

Abstract
There is a continued need to develop new selective human monoamine oxidase (hMAO) inhibitors that could be beneficial for the treatment of neurological diseases. However, hMAOs are closely related with high sequence identity and structural similarity, which hinders the development of selective MAO inhibitors. “Three-Dimensional Biologically Relevant Spectrum (BRS-3D)” method developed by our group has demonstrated its effectiveness in subtype selectivity studies of receptor and enzyme ligands. Here, we report a series of novel C7-substituted coumarins, either synthesized or commercially purchased, which were identified as selective hMAO inhibitors. Most of the compounds demonstrated strong activities with IC50 values (half-inhibitory concentration) ranging from sub-micromolar to nanomolar. Compounds, FR1 and SP1, were identified as the most selective hMAO-A inhibitors, with IC50 values of 1.5 nM (selectivity index (SI) < −2.82) and 19 nM (SI < −2.42), respectively. FR4 and FR5 showed the most potent hMAO-B inhibitory activity, with IC50 of 18 nM and 15 nM (SI > 2.74 and SI > 2.82). Docking calculations and molecular dynamic simulations were performed to elucidate the selectivity preference and SAR profiles.
Keywords: monoamine oxidase (MAO) inhibitors, BRS-3D, subtype selectivity, molecular dynamic simulations, in silico pharmacokinetic predictions
1. Introduction
The dynamic stability of amines is maintained by a reuptake mechanism and also the oxidation process by monoamine oxidases (MAOs) [1]. MAO is a flavin adenine dinucleotide (FAD) containing an enzyme bound in the mitochondrial outer membrane by a transmembrane helix [2]. It is responsible for regulation of biogenic and xenobiotic amines, which includes serotonin, tryptamine, dopamine, norepinephrine, epinephrine, β-phenylethylamine, tyramine and octopamine [3]. Two MAO subtypes were identified in mammals, namely MAO-A and MAO-B. They demonstrated different tissue distribution, substrate preference and inhibitor specificity [1,4]. In addition, different hMAO subtypes were associated with different neurological diseases, such as depression, Alzheimer’s disease, Parkinson’s disease and Huntington’s disease. Decreases in the expression level of hMAO-A with age were observed, which was proved to be the causes of aggressive behavior [5]. In contrast, a four-fold increase in the hMAO-B expression levels with aging was thought to be an important causative factor in the etiology of neurodegenerative diseases [6]. Therefore, hMAO-A and hMAO-B were essential targets for elucidating the mechanism of monoaminergic pathways and for the treatment of psychiatric disorders and neurodegenerative diseases [7].
Nevertheless, high sequence identity (72.6%) and structural similarity (superimposed root-mean-square deviation, RMSD = 0.94) between two hMAO subtypes hampered the discovery of selective MAO inhibitors (MAOIs) [4]. Efforts have been devoted for the development of subtype-selective MAO inhibitors over the last years [8,9,10]. For example, coumarin derivatives, which have many advantages such as the feasible synthesis and favorable bioavailability, were mainly designed as potential MAO inhibitors [9,11,12,13,14]. Various substitution patterns on the coumarin skeleton have been studied [14,15]. According to the orientation of pyrone ring of coumarin scaffold in the binding site, most coumarins can be categorized into two groups characterized by substitution positions: C3-substitution and C7-substitution. Of them, C3-substitution with an acyl function (e.g., carboxamide or carbohydrazide) or an aryl group, was the most promising [16]. For instance, 3-phenyl substituted coumarin derivatives displayed strong MAO-B inhibition [17,18,19] and the replacement of 3-phenyl with other heterocyclic motifs, such as thiophenyl or indolyl, also exhibited strong activity against MAO-B [9,20,21,22,23]. However, as shown in Figure 1, the development of hMAO-B selective inhibitors has attracted more attention than hMAO-A. There are very few highly potent hMAO-A inhibitors (pIC50 (−logIC50) > 7). As deposited in ChEMBL v24, 18 coumarins were reported to be selective to hMAO-B (pIC50 > 7 and SI > 2 (selectivity index = pIC50(MAO-B) − pIC50(MAO-A)) and only three coumarins were selective to hMAO-A with pIC50 > 7 and SI < 2 [24,25,26,27,28].
Figure 1.

Inhibition results of C3 and C7-subsituted coumarin derivatives against hMAO-A and hMAO-B (data collected from ChEMBL v24).
Discovery or design of subtype selective inhibitors is a challenging area due to the high sequential identity and small structural differences between the pockets of a conserved protein family (GPCRs, kinases, etc.). In such a case that protein subtypes have small structural differences, the dynamic profiles of the ligands’ conformation ensemble were believed to play an essential role in subtype selectivity. Our group recently developed a shape-similarity based protocol called “Three-Dimensional Biologically Relevant Spectrum (BRS-3D)” [29,30]. The method was validated in subtype selectivity studies of receptor and enzyme ligands [31,32,33]. A spectrum of C7-substituted coumarin analogues was discovered according to this protocol. The chemical space around these analogues were worthy to be further explored owing to their interesting structure-activity relationship (SAR) profiles. To this end, novel C7-substituted coumarin analogues were synthesized or commercially purchased and their inhibitory potencies were experimentally tested. The results revealed two C7-substituted coumarin compounds, FR1 and SP1, which gave the most potent hMAO-A inhibitory activities, with IC50 values of 1.5 nM and 19 nM, respectively (SI < −2.82 and SI < −2.42). FR4 and FR5 showed a selective hMAO-B inhibitory affinity, with IC50 value of 18 nM and 15 nM (SI > 2.74 and SI > 2.82). In addition, docking calculations and molecular dynamics (MD) simulations were performed to a better understand this selectivity and SAR profile. In silico pharmacokinetics predictions and drug-like properties of these molecules were also evaluated.
2. Results and Discussion
2.1. Qualitative SAR of Previously Identified Hits
In our previous study, BRS-3D method was introduced as a 3D shape similarity profile [29,30,32,33]. In BRS-3D, 300 diverse biologically relevant ligands were used as templates and their co-crystallized conformation was treated as the active conformation. Hence, BRS-3D was capable of reflecting the distribution of the objective compound in the known bioactive chemical space. For example, as shown in Figure 2, the different distribution of selective hMAO inhibitors in chemical space can be observed on the basis of three BRS-3D features (BRS12: 1cbq_RE9, BRS43: 3i8u_18B; BRS89: 1vj5_CIU). These characteristics make BRS-3D an important method for the study of subtype selectivity of receptor and enzyme ligands. Recently, we applied a BRS-3D based virtual screening protocol in the identification of selective hMAO inhibitors [31]. The experimental results demonstrated 70 compounds with MAO inhibition higher than 70% at a concentration of 10 μM, and 25 of them were potential hits with IC50 values less than 1 μM. Among the identified hits, several C7-substituted coumarin derivatives were worthy to be further explored owing to their interesting SAR profile. As illustrated in Figure 3, comparing with entries M30, M31 and M33, R5 group at C7-position was fixed, and installation of extended alkyl side chains at C4-position induced more robust MAO-A inhibition (M30, R2 = ethyl, 56% vs. M31, R2 = propyl, 92%) than the methyl homologue M33 (R2 = methyl, 41%). Comparing with entries M33 and M29, additional introduced methyl group at C8 position gave an improved MAO-A inhibition (M29, 75% vs. M33, 41%). Comparing with entries M33 and M34, the introduction of a Cl substitution at C6 position (M34) gave a nearly doubled decrease for MAO-A inhibition but a five-fold increase for MAO-B inhibition, suggesting C6-Cl substitution might be crucial to the MAO-B selectivity and binding affinity. It was also clear that the high MAO-B potency and selectivity of M32 was largely attributable to the C6-Cl group and the additional methyl substitution. Comparing with entries M33 and M43, modification of C7-substituion was clearly beneficial to both MAO-A and MAO-B inhibition. It is noteworthy that the activity of M31 and M32 against two hMAO subtypes was reversed with small changes in 3-, 4- and 6-positions.
Figure 2.

The distribution of selective MAO inhibitors on the basis of three BRS-3D features (BRS12: 1cbq_RE9, BRS43: 3i8u_18B; BRS89: 1vj5_CIU).
Figure 3.

Identified coumarin analogues through BRS-3D with hMAO inhibitory activities.
In addition, we can infer that hMAO-B binding site may prefer the small sized substituents at C4-position of the coumarin nucleus, and simultaneous substitutions at C3 and C4 position was beneficial to hMAO-B inhibition. From the SAR analysis, compound M43 with a 2-methoxy-5-phenyl-1,3,4-thiadiazole motif at C7-position had the most potent affinity towards both MAO-A and MAO-B subtypes, which was thereby taken as a lead to optimize its activity and selectivity by fragment replacement at the 3-, 4- and 8-positions.
2.2. MM/PBSA Binding Energy and Decomposition Analysis
Three compounds, M31, M32 and M43, were selected for docking simulations, with the co-crystallized ligand in 2V61 (C18) as reference. Based on the optimal docking models, eight systems were then investigated through MD simulations to elucidate the selectivity mechanism, four for each subtype. RMSD and RMSF (root mean square fluctuation) analysis was carried out to evaluate the structural stability. The stability of hydrogen bonds interactions between hMAO and C7 substituted coumarins, which are likely to contribute to the selectivity of MAOIs was also investigated. More details can be found in Supporting Information (Figures S1–S5, Table S1).
To further analyze the subtype selectivity mechanism, MM/PBSA method was employed to calculate the binding energy between hMAO subtypes and the discovered C7-substituted coumarins. Additionally, compound C18 bound in 2V61, was used as a reference. The predicted binding free energy and the inhibitory activity of each compound was summarized in Table 1. The calculated binding affinities correlated well with the experimental results. For hMAO-A selective system, the ΔG values of MAO-A-M31 complex and MAO-B-M31 complex were −127.034 kJ/mol and −113.631 kJ/mol with respect to their pIC50 values of 6.3 and 4.2, respectively. For hMAO-B selective system, the opposite results were obtained. The binding affinities of MAO-A-M32 complex and MAO-B-M32 complex were −112.539 kJ/mol and −122.606 kJ/mol corresponding to the pIC50 values of 4.1 and 6.2, respectively. The MM/PBSA binding energy contained four terms: the van der Waal contributions (ΔGvdW), the electrostatic contributions (ΔGele), the polar solvation contribution (ΔGpol) and the apolar solvation energy (ΔGapol). Then, the key terms responsible for binding were investigated separately. As listed in Table 2, taking M31 and M32 as an example, van der Waal contributions were the driving force for the binding in both selective systems and non-selective systems since the result of ΔGele and ΔGpol was positive. This demonstrated that in hMAO subtypes the unspecific binding activity was mainly attributed to hydrophobic interactions while selectivity to a specific subtype was driven by the above-mentioned H-bond interactions. In addition, the electrostatic terms to the solvation free energy ΔGpol were unfavorable for all systems. Compared to the polar solvation energy, the apolar solvation contributions were suggested to have a positive effect upon binding.
Table 1.
Experimental activities and MM/PBSA binding free energies.
| MAO-A | MAO-B | |||||
|---|---|---|---|---|---|---|
| Cmpd | Inh% | pIC50 | binding free energy | Inh% | pIC50 | binding free energy |
| M29 | 75 | 5.6 | −121.03 ± 0.92 | 12 | 4 | −109.72 ± 1.07 |
| M30 | 56 | ND | −116.90 ± 1.09 | 22 | ND | −102.79 ± 1.27 |
| M31 | 92 | 6.3 | −127.03 ± 1.28 | 16 | 4.2 | −113.63 ± 1.26 |
| M32 | 12 | 4.1 | −112.54 ± 1.12 | 90 | 6.2 | −122.61 ± 1.12 |
| M33 | 41 | ND | −115.25 ± 1.01 | 8 | ND | −105.59 ± 1.08 |
| M34 | 19 | ND | −107.67 ± 0.99 | 40 | ND | −111.18 ± 0.95 |
| M43 (7a) | 102 | 6.8 | −129.34 ± 1.10 | 96 | 6.4 | −135.59 ± 1.11 |
| C18 | - | 4.8 | −96.05 ± 1.31 | - | 7 | −144.57 ± 1.29 |
*ND: the IC50 values were not determined due to the low inhibitory activity. pIC50 = −log(IC50).
Table 2.
Analysis of calculated binding free energies of four systems.
| Terms | MAO-A vs. M31 | MAO-B vs. M31 | MAO-A vs. M32 | MAO-B vs. M32 |
|---|---|---|---|---|
| ΔGvdW | −211.794 ± 0.990 | −198.535 ± 1.147 | −214.799 ± 0.969 | −198.570 ± 1.187 |
| ΔGele | −38.523 ± 0.817 | −61.460 ± 0.704 | −31.376 ± 0.547 | −1.362 ± 0.727 |
| ΔGpol | 140.734 ± 0.539 | 164.645 ± 1.247 | 151.326 ± 0.787 | 144.800 ± 0.895 |
| ΔGnonpol | −17.431 ± 0.071 | −18.241 ± 0.073 | −17.715 ± 0.066 | −17.418 ± 0.065 |
| ΔG | −127.034 ± 1.281 | −113.631 ± 1.260 | −12.539 ± 1.116 | −122.598 ± 1.144 |
We then performed a residue-specific binding energy decomposition to identify key residues contributing to the subtype selectivity. The detailed inspection of per-residue energy contribution of the active site amino acids was shown in Table 3 and Table S2. Gln215 from α-helix region was calculated to be a key residue for hMAO-A selectivity. The energy contribution of Gln215 was −7.53 ± 0.24 kJ/mol, which is obviously larger than the contribution (−1.74 ± 0.22 kJ/mol) of the corresponding Gln206 residue in hMAO-B. Therefore, the decreased interaction with Gln206 reduced the activity to hMAO-B. Asn181 also played a critical role in hMAO-A selectivity since it was replaced by Cys172 in hMAO-B. Indeed, the corresponding energy contribution of Asn181 was −4.1478 ± 0.2633 kJ/mol for compound M31, which was larger than that of Cys172 in hMAO-B (0.2993 ± 0.124 kJ/mol). For M32, the Cl substitution at C6 position was thought to be crucial to hMAO-B selectivity. As can be seen in Table 3, the energy contribution of Cys172 (which was spatially close to C6-Cl) was −2.5912 ± 0.1582 kJ/mol, which was apparently larger than that of compound M31 (0.2993 ± 0.124 kJ/mol). Other residues important for selectivity were also identified through the binding energy decomposition analysis. For instance, the gatekeeper residue Phe208 in hMAO-A and Ile199 in hMAO-B may contribute to the subtype selectivity. Compared with Cys323 and Leu337 in hMAO-A, the corresponding residue (Thr314 and Leu328) in hMAO-B also displayed different energy contributions, which was supported by the previous research of Catto and coworkers [34].
Table 3.
Free energy contributions (kJ/mol) of hMAO-A and hMAO-B pocket residues.
| Residue | MAO-A vs. M31 | MAO-B vs. M31 | MAO-A vs. M32 | MAO-B vs. M32 | |
|---|---|---|---|---|---|
| MAO-A | MAO-B | Total Energy | Total Energy | Total Energy | Total Energy |
| Tyr69 | Tyr60 | −1.4631 ± 0.0443 | −3.5967 ± 0.0614 | −2.7071 ± 0.0511 | −3.0942 ± 0.0708 |
| Ala111 | Pro102 | 0.3332 ± 0.0714 | 0.8449 ± 0.1369 | 0.5828 ± 0.0985 | −0.0127 ± 0.0890 |
| Phe112 | Phe103 | −0.3019 ± 0.0205 | 0.4229 ± 0.0305 | −0.1243 ± 0.0153 | 0.0942 ± 0.0165 |
| Pro113 | Pro104 | −0.0764 ± 0.0091 | −0.0264 ± 0.0079 | 0.0414 ± 0.0055 | −0.0514 ± 0.0163 |
| Trp128 | Trp119 | −0.0145 ± 0.0064 | −0.0097 ± 0.0097 | 0.0893 ± 0.0033 | −0.1936 ± 0.0077 |
| Phe173 | Leu164 | −0.0364 ± 0.0108 | −0.6217 ± 0.0187 | −0.0662 ± 0.0114 | −0.4042 ± 0.0134 |
| Leu176 | Leu167 | −0.0701 ± 0.0213 | −0.0436 ± 0.0256 | 0.1918 ± 0.0137 | −0.2503 ± 0.0171 |
| Phe177 | Phe168 | −0.1931 ± 0.0494 | −0.4662 ± 0.026 | 0.6680 ± 0.0293 | −0.5791 ± 0.0819 |
| Ile180 | Leu171 | −5.3303 ± 0.1123 | −6.2815 ± 0.1427 | −2.4365 ± 0.2223 | −7.2863 ± 0.1543 |
| Asn181 | Cys172 | −4.1478 ± 0.2633 | 0.2993 ± 0.124 | 0.8583 ± 0.2391 | −2.5912 ± 0.1582 |
| Tyr197 | Tyr188 | 1.1053 ± 0.0829 | 1.7647 ± 0.076 | −4.1025 ± 0.1387 | 0.1502 ± 0.0949 |
| Ile207 | Ile198 | −3.9048 ± 0.1647 | −0.9873 ± 0.1489 | −3.1187 ± 0.1347 | −1.9079 ± 0.1410 |
| Phe208 | Ile199 | −6.1625 ± 0.2403 | −1.1994 ± 0.2342 | −2.2755 ± 0.2184 | 0.0638 ± 0.1966 |
| Gln215 | Gln206 | −7.5319 ± 0.236 | −1.7379 ± 0.2144 | −3.3143 ± 0.2539 | −1.3530 ± 0.2173 |
| Cys323 | Thr314 | −0.5525 ± 0.0492 | −2.5552 ± 0.0515 | −0.4392 ± 0.0685 | −2.3485 ± 0.0513 |
| Ile325 | Ile316 | −1.7599 ± 0.0637 | −3.1339 ± 0.0787 | −1.6549 ± 0.0398 | −4.0400 ± 0.0843 |
| Ile335 | Tyr326 | −4.5914 ± 0.1095 | −4.8286 ± 0.25 | −4.2492 ± 0.1381 | −5.9530 ± 0.1767 |
| Thr336 | Thr327 | −0.4413 ± 0.0476 | −0.3823 ± 0.0269 | −0.0784 ± 0.0246 | −0.5360 ± 0.0223 |
| Leu337 | Leu328 | −3.0775 ± 0.0836 | −1.0562 ± 0.026 | −2.2824 ± 0.0758 | −1.1358 ± 0.0269 |
| Met350 | Met341 | −0.168 ± 0.0143 | −0.438 ± 0.0118 | −0.6131 ± 0.0173 | −0.3265 ± 0.0107 |
| Phe352 | Phe343 | −0.501 ± 0.0391 | −2.0697 ± 0.1135 | −1.2125 ± 0.0482 | −2.5305 ± 0.0643 |
| Tyr407 | Tyr398 | −5.3629 ± 0.1462 | −6.0795 ± 0.1569 | −7.7790 ± 0.1624 | −8.5574 ± 0.1847 |
| Tyr444 | Tyr435 | −3.919 ± 0.2074 | −2.1508 ± 0.1615 | −7.4496 ± 0.1530 | −1.9394 ± 0.1860 |
2.3. Discovery, Synthesis and Biological Activity
The optimization of M43 analogues was performed to further validate the above conclusions and explore the chemical space around the hits. This series of compounds, along with others studied in this article, were listed in Figure 4. Detailed results were listed in Table 4 (we also synthesized several Esuprone analogs, shown in Table S3). To estimate the selectivity of inhibitors, the selectivity index (SI) was calculated as pIC50(MAO-B)–pIC50(MAO-A) [14]. For M43 derivatives, two out of six were experimentally identified as potent and selective hMAO-A inhibitors (FR1, SP1) with IC50 values of 1.5 nM (SI < –2.82) and 19 nM (SI < –2.42), respectively. Compared with M43, all M43 analogues showed an improved MAO-A inhibition except compound 7c, 7g. From an overall SAR profiles, a direct comparison can be made between compounds M43 and SP1, since the introduction of a methyl group at the C8 position in compound M43 increased hMAO-A selectivity. In contrast, simply changing the substituent at C7 position of compound M43 with other groups generally resulted in a significant decline in MAO-A selectivity (for example, compound M43 and FR5). However, the replacement of R5 group of 5d gave the most active hMAO-A inhibitors FR1 with an IC50 value of 1.5 nM. In addition, comparison of the activity values of compounds M43 and 7g revealed the steric hindrance at C4 position again, i.e., only substituents of small size and flexibility are tolerated in hMAO-A inhibitors. Based on the above experimental and theoretical results, the molecular basis of the SAR and SSR (structure-selectivity relationship) trends between hMAO subtypes and coumarin series were summarized in Figure 5.
Figure 4.

Strategy for the purchase or synthesis of C7 substituted coumarin analogues.
Table 4.
In vitro activities against hMAO subtypes of coumarin derivatives.
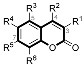
| Compounds | R1 | R2 | R4 | R5 | R6 | MAO-A a | MAO-B a | SI b |
|---|---|---|---|---|---|---|---|---|
| M29 | H | CH3 | H |
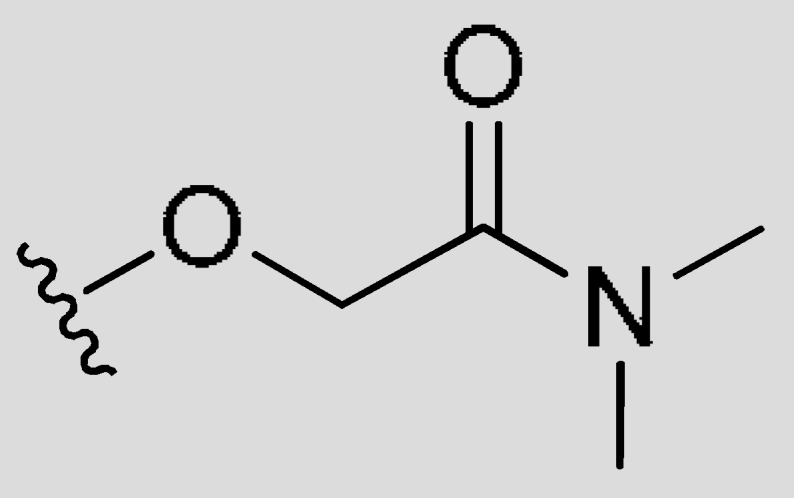
|
CH3 | 2.29 | 100 | −1.64 |
| M31 | H | (CH2)2CH3 | H |
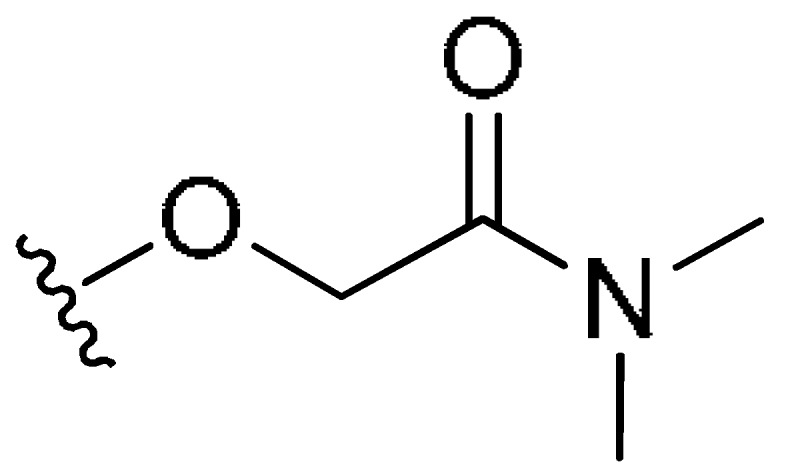
|
H | 0.50 | 64.57 | −2.11 |
| M32 | CH3 | CH3 | Cl |
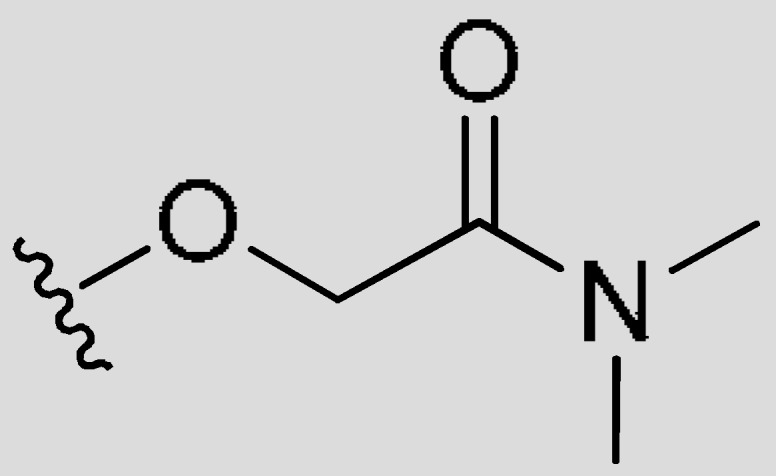
|
H | 81.28 | 0.60 | 2.13 |
| M43(7a) | H | CH3 | H |
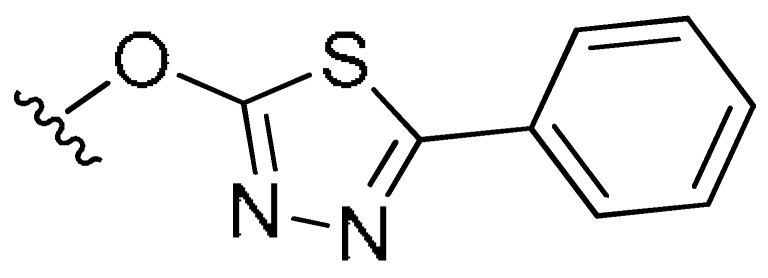
|
H | 0.16 | 0.36 | −0.35 |
| FR1 | –(CH2)3– | H |
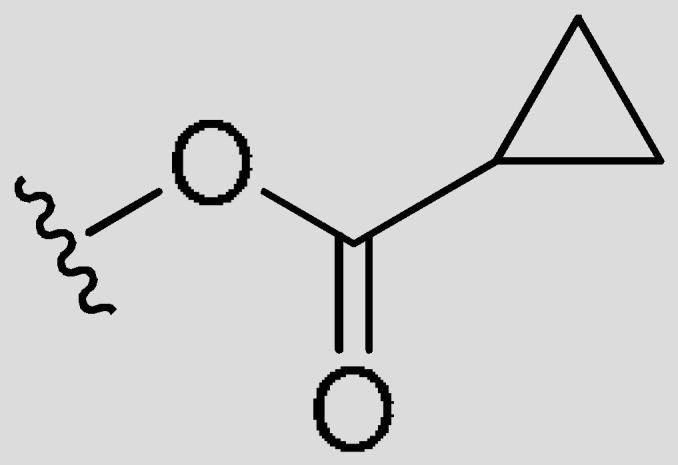
|
CH3 | 0.0015 | 75% (>1 uM) | <−2.82 | |
| FR2 | –(CH2)3– | H |
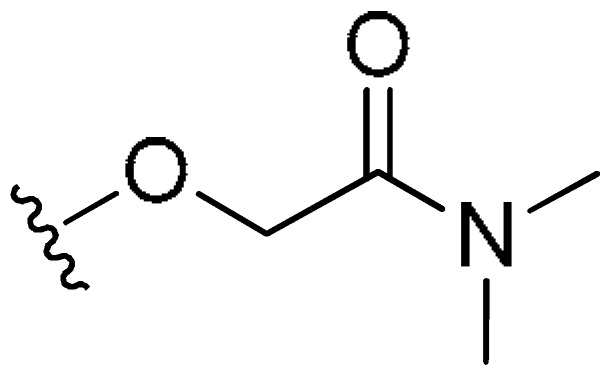
|
CH3 | 2.82 | 12% (>10 uM) | <−0.55 | |
| FR3 | –(CH2)4– | H |
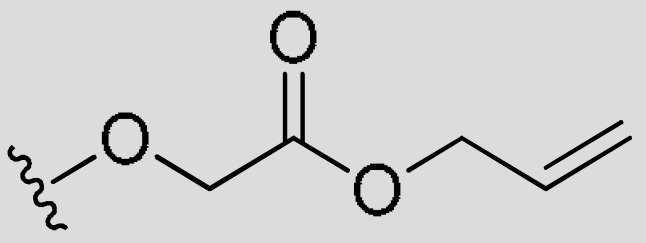
|
CH3 | 0.74 | 66% (>5 uM) | <−0.83 | |
| FR4 | –(CH2)3– | H |
|
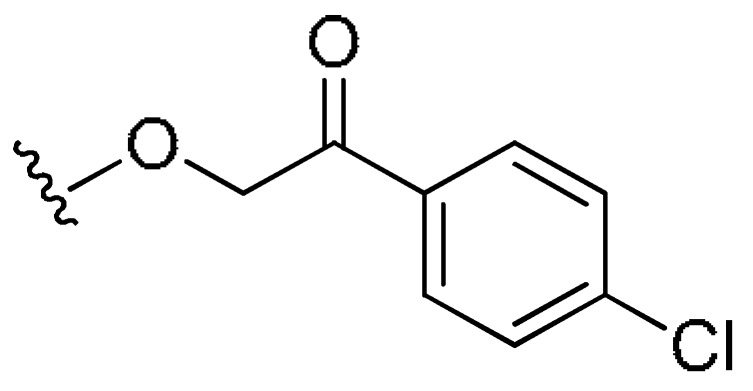
H | 23% (>10 uM) | 0.018 | >2.74 | |
| FR5 | H | CH3 | H |
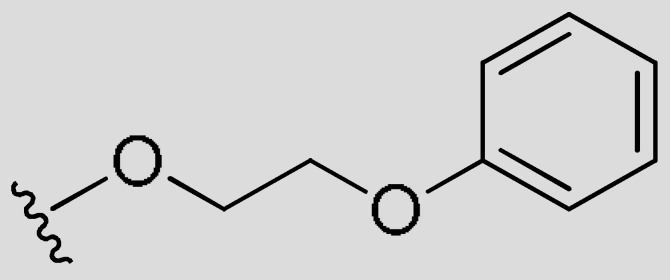
|
H | 28% (>10 uM) | 0.015 | >2.82 |
| SP1 | H | CH3 | H |
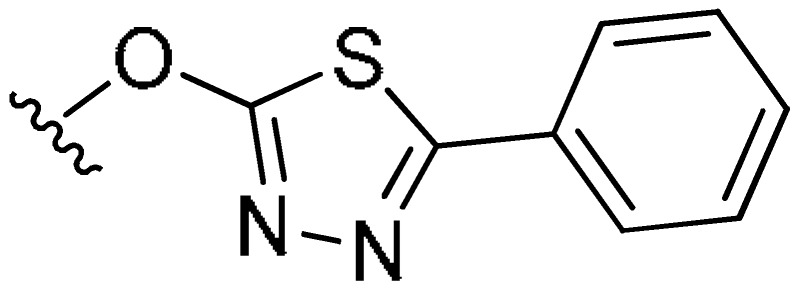
|
CH3 | 0.019 | 64% (>5 uM) | <−2.42 |
| 5d | –(CH2)3– | H |
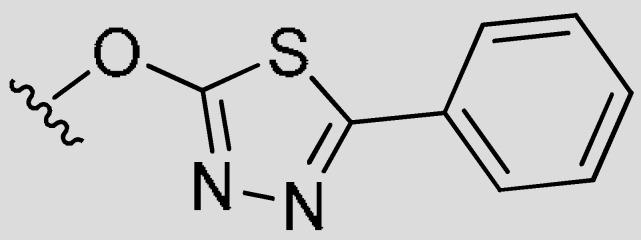
|
CH3 | 0.096 | 2.30 | −1.38 | |
| 7b | H | (CH2)2CH3 | H |
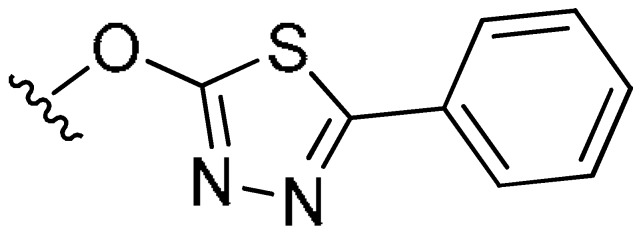
|
H | 0.024 | 0.01 | 0.38 |
| 7k | H | (CH2)2CH3 | H |
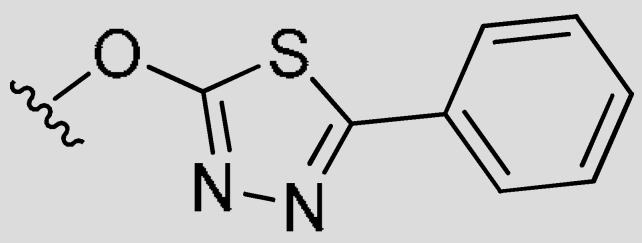
|
CH3 | 0.021 | 0.12 | −0.76 |
| 7c | CH3 | CH3 | Cl |
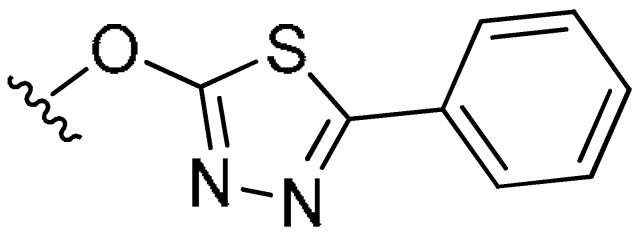
|
H | 0.81 | 0.44 | 0.27 |
| 7g | H |

|
H |
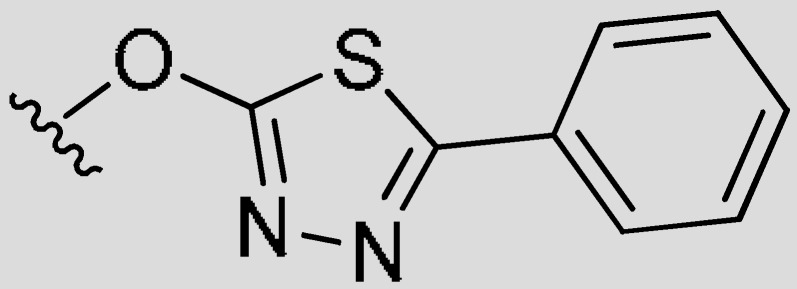
|
H | 2.00 | 6.20 | −0.49 |
a: IC50 values (μM) or inhibition activity at 10 μM against hMAO-A and hMAO-B. b: SI is the selectivity index expressed as pIC50(MAO-B)–pIC50(MAO-A).
Figure 5.

Structure-activity relationship (SAR)/ structure-selectivity relationship (SSR) profiles for the coumarins against hMAO subtypes based on compound M43.
Compounds M29, M31, M32 and M43 were obtained based on our BRS-3D virtual screening protocol. FR1-FR5 and SP1 were purchased from Specs. Compounds 5d, 7b, 7k, 7c and 7g were synthesized in this study.
2.4. In Silico Pharmacokinetic Properties
Additionally, in silico pharmacokinetic properties of all the tested C7-substituted coumarin derivatives were evaluated using ACD/percepta platform including lipophilicity logP, molecular weight, Lipinski’s rule of five, lead-like, Caco-2 and CNS properties. Detailed pharmacokinetic properties were presented in Table 5 [35]. According to the predicted properties, all these compounds complied with the Lipinski’s rule without any violation, which suggested that these new C7-substituted coumarin analogues might have good oral bioavailability. The Caco-2 permeability model was used to predict the absorption potential of oral drugs. The theoretical results suggested that most of these compounds possessed good intestinal permeability. A CNS penetration model was also applied suggesting the ability of these compounds to cross the blood–brain-barrier (BBB). These results suggested that the compounds might have the ability to pass the BBB, which is essential factor for MAO inhibition in human brain. In addition, a CYP isozymes inhibition model was applied for these derivatives using CypRules Server, which suggested that they do not inhibit the five CYP450 enzymes (CYP1A2, CYP2C19, CYP2C9, CYP2D6 and CYP3A4) [36].
Table 5.
Calculated physicochemical and pharmacokinetic parameters.
| Compounds | logP | MW | Lipinski | Lead-like | Caco-2 | CNS |
|---|---|---|---|---|---|---|
| M29 | 1.94 | 275.3 | 0 violations | 0 violations | Highly permeable | Penetrant |
| M31 | 2.21 | 289.33 | 0 violations | 0 violations | Highly permeable | Penetrant |
| M32 | 2.98 | 309.74 | 0 violations | 0 violations | Highly permeable | Penetrant |
| M43 (7a) | 3.79 | 336.37 | 0 violations | 0 violations | Highly permeable | Penetrant |
| FR1 | 3.3 | 284.31 | 0 violations | 0 violations | Highly permeable | Penetrant |
| FR2 | 2.32 | 301.34 | 0 violations | 0 violations | Highly permeable | Penetrant |
| FR3 | 4.33 | 328.36 | 0 violations | 1 violation | Highly permeable | Penetrant |
| FR4 | 4.57 | 354.78 | 0 violations | 1 violation | Highly permeable | Penetrant |
| FR5 | 3.77 | 296.32 | 0 violations | 0 violations | Highly permeable | Penetrant |
| SP1 | 3.97 | 350.39 | 0 violations | 0 violations | Highly permeable | Penetrant |
| 5d | 4.72 | 376.43 | 0 violations | 1 violation | Highly permeable | Penetrant |
| 7b | 4.49 | 364.42 | 0 violations | 1 violation | Highly permeable | Penetrant |
| 7k | 4.79 | 378.45 | 0 violations | 1 violation | Highly permeable | Penetrant |
| 7c | 4.99 | 384.84 | 0 violations | 1 violation | Highly permeable | Weak Penetrant |
3. Conclusions
An essential aspect in rational drug design or discovery against a conserved protein family is subtype selectivity. However, the small structural difference between subtypes makes it a huge challenge. In this study, based on our BRS-3D method and further SAR analysis, two selective hMAO-A inhibitors (FR1, SP1) and two selective hMAO-B inhibitors (FR4, FR5) were identified. Compound FR1 showed the best hMAO-A selectivity with an IC50 value of 1.5 nM and SI < −2.82. FR5 was the most active hMAO-B inhibitor with an IC50 value of 15 nM and SI > 2.82. Both docking and molecular dynamic simulations further demonstrated the details of MAO subtype selectivity at molecular level. Moreover, to evaluate drug-like properties of these molecules, in silico pharmacokinetic evaluation was carried out. All compounds were predicted to possess favorable pharmacokinetic profiles, and have good oral bioavailability. This analysis provided a better understanding of hMAO subtype selectivity mechanism using C7-subsituted coumarins as probes. In summary, the reported C7-substituted coumarins are lead compounds for developing new drugs against depression, Alzheimer’s disease and Parkinson’s disease. Furthermore, BRS-3D method mentioned in this paper can contribute to the discovery and rational design of subtype selective inhibitors for other protein families, such as GPCRs and kinases.
4. Materials and Methods
4.1. Docking Simulations
In this study, GOLD 5.2.2 program was used to dock the C7-substituted coumarin analogues into the binding site of two hMAO subtypes [37]. The 3D structures of C7-substituted coumarin analogues were first generated by CONCORD module and then energy minimization was performed to generate the initial conformation. The crystal structure of two hMAO subtypes was obtained from PDB (MAO-A: 2Z5X, MAO-B: 2V61), which were used for docking calculations. Each protein structure was optimized using the Sybyl X program in order to add hydrogen, remove the co-crystallized waters and ions. The binding area in the reference crystal structures was centered on the co-crystallized ligand with a radius of 6 Å and the Goldscore fitness function was applied to evaluate the binding affinities scores. For each ligand, 30 genetic algorithm runs were performed and the docking pose with the highest score was kept for the MD simulations.
4.2. Molecular Dynamic Simulations
MD simulations were performed using GROMACS 4.5.4. The AMBER ff99SB force field was selected to describe the MAO enzyme complexes [38]. The atomic parameters for the FAD cofactor were derived according to the restrained electrostatic potential (RESP) protocol at the level of HF/6-31(d) [39,40]. The force field parameters for the C7-substituted coumarins were described using the GAFF in antechamber module. The LINCS algorithm was used to keep the heavy atom H bonds in their correct lengths. Periodic boundary conditions with a 12 Å cutoff were applied in all directions for treating the non-bonded interaction. The long-range electrostatic interactions were calculated by the Particle–Mesh–Ewald (PME) method. An isothermal-isobaric ensemble (300 K, 1 atm) was employed during the MD simulation using the combination of Parrinello–Rahman method and modified Berendsen thermostat algorithm. The initial complex structure of each system was placed in an octahedral TIP3P water box. The final production run was performed for 20 ns with a restraint on the C-terminal of the protein. A time step of 2 fs was applied and the generated coordinates were saved every 100 ps.
4.3. MM/PBSA Calculations and Energy Decomposition Analysis
In this study, the MM/PBSA method was employed to evaluate the binding free energy between MAO subtypes and C7-subsituted coumarin analogues. The MM/PBSA binding energy was further decomposed per residue to identify key residues for subtype selectivity. The binding free energy calculations and the decomposition analysis was supported by g_mmpbsa tool [41]. For all simulated systems, the last 10 ns MD trajectory was used for analysis. In Equation (1), the first two terms, represented electrostatic interactions and van der Waals interactions, respectively. The sum of these terms was equal to the molecular mechanics potential energy part. is the solvation energy contribution, which here was calculated according to the PB (Poisson Boltzmann) model. term represented the apolar solvation energy, which was determined based on the SASA model in this paper. As described in Equation (2), the SASA was surface-accessible solvent area, γ was set to 0.02267 kJ/mol and the fitting parameter β was set to 3.84928 kJ/mol. The last term of Equation (1), was not considered in g_mmpbsa. Therefore, it gave the final MM/PBSA equation as presented in Equation (3).
| (1) |
| (2) |
| (3) |
4.4. Chemistry
The target 2,3-dihydrocyclopenta[c]chromen-4(1H)-one derivative (5) and 2H-chromen-2-one derivatives (7) were prepared according to the synthetic route outlined in Scheme 1 and Scheme 2, respectively.
Scheme 1.

The synthetic protocol for 2,3-dihydrocyclopenta[c]chromen-4(1H)-one derivatives (5). Reagents and conditions: (i) Hydrazinecarbothioamide, EtOH/H2O, r.t.; (ii) FeCl3⋅6H2O, EtOH, 80 °C; (iii) CuBr2, isoamyl nitrite, CH3CN, r.t; (iv) Conc. H2SO4, 2-methylbenzene-1,3-diol, 0 °C and (v) K2CO3, DMF, 80 °C.
Scheme 2.

Synthetic procedures of coumarin derivatives (7). Reagents and conditions: (i) Method 1. H2SO4, 0 °C and (ii) 2-Bromo-5-phenyl-1,3,4-thiadiazole (compound 3), K2CO3, DMF, 80 °C. Method 2. substituted sulfuryl chloride, TEA, DCM, r.t.
4.4.1. Synthesis of (E)-2-benzylidenehydrazinecarbothioamide (1)
Benzaldehyde (0.1 g, 0.942 mmol) was dissolved in warm anhydrous alcohol (3 mL) and a mixture of thiosemicarbazide (0.0945 g, 1.04 mmol) in warm water (3 mL) was added dropwise with continuous stirring. The mixture was allowed to react at room temperature until TLC (Thin Layer chromatography) showed the reaction has completed (PE/EA = 2:1). A large amount of white precipitate generated and filtered to give white solid (0.166 g, 0.93 mmol) yield: 98%; m.p. = 142–145 °C; 1H-NMR (400 MHz, DMSO-d6) δ 11.44 (s, 1H, NH), 8.28–8.14 (m, 1H, CH=N), 8.03 (d, J = 16.5 Hz, 2H, NH2), 7.80 (dd, J = 6.6, 2.9 Hz, 2H, ArH), 7.41 (dd, J = 5.2, 1.9 Hz, 3H, ArH); ESI-MS m/z: 180.2 [M + H]+.
4.4.2. Synthesis of 5-phenyl-1,3,4-thiadiazol-2-amine (2)
Thiosemicarbazone (0.25 g, 1.39 mmol) was suspended in 5 mL of ethanol, FeCl3⋅6H2O (1.13 g, 4.18 mmol) was slowly added with constant stirring. The resulting solution was stirred for 1.5 h at 80 °C until TLC showed the reaction has completed (PE/EA = 1:1). The reaction mixture was allowed to cool to room temperature and quenched by adding citric acid (3.2 g). After neutralized with 10% ammonia solution, the crude product was precipitated and filtered to afford a white solid (0.18 g, 1.02 mmol). Yield: 72%; m.p. = 220–223 °C; 1H-NMR (400 MHz, DMSO-d6): δ 7.76 (dd, J = 7.9, 1.7 Hz, 2H, NH2), 7.53–7.36 (m, 5H, ArH); ESI-MS m/z: 175.8 [M + H]−.
4.4.3. Preparation of 2-bromo-5-phenyl-1,3,4-thiadiazole (3)
The copper bromide (0.116 g, 0.520 mmol) and isoamyl nitrite (0.061 g, 0.520 mmol) were added successively in CH3CN (10 mL). The resulting mixture was stirred at room temperature for 10 min, then 5-phenyl-1,3,4-thiadiazol-2-amine (2, 0.04 g, 0.226 mmol) was added in one portion. The reaction mixture was allowed to react at 25 °C for 1 h until completion, as determined by TLC. The solvent was subsequently removed under reduced pressure, and the residue was washed with ethyl acetate (3 × 20 mL). The organic phase was extracted successively with 2N HCl (3 × 25 mL) and brine (25 mL). The combined organic phase was dried over MgSO4 and filtered. All the solvent was removed under vacuum and the resulting residue was purified by flash column chromatography (PE/EA = 1:1, v:v) to afford light yellow solid (0.049 g, 0.20 mmol). Yield: 90.7%; m.p. = 82–84 °C; 1H-NMR (400 MHz, DMSO-d6): δ 8.10–7.77 (m, 2H, ArH), 7.71–7.44 (m, 3H, ArH); 13C-NMR (100 MHz, CDCl3): δ 172.16, 137.99, 131.79, 129.37, 127.85; ESI-MS m/z: 240.7 [M + H]+.
4.4.4. General Procedure for the Synthesis of 7-hydroxy-6-methyl-2,3-dihydrocyclopenta[c]chromen- 4(1H)-one (4)
To a solution of 2-methylbenzene-1,3-diol (0.1 g) in alkylated ethyl acetoacetate, concentrated sulfuric acid (1 mL) was slowly added dropwise at 0 °C in an ice bath. The resulting solution was allowed to react at 0 °C for 5–6 h until the disappearance of starting material, as monitored by TLC (MeOH/DCM = 100:1). The reaction was quenched by adding 50 mL of ice water. The obtained crude solid was filtered, washed with water, dried, and recrystallized by 95% ethanol to give the desired compound (4) as a light yellow solid. Yield: 78%; m.p. = 243–244 °C; 1H-NMR (400 MHz, DMSO-d6): δ 10.32 (s, 1H, OH), 7.25 (d, J = 8.0 Hz, 1H, ArH), 6.84 (d, J = 8.0 Hz, 1H, ArH), 3.00 (t, J = 6.9 Hz, 2H, CH2CH2CH2), 2.71 (t, J = 6.8 Hz, 2H, CH2CH2CH2), 2.16 (s, 3H, CH3), 2.08–2.06 (m, 2H, CH2CH2CH2); ESI-MS m/z: 215.8 [M + H]−.
4.4.5. Preparation of 7-hydroxyl Substituted Coumarin Derivatives (6a–e)
The 7-hydroxyl substituted coumarin derivatives (6a–e) were accomplished according to the similar procedures as that of compound 4.
1Hydroxy-4-methyl-2H-chromen-2-one (6a). Light yellow solid, yield: 56%; m.p. = 180–182 °C; 1H-NMR (400 MHz, DMSO-d6): δ 10.53 (s, 1H, OH), 7.60 (d, J = 8.7 Hz, 1H, ArH), 6.81 (dd, J = 8.7, 2.4 Hz, 1H, ArH), 6.71 (d, J = 2.4 Hz, 1H, ArH), 6.13 (d, J = 1.4 Hz, 1H, C=CH), 2.37 (s, 3H, CH3); ESI-MS m/z: 174.5 [M + H]−.
7-Hydroxy-4-propyl-2H-chromen-2-one (6b). Light yellow solid, yield: 76.6%; m.p. = 128–130 °C; 1H-NMR (400 MHz, DMSO-d6) δ 10.55 (s, 1H, OH), 7.81–7.11 (m, 1H, ArH), 6.91–6.57 (m, 2H, ArH), 6.08 (s, 1H, C=CH), 2.71 (t, J = 7.8 Hz, 2H, CH2CH2CH3), 1.78–1.46 (m, 2H, CH2CH2CH3), 1.12–0.79 (m, 3H, CH2CH2CH3); ESI-MS m/z: 203 [M + H]−.
6-Chloro-7-hydroxy-3,4-dimethyl-2H-chromen-2-one (6c). Light yellow solid, yield: 81.1%; m.p. = 258–260 °C; 1H-NMR (400 MHz, DMSO-d6): δ 11.19 (s, 1H, OH), 7.75 (s, 1H, ArH), 6.87 (s, 1H, ArH), 2.34 (s, 3H, CH3), 2.06 (s, 3H, CH3); ESI-MS m/z: 225 [M + H]+.
4-Cyclopropyl-7-hydroxy-2H-chromen-2-one (6d). Light yellow solid, yield: 86%; m.p. = 199–200 °C; 1H-NMR (400 MHz, DMSO-d6): δ 10.54 (s, 1H, OH), 7.90 (d, J = 8.7 Hz, 1H, ArH), 6.83 (dd, J = 8.7, 2.4 Hz, 1H, ArH), 6.71 (d, J = 2.4 Hz, 1H, ArH), 5.82 (s, 1H, C=CH), 2.24 (dd, J = 8.5, 4.2 Hz, 1H, CHCH2CH2), 1.17–1.00 (m, 2H, CHCH2CH2), 0.98–0.76 (m, 2H, CHCH2CH2); ESI-MS m/z: 202.9 [M + H]+.
7-Hydroxy-8-methyl-4-propyl-2H-chromen-2-one (6e). Light yellow solid, yield: 72%; m.p. = 160 °C; 1H-NMR (400 MHz, DMSO-d6): δ 10.42 (s, 1H, OH), 7.50 (d, J = 8.7 Hz, 1H, ArH), 6.86 (d, J = 8.7 Hz, 1H, ArH), 6.08 (s, 1H, C=CH), 2.70 (t, J = 7.6 Hz, 2H, CH2CH2CH3), 2.15 (s, 3H, CH3), 1.67–1.62 (m, 2H, CH2CH2CH3), 0.96 (t, J = 7.3 Hz, 3H, CH2CH2CH3); ESI-MS m/z: 217.8 [M + H]−.
4.4.6. General Procedure for the Synthesis of Target Compounds (5d, 7a–c, 7g and 7k)
Appropriate coumarin analogues (0.5 mmol) and 2-bromo-5-phenyl-1,3,4-thiadiazole (3, 0.8 mmol) were dissolved in anhydrous DMF (10 mL), and K2CO3 (2.0 mmol) was added in one portion. The mixture was then stirred at 100 °C for 6–7 h under an inert nitrogen atmosphere. Once the completion of the reaction was detected by TLC, the reaction mixture was cooled to room temperature. The reaction mixture was extracted with ethyl acetate (3 × 20 mL) and the combined organic layer was washed with brine (3 × 25 mL) and dried over anhydrous sodium sulfate and filtered. The solvent was removed under vacuum and the crude product was purified by flash chromatography (silica gel, PE/EA = 10:3) to afford the compound 5d as white powder. Yield: 56%; m.p. = 140 °C; 1H-NMR (400 MHz, DMSO-d6): δ 7.92–7.83 (m, 2H, ArH), 7.62 (d, J = 8.6 Hz, 1H, ArH), 7.58–7.47 (m, 4H, ArH), 3.13 (t, J = 7.7 Hz, 2H,CH2CH2CH2), 2.81 (t, J = 7.6 Hz, 2H, CH2CH2CH2), 2.34 (s, 3H, CH3), 2.23–1.99 (m, 2H, CH2CH2CH2); 13C-NMR (100 MHz, CDCl3): δ 173.55, 164.27, 159.83, 155.91, 155.03, 153.43, 131.11, 130.32, 129.17, 127.51, 127.21, 123.00, 118.71, 117.01, 116.18, 32.20, 30.62, 22.51, 9.38; ESI-MS m/z: 377.0 [M + H]+.
4-Methyl-7-((5-phenyl-1,3,4-thiadiazol-2-yl)oxy)-2H-chromen-2-one (7a). White powder, yield: 42.8%; m.p. = 124–125 °C; 1H-NMR (400 MHz, DMSO-d6): δ 7.94–7.90 (m, 3H), 7.66 (d, J = 2.5 Hz, 1H, ArH), 7.62–7.47 (m, 4H, ArH), 6.45 (d, J = 1.4 Hz, 1H, C=CH), 2.47 (s, 3H, CH3); 13C-NMR (100 MHz, CDCl3): δ 172.31, 164.90, 160.29, 157.08, 154.48, 151.76, 131.27, 130.19, 129.22, 127.31, 126.06, 117.93, 115.67, 114.66, 108.16, 18.81; ESI-MS m/z: 336.9 [M + H]+.
7-((5-Phenyl-1,3,4-thiadiazol-2-yl)oxy)-4-propyl-2H-chromen-2-one (7b). White powder, yield: 48.3%; m.p. = 150 °C; 1H-NMR (400 MHz, DMSO-d6): δ 7.99 (d, J = 8.8 Hz, 1H, ArH), 7.95–7.88 (m, 2H, ArH), 7.66 (d, J = 2.5 Hz, 1H, ArH), 7.60–7.53 (m, 3H, ArH), 7.50 (dd, J = 8.8, 2.5 Hz, 1H, ArH), 6.39 (s, 1H, C=CH), 2.82 (t, J = 7.6 Hz, 2H, CH2CH2CH3), 1.72–1.63 (m, 2H, CH2CH2CH3), 1.02 (t, J = 7.3 Hz, 3H, CH2CH2CH3); 13C-NMR (100 MHz, CDCl3): δ 172.34, 164.88, 160.55, 156.91, 155.47, 154.71, 131.27, 130.37, 129.22, 127.31, 125.84, 117.31, 115.64, 113.52, 108.36, 33.81, 21.30, 13.94; ESI-MS m/z: 364.9 [M + H]+.
6-Chloro-3,4-dimethyl-7-((5-phenyl-1,3,4-thiadiazol-2-yl)oxy)-2H-chromen-2-one (7c). White powder, yield: 51.3%; m.p. = 171–173 °C; 1H-NMR (400 MHz, DMSO-d6): δ 8.12 (s, 1H, ArH), 7.94–7.88 (m, 3H, ArH), 7.60–7.52 (m, 3H, ArH), 2.45 (s, 3H, CH3), 2.15 (s, 3H, CH3); 13C-NMR (100 MHz, CDCl3): δ 172.32, 164.93, 161.01, 151.21, 150.98, 144.32, 131.24, 130.19, 129.20, 127.29, 126.07, 123.37, 121.59, 119.96, 110.64, 15.32, 13.67; ESI-MS m/z: 385.0 [M + H]+.
4-Cyclopropyl-7-((5-phenyl-1,3,4-thiadiazol-2-yl)oxy)-2H-chromen-2-one (7g).White powder, yield: 89%; m.p. = 158–160 °C; 1H-NMR (400 MHz, CDCl3): δ 7.99 (d, J = 8.6 Hz, 1H, ArH), 7.87 (dd, J = 7.5, 2.2 Hz, 2H, ArH), 7.54–7.44 (m, 3H, ArH), 7.43–7.35 (m, 2H, ArH), 6.06 (s, 1H, C=CH), 2.14–1.97 (m, 1H, CH), 1.22–1.11 (m, 2H, CH2CH2), 0.94–0.80 (m, 2H, CH2CH2); 13C-NMR (100 MHz, CDCl3): δ 172.36, 165.43, 160.91, 157.07, 157.06, 154.36, 131.24, 130.22, 129.21, 127.31, 126.24, 118.21, 115.61, 110.05, 108.14, 12.17, 8.07; ESI-MS m/z: 362.8 [M + H]+.
8-Methyl-7-((5-phenyl-1,3,4-thiadiazol-2-yl)oxy)-4-propyl-2H-chromen-2-one (7k). White powder, yield: 46.8%; m.p. = 110–112 °C; 1H-NMR (400 MHz, DMSO-d6): δ 7.92–7.86 (m, 2H, ArH), 7.84 (d, J = 8.9 Hz, 1H, ArH), 7.60–7.52 (m, 3H, ArH), 7.49 (d, J = 8.8 Hz, 1H, ArH), 6.40 (s, 1H, C=CH), 2.81 (t, J = 7.6 Hz, 2H, CH2CH2CH3), 2.32 (s, 3H, CH3), 1.67 (q, J = 7.5 Hz, 2H, CH2CH2CH3), 1.00 (t, J = 7.3 Hz, 3H, CH2CH2CH3); 13C-NMR (100 MHz, CDCl3): δ 173.28, 164.40, 160.75, 155.89, 155.41, 153.13, 131.15, 130.28, 129.18, 127.23, 122.69, 118.95, 117.51, 116.05, 113.41, 33.93, 21.41, 13.94, 9.25; ESI-MS m/z: 377.0 [M + H]−.
4.4.7. Synthesis of the Coumarin Sulfonates (5a–c, 7d–f and 7h–j)
A solution of the appropriate coumarin analogues (0.1 mmol) and triethylamine (0.2 mmol) in dry dichloromethane (5 mL) was cooled in an ice bath. A solution of an appropriate sulfonyl chloride derivative (0.12 mmol) in dry dichloromethane (2 mL) was subsequently added dropwise at the same temperature. The reaction mixture was allowed to warm to room temperature and stirred for additional 0.5 h until the disappearance of starting material, as monitored by TLC. The reaction mixture was extracted with ethyl acetate (3 × 20 mL), and the combined organic phase was washed with brine (3 × 25 mL) and dried over anhydrous sodium sulfate. The solvent was evaporated under reduced pressure and the crude residue was purified by flash column chromatography (silica gel, PE/EA = 10:3 to 10:4, v:v) to afford the desired compounds.
6-Methyl-4-oxo-1,2,3,4-tetrahydrocyclopenta[c]chromen-7-yl ethanesulfonate (5a). White powder. Yield: 86%; m.p. = 150–151 °C; 1H-NMR (400 MHz, DMSO-d6) δ 7.55 (d, J = 8.6 Hz, 1H, ArH), 7.34 (d, J = 8.7 Hz, 1H, ArH), 3.71 (q, J = 7.3 Hz, 2H, CH3CH2), 3.10 (t, J = 8.4 Hz,2H, CH2CH2CH2), 2.79 (t, J = 7.4 Hz, 2H, CH2CH2CH2), 2.35 (s, 3H, CH3), 2.18–2.09 (m, 2H, CH2CH2CH2), 1.43 (t, J = 7.3 Hz, 3H, CH3CH2); 13C-NMR (100 MHz, CDCl3): δ 159.66, 155.75, 153.24, 148.50, 127.81, 122.56, 120.51, 118.16, 117.39, 46.37, 32.15, 30.62, 22.47, 9.89, 8.35; ESI-MS m/z: 309.4 [M + H]+.
6-Methyl-4-oxo-1,2,3,4-tetrahydrocyclopenta[c]chromen-7-yl 4-methylbenzenesulfonate (5b). White powder, yield: 81%; m.p. = 152 °C; 1H-NMR (400 MHz, CDCl3): δ 7.73 (d, J = 8.2 Hz, 2H, ArH), 7.33 (d, J = 8.1 Hz, 2H, ArH), 7.27 (d, J = 3.5 Hz, 1H, ArH), 7.13 (d, J = 8.6 Hz, 1H, ArH), 3.06 (dd, J = 8.2, 1.9 Hz, 2H, CH2CH2CH2), 3.00–2.78 (m, 2H, CH2CH2CH2), 2.47 (s, 3H, CH3), 2.27–2.14 (m, 2H, CH2CH2CH2), 2.07 (s, 3H, CH3); 13C-NMR (100 MHz, CDCl3): δ 159.75, 155.84, 153.00, 149.32, 145.89, 132.56, 130.01, 128.39, 127.77, 122.38, 120.67, 118.61, 117.29, 32.15, 30.59, 22.48, 21.80, 9.49; ESI-MS m/z: 370.8 [M + H]+.
6-Methyl-4-oxo-1,2,3,4-tetrahydrocyclopenta[c]chromen-7-yl 4-nitrobenzenesulfonate (5c). White powder, yield: 79%; m.p. = 192–194 °C; 1H-NMR (400 MHz, DMSO-d6): δ 8.59–8.36 (m, 2H, ArH), 8.31–8.08 (m, 2H, ArH), 7.51 (d, J = 8.6 Hz, 1H, ArH), 7.07 (d, J = 8.6 Hz, 1H, ArH), 3.07 (t, J = 7.7 Hz, 2H, CH2CH2CH2), 2.77 (t, J = 7.5 Hz, 2H, CH2CH2CH2), 2.13 (t, J = 7.6 Hz, 2H, CH2CH2CH2), 2.08 (s, 3H, CH3); 13C-NMR (100 MHz, DMSO-d6): δ 158.87, 156.38, 152.87, 151.76, 148.68, 140.03, 130.47, 128.08, 125.72, 124.24, 119.84, 118.50, 117.84, 32.21, 30.71, 22.43, 9.91; ESI-MS m/z: 402.1 [M + H]+.
4-Cyclopropyl-2-oxo-2H-chromen-7-yl ethanesulfonate (7d). White powder, yield: 81%; m.p. = 92 °C; 1H-NMR (400 MHz, DMSO-d6): δ 8.19 (d, J = 8.8 Hz, 1H, ArH), 6.16 (s, 1H, C=CH), 3.63 (q, J = 7.3 Hz, 2H, CH3CH2), 2.35–2.29 (m, 1H, CH), 1.40 (t, J = 7.3 Hz, 3H, CH3CH2), 1.11 (dt, J = 8.3, 3.3 Hz, 2H, CH2CH2), 0.99–0.84 (m, 2H, CH2CH2); 13C-NMR (100 MHz, CDCl3): δ 160.63, 156.90, 154.07, 150.87, 126.19, 119.21, 118.19, 110.69, 110.65, 45.76, 12.17, 8.28, 8.03; ESI-MS m/z: 294.7 [M + H]+.
4-Cyclopropyl-2-oxo-2H-chromen-7-yl 4-methylbenzenesulfonate (7e). White powder, yield: 86%; m.p. = 132–134 °C; 1H-NMR (400 MHz, DMSO-d6): δ 8.10 (d, J = 8.8 Hz, 1H, ArH), 7.88–7.68 (m, 2H, ArH), 7.50 (d, J = 8.1 Hz, 2H, ArH), 7.14 (d, J = 2.3 Hz, 1H, ArH), 7.09 (dd, J = 8.8, 2.4 Hz, 1H, ArH), 6.12 (s, 1H, C=CH), 2.44 (s, 4H), 2.32–2.12 (m, 1H, CH), 1.08 (dt, J = 8.3, 3.3 Hz, 2H, CH2CH2), 0.92–0.88 (m, 2H, CH2CH2); 13C-NMR (100 MHz, CDCl3): δ 160.72, 157.01, 153.73, 151.52, 146.03, 132.22, 130.07, 128.47, 125.91, 119.12, 118.82, 110.88, 110.48, 21.81, 12.13, 8.10; ESI-MS m/z: 356.7 [M + H]+.
4-Cyclopropyl-2-oxo-2H-chromen-7-yl 4-nitrobenzenesulfonate (7f). White powder, yield: 88%; m.p. = 192–194 °C; 1H-NMR (400 MHz, DMSO-d6): δ 8.59–8.39 (m, 2H, ArH), 8.31–8.18 (m, 2H, ArH), 8.12 (d, J = 8.8 Hz, 1H, ArH), 7.26 (d, J = 2.4 Hz, 1H, ArH), 7.16 (dd, J = 8.8, 2.4 Hz, 1H, ArH), 6.14 (s, 1H, C=CH), 2.33–2.16 (m, 1H, CH), 1.09 (dt, J = 8.3, 3.2 Hz, 2H, CH2CH2), 0.91 (dt, J = 6.8, 3.4 Hz, CH2CH2); 13C-NMR (100 MHz, CDCl3): δ 160.32, 156.70, 153.90, 151.23, 150.78, 140.61, 129.88, 126.27, 124.63, 119.67, 118.14, 111.01, 110.80, 12.15, 8.10; ESI-MS m/z: 388.0 [M+H]+.
8-Methyl-2-oxo-4-propyl-2H-chromen-7-yl ethanesulfonate (7h). White powder, yield: 71%; m.p. = 78–80 °C; 1H-NMR (400 MHz, DMSO-d6): δ 7.79 (d, J = 8.8 Hz, 1H, ArH), 7.35 (d, J = 8.8 Hz, 1H, ArH), 6.39 (s, 1H, C=CH), 3.72 (q, J = 7.3 Hz, 2H, CH3CH2), 2.79 (t, J = 7.6 Hz, 2H, CH2CH2CH3), 2.33 (s, 3H, CH3), 1.70–1.61 (m, 2H, CH2CH2CH3), 1.43 (t, J = 7.3 Hz, 3H, CH3CH2), 0.99 (t, J = 7.3 Hz, 3H, CH2CH2CH3); 13C-NMR (100 MHz, CDCl3): δ 160.55, 155.75, 152.95, 149.03, 122.30, 120.79, 118.05, 117.95, 113.68, 46.42, 33.87, 21.38, 13.89, 9.74, 8.34; ESI-MS m/z: 310.7[M + H]+.
8-Methyl-2-oxo-4-propyl-2H-chromen-7-yl 4-methylbenzenesulfonate (7i). White powder, yield: 83%; m.p. = 144–146 °C; 1H-NMR (400 MHz, DMSO-d6): δ 7.81 (d, J = 8.2 Hz, 2H, ArH), 7.75 (d, J = 8.9 Hz, 1H, ArH), 7.52 (d, J = 8.1 Hz, 2H, ArH), 7.03 (d, J = 8.8 Hz, 1H, ArH), 6.37 (s, 1H, C=CH), 2.76 (t, J = 7.6 Hz, 2H, CH2CH2CH3), 2.45 (s, 3H, CH3), 2.01 (s, 3H, CH3), 1.66–1.60 (m, 2H, CH2CH2CH3), 0.97 (t, J = 7.3 Hz, 3H, CH2CH2CH3); 13C-NMR (100 MHz, CDCl3): δ 160.61, 155.77, 152.73, 149.87, 145.92, 132.66, 130.04, 128.37, 122.06, 120.97, 118.47, 117.86, 113.61, 33.86, 21.79, 21.34, 13.91, 9.35; ESI-MS m/z: 372.8 [M + H]+.
8-Methyl-2-oxo-4-propyl-2H-chromen-7-yl 4-nitrobenzenesulfonate (7j). White powder, yield: 86%; m.p. = 171–174 °C; 1H-NMR (400 MHz, DMSO-d6): δ 8.49 (d, J = 8.7 Hz, 2H, ArH), 8.23 (d, J = 8.6 Hz, 2H, ArH), 7.75 (d, J = 8.9 Hz, 1H, ArH), 7.07 (d, J = 8.8 Hz, 1H, ArH), 6.40 (s, 1H, C=CH), 2.77 (t, J = 7.6 Hz, 2H, CH2CH2CH3), 2.07 (s, 3H, CH3), 1.68–1.61 (m, 2H, CH2CH2CH3), 0.98 (t, J = 7.3 Hz, 3H, CH2CH2CH3); 13C-NMR (100 MHz, CDCl3): δ 160.22, 155.50, 152.92, 151.21, 149.28, 141.33, 129.78, 124.63, 122.40, 120.90, 118.38, 117.79, 114.06, 33.84, 21.32, 13.90, 9.69; ESI-MS m/z: 402.0 [M + H]−.
4.5. Biological Assays
The inhibition and IC50 values of the identified C7-substituted coumarin analogues were assayed by Shanghai ChemPartner Co. Ltd., using MAO-GloTM assay kit (Promega Corporation, V1402). Two human MAO enzymes (Cat#31502, Cat#31503) were purchased from Active Motif. Clorgyline and R(-)-deprenyl were purchased as reference drugs from Sigma (Cat. No. M3778) and Abcam (Cat. No. ab120604), respectively. The MAO inhibition assays were carried out in modified HEPES buffer in light of the manufacturer’s protocol. Firstly, the tested compounds in DMSO were transferred to a 384-well plate by Echo. The enzyme solutions were then added into the plate and the plate was incubated for 15 min at room temperature (r.t.). Secondly, the luciferin derivative substrate was added to initiate the reaction. After 60 min incubation at r.t., the Luciferin detection reagent was added to each well to terminate the reaction and generate the luminescent signal. Before reading the signal, the plate was incubated for 20 min at r.t. Finally, plate reader was used to measure and record the luminescent signal and corresponding data were processed using GraphPad Prism 5.
Supplementary Materials
The following are available online at https://www.mdpi.com/1420-3049/24/21/4003/s1, Figure S1: The residue differences of binding pockets between hMAO-A and hMAO-B. FAD cofactor is shown as space-filling model, MAO-A ligand as green sticks and MAO-B ligand as orange sticks. Figure S2: Predicted binding modes of M31, M32 and M43 with hMAO-A and hMAO-B active site. FAD cofactor is shown as space-filling model and compound as green sticks. The H-bond receptor surfaces are shown in pink and the H-bond acceptor surfaces are shown in green. Figure S3: RMSD plots of M31, M32, M43 and C18 with different hMAO subtypes. Figure S4: Per-residue RMSF curves of the hMAO-A and hMAO-B systems. Figure S5: The labeled key motifs of hMAO. Loop area is colored red. Alpha helix is colored magenta. Beta sheet is colored orange. Figure S6: RMSD plots for 16 systems. Table S1: Hydrogen bond analysis of four systems according to MD trajectories. Table S2: Free energy decomposition results (kJ/mol) of hMAO-A and hMAO-B pocket residues. Table S3: pIC50 values of synthesized Esuprone derivatives.
Author Contributions
Conceptualization, D.W. and D.-X.K.; Data curation, D.W., R.-Y.H., M.G. and N.C.; Funding acquisition, D.-X.K.; Investigation, D.W., M.G., Y.L. and N.C.; Methodology, R.-Y.H., X.L. and D.-X.K.; Writing—original draft, D.W.; Writing—review and editing, X.L. and D.-X.K.
Funding
This research was funded by Fundamental Research Funds for the Central Universities grant number 2662017PY117, National Natural Science Foundation of China grant number 21275061, 21977033 and Natural Science Foundation of Shandong Province grant number ZR2018MH042. The APC was funded by 21275061.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds are not available from the authors.
References
- 1.Tripathi A.C., Upadhyay S., Paliwal S., Saraf S.K. Privileged scaffolds as MAO inhibitors: Retrospect and prospects. Eur. J. Med. Chem. 2018;145:445–497. doi: 10.1016/j.ejmech.2018.01.003. [DOI] [PubMed] [Google Scholar]
- 2.Entzeroth M., Ratty A.K. Monoamine Oxidase Inhibitors—Revisiting a Therapeutic Principle. Open J. Depress. 2017;6:31–68. doi: 10.4236/ojd.2017.62004. [DOI] [Google Scholar]
- 3.Youdim M.B.H., Edmondson D., Tipton K.F. The therapeutic potential of monoamine oxidase inhibitors. Nature Rev. Neurosci. 2006;7:295–309. doi: 10.1038/nrn1883. [DOI] [PubMed] [Google Scholar]
- 4.Grimsby J., Chen K., Wang L.J., Lan N.C., Shih J.C. Human monoamine oxidase A and B genes exhibit identical exon-intron organization. Proc. Natl. Acad. Sci. USA. 1991;88:3637–3641. doi: 10.1073/pnas.88.9.3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eisner P., Klasen M., Wolf D., Zerres K., Eggermann T., Eisert A., Zvyagintsev M., Sarkheil P., Mathiak K.A., Zepf F., et al. Cortico-limbic connectivity in MAOA-L carriers is vulnerable to acute tryptophan depletion. Hum. Brain Mapp. 2017;38:1622–1635. doi: 10.1002/hbm.23475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Edmondson D.E., Binda C., Wang J., Upadhyay A.K., Mattevi A. Molecular and mechanistic properties of the membrane-bound mitochondrial monoamine oxidases. Biochemistry. 2009;48:4220–4230. doi: 10.1021/bi900413g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Narayanaswami V., Drake L.R., Brooks A.F., Meyer J.H., Houle S., Kilbourn M.R., Scott P.J.H., Vasdev N. Classics in Neuroimaging: Development of PET Tracers for Imaging Monoamine Oxidases. ACS Chem. Neurosci. 2019;10:1867–1871. doi: 10.1021/acschemneuro.9b00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Is Y.S., Durdagi S., Aksoydan B., Yurtsever M. Proposing Novel MAO-B Hit Inhibitors Using Multidimensional Molecular Modeling Approaches and Application of Binary QSAR Models for Prediction of Their Therapeutic Activity, Pharmacokinetic and Toxicity Properties. ACS Chem. Neurosci. 2018;9:1768–1782. doi: 10.1021/acschemneuro.8b00095. [DOI] [PubMed] [Google Scholar]
- 9.Matos M.J., Teran C., Perez-Castillo Y., Uriarte E., Santana L., Vina D. Synthesis and study of a series of 3-arylcoumarins as potent and selective monoamine oxidase B inhibitors. J. Med. Chem. 2011;54:7127–7137. doi: 10.1021/jm200716y. [DOI] [PubMed] [Google Scholar]
- 10.Blair H.A., Dhillon S. Safinamide: A Review in Parkinson’s Disease. CNS Drugs. 2017;31:169–176. doi: 10.1007/s40263-017-0408-1. [DOI] [PubMed] [Google Scholar]
- 11.Pisani L., Farina R., Nicolotti O., Gadaleta D., Soto-Otero R., Catto M., Di Braccio M., Mendez-Alvarez E., Carotti A. In silico design of novel 2H-chromen-2-one derivatives as potent and selective MAO-B inhibitors. Eur. J. Med. Chem. 2015;89:98–105. doi: 10.1016/j.ejmech.2014.10.029. [DOI] [PubMed] [Google Scholar]
- 12.Huang M., Xie S.S., Jiang N., Lan J.S., Kong L.Y., Wang X.B. Multifunctional coumarin derivatives: Monoamine oxidase B (MAO-B) inhibition, anti-beta-amyloid (Abeta) aggregation and metal chelation properties against Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2015;25:508–513. doi: 10.1016/j.bmcl.2014.12.034. [DOI] [PubMed] [Google Scholar]
- 13.Joubert J., Foka G.B., Repsold B.P., Oliver D.W., Kapp E., Malan S.F. Synthesis and evaluation of 7-substituted coumarin derivatives as multimodal monoamine oxidase-B and cholinesterase inhibitors for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2017;125:853–864. doi: 10.1016/j.ejmech.2016.09.041. [DOI] [PubMed] [Google Scholar]
- 14.Pisani L., Catto M., Nicolotti O., Grossi G., Di Braccio M., Soto-Otero R., Mendez-Alvarez E., Stefanachi A., Gadaleta D., Carotti A. Fine molecular tuning at position 4 of 2H-chromen-2-one derivatives in the search of potent and selective monoamine oxidase B inhibitors. Eur. J. Med. Chem. 2013;70:723–739. doi: 10.1016/j.ejmech.2013.09.034. [DOI] [PubMed] [Google Scholar]
- 15.Fonseca A., Reis J., Silva T., Matos M.J., Bagetta D., Ortuso F., Alcaro S., Uriarte E., Borges F. Coumarin versus Chromone Monoamine Oxidase B Inhibitors: Quo Vadis? J. Med. Chem. 2017;60:7206–7212. doi: 10.1021/acs.jmedchem.7b00918. [DOI] [PubMed] [Google Scholar]
- 16.Chimenti F., Secci D., Bolasco A., Chimenti P., Bizzarri B., Granese A., Carradori S., Yanez M., Orallo F., Ortuso F., et al. Synthesis, molecular modeling, and selective inhibitory activity against human monoamine oxidases of 3-carboxamido-7-substituted coumarins. J. Med. Chem. 2009;52:1935–1942. doi: 10.1021/jm801496u. [DOI] [PubMed] [Google Scholar]
- 17.Secci D., Carradori S., Bolasco A., Chimenti P., Yanez M., Ortuso F., Alcaro S. Synthesis and selective human monoamine oxidase inhibition of 3-carbonyl, 3-acyl, and 3-carboxyhydrazido coumarin derivatives. Eur. J. Med. Chem. 2011;46:4846–4852. doi: 10.1016/j.ejmech.2011.07.017. [DOI] [PubMed] [Google Scholar]
- 18.Matos M.J., Vina D., Picciau C., Orallo F., Santana L., Uriarte E. Synthesis and evaluation of 6-methyl-3-phenylcoumarins as potent and selective MAO-B inhibitors. Bioorg. Med. Chem. Lett. 2009;19:5053–5055. doi: 10.1016/j.bmcl.2009.07.039. [DOI] [PubMed] [Google Scholar]
- 19.Matos M.J., Vina D., Quezada E., Picciau C., Delogu G., Orallo F., Santana L., Uriarte E. A new series of 3-phenylcoumarins as potent and selective MAO-B inhibitors. Bioorg. Med. Chem. Lett. 2009;19:3268–3270. doi: 10.1016/j.bmcl.2009.04.085. [DOI] [PubMed] [Google Scholar]
- 20.Matos M.J., Vilar S., Garcia-Morales V., Tatonetti N.P., Uriarte E., Santana L., Vina D. Insight into the functional and structural properties of 3-arylcoumarin as an interesting scaffold in monoamine oxidase B inhibition. ChemMedChem. 2014;9:1488–1500. doi: 10.1002/cmdc.201300533. [DOI] [PubMed] [Google Scholar]
- 21.Matos M.J., Rodriguez-Enriquez F., Vilar S., Santana L., Uriarte E., Hripcsak G., Estrada M., Rodriguez-Franco M.I., Vina D. Potent and selective MAO-B inhibitory activity: Amino- versus nitro-3-arylcoumarin derivatives. Bioorg. Med. Chem. Lett. 2015;25:642–648. doi: 10.1016/j.bmcl.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 22.Delogu G., Picciau C., Ferino G., Quezada E., Podda G., Uriarte E., Vina D. Synthesis, human monoamine oxidase inhibitory activity and molecular docking studies of 3-heteroarylcoumarin derivatives. Eur. J. Med. Chem. 2011;46:1147–1152. doi: 10.1016/j.ejmech.2011.01.033. [DOI] [PubMed] [Google Scholar]
- 23.Costas-Lago M.C., Besada P., Rodriguez-Enriquez F., Vina D., Vilar S., Uriarte E., Borges F., Teran C. Synthesis and structure-activity relationship study of novel 3-heteroarylcoumarins based on pyridazine scaffold as selective MAO-B inhibitors. Eur. J. Med. Chem. 2017;139:1–11. doi: 10.1016/j.ejmech.2017.07.045. [DOI] [PubMed] [Google Scholar]
- 24.Rendenbach-Müller B., Schlecker R., Traut M., Weifenbach H. Synthesis of coumarins as subtype-selective inhibitors of monoamine oxidase. Bioorg. Med. Chem. Lett. 1994;4:1195–1198. doi: 10.1016/S0960-894X(01)80328-0. [DOI] [Google Scholar]
- 25.Santana L., Gonzalez-Diaz H., Quezada E., Uriarte E., Yanez M., Vina D., Orallo F. Quantitative structure-activity relationship and complex network approach to monoamine oxidase A and B inhibitors. J. Med. Chem. 2008;51:6740–6751. doi: 10.1021/jm800656v. [DOI] [PubMed] [Google Scholar]
- 26.Abdelhafez O.M., Amin K.M., Ali H.I., Abdalla M.M., Batran R.Z. Synthesis of new 7-oxycoumarin derivatives as potent and selective monoamine oxidase A inhibitors. J. Med. Chem. 2012;55:10424–10436. doi: 10.1021/jm301014y. [DOI] [PubMed] [Google Scholar]
- 27.Abdelhafez O.M., Amin K.M., Ali H.I., Abdalla M.M., Batran R.Z. Monoamine oxidase A and B inhibiting effect and molecular modeling of some synthesized coumarin derivatives. Neurochem. Int. 2013;62:198–209. doi: 10.1016/j.neuint.2012.11.005. [DOI] [PubMed] [Google Scholar]
- 28.Gnerre C., Catto M., Leonetti F., Weber P., Carrupt P.A., Altomare C., Carotti A., Testa B. Inhibition of monoamine oxidases by functionalized coumarin derivatives: Biological activities, QSARs, and 3D-QSARs. J. Med. Chem. 2000;43:4747–4758. doi: 10.1021/jm001028o. [DOI] [PubMed] [Google Scholar]
- 29.Deng Z.L., Du C.X., Li X., Hu B., Kuang Z.K., Wang R., Feng S.Y., Zhang H.Y., Kong D.X. Exploring the biologically relevant chemical space for drug discovery. J. Chem. Inf. Model. 2013;53:2820–2828. doi: 10.1021/ci400432a. [DOI] [PubMed] [Google Scholar]
- 30.Hu B., Kuang Z.K., Feng S.Y., Wang D., He S.B., Kong D.X. Three-Dimensional Biologically Relevant Spectrum (BRS-3D): Shape Similarity Profile Based on PDB Ligands as Molecular Descriptors. Molecules. 2016;21:1554. doi: 10.3390/molecules21111554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang D., Li Z., Liu Y., Chen M., Chen N., Zuo Z., Kong D.X. Identification of novel monoamine oxidase selective inhibitors employing a hierarchical ligand-based virtual screening strategy. Future Med. Chem. 2019;11:801–816. doi: 10.4155/fmc-2018-0596. [DOI] [PubMed] [Google Scholar]
- 32.He S.B., Ben H., Kuang Z.K., Wang D., Kong D.X. Predicting Subtype Selectivity for Adenosine Receptor Ligands with Three-Dimensional Biologically Relevant Spectrum (BRS-3D) Sci. Rep. 2016;6:36595. doi: 10.1038/srep36595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuang Z.K., Feng S.Y., Hu B., Wang D., He S.B., Kong D.X. Predicting subtype selectivity of dopamine receptor ligands with three-dimensional biologically relevant spectrum. Chem. Biol. Drug Des. 2016;88:859–872. doi: 10.1111/cbdd.12815. [DOI] [PubMed] [Google Scholar]
- 34.Catto M., Nicolotti O., Leonetti F., Carotti A., Favia A.D., Soto-Otero R., Mendez-Alvarez E., Carotti A. Structural insights into monoamine oxidase inhibitory potency and selectivity of 7-substituted coumarins from ligand- and target-based approaches. J. Med. Chem. 2006;49:4912–4925. doi: 10.1021/jm060183l. [DOI] [PubMed] [Google Scholar]
- 35.ACD/Percepta Platform . Advanced Chemistry Development. Advanced Chemistry Development, Inc.; Toronto, ON, Canada: [(accessed on 26 May 2019)]. Available online: www.acdlabs.com. [Google Scholar]
- 36.Shao C.-Y., Su B.-H., Tu Y.-S., Lin C., Lin O.A., Tseng Y.J. CypRules: A rule-based P450 inhibition prediction server. Bioinformatics. 2015;31:1869–1871. doi: 10.1093/bioinformatics/btv043. [DOI] [PubMed] [Google Scholar]
- 37.Jones G., Willett P., Glen R.C., Leach A.R., Taylor R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 38.Berendsen H.J.C., van der Spoel D., van Drunen R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995;91:43–56. doi: 10.1016/0010-4655(95)00042-E. [DOI] [Google Scholar]
- 39.Bayly C.I., Cieplak P., Cornell W., Kollman P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993;97:10269–10280. doi: 10.1021/j100142a004. [DOI] [Google Scholar]
- 40.Gaussian 09 . Revision E. 01. Gaussian, Inc.; Wallingford, CT, USA: 2009. [Google Scholar]
- 41.Kumari R., Kumar R., Lynn A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014;54:1951–1962. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.