

Abstract
IL-25, a member of the IL-17 family of cytokines, is known to enhance type 2 immune responses, but suppress type 3 (IL-17A)-mediated immune responses. Mice deficient in IL-1 receptor antagonist (Il1rn−/− mice) have excessive IL-1 signaling, resulting in spontaneous development of IL-1–, TNF– and IL-17A–dependent aortitis. We found that expression of II25 mRNA was increased in the aortae of Il1rn−/− mice, suggesting that IL-25 may suppress development of IL-1–, TNF– and IL-17A–dependent aortitis in Il1rn−/− mice by inhibiting type 3-mediated immune responses. However, we unexpectedly found that Il25−/−Il1rn−/− mice showed attenuated development of aortitis, accompanied by reduced accumulation of inflammatory cells such as dendritic cells, macrophages and neutrophils and reduced mRNA expression of Il17a and Tnfa—but not Il4 or Il13—in local lesions compared with Il1rn−/− mice. Tissue–, but not immune cell–, derived IL-25 was crucial for development of aortitis. IL-25 enhanced IL-1β and TNF production by IL-25 receptor–expressing dendritic cells and macrophages, respectively, at inflammatory sites of aortae of Il1rn−/− mice, contributing to exacerbation of development of IL-1–, TNF– and IL-17A–dependent aortitis in those mice. Our findings suggest that neutralization of IL-25 may be a potential therapeutic target for aortitis.
Subject terms: Interleukins, Cardiovascular diseases
Introduction
Giant cell arteritis (GCA) and Takayasu arteritis (TAK) are the most common large-vessel vasculitides (LVV). They are infrequent, but potentially fatal, diseases. The pathologic hallmarks of LVV are focal granulomatous vasculitides, characterized by lymphomonocytic infiltration and occasional multinucleate giant cells1. Those lesions lead to intimal hyperplasia that predisposes to luminal obstruction, arterial wall remodeling with disruption of the elastic laminae, excessive deposition of extracellular matrix, and fibrosis2. Consequently, vasculitis involving the aorta, subclavian-axillary bed, carotid branches and arteries of all sizes can lead to vision loss, aortic arch syndrome, aortic dissection, and aortic aneurysms and rupture3. Although glucocorticoids are very effective for treatment of both GCA and TAK, relapse is common following reduction of the dose4,5. In such cases, methotrexate, an immunosuppressive drug, is used for sparing of glucocorticoids while preventing relapse6–8. Newer biologics, such as neutralizing antibodies for TNF-α, IL-6, and/or IL-12/IL-23p40, have also been developed for GCA and TAK9. Anti-TNF-α agents are effective for treatment of TAK, but not GCA10–14. Increased expression of IL-1β was observed in specimens from patients with LVV such as TAK15 and GCA,16,17 suggesting that this cytokine is responsible for the development of such diseases. In support of that notion, associations were reported between polymorphisms of IL-1B and IL-1RN genes and susceptibility to TAK18 and GCA19 in certain populations. In addition, mice deficient in IL-1 receptor antagonist (Il1rn−/− mice) had excessive IL-1 signaling, resulting in spontaneous development of aortitis accompanied by infiltration of predominantly lymphocytes, which contributed to destruction of the elastic laminae with fibrosis, resembling LVV such as TAK and GCA20,21. The spontaneous development of aortitis in those Il1rn−/− mice was dependent on T cells, whereas it was suppressed by deficiency of TNF-α or IL-17A, but not IL-622,23. Based on those findings in Il1rn−/− mice, a therapeutic trial of Anakinra (recombinant human IL-1RN) in GCA patients found that its blockade of IL-1 activity was effective for treatment of the disease24.
Using that same Il1rn−/− mouse model, we found that the expression level of IL-25 (also called IL-17E), which is a member of the IL-17 family of cytokines, was increased in the aorta. IL-25 binds to IL-25 receptor (IL-25R), which consists of IL-17RA and IL-17RB. IL-25 is preferentially produced by epithelial cells and such immune cells as macrophages, mast cells, basophils, eosinophils and T cells25,26. IL-25 induces production of type 2 cytokines by various types of cells, including Th2 cells, Th9 cells, invariant NKT cells and group 2 innate lymphoid cells27,28. Those cytokines are involved in such type 2 immune responses as protection from nematode infection and development of allergic disorders26–29. In addition, IL-25 plays dual roles in type 3 immune responses: it can suppress IL-17A–mediated autoimmune diseases25,30,31, but it enhances IL-17A–mediated dermatitis32,33. However, the role of IL-25 in IL-1–, TNF– and IL-17A–mediated aortitis in Il1rn−/− mice has been unclear. Here, we demonstrate that IL-25 plays a facilitative role in the development of IL-1–, TNF– and IL-17A–mediated aortitis in Il1rn−/− mice.
Results
Identification of IL-1β– and IL-1Ra–producing cells in lesions of aortitis
As reported previously20,22,23, Il1rn−/− mice, but not wild–type mice or Il25−/− mice (data not shown), developed aortitis accompanied by infiltration of immune cells into the adventitia, destruction of the elastic laminae, adventitial thickening with replacement by fibrous tissue, and neointimal thickening (Fig. 1a,b). CD3+ T cells, CD11c+ DCs, Gr1+ neutrophils, Mac2+ macrophages and tryptase+ mast cells had infiltrated the lesions of aortitis in Il1rn−/− mice (Fig. 1c). In particular, CD8+ T cells and GL3+ γδT cells, but not CD4+ T cells, are seen (Fig. 1c). However, it remains unclear which types of cells produce IL-1Ra and IL-1β in the lesions of aortitis in Il1rn−/− mice. For detection of IL-1Ra, we newly generated IL-1Ra–reporter mice (Il1rngfp/gfp mice; Fig. 2a), which express EGFP under the promoter of Il1rn genes instead of IL-1Ra (Fig. 2b) and also develop aortitis spontaneously. We found that CD11c+ dendritic cells (DCs), Gr1+ neutrophils, and GL3+ γδT cells—but not CD4+ T cells, CD8+ T cells or Mac2+ macrophages—were IL-1Ra–producing EGFP+ cells in the lesions of aortitis (Fig. 3a, and data not shown). On the other hand, CD11c+ dendritic cells (DCs), Gr1+ neutrophils and Mac2+ macrophages—but not CD4+ T cells, CD8+ T cells or GL3+ γδT cells—were IL-1β–producing cells (Fig. 3b, and data not shown). Neither of those cytokines was produced by non-immune cells such as endothelial cells, epithelial cells, vascular smooth muscle cells or fibroblasts (data not shown).
Figure 1.

Characterization of infiltrating types of immune cells in aortitis in Il1rn−/− mice. (a) H&E and (b) EVG staining of the aortae of wild–type and Il1rn−/− mice (12 weeks old). Scale bars = 300 μm. (c) IHC using anti-CD3, CD4, CD8, CD11c, GL1, Gr1, Mac2 and tryptase Abs for local lesions of aortitis in Il1rn−/− mice (12 weeks old). Scale bars = 20 μm. IHC, immunohistochemistry.
Figure 2.

Generation of Il1rngfp/gfp mice. (a) IL-1Ra gene–targeting strategy. The region from a part of the sequence behind the start codon of exon 1 to a part of the intron behind exon 2 of the II1rn locus was replaced with a cassette consisting of IRES-EGFP and a neomycin resistance gene (Neor), flanked by loxP sequences. Then the neomycin resistance gene (Neor), flanked by loxP sequences, was depleted with Cre. (b) Expression of Il1rn and Gfp mRNA in the skin of Il1rngfp/gfp mice (n = 5, 12 weeks old), determined by qPCR. Data show the mean + SD. *p < 0.05 and **p < 0.01.
Figure 3.

Identification of IL-1Ra– and IL-1β–producing cells in aortitis in Il1rn−/− mice. (a) Expression of IL-1Ra (GFP) in Gr1+, CD11c+ and GL3+ cells and (b) expression of IL-1β in CD11c+, GL3+ and Mac2+ cells in local lesions of aortitis in Il1rn−/− mice (12 weeks old). Scale bars = 20 μm. Arrowheads = Cell surface marker-positive GFP+ cells (a) and cell surface marker-positive IL-1β+ cells.
Exacerbation of aortitis by IL-25
The development of aortitis in Il1rn−/− mice is dependent mainly on IL-1 and TNF, and at least in part on IL-17A22,23. IL-25 can suppress or enhance certain IL-17A–mediated diseases25,30,32,33. We found that expression of Il25 mRNA was significantly increased in the aortae of Il1rn−/− mice compared with wild–type mice and Il25−/−Il1rn−/− mice (as a negative control) (Fig. 4a), suggesting involvement of IL-25 in the development of aortitis. Indeed, the incidence of aortitis in Il25−/−Il1rn−/− mice was significantly lower than in Il1rn−/− mice (Fig. 4b). In association with this, the aortitis severity score and the aortic inflamed area were significantly smaller in Il25−/−Il1rn−/− mice compared with Il1rn−/− mice (Fig. 4c,d). In addition, the numbers of infiltrating inflammatory cells such as CD3+ T cells, CD11c+ DCs, Gr1+ neutrophils and Mac2+ macrophages—but not tryptase+ mast cells—in the aortae were also significantly less in Il25−/−Il1rn−/− mice compared with Il1rn−/− mice (Fig. 4e). These results indicate that IL-25 can exacerbate the development of aortitis.
Figure 4.

IL-25 exacerbates aortitis in Il1rn−/− mice. (a) Relative expression levels of Il25 mRNA in the aortae of wild–type (n = 8), Il1rn−/− (n = 10) and Il25−/−Il1rn−/− (n = 8) mice. (b) Incidence of aortitis. (c) Aortic inflamed area. (d) Aortitis severity score. (e) Numbers of CD3+, CD11c+, Mac2+, Gr1+, Mac2+ and tryptase+ cells in the aortitis of Il1rn−/− (n = 8) and Il25−/−Il1rn−/− (n = 8) mice (12 weeks old). Data show the mean + SD. *p < 0.05 and ***p < 0.01.
IL-25 enhances type 3–related cytokines, but not type 2 cytokines, in aortitis
In the aortae of Il1rn−/− mice, the expression level of Ifng mRNA was comparable—but the levels of Il4 and Il13 mRNA were lower—compared with in wild–type mice (Fig. 5). Regarding type 3 and type 3–related cytokines such as IL-17A, IL-6, IL-23p19 and TNF, the expression levels of Il17a and Tnfa mRNA were significantly higher, while those of Il6 and Il23p19 mRNA were comparable, in the aortae of Il1rn−/− mice compared with wild–type mice (Fig. 4a). Although IL-25 is known to induce production of type 2 cytokines by various types of cells27,28, the expression levels of Il4 and Il13 mRNA were comparable in the aortae of Il25−/−Il1rn−/− mice and Il1rn−/− mice (Fig. 5), suggesting that IL-25 is not crucial for induction of such type 2 cytokines in the lesions of aortitis. In addition, the expression levels of Ifng, Il6 and Il23p19 mRNA were comparable in the aortae of Il1rn−/− mice and Il25−/−Il1rn−/− mice (Fig. 5). On the other hand, the expression levels of Il6, Il17a and Tnfa mRNA were significantly reduced in Il25−/−Il1rn−/− mice compared with Il1rn−/− mice (Fig. 5). IL-25 was reported to inhibit IL-13–dependent Th17 cell differentiation, thereby contributing to suppression of Th17–mediated autoimmune diseases25,30. On the other hand, our results suggest that IL-25 may enhance production of IL-6, IL-17A and TNF, but not IL-23, thereby contributing to development of IL-1–, TNF– and IL-17A–dependent aortitis in Il1rn−/− mice.
Figure 5.

IL-25 is involved in IL-17A, IL-6 and TNF expression in aortitis in Il1rn−/− mice. Relative mRNA expression levels of Il17rb, and types 1, 2, 3 and related cytokines in the aortae of wild–type (WT; n = 8), Il1rn−/− (n = 8) and Il25−/−Il1rn−/− (n = 8) mice (12 weeks old). Data show the mean + SD. *p < 0.05, **p < 0.01 and ***p < 0.005 vs WT mice, and † < 0.05, ††p < 0.01 and †††p < 0.005 vs Il1rn−/− mice.
Identification of IL-25R-expressing cells in lesions of aortitis
mRNA expression of IL-17RB, which is a component of IL-25R, was increased in the aortae of Il1rn−/− mice compared with wild–type mice (Fig. 5). In addition, IL-17RB was expressed on some, but not all, CD11c+ DCs, Mac2+ macrophages, B220+ B cells and GL3+ γδT cells in the aortae of Il1rn−/− mice (Fig. 6), while it was not detected on CD3+ T cells or Gr1+ neutrophils (Fig. 6). Among macrophages, IL-17RB was not expressed on CD206+ M2 macrophages or CD86+ M1 macrophages in the aortae of Il1rn−/− mice (Fig. 6).
Figure 6.
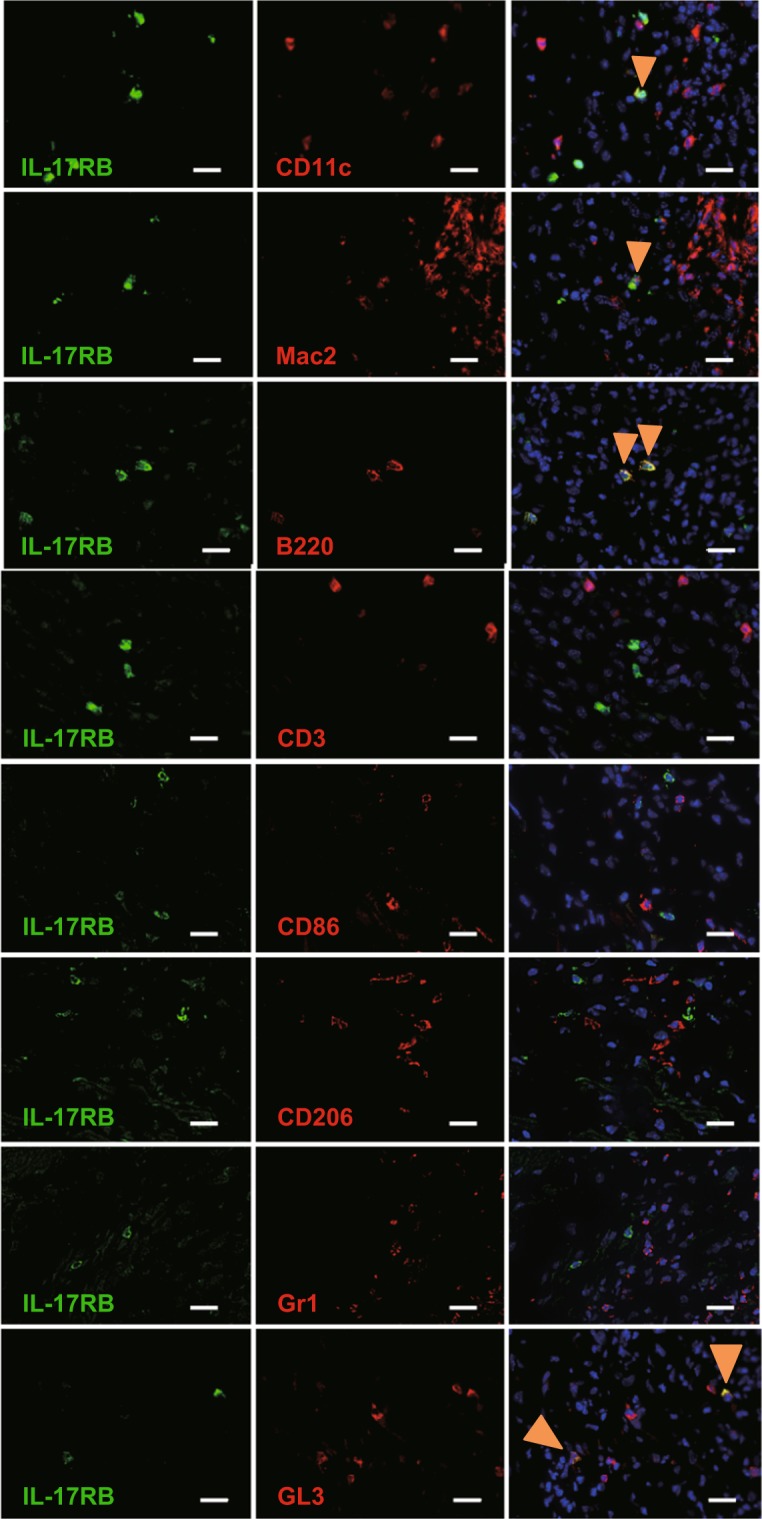
Identification of IL-25 receptor–expressing cells in aortitis in Il1rn−/− mice. IL-17RB was expressed in CD11c+, Mac2+, B220+ and GL3+ cells—but not CD3+, CD86+, CD206+ or Gr1+cells—in a local lesion of aortitis in Il1rn−/− mice (12 weeks old). Scale bars = 20 μm. Arrowheads = Cell surface marker-positive IL-17RB+ cells
Importance of non-immune cell-derived IL-25 for development of aortitis
Although Il25 mRNA expression was significantly increased in the aortae of Il1rn−/− mice (Fig. 3a), IL-25 proteins were below the limit of detection by immunohistochemical analysis of those aortic specimens (data not shown). To identify the cells producing IL-25 in the aortae of Il1rn−/− mice, we performed bone marrow (BM) cell transfer analysis. Il1rn−/− mice transferred with Il25−/−Il1rn−/− BM cells developed aortitis similarly to Il1rn−/− mice transferred with Il1rn−/− BM cells (Fig. 7a), indicating that IL-25 produced by BM cell–derived immune cells was not essential for development of aortitis in Il1rn−/− mice. On the other hand, like Il25−/−Il1rn−/− mice transferred with Il25−/−Il1rn−/− BM cells, Il25−/−Il1rn−/− mice transferred with Il1rn−/− BM cells showed reduced development of aortitis compared with Il1rn−/− mice transferred with Il1rn−/− BM cells (Fig. 7a), indicating that IL-25 produced by non-immune cells is crucial for the setting.
Figure 7.

IL-25 responder and producer cells in aortitis in Il1rn−/− mice. (a) Incidence and severity score of aortitis in Il1rn−/− mice transferred with Il1rn−/− BM cells (n = 13) or Il25−/−Il1rn−/− BM cells (n = 19) and in Il25−/−Il1rn−/− mice transferred with Il1rn−/− BM cells (n = 14) or Il25−/−Il1rn−/− BM cells (n = 5). Data show the mean + SD. *p < 0.05 vs. Il1rn−/− mice transferred with Il1rn−/− BM cells. (b,c) Expression of IL-1β in CD11c+ DCs and (d,e) expression of TNF in Mac2+ macrophages prepared from the aortae of Il1rn−/− mice after IL-25 stimulation. Data show the mean + SD (n = 130 fields; c and e). *p < 0.001 vs medium alone. Scale bars = 20 μm.
IL-25 enhances production of IL-1β by dendritic cells and TNF by macrophages
We next investigated the roles of IL-25 in production of IL-1β by DCs, which were shown to produce IL-1β and to express IL-17RB in the aortae of Il1rn−/− mice (Figs. 3b and 6), and in production of TNF by M2 macrophages, which express IL-17RB and produce TNF in response to IL-25 in humans34. Therefore, we isolated single cells from the aortae of Il1rn−/− mice and cultured them in the presence and absence of IL-25. Immunohistochemical analysis showed increased expression of IL-1β in CD11c+ DCs and of TNF in Mac2+ macrophages derived from the aortae of Il1rn−/− mice after IL-25 stimulation (Fig. 7b–e). These observations suggest that non-immune cell–derived IL-25 is crucial for production of IL-1β by IL-17RB+ CD11c+ DCs and production of TNF by IL-17RB+ Mac2+ macrophages, and that it contributes to development of aortitis in Il1rn−/− mice.
Discussion
Il1rn−/− mice spontaneously develop aortitis resembling such LVV as TAK and GCA; that is, infiltration of inflammatory cells into all aortic layers, necrosis of vascular smooth muscle cells, disruption of elastic fibers, and vascular remodeling, including stenosis of the vascular lumen and aortic dilatation1 (Fig. 1a). It was reported that the development of aortitis was diminished in Il1r1−/− Il1rn−/− mice and Tnfa−/− Il1rn−/− mice, whereas it was normal in Il6−/− Il1rn−/− mice21,22, indicating that IL-1α and/or IL-1β, and TNF are crucial for development of aortitis in Il1rn−/− mice. T cells and IL-17A are also known to be important for development of aortitis in Il1rn−/− mice22,23. Since IL-1 and/or TNF can activate Th17 cells35, it is thought that IL-1– and/or TNF–induced T cell–derived IL-17 may be involved in development of aortitis in Il1rn−/− mice. In the present study, we demonstrated that IL-25 is crucial for development of IL-1–, TNF– and IL-17–mediated aortitis in Il1rn−/− mice.
We found that expression of Il25 mRNA was significantly increased in the aortae of Il1rn−/− mice compared with wild–type mice (Fig. 4a). It has been reported that the source of IL-25 is resident microglia and/or brain capillary endothelial cells in EAE and/or MS30,36. In addition, IL-25 was expressed in lung and/or nasal epithelial cells of mice and/or humans37–39, in B cells, smooth muscle cells and endothelial cells in atherosclerotic arteries of humans40, in keratinocytes of patients with atopic dermatitis41, and in subepithelial macrophage-like cells and epithelial cells in the human colon42. We previously reported that IL-25 is crucial for development of allergic contact dermatitis as well as asthma in mice32,38. Although IL-25 proteins were detectable in the lung during asthma38, they were hardly detectable in the skin during allergic contact dermatitis32. Like in allergic contact dermatitis, IL-25 proteins were hardly detectable in aortae of Il1rn−/− mice (12 weeks) by immunofluorescence using anti-IL-25 Ab (data not shown). In addition to such non-immune cells as epithelial cells, endothelial cells, fibroblasts and tuft cells, immune cells such as mast cells, eosinophils and alveolar macrophages (M2) are known to be sources of IL-2543–45. Although there was macrophage infiltration in the aortitis of Il1rn−/− mice (Fig. 1c), CD206+ M2 macrophages were not observed among them (Fig. 6). Infiltrating granulocytes in the aortitis of Il1rn−/− mice were overwhelmingly neutrophils (Fig. 1c), not eosinophils (data not shown). Mast cells were observed in the aortitis of Il1rn−/− mice, but their number was relatively small compared with macrophages and neutrophils (Figs. 1c and 4e). Regarding this, using bone marrow cell transfer analysis, we showed that IL-25 derived from non-hematopoietic cells rather than immune cells was crucial for development of IL-1–, TNF– and IL-17–mediated aortitis in Il1rn−/− mice. Since the levels of IL-25 proteins in bronchoalveolar lavages from mice with ILC2–dependent innate–type airway inflammation and the levels of Il25 mRNA in the skin of mice with imiquimod-induced psoriatic dermatitis peaked at one hour after antigen challenge46,47, IL-25 production in the onset phase of aortitis in Il1rn−/− mice (i.e., 4 weeks) may be important for development of the disease.
IL-25 can induce such type 2 cytokines as IL-4, IL-5 and IL-13 by various types of cells, including Th2 cells27,28. It is well known that Th2 cells suppress Th1 cell– and Th17 cell–mediated immune responses. In addition, IL-25 can inhibit IL-13–dependent Th17 cell differentiation, contributing to suppression of Th17–mediated autoimmune diseases25,30,31. Since accumulation of Th2 cells as well as Th1 cells was observed in inflamed lesions of aortitis in Il1rn−/− mice21, we suspected that IL-25 may be involved in suppression of IL-1–, TNF– and IL-17–mediated aortitis in Il1rn−/− mice by enhancing Th2 cell activation. However, Il25−/− Il1rn−/− mice showed attenuated development of aortitis compared with Il1rn−/− mice (Fig. 4b), indicating that IL-25 contributed to exacerbation—not suppression—of IL-1–, TNF– and IL-17–mediated aortitis in Il1rn−/− mice. Intraperitoneal IL-25 administration to mice resulted in induction of such type 2 cytokines as IL-4, IL-5 and/or IL-13 in the spleen, stomach, small intestine, kidney and liver48. On the other hand, IL-25 injection into the skin of mice resulted in induction of type 3–related cytokines such as IL-1β, IL-6, and TNF and neutrophil chemoattractant factors32,47. In our model, IL-25 deficiency resulted in reduced expression of Il17a and Tnfa mRNA in the aortae of Il1rn−/− mice, suggesting that IL-25 somehow enhanced the development of IL-1–, TNF– and IL-17–mediated aortitis in Il1rn−/− mice.
As shown in Fig. 6, we demonstrated that type 2 immune cells (CD3+ T cells, including Th2 cells, and CD206+ M2-macrophages) did not express IL-25 receptor (IL-17RB). Thus, it seemed that IL-25 cannot activate such immune cells to induce type 2 immune responses in the aortae of Il1rn−/− mice. On the other hand, in the local lesions of aortitis in Il1rn−/− mice, we identified CD11c+ DCs, Mac2+ macrophages, γδ T cells and B cells as IL-25 receptor (IL-17RB)–expressing cells (Fig. 6). We cultured GM-CSF–induced and GM-CSF plus IL-4–induced BM cell–derived DCs (BMDCs) and TNF– and LPS–treated BMDCs in the presence of IL-25 in vitro. However, we found that IL-25 did not induce production of IL-1β or TNF by those cells (data not shown), because Il17rb mRNA was hardly detected by qPCR (data not shown). In addition, IL-17RB was expressed on M2—but not M0 or M1—macrophages derived from human PBMCs, and the M2 macrophages produced TNF, IL-6 and certain chemokines in response to IL-2534. We generated M2 and M0 macrophages from BM cells in the presence of rmM-CSF and rmIL-4, respectively, and then stimulated them in vitro with IL-25. However, we found that IL-25 did not induce TNF production by either of those macrophages because their expression of Il17rb mRNA was below the limit of detection by qPCR (data not shown). Consistent with this, IL-17RB was not expressed on CD206+ M2 macrophages or CD86+ M1 macrophages from the aortae of Il1rn−/− mice (Fig. 6). Thus, IL-17RB+ Mac2+ macrophages seen in the aortae of Il1rn−/− mice were a distinct population from the CD206+ M2 and CD86+ M1 macrophages. Likewise, γδ T cells purified from the peritoneal cavity of naïve wild–type mice did not produce IL-17A in response to IL-25, since expression of Il17rb mRNA was below the limit of detection by qPCR (data not shown). Further study is needed to elucidate the contribution of IL-17RB+ γδ T cells in local lesions to development of aortitis in Il1rn−/− mice. Since no CD3+ T cells, including Th2 cells, in the aortae expressed IL-25R, Th2 cells apparently do not produce IL-13 in the aortae of Il1rn−/− mice. On the other hand, IL-25R-expressing DCs and macrophages can produce IL-1β and TNF, respectively, in response to IL-25 (Fig. 7). DC-derived IL-1β and macrophage-derived TNF are thought to subsequently activate Th17 cells to induce IL-17, contributing to development of aortitis in Il1rn−/− mice. Therefore, the function of IL-25 is reflected by the types of IL-25R-expressing cells in the local site.
As shown in Fig. 4b, the development of aortitis was attenuated, but not completely abrogated, in Il25−/− Il1rn−/− mice compared with Il1rn−/− mice. Recently, it was reported that IL-17B, which (like IL-25) is a ligand for IL-17RB, is crucial for development of bleomycin-induced pulmonary fibrosis in mice by promoting type 3 immune responses in mice49. Therefore, IL-17B may have a compensatory role in induction of type 3 immune responses in the aortitis of Il25−/− Il1rn−/− mice.
In the present study, we identified the steps of a new function of IL-25: 1. Non-immune cell–derived IL-25 can induce production of IL-1β and TNF by IL-17RB–expressing DCs and macrophages. 2. DC-derived IL-1β and macrophage-derived TNF can activate Th17 cells to produce IL-17A. And 3. Th17 cell-derived IL-17A contributes to development of aortitis in Il1rn−/− mice. Our findings improve our understanding of the molecular mechanisms involved in development of aortitis and suggest that neutralization of IL-25 may be a potential therapeutic target for aortitis.
Methods
Mice
BALB/cNcr–wild-type mice were purchased from Japan SLC, Inc. (Shizuoka, Japan). Il1rn−/− mice and Il25−/− mice on the BALB/c background were generated as described previously50,51, and they were crossed to generate Il25−/−Il1rn−/− mice. All mice were housed under specific–pathogen–free conditions in an environmentally–controlled animal room and fed a normal chow diet at the Institute of Medical Science, The University of Tokyo. The animal protocols for experiments were approved by the Institutional Review Board of the Institute (A16–23 and A16–30), and all experiments were conducted according to the ethical and safety guidelines of the Institute.
Generation of Il1rngfp/gfp mice
The Il1rn gene (NM_001039701) was disrupted by replacement of the region from a part of the sequence behind the start codon of exon 1 to a part of the intron behind exon 2 with a cassette consisting of IRES-EGFP and a neomycin resistance gene, flanked by loxP sequences. Homologous regions were amplified by PCR using the following primers: 5′-CTGAAAGAAGGAATCAGAAACAGC-3′ and 5′- GGGTCTTTTCCCAGAAGGGCGGCAGG-3′ to generate an 8-kb fragment, and 5′-GACATAGAGTCCTTTGCCCTGCTC-3′, and 5′-GAAGGTAGGCTCAACTGGTTTAGG-3′ to generate a 1.5-kb fragment. The targeting vector was electroporated into BALB/c ES cells (SCC052, Merck). Male chimeric mice were obtained from two distinct targeted clones and mated with BALB/cNcr female mice. Genotyping of Il1rn−/− mice was performed by PCR using the following primers: common (5′-AATGAGGACATCCCACCTCCAGGC-3′), WT (5′-ACTATAGGATGTGCTTGCATCGCC-3′), and MT (5′-GACGTGCTACTTCCATTTGTCACG-3′). The common and WT primers were used for detection of wild–type alleles (479 bp), and the common and MT primers were used for detection of mutant alleles (383 bp). Then a plasmid carrying Cre cDNA (pCAG-Cre; kindly provided by Dr. Jun-ichi Miyazaki, Osaka University) was injected into fertilized eggs from the Il1rn−/− mice to deplete the neomycin resistance gene, and it was flanked by loxP sequences. The eggs were then transferred into pseudopregnant female BALB/c mice. Genotyping of Il1rngfp/gfp mice was performed by PCR using the following primers: common 2 (5′-GGAACGGAATGACAGCAGCACAGG-3′), WT2 (5′-TATATCTCCTATTCCTGCATATGCTC-3′), and MT2 (5′-CGACACCGGCCTTATTCCAAGCGG-3′). The common 2 and WT2 primers were used for detection of wild-type alleles (325 bp), and the common 2 and MT2 primers were used for detection of mutant alleles (269 bp).
Scoring of aortitis
Mice were anesthetized with sevoflurane (Pfizer, Japan) and then perfused with phosphate–buffered saline (PBS). The aortae and hearts were then embedded in Optimal Cutting Temperature (OCT) compound (Tissue-Tek; Sakura Finetek Japan Co., Tokyo, Japan) and frozen in liquid nitrogen and an isopentane slurry. Then frozen coronal sections (7-μm thickness) were prepared with Cryostat (Leica) and stored at −80 °C until use. The sections were stained with hematoxylin and eosin (H&E) according to the standard protocol and with Elastica van Gieson (EVG) according to the manufacturer’s recommendations (Muto Pure Chemicals, Japan). The aortic inflamed area was evaluated using a BZ-X710 microscope and software (KEYENCE, Japan). The severity of aortitis was scored as follows: 0, no inflammation; 1, rare inflammatory cells infiltrating the intimal layer or tunica adventitia layer; 2, infiltration into the tunica media layer; and 3, diffuse infiltration by inflammatory cells.
For immunohistochemistry (IHC), the frozen coronal sections were fixed in 4% paraformaldehyde (PFA) at room temperature (RT) for 10 minutes. The sections were blocked with Blocking One Histo (Nacalai, Japan) to prevent non-specific Ab binding at RT for 10 min, followed by incubation with primary Abs: rabbit anti-mouse CD3 pAb (ab5690; Abcam plc.); rabbit anti-mouse CD4 mAb (25229; Cell Signaling Technology); rabbit anti-CD8 mAb (98941; Cell Signaling Technology); rabbit anti-mouse CD11c pAb (97585; Cell Signaling Technology); rat anti-mouse Ly-6G/-6C mAb (ab2557; Abcam plc.); rat anti-mouse Mac-2 (CL8942AP; Cedarlane); hamster anti-mouse GL3 mAb (118101; Biolegend); rabbit anti-tryptase mAb (ab134932; Abcam plc.); rabbit anti-mannose pAb (ab64693; Abcam plc); rat anti-CD86 mAb (ab119857; Abcam plc); and rat anti-CD45R mAb (550286; BD Pharmingen). The sections were then incubated at 4 °C overnight in a PBS–humidified staining box. The sections were next blocked with a 0.3% hydrogen peroxide solution and 0.1% sodium azide at RT for 10 minutes to inhibit endogenous peroxidase activities. After washing, the specimens were incubated at RT for 30 minutes with secondary Abs: N-Histofine Simple Stain MAX PO Rat (414311; Nichirei Biosciences Inc.); Dako EnVision + System-HRP Labeled Polymer Anti-rabbit (K4002; Dako); N-Histofine Simple Stain MAX PO Goat (414351; Nichirei Biosciences Inc.); and Polink-2 Plus HRP Armenian Hamster with DAB kit (D87–6; Golden Bridge International). The specimens were developed using ImmPACT™ DAB (SK-4105; Vector Laboratories, Burlingame, CA). The sections were counterstained with Mayer’s hematoxylin at RT for 1 minute. Samples were mounted on slides using Malinol (20092; Muto Pure Chemicals, Japan) and imaged using a BZ-X710 (KEYENCE, Japan). Cells (Ab-positive cells per section) in the cross-section of the aortae were counted.
For immunofluorescence (IF) of aortae, the frozen coronal sections were fixed in 4% PFA at RT for 10 minutes. The sections were blocked at RT for 10 min with Blocking One Histo (Nacalai, Japan) to prevent non-specific Ab binding, followed by incubation overnight at 4 °C with primary Ab (the Abs for cell surface markers described above), chicken anti-GFP pAb (ab13970; Abcam plc.), biotin-conjugated anti-mouse IL-25 pAb (BAF1399; R&D Systems), biotin-conjugated anti-mouse IL-17RB pAb (BAF1040; R&D Systems), and goat anti-IL-1β pAb (AF401; R&D Systems). After washing, the samples were incubated with secondary Abs at RT for 45 minutes: Alexa Fluor® 488-conjugated streptavidin (S11223; Thermo Fisher Scientific); Alexa Fluor® 488-conjugated donkey anti-rat (ab150153; Abcam plc.); Alexa Fluor® 594-conjugated donkey anti-rabbit (ab150064; Abcam plc.); Alexa Fluor® 594-conjugated donkey anti-rat (ab150156; Abcam plc.); Alexa Fluor® 594-conjugated goat anti-hamster (A21113; Thermo Fisher Scientific); Alexa Fluor® 488-conjugated goat anti-chicken (ab150169, Abcam plc); and Alexa Fluor® 647-conjugated donkey anti-goat (ab150135; Abcam plc). For biotinylated antibodies, endogenous biotin was blocked according to the manufacturer’s protocol (SP-2001; Vector Laboratories, Inc.). The cell nuclei were counter-stained using NucBlue Fixed Cell Stain ReadyProbes reagent (1890342; Thermo Fisher Scientific) at RT for 30 minutes. Then the samples were mounted on slides using Dako Fluorescence Mounting Medium (S3023; Dako, Japan) and analyzed with a BZ-X710 microscope (4×, 20×, 40×, 80× original magnification; KEYENCE, Japan) and software (BZ-X800 analyzer; KEYENCE, Japan).
For IF of aortic cells, Il1rn−/− mice were deeply anesthetized with isoflurane and 100% oxygen and perfused intracardially with ice-cold PBS to remove blood cells. Then the aortae (pooled from 10 mice) were dissected and enzymatically digested in HBSS containing 1 mg/ml type II collagenase (Worthington Biochemical Corporation, NJ), 4.5 units of elastase (Worthington Biochemical Corporation) and 30 μg/ml DNase I (Sigma-Aldrich) at 37 °C for 1 hour on a shaker at 60 rpm. The cell suspensions of the digested aortae were then passed through a 40-μm–pore cell strainer (Greiner Bio-One). The cells were cultured in RPMI1640 medium containing 10% FBS and penicillin/streptomycin in the presence and absence of 100 ng/ml rmIL-25 (R&D Systems) at 37 °C for 6 hours. After cultivation, the cells were collected and adhered to microscope slides by centrifugation with Cytospin (Thermo Scientific) at 400 rpm for 5 min. The specimens were fixed in Phosflow (BD Bioscience) at RT for 15 min and blocked with Blocking One Histo (Nacalai) at RT for 10 min. After washing, the specimens were permeabilized with 0.1% Tween 20 at RT for 5 min, followed by incubation with primary Abs (rabbit anti-mouse CD11c pAb, and goat anti-IL-1β pAb) and secondary Abs (Alexa Fluor® 488-conjugated donkey anti-goat Ig, and Alexa Fluor® 594-conjugated donkey anti-rabbit Ig), as described above. The cell nuclei were counter-stained using NucBlue Fixed Cell Stain ReadyProbes reagent (Thermo Fisher Scientific) at RT for 30 minutes. Then the samples were mounted on slides using Dako Fluorescence Mounting Medium (Dako, Japan) and analyzed with a BZ-X710 microscope, as described above.
BM cell transplantation
BM cells were collected from the femurs, tibias and pelvises of donor mice (5 to 6 weeks old). Recipient mice (4 weeks old) were lethally irradiated with one dose of 7.5 Gy X-rays and then injected retro-orbitally with the BM cells (2.0 × 107 cells/mouse) in 0.15 ml of PBS under anesthesia by continuous inhalation of isoflurane (Pfizer, Japan).
Quantitative polymerase chain reaction (qPCR)
NucleoSpin RNA Plus XS (Takara, Japan) was used to prepare total RNA from the residual frozen aortae that had been embedded in OCT compound for histological analysis, as described above. cDNA was transcribed from purified RNA using a ReverTra Ace qPCR RT Master Mix (TOYOBO, Japan). qPCR was performed using SYBR Premix Dimer Eraser (Takara) and a CFX 384 Touch Real-time PCR Detection System (Bio-Rad, Hercules, CA) according to the manufacturers’ instructions. For detection of Il25 mRNA, qPCR analysis was performed twice, as follows. After the first round of qPCR, the PCR products were purified by NucleoSpin Gel and PCR Clean-up (Takara). Then a second round of qPCR was performed using the purified PCR products. Amplification reactions were performed in duplicate, and all mRNA expression levels were calculated using the comparative CT method formula 2-ΔΔct. The data were normalized to the level of Gapdh mRNA. The designed primers are shown in Table 1.
Table 1.
Sequences of primers.
| Gene | Forward (5′-3′) | Reverse (5′-3′) |
|---|---|---|
| Ifng | GAACTGGCAAAAGGATGGTGA | TGTGGGTTGTTGACCTCAAAC |
| Il1b | CAACCAACAAGTGATATTCTCCATG | GATCCACACTCTCCAGCTGCA |
| Il1rn | GCTCATTGCTGGGTACTTACAA | CCAGACTTGGCACAAGACAGG |
| Il4 | TCCAAGGTGCTTCGCATATTTT | CAGCTTATCGATGAATCCAGGC |
| Il6 | GAGGATACCACTCCCAACAGACC | AAGTGCATCATCGTTGTTCATACA |
| Il13 | GGCAGCAGCTTGAGCACATT | GGCATAGGCAGCAAACCATG |
| Il17a | CCGCAATGAAGACCCTGATAGAT | AGAATTCATGTGGTGGTCCAGC |
| Il17rb | GGCTGCCTAAACCACGTAATG | CCCGTTGAATGAGAATCGTGT |
| Il23 | GGTGGCATCGAGAAACTGTGA | GCTGTCTGGAGTACTGTGCATCTG |
| Il25 | AAGTGGAGCTCTGCATCTGT | CGATTCAAGTCCCTGTCCAA |
| Tnfa | GCCTCCCTCTCATCAGTTCT | CACTTGGTGGTTTGCTACGA |
| Gapdh | CCCACTCTTCCACCTTCGATG | AGGTCCACCACCCTGTTGCT |
| Egfp | CAGCTCGCCGACCACTACC | TTACTTGTACAGCTCGTCCATG |
Statistical analysis
All values were calculated as the mean ± SD except where indicated otherwise. Fisher’s exact test was used for evaluation of the incidence of aortitis between unpaired groups. Unless otherwise specified, the unpaired Student’s t test, two-tailed, were used.
Acknowledgements
We thank Ayano Yamauchi for her skilled technical assistance and Dr. Yoichiro Iwakura (Tokyo University of Biomedial Sciences, Chiba, Japan) for providing Il1rn−/− mice. We also thank Lawrence W. Stiver (Quality Translation Co., Ltd.; Tokyo, Japan; qualityt@gol.com) for his critical reading of the manuscript. This work was supported by a Grant for Joint Research Project of the Institute of Medical Science, the University of Tokyo (NK), Grants-in-Aid for Scientific Research (16K10666 to SI, 18H02847 to SN) from Japan Society for the Promotion of Science, Japan, and Precursory Research for Embryonic Science and Technology, Japan Science and Technology Agency (SN).
Author contributions
T.Y. and S.N. designed the study. T.Y., S.Y. and T.N. conducted the experiments and acquired data. T.Y., S.Y., T.N. and S.N. analyzed the data. S.Y., A.N., K.S. and S.N. generated Il1ragfp/gfp mice. S.I., N.K., H.S., K.O. and A.Y. provided reagents/mice. T.Y. and S.N. drafted the manuscript, with final editing by all authors.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Stone JR, et al. Consensus statement on surgical pathology of the aorta from the Society for Cardiovascular Pathology and the Association for European Cardiovascular Pathology: I. Inflammatory diseases. Cardiovascular pathology: the official journal of the Society for Cardiovascular Pathology. 2015;24:267–278. doi: 10.1016/j.carpath.2015.05.001. [DOI] [PubMed] [Google Scholar]
- 2.Miller DV, Maleszewski JJ. The pathology of large-vessel vasculitides. Clinical and experimental rheumatology. 2011;29:S92–98. [PubMed] [Google Scholar]
- 3.de Boysson H, et al. Large-vessel involvement and aortic dilation in giant-cell arteritis. A multicenter study of 549 patients. Autoimmun Rev. 2018;17:391–398. doi: 10.1016/j.autrev.2017.11.029. [DOI] [PubMed] [Google Scholar]
- 4.Mazlumzadeh M, et al. Treatment of giant cell arteritis using induction therapy with high-dose glucocorticoids: a double-blind, placebo-controlled, randomized prospective clinical trial. Arthritis Rheum. 2006;54:3310–3318. doi: 10.1002/art.22163. [DOI] [PubMed] [Google Scholar]
- 5.Mukhtyar C, et al. EULAR recommendations for the management of large vessel vasculitis. Ann Rheum Dis. 2009;68:318–323. doi: 10.1136/ard.2008.088351. [DOI] [PubMed] [Google Scholar]
- 6.Hoffman GS, et al. Treatment of glucocorticoid-resistant or relapsing Takayasu arteritis with methotrexate. Arthritis Rheum. 1994;37:578–582. doi: 10.1002/art.1780370420. [DOI] [PubMed] [Google Scholar]
- 7.Mahr AD, et al. Adjunctive methotrexate for treatment of giant cell arteritis: an individual patient data meta-analysis. Arthritis Rheum. 2007;56:2789–2797. doi: 10.1002/art.22754. [DOI] [PubMed] [Google Scholar]
- 8.Ohigashi H, et al. Effects of immunosuppressive and biological agents on refractory Takayasu arteritis patients unresponsive to glucocorticoid treatment. J Cardiol. 2017;69:774–778. doi: 10.1016/j.jjcc.2016.07.009. [DOI] [PubMed] [Google Scholar]
- 9.Samson M, et al. Biological treatments in giant cell arteritis & Takayasu arteritis. Eur J Intern Med. 2018;50:12–19. doi: 10.1016/j.ejim.2017.11.003. [DOI] [PubMed] [Google Scholar]
- 10.Comarmond C, et al. Anti TNF-alpha in refractory Takayasu’s arteritis: cases series and review of the literature. Autoimmun Rev. 2012;11:678–684. doi: 10.1016/j.autrev.2011.11.025. [DOI] [PubMed] [Google Scholar]
- 11.Clifford A, Hoffman GS. Recent advances in the medical management of Takayasu arteritis: an update on use of biologic therapies. Current opinion in rheumatology. 2014;26:7–15. doi: 10.1097/BOR.0000000000000004. [DOI] [PubMed] [Google Scholar]
- 12.Hoffman GS, et al. Infliximab for maintenance of glucocorticosteroid-induced remission of giant cell arteritis: a randomized trial. Ann Intern Med. 2007;146:621–630. doi: 10.7326/0003-4819-146-9-200705010-00004. [DOI] [PubMed] [Google Scholar]
- 13.Martinez-Taboada VM, et al. A double-blind placebo controlled trial of etanercept in patients with giant cell arteritis and corticosteroid side effects. Ann Rheum Dis. 2008;67:625–630. doi: 10.1136/ard.2007.082115. [DOI] [PubMed] [Google Scholar]
- 14.Seror R, et al. Adalimumab for steroid sparing in patients with giant-cell arteritis: results of a multicentre randomised controlled trial. Ann Rheum Dis. 2014;73:2074–2081. doi: 10.1136/annrheumdis-2013-203586. [DOI] [PubMed] [Google Scholar]
- 15.Noris M. Pathogenesis of Takayasu’s arteritis. Journal of nephrology. 2001;14:506–513. [PubMed] [Google Scholar]
- 16.Blain H, et al. Arterial wall production of cytokines in giant cell arteritis: results of a pilot study using human temporal artery cultures. J Gerontol A Biol Sci Med Sci. 2002;57:M241–245. doi: 10.1093/gerona/57.4.m241. [DOI] [PubMed] [Google Scholar]
- 17.Hernandez-Rodriguez J, et al. Tissue production of pro-inflammatory cytokines (IL-1beta, TNFalpha and IL-6) correlates with the intensity of the systemic inflammatory response and with corticosteroid requirements in giant-cell arteritis. Rheumatology. 2004;43:294–301. doi: 10.1093/rheumatology/keh058. [DOI] [PubMed] [Google Scholar]
- 18.Soto Lopez ME, et al. The interleukin-1 gene cluster polymorphisms are associated with Takayasu’s arteritis in Mexican patients. J Interferon Cytokine Res. 2013;33:369–375. doi: 10.1089/jir.2012.0126. [DOI] [PubMed] [Google Scholar]
- 19.Alvarez-Rodriguez L, et al. Interleukin-1RN gene polymorphisms in elderly patients with rheumatic inflammatory chronic conditions: Association of IL-1RN*2/2 genotype with polymyalgia rheumatica. Hum Immunol. 2009;70:49–54. doi: 10.1016/j.humimm.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 20.Nicklin MJ, Hughes DE, Barton JL, Ure JM, Duff GW. Arterial inflammation in mice lacking the interleukin 1 receptor antagonist gene. J Exp Med. 2000;191:303–312. doi: 10.1084/jem.191.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shepherd J, Nicklin MJ. Elastic-vessel arteritis in interleukin-1 receptor antagonist-deficient mice involves effector Th1 cells and requires interleukin-1 receptor. Circulation. 2005;111:3135–3140. doi: 10.1161/circulationaha.104.519132. [DOI] [PubMed] [Google Scholar]
- 22.Matsuki T, et al. Involvement of tumor necrosis factor-alpha in the development of T cell-dependent aortitis in interleukin-1 receptor antagonist-deficient mice. Circulation. 2005;112:1323–1331. doi: 10.1161/CIRCULATIONAHA.105.564658. [DOI] [PubMed] [Google Scholar]
- 23.Ishigame, H. et al. The role of TNFalpha and IL-17 in the development of excess IL-1 signaling-induced inflammatory diseases in IL-1 receptor antagonist-deficient mice. Ernst Schering Research Foundation workshop, 129–153 (2006). [DOI] [PubMed]
- 24.Ly KH, et al. Interleukin-1 blockade in refractory giant cell arteritis. Joint Bone Spine. 2014;81:76–78. doi: 10.1016/j.jbspin.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 25.Zaph C, et al. Commensal-dependent expression of IL-25 regulates the IL-23-IL-17 axis in the intestine. J Exp Med. 2008;205:2191–2198. doi: 10.1084/jem.20080720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paul WE, Zhu J. How are T(H)2-type immune responses initiated and amplified? Nature reviews. Immunology. 2010;10:225–235. doi: 10.1038/nri2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reynolds, J. M., Angkasekwinai, P. & Dong, C. IL-17 family member cytokines: regulation and function in innate immunity. Cytokine Growth Factor Rev, 21, 413–423, doi:S1359-6101(10)00069-9 [pii] 10.1016/j.cytogfr.2010.10.002 [doi] (2010). [DOI] [PMC free article] [PubMed]
- 28.Klose CS, Artis D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat Immunol. 2016;17:765–774. doi: 10.1038/ni.3489. [DOI] [PubMed] [Google Scholar]
- 29.Saenz SA, Taylor BC, Artis D. Welcome to the neighborhood: epithelial cell-derived cytokines license innate and adaptive immune responses at mucosal sites. Immunol Rev. 2008;226:172–190. doi: 10.1111/j.1600-065X.2008.00713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kleinschek, M. A. et al. IL-25 regulates Th17 function in autoimmune inflammation. J Exp Med, 204, 161–170, doi:jem.20061738 [pii] 10.1084/jem.20061738 [doi] (2007). [DOI] [PMC free article] [PubMed]
- 31.Liu D, et al. IL-25 attenuates rheumatoid arthritis through suppression of Th17 immune responses in an IL-13-dependent manner. Sci Rep. 2016;6:36002. doi: 10.1038/srep36002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suto, H. et al. IL-25 enhances TH17 cell-mediated contact dermatitis by promoting IL-1beta production by dermal dendritic cells. J Allergy Clin Immunol, 142, 1500–1509 e1510, doi:S0091-6749(18)30326-9 [pii] 10.1016/j.jaci.2017.12.1007 [doi] (2018). [DOI] [PubMed]
- 33.Xu M, et al. An interleukin-25-mediated autoregulatory circuit in keratinocytes plays a pivotal role in psoriatic skin inflammation. Immunity. 2018;48:787–798 e784. doi: 10.1016/j.immuni.2018.03.019. [DOI] [PubMed] [Google Scholar]
- 34.Senra L, et al. Keratinocyte-derived IL-17E contributes to inflammation in psoriasis. J Invest Dermatol. 2016;136:1970–1980. doi: 10.1016/j.jid.2016.06.009. [DOI] [PubMed] [Google Scholar]
- 35.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 36.Sonobe Y, et al. Interleukin-25 expressed by brain capillary endothelial cells maintains blood-brain barrier function in a protein kinase Cepsilon-dependent manner. J Biol Chem. 2009;284:31834–31842. doi: 10.1074/jbc.M109.025940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Angkasekwinai P, et al. Interleukin 25 promotes the initiation of proallergic type 2 responses. J Exp Med. 2007;204:1509–1517. doi: 10.1084/jem.20061675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Suzukawa, M. et al. Epithelial cell-derived IL-25, but not Th17 cell-derived IL-17 or IL-17F, is crucial for murine asthma. J Immunol, 189, 3641–3652, doi:jimmunol.1200461 [pii] 10.4049/jimmunol.1200461 [doi] (2012). [DOI] [PMC free article] [PubMed]
- 39.Nakanishi W, et al. IL-33, but not IL-25, is crucial for the development of house dust mite antigen-induced allergic rhinitis. PLoS One. 2013;8:e78099. doi: 10.1371/journal.pone.0078099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Boer OJ, et al. Differential expression of interleukin-17 family cytokines in intact and complicated human atherosclerotic plaques. J Pathol. 2010;220:499–508. doi: 10.1002/path.2667. [DOI] [PubMed] [Google Scholar]
- 41.Hvid, M. et al. IL-25 in atopic dermatitis: a possible link between inflammation and skin barrier dysfunction? J Invest Dermatol, 131, 150–157, doi:S0022-202X(15)34994-0 [pii] 10.1038/jid.2010.277 [doi] (2011). [DOI] [PubMed]
- 42.Caruso R, et al. Interleukin-25 inhibits interleukin-12 production and Th1 cell-driven inflammation in the gut. Gastroenterology. 2009;136:2270–2279. doi: 10.1053/j.gastro.2009.02.049. [DOI] [PubMed] [Google Scholar]
- 43.Xu M, Dong C. IL-25 in allergic inflammation. Immunol Rev. 2017;278:185–191. doi: 10.1111/imr.12558. [DOI] [PubMed] [Google Scholar]
- 44.Wang W, et al. Bronchial Allergen Challenge of Patients with Atopic Asthma Triggers an Alarmin (IL-33, TSLP, and IL-25) Response in the Airways Epithelium and Submucosa. J Immunol. 2018;201:2221–2231. doi: 10.4049/jimmunol.1800709. [DOI] [PubMed] [Google Scholar]
- 45.Schneider C, O’Leary CE, Locksley RM. Regulation of immune responses by tuft cells. Nature reviews. Immunology. 2019;19:584–593. doi: 10.1038/s41577-019-0176-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Van Dyken SJ, et al. Chitin activates parallel immune modules that direct distinct inflammatory responses via innate lymphoid type 2 and gammadelta T cells. Immunity. 2014;40:414–424. doi: 10.1016/j.immuni.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Senra L, et al. IL-17E (IL-25) Enhances Innate Immune Responses during Skin Inflammation. J Invest Dermatol. 2019 doi: 10.1016/j.jid.2019.01.021. [DOI] [PubMed] [Google Scholar]
- 48.Fort, M. M. et al. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity, 15, 985–995, doi:S1074-7613(01)00243-6 [pii] (2001). [DOI] [PubMed]
- 49.Yang D, et al. Dysregulated Lung Commensal Bacteria Drive Interleukin-17B Production to Promote Pulmonary Fibrosis through Their Outer Membrane Vesicles. Immunity. 2019;50:692–706.e697. doi: 10.1016/j.immuni.2019.02.001. [DOI] [PubMed] [Google Scholar]
- 50.Horai R, et al. Production of Mice Deficient in Genes for Interleukin (IL)-1, IL-1, IL-1 /, and IL-1 Receptor Antagonist Shows that IL-1 Is Crucial in Turpentine-induced Fever Development and Glucocorticoid Secretion. J Exp Med. 1998;187:1463–1475. doi: 10.1084/jem.187.9.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ishii A, et al. Development of IL-17-mediated delayed-type hypersensitivity is not affected by down-regulation of IL-25 expression. Allergol Int. 2010;59:399–408. doi: 10.2332/allergolint.10-OA-0218. [DOI] [PubMed] [Google Scholar]