

Abstract
Huntington’s disease (HD) is a deadly neurodegenerative disease with abnormal expansion of CAG repeats in the huntingtin gene. Mutant Huntingtin protein (mHTT) forms abnormal aggregates and intranuclear inclusions in specific neurons, resulting in cell death. Here, we tested the ability of a natural heat-shock protein 90 inhibitor, Gedunin, to degrade transfected mHTT in Neuro-2a cells and endogenous mHTT aggregates and intranuclear inclusions in both fibroblasts from HD patients and neurons derived from induced pluripotent stem cells from patients. Our data showed that Gedunin treatment degraded transfected mHTT in Neuro-2a cells, endogenous mHTT aggregates and intranuclear inclusions in fibroblasts from HD patients, and in neurons derived from induced pluripotent stem cells from patients in a dose- and time-dependent manner, and its activity depended on the proteasomal pathway rather than the autophagy route. These findings also showed that although Gedunin degraded abnormal mHTT aggregates and intranuclear inclusions in cells from HD patient, it did not affect normal cells, thus providing a new perspective for using Gedunin to treat HD.
Electronic supplementary material
The online version of this article (10.1007/s12264-019-00421-5) contains supplementary material, which is available to authorized users.
Keywords: Huntington’s disease, Gedunin, Degradation, Mutant Huntingtin protein
Introduction
Huntington’s disease (HD), a devastating and rare monogenic neurodegenerative disease, is caused by an expansion of the polyglutamine (polyQ) coding sequence in the huntingtin gene (HTT) [1–3]. HD patients endure slow and relentless deterioration of cognitive, motor, and psychiatric abilities from the onset of disease [4, 5]. HD progresses for 15 to 30 years, imposing a heavy mental and economic burden on patients and their families [6, 7]. Mutant Huntingtin protein (mHTT) containing > 37 polyQ repeats is prone to misfolding and aggregates to form tangles and dislocates to the nucleus [8–10]. mHTT causes abnormal protein interactions in cells and is sequestrated with vital cellular components, resulting in the loss of normal functions and gain of toxic functions [9, 11–13]. Abnormal aggregation of mHTT and intranuclear inclusions of mHTT lead to the selective loss of striatal GABAergic neurons, causing devastating uncontrolled movements [14]. Small molecules provide a promising strategy to degrade or correct the abnormal aggregates and intranuclear inclusion of mHTT in HD patients.
Heat-shock protein 90 (Hsp90), a chaperone protein, maintains the stability and survival of various cells under stress [15, 16]. It preserves the function of aberrant proteins in neurons, resulting in the accumulation of abnormal protein aggregates in neurons such as mHTT, α-synuclein, and tau [17, 18]. Hsp90 binds to the N-terminus of mHTT and interacts with ubiquitin-specific protease 19 (USP19), a de-ubiquitinating enzyme [19]. The binding of USP19 with Hsp90 promotes mHTT aggregation, and disrupting the binding reduces the effect of USP19 on mHTT-N90 [19]. Previous research has shown that 17-AAG, an Hsp90 inhibitor, inhibits the formation of the stable Hsp90 complex and changes the stable form of Hsp90 to a proteasome-targeting form, resulting in proteasomal degradation [20]. NVP-AUY922, another Hsp90 inhibitor, reduces the mHTT levels in different types of cells, including embryonic stem cell-derived neurons, by disrupting the association of Hsp90 with other proteins or by decreasing mHTT RNA [20]. Gedunin, a natural Hsp90 inhibitor, is isolated from plants of the Meliaceae family and has been used to treat malaria and other infectious diseases in traditional Indian medicine [21]. Thus, it is reasonable to speculate that Gedunin may have the potential to degrade abnormal mHTT aggregates. As Gedunin is already known to be safe, an ability to degrade mHTT aggregates would make it a promising candidate for treatment of HD.
Therefore, we set out to test the ability of Gedunin to degrade the mHTT aggregates and intranuclear inclusion in fibroblasts and neurons from HD patients and in transfected cells, and determine by which way Gedunin degrades in this study.
Materials and Methods
Culture of Fibroblasts from HD Patients and Healthy Siblings, and Human Induced Pluripotent Stem Cells (hiPSCs)
The study was approved (No. 28) by the Ethics Committee of the Institutes of Biomedical Sciences at Fudan University. All members of the HD families gave informed consent. We used cells from four HD patient and two healthy siblings. Fibroblasts were obtained from the skin of HD patients and healthy siblings and grown in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS) (Life Technologies, USA). The fibroblasts were passaged for 4–5 days. We generated hiPSCs from fibroblasts using transfected Yamanaka’s factors (Oct4, Sox2, Klf4, and c-Myc) with Sendai virus in feeder-free defined culture conditions (data not shown). All hiPSCs were maintained on Matrigel in E8 medium.
Plasmid Transfection
We plated Neuro-2a cells in 12-well dishes and transfected each dish with 2 μg of the vector containing EGFP-Q74 that included the first exon of an mHTT gene that harbored 74 CAG repeats using Lipofectamine 2000 according to the manufacturer’s instructions and cultured the cells for another 3 h.
Gedunin Dosage and Treatment
We prepared a 100 mmol/L stock solution of Gedunin in DMSO and used fresh medium to dilute it to the working concentrations of 5 μmol/L–20 μmol/L. To show effects on the mHTT-p23 complex, we treated the transfected cells with 20 μmol/L Gedunin for 24 h after transfection. In the proteasome-inhibition study, we treated the transfected cells with 20 µmol/L Gedunin for 6 h after transfection and then added 5 μmol/L or 10 μmol/L MG132.
Cell Line Culture
The Neuro-2a cells were incubated at 37 °C and cultured in DMEM (Life Technologies) with 10% (v/v) FBS (Life Technologies).
Protein Expression Analysis
Cells were lysed in CelLytic-M Mammalian Cell Lysis/Extraction Reagent (Sigma) and centrifuged at 12,000 g for 10 min at 4 °C. The primary antibodies were as follows: Htt-specific antibody MW1; Hsp70-specific antibody; Hsp90-specific antibody; p23-specific antibody (Thermo Scientific); and α-tubulin-specific antibody (Sigma). We used ImageJ software to measure expression levels. Densitometry values for Htt, Hsp90, Hsp70, and p23 were normalized to that of endogenous α-tubulin (Table 1).
Table 1.
Information of primary antibodies.
| Antibody | Host | Dilution | Source |
|---|---|---|---|
| Anti-MW1 | Mouse | WB 1:1000 | Developmental studies hybridoma |
| Anti-Hsp70 | Mouse | WB 1:1000 | Cell technology |
| Anti- Hsp90 | Mouse | WB 1:1000 | Santa Cruz |
| Anti-p23 | Rabbit | WB 1:1000 | Thermo scientific |
| Anti-α-tubulin | Mouse | WB 1:1000 | Sigma |
| Anti-3B5H10 | Mouse | WB 1:5000 | Sigma |
| Anti-Tuj1 | Rabbit | WB 1:10000 | Covance |
Total mRNA Extraction and Real-Time (RT) PCR
Total RNA was extracted from the cells using TRIzol reagent (Life Technologies). cDNA was prepared following the standard protocol of the provider (SuperScript IV, ThermoFisher). RT-PCR was done in an ABI Prism 7000 sequence detection system using SYBR Green PCR Master Mix. The PCR cycling conditions for the mHTT sequence RT-PCR included an initial denaturation step at 95 °C for 1.5 min, followed by 40 cycles of 95 °C for 5 s, 60 °C for 30 s, and 72 °C for 10 s. Final extension of the PCR products took place at 72 °C for 2 min. The sequences of the primer for RT-PCR were: Forward: 5′-CTGCACGGCATCCTCTATGT-3′; Reverse: 5′-TGTTCACGCAGTGGGCTATT-3′.
Differentiation of hiPSCs
Neuronal differentiation from hiPSCs was as previously described [22]. In brief, hiPSCs were differentiated to Pax6-expressing primitive neuroepithelial cells for 10–12 days in a neural induction medium, and sonic hedgehog (200 ng/mL) was added on days 10–25 to induce ventral progenitors. Neural progenitor clusters were dissociated and placed onto coverslips double-coated with poly-ornithine and laminin on day 26 in Neurobasal medium (Life Technologies) to obtain neurons, followed by addition of the trophic factors glial-derived neurotrophic factor (10 ng/mL), brain-derived neurotrophic factor (20 ng/mL), insulin-like growth factor 1 (10 ng/mL), and cAMP (1 μmol/L) (all from R&D Systems, USA). All cells were maintained in an incubator at 37 °C under 5% CO2.
Fluorescence Imaging
Cells grown on coverslips in 24-well plates were fixed in 4% paraformaldehyde (PFA) for 10 min at room temperature and then rinsed with phosphate-buffered saline (PBS). Cells were incubated with 0.1 µg/mL–1 µg/mL DAPI (DNA stain) for 1 min. Slides were mounted with Fluoromount-G (SouthernBiotech, Birmingham, AL) and examined by fluorescence microscopy. All images were captured with a laser scanning confocal microscope (Leica SP8, Germany). We counted the number of GFP-positive transfected cells using ImageJ.
Immunostaining of Cells and Measuring Tangle Size and Intranuclear Inclusions
Fibroblasts or neurons were rinsed once with PBS and fixed in 4% PFA for 30 min. After washing three times with PBS, cells were incubated in blocking buffer (10% donkey serum and 0.2% Triton X-100 in PBS) for 45 min at room temperature, and then in primary antibody solution (5% donkey serum and 0.2% Triton X-100 in PBS) with primary antibodies (Table 1) for overnight at 4 °C. Next, the coverslips were rinsed three times in PBS, followed by 30-min incubation in the appropriate secondary antibody solution containing DAPI at room temperature and then washed three times in PBS. Next, the immunostained coverslips were mounted on slides with an aqueous mounting medium for confocal scanning.
All mHTT and HTT tangles and intranuclear inclusions were captured under a laser scanning confocal microscope with a 63 × objective (oil). All neurons were scanned at sufficient z-stack depth to include the entire structure of tangles. After scanning, we stacked the images by maximum intensity, separated the color image by the color-splitting channel, skeletonized the whole tangle structures to make them easy to measure, and measured the tangle size using the measure panel of the Fiji ImageJ software (Fig. S1). The intranuclear inclusions were counted using the cell counter panel of the Fiji ImageJ software.
Statistical Analysis
We assessed the statistical significance of the dose- or time-dependency using the one-way or two-way ANOVA test in GraphPad.
Results
Gedunin Degrades mHTT in a Dose- and Time-Dependent Manner
To test whether Gedunin promotes the degradation of mHTT, we transfected Neuro-2a cells with a high-expression vector (EGFP-HD-Q74) containing the first exon of an mHTT gene with 74 CAG repeats [23]. We then treated the transfected cells with a gradient of Gedunin doses (5 μmol/L, 10 μmol/L, 15 μmol/L, and 20 μmol/L). As a control, we treated cells with DMSO. We used an MW1 antibody that binds specifically to mHTT but not HTT to evaluate the changes in mHTT abundance in transfected cells [24, 25]. Immunoblotting data showed that Gedunin degraded mHTT in a dose-dependent manner. Gedunin did not significantly reduce the chaperone complex proteins Hsp70, p23, and Hsp90 (Fig. 1A, B). The highest mHTT protein degradation in the transfected cells occurred at the 20 μmol/L dose (Fig. 1A, B). Consistent with the immunoblotting results, fluorescence images of transfected cells showed that the EGFP fluorescence was significantly lower after treatment with 20 μmol/L Gedunin for 24 h than in the untreated group (Fig. 1C). To test whether a longer exposure to Gedunin enhances the degradation of mHTT, we treated transfected cells with 20 μmol/L Gedunin for 8 h, 16 h, and 24 h and then harvested cells to evaluate mHTT abundance. The data showed that longer exposure to Gedunin enhanced the degradation of mHTT in transfected cells. The degradation was especially marked at 16 h and 24 h compared to 8 h (Fig. 1D). Most Hsp90 inhibitors only target the chaperone complex and not mHTT RNA levels in cells [17]. To test whether Gedunin only affects mHTT protein and not mHTT RNA, we assessed the mHTT RNA level by RT-PCR after treatment with Gedunin. We found no reduction in mHTT RNA after Gedunin treatment (Fig. 1E). Collectively, our data showed that Gedunin decreased the abundance of mHTT protein but had no effect on transcription of the mHTT gene in transfected cells.
Fig. 1.

Effects of Gedunin on mHTT and Hsp90 chaperones in vitro. A, B Immunoblots (A) and statistics (B) showing a dose-dependent decrease of mHTT expression after treatment with Gedunin (5 μmol/L, 10 μmol/L, 15 μmol/L, and 20 μmol/L) in Neuro-2a cells transfected with EGFP-HD-Q74. The levels of Hsp70, p23 and Hsp90 were slightly decreased but the difference was not statistically significant (n = 3; mean ± SEM; P < 0.05, one-way ANOVA). C Representative images showing the level of EGFP-Q74 in transfected Neuro-2a cells after treatment with 20 μmol/L Gedunin for 24 h. Scale bar, 50 μm. D Immunoblots and results for MW1 antibody showing a time-dependent decrease of mHTT in EGFP-HD-Q74 transfected Neuro-2a cells after treatment with 20 μmol/L Gedunin (n = 3; mean ± SEM; P < 0.05, one-way ANOVA). E Treatment with Gedunin did not affect mHTT RNA in EGFP-Q74-transfected Neuro-2a cells.
Degradation Effects of Gedunin on mHTT Depend on the Proteasomal Pathway
Eukaryotic cells degrade cytoplasmic proteins through the ubiquitin-proteasome or autophagy-lysosome systems [26, 27]. The ubiquitin-proteasome system degrades misfolded proteins such as mHTT, α-synuclein, and tau [26]. To determine whether the Gedunin-mediated mHTT degradation is proteasomal, we used the proteasome inhibitor MG132 and the lysosome inhibitor Bafilomycin A1 to inhibit the proteasomal and autophagy pathways, respectively [28, 29]. Based on the effects of Geduinin on mHTT in transfected cells, we transfected Neuro-2a cells with EGFP-HD-Q74 again and added 20 μmol/L Gedunin or DMSO (control) after transfection. After incubating with Gedunin for 16 h, we added MG132 (5 μmol/L and 10 μmol/L) or Bafilomycin A1 (0.5 μmol/L and 1 μmol/L). These cells were incubated for another 6 h before harvesting for immunoblotting or fixation for fluorescence imaging. In the transfected cells, MG132 reduced the Gedunin-mediated degradation of mHTT but Bafilomycin A1 did not (Fig. 2A, B). Consistent with the immunoblotting results, the fluorescence images showed that Gedunin significantly reduced the cellular EGFP levels. Blocking the proteasomal pathway with MG132 (10 μmol/L) rescued the effects of Gedunin on the transfected cells (Fig. 2C, D). These findings showed that MG132 inhibits the effect of Gedunin on mHTT degradation and thus suggest that the effect occurs via the proteasomal pathway.
Fig. 2.

Gedunin degrades mHTT via the proteasomal pathway. A, B Gedunin promotes the proteasome clearance of mHTT. Immunoblots of cells treated with the proteasome inhibitor MG132 (5 μmol/L and 10 μmol/L) (A), or the autophagy inhibitor bafilomycin A1 (BafA1; 0.5 μmol/L and 1 μmol/L) (B) for 16 h. C, D Fluorescence images (C) and analysis (D) of EGFP-HD-Q74-transfected Neuro-2a cells incubated with Gedunin (20 μmol/L) and MG132 (10 μmol/L) for 16 h. Data are presented as the mean ± SD of three independent experiments. P < 0.05. Scale bar, 50 µm; Magnification, × 40.
Gedunin Degrades Endogenous mHTT Aggregates and Intranuclear Inclusions in Fibroblasts from HD Patients via the Proteasomal Pathway but not HTT in Normal Fibroblasts
Human HTT is a large 3144 amino-acid protein [9]. The polyQ expansion mutation in HD patients increases the aggregation and intranuclear inclusions of mHTT [9]. These aggregates and intranuclear inclusions are correlated with the toxicity and onset of HD. To test whether Gedunin is able to degrade the endogenous mHTT aggregates and intranuclear inclusions in cells from HD patients, we treated fibroblasts from three patients, each with a different CAG expansion (55, 47, and 59 repeats). In addition, we treated fibroblasts from two healthy siblings (19 CAG repeats). All cells were treated with 5 μmol/L or 10 μmol/L Gedunin. To evaluate the changes in mHTT or HTT aggregates, we stained them with 3B5H10 antibody, which recognizes both mHTT and HTT [30]. We imaged the aggregates using confocal microscopy and reconstructed them using a z-projection. The size of the tangle in each cell was calibrated using ImageJ (Figs. S1 and S2). Our data showed that treating fibroblasts with 5 μmol/L Gedunin reduced the size of mHTT tangles in HD fibroblasts but did not significantly decrease the intranuclear inclusions (Fig. 3A). We found that treating cells with 5 μmol/L or 10 μmol/L Gedunin did not significantly decrease the size of HTT aggregates and did not change their shape in healthy sibling fibroblasts (Fig. 3A). Treating the HD fibroblasts with 10 μmol/L Gedunin not only reduced the size of mHTT aggregates but also decreased the intranuclear inclusions of mHTT (Fig. 3A–C). Surprisingly, we found that the larger mHTT aggregates in HD fibroblasts degraded into smaller fragments after treatment with 10 μmol/L Gedunin for 24 h. Contrary to the HD fibroblasts, treating the healthy sibling fibroblasts with 10 μmol/L Gedunin did not significantly degrade HTT aggregates to smaller fragments (Fig. 3D). Our findings showed that Gedunin decreased the size and intranuclear inclusions of mHTT aggregates in fibroblasts from HD patients, but had no effect on normal HTT protein aggregates in those from healthy individuals. In addition, the effects of Gedunin on endogenous mHTT aggregates in HD fibroblasts were dose-dependent.
Fig. 3.

Gedunin degrades endogenous mHTT in fibroblasts from HD patients. A Representative images of mHTT and HTT aggregates in fibroblasts from HD patients and healthy siblings after treatment with Gedunin (white arrowheads, smaller mHTT fragments; green arrowheads, intranuclear inclusions). B Change in mHTT aggregate size in the Gedunin-treated group compared with control (mean ± SD; **P < 0.001, one-way ANOVA, non-parametric and multiple comparisons). C Intranuclear inclusions of mHTT in the Gedunin-treated group compared with control (mean ± SD; **P < 0.001, non-parametric one-way ANOVA, multiple comparisons). D Changes in HTT aggregates in the Gedunin-treated group compared with control.
To confirm that the Gedunin-mediated degradation of endogenous mHTT aggregates and intranuclear inclusions in fibroblasts from HD patients is also proteasomal, we treated fibroblasts from one HD patient that harbored 55 CAG repeats with 10 μmol/L Gedunin for 12 h, and then incubated the cells with 10 μmol/L MG132 for another 12 h to inhibit the proteasomal pathway. After treatment, we fixed these cells in 4% PFA and stained mHTT protein with 3B5H10 antibody. We imaged the mHTT aggregates using confocal microscopy and reconstructed them using z-projection. The size of the aggregates in each cell was measured using ImageJ. In HD fibroblasts, MG132 treatment not only decreased the Gedunin-mediated degradation of endogenous mHTT aggregates and slowed the Gedunin-mediated degradation of large aggregates into smaller mHTT fragments, but also rescued the Gedunin-mediated decreases of mHTT intranuclear inclusions (Fig. 4A–C). Our data showed that MG132 rescued the Gedunin-mediated degradation of endogenous mHTT aggregates and intranuclear inclusions in HD fibroblasts, implying that Gedunin promotes the degradation of endogenous mHTT aggregates and intranuclear inclusion in HD fibroblasts by the proteasomal pathway.
Fig. 4.
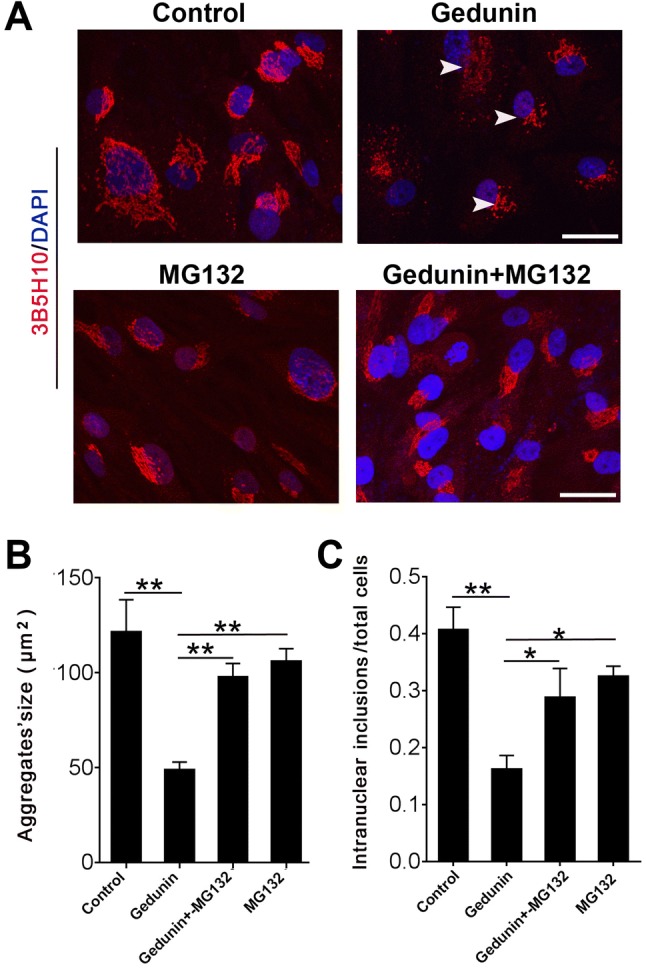
MG132 rescues the Gedunin-mediated degradation of endogenous mHTT aggregates and intranuclear inclusions in fibroblasts from HD patients. A Representative images of mHTT aggregates and intranuclear inclusions in controls and HD fibroblasts treated with 10 μmol/L Gedunin, or 10 µmol/L MG132, or both. Scale bar, 40 µm. B Size of mHTT aggregates in controls and HD fibroblasts treated with 10 µmol/L MG132 or 10 µmol/L Gedunin or both (**P < 0.001, one-way ANOVA, non-parametric and multiple comparisons). C Intranuclear inclusions of mHTT aggregates in control and HD fibroblasts treated with 10 μmol/L MG132 or 10 µmol/L Gedunin, or both (**P < 0.001, one-way ANOVA, non-parametric and multiple comparisons).
Gedunin Degrades mHTT Aggregates and Intranuclear Inclusion in Neurons Derived from iPSCs of HD Patients
Abnormal mHTT aggregates and intranuclear inclusion are the hallmark of HD neuropathology [11, 31]; they induce neuronal apoptosis in the HD brain, particularly in neurons of the striatum [8, 32]. To determine whether Gedunin can also degrade endogenous mHTT aggregates in the neurons of HD patients, we differentiated iPSCs from an HD patient with 47 CAG repeats into neurons using previously described methods [22]. Briefly, the iPSCs passed through four stages: embryoid body, rosette, neurosphere, and neuron (Fig. 5A, B). On day 47, we fixed the neurons and co-immunostained them with Tuj1 antibody, a neuronal marker, and GABA antibody, a marker of GABAergic neurons. The results showed that the Tuj1-positive neurons expressed GABA (Fig. 5C). By counting, we found that the Tuj1-positive neurons accounted for > 90% of the total cell population, and almost 88% of the Tuj1-positive neurons expressed GABA (Fig. 5D). We treated these neurons on day 47 with 5 μmol/L Gedunin for 24 h; and continued to culture them. We fixed neurons on days 50, 57, and 62, and stained them with the 3B5H10 antibody and the neuronal marker Tuj1. We reconstructed the mHTT aggregates using confocal microscopy and measured the size of aggregates using ImageJ. Our data showed that treating these neurons with 5 μmol/L Gedunin did not affect their morphology (Fig. 6A). The size of mHTT aggregates and intranuclear inclusions steadily increased in the untreated neurons from days 50 to 62 (Fig. 6B, C). However, these aggregates and inclusions in Gedunin-treated neurons on day 57 were smaller than those on day 50 and those in untreated HD neurons on day 57 (Fig. 6B, C). On day 62, although the aggregates and inclusions in the Gedunin-treated neurons were larger than those on days 50 and 57, they were significantly smaller than those of untreated HD neurons on day 62 (Fig. 6B, C). Our findings further demonstrated that Gedunin is able to induce degradation of mHTT aggregates and intranuclear inclusions in HD neurons.
Fig. 5.

Differentiation of iPSCs from an HD patient into ventral GABAergic neurons. A Steps of differentiation of iPSCs. B Typical cellular morphology at the embryoid body (EB), rosette, neurosphere, and neuron stages. Scale bars, 500 µm (iPSC stage) and 200 µm (EB, Rosette, Neurosphere and Neuron stages). C Representative co-immunostaining images of cells stained with Tuj1 and GABA antibodies. Scale bar, 50 µm. D Counts of Tuj1-positive neurons or Tuj1 and GABA double-positive neurons (mean ± SD).
Fig. 6.

Gedunin degrades endogenous mHTT in neurons derived from iPSCs from an HD patient. A Representative images of mHTT aggregates and intranuclear inclusions in HD neurons on days 50, 57, and 62 after treatment with a single dose of 5 µmol/L Gedunin on day 47, and untreated controls. Scale bar, 40 µm. (i, ii and iii showed the typical morphology of mHTT aggregates in Gedunin treated cells and control cells). B Size of mHTT aggregates in Gedunin-treated neurons compared with untreated neurons on days 50, 57, and 62 after treatment with a single dose of 5 µmol/L Gedunin on day 47 (mean ± SEM; two-way ANOVA, multiple comparisons). C Intranuclear inclusions of mHTT aggregates in Gedunin-treated neurons compared with untreated neurons on days 50, 57, and 62 after treatment with a single dose of 5 µmol/L Gedunin on day 47 (mean ± SD; *P < 0.05, two-way ANOVA, multiple comparisons).
Discussion
Abnormal mHTT aggregates and intranuclear inclusion in the brain of HD patients are hallmarks of HD neuropathology and cause the early death of affected neurons [5, 11]. We showed that Gedunin, a natural Hsp90 inhibitor, degraded mHTT protein in a dose- and time-dependent manner in transfected cells. Gedunin also degraded these aggregates and inclusions in fibroblasts from HD patients and in neurons derived from the iPSCs of an HD patient. Hsp90 inhibitors disrupt the association of Hsp90 with p23 or other chaperones and result in mHTT degradation via the proteasomal pathway [15, 17]. Consistent with the mechanism of Hsp90 inhibitors, we demonstrated that Gedunin also induces the degradation of mHTT via the proteasomal pathway and not the autophagy pathway.
Despite extensive research for more than two decades, no therapeutics for degrading mHTT protein aggregates and intranuclear inclusions or for decreasing mHTT RNA are clinically available. Targeting mHTT RNA using small interfering RNA (siRNA) is a promising treatment strategy for HD patients [33]. Targeted siRNA or antisense oligonucleotides reduced mHTT RNA in animal models [34]. However, the biggest challenges of siRNA therapy are stability, safety, and off-target effects [34, 35]. CRISPR/Cas9-mediated gene-editing has shown promise in depleting HTT aggregates and attenuating early neuropathology in mouse HD models [36]. However, the safety of this method still needs to be tested [35, 37]. Gedunin is isolated from a traditional Indian drug [21], so it has a safety advantage over other small molecules or siRNA. Our present data demonstrated that Gedunin degrades mHTT protein in a dose- and time-dependent manner in transfected cells. Consistent with the role of an Hsp90 inhibitor, Gedunin may promote the dissociation of p23 from Hsp90 chaperone protein complexes in transfected cells and not affect the individual proteins in this complex. Based on our data, we speculate that a combination of Gedunin with small molecules that enhance proteasomal degradation may result in a better outcome in degrading mHTT in future.
The formation of abnormal large mHTT aggregates and intranuclear inclusions is the neuropathological hallmark of the HD brain and the major cause of striatal neuronal death [38, 39]. HTT is a large protein that plays a critical role in cellular survival [39]. Gedunin degraded endogenous mHTT aggregates and intranuclear inclusions in fibroblasts derived from all three HD patients but not in normal cells. Doses of 5 µmol/L and 10 µmol/L Gedunin did not induce large levels of cell death in fibroblasts from HD patient or normal fibroblasts. These data suggest that Gedunin is able to enhance the degradation of endogenous mHTT and specifically target mHTT tangles but not normal HTT aggregates by the proteasomal pathway. In addition, 5 µmol/L and 10 µmol/L are tolerable by HD fibroblasts. Cultured neurons and iPSC-derived neurons, especially those that carry a disease-causing mutation, are vulnerable to stress [40]. The neurons derived from the iPSCs of an HD patient tolerated 5 μmol/L Gedunin, and a single 5 μmol/L dose significantly decreased the size of mHTT aggregates and intranuclear inclusions in these neurons 8 days after treatment. This finding indicates that Gedunin has a long-term ability to degrade mHTT aggregates and intranuclear inclusions in HD neurons. Thus, it is tempting to test the effects of Gedunin on the endogenous mHTT in animal models in the future. If Gedunin degradation of mHTT is replicated in GABAergic neurons and mouse models, it may become a promising candidate target for HD therapy.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We thank all members of the HD families for trusting us and supporting us to the end of this work. We also thank Dr. Su-Chun Zhang for helping with this project. This work was supported by the National Key Research and Development Program of China (2018YFA0108004) and the National Natural Science Foundation of China (81271259).
Conflict of interest
All authors claim that there are no conflicts of interest.
Footnotes
Weiqi Yang, Jingmo Xie, Qiang Qiang and Li Li have contributed equally to this work.
Contributor Information
Hexige Saiyin, Email: saiyin@fudan.edu.cn.
Lixiang Ma, Email: lxma@fudan.edu.cn.
References
- 1.MacDonald ME, Ambrose CM, Duyao MP, Myers RH, Lin C, Srinidhi L. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-E. [DOI] [PubMed] [Google Scholar]
- 2.Kieburtz K, Reilmann R, Olanow CW. Huntington’s disease: current and future therapeutic prospects. Mov Disord. 2018;33:1033–1041. doi: 10.1002/mds.27363. [DOI] [PubMed] [Google Scholar]
- 3.Squitieri F, Griguoli A, Capelli G, Porcellini A, D’Alessio B. Epidemiology of Huntington disease: first post-HTT gene analysis of prevalence in Italy. Clin Genet. 2016;89:367–370. doi: 10.1111/cge.12574. [DOI] [PubMed] [Google Scholar]
- 4.Louis ED, Lee P, Quinn L, Marder K. Dystonia in Huntington’s disease: prevalence and clinical characteristics. Mov Disord. 1999;14:95–101. doi: 10.1002/1531-8257(199901)14:1<95::AID-MDS1016>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 5.Walker FO. Huntington’s disease. Semin Neurol. 2007;27:143–150. doi: 10.1055/s-2007-971176. [DOI] [PubMed] [Google Scholar]
- 6.Slaughter JR, Martens MP, Slaughter KA. Depression and Huntington’s disease: prevalence, clinical manifestations, etiology, and treatment. CNS Spectr. 2001;6:306–326. doi: 10.1017/S109285290002201X. [DOI] [PubMed] [Google Scholar]
- 7.Orth M, Handley OJ, Schwenke C, Dunnett SB, Craufurd D, Ho AK, et al. Observing Huntington’s disease: the European Huntington’s disease network’s REGISTRY. PLoS Curr. 2010;2:RRN1184. doi: 10.1371/currents.RRN1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aylward EH, Nopoulos PC, Ross CA, Langbehn DR, Pierson RK, Mills JA, et al. Longitudinal change in regional brain volumes in prodromal Huntington disease. J Neurol Neurosurg Psychiatry. 2011;82:405–410. doi: 10.1136/jnnp.2010.208264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saudou F, Humbert S. The biology of Huntingtin. Neuron. 2016;89:910–926. doi: 10.1016/j.neuron.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 10.Li HL, Zhang YB, Wu ZY. Development of research on Huntington disease in China. Neurosci Bull. 2017;33:312–316. doi: 10.1007/s12264-016-0093-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, et al. Aggregation of Huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 12.Lee JM, Ramos EM, Lee JH, Gillis T, Mysore JS, Hayden MR, et al. CAG repeat expansion in Huntington disease determines age at onset in a fully dominant fashion. Neurology. 2012;78:690–695. doi: 10.1212/WNL.0b013e318249f683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang WY, Gu ZL, Liang ZQ, Qin ZH. Mitochondrial dysfunction and Huntington disease. Neurosci Bull. 2006;22:129–136. [PubMed] [Google Scholar]
- 14.Fisher ER, Hayden MR. Multisource ascertainment of Huntington disease in Canada: prevalence and population at risk. Mov Disord. 2014;29:105–114. doi: 10.1002/mds.25717. [DOI] [PubMed] [Google Scholar]
- 15.Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol. 2010;11:515–528. doi: 10.1038/nrm2918. [DOI] [PubMed] [Google Scholar]
- 16.Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 17.Baldo B, Weiss A, Parker CN, Bibel M, Paganetti P, Kaupmann K. A Screen for enhancers of clearance identifies Huntingtin as a heat shock protein 90 (Hsp90) client protein. J Biol Chem. 2012;287:1406–1414. doi: 10.1074/jbc.M111.294801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luo WJ, Sun WL, Taldone T, Rodina A, Chiosis G. Heat shock protein 90 in neurodegenerative diseases. Mol Neurodegener. 2010;5:24. doi: 10.1186/1750-1326-5-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He WT, Xue W, Gao YG, Hong JY, Yue HW, Jiang LL, et al. HSP90 recognizes the N-terminus of huntingtin involved in regulation of huntingtin aggregation by USP19. Sci Rep. 2017;7:14797. doi: 10.1038/s41598-017-13711-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tokui K, Adachi H, Waza M, Katsuno M, Minamiyama M, Doi H, et al. 17-DMAG ameliorates polyglutamine-mediated motor neuron degeneration through well-preserved proteasome function in an SBMA model mouse. Hum Mol Genet. 2009;18:898–910. doi: 10.1093/hmg/ddn419. [DOI] [PubMed] [Google Scholar]
- 21.Patwardhan CA, Fauq A, Peterson LB, Miller C, Blagg BS, Chadli A. Gedunin inactivates the co-chaperone p23 protein causing cancer cell death by apoptosis. J Biol Chem. 2013;288:7313–7325. doi: 10.1074/jbc.M112.427328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma L, Hu B, Liu Y, Vermilyea Scott C, Liu H, Gao L, et al. Human embryonic stem cell-derived gaba neurons correct locomotion deficits in quinolinic acid-lesioned mice. Cell Stem Cell. 2012;10:455–464. doi: 10.1016/j.stem.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Hagen M, Piebes DGE, de Leeuw WC, Vuist IM, van Roon-Mom WMC, Moerland PD, et al. The dynamics of early-state transcriptional changes and aggregate formation in a Huntington’s disease cell model. BMC Genomics. 2017;18:373. doi: 10.1186/s12864-017-3745-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Legleiter J, Lotz GP, Miller J, Ko J, Ng C, Williams GL, et al. Monoclonal antibodies recognize distinct conformational epitopes formed by polyglutamine in a mutant huntingtin fragment. J Biol Chem. 2009;284:21647–21658. doi: 10.1074/jbc.M109.016923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weiss A, Grueninger S, Abramowski D, Giorgio FP, Lopatin MM, Rosas HD, et al. Microtiter plate quantification of mutant and wild-type huntingtin normalized to cell count. Anal Biochem. 2011;410:304–306. doi: 10.1016/j.ab.2010.11.044. [DOI] [PubMed] [Google Scholar]
- 26.Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443:780–786. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- 27.Button RW, Luo SQ, Rubinsztein DC. Autophagic activity in neuronal cell death. Neuroscience Bulletin. 2015;31:382–394. doi: 10.1007/s12264-015-1528-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee DH, Goldberg AL. Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 1998;8:397–403. doi: 10.1016/S0962-8924(98)01346-4. [DOI] [PubMed] [Google Scholar]
- 29.Shacka JJ, Klocke BJ, Roth KA. Autophagy, bafilomycin and cell death: the “a-B-cs” of plecomacrolide-induced neuroprotection. Autophagy. 2006;2:228–230. doi: 10.4161/auto.2703. [DOI] [PubMed] [Google Scholar]
- 30.Brooks E, Arrasate M, Cheung K, Finkbeiner SM. Using antibodies to analyze polyglutamine stretches. Methods Mol Biol. 2004;277:103–128. doi: 10.1385/1-59259-804-8:103. [DOI] [PubMed] [Google Scholar]
- 31.Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R, et al. Nuclear and neuropil aggregates in Huntington’s disease: relationship to neuropathology. J Neurosci. 1999;19:2522–2534. doi: 10.1523/JNEUROSCI.19-07-02522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gasset-Rosa F, Chillon-Marinas C, Goginashvili A, Atwal RS, Artates JW, Tabet R, et al. Polyglutamine-expanded huntingtin exacerbates age-related disruption of nuclear integrity and nucleocytoplasmic transport. Neuron. 2017;94(48–57):e44. doi: 10.1016/j.neuron.2017.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rodriguez-Lebron E, Paulson HL. Allele-specific RNA interference for neurological disease. Gene Ther. 2006;13:576–581. doi: 10.1038/sj.gt.3302702. [DOI] [PubMed] [Google Scholar]
- 34.Aguiar S, van der Gaag B, Cortese FAB. RNAi mechanisms in Huntington’s disease therapy: siRNA versus shRNA. Transl Neurodegener. 2017;6:30. doi: 10.1186/s40035-017-0101-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ihry RJ, Worringer KA, Salick MR, Frias E, Ho D, Theriault K, et al. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat Med. 2018;24:939–946. doi: 10.1038/s41591-018-0050-6. [DOI] [PubMed] [Google Scholar]
- 36.Yang S, Chang R, Yang H, Zhao T, Hong Y, Kong HE, et al. CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington’s disease. J Clin Invest. 2017;127:2719–2724. doi: 10.1172/JCI92087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haapaniemi E, Botla S, Persson J, Schmierer B, Taipale J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat Med. 2018;24:927. doi: 10.1038/s41591-018-0049-z. [DOI] [PubMed] [Google Scholar]
- 38.Becher MW, Kotzuk JA, Sharp AH, Davies SW, Bates GP, Price DL, et al. Intranuclear neuronal inclusions in Huntington’s disease and dentatorubral and pallidoluysian atrophy: correlation between the density of inclusions and IT15 CAG triplet repeat length. Neurobiol Dis. 1998;4:387–397. doi: 10.1006/nbdi.1998.0168. [DOI] [PubMed] [Google Scholar]
- 39.Duyao MP, Auerbach AB, Ryan A, Persichetti F, Barnes GT, McNeil SM, et al. Inactivation of the mouse Huntington’s disease gene homolog Hdh. Science. 1995;269:407–410. doi: 10.1126/science.7618107. [DOI] [PubMed] [Google Scholar]
- 40.Flierl A, Oliveira LM, Falomir-Lockhart LJ, Mak SK, Hesley J, Soldner F, et al. Higher vulnerability and stress sensitivity of neuronal precursor cells carrying an alpha-synuclein gene triplication. PLoS One. 2014;9:e112413. doi: 10.1371/journal.pone.0112413. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.