

Abstract
Glioblastoma (GBM) is the most common malignant primary tumor of the central nervous system. With no effective therapy, the prognosis for patients is terrible poor. It is highly heterogeneous and EGFR amplification is its most frequent molecular alteration.
In this light, we aimed to examine the genetic heterogeneity of GBM and to correlate it with the clinical characteristics of the patients. For that purpose, we analyzed the status of EGFR and the somatic copy number alterations (CNAs) of a set of tumor suppressor genes and oncogenes.
Thus, we found GBMs with high level of EGFR amplification, low level and with no EGFR amplification. Highly amplified tumors showed histological features of aggressiveness. Interestingly, accumulation of CNAs, as a measure of tumor mutational burden, was frequent and significantly associated to shortened survival. EGFR-amplified GBMs displayed both a higher number of concrete CNAs and a higher global tumor mutational burden than their no EGFR-amplified counterparts. In addition to genetic changes previously described in GBM, we found PARK2 and LARGE1 CNAs associated to EGFR amplification. The set of genes analyzed allowed us to explore relevant signaling pathways on GBM. Both PARK2 and LARGE1 are related to receptor tyrosine kinase/PI3K/PTEN/AKT/mTOR-signaling pathway. Finally, we found an association between the molecular pathways altered, EGFR amplification and a poor outcome.
Our results underline the potential interest of categorizing GBM according to their EGFR amplification level and the usefulness of assessing the tumor mutational burden. These approaches would open new knowledge possibilities related to GBM biology and therapy.
Abbreviations: ANOVA, ANalysis Of VAriance; BCL2, B-cell cll/ lymphoma 2; CDH1, Cadherin 1; CHD7, Chromodomain Helicase DNA Binding Protein 7; CDK6, cyclin-dependent kinase 6; CDKN1B, cyclin-dependent kinase inhibitor 1B; CDKN2A, cyclin-dependent kinase inhibitor 2A; CDKN2B, cyclin-dependent kinase inhibitor 2B; CNAs, copy number algerations; CNS, central nervous system; CNV-load, load of copy number variations; CREM, cAMP response element modulator; DAPI, 4′,6-diamidino-2-phenylindole; EGFR, epidermal growth factor receptor; EGFRvIII, epidermal growth factor receptor variant number III; FISH, fluorescence in situ hibridization; GBM, glioblastoma, IDH-wildtype (glioblastoma multiforme, primary glioblastoma).; GFAP, glial fibrillary acidic protein; H-amp GB, EGFR-high amplified glioblastoma; HIC1, hypermethylated in cancer 1; IDH1, isocitrate dehydrogenase 1; IDH2, isocitrate dehydrogenase 2; IHC, immunohistochemistry; LARGE1, acetylglucosaminyltransferase-like protein 1; MLPA, multiplex ligation-dependent probe amplification; MYBPC3, myosin-binding protein C; L-amp GB, EGFR-low amplified glioblastoma; N-amp GB, EGFR-no amplified glioblastoma; p53-pat, p53 signaling pathway; PAH, phenylalanine hydroxylase; PARK2, parkin; PTEN, phosphatase and tensin homolog; RARB, retinoic acid receptor beta; RB-pat, RB signaling pathway; TKR-pat, tyrosine-kinase receptors signaling pathway; TCGA, The Cancer Genome Atlas; TMA, tissue microarray; wt, wildtype; WHO, World Health Organization
Introduction
Glioblastoma, IDH-wildtype (GBM) is the most frequent malignant neoplasm of the human central nervous system (CNS). Although scientists and clinicians all over the world have made countless efforts over the past decades to improve the therapies for GBM patients, tumor tends to spread rapidly and to return after treatment, resulting in a very short median survival. The 2016-revised WHO classification has incorporated valuable molecular features for brain tumor classification [1]. However, while detection of IDH1 mutation is now used in diagnosis for its prognostic meaning in glioma, the prognostic value of other common genetic characteristics, as EGFR amplification in GBM, remains unclear [1], [2]. Recent research demonstrated an enhanced migratory behavior of cells within EGFR-amplified tumors, [2] supporting a relationship between EGFR status and the clinical course. This fact underlines the interest of deepen in the role of EGFR considering also the existence of different levels of EGFR amplification, as previous works have delineated [3], [4], [5], [6].
GBM is characterized by both, inter- and intra-tumor heterogeneity, with great variations at the histological and the molecular levels [1], [7]. This heterogeneity is responsible in part of drug resistance and treatment failure [8], [9]. GBM heterogeneity reach levels that, even regarding EGFR, several variants have been described; among them, variant III (EGFRvIII) promotes cell proliferation, angiogenesis and invasion in different models, [10], [11], [12] making worthy to give it special attention. The identification of differential targets among GBM IDH-wildtype genetic-subgroups could led to reach better approaches to GBM management.
Among the multiple signaling pathways deregulated in cancer, GBM stands out by alterations in the receptor tyrosine kinase/PI3K/PTEN/AKT/mTOR-signaling pathway. In addition, both the CDKN2A/CDK4/6/retinoblastoma and the p53/MDM2/p14ARF molecular pathways are also widely affected [1], [13], [14], [15]. Those signaling alterations seem to be a core requirement for GBM pathogenesis and they are associated with poor prognosis [16], [17]. Many groups have used high-throughput techniques for the genomic analysis of these pathways in GBM [18], [19], [20]. However, the enormous complexity of the results (in part because of tumor heterogeneity) usually leads to sum up the data in relation to the chromosomal loci affected, more than gene-by-gene detail with exemption of a little number of well-known genes [1], [18]. A novel approach to better understand the genetic results in cancer, considers the global extent of somatic copy number alterations (CNAs), introducing the term of “tumor mutational burden” [21], [22], [23] or CNV-load [24]. Despite different definitions according to the experimental design, this concept may be important in GBM, as it is a genetic feature that in several tumor types correlates with response to immune-checkpoint inhibitors [21], [23], [24]. Multiplex ligation-dependent probe amplification (MLPA) seems to be appropriate to explore concrete genetic changes but also the accumulation of alterations per case, as tumor mutational burden [25], [26].
The aim of the present work is to characterize in a semi-guided way the genetic landscape of fresh primary GBM, IDH-wildtype, with different EGFR amplification status; we want to identify potential biological targets differentially distributed according to EGFR, in order to improve, in the near future, the prognostic and therapy of this heterogeneous tumor.
Material and methods
Patient samples and histological analysis
This work included 46 tumor samples from patients that underwent surgery in the Hospital Clínico Universitario in Valencia. The study was reviewed and approved by the clinical investigation ethics committee at the Hospital Clínico Universitario (CEIC). Tumor samples were fixed in neutral-buffered formalin, embedded in paraffin, sectioned, and stained with hematoxylin-eosin. They were diagnosed according to the WHO classification criteria [1] as primary GBM by two different neuropathologists. Immunohistochemistry analysis (IHC) was performed on paraffin-embedded sections using the avidin-biotin peroxidase method. IHC was carried out using antibodies directed against glial fibrillary acidic protein (GFAP), Ki-67 (MIB1) and EGFR -clone H11 (all from Dako, Glostrup, Denmark). The proliferation rate was calculated as the percentage of MIB1 immunopositive nuclei. GFAP and EGFR expression were scored according to the staining intensity and the number of stained cells using previously described criteria: 0, no staining; 1, light or focal staining; 2, moderate staining present in 50% to 75% of the sample and 3, strong staining, present in more than 75% of the sample. For EGFR IHC analysis, 0–1 were defined as non-overexpression and 2–3 were considered overexpression of EGFR [27].
DNA extraction and DNA sequencing for IDH1/2 and TP53 mutations
Genomic DNA was extracted from fresh tissue samples using a QIAamp DNA Mini Kit (Qiagen, Inc., Valencia, CA, USA) according to the manufacturer’s instructions. We analyzed by direct sequencing the genomic regions spanning wild-type R132 of IDH1 and wild-type R172 of IDH2. We also carried out TP53 sequencing in four different PCR amplification reactions to analyze exons 5–8. PCR was performed using standard buffer conditions, 200 ng of DNA and an AmpliTaq Gold Master Mix (Thermo Fisher Scientific). PCR products were purified with Centricon columns (Amicon, Beverly, MA, USA) and they were analyzed on an ABI 310 Sequencer (Applied Biosystems, Foster City, CA, USA). Primer sequences forward (fw) and reverse (rv) were as follow: IDH1 fw 5′-ACCAAATGGCACCATACGAA, IDH1 rv 5′-TCACA TTATTGCCAACATGACTT, IDH2 fw 5′-CCAATGGAACTATCCGGAAC, IDH2 rv 5′-CCTCTCCACCCTGGC CTAC, TP53(e5) fw 5′-CAGCCCTGTCGTCTCTCCAG, TP53(e5) rv 5′-TTCAACTCTG TCTCCTTCCT, TP53(e6) fw 5′-GTCTGGCCCCTCCTC AGCAT, TP53(e6) rv 5′-GTCTGGCCCCTCCTCAGCAT, TP53(e7) fw 5′-CTCATCTTG GGCCTGTGTTA, TP53(e7) rv, 5′-AGTGTGCAGGGTGGCAAGTG, TP53(e8) fw 5′-ACCTGATTTCC TTACTGCCTCTTGC and TP53(e8) rv 5′-GTCCTGCTTGCTTACCTC GCTTAGT.
Fluorescence in situ hybridization analysis (FISH)
To evaluate the EGFR gene amplification status in this series, we used FISH on tissue microarrays (TMAs) that included representative samples from de 46 tumors studied. Firstly, we build eight TMA using a Manual Tissue arrayer I (Beecher Instruments, Sun Prairie, WI, USA). Briefly, we removed four 0.6-mm cores from selected tumor areas of the paraffin blocks in each case. Afterwards, the paraffin-embedded TMA blocks were sectioned at 5 μm and mounted on Superfrost Plus microscope slides (Microm International, Walldorf Germany). Dual-color fluorescence in-situ hybridizations (FISH) were performed using the LSR-EGFR Spectrum Orange/CEP-7 Spectrum Green Probe from Vysis (Abbott Laboratories, Downers Grove, IL, USA) according to the manufacturer's instructions and the nuclei were counterstained with DAPI. Fluorescent signals were detected using a Leica DM400B photomicroscope equipped with an appropriate filter set (Leica microsystems AG, Wetzlar, Alemania). Signals were counted in 100–150 non-overlapping tumor cell nuclei and their values were used to calculate the EGFR/CEP-7 signal ratio. The EGFR gene was considered to be amplified when the EGFR/CEP-7 signal ratio was > 2. The exact ratio was not calculated in cases with high amplification levels [28]. Cases were subclassified according to previous descriptions as H-amp GBMs when more than 20% of the cells showed more than 20 copies of EGFR; L-amp GBMs included cases with 5–20% of cells with 3–12 copies of EGFR and cases with 2 copies of EGFR composed the N-amp group of GBMs [3].
Multiplex ligation-dependent probe amplification analysis (MLPA).
MLPA was performed to determine somatic CNAs of multiple oncogenes and tumor-suppressor genes simultaneously. Among them, probes for all the exons of EGFR were included, allowing us to determine a wildtype status (wt) or the presence of the mutant form EGFRvIII, which losses exons 2–7, [29] in addition to CNAs. Two different MLPA kits (Salsa MLPA kit P105-C1 and Salsa MLPA KIT ME001-C2) were used following the manufacturer’s instructions (MRC-Holland, Amsterdam, Netherlands). Both kits are approved for investigational-use only. Amplification fragments were separated by capillary electrophoresis in an ABI 310 Sequencer (Applied Biosystems) and data analysis was performed using the Coffalyser excel-based MLPA analysis software (MRC-Holland). The thresholds established to classify losses and gains of genetic material were set at 0.75 and 1.3, respectively. The set of probes included in these assays covered the following genes: APC, ATM, BCL2, BRCA1, BRCA2, CASP8, CASR, CD27, CD44, CDH1, CDH13, CDK4, CDK6, CDKN1B, CDKN2A, CDKN2B, CHD7, CHFR, CREM, CTNNB1, DAPK1, EGFR, ESR1, FHIT, GSTP1, HIC1, HIRIP3, IGSF4, IL4, KLK3, LARGE1, MDM2, MIR26, MLH1, MLH3, MYBPC3, MYO5B, NFKBIA, OCA2, PAH, PARK2, PDGFRA, PKHD1, PTEN, RAB7, RARB, RASSF1, RYBP, SAMHD1, SCN1A, TIMP3, TP53, TSC2 and VHL. The collection of genes included allowed us to estimate the implication of the receptor tyrosine kinase/PI3K/PTEN/AKT/mTOR-signaling pathway, the CDKN2A/ CDK4/6/ retinoblastoma-signaling pathway and the p53/MDM2/p14ARF signaling pathway. Considering MLPA results we have established different thresholds to assess the tumor genetic burden as accumulation of CNAs. In order to be able to take into account the dispersion of the data within the three groups formed on the basis of FISH-EGFR status, we have determined thresholds for losses, gains and global CNAs. This formula was the product of the maximum value and the average value, divided by the product of the variance and the asymmetry.
Analysis of TCGA dataset
To validate the association between EGFR amplification status and somatic CNAs in the genes mentioned above, we obtained the data for GBM samples from The Cancer Genome Atlas (TCGA) by using cBioPortal for Cancer Genomics (www.cbioportal.org) [20], [30], [31]. We studied the Genomic Profile “Putative copy-number alterations from GISTIC” for latest dataset available in cBioportal for GBM (TCGA, Provisional 604 samples). Copy number alteration data from 577 cases were obtained and further analyzed. A “User-defined List” including EGFR, BCL2, CDK6, CDKN1B, CDKN2B, ER1, LARGE1, MYBPC3, PARK2 and PAH was entered into the “Enter Gene” box. Samples were classified for each gene into 5 groups according to their putative copy number variation calculated by GISTIC with default cBioportal thresholds [30]. The groups were Diploid (0), Shallow Deletion (-1), Deep deletion (-2), Gain (1) and Amplification (2). The associations between EGFR amplification and the copy number profile were analyzed using the “Plots” tool and retrieving the raw data. A contingency table was built for expressing the distribution among groups of all samples according to the EGFR amplification status. A Fisher’s exact test was used for calculating statistical significance. Statistical significance for amplification vs diploid and deletion (shallow or deep) vs diploid for each gene was calculated.
Statistical analysis
The statistical analysis of the different analyzed parameters was carried out according to the type of variable. Quantitative variables (age, Karnosky index, tumor size, survival and Ki-67) were evaluated using the Kolmogorov-Smirnov and Levene tests; depending on their results and their characteristics, two-tailed Student’s t-test, ANOVA, Mann-Whitney U test or Kruskcal-Wallis test were performed. For comparisons among categorical variables, Fisher exact test calculator was used for 2 × 2 tables and Chi-square test was used for tables with higher number or rows/columns. We also carried out survival analyses using the Kaplan-Meier method. The statistical significance of these survival curves was calculated using the log-rank (Mantel-Cox) test. Significance was accepted at least at p < 0.05 level. To facilitate understanding, in addition to p value, symbols were added to significant results (* when p < 0.05), very significant results (**p ≤ 0.01) and highly significant results (***p ≤ 0.001). Data were analyzed with SPSS (version 22) software (IBM, Madrid, Spain).
Results
Patient population and histopathological study
We studied a series of 46 primary GBM from adult patients; 29 patients were men and 17 women, reflecting the GBM male predominance. The patients’ ages ranged from 24-75, with a mean of 59 years. The tumor size ranged between one and eight cm3. Tumor location affected the frontal lobe in 12 cases, temporal lobe in 18 cases, parietal lobe in 13 cases, occipital lobe in two cases and one case in callosa commissure. The patients received, in addition to the surgical treatment, chemotherapy in 2 cases, radiotherapy (RT) in 18 cases, both RT and chemotherapy in 23 cases and 3 cases only received palliative care. Five patients that did not received RT tend to be older than the other 41 patients (68.4 ± 4.0 and 57.27 ± 1.3, p = 0.058). Overall survival for these patients averaged 13.2 months, without differences between women and men. Histologically, all the tumors demonstrated features of GBM with pleomorphic, astrocytic tumor cells, prominent microvascular proliferation, and necrosis (Figure 1A–C). In every case, GFAP expression was confirmed in neoplastic cells. Ki-67 indicated a high proliferation potential with a mean value of 15% (Figure 1D). EGFR expression was variable both among tumors and among different regions of the same tumor. In 24 cases, a level 2–3 was determined and 8 cases scored level 1 (Figure 1E–F). The remaining 14 cases were negative for EGFR expression. The clinical data and the histopathological results are summarized in Table 1.
Figure 1.

Histopathology, immunohistochemistry and fluorescence in situ hybridization (FISH) analysis of EGFR in primary GBM. (A) Pleomorphic, astrocytic tumor cells, (B) prominent microvascular proliferation and (C) presence of necrosis. (D) Ki-67 positive cells ratio indicated a higher proliferation range. (E) EGFR overexpression with a level 3 and (F) with a level 2 was present. (G) Tumor cells showing a high level of EGFR amplification, (H) cells with a low level of EGFR amplification and (I) cells without EGFR amplification, determined by FISH.
Table 1.
Clinical and histopathological data and genetic status of EGFR in 46 GB cases. T: temporal; F: frontal; P: parietal; O: occipital; CC: callosa commissure; RT: radiotherapy; CH: chemotherapy; amp: amplification; wt: wild type; EGFRvIII: EGFR mutant form.
| Case | Age/Sex | Location | Size (cm) | Treatment | Survival (months) | Ki-67 (%) | EGFR expression | EGFRampl. | EGFRwt/mutant | |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 56/F | T | 2 | RT + CH | 23 | 5 | 3 | 2 | EGFRwt | |
| 2 | 72/F | F | 6 | RT + CH | 8 | 20 | 3 | 3 | EGFRvIII | |
| 3 | 63/F | T | 7 | RT + CH | 11 | 8,5 | 3 | 3 | EGFRvIII | |
| H-amp | 4 | 69/M | T | 6 | RT | 7 | 15 | 3 | 2 | EGFRvIII |
| 5 | 48/M | T | 5,3 | RT + CH | 11 | 3 | 3 | 2 | EGFRwt | |
| 6 | 55/F | CC | 4 | CH | 4 | 14,5 | 3 | 3 | EGFRvIII | |
| 7 | 59/F | P | 2,6 | RT | 18 | 40 | 3 | 3 | EGFRvIII | |
| 8 | 58/F | T | 6 | RT + CH | 23 | 35 | 3 | 2 | EGFRvIII | |
| 9 | 61/M | T | 8 | RT + CH | 12 | 4 | 3 | 3 | EGFRwt | |
| 10 | 59/F | P | 2 | RT + CH | 20 | 30 | 3 | 3 | EGFRwt | |
| 11 | 66/M | P | 4 | RT + CH | 6 | 19,7 | 2 | 3 | EGFRwt | |
| L-amp | 12 | 55/M | T | 3 | RT + CH | 5 | 6 | 2 | 2 | EGFRwt |
| 13 | 66/F | F | 4 | RT | 36 | 8 | 2 | 2 | EGFRwt | |
| 14 | 69/M | T | 6 | RT + CH | 12 | 50 | 2 | 3 | EGFRvIII | |
| 15 | 66/M | F | 2 | RT | 5 | 45 | 3 | 3 | EGFRvIII | |
| 16 | 61/F | F | 6 | RT | 2 | 22,5 | 3 | 3 | EGFRvIII | |
| 17 | 58/F | O | 2 | RT + CH | 20 | 5 | 2 | 2 | EGFRwt | |
| 18 | 57/M | F | 3,5 | RT | 17 | 12 | 2 | 3 | EGFRwt | |
| 19 | 71/M | F | 2 | RT + CH | 3 | 20 | 2 | 3 | EGFRwt | |
| 20 | 63/M | T | 2 | RT | 5 | 43 | 2 | 2 | EGFRwt | |
| 21 | 45/M | T | 7 | RT | - | 3 | 1 | 1 | EGFRwt | |
| 22 | 24/M | T | 4 | RT | 2 | 30 | 1 | 1 | EGFRwt | |
| 23 | 67/M | T | 5 | RT + CH | 7 | 43 | 0 | 1 | EGFRwt | |
| 24 | 73/M | P | 2,5 | RT | 5 | 22 | 0 | 1 | EGFRwt | |
| 25 | 45/M | P | 2,5 | RT + CH | 10 | 6,8 | 2 | 1 | EGFRwt | |
| 26 | 42/M | P | 2 | RT | - | 3 | 0 | 1 | EGFRwt | |
| 27 | 60/M | P | 3,7 | RT | 5 | 2 | 1 | 1 | EGFRwt | |
| 28 | 31/F | F | 2 | RT | - | 3 | 1 | 1 | EGFRwt | |
| N-amp | 29 | 73/F | P | 6 | RT + CH | 26 | 1 | 1 | 1 | EGFRwt |
| 30 | 35/M | T | 2 | RT | 38 | 30 | 1 | 1 | EGFRwt | |
| 31 | 74/F | F | 4 | NONE | 1 | 12 | 0 | 0 | EGFRwt | |
| 32 | 66/M | T | 4 | RT + CH | 11 | 5 | 0 | 0 | EGFRwt | |
| 33 | 35/F | P | 1 | RT | - | 2 | 0 | 0 | EGFRwt | |
| 34 | 54/M | T | 4 | RT + CH | 5 | 26 | 0 | 0 | EGFRwt | |
| 35 | 63/M | F | 4 | RT + CH | 36 | 23 | 0 | 0 | EGFRwt | |
| 36 | 75/M | P | 3,5 | NONE | 6 | 44 | 2 | 0 | EGFRwt | |
| 37 | 55/M | F | 2 | RT + CH | 26 | 3 | 1 | 0 | EGFRwt | |
| 38 | 73/M | O | 6,6 | RT + CH | 11 | 2 | 0 | 0 | EGFRwt | |
| 39 | 63/F | P | 4 | CH | 9 | 2 | 1 | 0 | EGFRwt | |
| 40 | 67/M | P | 3 | RT + CH | 21 | 7,5 | 2 | 0 | EGFRwt | |
| 41 | 75/F | F | 1 | NONE | 2 | 2 | 0 | 0 | EGFRwt | |
| 42 | 60/M | F | 6 | RT + CH | 2 | 1 | 0 | 0 | EGFRwt | |
| 43 | 50/M | T | 3,5 | RT | 10 | 5 | 0 | 0 | EGFRwt | |
| 44 | 65/F | T | 1 | RT + CH | 22 | 1 | 2 | 0 | EGFRwt | |
| 45 | 38/M | P | 1 | RT | 30 | 1 | 0 | 0 | EGFRwt | |
| 46 | 52/M | T | 2 | RT | 23 | 2 | 0 | 0 | EGFRwt |
Sequencing studies of IDH1/IDH2 and TP53
Genomic analysis encompassing the R132 region of IDH1 and the R172 region of IDH2 revealed no mutations in any of the 46 cases studied. This fact confirmed their classification as IDH-wildtype GBMs, according to the most recent WHO classification [1]. Exons 5, 6, 7 and 8 on TP53 were also studied. TP53 mutations were found in case 29 (heterozygous substitution-missense mutation p.C141R, in exon 5) and in case 33 (heterozygous substitution-missense mutation p.R273C, in exon 8).
EGFR characterization: amplification status is associated to EGFR protein expression and to EGFRvIII form
EGFR copy number analysis by FISH was successfully assessed in the 46 primary GBMs and revealed EGFR amplification in 30 cases (65.2%). Based on this analysis cases 1–20 showed a high level of EGFR amplification (H-amp group, Figure 1G), cases 21–30 showed a low level of EGFR amplification (L-amp group, Figure 1H) and cases 31–46 displayed no EGFR amplification (N-amp group, Figure 1I). Immunohistochemical analysis of EGFR protein expression is shown in Table 1. It revealed a strong statistical association between the genetic status of EGFR determined by FISH and the protein levels detected by IHC (p = 4.3E-6***). The receptor was overexpressed in every case from the H-amp group (levels 2–3). One case showed EGFR protein overexpression in L-amp group and only three cases from the N-amp group displayed EGFR protein overexpression. These results agreed with previous reports that confirm the appropriateness of FISH for the classification of EGFR amplification [3]. MLPA study showed the presence of the mutant form of EGFR gene, EGFRvIII, in 9 cases. All of them were also H-amp GBMs (p = 0.001***). All these data are summarized in Table 1.
EGFR genetic status correlates with morphological and histological features of aggressiveness
The frequency of men was higher in both L-amp (8 men/ 2 women) and N-amp groups (11 men/ 5 women) whereas that difference disappeared in the H-amp group (10 men/ 10 women). The average size of the tumors was the greatest in the H-amp group (4.2 cm3), followed by the L-amp group (3.7 cm3) and the N-amp group (3.2 cm3) but these differences did not reach statistical significance (supplementary file 1). EGFRvIII cases showed an average of 5.1 cm3, which was significantly higher than 3.4 cm3 in EGFR wildtype (wt) cases (p = 0.013*) but these differences disappear if we consider exclusively variant III and wild-type cases among EGFR H-amp GBMs. Overall survival of the patients was similar among the different groups (12–14 months). Regarding Ki-67, the H-amp group presented a mean value of 20.3%. It was statistically higher than the average Ki-67 of the L-amp group and the N-amp group, which showed 14.4% and 8.7%, respectively (p = 0.020*). In addition, EGFRvIII cases showed 27.8% Ki-67 compared to 11.8% in EGFRwt cases (p = 0.013*).
Somatic copy number alterations are frequent in GBM and their accumulation is associated to shortened survival
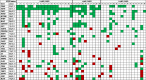
Somatic copy number alteration (CNA) analysis showed losses and/or gains in, at least, one of the regions explored in all the tumor samples assayed (Table 2). CNAs were frequent in our series showing an average of 12.6 ± 8.9 CNAs per patient. CNAs were statistically higher in the 41 patients that received RT than in the other 5 patients (5.8 ± 0.8 and 2.8 ± 0.9, respectively, p = 0.025). From the 54 genes studied, 12 presented losses in more than 20% of cases: CDKN2A (54.3%), CDKN2B (54.3%), PTEN (41.3%), CDH1 (37.0%), TP53 (34.8%), LARGE1 (34.8%), CREM (32.6%), MYBPC3 (32.6%), VHL (32.6%), GSTP1 (28.3%), SAMHD1 (28.3%) and PAH (23.9%). The losses for CDKN2A and CDKN2B were mostly in homozygosis, and they were confirmed by probes included into the two kits assayed. Likewise, six genes showed gains with that frequency: EGFR (60.9%), CDK6 (60.9%), CHD7 (39.1%), RARB (28.3%), CDKN1B (26.1%), HIC1 (23.9%) and PARK2 (21.7%). These alterations were located on chromosomes 3, 6, 7, 8, 9, 10, 11, 12, 16, 17, 20 and 22 (Figure 2).
Table 2.
Somatic copy number alteration analysis. Individual results (part I).
 |
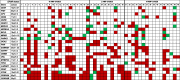 |
The copy number alterations are color-coded. Gene losses are shown in red. Gene gains are shown in green. Abbreviations: H-AMP, high-EGFR amplification level; L-AMP, low-EGFR amplification level; N-AMP, no EGFR amplification.
Figure 2.

Chromosomal location of the genes that displayed somatic copy number alterations within the 46 studied primary glioblastomas. Dark circles represent gains and clear circles represent losses.
Tumors displaying CDK6 gains were significantly bigger, with a mean value of 4.2 cm3, compared to 3.0 cm3 in CDK6wt tumors (p = 0.038*). Furthermore, patients’ tumors with losses in LARGE1 tended to present statistically higher Ki-67, with an average of 24.0% compared to 10.3% in LARGE1wt tumors (p = 0.040*).
Each CNA had no impact on survival independently. However, overall data showed that survival was strongly associated to global CNAs. Threshold for losses was 10, for gains was 4 and for global CNAs was 7 (supplementary file 2). On one hand, survival time statistically fell from 10 losses/patient from 452.7 days to 193.3 days (p = 0.001***). On the other hand, it also fell from 4 gains/patient, since 477.6 to 278.8 days (p = 0.025*). On the whole, cases under 7 CNAs showed 556.0 days while from that threshold it fell to 308.9 (p = 0.015*) although considering together losses and gains seems to lose biological perspective. Kaplan-Meier survival analysis showed significant associations through Long Rank (Mantel-Cox) statistic for both, >4 gains/patient (p = 0.036*) and > 10 losses / patient (p = 0.009**) (Figure 3A, B). The statistical strength reduced when we consider together losses and gains, and Kaplan-Meier survival analysis offered significance through Breslow approach. It is remarkable that all the cases above 10 copy number losses exhibited simultaneously more than 4 copy number gains among different loci.
Figure 3.

Accumulation of somatic copy number alterations (CNAs) associates to overall survival. (A) Green line represents survival in patients which tumors showed more than 4 gene gains (n = 17, mean = 278.8 days) and blue line represents survival in patients which tumors showed 4 or less gene gains (n = 25, mean = 477.6 days), reflecting a statistical association (p = 0.025*). (B) Green line represents survival in patients which tumors showed more than 10 gene losses (n = 15, mean = 193.3 days) and blue line represents survival in patients which tumors showed 10 or less gene losses (n = 27, mean = 452.7 days), reflecting an even stronger statistical association (p = 0.009**). In two cases was impossible to achieve survival data. (C) Somatic CNAs that were differentially found among the EGFR groups. CDKN2A and CDKN2B were significantly more altered in H-amp GBs compared with N-amp GBs (p = 0.030* in both cases). LARGE1, PARK2 and CDK6 were significantly more altered in both H-amp and L-amp GBMs than in N-amp GBs (p = 0.009**, p = 0.046* and p = 0.046*, respectively). Definitions: *p < 0.05 and **p < 0.01; CNAs, copy number alterations; H-amp, glioblastomas with a high number of extra copies of EGFR; L-amp, glioblastomas with a low number of additional copies of EGFR; N-amp, glioblastomas without EGFR amplification.
EGFR amplified GBMs display a high number of concrete genetic copy number alterations
We analyzed the distribution of CNAs among the three EGFR amplification groups. Five genes presented significant CNA changes among them (Figure 3C). CDKN2A (p = 0.030*), CDKN2B (p = 0.030*), LARGE1 (p = 0.009**), PARK2 (p = 0.046*) and CDK6 (p = 0.046*). All these genes were statistically more affected in the H-amp group or both in H-amp and L-amp, compared to N-amp group (Figure 3C). CDKN2A, CDKN2B and LARGE1 displayed mainly losses, CDK6 showed gains and PARK2 exhibited both losses and gains. Taken together all the genetic changes detected, we found that somatic CNAs were lower in N-amp group, with 9.3 ± 7.6 CNAs, compared to L-amp and H-amp which accounted 14.8 ± 6.6 and 14.1 ± 10.1 CNAs, respectively (p > 0.050).
Genetic landscape of L-amp GBMs partially overlaps with both H-amp GBMs and N-amp GBMs
Then, we made an overview of the most relevant genes based on the frequency they were altered. We considered the five aforementioned genes and others that showed CNAs affecting at least 40% of the cases within an EGFR amplification group although they did not show statistical differences among all the three subgroups (MYBPC3, PTEN, BCL2, PAH, CREM, CDH1, CHD7, CDKN1B, RARB, PARK2 and HIC1). H-amp group exhibited five genes reaching the 40% of cases with loss, L-amp group presented eight genes and N-amp only one gene (Figure 4A). These results reflect an increasing affectation from N-amp to H-amp and an overlapping profile between L-amp and H-amp. Regarding to the gains, H-amp group presented two genes reaching this threshold, L-amp group showed six genes and N-amp two genes (Figure 4B). The analysis of TCGA putative copy number alterations data for GBM showed that there are statistically significant associations between EGFR amplification and CDK6 amplification, CDKN2A and CDKN2B deletion, MYBPC3 deletion and PAH deletion (Table 3). CDKN1B, ESR1, PARK2 or LARGE1 copy number alterations are published in the TCGA collection but their association to EGFR amplification were not statistically significant.
Figure 4.

Genes analyzed above the established threshold of somatic CNAs. The figures show the proportion of cases with alterations in each gene within each EGFR amplification group. (A) With respect to the losses, H-amp group exhibited five genes reaching the 40% of cases with losses, L-amp group presented eight genes and N-amp only one gene. (B) With respect to the gains, H-amp group presented two genes reaching the 40% of cases with losses, L-amp group presented six and N-amp two. Definitions: CNAs, copy number alterations; H-amp, glioblastomas with a high number of extra copies of EGFR; L-amp, glioblastomas with a low number of additional copies of EGFR; N-amp, glioblastomas without EGFR amplification.
Table 3.
Associations between EGFR amplification and SCNAs in other genes (TCGA analysis).
| EGFR diploid |
EGFR amplification |
|||||||
|---|---|---|---|---|---|---|---|---|
| Dip | Amp | Del | Dip | Amp | Del | p-value (1) | p-value (2) | |
| CDK6 | 56 | 2 | 1 | 25 | 15 | 0 | 1.5E-05**** | 1.0E + 00 |
| CDKN1B | 47 | 1 | 9 | 207 | 1 | 26 | 3.4E-01 | 3.6E-01 |
| CDKN2A | 22 | 1 | 39 | 35 | 0 | 215 | 4.0E-01 | 1.8E-04# # # |
| CDKN2B | 22 | 1 | 39 | 35 | 0 | 215 | 4.0E-01 | 1.8E-04# # # |
| ESR1 | 46 | 0 | 14 | 173 | 0 | 72 | 1.0E + 00 | 4.2E-01 |
| LARGE1 | 41 | 0 | 19 | 179 | 0 | 69 | 1.0E + 00 | 6.3E-01 |
| MYBPC3 | 46 | 0 | 17 | 216 | 0 | 26 | 1.0E + 00 | 2.0E-03# # |
| PAH | 50 | 1 | 11 | 222 | 0 | 14 | 1.9E-01 | 7.2E-03# # |
| PARK2 | 45 | 4 | 15 | 155 | 7 | 91 | 2.9 E-01 | 9.61E-02 |
(1)Statistical significance for the comparison between the ratio of amplifications for the target gene in EGFR amplified samples vs EGFR diploid samples. ****p < 0.0001.
(2)Statistical significance for the comparison between the ratio of deletions for the target gene in EGFR amplified samples vs EGFR diploid samples. # #p < 0.01, # # #p < 0.001.
Relevant signaling pathways on GBM are altered in EGFR amplified tumors and related to a poor outcome
From the 46 GBM studied cases, 84% presented CNAs in genes involved in the receptor tyrosine kinase/PI3K/PTEN/AKT/mTOR-signaling pathway (TKR-pat), 43.5% had gene alterations in genes from p53-pathway (p53-pat) and 65% in genes associated with RB-signaling pathway (RB-pat). CNAs in genes from TKR-pat were detected in all the samples belonging to H-amp group and in 90% of the samples in L-amp group, but only in 37.5% of N-amp cases (p = 0.001***). Similar frequencies were observed for CNAs in genes from RB-Pat, but with closer percentages (90% in H-amp, 80% in L-amp and 56.3% in N-amp, p = 0.059). Affectation of genes from RB-pat, was similar among the three EGFR amplification groups, with values ranging 30–50%.
Regarding the clinical characteristics of the cases, we found that patients that showed alterations on genes in TRK-pat and RB-pat presented bigger tumors: tumor size was 2.7 cm3 and 2.5 cm3 when TRK-pat or RB-pat were not altered and it was 4.1 cm3 when any of them were affected (p = 0.019* and p = 0.013*, respectively). In addition, Kaplan-Meier analysis showed that survival was significantly shortened when we found alterations in genes belonging to the three different pathways simultaneously in a case: it was 584 days in cases which alterations belong to only one pathway, 411 when two pathways were altered and 234 days when genes from the three pathways were implicated (p = 0.016*). Although it did not reach significant meaning, 45% of H-amp GBMs showed the three pathways altered compared with 20.0% of L-amp and 18.8% of N-amp.
Discussion
EGFR amplification has been identified as a genetic hallmark of primary GBM and occurs in approximately 40–60% of primary GBMs, but rarely in secondary GBMs; [28], [32], [33] this frequency is similar to the one detected in our series. EGFR genetic status was studied by two complementary techniques: FISH and MLPA. MLPA is an excellent tool to study EGFR amplification but also it has been used to demonstrate that EGFRvIII affects up to 67% of the GBMs with EGFR amplification [13], [34], [35]. However, it is not able to discriminate low levels of EGFR amplification, because of trisomies and polysomies of chromosome 7 [5]. For that reason, FISH was chosen to distinguish L-amp GBMs. A previous report from our group showed that high amplification presented as double minutes whereas low amplification presented as insertions into different loci on chromosoma 7 [3]. EGFR genetic status use to correlate to the EGFR protein expression level [28], [33], [36], fact that is broadly supported by our results with a high statistical meaning. In our study, all the H-amp GBMs presented overexpression of the protein. EGFR variable expression among L-amp GBMs indicates that there are cases closer to the H-amp group and others closer to the N-amp group. The presence of a little subgroup of N-amp cases with high EGFR protein levels suggests the existence of mechanisms for its expression, which would be independent to DNA amplification, as previous works have investigated and underlines the important role of EGFR in GBM [37], [38].
The use of MLPA and FISH allowed as to confirm the high frequency of EGFR amplification, both as L-amp and as H-amp, and the presence of EGFRvIII limited to H-amp GBMs, in agreement with previous reports [11], [12]. We found statistically increased proliferation and tumor size in patients that presented EGFRvIII in concordance with previous works [10], [11], [12]; our association among H-amp/EGFRvIII/tumor size/Ki-67 goes in favor of descriptions that propose a direct role of EGFRvIII on aggressiveness [11], [12], [29], [39].
In the present work, we show a semi-guided genetic analysis of many TSGs and oncogenes that are important in GBM, and we correlate these analyses to the EGFR amplification status. Although it would be desirable to confirm our results in larger series, the genetic study of fresh tumor specimens provides high quality data with low background making easy to analyze the information obtained by this technical approach.
CNAs were, as previously described [18], [40], frequent in our series supporting the classical idea of a high chromosomal and genetic instability, and they are also increased in response to RT [7], [14], [41]. The global incidence of CNAs was higher in both groups of tumors with EGFR amplification but even higher in EGFR H-amp GBMs than in L-amp tumors. In addition, the high frequency of concrete genes altered in the EGFR amplified GBMs, and especially in the EGFR H-amp group, indicated they would be directly involved in the genesis and progression of GBM.
CDK6 gains and LARGE1 losses were significantly associated to tumor size and Ki-67. Both genes, plus CDKN2A/B were statistically more altered in H-amp GBMs and all them were validated with TCGA cohort data [30]. CDK6 and CDKN2A/B are well-known gens in GBM. It is necessary to remark the potential influence of LARGE1 and PARK2 in GBM related pathways (Figure 5). There is little described about them on GBM literature but both resulted significantly altered in EGFR amplified GBMs in this study. LARGE1 encodes a glycosyltransferase that participates in glycosylation of alpha-dystroglycan. Mutations in this gene cause diverse forms of congenital muscular dystrophies which include brain damage among their characteristics [42], [43]. Dystroglycan alterations have also been related to different cancers, as RMS, prostate, colon or breast cancer [42], [44], [45]. Dystroglycan requires a correct function of LARGE1 gene and interacts with MEK and ERK components of the MAP kinase cascade, and thus, with TRK-pat [46]. PARK2 has also recently demonstrated a role in this pathway and it is one of the most frequently altered tumor suppressor genes in cancer [47]. Its depletion leads to PTEN inactivation and the co-occurrence of PARK2 and PTEN losses promotes tumorigenesis in vivo [47]. All these facts highlight the potential role of these genes in GBM biopathology and particularly in EGFR-amplified tumors. In addition, PARK2 is located on 6q, near to MAP3K4, reinforcing the influence of MAP kinases in GBM biopathology. As we showed in a previous work, MAP kinase cascade is intensely activated through ERK1/2 in GBM that display EGFR amplification [48]. Conversely, QKI is another gene in that region which acts as a target of miR200. Recently another work demonstrated that miR200c is under expressed on EGFR-amplified tumors [49]. This fact would mean that in 2/3 of our samples QKI could not develop its role. In line with these considerations of chromosomal location more than concrete gene, it is to mention that CCND2 is a cyclin frequently amplified in gliomas and in about 3% of GBM. Although we did not studied it, near, in 12p13 we can find CDNK1B, which showed similar frequency of CNA in our series. Thus, both 6q and 12p segments seem to contain genes whose knowledge is necessary to better understand GBM aggressiveness. Interestingly, tumor mutational burden resulted more predictive than any individual alteration: high values of both losses and/or gains statistically correlated to shortened survival in GBM patients. This fact agrees with recent published work on gliomas IDH-mutant and other neoplasia as lung or breast cancer and supports the potential usefulness of immunotherapy for GBM patients [23], [24], [50], [51], [52].
Figure 5.

Relevant signaling pathways deregulated in glioblastoma: receptor tyrosine kinase/PI3K/PTEN/AKT/mTOR pathway is shown in blue, CDKN2A/CDK4/6/ retinoblastoma is shown in green and p53/MDM2/p14ARF signaling pathway is shown in red. Targets for LARGE1 and PARK2 novel alerations are indicated. Modified from Ohgaki et al. [15].
The genetic landscape of GBMs was widely varied in our series, in line with previous studies that demonstrated the coexistence of multiple clones in a manner that resemble admixtures of molecular distinct tumors rather than a uniform disease [41], [53]. Our results showed also common genetic alterations that are independent to EGFR status. This fact could explain the varied intrinsic biological strategies related to tumor progression that make such complicated to achieve therapeutic improvements; however, our data also reflected the presence of specific differential cell clones in H-amp GBMs or in N-amp GBMs. Interestingly, L-amp GBM genetics partially overlapped with that of H-amp and L-amp groups. These findings imply that the therapeutic actions on these three groups would not cause the same clinical results and remarks the importance of establishing genetic GBM subgroups based on EGFR amplification levels.
The deregulation of molecular signaling pathways has impact on cell proliferation (Figure 5) and it is associated with a bad prognosis in GBM [17], [54]. The proportion of patients in this work with alterations in the RTK-pat agrees with previous studies that indicate ranges between 50–88% [40], [55]. Interestingly, in the H-amp group we found a number of PTEN alterations that is two-fold compared with L-amp. It is worth mentioning that PTEN is currently subject of therapeutic interventions in GBM [38]. Related to P53-pat, TP53 and MDM2 showed similar alterations to previous descriptions although the pathway global affectation was lower than the previously published data [1], [17], [40], [55]. This fact may be explained because this pathway could be more EGFR-independent than the others; it also suggests that N-amp GBMs could genetically resemble secondary GB, in which p53 alterations are more frequent [1]. Regarding RB-pat, the frequency of changes is concordant with the published frequencies too [17], [40], [55]. Our results show that the genetic burden affecting this pathway increases progressively from N-amp to H-amp. Interestingly, all the cases with simultaneous alteration of CDKN2A/B and CDK6 belonged to H-amp group. Globally, our series showed that, p53-pat and RB-pat deregulation in GBM happens more frequently because of MDM2 and CDKN2A changes respectively, than because of TP53 and RB1 alterations, in line with previous approaches [1], [13], [14].
In GBM, near 80% of the cases show deregulation of more than one pathway simultaneously [56], similarly to our data. The simultaneous alteration of genes from the three pathways was higher in H-amp GBMs, and interestingly, it was statistically associated to a shortened survival, which suggests, in an indirect manner, a poorer outcome when EGFR is highly amplified although larger series need to confirm these findings.
Conclusions
The accumulation of genomic alterations in the studied GBMs remarks the high genomic instability of this tumor. It has been shown an interesting relationship between high CNAs in GBM and patients’ survival, more prominent in EGFR H-amp and L-amp tumors, which may indicate the convenience of classifying GBMs depending on EGFR status in order to better understand the behavior of this highly heterogeneous entity. The association of high CNAs with patients’ survival underlines the influence of the tumor mutational burden in the progression of GBM. The current study highlights the prognostic strength of the combination of MLPA/FISH, which are technologies completely implemented in most of the hospitals. For sure, further studies are necessary to follow improving the knowledge on the molecular mechanisms of GBM pathogenesis and progression in order to find better therapeutic approaches.
Authors' contributions
Concept and design: CL-G and MC-N. Acquisition of data: LM-H, JM, TS-M, Analysis and interpretation of data: LM-H, DM, CL-G, JM, P-R and TS-M. Writing and review of manuscript: CL-G, LM-H, JM and MC-N. Study supervision: JM, TS-M, CL-G and MC-N.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by grants PROMETEO 2011-11/83 and GV/2018/130 from the Conselleria d’Educació, Investigació, Cultura I Esport (Generalitat Valencina-Spain), PI14/01669, SAF2014-52875R and PCIN2017-117 from the Ministerio de Economía y Competividad-Spain (Instituto de Salud Carlos III), and GUTMOM from the Joint Programming Initiative HDHL.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.neo.2019.09.001.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Supplementary file 1.
References
- 1.Louis D.N., Ohgaki H., Wiestier O.D. Fourth edition. World Health Organization; Lyon: 2016. WHO classification of tumours of the central nervous system. [Google Scholar]
- 2.Parker J.J., Canoll P., Niswander L. Intratumoral heterogeneity of endogenous tumor cell invasive behavior in human glioblastoma. Sci Rep. 2018;8:18002. doi: 10.1038/s41598-018-36280-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lopez-Gines C., Gil-Benso R., Ferrer-Luna R. New pattern of EGFR amplification in glioblastoma and the relationship of gene copy number with gene expression profile. Mod Pathol. 2010;23:856–865. doi: 10.1038/modpathol.2010.62. [DOI] [PubMed] [Google Scholar]
- 4.Stichel D., Ebrahimi A., Reuss D. Distribution of EGFR amplification, combined chromosome 7 gain and chromosome 10 loss, and TERT promoter mutation in brain tumors and their potential for the reclassification of IDHwt astrocytoma to glioblastoma. Acta Neuropathol (Berl) 2018;136:793–803. doi: 10.1007/s00401-018-1905-0. [DOI] [PubMed] [Google Scholar]
- 5.Navarro L., Gil-Benso R., Megías J. Alteration of major vault protein in human glioblastoma and its relation with EGFR and PTEN status. Neuroscience. 2015;297:243–251. doi: 10.1016/j.neuroscience.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 6.An Z., Aksoy O., Zheng T. Epidermal growth factor receptor (EGFR) and EGFRvIII in glioblastoma (GBM): signaling pathways and targeted therapies. Oncogene. 2018;37:1561–1575. doi: 10.1038/s41388-017-0045-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nathanson D.A., Gini B., Mottahedeh J. Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science. 2014;343:72–76. doi: 10.1126/science.1241328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jue T.R., McDonald K.L. The challenges associated with molecular targeted therapies for glioblastoma. J Neurooncol. 2016;127:427–434. doi: 10.1007/s11060-016-2080-6. [DOI] [PubMed] [Google Scholar]
- 9.Miranda A., Blanco-Prieto M., Sousa J. Breaching barriers in glioblastoma. Part I: molecular pathways and novel treatment approaches. Int J Pharm. 2017;531:372–388. doi: 10.1016/j.ijpharm.2017.07.056. [DOI] [PubMed] [Google Scholar]
- 10.Pedersen M.W., Tkach V., Pedersen N. Expression of a naturally occurring constitutively active variant of the epidermal growth factor receptor in mouse fibroblasts increases motility. Int J Cancer. 2004;108:643–653. doi: 10.1002/ijc.11566. [DOI] [PubMed] [Google Scholar]
- 11.Feng H., Hu B., Vuori K. EGFRvIII stimulates glioma growth and invasion through PKA-dependent serine phosphorylation of Dock180. Oncogene. 2014;33:2504–2512. doi: 10.1038/onc.2013.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keller S., Schmidt M.H.H. EGFR and EGFRvIII promote angiogenesis and cell invasion in glioblastoma: combination therapies for an effective treatment. Int J Mol Sci. 2017;18:1295. doi: 10.3390/ijms18061295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hochberg F.H., Atai N.A., Gonda D. Glioma diagnostics and biomarkers: an ongoing challenge in the field of medicine and science. Expert Rev Mol Diagn. 2014;14:439–452. doi: 10.1586/14737159.2014.905202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Verhaak R.G.W., Hoadley K.A., Purdom E. An integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR and NF1. Cancer Cell. 2010;17:98. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ohgaki H., Kleihues P. Genetic alterations and signaling pathways in the evolution of gliomas. Cancer Sci. 2009;100:2235–2241. doi: 10.1111/j.1349-7006.2009.01308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huse JT (2014) Elucidating the oncogenic role of ATRX deficiency in glioma. Neuro-Oncol 16:iii45. doi:10.1093/neuonc/nou209.14.
- 17.Ohgaki H., Kleihues P. Genetic pathways to primary and secondary glioblastoma. Am J Pathol. 2007;170:1445–1453. doi: 10.2353/ajpath.2007.070011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boisselier B., Dugay F., Belaud-Rotureau M.-A. Whole genome duplication is an early event leading to aneuploidy in IDH-wild type glioblastoma. Oncotarget. 2018;9:36017–36028. doi: 10.18632/oncotarget.26330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mao X., Hamoudi R.A. Molecular and cytogenetic analysis of glioblastoma multiforme. Cancer Genet Cytogenet. 2000;122:87–92. doi: 10.1016/s0165-4608(00)00278-8. [DOI] [PubMed] [Google Scholar]
- 20.The TCGA research network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Indraccolo S., Lombardi G., Fassan M. Genetic, epigenetic, and immunologic profiling of MMR-deficient relapsed glioblastoma. Clin Cancer Res. 2019;25:1828–1837. doi: 10.1158/1078-0432.CCR-18-1892. [DOI] [PubMed] [Google Scholar]
- 22.Nesseler J.P., Schaue D., McBride W.H. Irradiation to improve the response to immunotherapeutic agents in glioblastomas. Adv Radiat Oncol. 2018;4:268–282. doi: 10.1016/j.adro.2018.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berland L., Heeke S., Humbert O. Current views on tumor mutational burden in patients with non-small cell lung cancer treated by immune checkpoint inhibitors. J Thorac Dis. 2019;11:S71–S80. doi: 10.21037/jtd.2018.11.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shirahata M., Ono T., Stichel D. Novel, improved grading system(s) for IDH-mutant astrocytic gliomas. Acta Neuropathol (Berl) 2018;136:153–166. doi: 10.1007/s00401-018-1849-4. [DOI] [PubMed] [Google Scholar]
- 25.Trabelsi S., Mama N., Ladib M. MGMT methylation assessment in glioblastoma: MS-MLPA versus human methylation 450K beadchip array and immunohistochemistry. Clin Transl Oncol. 2016;18:391–397. doi: 10.1007/s12094-015-1381-0. [DOI] [PubMed] [Google Scholar]
- 26.San-Miguel T., Navarro L., Megías J. Epigenetic changes underlie the aggressiveness of histologically benign meningiomas that recur. Hum Pathol. 2019;84:105–114. doi: 10.1016/j.humpath.2018.07.035. [DOI] [PubMed] [Google Scholar]
- 27.Shinojima N., Tada K., Shiraishi S. Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res. 2003;63:6962–6970. [PubMed] [Google Scholar]
- 28.Layfield L., Willmore C., Tripp S. Epidermal growth factor receptor gene amplification and protein expression in glioblastoma multiforme. Appl Immunohistochem Mol Morphol. 2006;14:91–96. doi: 10.1097/01.pai.0000159772.73775.2e. [DOI] [PubMed] [Google Scholar]
- 29.Gan H.K., Kaye A.H., Luwor R.B. The EGFRvIII variant in glioblastoma multiforme. J Clin Neurosci. 2009;16:748–754. doi: 10.1016/j.jocn.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 30.Cerami E., Gao J., Dogrusoz U. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brennan C.W., Verhaak R.G.W., McKenna A. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ohgaki H., Dessen P., Jourde B. Genetic pathways to glioblastoma: a population-based study. Cancer Res. 2004;64:6892–6899. doi: 10.1158/0008-5472.CAN-04-1337. [DOI] [PubMed] [Google Scholar]
- 33.Houillier C., Lejeune J., Benouaich-Amiel A. Prognostic impact of molecular markers in a series of 220 primary glioblastomas. Cancer. 2006;106:2218–2223. doi: 10.1002/cncr.21819. [DOI] [PubMed] [Google Scholar]
- 34.Frederick L., Wang X.-Y., Eley G., James C.D. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res. 2000;60:1383–1387. [PubMed] [Google Scholar]
- 35.Bleeker F.E., Molenaar R.J., Leenstra S. Recent advances in the molecular understanding of glioblastoma. J Neurooncol. 2012;108:11–27. doi: 10.1007/s11060-011-0793-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sauter G., Maeda T., Waldman F.M. Patterns of epidermal growth factor receptor amplification in malignant gliomas. Am J Pathol. 1996;148:1047–1053. [PMC free article] [PubMed] [Google Scholar]
- 37.Liu K.-J., Chen C.-T., Hu W.S. Expression of cytoplasmic-domain substituted epidermal growth factor receptor inhibits tumorigenicity of EGFR-overexpressed human glioblastoma multiforme. Int J Oncol. 2004;24:581–590. [PubMed] [Google Scholar]
- 38.Varela M., Ranuncolo S.M., Morand A. EGF-R and PDGF-R, but not bcl-2, overexpression predict overall survival in patients with low-grade astrocytomas. J Surg Oncol. 2004;86:34–40. doi: 10.1002/jso.20036. [DOI] [PubMed] [Google Scholar]
- 39.Pelloski C.E., Ballman K.V., Furth A.F. Epidermal growth factor receptor variant III status defines clinically distinct subtypes of glioblastoma. J Clin Oncol. 2007;25:2288–2294. doi: 10.1200/JCO.2006.08.0705. [DOI] [PubMed] [Google Scholar]
- 40.Rao S.K., Edwards J., Joshi A.D. A survey of glioblastoma genomic amplifications and deletions. J Neurooncol. 2010;96:169–179. doi: 10.1007/s11060-009-9959-4. [DOI] [PubMed] [Google Scholar]
- 41.Sottoriva A., Spiteri I., Piccirillo S.G.M. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci U S A. 2013;110:4009–4014. doi: 10.1073/pnas.1219747110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beltrán D., Anderson M.E., Bharathy N. Exogenous expression of the glycosyltransferase LARGE1 restores α-dystroglycan matriglycan and laminin binding in rhabdomyosarcoma. Skelet Muscle. 2019;9:11. doi: 10.1186/s13395-019-0195-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meilleur K.G., Zukosky K., Medne L. Clinical, pathological and mutational spectrum of dystroglycanopathy due to LARGE mutations. J Neuropathol Exp Neurol. 2014;73:425–441. doi: 10.1097/NEN.0000000000000065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sgambato A., Migaldi M., Montanari M. Dystroglycan expression is frequently reduced in human breast and colon cancers and is associated with tumor progression. Am J Pathol. 2003;162:849–860. doi: 10.1016/S0002-9440(10)63881-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de Bernabé D.B.-V., Inamori K.-I., Yoshida-Moriguchi T. Loss of alpha-dystroglycan laminin binding in epithelium-derived cancers is caused by silencing of LARGE. J Biol Chem. 2009;284:11279–11284. doi: 10.1074/jbc.C900007200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Spence H.J., Dhillon A.S., James M., Winder S.J. Dystroglycan, a scaffold for the ERK–MAP kinase cascade. EMBO Rep. 2004;5:484–489. doi: 10.1038/sj.embor.7400140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.PARK2 Depletion Connects Energy and Oxidative Stress to PI3K/Akt Activation via PTEN S-Nitrosylation. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5426642/. Accessed 22 May 2019. [DOI] [PMC free article] [PubMed]
- 48.Lopez-Gines C., Gil-Benso R., Benito R. The activation of ERK1/2 MAP kinases in glioblastoma pathobiology and its relationship with EGFRamplification. Neuropathology. 2008;28:507–515. doi: 10.1111/j.1440-1789.2008.00911.x. [DOI] [PubMed] [Google Scholar]
- 49.Kim EJ, Kim SS, Lee S, et al QKI, a miR‐200 target gene, suppresses epithelial‐to‐mesenchymal transition and tumor growth. Int J Cancer 145:1585–1595. doi:10.1002/ijc.32372. [DOI] [PubMed]
- 50.Zhang L., Feizi N., Chi C., Hu P. Association analysis of somatic copy number alteration burden with breast cancer survival. Front Genet. 2018;9 doi: 10.3389/fgene.2018.00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Richardson T.E., Sathe A.A., Kanchwala M. Genetic and Epigenetic Features of Rapidly Progressing IDH-Mutant Astrocytomas. J Neuropathol Exp Neurol. 2018;77:542–548. doi: 10.1093/jnen/nly026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Umehara T., Arita H., Yoshioka E. Distribution differences in prognostic copy number alteration profiles in IDH-wild-type glioblastoma cause survival discrepancies across cohorts. Acta Neuropathol Commun. 2019;7:99. doi: 10.1186/s40478-019-0749-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li C, Wang S, Yan J-L, et al Intratumoral heterogeneity of glioblastoma infiltration revealed by joint histogram analysis of diffusion tensor imaging. Neurosurgery. doi:10.1093/neuros/nyy388. [DOI] [PubMed]
- 54.Huttner A. Overview of primary brain tumors: pathologic classification, epidemiology, molecular biology, and prognostic markers. Hematol Oncol Clin North Am. 2012;26:715–732. doi: 10.1016/j.hoc.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 55.Vogelstein B., Kinzler K.W. Cancer genes and the pathways they control. Nat Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 56.Eder K., Kalman B. Molecular heterogeneity of glioblastoma and its clinical relevance. Pathol Oncol Res. 2014;20:777–787. doi: 10.1007/s12253-014-9833-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.