

Abstract
Photodynamic therapy attracted significant attention due to its localized treatment advantage. However, the non-specific distribution of photosensitizer and subsequent potential toxicity caused by sunshine exposure hinders its wild adoption in cancer treatment. To minimize this unwanted effect and improve its efficacy, we developed a bioactivatable self-quenched nanogel, which remains in its inactivated state in healthy tissues. Anti-EGFR Affibody decorated nanogels can effectively target head and neck cancer and liberate activated Pheophorbide A in a reducing environment, such as a tumor stroma and cytoplasm. Consequently, the EGFR targeted nanogel coupled with NIR irradiation alleviate tumor burden by 94.5% while not inducing systemic toxicity.
Graphical Abstract
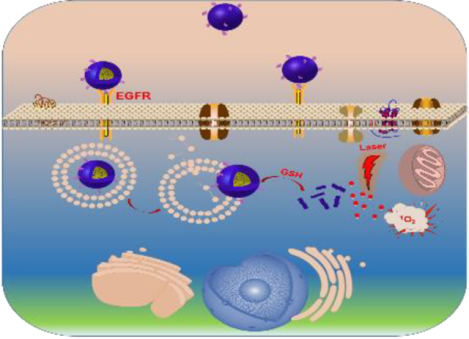
An EGFR-targeted intracellular activatable nanogel effectively inhibits head and neck cancer progression through photodynamic therapy while not inducing systemic toxicity.
Introduction
Photodynamic therapy (PDT) has been extensively studied in the past two decades as an alternative or adjunctive therapy to conventional cancer treatment modalities.1 It involves three components: photosensitizer (PS), PS-activating light, and oxygen.2 During PDT, PS is localized to a tumor, and followed by the illuminating the tumor with a light of a specific wavelength to activate the PS. Subsequently, the activated PS transfers its energy to molecular oxygen to generate cytotoxic reactive oxygen species (ROS), which ultimately destruct tumor vasculature and kill cancer cells.3, 4 As all three components are necessary for PDT to take effect, just the nonspecific accumulation of PS without light illumination will not cause systemic toxicity. Similarly, irradiating healthy tissue with a PS corresponding activating light without the accumulation of PS is not harmful either. Thus, in the aspect of side effects, PDT shows great advantages over traditional chemotherapy and radiotherapy.5
PDT has been proven to be effective in treating many superficial cancers, such as Barrett’s esophagus,6 skin cancer,7 and head and neck cancers,8 to which PS activating light is easily accessible. It still has yet to be clinically accepted as a first-line oncological intervention due to several limitations, including the poor aqueous solubility and low level tumor accumulation of PS. The application of nanoparticles, including polymer conjugates,9, 10 micelles,11, 12 dendrimers,13 mesoporous silica nanoparticles,14, 15 and nanogels,16 significantly drive the development of PDT through overcoming some of those challenges. Encapsulating PS into nanocarriers could improve the water solubility and serum stability of a PS, making it suitable for systemic application. Furthermore, the tumor-specific accumulation of nanoparticles through either passive or active targeting effects can increase the local concentration of the PS in the tumor, which ultimately yields a high PDT efficacy while reducing adverse effects.17
PDT associated photosensitivity reaction can cause severe skin and eye damage due to the activation of photosensitizers after exposure of these areas to sunlight.18 Patients who have received PDT have to stay away from light for several months. To avoid the PS-induced photosensitivity reaction, PSs have been encapsulated into nanoparticles to reduce phototoxicity through an aggregation-induced photo-quenching effect, similar to the fluorescence resonance energy transfer (FRET) effect. Therefore, less singlet oxygen (1O2), one of the cellular toxic reactive oxygen species,19 will be generated. However, due to the incomplete release of PSs from nanoparticles in the tumor site, the nano-encapsulation strategy significantly reduces the PDT efficacy.20 To overcome these obstacles, many stimuli-responsive nanoparticles have been developed, which can release PSs efficiently at tumor sites under the trigger of pH,21 glutathione (GSH),22 and enzymes,23 and therefore restore the PDT efficacy of PSs. Among them, GSH responsive nanoparticles have received the most attention due to their quick response. GSH is a tripeptide which has a high intracellular concentration (2–10 mM)24 and can be used as a biological trigger to induce drug release from nanoparticles through the cleavage of disulfide bonds. Huh et al. reported a polymer conjugate which used disulfide bonds to link glycol chitosan and Pheophorbide A (PhA), a PS of the second generation.25 The polymer-PhA conjugate could form micelles by self-assembly and exhibited a reduction of phototoxicity at low GSH condition due to the aggregation of PhA, while its PDT effect could be restored upon cellular uptake because of the release of PhA at high intracellular GSH condition. Na et al. also reported a nanoparticle-based on disulfide-linked pullulan-PhA conjugate and revealed the GSH-induced PDT effect.26 However, the serum stability of these nanoparticles is a concern since they are either micelles or physically crosslinked colloids which may break up and release free PhA during blood circulation, which subsequently induce photosensitivity reactions and damage healthy tissues. Alternative nanoparticles with great serum stability combined with GSH responsiveness are highly demanded.
Our group previously developed a series of GSH responsive nanogels based on poly[(2-(pyridin-2-yldisulfanyl) ethyl acrylate)-co-[poly(ethylene glycol)]] (PDA-PEG) polymer.27, 28 The PDA-PEG polymer contains abundant 2-mercaptopyridine segments which can be easily replaced with other molecules to form a polymer-drug conjugate through disulfide bond linkage. The polymer-drug conjugate can be easily fabricated into disulfide crosslinked nanogel by simply adding a small amount of reducing agent. These nanogels have been proven to be very stable in bloodstream condition while rapidly releasing drug after exposure to intracellular GSH, making the polymer an ideal carrier material to fabricate an activatable nanogel for PDT.
Herein, we developed a bioactivatable self-quenched nanogel for targeted photodynamic therapy (Scheme 1). PhA was first conjugated onto PDA-PEG polymer through a disulfide bond and then fabricated into nanogel by disulfide crosslinking. The formation of the nanogel would induce the fluorescence quenching of PhA due to its self-aggregation, and subsequently inhibit the generation of 1O2. The nanogel was further decorated with an anti-EGFR affibody to improve its tumor homing ability in a head and neck tumor mouse model. After systemic administration, the nanogel would stay in its quenched state in the bloodstream, where the GSH concentration is low (2–20 μM),24 and greatly reduce the phototoxicity to normal tissues. Upon the nanogel accumulating in tumor tissues by the leaking structure of the blood capillary and efficiently entered cancer cells through ligand-receptor interactions, the nanogel would be rapidly and fully activated through the elevated GSH concentration (~10 mM), and produce a high level of 1O2 for tumor destruction when coupled with laser irradiation.
Scheme 1.

Schematic illustration of the bioactivatable self-quenched nanogel for targeted photodynamic therapy.
Results and discussion
In order to prepare the PhA encapsulated nanogel, two polymers were synthesized and post-modified. Poly[(2-(pyridin-2-yldisulfanyl) ethyl acrylate)-co-[poly(ethylene glycol)]] (PDA-PEG) was first polymerized according to our previous reports16, 27, 29 and subsequently modified by replacing 60% of the PDA segment of the polymer with cysteamine through a thiol-disulfide exchange reaction to incorporate the amine functional group to yield PDA-PEG-NH2. PhA was connected to PDA-PEG-NH2 by an amide bond through a coupling reaction between amine and carboxyl groups (Scheme 2A). The second polymer, poly[(2-(pyridin-2-yldisulfanyl) ethyl acrylate)-co-[poly(ethylene glycol)]-co-[poly(2-aminoethyl methacrylate)]] (PDA-PEG-AEME) was synthesized the same as PDA-PEG but including 10% 2-aminoethyl methacrylate monomer in the initial monomer solution (Scheme 2B). Both polymers’ structures were verified by 1H-NMR (Fig. S1). 1H-NMR result proved the successful conjugation of PhA to the polymer since the polymer conjugate (PDA-PEG-PhA) included peaks at 9.65 ppm, 9.19 ppm, 8.71 ppm, and 6.21 ppm, which were the characteristic peaks belonging to PhA, and peaks at 8.50 ppm, 7.79 ppm, and 7.29 ppm, which were the characteristic peaks of pyridine ring from the PDA segment of PDA-PEG (Fig. S1A).
Scheme 2.

Illustration of the synthesis of polymers and the fabrication of PhA-ENG nanogels.
PhA nanogel (PhA-NG) was formed through disulfide cross-linking, which was generated by the addition of a predetermined amount of tris(2-carboxyethyl)phosphine (TCEP) into the PDA-PEG-PhA and PDA-PEG-AEME polymer mixture (Scheme 2C). The addition of PDA-PEG-AEME endowed PhA-NG containing amine functional group on its surface, making post-modification of the nanogel much easier. Zetasizer analysis revealed that the nanogel had a hydrodynamic size of 129.3 ± 1.1 nm (Fig. 1A) and carried a nearly neutral surface charge (0.22 ± 0.83 mV). Transmission electron microscopy (TEM) indicated a spherical morphology of the nanogel with a size of around 50 nm (Fig. 1C). We further tested the responsiveness of the PhA-nanogel to redox potential since we previously had reported that disulfide crosslinked nanogel was sensitive to redox potential.16, 27 To mimic the intracellular reducing environment, the nanogel was dispersed in 10 mM GSH solution. The broken expanded nanogels and aggregated nanogels in Fig. 1D evidenced the breakage of disulfide bonds and its reconstruction, respectively, proving the responsiveness of the nanogel to elevated redox potential. We first tested the serum stability of the nanogel in 10% FCS by DLS for up to 7 days. Fig. 1E shows that the nanogel was very stable and only had a slight size increase.
Fig. 1.

Size distribution and TEM images of PhA-NG and PhA-ENG nanogels. Size distribution of PhA-NG (A) and PhA-ENG (B) nanogels measured by Zetasizer based on dynamic light scattering (DLS). Representative TEM images of PhA-NG before (C) and after adding 10 mM GSH (D). Scale bars are 100 nm. (E) Size change of PhA-NG in the in PBS supplemented with 10% FBS.
To improve its cellular uptake by cancer cells, PhA-NG was decorated with an EGFR targeting ligand. Since protruding ligands from nanoparticle’s surface could achieve much better targeting effect due to the relative high opportunity for the ligand to interact with its corresponding receptors,30 a bifunctional PEG, Mal-PEG-COOH, with both carboxyl and maleimide functional groups on its ends was employed. The carboxyl group of Mal-PEG-COOH was first reacted with the amine group of the nanogel, introducing maleimide to the nanogel’s surface. Then a thoil containing targeting ligand was anchored to the nanogel by thiol-maleimide Michael addition reaction. Anti-EGFR Affibody® molecule was a specific affinity ligand selected against the extracellular domain of the Epidermal Growth Factor Receptor (EGFR) which highly overexpressed in many cancers including the head and neck cancer.31 Thus, Anti-EGFR Affibody® molecule was selected as the targeting ligand for our study. The affibody was initially activated to expose the thiol functional group and then conjugated to the nanogel (Scheme 2C). The EGFR targeting nanogel (PhA-ENG) showed an increased hydrodynamic size (178.8 ± 4.8 nm) (Fig. 1B)compared to PhA-NG with a slightly negatively charged surface (−7.95 ± 4.04 mV).
It has been widely reported that the aggregation of PhA in nanoparticle would induce fluorescence quenching.20 Consequently, it could affect the photodynamic activity of a photosensitizer and thus reduce PDT associated side effects. Therefore, we further investigated the fluorescence properties of PhA-NG. Fluorescence spectroscopy results revealed that PhA-NG had very low fluorescence intensity in PBS buffer due to the aggregation-induced quenching (Fig. 2A, gray line). Since GSH could break down the nanogel, liberate the PhA from the nanogel, and recover PhA’s fluorescence, we further tested the effect of GSH of physiologically relevant levels on the fluorescence intensity of the PhA-NG. Because free PhA itself forms dimers in aqueous condition, which leads to low fluorescence,32 1% Tween 80 was supplemented in the nanogel solution to inhibit the dimeric form of PhA after the addition of GSH. As shown in Fig. 2A, the addition of 1% Tween 80 increased the fluorescence of the nanogel, probably because of the disturbance of Tween 80 reduced hydrophobic interaction and π-π stacking among PhA molecules inside the nanogel. However, due to the restriction of PhA within the nanogel, its fluorescence was still significantly quenched. It was revealed that the recovery of PhA fluorescence by GSH in the nanogel was concentration dependent (Fig. 2A). At low GSH concentration such as 10 μM, 100 μM and 1 mM, no or very little fluorescence increase was observed, probably due to a small amount of GSH diffusing into the nanogel to attack the disulfide bonds. When the GSH concentration increased to 10 mM, a sharp fluorescence increase was observed and continued growing as GSH concentration was increased to 25 mM. Fluorescence imaging also confirmed the spectroscopy result that GSH could activate PhA in the nanogel. As shown in Fig. 2B at low GSH concentrations (<1 mM), very dim images were exhibited, revealing that PhA was in its quenched state. As GSH increased to 10 mM, bright fluorescence images were achieved, indicating the release of PhA from the nanogel transitioning into its activated form. The GSH-mediated activation of PhA will be extremely important for the nanogel to be used for PDT since the nanogel will remain in its inactivated state and thus reduce non-specific light irradiation to normal tissues (e.g. blood, skin, and eye) due to the low GSH level there. Upon accumulation in tumor tissue through EPR effect, the nanogel will turn to its activated form under the influence of high GSH level and be ready for PDT.
Fig. 2.

Fluorescence and photoactivity of the nanogel. (A) Fluorescence emission spectra of PhA-NG in different condition. (B) Fluorescence images of PhA-NG after being treated with 0–50 mM GSH for 1 h in PBS 7.4 supplemented with 1% Tween 80.
Ideally, a nanogel should be stable enough during the circulation period after systemic administration while rapidly being activated by GSH as it locates to the tumor. In general, the GSH level in tumor tissue or cancer cells is in the range of millimolar (2–20 mM).24 Therefore, we further studied the kinetical process of fluorescence activation of PhA-NG in 1% Tween PBS in the presence of 10 mM GSH to mimic the tumor environment. To mimic the bloodstream condition, the nanogel was directly dispersed in 10% serum. As shown in Fig. 3A, the nanogel’s fluorescence increased rapidly after the addition of 10 mM GSH, and almost 90% of PhA fluorescence had been recovered at 4 h. The fluorescence intensity slowly proceeded to a plateau to get a 4.3 fold fluorescence increase compared to the nanogel in 1% Tween 80 without GSH. In contrast, the fluorescence intensity from the nanogel had no apparent increase even after 48 h in the presence of serum, indicating that the nanogel could maintain its self-quenched status during the blood circulation process. Inset in Fig. 3A shows the fluorescence images of the nanogel at different time points after the treatment of GSH. The nanogel exhibited an increase of image brightness as time went on, which further confirmed that the nanogel was rapidly and continuously activated by high GSH in the tumor.
Fig. 3.

Fluorescence and ROS change over time in reducing condition. (A) GSH effect on the fluorescence intensity change of PhA-NG. Inset was the representative images visually exhibiting the fluorescence change of PhA-NG as the incubation time increasing in the presence of 10 mM GSH. (B) Change of DMA fluorescence signal due to the generation of singlet oxygen by PhA-NG (Ex=370 nm, Em=530 nm).
The activation of PhA-NG by GSH is expected to be applicable for PDT since the monomerized PhA could generate 1O2 under laser irradiation. The singlet oxygen is one type of reactive oxygen species (ROS) that governs the PDT efficacy of PhA. By comparing the capacity of 1O2 generation of PhA-NG with that of free PhA, the PDT potential of the nanogel could be initially assessed before proceeding to in vitro and in vivo evaluation. The generation of 1O2 could be determined by calculating the 1O2 quantum yield (SOQ, ∅Δ) by using DMA as the 1O2 trap. DMA was a fluorescence dye and its fluorescence would be lost by reacting with 1O2 to form a nonfluorescent endoperoxide. As shown in Fig. 3B, a slight fluorescence recovery of PhA-NG in 1% Tween 80 could not effectively generate 1O2 since only a minor fluorescence loss of DMA occurred. A similar result was also observed when adding 10% serum to the nanogel because of the low activation of the nanogel. In contrast, after incubation of the nanogel with GSH (10 mM) for 1 h, an increased loss of fluorescence of DMA was observed and the loss was continuously enhanced as the elongation of incubation time, which produced more activated PhA. After 5 h of incubation which accounted for more than 90% PhA activation, the nanogel reached a very similar 1O2 generation kinetics as free PhA, with a SOQ equal to 0.49 which was about 94% SOQ of the free PhA (0.52), indicating that the nanogel could rapidly induce high PDT effect at tumor site where GSH level was elevated.
Due to the short lifetime of 1O2,33 it needs to be intracellularly generated to take PDT into effect in treating cancer cells. This requires PhA-NG to have high cellular uptake. In order to improve it, the nanogel was decorated with a targeting ligand. Here, we chose head and neck squamous cell carcinoma (HNSCC) to explore the PDT efficacy of the nanogel because most HNSCC cases are localized, making a PDT laser easily reachable to the tumor. Additionally, PDT is advantageous over surgery in treating HNSCC since it causes minimal damage to cosmetic appearance and tissue function.
Because EGFR receptors are highly overexpressed in HNSCC,34 Anti-EGFR Affibody® molecules were selected as targeting ligands to modify PhA-NG due to its relatively small size compared to Anti-EGFR antibodies and easy preparation for nanogel modification. The affibody could be facilely activated by dithiothreitol to expose cysteine residue and reacted with PhA-NG by thiol-maleimide Michael addition reaction. The reacting ratio between the affibody and PhA-NG were varied in order to obtain PhA-ENG with different surface ligand densities. The cellular uptake efficiency of the PhA-ENG was first evaluated using flow cytometry in UMSCC 22A cells, a head and neck cancer cell line which has been extensively reported to have an overexpression of EGFR.35 As shown in Fig. 4A, free PhA had the highest uptake because it could easily pass through cell membrane via simple diffusion.36 For nanogels, their cellular uptake efficiency was dramatically different. Compared to the relatively low uptake of PhA-NG, all three targeting nanogels exhibited enhanced uptake efficiency due to the facilitation of ligand-receptor interactions. However, the affibody modification density on the nanogel significantly determined their uptake behaviors. Only PhA-ENG modified with an intermediate density of affibody (5×) showed the highest uptake efficiency. Neither higher (25×) or lower (1×) targeting density would exhibit the highest capability for nanogels to enter the cells because of the too strong or too weak interaction between ligand and receptors.37 Confocal laser scanning microscopy (CLSM) was further used to study the uptake behavior of the nanogels. CLSM data reconfirmed that Anti-EGFR Affibody® molecule could effectively improve the cellular uptake of PhA-ENG (Fig. 4B). In the following experiments, PhA-ENG represented the Anti-EGFR Affibody modified nanogel with the intermediate ligand density, otherwise noted.
Fig. 4.

Cellular uptake and intracellular localization of nanogels. (A) Flow cytometry spectra of PhA, PhA-NG, and PhA-ENG with different ligand densities. (B) Confocal images of PhA, PhA-NG, and PhA-ENG (5×), UMSCC 22A cells were incubated with different treatments for 3 h at 800 nM PhA. Scale bars are 50 μm. (C) Co-localization of PhA-ENG (5×) with mitochondria. Mitochondria were stained with MitoTracker Green (MTG). red: nanogel, Green: MTG. Scale bar=20 μm. (D) Co-localization of PhA-ENG (5×) with endosomes/lysosomes. Endosomes/lysosomes were stained with Lysotracker Green (LTG). Red: nanogel, Green: LTG, Blue: nuclei. Scale bars are 20 μm.
As the PDT efficacy of chlorophyllide derivatives was reported relevant to the extent of co-localization with mitochondria,38, 39 we subsequently investigated whether the nanogel could also deliver PhA to mitochondria. Surprisingly, the nanogel could not localize to mitochondria, as shown in Fig. 4C, since no overlay between green (mitochondria) and red color (nanogel) was observed. Also no PhA nuclear localization was observed since no blue color showed up in the nuclei areas (black voids surrounded by green color in Fig. 4C). Further experiments found that some nanogels were located in endosomes/lysosomes as the nanogel’s fluorescence (red) coincided with endosomes/lysosomes labeled with LysoTracker (green), revealed by CLSM in Fig. 4D, while some nanogels have escaped from the lysosome as evidenced by the standing alone red dots. These findings were in line with several early reports which also found that pheophorbide A derivatives preferred to stay in intracellular membranes and lysosomes.38, 40
To evaluate whether the enhanced cellular uptake and endosomes/lysosomes co-localization of nanogels would induce efficient PDT effect in cell killing, MTT assay was carried out to assess the cell viability after PDT in UMSCC 22A cells. Before irradiation with 670 nm laser (800 mJ/cm2), UMSCC 22A cells were incubated with free PhA, PhA-NG, and PhA-ENG for 24 h. Cells treated with the same dose of free drug and nanogels but without laser irradiation were included as control. As shown in Fig. 5A, without laser irradiation, nanogels had no obvious toxicity to cells since negligible cytotoxicity was observed at all doses for the control cells, suggesting that the nanogel itself was safe. When the cells received laser irradiation (800 mJ/cm2), dose-dependent cytotoxicity was observed (Fig. 5B). PhA-ENG exhibited enhanced cytotoxicity at almost all tested doses compared to non-targeted nanogel, which had limited cell-killing effect even at the highest PhA concentration due to its difficulty in entering the cells. PhA-ENG also showed a similar cell-killing effect (>95%) to free PhA at 800 nM PhA equivalent concentration, proving that enhanced cellular uptake could be translated to higher PDT efficacy. To be noted, the laser power we used here (800 mJ/cm2) was less than others reported,38 which further provided the evidence of the high PDT efficacy of PhA-ENG nanogel.
Fig. 5.

In vitro cytotoxicity of PhA, PhAma-NG and PhA-ENG for UMSCC 22A cells with the absence (A) and presence (B) of a 670 nm laser (800 mJ/cm2). *P <0.05, #P <0.01.
As in vitro data showed that PhA-ENG had high cellular uptake and PDT efficacy, its PDT efficacy and tumor homing ability were further evaluated in vivo. The in vivo PDT effect of PhA nanogels was then studied in an HNSCC xenograft mice model which was established by inoculating UMSCC 22A cells in both flanks of athymic nude mice. When the tumor grew to 50–150 mm3, free PhA, PhA-NG, and PhA-ENG were administered to the mice by retro-orbital injection (PhA dose equal to 1 mg/kg). The biodistribution of nanogels and free PhA were visualized using a non-invasive fluorescence imaging system. As shown in Fig. 6A, compared to free PhA, both nanogels exhibited higher fluorescence intensity in tumor sites at 24 h after injection, indicating that the nanogels could accumulate in the tumor by the leaking structure of the blood capillary and they were activated to restore high fluorescence in a tumor-specific, high GSH environment. PhA-ENG had even higher fluorescence intensity than its non-targeting counterpart, proving that the targeting ligand facilitated the tumor accumulation of the nanogel in vivo. To be noted, free PhA also showed slight localization to the tumor, probably because a portion of the free drug aggregated or bound to serum proteins and formed particles in a circulatory system, which also entered the tumor due to its leaking blood vessels. However, the free PhA signal quickly decreased in the mouse 24 h post-treatment. On the contrary, the nanogel showed high fluorescence intensity throughout the entire mouse’s body, indicating the long circulation time for the nanogels.
Fig. 6.

(A) Biodistribution of PhA, PhA-NG, and PhA-ENG. UMSCC 22A xenograft mice were administered with PhA, PhA-NG, and PhA-ENG by retro-orbital injection. Mice were imaged 24 h post-injection under IVIS Lumina III In Vivo Imaging System (Ex=620 nm, Em=670 nm). Circle reveals the location of the tumor. White circles indicate a tumor is receiving PDT. The laser power was 11.4 J/cm2. (B) Tumor growth curves after PDT treatment. Inset showed the tumor sizes of different treatments 21 days after PDT. (C) The effect of different treatments on mouse body weight (bars represent ± SD).
At 24 h post injection, the tumor was exposed to a 670 nm laser (11.4 J/cm2) once to produce PDT effect. The size of the tumor was monitored for 21 days. As shown in Fig. 6B, free PhA coupled with laser irradiation inhibited the growth of tumor by 73.2%. Both PhA loaded nanogels exhibited better tumor growth inhibitory effects than free Pha, while PhA-ENG had the highest PDT efficacy. Non-targeted PhA-NG and EGFR-targeted PhA-ENG reduced the tumor size by 86.8% and 94.5%, respectively. This was probably because of the collective effect of the substantially high accumulation of PhA-ENG in the tumor boosted by the targeting ligand, rapid activation of its photoactivity by the high GSH level in the tumor, and subsequently efficient generation of 1O2 to kill cancer cells.
The in vivo toxicity of the nanogels was also investigated. Within the duration of 21 days after PDT treatment, no apparent bodyweight change was observed for all four groups (Fig. 6C). Organs including liver, lung, kidney, and spleen were also harvested 21 days after treatment for H&E staining histological analysis. No obvious signs of toxic side effects were found in these organs (Fig. 7). All these results suggested that the PhA nanogel was safe and effective for head and neck cancer therapy.
Fig. 7.

Representative images of H&E stained liver, kidney, lung, and spleen tissue sections.
Conclusions
A bioactivatable self-quenching nanogel was designed and synthesized for targeted photodynamic therapy. The nanogel was fabricated by crosslinking PhA conjugated polymer through disulfide bonds. The aggregation of PhA in the nanogel resulted in a fluorescence quenching and the reduced 1O2 generation. Due to its responsiveness to redox potential, both the fluorescence intensity and 1O2 generation capacity can be restored in the tumor stroma and cytosol. The GSH mediated activation of photoactivity made the nanogel very safe for in vivo application since the nanogel would remain in its inactivated state and inhibit its phototoxicity to normal tissues due to the low GSH level there. While in high GSH level environment such as a tumor, the photoactivity of the nanogel would be activated and efficiently produce 1O2 for PDT. Due to the overexpression of EGFR in the tumor, Anti-EGFR Affibody decorated nanogel showed better tumor targeting efficiency and PDT effect in HNSCC. Furthermore, thanks to its self-quenched property in non-targeted tissues, no toxic side effect was observed. Thus, the EGFR targeted nanogel is a safe and effective PS carrier for PDT of head and neck cancer.
Supplementary Material
Acknowledgements
The authors want to thank the National Institutes of Health (1R15CA188847-01A1 and 1R01AG054839-01A1) for financial support of the research.
Footnotes
Electronic Supplementary Information (ESI) available: Details of synthesis and characterization of polymer, nanoparticles, and cell based in vitro assays. See DOI: 10.1039/x0xx00000x
References
- 1.Lucky SS, Soo KC and Zhang Y, Chem Rev, 2015, 115, 1990–2042. [DOI] [PubMed] [Google Scholar]
- 2.Chatterjee DK, Fong LS and Zhang Y, Adv Drug Deliv Rev, 2008, 60, 1627–1637. [DOI] [PubMed] [Google Scholar]
- 3.Dolmans DE, Fukumura D and Jain RK, Nat.Rev.Cancer, 2003, 3, 380–387. [DOI] [PubMed] [Google Scholar]
- 4.Agostinis P, Berg K, Cengel KA, Foster TH, Girotti AW, Gollnick SO, Hahn SM, Hamblin MR, Juzeniene A, Kessel D, Korbelik M, Moan J, Mroz P, Nowis D, Piette J, Wilson BC and Golab J, CA Cancer J.Clin, 2011, 61, 250–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Master A, Livingston M and Sen Gupta A, J Control Release, 2013, 168, 88–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hur C, Nishioka NS and Gazelle GS, Digestive diseases and sciences, 2003, 48, 1273–1283. [DOI] [PubMed] [Google Scholar]
- 7.Kostovic K, Pastar Z, Ceovic R, Mokos ZB, Buzina DS and Stanimirovic A, Collegium antropologicum, 2012, 36, 1477–1481. [PubMed] [Google Scholar]
- 8.Jerjes W, Upile T, Akram S and Hopper C, Clin Oncol (R Coll Radiol), 2010, 22, 785–791. [DOI] [PubMed] [Google Scholar]
- 9.Brasseur N, Ouellet R, La Madeleine C and van Lier JE, Br J Cancer, 1999, 80, 1533–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malanga M, Seggio M, Kirejev V, Fraix A, Di Bari I, Fenyvesi E, Ericson MB and Sortino S, Biomaterials Science, 2019, 7, 2272–2276. [DOI] [PubMed] [Google Scholar]
- 11.Rijcken CJ, Hofman JW, van Zeeland F, Hennink WE and van Nostrum CF, J Control Release, 2007, 124, 144–153. [DOI] [PubMed] [Google Scholar]
- 12.Wang Q, Li JM, Yu H, Deng K, Zhou W, Wang CX, Zhang Y, Li KH, Zhuo RX and Huang SW, Biomaterials Science, 2018, 6, 3096–3107. [DOI] [PubMed] [Google Scholar]
- 13.Kojima C, Toi Y, Harada A and Kono K, Bioconjug Chem, 2007, 18, 663–670. [DOI] [PubMed] [Google Scholar]
- 14.Yang H, Chen Y, Chen ZY, Geng Y, Xie XX, Shen X, Li TT, Li S, Wu CH and Liu YY, Biomaterials Science, 2017, 5, 1001–1013. [DOI] [PubMed] [Google Scholar]
- 15.Lin AL, Li SZ, Xu CH, Li XS, Zheng BY, Gu JJ, Ke MR and Huang JD, Biomaterials Science, 2019, 7, 211–219. [DOI] [PubMed] [Google Scholar]
- 16.He H, Cattran AW, Nguyen T, Nieminen A-L and Xu P, Biomaterials, 2014, 35, 9546–9553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng Y, A CS, Meyers JD, Panagopoulos I, Fei B and Burda C, J Am Chem Soc, 2008, 130, 10643–10647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dougherty TJ, Cooper MT and Mang TS, Lasers Surg Med, 1990, 10, 485–488. [DOI] [PubMed] [Google Scholar]
- 19.Ogawa K and Kobuke Y, Anti-cancer agents in medicinal chemistry, 2008, 8, 269–279. [DOI] [PubMed] [Google Scholar]
- 20.Hackbarth S, Horneffer V, Wiehe A, Hillenkamp F and Roder B, Chem Phys, 2001, 269, 339–346. [Google Scholar]
- 21.Meng LB, Zhang WY, Li DQ, Li Y, Hu XY, Wang LY and Licd GG, Chemical Communications, 2015, 51, 14381–14384. [DOI] [PubMed] [Google Scholar]
- 22.Li L, Nurunnabi M, Nafiujjaman M, Jeong YY, Lee YK and Huh KM, J Mater Chem B, 2014, 2, 2929–2937. [DOI] [PubMed] [Google Scholar]
- 23.Jang B and Choi Y, Theranostics, 2012, 2, 190–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen W, Zhong P, Meng FH, Cheng R, Deng C, Feijen J and Zhong ZY, Journal of Controlled Release, 2013, 169, 171–179. [DOI] [PubMed] [Google Scholar]
- 25.Oh IH, Min HS, Li L, Tran TH, Lee YK, Kwon IC, Choi K, Kim K and Huh KM, Biomaterials, 2013, 34, 6454–6463. [DOI] [PubMed] [Google Scholar]
- 26.Bae BC and Na K, Biomaterials, 2010, 31, 6325–6335. [DOI] [PubMed] [Google Scholar]
- 27.Bahadur K. C R and Xu P, Advanced Materials, 2012, 24, 6479–6483. [DOI] [PubMed] [Google Scholar]
- 28.He H, Altomare D, Ozer U, Xu H, Creek K, Chen H and Xu P, Biomaterials Science, 2016, DOI: 10.1039/C5BM00325C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.K CR, Thapa B and Xu P, Mol Pharm, 2012, 9, 2719–2729. [DOI] [PubMed] [Google Scholar]
- 30.Gregori M, Orlando A, Re F, Sesana S, Nardo L, Salerno D, Mantegazza F, Salvati E, Zito A, Malavasi F, Masserini M and Cazzaniga E, J Pharm Sci-Us, 2016, 105, 276–283. [DOI] [PubMed] [Google Scholar]
- 31.Nicholson RI, Gee JMW and Harper ME, European Journal of Cancer, 2001, 37, S9–S15. [DOI] [PubMed] [Google Scholar]
- 32.Eichwurzel I, Stiel H and Roder B, J Photoch Photobio B, 2000, 54, 194–200. [DOI] [PubMed] [Google Scholar]
- 33.Ogilby PR, Chemical Society reviews, 2010, 39, 3181–3209. [DOI] [PubMed] [Google Scholar]
- 34.Hansen AR and Siu LL, Journal of Clinical Oncology, 2013, 31, 1381–1383. [DOI] [PubMed] [Google Scholar]
- 35.Weigum SE, Floriano PN, Christodoulides N and McDevitt JT, Lab Chip, 2007, 7, 995–1003. [DOI] [PubMed] [Google Scholar]
- 36.Rancan F, Helmreich M, Molich A, Jux N, Hirsch A, Roder B, Witt C and Bohm F, J Photoch Photobio B, 2005, 80, 1–7. [DOI] [PubMed] [Google Scholar]
- 37.Elias DR, Poloukhtine A, Popik V and Tsourkas A, Nanomed-Nanotechnol, 2013, 9, 194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jakubowska M, Szczygiel M, Michalczyk-Wetula D, Susz A, Stochel G, Elas M, Fiedor L and Urbanska K, Photodiagnosis and photodynamic therapy, 2013, 10, 266–277. [DOI] [PubMed] [Google Scholar]
- 39.MacDonald IJ, Morgan J, Bellnier DA, Paszkiewicz GM, Whitaker JE, Litchfield DJ and Dougherty TJ, Photochemistry and photobiology, 1999, 70, 789–797. [PubMed] [Google Scholar]
- 40.Matroule JY, Bonizzi G, Morliere P, Paillous N, Santus R, Bours V and Piette J, Journal of Biological Chemistry, 1999, 274, 2988–3000. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.