

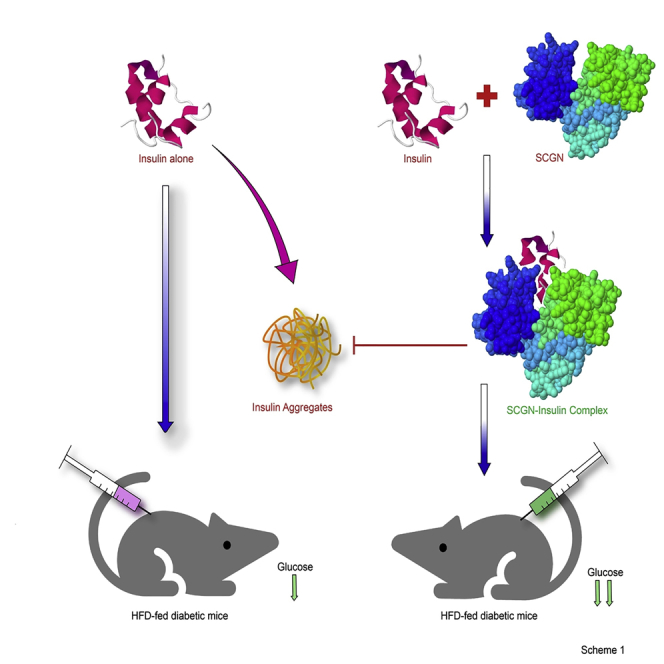
Summary
Secretagogin (SCGN) is a β-cell enriched, secretory/cytosolic Ca2+-binding protein with unknown secretory regulation and functions. Recent findings suggest that SCGN deficiency correlates with compromised insulin response and diabetes. However, the (patho)physiological SCGN-insulin nexus remains unexplored. We here report that SCGN is an insulin-interacting protein. The protein-protein interaction between SCGN and insulin regulates insulin stability and increases insulin potency in vitro and in vivo. Mutagenesis studies suggest an indispensable role for N-terminal domain of SCGN in modulating insulin stability and function. SCGN supplementation in diabetogenic-diet-fed mice preserves physiological insulin responsiveness while relieving obesity and cardiovascular risk. SCGN-insulin interaction mediated alleviation of hyperinsulinemia by increased insulin internalization, which translates to reduced body fat and hepatic lipid accumulation, emerges as a plausible mechanism for the preservation of insulin responsiveness. These findings establish SCGN as a functional insulin-binding protein (InsBP) with therapeutic potential against diabetes.
Subject Areas: Molecular Interaction, Endocrinology, Diabetology, Specialized Functions of Cells
Graphical Abstract
Highlights
-
•
SCGN is an insulin-interacting calcium sensor protein
-
•
SCGN binding protects insulin from aggregation
-
•
SCGN potentiates insulin action in vivo
-
•
SCGN administration into HFD-fed animals impedes insulin resistance
Molecular Interaction; Endocrinology; Diabetology; Specialized Functions of Cells
Introduction
Peptide-hormone-binding proteins (such as CRH-BPs and insulin-like growth factor-binding proteins [IGFBPs]) bind to the vulnerable peptide hormones, regulating their serum concentration, stability, and activity (Clemmons, 1993, Behan et al., 1995). Insulin is a delicate hormone with aggregation-prone β-chain and redox-sensitive inter/intrachain disulfide bonds (Baker et al., 1988, Hua et al., 1996, Hua et al., 2002). However, in the absence of a bona fide functional insulin-binding protein (InsBP), the understanding of insulin stabilization and activity modulation remains limited. Unlike insulin, IGF (which shares large structural similarities with insulin) signaling employs a dedicated family of IGFBPs) (Clemmons, 1993, Bach, 2015). Thus, as IGFs, insulin intuitively requires a stabilizer/chaperone in the extracellular environment. Several observations prompted us to hypothesize that secretagogin (SCGN) is an InsBP: (1) the existence of SCGN in circulation, (2) abundant expression and partial colocalization with insulin in pancreatic β-cells, and (3) identification of insulin as an interacting partner in our mass-spectroscopy-assisted pull-down of β-cell-specific SCGN-interacting proteins (which we report herein).
Impairment in insulin synthesis, secretion, stability, or sensitivity is responsible for the progression of type 2 diabetes (T2D). Various reports suggest the regulatory role of SCGN in insulin expression and secretion (Wagner et al., 2000, Yang et al., 2016, Kobayashi et al., 2016). Besides, type 2 diabetes patients show reduced SCGN expression (Malenczyk et al., 2017), whereas feeding a diabetogenic high-fat diet (HFD) to mice causes downregulation of SCGN (Hasegawa et al., 2013), implicating SCGN in the pathophysiology of T2D. The reduced SCGN levels in cerebrospinal fluid of insulin-resistant subjects (Westwood et al., 2017) reiterate the involvement of SCGN in insulin resistance. Consistently, the SCGN knockout mice develop glucose intolerance at an early life-stage, which deteriorates progressively to hyperglycemia (Malenczyk et al., 2017), suggesting a confounding role of SCGN in preserving euglycemia. These observations point toward a perplexing correlation between SCGN deficiency and diabetes. Because SCGN deficiency correlates with the incidence of diabetes (Malenczyk et al., 2017, Hasegawa et al., 2013, Westwood et al., 2017), we hypothesized that the exogenous SCGN supplementation would have an anti-diabetic effect in vivo.
In this study, using various biophysical and biochemical approaches, we report that SCGN is an insulin-binding protein protecting insulin from physical and chemical stressor-induced aggregation. SCGN also potentiates insulin signaling and hence augments the glucose-lowering activity of insulin in vivo. The N-terminal domain of SCGN seems to play an important role in binding and modulating insulin function. The downregulation of SCGN in diabetes led us to investigate if exogenous rSCGN supplementation could rescue from insulin resistance in HFD-fed animals. Our data demonstrate that chronic rSCGN administration preserves the insulin sensitivity in HFD-fed (DIO) animals by promoting insulin clearance, hence alleviating the hyperinsulinemia by the virtue of SCGN-insulin interaction. The insulin-sensitizing effects of rSCGN are detached from the side effects, such as obesity or cholesterol metabolism deregulation. Insulin potentiation and diabetes prevention are the consequences of insulin binding to SCGN. Our data establish SCGN as a functional insulin-binding protein and suggest the translational potential of recombinant SCGN as an anti-diabetic therapy.
Results
SCGN Readily Interacts with Insulin: The Synergy of Ca2+ and Insulin Binding
In search of novel SCGN interactions and function, we performed a mass-spectroscopy-based pull-down assay from MIN6 cell lysate. We identified several Ca2+ binding/chaperone proteins as SCGN interactors. These proteins—binding immunoglobulin protein/78 kDa glucose-regulated protein (BiP/GRP78), protein disulfide isomerase (PDI), 94 kDa glucose-regulated protein (GRP94)—are implicated in the insulin maturation process (Figure 1A). A compelling observation was that, in addition to the aforesaid chaperones, insulin was also captured as an SCGN-interactor. Consolidating previously reported occasional colocalization of SCGN with insulin (Wagner et al., 2000, Gartner et al., 2007), we observed that endogenous SCGN (red) and insulin (green) colocalize (yellow) to a substantial magnitude in insulin-expressing MIN6 cells, which is a pancreatic β-cell line (Figure 1B).
Figure 1.

SCGN Interacts with Insulin in vitro and ex vivo
(A) List of SCGN interacting partners identified in mass-spectrometry-assisted pull-down assay.
(B) Microscopy images depicting colocalization (yellow) of SCGN (red) and insulin (green) immunostained with Alexa Fluor 594 (red) and Alexa Fluor 488 (green) respectively in MIN6 cells. Scale bar, 20 μm.
(C) BioLayer interferometry (BLI) binding curve of SCGN-insulin interaction. The data were fit in 1:1 model [R2 for apo-SCGN-insulin experiment is 0.9547 (Chi0 value is 0.004) and for holo-SCGN-insulin binding is 0.9626 (Chi2 value being 0.0126)].
(D) Protein overlay assay to assess the SCGN-insulin interaction.
(E and F) CoIP from animal plasma and MIN6 cell lysate showing in vivo interaction of SCGN with insulin in both cellular and extra-cellular milieu.
(G) Analytical size-exclusion chromatography of SCGN, insulin, and SCGN-insulin mixture in 1:1 and 1:2 stoichiometric ratios in the presence of Ca2+ representing a distinct complex formation between SCGN and insulin.
SCGN being a Ca2+ sensor (Rogstam et al., 2007), we evaluated the effects of Ca2+ on insulin binding using bio-layer interferometry (BLI) analysis (Figure 1C). Recombinant mouse SCGN was prepared as described earlier (Sharma et al., 2015, Sharma et al., 2019a, Sharma et al., 2019b). We found that apo-SCGN (Ca2+-free SCGN) binds insulin with an affinity in the lower micromolar range [Kd = 1.8 μM (±5 × 10−8)], whereas in the presence of Ca2+ the overall binding affinity is marginally higher [Kd = 1.4 μM (±5 × 10−8)]. The rate of SCGN-insulin complex formation (kon) is of the same order irrespective of the presence of Ca2+ [kon(M−1s−1) for apo-SCGN= 7.2 × 102 for holo-SCGN= 4.6 × 102]. The rate of dissociation in the presence of Ca2+ is decreased slightly [koff(s−1) for apo-SCGN = 1 × 10−3, for holo-SCGN = 6 × 10−4], suggesting a strong and specific binding. In the biological context where cytosol has nM [Ca2+, whereas the extracellular milieu has mM [Ca2+, the apparent lack of Ca2+ dependency would make the interaction pervasive. Although more comprehensive titration studies for affinity measurement are required to draw insight, a decreased rate of dissociation in the Ca2+-bound form may suggest increased stability of the complex in the Ca2+-rich extracellular milieu.
To further validate SCGN-insulin interaction in the extracellular milieu, we performed a protein overlay assay and observed that SCGN binds insulin, irrespective of the presence of Ca2+ (Figure 1D). Even partially unfolded insulin (insulin-treated with 4 M urea prior to immobilization) binds to SCGN, suggesting a possibility of SCGN acting as a chaperone for insulin. To examine the physiological occurrence of SCGN-insulin interaction, we performed CoIP experiments from mouse plasma and MIN6 cell lysate/spent media. We found that SCGN, in the animal serum, remains bound to insulin independently of the glycemic status of animals because SCGN was co-precipitated with insulin from the fed/fasting animal serum (Figure 1E). Consistently, in MIN6 cells, we observed a co-precipitation of SCGN with insulin in both the cell lysate and spent media fraction, suggesting an intracellular and extracellular interaction (Figure 1F). Results suggest a universal existence of SCGN-insulin complex in intracellular and extracellular milieu (Figures 1E and 1F). To further affirm if insulin and SCGN form a complex in solution, we performed analytical gel filtration. The holo-SCGN-insulin mixture elutes prior to the holo-SCGN peak (Figure 1G) but not the apo-SCGN (Figure S1), suggesting the formation of a ternary complex between Ca2+, SCGN, and insulin. These observations cumulatively establish a pervasive interaction of SCGN with insulin within and beyond pancreatic β-cells.
To gain insight into the effect of Ca2+ on SCGN-insulin binding, we performed insulin titration to SCGN solution and examined the change in Trp fluorescence to quantify local changes and monitored tertiary CD fingerprints to analyze global effects. Insulin binding to holo-SCGN leads to a more prominent reduction in the fluorescence intensity than to apo-SCGN (Figures S2A and S2B), suggesting a positive modulation by Ca2+. Because the blue-shifted spectrum is a hallmark of Ca2+-bound SCGN (Rogstam et al., 2007), the apparent absence of insulin-induced shift in λmax in holo-SCGN suggests that Ca2+ is neither replaced nor removed allosterically by insulin. The mutual independence of Ca2+ and insulin binding to SCGN is further supported by near-UV circular dichroism spectral analysis. Insulin binding induces conformational changes similar to Ca2+. Insulin also induces similar changes in near-UV CD spectrum of Ca2+-SCGN producing similar conformational changes irrespective of the order of the ligand (Figures S2C and S2D). These results establish SCGN as an insulin-binding protein with scope for positive modulation by Ca2+.
SCGN Protects Insulin from Aggregation and Fibrillation and Alleviates Fibril Toxicity
The major regulation of insulin action is supposed to be achieved at the insulin secretion level. Structural stabilization and activity modulation of circulatory insulin, however, remain poorly understood. Because insulin has a soaring propensity to form cytotoxic fibrils and because SCGN was found to interact even with denatured insulin suggestive of chaperone property (Figure 1D), we speculated that SCGN would protect insulin from detrimental misfolding and aggregation. Consistently, we observed that SCGN impedes heat-induced fibrillation of insulin (Figure 2). The inhibition of the nucleation of fibrillation is largely independent of Ca2+ because the apo- as well as holo-SCGN inhibit insulin fibrillation (Figure 2A; vertical panels 1 and 2). Unexpectedly, SCGN could also dissolve preformed fibrils within an hour under the given experimental conditions (Figure 2A; last panel). Measurements of the rate of fibril formation using the fluorescent probe thioflavin T (ThT) show that apo-SCGN exerts ∼50% protection against heat-induced amyloidogenesis of insulin (Figure 2B), whereas holo-SCGN completely abolishes the fibrillation of insulin (Figure 2C).
Figure 2.

SCGN Protects Insulin from Aggregation and Fibrillation and Reduces Cytotoxicity ex-vivo
(A) Transmission electron microscope (TEM) images depicting the inhibition of insulin fibrillation by SCGN.
(B and C) Time scan spectra representing the change in the emission of ThT fluorescence of insulin fibrillation in the presence of SCGN (1:2 ratio) in its apo- or holo-form. Scale bar, 0.6 μm.
(D) MTT cell viability assay demonstrating the role of SCGN in alleviating cell cytotoxicity caused due to insulin fibrils in MIN6 cells.
Error bars represent mean ± SEM (fibrils vs (fibril + SCGN) *p value = 0.034).
The protection exerted by SCGN is also visible in the disulfide-bond reduction-induced aggregation of insulin. Apo-SCGN rescues insulin from aggregation in a dose-dependent manner (Figure S3), implying that apo-SCGN stabilizes insulin, likely by providing a niche to protect the exposed disulfide bonds involved in interchain disulfide bridge formation. These results demonstrate that SCGN prevents insulin misfolding and aggregation and shears existing fibrils. Because of their cytotoxicity, fibrils have been implicated in various neurodegenerative diseases and in diabetes (Schneider et al., 1980, Maloy et al., 1981, Höppener et al., 2000, Ross and Poirier, 2004, Muchowski and Wacker, 2005, Wang et al., 2010). We verified if SCGN alleviates cytotoxicity caused by insulin fibrils ex vivo. Consistently, exogenous SCGN reduces insulin fibril toxicity significantly in MIN6 cells (Figure 2D). BSA, which was included as a control, did not exert any protection. These results suggest that SCGN effectively inhibits the fibrillation of insulin, dissolves the preformed fibrils, and alleviates the fibril-associated cytotoxicity. The chaperone interactome (Figure 1A) and the observed anti-amyloid properties of SCGN suggest that SCGN is a molecular chaperone.
SCGN Improves Insulin Signaling
Having established the SCGN-insulin interaction, we next studied the effect of SCGN on physiological insulin signaling. We hypothesized that if the described results were a consequence of a biologically relevant and functional SCGN-insulin interaction, SCGN would increase insulin potency in vivo. To test the hypothesis, we performed an insulin tolerance test (ITT) in healthy mice (Figure 3A). Each group of animals received one of the following treatments: (1) recombinant SCGN (rSCGN), (2) insulin, (3) SCGN-insulin combination, or (4) insulin into the rSCGN-pretreated animals. rSCGN injections were inconsequential, whereas administration of SCGN-insulin combination was more effective in clearing blood glucose than insulin (Figure 3A). Owing to the apparent lack of glucose clearance by SCGN alone, the enhanced potency of SCGN-insulin combination is attributed to SCGN-insulin interaction. To validate the data, we analyzed the ability of insulin or insulin-SCGN combination to induce Akt phosphorylation in two relevant cell lines (HepG2 and C2C12 myotubes). We found statistically increased Akt phosphorylation in C2C12 myotubes treated with SCGN-insulin combination when compared with cells treated with insulin alone (Figure 3B). However, in the case of HepG2, the effect is insignificant, which may suggest a need for additional cofactors or a tissue-specific modulation of insulin action by SCGN. These results demonstrate that SCGN enhances insulin signaling consistent with in vivo data.
Figure 3.

SCGN Potentiates Insulin Signaling
(A) ITT of BALB/c mice injected with either insulin, SCGN, or combination of insulin and SCGN after 6-h fasting.
(B) Western blots and corresponding densitometry analysis showing the increased insulin-induced phosphorylation of Akt in the presence of SCGN in differentiated C2C12 and HepG2 cells.
(C) ITT in STZ animals after injection of SCGN or insulin or a combination.
(D) Serum SCGN concentration of C57BL/6 mice maintained on normal chow-diet (blue bars) or HFD (pink bars) as measured by ELISA at different time points in OGTT paradigm.
Error bars represent mean ± SEM. *= p<0.05, **= p<0.01.
Secretagogin-induced lowering of glucose in mice compelled us to explore if SCGN wields insulin mimic activity in vivo. To check this, we injected SCGN to β-cell-deficient streptozotocin (STZ)-treated mice. rSCGN injection does not elicit any glucose-lowering effect, thus ruling out the possibility of insulin-mimetic activity of SCGN (Figure 3C). Even in STZ animals, substantiating the physiological importance of SCGN-insulin interaction, the SCGN-insulin combination was found to be more potent than insulin. These findings established that SCGN could increase the potency of exogenous insulin. Our data may offer an explanation to the cause of compromised insulin response in the SCGN knockout upon glucose challenge (Malenczyk et al., 2017). These results suggest that SCGN is a physiological modulator of insulin signaling. Consistently, SCGN was always present in the serum of healthy animals (Figure 3D). In the oral glucose tolerance test (OGTT) paradigm, fasting SCGN concentration was found to be higher. An oral glucose gavage led to a reduction in SCGN concentration, which significantly drops after 120 min (Figure 3D). However, consistent with the reported downregulation of SCGN in diabetic subjects (Malenczyk et al., 2017), we noted a significant reduction in serum SCGN concentration of HFD-fed mice at all time-points in OGTT paradigm (Figure 3D). This may suggest that at hypoglycemic levels, SCGN concentration remains high. Once circulatory glucose levels increase, abundant SCGN binds insulin to help it function appropriately. As the insulin levels subside after 120 min of glucose administration, SCGN levels also drop correspondingly. This suggests that there is an intricate coherence in SCGN secretion, which is disturbed in pathophysiology.
SCGN N-terminal Domain Is Essential for Functional Modulation of Insulin
We then directed our efforts to map the individual domains responsible for insulin binding and the biological properties. We first modeled the SCGN-insulin complex using Patchdoc software (Schneidman-Duhovny et al., 2005). The initial analysis suggests that all the three lobes of SCGN participate in insulin binding, thus complicating the experimental analysis of the precise binding site (Figure 4A). Based on this information, two truncated versions of SCGN lacking either N-terminal (ΔNTD) or C-Terminal (ΔCTD) domain were designed (Figure 4B). The insulin binding examined by protein overlay assay suggested a preserved (but weaker) insulin binding to both the truncations (Figure 4C), which is anticipated from the large contact area in SCGN-insulin complex. Similarly, in vitro insulin potentiation was unaffected by deletions (Figure S4), suggesting that middle domain of SCGN may have a primary role in establishing insulin contact and modulating insulin functions. Nonetheless, in Trp fluorescence, we observed that addition of insulin brought more prominent changes in ΔCTD than ΔNTD, suggesting a stronger binding to ΔCTD (Figure S5). Similarly, when we monitored anti-fibrillar activity of truncated proteins for insulin by TEM, ΔNTD showed compromised protection of insulin from aggregation while ΔCTD is unaffected (Figures2A and 4D). This suggests that N-terminal domain of SCGN is important for a tight insulin binding and thereby for inhibition of fibrillation. Consistently, ThT fluorescence also demonstrated compromised protection of insulin by ΔNTD (both the apo- and holo-forms), whereas ΔCTD showed enhanced protection in the presence of Ca2+, which was comparable to full-length SCGN (Figures 4E and 4F). We performed these studies at three different SCGN concentrations to examine dose-response and found that 20 μM ΔCTD (as full-length SCGN) showed complete protection while ΔNTD was relatively ineffective (Figure S6). Although at high concentrations BSA also inhibits insulin fibrillation (Finn et al., 2012) (Figures 4E and S7), the effect of SCGN was much more prominent even at lower concentration (1:10 protein:insulin stoichiometry), suggesting a specificity of insulin chaperoning by SCGN. Moreover, TEM imaging suggested that the inhibition by BSA was partial as visible insulin fibrils were present in the presence of BSA (Figure S7). The ThT fluorescence or the fibrils visualized by TEM were not due to protein, as alone protein did not exhibit any ThT fluorescence or visible fibrils on TEM (Figures S6 and S8).
Figure 4.

SCGN N-terminal Domain Imparts Specificity and Functionality
(A) SCGN-insulin-complex model showing a large surface contact area between SCGN and insulin.
(B) Truncation strategy to generate N-terminal or C-terminal deleted SCGN.
(C) Protein-overlay assay suggesting a preservation of insulin binding to domain deleted mutants of SCGN.
(D) TEM images depicting a compromised chaperoning of insulin by ΔNTD but the upholding of the same in ΔCTD. Scale bar, 0.2 μm.
(E and F) ThT fluorescence (intermittent readings) representing a compromised protection by ΔNTD but a better protection by ΔCTD under given conditions. Please note that same insulin aggregation profile is used in both panels (and associated supplementary information) as the data are derived from the same experiment.
(G) ITT with truncated SCGN showing a loss of insulin potentiation by ΔNTD, whereas the ΔCTD is as effective as full-length SCGN, (H) blood glucose levels after administration of truncated SCGN showing no reduction in glucose level. Error bars represent mean ± SEM. *= p<0.05.
We then sought to check if in vivo effects of SCGN on insulin potentiation are preserved in truncated proteins. Intriguingly (and consistent with anti-fibrillation results), ΔCTD was as effective as full-length SCGN in potentiating insulin signaling when co-administered with insulin (Figure 4G). In contrary, the deletion of the N-terminal domain completely obliterates SCGN's ability to potentiate insulin (Figure 4G). These effects were insulin-dependent, as no reduction in blood glucose was seen in animals injected with only SCGN protein (Figure 4H). The maintenance of in vitro insulin modulatory activity of ΔNTD is likely due to the slack binding of insulin and simplicity of the system. Contrary to inclined cellular system, when ΔNTD is co-administered with insulin in vivo, the changing microenvironment and absorption process leads to dissociation of the weak ΔNTD-insulin complex, thus the interaction becomes inconsequential. These results demonstrate specificity of insulin binding to SCGN with an imperative role for the N-terminal domain of SCGN for efficacious insulin binding while C-terminal domain is dispensable.
Chronic SCGN Administration Preserves Insulin and Glucose Tolerance and Lowers Hyperglycemia in an Insulin-Dependent Manner
Insulin resistance is a physiological state where insulin, despite its (abundant) presence, cannot induce the clearance of circulating glucose. To ascertain if the exogenous rSCGN administration could rescue HFD-fed animals from systemic insulin under-responsiveness and to explore the therapeutic potential of these findings in diabetes, we systematically studied the effect of rSCGN administration in insulin-resistant animals. Because the diet-induced obese (DIO) animal model mimics the multifactorial human lifestyle-associated diabetes better than monogenic knockout animal models, we utilized DIO mice to examine the insulin-sensitizing activity of exogenous SCGN. We designed an ab initio SCGN-treatment protocol, wherein the treatment group received a subcutaneous rSCGN injection every other day from day 1 of exposure to the confounding factor (i.e., HFD), whereas control groups (HFD and normal-chow fed) received phosphate buffered saline (PBS) injections. After 12 weeks of exposure to HFD and concurrent treatment, the effect of rSCGN administration on insulin and glucose tolerance was assessed (Figures 5A and 5B upper and lower panels). Upon exogenous insulin injection, rSCGN-treated HFD-fed animals cleared blood glucose more efficiently than control HFD despite the adversity of higher initial blood glucose level (t = 0 in ITT) (Figure5A). However, the statistically significant glucose lowering was achieved only after 60 min of insulin injection. Therefore, SCGN-mediated increase in glucose clearance might not reflect a direct effect on insulin action, and additional work is required to understand the potential impact of SCGN on insulin sensitivity. To check if the same is true for endogenous insulin, we performed an oral glucose tolerance test (OGTT). Although the initial blood glucose levels of both treated and untreated HFD-fed animals were high, rSCGN-treated animals efficiently cleared excess glucose from the blood and achieved a normoglycemia comparable to the normal-diet-fed animals (Figure 5B).
Figure 5.

Chronic SCGN Administration Lessens Insulin Resistance in DIO Animals
(A) ITT of chow-/HFD-fed animals after 12 weeks of exposure to HFD.
(B) OGTT of the animals after 12 weeks. ITT and OGTT were repeated after an additional two months of SCGN treatment (or vehicle injection).
(C–E) (C) ITT; (D) OGTT; (E) fasting blood glucose levels of DIO mice (0-min glucose values in last OGTT).
Error bars represent mean ± SEM. *= p<0.05, **= p<0.01, ***= p<0.001.
Encouraged by these results, we continued HFD and concomitant rSCGN administration for an additional two months to assess the efficacy of treatment on the severity of insulin resistance and hyperglycemia. At the conclusion of the experimental period, we found that rSCGN-treated HFD-fed animals had complete preservation of insulin sensitivity, comparable to the chow-fed control animals (Figure 5C). Glucose was better cleared in rSCGN-treated HFD-fed animals when compared with the untreated HFD group (Figure 5D). These results are better reflected in fasting blood glucose (derived from t = 0 of OGTT) levels, where the rSCGN-treated animals have lower blood glucose levels reflecting better glycemic control and sustenance of normoglycemia (Figure5E). These results suggest that SCGN supplementation improves insulin response and point toward the anti-diabetic potential of SCGN.
SCGN-mediated Alleviation of Hyperinsulinemia Prevents Insulin Resistance
Pronounced preservation of insulin sensitivity in rSCGN-treated HFD-fed mice intrigued us to explore the mechanism of persistent insulin responsiveness. We found that the control chow group had expected normal insulin concentration. However, untreated HFD-fed animals had much higher circulating insulin level at different time points in the OGTT paradigm, whereas rSCGN-injected animals showed reduced insulin levels (Figure 6A). Because sustained hyperinsulinemia is an established determinant of insulin resistance (Czech, 2017), and because SCGN is already shown to positively regulate the expression and secretion of insulin (Wagner et al., 2000, Malenczyk et al., 2017), we were perplexed with the reduction of insulin levels on SCGN administration. We thus examined if SCGN binding could induce the clearance of circulatory insulin post-secretion. For this purpose, we designed an in vitro insulin clearance assay wherein fluorescently labeled insulin was incubated with physiologically relevant hepatic cell line, HepG2. Consistent with our post-secretion clearance hypothesis, SCGN increases the FITC-insulin clearance to a visually appreciable extent seen as cell-internalized fluorescence (Figure 6B). In addition, in the presence of SCGN, there were more visible puncta or granular structures, pointing toward improved vesicular endocytosis (of the insulin-IR complex) by SCGN. We validated microscopy data with FACS-assisted quantification of FITC-insulin internalization (Figure 6C). The intensity of the internalized FITC-insulin was higher in the presence of SCGN than in the absence thereof. The increased insulin internalization was also seen in ELISA mediated insulin clearance using hepatic cell line but not in non-hepatic N2A neuroblastoma cells (Figures S9A and S9B). Nonetheless, it is the anatomic design that liver is the first organ exposed to secreted insulin. Thus, even though other organs/cells may be capable of insulin degradation, they do not encounter high insulin concentration in healthy conditions. These results suggest that SCGN facilitates the hepatic clearance of circulatory insulin, thus retarding the progression of insulin resistance.
Figure 6.

SCGN Administration Moderates Hyperinsulinemia and Body/Liver Fat Deposition
(A) Serum insulin concentration measured by ELISA in the same cohort of animals at different time points in OGTT paradigm.
(B) Microscopic images showing increased internalization of FITC-insulin conjugate into HepG2 cells in the presence of unlabeled SCGN. FITC-labeled insulin (10 μM) and 10-μM FITC-insulin mixed with unlabeled SCGN were incubated for an hour followed by acid buffer wash, fixation by formaldehyde, and subsequently mounted on slides, Scale bar (right bottom), 20 μm.
(C) FACS-mediated quantification of FITC-insulin conjugate internalization in the presence and absence of SCGN.
(D) Change in body weight in the same cohort of animals over a period of 20 weeks.
(E) Total body fat measured by echoMRI.
(F) Lean mass (differences between the groups are insignificant).
(G) Insulin sensitivity index (ISI0, 120).
(H) Oil Red O staining of liver cryosections. Scale bar, 100 μm.
Error bars represent mean ± SEM. *= p<0.05, **= p<0.01.
Hyperinsulinemia underwrites the weight gain (Pories and Dohm, 2012, Corkey, 2012, Vernochet et al., 2012, Pedersen et al., 2015, Templeman et al., 2015, D'souza et al., 2016), and resulting obesity is a major risk factor for lifestyle-associated diabetes and insulin resistance (Samuel and Shulman, 2012). Thus, we monitored weight gain in animals by HFD. rSCGN-treated animals had an insignificant reduction in weight gain than untreated HFD-fed animals (Figure 6D). Nonetheless, the rSCGN-treated animals had significantly lower body fat than untreated HFD-fed mice while the lean mass was comparable (Figures 6E and 6F). These results suggest that SCGN-mediated insulin clearance alleviates hyperinsulinemia in HFD-fed animals, which transcribes into the reduced body fat. To test if SCGN induced preservation of metabolic features specific to the diabetogenic diet-fed state, we performed a short control study on another cohort of mice fed either a normal chow or HFD (Figure S10). The SCGN administration to normal chow-fed animals was largely inconsequential (Figure S10). After six weeks of SCGN treatment, the ITT, OGTT, and weight gain were same in treated vs untreated normal chow-fed animals (Figures S10A–S10C), whereas the HFD-fed mice treated with SCGN showed a better insulin tolerance than untreated HFD mice (Figure S10D). This suggests that SCGN administration does not cause a negative effect in healthy animals but imparts beneficial effects under diabetogenic conditions.
To calculate the extent of insulin sensitivity in control HFD group and to find the extent of protection exerted by SCGN administration, we calculated the insulin sensitivity index, ISI(0, 120) (Gutt et al., 2000). The normal chow-fed animals had the best insulin sensitivity; rSCGN-treated mice had significantly higher ISI(0, 120), suggesting a better insulin sensitivity than in untreated HFD-fed animals (Figure 6G). However, further work needs to be done to understand whether SCGN potentiates/preserves/improves insulin action/sensitivity directly by modulating insulin signaling in probably a tissue-specific manner (as demonstrated in Figure 3) or indirectly through its effects on parameters such as body weight and fat mass composition (Figure 6).
In addition to reduced body fat, Oil red O staining demonstrated reduced hepatic triglycerides and lipid levels in rSCGN-treated HFD animals when compared with that of the HFD control group (Figure 6H). SCGN-treated HFD-fed animals exhibited significantly reduced Oil red O staining, demonstrating reduced hepatic triglyceride/lipid accumulation (Figure S9C). Hepatic steatosis symptoms were alleviated in SCGN-treated animals, with smaller lipid droplets and reduced necrotic areas (Figure 6H). Growing results suggest that hepatic lipid accumulation profoundly contributes to insulin resistance (Perry et al., 2014). Thus, SCGN administration induces the lowering of hepatic lipid deposition, which also contributes to the sustenance of insulin sensitivity.
Systemic Protection in DIO Mice after SCGN Administration
Altered body fat composition leads to adjusted lipid metabolism, which precipitates the risk of cardiovascular diseases (Van Gaal et al., 2006). We tested the effect of rSCGN treatment on serum lipid profile and other related factors (Figures 7A–7E). rSCGN administration marginally reduced total serum triglyceride level, whereas cholesterol levels were significantly reduced when compared with untreated HFD-fed animals (Figures 7A and 7B). The reduction in total cholesterol is likely due to a reduction in low-density lipoprotein (LDL) concentration because high-density lipoprotein level remains comparable to untreated animals (Figures 7C and 7D), suggesting that rSCGN treatment mitigates the obesity-associated risk of cardiovascular diseases. Because SCGN is a redox-responsive Ca2+ sensor (Rogstam et al., 2007, Khandelwal et al., 2017) and altered serum Ca2+ concentration is a marker for early stage insulin resistance (Thalassinos et al., 1993, Becerra-Tomás et al., 2014, Rooney et al., 2016, Holmes, 2016), we also measured the serum Ca2+ concentration. This is also important to validate that rSCGN administration does not quench the plasma Ca2+ that may affect the systemic physiology negatively. The untreated HFD-fed mice had morbidly higher serum Ca2+ levels, whereas rSCGN-treated animals had normalized levels comparable to normal chow-fed group, reconfirming preserved insulin sensitivity in rSCGN-treated HFD-fed animals (Figure 7E).
Figure 7.

SCGN Administration Elicits Systemic Protection
Serum and tissues were collected at the end of study.
Serum analysis of (A) total triglyceride; (B) cholesterol; (C) HDL cholesterol; and (D) LDL cholesterol; (E) Ca2+; estimation was performed as as per kit guidelines.
(F) H&E staining of pancreas, liver, and kidney sections was performed with formalin fixed tissue following standard protocol. Scale bar, 200 μm.
Error bars represent mean ± SEM. *= p<0.05, **= p<0.01.
We next compared the histology of selected tissues of the animals. Because SCGN is highly expressed in pancreatic β-cells, we examined pancreatic histology to rule out negative effects of exogenous rSCGN (e.g., by immune activation, that will be reflected in islet loss) on islet integrity. rSCGN-treated animals had increased islet density and islet area than in corresponding controls (Figure 7F). This observation extends recent findings that SCGN promotes islet integrity and β-cell mass (Malenczyk et al., 2017). The liver of untreated animals had prominent hepatic necrosis along with large fat bodies (as also visualized in Oil Red O-stained liver sections; Figure S9C), whereas treated animals were largely protected from necrosis and fat deposition. Because chronic metabolic stress leads to renal dysfunction, and the rSCGN administration was a relatively long-term study, we confirmed that excretory system is not stressed due to rSCGN administration. Consistently, due to long-term HFD-induced metabolic stress, kidneys of untreated HFD-fed animals were severely degenerated, with large necrotic areas devoid of cells. Glomerular density was also reduced in untreated HFD-fed animals. In rSCGN-treated animals, these features were less prominent, likely due to a reduced glycemic and metabolic load. These observations suggest that rSCGN administration not only improves glucose and lipid metabolism but also helps in maintaining the metabolic organs healthy.
SCGN Administration Causes Pancreatic β-cell Regeneration
Based on our observations that rSCGN administration increases β-cell proliferation in HFD-fed animals, we wondered if it would exert a similar effect in the β-cell-deficient STZ animals. After four weeks of rSCGN injections (10 mg/kg, on alternate days), STZ mice had increased islet frequency and islet area in pancreatic sections (Figure 8A) when compared with control mice. Although it is not clear if exogenous SCGN mimics endogenous molecular function in maintaining β-cell mass, or if it embraces a new pathway, our results suggest a prospective application of SCGN in β-cell regeneration. Although it is interesting to notice therapeutically relevant islet regenerative activity of SCGN, it will be useful to discern an active minimal peptide with enhanced efficacy and specificity at lower doses.
Figure 8.

SCGN-Induced Pancreatic β-cell Proliferation and Gene Expression in STZ Mice
(A) H&E staining of formalin-fixed pancreas sections of STZ-treated BALB/c mice upon SCGN treatment. Scale bar, 200 μm.
(B–G) Transcriptional reprogramming of pancreatic genes in STZ-untreated and SCGN-treated animals as measured by qRT-PCR.
Error bars represent mean ± SEM. *= p<0.05.
To validate if the enhanced islets in SCGN-treated STZ animals harbor functional β-cells, we characterized the molecular expression of β-cell-specific genes by qRT-PCR analysis (Figures 8B–8G). A substantial increase was seen in the expression of insulin-signaling-related genes in the pancreas, suggesting an increase in total β-cell mass. Both isoforms of insulin (INS1 and INS2) were upregulated in the pancreas of rSCGN-treated animals, suggesting a true increase in β-cell content (Figures 8B and 8C). In addition, insulin receptor (INSR) and prohormone convertase (PC1/3 and PC2) were significantly upregulated (Figures 8D–8F). Moreover, SCGN was also upregulated in treated animals (Figure 8G). Increased β-cell mass and enhanced insulin expression suggest that rSCGN treatment can increase the insulin-producing capacity of insulin-deficient animals, whereas increased IR expression provides a way to achieve increased insulin sensitivity. Prohormone convertase (PC1/3) has a critical role in insulin maturation, and its upregulation fulfills the obvious requirement of insulin maturation. Interestingly, PC2, which is primarily involved in glucagon maturation, was also upregulated in SCGN-treated animals. Although we did not explore the possibility, this suggests that SCGN's functional role may extend to α-cell biology and glucagon secretion from the pancreas. To establish the organ specificity of SCGN action on transcriptional modulation, we quantified the expression of authentic hepatic genes. Except insulin receptor (which was significantly upregulated), other tested genes' expression was unaltered (Figure S11), suggesting a tissue-specific modulation of specific genes by circulatory SCGN.
Discussion
Insulin, secreted from pancreatic β-cells, acts on several organs, such as liver, muscle, and fat tissue to maintain normoglycemia. To perform these functions, insulin travels long distances, encountering harsh, multicomponent circulatory environment, which occasionally causes aggregation of insulin leading to diabetes (Baker et al., 1988, Hua et al., 1996, Hua et al., 2002). Because insulin is a vulnerable hormone, it forms Zn2+-mediated tetramer/hexamer to attain stability in pancreatic β-cells (Blundell et al., 1972, Blundell et al., 1972a, Emdin et al., 1980). However, once the complex reaches the circulation, Zn2+ dissociates from insulin, which returns to the monomeric, biologically active form. Undesirably, the monomeric insulin is more prone to aggregation, fibrillation, and other anomalies (Gold and Grodsky, 1984, Formby et al., 1984, Li, 2014, Robertson et al., 2011). These anomalies impose a fundamental question on insulin stabilization and add to pharmaceutical difficulties in insulin manufacture and therapy (Brange, 1987, Brange et al., 1997, Vajo et al., 2001). Insulin-like growth factors (IGFs) are insulin-related hormones with similar structural features but different functions. To regulate the function and half-life of IGFs, there is a dedicated family of proteins called IGFBPs (Clemmons, 1993, Bach, 2015). However, for insulin, despite being an important hormone of primary importance, a circulatory InsBP has not been identified. The aggregation-prone disposition of insulin intuitively necessitates a structural stabilizer while in circulation. Although earlier studies suggested the existence of cytosolic insulin-binding proteins (CIBP) (Harada et al., 1995, Harada et al., 1996, Lokhov et al., 2004), to the best of our knowledge, circulatory InsBP remain an abstract idea. Harada et al. (Harada et al., 1996), in addition to several other proteins, described a 32 kDa CIBP, which was found to interact with insulin in the serum or in insulin-treated cells but not in serum-deprived cells. Coincidentally, SCGN is a 32 kDa protein and has been shown to exhibit serum/insulin responsive expression (Maj et al., 2012), suggesting that the reported 32 kDa CIBP could be SCGN. Moreover, SCGN seems to be synchronously secreted with insulin upon cAMP stimulation (Gartner et al., 2007). Likewise, a measurable fraction of SCGN dwells in circulation (Gartner et al., 2001, Tan et al., 2012), but the biological functions and the insulin-binding propensity of SCGN remain unexplored. Based on these interesting possibilities and our pull-down results, we argued that, owing to its localization and secretory properties, SCGN ought to be a candidate protein acting as InsBP. Here, we report that SCGN is an InsBP.
Protein fibrillation (including that of insulin) is a prominent anomaly associated with diabetes and neurodegenerative diseases (Schneider et al., 1980, Maloy et al., 1981, Höppener et al., 2000, Ross and Poirier, 2004, Muchowski and Wacker, 2005, Wang et al., 2010). Insulin has been used as a model to study protein fibrillation in vitro. There are several macroscopic and microscopic mechanisms described for the formation of insulin fibrils (Hua and Weiss, 2004, Ivanova et al., 2009, Arosio et al., 2016). A prevailing model suggests that a well-folded insulin molecule, under amyloidogenic conditions, adopts a conformation of protein-folding intermediate, which is prone to aggregation and self-association (Hua and Weiss, 2004). For chaperone-mediated protection of an amyloidogenic protein, it is suggested that although the microscopic mechanisms may differ from one chaperone/client to other, at macroscopic level chaperone proteins help a client protein to conserve its physiological, less aggregation-prone conformation (Arosio et al., 2016). In the case of SCGN-insulin, most plausible mechanism of protection is apparently the preservation of a soluble monomeric form where the aggregation-prone β-chain of insulin is protected by SCGN binding. This hypothesis is supported by the extended lag phase of insulin fibrillation as seen in ThT fluorescence and is also backed by the prevention of DTT-induced insulin aggregation, which directly indicates protection of β-chain of insulin. SCGN has earlier been reported to exert a possible protective effect against the Alzheimer disease (Attems et al., 2008, Maj et al., 2010) Thus, SCGN-mediated inhibition of fibrillation could provide a plausible explanation of the previously observed conspicuous protection against the disease. Moreover, it also suggests that SCGN is a common factor involved in diabetes and Alzheimer disease.
Besides exerting protection from fibrillation, SCGN also modulates insulin signaling. An SCGN-induced increase in insulin response in healthy animals prompted us to explore if the same would be true in diabetic animals as well. We found that prolonged SCGN treatment is required for sustained insulin sensitization and glucose lowering. The insulin dependence of SCGN action was further validated in STZ mice, a model for insulin deficiency. In both the animal models, we found that subcutaneous injection is more effective for sustained action when compared with intraperitoneal injection. We also studied the impact of rSCGN on hepatic fat deposition and degree of insulin resistance to discover the mechanism of sustained insulin sensitivity. Persistent hyperinsulinemia is directly implicated in obesity and insulin resistance (Pories and Dohm, 2012, Corkey, 2012, Vernochet et al., 2012, Pedersen et al., 2015, Templeman et al., 2015, D'souza et al., 2016). We observed that rSCGN treatment prevents hyperinsulinemia. Considering the previous findings that hyperinsulinemia is a crucial factor for the weight gain in DIO animals (Templeman et al., 2015, D'souza et al., 2016), reduced weight gain in SCGN-treated DIO animals is attributed to the reduction in hyperinsulinemia. Emerging evidence suggests that hepatic lipid accumulation is a prominent factor in insulin resistance and diabetes (Perry et al., 2014). We found that SCGN-administered animals had appreciably lower hepatic lipid accumulation, which conceivably is a consequence (and in a feedback loop, a contributor) of improved insulin sensitivity. The SCGN-mediated hepatic insulin clearance likely contributes to a reduction in circulating insulin levels and reduced weight gain in SCGN-treated DIO animals. Because diabetes is associated with increased risk of cardiovascular disease (Grundy et al., 1999, Resnick and Howard, 2002), the reduction of LDL by SCGN suggests that the SCGN administration could also reduce the risk of cardiac problems.
While examining the histological integrity of pancreas, liver, and kidney, we observed a positive systemic influence on key metabolic organs, which is ultimately reflected in better glycemic control in rSCGN-treated animals. In the backdrop of a recent study reporting the role of intracellular SCGN in maintaining β-cell mass (Malenczyk et al., 2017), our results showing that exogenous SCGN administration exerts a positive effect on islet regeneration are encouraging. These observations also corroborate with a recent study on the essentiality of SCGN for β-cell maturation and maintenance (Malenczyk et al., 2018). Moreover, owing to its insulin-sensitizing effect, SCGN emerges as a potential candidate against T2D. Future studies are needed to explore and optimize the therapeutic application of SCGN-induced β-cell regeneration. Being a relatively large protein, identifying individual domain(s) of SCGN responsible for specific function, such as β-cell regeneration or insulin sensitization, would be a fruitful exercise.
A recent report suggested that type 2 diabetes is a state of SCGN deficiency (Malenczyk et al., 2017). Similarly, SCGN knockout animals showed significant glucose intolerance (Malenczyk et al., 2017), pointing toward compromised insulin signaling. Our observations of synchronous secretion of SCGN and insulin compelled us to hypothesize that SCGN is a positive regulator of insulin action and this is why the SCGN knockout animals have compromised insulin function. Further, we demonstrate that exogenous SCGN supplementation can be used as preventive medicine in pre-diabetic animals. SCGN administration preserves insulin sensitivity and lowers systemic diabetes-associated complications in model systems. Thus, SCGN can also be used in combination with insulin (in type 1 diabetes) to increase insulin efficacy while promoting pancreatic regeneration over the course of treatment. Available anti-diabetic drugs have associated side effects such as weight gain, liver problems, and cardiovascular risk (Lehrke and Lazar, 2005, Stein et al., 2013, Inzucchi et al., 2014). In our study, we did not observe any prominent side effects even after five months of the rSCGN administration. Rather, rSCGN exerted beneficial effects, such as reduced serum Ca2+, low-density lipoprotein, and hepatic steatosis.
In summary, our data establish SCGN as a functional insulin-binding protein. We demonstrate that insulin-binding ability bestows SCGN with competence to regulate the concentration, stability, and function of insulin beyond pancreas. As proposed recently (Sharma et al., 2019a, Sharma et al., 2019b), SCGN administration potentially preserves insulin sensitivity and lowers systemic diabetes-associated complications in model systems (Figure 9). Thus, SCGN appears to be an effective preventive therapy against insulin resistance. Nonetheless, further work is required to derive mechanistic insights into the mode of action of SCGN and its potential use.
Figure 9.

Working Model
SCGN positively regulates insulin expression and secretion (Wagner et al., 2000, Yang et al., 2016, Kobayashi et al., 2016). SCGN insulin interaction potentiates insulin stability and action and hence reducing the required effective concentration. Because increased circulatory insulin is also a founding factor for insulin resistance and diabetes, it suggests that insulin expression and functional regulation by SCGN is a mechanism for SCGN-mediated prevention of diabetes. The hypothesis is supported by a recent observation that the absence of SCGN leads to a compromised insulin action (Malenczyk et al., 2017).
Limitation of the Study
The physiological effect of exogenously SCGN administration on insulin action is examined in the HFD-fed wild-type mouse model. To discern the effect of exogenous SCGN from the endogenous SCGN, similar experiments carried out in SCGN knockout mouse model would provide further insight. It would not only validate the role of SCGN in diabetic pathophysiology but would also help in delineating the site of action of exogenous SCGN and provide mechanistic insight into pathways regulated by SCGN. Given the anticipated importance of SCGN in insulin potentiation, identifying the specific residues in the insulin-binding pocket of SCGN using NMR experiments would provide mechanistic insight and aid in developing the minimal SCGN peptide for potential therapeutic uses. Moreover, further studies with genetic models of diabetes are needed to derive mechanistic insight into the mode of SCGN action.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
Authors thank Miss Swathi Chadalawada for technical help. We are grateful to Drs. T Ramakrishna Murti and B Raman for help in biolayer interferometry. Mr Prashant and Mrs Jyothi have been helpful in a few animal experiments and Mr Harikrishna helped in TEM imaging. They thank Syed Sayeed Abdul for excellent laboratory assistance and Dr. Sushil Chandani for discussion and critical reading of the manuscript. In addition, discussions with Dr. G R Chandak were useful in experimental design. J. C Bose National Fellowship (SERB), CSIR, and DST are acknowledged for grants to Y.S.. H.C.A. and R.K. are the recipients of ICMR and CSIR-GATE research fellowships, respectively.
Author Contributions
Inception: A.K.S.; Experimental design: A.K.S., R.K., J.M.K., Y.S.; Protein purification: A.K.S., R.K., A.H.C.; Data acquisition: A.K.S., R.K., S.R.; Histology slide preparation: A.R.; Fibrillation experiments and TEM sample preparation: A.K.S., R.K., A.H.C.; Data analysis: A.K.S., R.K., J.M.K., Y.S.; Resources: Y.S.; Writing—original draft: A.K.S., R.K., Y.S.; Writing—review and editing: A.K.S., R.K., Y.S.; Supervision, project administration and funding acquisition: Y.S.
Declaration of Interests
The authors declare that they have no conflict of interest with the content of this article. Current affiliation of AKS is Laboratory of Translational Nutrition Biology, DHEST, ETH Zurich (email id: anand.sharma@hest.ethz.ch).
Published: November 22, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.10.066.
Supplemental Information
References
- Arosio P., Michaels M.C.T., Linse S., Månsson C., Emanuelsson C., Presto J., Johansson J., Vendruscolo M., Dobson C.M., Knowles T.P.J. Kinetic analysis reveals the diversity of microscopic mechanisms through which molecular chaperones suppress amyloid formation. Nat. Commun. 2016;7:10948. doi: 10.1038/ncomms10948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attems J., Preusser M., Grosinger-Quass M., Wagner L., Lintner F., Jellinger K. Calcium-binding protein secretagogin-expressing neurons in the human hippocampus are largely resistant to neurodegeneration in Alzheimer's disease. Neuropathol. Appl. Neurobiol. 2008;34:23–32. doi: 10.1111/j.1365-2990.2007.00854.x. [DOI] [PubMed] [Google Scholar]
- Baker E.N., Blundell T.L., Cutfield J.F., Cutfield S.M., Dodson E.J., Dodson G.G., Hodgkin D.M., Hubbard R.E., Isaacs N.W., Reynolds C.D. The structure of 2Zn pig insulin crystals at 1.5 A resolution. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1988;319:369–456. doi: 10.1098/rstb.1988.0058. [DOI] [PubMed] [Google Scholar]
- Bach L.A. Insulin-like growth factor binding proteins–an update. Pediatr. Endocrinol. Rev. 2015;13:521–530. [PubMed] [Google Scholar]
- Becerra-Tomás N., Estruch R., Bulló M., Casas R., Díaz-López A., Basora J., Fitó M., Serra-Majem L., Salas-Salvadó J. Increased serum calcium levels and risk of type 2 diabetes in individuals at high cardiovascular risk. Diabetes Care. 2014;37:3084–3091. doi: 10.2337/dc14-0898. [DOI] [PubMed] [Google Scholar]
- Behan D.P., De Souza E.B., Lowry P.J., Potter E., Sawchenko P., Vale W.W. Corticotropin releasing factor (CRF) binding protein: a novel regulator of CRF and related peptides. Front. Neuroendocrinol. 1995;16:362–382. doi: 10.1006/frne.1995.1013. [DOI] [PubMed] [Google Scholar]
- Blundell T.L., Cutfield J.F., Cutfield S.M., Dodson E.J., Dodson G.G., Hodgkin D.C., Mercola D.A. Three-dimensional atomic structure of insulin and its relationship to activity. Diabetes. 1972;21:492–505. doi: 10.2337/diab.21.2.s492. [DOI] [PubMed] [Google Scholar]
- Blundell T.L., Cutfield J.F., Dodson E.J., Dodson G.G., Hodgkin D.C., Mercola D.A. The crystal structure of rhombohedral 2 zinc insulin. Cold Spring Harb. Symp. Quant. Biol. 1972;36:233–241. doi: 10.1101/sqb.1972.036.01.031. [DOI] [PubMed] [Google Scholar]
- Brange J. Springer-Verlag; 1987. Galenics of Insulin: The Physico-Chemical and Pharmaceutical Aspects of Insulin and Insulin Preparations. [Google Scholar]
- Brange J., Andersen L., Laursen E.D., Meyn G., Rasmussen E. Toward understanding insulin fibrillation. J. Pharm. Sci. 1997;86:517–525. doi: 10.1021/js960297s. [DOI] [PubMed] [Google Scholar]
- Clemmons D.R. IGF binding proteins and their functions. Mol. Reprod. Dev. 1993;35:368–375. doi: 10.1002/mrd.1080350409. [DOI] [PubMed] [Google Scholar]
- Corkey B.E. Banting lecture 2011: hyperinsulinemia: cause or consequence? Diabetes. 2012;61:4–13. doi: 10.2337/db11-1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czech M.P. Insulin action and resistance in obesity and type 2 diabetes. Nat. Med. 2017;23:804–814. doi: 10.1038/nm.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'souza A.M., Johnson J.D., Clee S.M., Kieffer T.J. Suppressing hyperinsulinemia prevents obesity but causes rapid onset of diabetes in leptin-deficient Lepob/ob mice. Mol. Metab. 2016;5:1103–1112. doi: 10.1016/j.molmet.2016.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emdin S.O., Dodson G.G., Cutfield J.M., Cutfield S.M. Role of zinc in insulin biosynthesis. Some possible zinc-insulin interactions in the pancreatic β-cell. Diabetologia. 1980;19:174–182. doi: 10.1007/BF00275265. [DOI] [PubMed] [Google Scholar]
- Finn T.E., Nunez A.C., Sunde M., Easterbrook-Smith S.B. Serum albumin prevents protein aggregation and amyloid formation and retains chaperone-like activity in the presence of physiological ligands. J. Biol. Chem. 2012;287:21530–21540. doi: 10.1074/jbc.M112.372961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formby B., Schmid-Formby F., Grodsky G.M. Relationship between insulin release and 65zinc efflux from rat pancreatic islets maintained in tissue culture. Diabetes. 1984;33:229–234. doi: 10.2337/diab.33.3.229. [DOI] [PubMed] [Google Scholar]
- Gartner W., Lang W., Leutmetzer F., Domanovits H., Waldhäusl W., Wagner L. Cerebral expression and serum detectability of secretagogin, a recently cloned EF-hand Ca2+-binding protein. Cereb. Cortex. 2001;11:1161–1169. doi: 10.1093/cercor/11.12.1161. [DOI] [PubMed] [Google Scholar]
- Gartner W., Vila G., Daneva T., Nabokikh A., Koc-Saral F., Ilhan A., Majdic O., Luger A., Wagner L. New functional aspects of the neuroendocrine marker secretagogin based on the characterization of its rat homolog. Am. J. Physiol. Endocrinol. Metab. 2007;293:347–354. doi: 10.1152/ajpendo.00055.2007. [DOI] [PubMed] [Google Scholar]
- Gold G., Grodsky G.M. Kinetic aspects of compartmental storage and secretion of insulin and zinc. Experientia. 1984;40:1105–1114. doi: 10.1007/BF01971458. [DOI] [PubMed] [Google Scholar]
- Grundy S.M., Benjamin I.J., Burke G.L., Eckel A.C.R.H., Howard B.V., Mitch W., Smith S.C., Sowers J.R. Diabetes and cardiovascular disease. Circulation. 1999;100:1134–1146. doi: 10.1161/01.cir.100.10.1134. [DOI] [PubMed] [Google Scholar]
- Gutt M., Davis C.L., Spitzer S.B., Llabre M.M., Kumar M., Czarnecki E.M., Schneiderman N., Skyler J.S., Marks J.B. Validation of the insulin sensitivity index (ISI(0,120)): comparison with other measures. Diabetes Res. Clin. Pract. 2000;47:177–184. doi: 10.1016/s0168-8227(99)00116-3. [DOI] [PubMed] [Google Scholar]
- Harada S., Smith R.M., Smith J.A., Shah N., Jarett L. Demonstration of specific insulin binding to cytosolic proteins in H35 hepatoma cells, rat liver and skeletal muscle. Biochem. J. 1995;306:21–28. doi: 10.1042/bj3060021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada S., Smith R.M., Hu D.Q., Jarett L. Dexamethasone inhibits insulin binding to insulin-degrading enzyme and cytosolic insulin-binding protein p82. Biochem. Biophys. Res. Commun. 1996;218:154–158. doi: 10.1006/bbrc.1996.0027. [DOI] [PubMed] [Google Scholar]
- Hasegawa K., Wakino S., Kimoto M., Minakuchi H., Fujimura K., Hosoya K., Komatsu M., Kaneko Y., Kanda T., Tokuyama H. The hydrolase DDAH2 enhances pancreatic insulin secretion by transcriptional regulation of secretagogin through a Sirt1-dependent mechanism in mice. FASEB J. 2013;27:2301–2315. doi: 10.1096/fj.12-226092. [DOI] [PubMed] [Google Scholar]
- Holmes D. Adipose tissue: angiogenic factor regulates beiging. Nat. Rev. Endocrinol. 2016;12:626. doi: 10.1038/nrendo.2016.144. [DOI] [PubMed] [Google Scholar]
- Höppener J.W.M., Ahrén B., Lips C.J.M. Islet amyloid and type 2 diabetes mellitus. N. Engl. J. Med. 2000;343:411–419. doi: 10.1056/NEJM200008103430607. [DOI] [PubMed] [Google Scholar]
- Hua Q.X., Hu S.Q., Frank B.H., Jia W., Chu Y.C., Wang S.H., Burke G.T., Katsoyannis P.G., Weiss M.A. Mapping the functional surface of insulin by design: structure and function of a novel A-chain analogue. J. Mol. Biol. 1996;264:390–403. doi: 10.1006/jmbi.1996.0648. [DOI] [PubMed] [Google Scholar]
- Hua Q.X., Chu Y.C., Jia W., Phillips N.F.B., Wang R.Y., Katsoyannis P.G., Weiss M.A. Mechanism of insulin chain combination. Asymmetric roles of A-chain alpha-helices in disulfide pairing. J. Biol. Chem. 2002;277:43443–43453. doi: 10.1074/jbc.M206107200. [DOI] [PubMed] [Google Scholar]
- Hua Q.X., Weiss M.A. Mechanism of insulin fibrillation: the structure of insulin under amyloidogenic conditions resembles a protein-folding intermediate. J. Biol. Chem. 2004;279:21449–21460. doi: 10.1074/jbc.M314141200. [DOI] [PubMed] [Google Scholar]
- Inzucchi S.E., Lipska K.J., Mayo H., Bailey C.J., McGuire D.K. Metformin in patients with type 2 diabetes and kidney disease: a systematic review. JAMA. 2014;312:2668–2675. doi: 10.1001/jama.2014.15298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova M.I., Sievers S.A., Sawaya M.R., Wall J.S., Eisenberg D. Molecular basis for insulin fibril assembly. Proc. Natl. Acad. Sci. USA. 2009;106:18990–18995. doi: 10.1073/pnas.0910080106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandelwal R., Sharma A.K., Chadalawada S., Sharma Y. Secretagogin is a redox-responsive Ca2+ sensor. Biochemistry. 2017;56:411–420. doi: 10.1021/acs.biochem.6b00761. [DOI] [PubMed] [Google Scholar]
- Kobayashi M., Yamato E., Tanabe K., Tashiro F., Miyazaki S., Miyazaki J. Functional analysis of novel candidate regulators of insulin secretion in the MIN6 mouse pancreatic β cell line. PLoS One. 2016;11:e0151927. doi: 10.1371/journal.pone.0151927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehrke M., Lazar M.A. The many faces of PPARγ. Cell. 2005;123:993–999. doi: 10.1016/j.cell.2005.11.026. [DOI] [PubMed] [Google Scholar]
- Li Y.V. Zinc and insulin in pancreatic beta-cells. Endocrine. 2014;45:178. doi: 10.1007/s12020-013-0032-x. [DOI] [PubMed] [Google Scholar]
- Lokhov P.G., Moshkovskii S.A., Ipatova O.M., Prozorovskii V.N. Cytosolic insulin-binding proteins of mouse liver cells. Protein Pept. Lett. 2004;11:29–33. doi: 10.2174/0929866043478356. [DOI] [PubMed] [Google Scholar]
- Maj M., Gartner W., Ilhan A., Neziri D., Attems J., Wagner L. Expression of TAU in insulin-secreting cells and its interaction with the calcium-binding protein secretagogin. J. Endocrinol. 2010;205:25–36. doi: 10.1677/JOE-09-0341. [DOI] [PubMed] [Google Scholar]
- Maj M., Milenkovic I., Bauer J., Berggård T., Veit M., Ilhan-Mutlu A., Wagner L., Tretter V. Novel insights into the distribution and functional aspects of the calcium binding protein Secretagogin from studies on rat brain and primary neuronal cell culture. Front. Mol. Neurosci. 2012;5:84. doi: 10.3389/fnmol.2012.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenczyk K., Girach F., Szodorai E., Storm P., Segerstolpe Å., Tortoriello G., Schnell R., Mulder J., Romanov R.A., Borók E. A TRPV1-to-secretagogin regulatory axis controls pancreatic β-cell survival by modulating protein turnover. EMBO J. 2017;36:2107–2125. doi: 10.15252/embj.201695347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenczyk K., Szodorai E., Schnell R., Lubec G., Szabó G., Hökfelt T., Harkany T. Secretagogin protects Pdx1 from proteasomal degradation to control a transcriptional program required for ß cell specification. Mol. Metab. 2018;14:108–120. doi: 10.1016/j.molmet.2018.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloy A.L., Longnecker D.S., Greenberg E.R. The relation of islet amyloid to the clinical type of diabetes. Hum. Pathol. 1981;12:917–922. doi: 10.1016/s0046-8177(81)80197-9. [DOI] [PubMed] [Google Scholar]
- Muchowski P.J., Wacker J.L. Modulation of neurodegeneration by molecular chaperones. Nat. Rev. Neurosci. 2005;6:11–22. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- Pedersen D.J., Guilherme A., Danai L.V., Heyda L., Matevossian A., Cohen J., Nicoloro S.M., Straubhaar J., Noh H.L., Jung D. A major role of insulin in promoting obesity-associated adipose tissue inflammation. Mol. Metab. 2015;4:507–518. doi: 10.1016/j.molmet.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry R.J., Samuel V.T., Petersen K.F., Shulman G.I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature. 2014;510:84–91. doi: 10.1038/nature13478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pories W.J., Dohm G.L. Diabetes: have we got it all wrong? Hyperinsulinism as the culprit: surgery provides the evidence. Diabetes Care. 2012;35:2438–2442. doi: 10.2337/dc12-0684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnick H.E., Howard B.V. Diabetes and cardiovascular disease. Annu. Rev. Med. 2002;53:245–267. doi: 10.1146/annurev.med.53.082901.103904. [DOI] [PubMed] [Google Scholar]
- Robertson R.P., Zhou H., Slucca M. A role for zinc in pancreatic islet beta-cell cross-talk with the alpha-cell during hypoglycaemia. Diabetes Obes. Metab. 2011;13:106–111. doi: 10.1111/j.1463-1326.2011.01448.x. [DOI] [PubMed] [Google Scholar]
- Rogstam A., Linse S., Lindqvist A., James P., Wagner L., Berggård T. Binding of calcium ions and SNAP-25 to the hexa EF-hand protein secretagogin. Biochem. J. 2007;401:353–363. doi: 10.1042/BJ20060918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney M.R., Pankow J.S., Sibley S.D., Selvin E., Reis J.P., Michos E.D., Lutsey P.L. Serum calcium and incident type 2 diabetes: the Atherosclerosis risk in communities (ARIC) study. Am. J. Clin. Nutr. 2016;104:1023–1029. doi: 10.3945/ajcn.115.130021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross C.A., Poirier M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004;10:S10–S17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- Samuel V.T., Shulman G.I. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148:852–871. doi: 10.1016/j.cell.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider H.M., Störkel F.S., Will W. The influence of insulin on local amyloidosis of the islets of Langerhans and insulinoma. Pathol. Res. Pract. 1980;170:180–191. doi: 10.1016/S0344-0338(80)80165-8. [DOI] [PubMed] [Google Scholar]
- Schneidman-Duhovny D., Inbar Y., Nussinov R., Wolfson H.J. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 2005;33:W363–W367. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A.K., Khandelwal R., Sharma Y. Veiled potential of secretagogin in diabetes: correlation or coincidence? Trends Endocrinol. Metab. 2019;30:234–243. doi: 10.1016/j.tem.2019.01.007. [DOI] [PubMed] [Google Scholar]
- Sharma A.K., Khandelwal R., Sharma Y. Secretagogin purification and quality control strategies for biophysical and cell biological studies. Methods Mol. Biol. 2019;1929:551–566. doi: 10.1007/978-1-4939-9030-6_34. [DOI] [PubMed] [Google Scholar]
- Sharma A.K., Khandelwal R., Sharma Y., Rajnikanth V. Secretagogin, a hexa EF-hand CaBP: high level, one-step bacterial purification, and properties. Protein Expr. Purif. 2015;109:113–119. doi: 10.1016/j.pep.2015.02.011. [DOI] [PubMed] [Google Scholar]
- Stein S.A., Lamos E.M., Davis S.N. A review of the efficacy and safety of oral antidiabetic drugs. Expert Opin. Drug Saf. 2013;12:153–175. doi: 10.1517/14740338.2013.752813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan W.S., Lee J.J., Satish R.L., Ang E.T. Detectability of secretagogin in human erythrocytes. Neurosci. Lett. 2012;526:59–62. doi: 10.1016/j.neulet.2012.08.006. [DOI] [PubMed] [Google Scholar]
- Templeman N.M., Clee S.M., Johnson J.D. Suppression of hyperinsulinaemia in growing female mice provides long-term protection against obesity. Diabetologia. 2015;58:2392–2402. doi: 10.1007/s00125-015-3676-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thalassinos N.C., Hadjiyanni P., Tzanela M., Alevizaki C., Philokiprou D. Calcium metabolism in diabetes mellitus: effect of improved blood glucose control. Diabet. Med. 1993;10:341–344. doi: 10.1111/j.1464-5491.1993.tb00076.x. [DOI] [PubMed] [Google Scholar]
- Vernochet C., Mourier A., Bezy O., Macotela Y., Boucher J., Rardin M.J., An D., Lee K.Y., Ilkayeva O.R., Zingaretti C.M. Adipose-specific deletion of TFAM increases mitochondrial oxidation and protects mice against obesity and insulin resistance. Cell Metab. 2012;16:765–776. doi: 10.1016/j.cmet.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gaal L.F., Mertens I.L., De Block C.E. Mechanisms linking obesity with cardiovascular disease. Nature. 2006;444:875–880. doi: 10.1038/nature05487. [DOI] [PubMed] [Google Scholar]
- Vajo Z., Fawcett J., Duckworth W.C. Recombinant DNA technology in the treatment of diabetes: insulin analogs. Endocr. Rev. 2001;22:706–717. doi: 10.1210/edrv.22.5.0442. [DOI] [PubMed] [Google Scholar]
- Wagner L., Oliyarnyk O., Gartner W., Nowotny P., Groeger M., Kaserer K., Waldhausl W., Pasternack M.S. Cloning and expression of secretagogin, a novel neuroendocrine- and pancreatic islet of Langerhans-specific Ca2+-binding protein. J. Biol. Chem. 2000;75:24740–24751. doi: 10.1074/jbc.M001974200. [DOI] [PubMed] [Google Scholar]
- Wang S.S., Liu K.N., Han T.C. Amyloid fibrillation and cytotoxicity of insulin are inhibited by the amphiphilic surfactants. Biochim. Biophys. Acta. 2010;1802:519–530. doi: 10.1016/j.bbadis.2010.02.008. [DOI] [PubMed] [Google Scholar]
- Westwood S., Liu B., Baird A.L., Anand S., Nevado-Holgado A.J., Newby D., Pikkarainen M., Hallikainen M., Kuusisto J., Streffer J.R. The influence of insulin resistance on cerebrospinal fluid and plasma biomarkers of Alzheimer's pathology. Alzheimers Res. Ther. 2017;9:31. doi: 10.1186/s13195-017-0258-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S.Y., Lee J.J., Lee J.H., Lee K., Oh S.H., Lim Y.M., Lee M.S., Lee K.J. Secretagogin affects insulin secretion in pancreatic β-cells by regulating actin dynamics and focal adhesion. Biochem. J. 2016;473:1791–1803. doi: 10.1042/BCJ20160137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.