;109:15594–15599. doi: 10.1073/pnas.1120757109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gloaguen F, Rauchfuss TB. Small molecule mimics of hydrogenases: Hydrides and redox. Chem. Soc. Rev. 2009;38:100–109. doi: 10.1039/B801796B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaur-Ghumann S, Schwartz L, Lomoth R, Stein M, Ott S. Catalytic hydrogen evolution from mononuclear iron(II) carbonyl complexes as minimal functional models of the [FeFe] hydrogenase active site. Angew. Chem. Int. Ed. 2010;49:8033–8036. doi: 10.1002/anie.201002719. [DOI] [PubMed] [Google Scholar]
- 18.Fourmond V, Jacques PA, Fonecave M, Artero V. H2 Evolution and molecular electrocatalysts: determination of overpotentials and effect of homoconjugation. Inorg. Chem. 2010;49:10338–10347. doi: 10.1021/ic101187v. [DOI] [PubMed] [Google Scholar]
- 19.Tard C, et al. Synthesis of the H-cluster framework of iron only hydrogenase. Nature. 2005;433:610–613. doi: 10.1038/nature03298. [DOI] [PubMed] [Google Scholar]
- 20.Best SP. Spectroelectrochemistry of hydrogenase enzymes and related compounds. Coord. Chem. Rev. 2005;249:1536–1554. doi: 10.1016/j.ccr.2005.01.012. [DOI] [Google Scholar]
- 21.Gloaguen F, Lawrence JD, Rauchfuss TB. Biomimetic hydrogen evolution catalyzed by an iron carbonyl thiolate. J. Am. Chem. Soc. 2001;123:9476–9477. doi: 10.1021/ja016516f. [DOI] [PubMed] [Google Scholar]
- 22.Mejia-Rodriguez R, Chong D, Reibenspies JH, Soriaga MP, Darensbourg MY. The hydrophilic phosphatriazaadamantane ligand in the development of H2 production electrocatalysts: iron hydrogenase model complexes. J. Am. Chem. Soc. 2004;126:12004–12014. doi: 10.1021/ja039394v. [DOI] [PubMed] [Google Scholar]
- 23.Ott S, Kritikos M, Akermark B, Sun L, Lomoth R. Biomimetic Pathway for Hydrogen Evolution from a Model of the Iron Hydrogenase Active Site. Angew. Chem. Int. Ed. 2004;43:1006–1009. doi: 10.1002/anie.200353190. [DOI] [PubMed] [Google Scholar]
- 24.Borg SJ, et al. Electron Transfer at a Dithiolate-Bridged Diiron Assembly: Electrocatalytic Hydrogen Evolution. J. Am. Chem. Soc. 2004;126:16988–16999. doi: 10.1021/ja045281f. [DOI] [PubMed] [Google Scholar]
- 25.Wang D, Astruc D. The recent development of efficient Earth-abundant transition-metal nanocatalysts. Chem. Soc. Rev. 2017;46:816–854. doi: 10.1039/C6CS00629A. [DOI] [PubMed] [Google Scholar]
- 26.Zou X, Zhang Y. Noble metal-free hydrogen evolution catalysts for water splitting. Chem. Soc. Rev. 2015;44:5148–5180. doi: 10.1039/C4CS00448E. [DOI] [PubMed] [Google Scholar]
- 27.Zhu C, Li H, Fu S, Du D, Lin Y. Highly efficient nonprecious metal catalysts towards oxygen reduction reaction based on three-dimensional porous carbon nanostructures. Chem. Soc. Rev. 2016;45:517–531. doi: 10.1039/C5CS00670H. [DOI] [PubMed] [Google Scholar]
- 28.Zeng Z, et al. Unraveling cooperative synergy of graphene quantum dots and metal nanocrystals at zero-dimension enabled by layer-by-layer assembly. J. Mater. Chem. A. 2018;6:1700–1713. doi: 10.1039/C7TA09119B. [DOI] [Google Scholar]
- 29.Zeng Zhiping, Li Tao, Li Yu-Bing, Dai Xiao-Cheng, Huang Ming-Hui, He Yunhui, Xiao Guangcan, Xiao Fang-Xing. Plasmon-induced photoelectrochemical water oxidation enabled by in situ layer-by-layer construction of cascade charge transfer channel in multilayered photoanode. Journal of Materials Chemistry A. 2018;6(48):24686–24692. doi: 10.1039/C8TA08841A. [DOI] [Google Scholar]
- 30.Li T, et al. Ligand-Triggered Tunable Charge Transfer Toward Multifarious Photoreduction Catalysis. J. Phys. Chem. C. 2019;123:4701–4714. doi: 10.1021/acs.jpcc.8b11363. [DOI] [Google Scholar]
- 31.Begum A, Sheikh AH, Moula G, Sarkar S. Fe4S4 Cubane Type Cluster Immobilized on a Graphene Support: A High Performance H2 Evolution Catalysis in Acidic Water. Sci. Rep. 2017;7:16948. doi: 10.1038/s41598-017-17121-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Begum A, Moula G, Sarkar S. Nickel(II)–sulfur-based radical-ligand complex as a functional model of hydrogenase. Chem. Eur. J. 2010;16:12324–12327. doi: 10.1002/chem.201001812. [DOI] [PubMed] [Google Scholar]
- 33.Begum, A. & Sarkar S. An iron(III) dithiolene complex as a functional model of iron hydrogenase. Eur. J. Inorg. Chem. 40–43 (2012).
- 34.Begum A, Moula G, Bose M, Sarkar S. Super reduced Fe4S4 cluster of Balch’s dithiolene series. Dalton. Trans. 2012;41:3536–3540. doi: 10.1039/c2dt12184k. [DOI] [PubMed] [Google Scholar]
- 35.Begum A, Sonkar SK, Saxena M, Sarkar S. Nanocomposites of carbon quantum dots-nickel(II) dithiolene as nanolights. J. Mater. Chem. 2011;21:19210–19213. doi: 10.1039/c1jm14073f. [DOI] [Google Scholar]
- 36.Begum A, Saxena M, Sonkar SK, Sarkar S. From molecular to microstructure via nanostructure of a nickel(II) dithiolene complex. Indian J. Chem. 2011;50A:1257–1262. [Google Scholar]
- 37.Tian N, Zhou ZY, Sun SG, Ding Y, Wang ZL. Synthesis of Tetrahexahedral Platinum Nanocrystals with High-Index Facets and High Electro-Oxidation Activity. Science. 2007;316:732–735. doi: 10.1126/science.1140484. [DOI] [PubMed] [Google Scholar]
- 38.Stamenkovic VR, et al. Improved Oxygen Reduction Activity on Pt3Ni(111) via Increased Surface Site Availability. Science. 2007;315:493–497. doi: 10.1126/science.1135941. [DOI] [PubMed] [Google Scholar]
- 39.Wang C, et al. Redox synthesis and high catalytic efficiency of transition-metal nanoparticle-graphene oxide nanocomposites. J. Mater. Chem. A. 2017;5:21947–21954. doi: 10.1039/C7TA06182J. [DOI] [Google Scholar]
- 40.Zhang, N., Yang, M. -Q., Liu, S., Sun, Y. & Xu, Y. -J. Waltzing with the versatile platform of graphene to synthesize composite photocatalysts. Chem. Rev. 115, 10307–10377. [DOI] [PubMed]
- 41.Zhang Y, Zhang N, Tang Z-R, Xu Y-J. Graphene Oxide as a Surfactant and Support for In-Situ Synthesis of Au−Pd Nanoalloys with Improved Visible Light Photocatalytic Activity. J. Phys. Chem. C. 2014;118:5299–5308. doi: 10.1021/jp410911j. [DOI] [Google Scholar]
- 42.Yuana L, Hana C, Yanga M-Q, Xua Y-J. Photocatalytic water splitting for solar hydrogen generation: fundamentals and recent advancements. Int. Rev. Phys. Chem. 2016;35:1–36. doi: 10.1080/0144235X.2015.1127027. [DOI] [Google Scholar]
- 43.Suen N-T, et al. Electrocatalysis for the oxygen evolution reaction: Recent development and future perspectives. Chem. Soc. Rev. 2017;46:337–365. doi: 10.1039/C6CS00328A. [DOI] [PubMed] [Google Scholar]
- 44.Chen H, Mueller MB, Gilmore KJ, Wallace GG, Li D. Mechanically strong, electrically conductive and biocompatible graphene paper. Adv. Mater. 2008;20:3557–3561. doi: 10.1002/adma.200800757. [DOI] [Google Scholar]
- 45.Gao J, Zou J, Zeng X, Ding W. Carbon supported nano Pt-Mo alloy catalysts for oxygen reduction in magnesium air-batteries. RSC. Adv. 2016;6:83025–83030. doi: 10.1039/C6RA16142A. [DOI] [Google Scholar]
- 46.Balch, A. L. & Olmstead, M. M. Reactions of transition metal complexes with fullerenes (C60,C70, etc.) and related materials. Chem. Rev.98, 2123–2165 (1998). [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.