

Abstract
Parasitic diseases have serious health, social, and economic impacts, especially in the tropical regions of the world. Diseases caused by protozoan parasites are responsible for considerable mortality and morbidity, affecting more than 500 million people worldwide. Globally, the burden of protozoan diseases is increasing and is been exacerbated because of a lack of effective medication due to the drug resistance and toxicity of current antiprotozoal agents. These limitations have prompted many researchers to search for new drugs against protozoan parasites. In this review, we have compiled the latest information (2012–2017) on the structures and pharmacological activities of newly developed organic compounds against five major protozoan diseases, giardiasis, leishmaniasis, malaria, trichomoniasis, and trypanosomiasis, with the aim of showing recent advances in the discovery of new antiprotozoal drugs.
Keywords: protozoan diseases, parasitic, giardiasis, leishmaniasis, malaria, trichomoniasis, trypanosomiasis
1. Introduction
It is well known that parasitic diseases are a serious health problem, that has a deep impact on the global human population [1]. Among parasites, protozoan parasites, such as Trypanosoma cruzi, Leishmania mexicana, Plasmodium falciparum, Giardia intestinalis, and Trichomonas vaginalis, are the major disease-causing organisms. They are responsible for spreading infections worldwide, especially in undeveloped countries, where a tropical or temperate climate and poor sanitary and hygiene conditions are common [2,3,4]. Protozoan parasites are single-celled eukaryotes characterized as a diverse polyphyletic group. The infections caused by these parasites are responsible for 500 million deaths worldwide [5,6,7,8]. These infections are considered neglected because relatively little attention has been devoted to their surveillance, prevention, and treatment [9]. According to the global burden of disease analysis report by the World Health Organization in 2008, around 17% of deaths worldwide are caused by neglected tropical diseases [10]. These neglected protozoan diseases are a group of tropical infections that are particularly dominant in low-income majority populations, and affect millions of people and animals [6,11]. The major neglected protozoan diseases are Chagas disease, leishmaniasis, trichomoniasis, amebiasis, and giardiasis [9,12,13].
The modes of transmission of these protozoan parasites differ from each other. Some of them are transmitted by insects that are vectors of Plasmodium species (malaria), T. brucei (human African trypanosomiasis, HAT), T. cruzi (Chagas disease), and Leishmania species (leishmaniasis). In addition, E. histolytica (amebiasis), Cryptosporidium parvum (cryptosporidiosis), Cyclospora cayetanensis (cyclosporiasis), and Giardia lamblia (giardiasis) are transmitted through food and water contaminated with fecal matter [12]. The biochemistry of a large number of protozoan parasites has been studied during previous studies. According to the mode of infection of the parasites, many organic molecules have been manufactured in the last 50 years for the development of new antiprotozoal agents. Some of these agents, e.g., metronidazole and tinidazole (for amebiasis, trichomoniasis, and giardiasis), sodium stibogluconate (for leishmaniasis), chloroquine (for malaria and trichomoniasis), pentamidine and melarsoprol (for HAT), and benznidazole (for American trypanosomiasis) are very effective and are currently used in medical practice. However, the available drugs are not the final solution because of their existing resistance and toxicity [14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30]. The above-mentioned shortcomings promote the ongoing drug research effort to identify mechanistically novel, nontoxic, and cost-effective chemotherapies for the treatment of these neglected protozoan problems [31].
As a continuation of our studies on antiprotozoal drug development, we present the latest information with respect to recent drug developments against five major protozoan diseases, giardiasis, leishmaniasis, malaria, trichomoniasis, and trypanosomiasis in the form of a review. Notably, during the literature review, we found many articles on antiprotozoal drug discovery published between 2012 and 2017 [32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47]. Therefore, keeping their findings in mind, we wrote this review in a different style; we have described the five major protozoan diseases and compiled their latest synthetic and pharmacological data individually from 2012 to 2017 in Table 1, Table 2, Table 3, Table 4 and Table 5. This tabular form of data has not been reported in previously published reviews.
Table 1.
Selected data of reported antigiardial agents.
| Compound | Activity | Ref. | |
|---|---|---|---|
|
G. lamblia [IC50 (µg/mL)] |
[61] | |
| 7 | 4.62 ± 0.12 | ||
| MTZ | 0.86 ± 0.03 | ||
|
G. lamblia [IC50 (µg/mL)] |
[62] | |
| 8 | 84.2 | ||
| MTZ | 1.22 | ||
|
G. intestinalis [IC50 (µM)] |
[63] | |
| 9a | 0.122 | ||
| 9b | 0.151 | ||
| Aminitrozole | 0.490 | ||
| MTZ | 5.360 | ||
|
G. lamblia [IC50 (µM)] |
[64] | |
| 10 | 19.58 | ||
| MTZ | 62.48 | ||
|
G. lamblia [IC50 (µg/mL)] |
[65] | |
| 11 | 30.6 | ||
| MTZ | 0.21 | ||

|
G. intestinalis [IC50 (µM)] |
[66] | |
| 12a | 0.027 ± 0.002 | ||
| 12b | 0.022 ± 0.001 | ||
| 12c | 0.027 ± 0.002 | ||
| 12d | 0.021 ± 0.005 | ||
| Nitazoxamide | 0.015 ± 0.002 | ||
| Mebendazole | 0.046 ± 0.009 | ||
| MTZ | 1.224 ± 0.021 | ||
|
G. duodenalis [IC50 (µg/mL)] |
[67] | |
| 13a | 14.3 | ||
| 13b | 8.2 | ||
| MTZ | 11.4 | ||
|
G. intestinalis [IC50 (µM)] |
[68] | |
| 14a | 0.87 | ||
| 14b | 0.39 | ||
| MTZ | 1.40 | ||
|
G. lamblia [IC50 (µM)] |
[69] | |
| Disulfiram (15) | 6.6 | ||
Table 2.
Selected data of reported antileishmanial agents.
| Compound | Activity | Ref. | ||
|---|---|---|---|---|
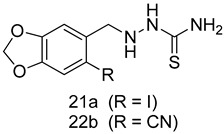
|
L. amazonensis (Promastigotes) [IC50 (µM)] |
L. amazonensis (Amastigotes) [IC50 (µM)] |
[94] | |
| 21a | 20.74 ± 0.5 | 22.0 ± 0.1 | ||
| 22b | 16.4 ± 1.1 | 17.0 ± 1.1 | ||
| Pentamidine | 4.8 ± 0.1 | 1.9 ± 0.1 | ||
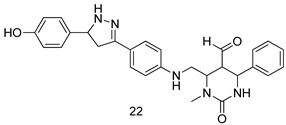
|
L. major [IC50 (µg/mL)] |
L. donovani [IC50 (µg/mL)] |
[95] | |
| 22 | 0.47 ± 0.02 | 1.5 ± 0.17 | ||
| AmB | 0.56 ± 0.01 | - | ||
| SSG | - | 2.98 | ||
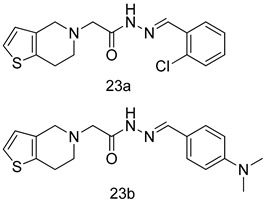
|
L. donovani [IC50 (µg/mL)] | [96] | ||
| 23a | 98.75 | |||
| 23b | 93.75 | |||
| SSG | 490 | |||
| Pentamidine | 5.5 | |||
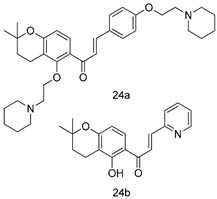
|
L. donovani (Amastigotes) [IC50 (µM)] |
[97] | ||
| 24a | 2.8 | |||
| 24b | 2.0 | |||
| SSG | 49.7 | |||
| Miltefosine | 8.4 | |||
|
L. donovani [IC50 (µM)] | [98] | ||
| Promastigotes | ||||
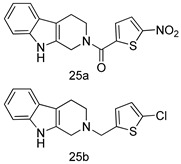
| 25a | 12.7 | |||
| 25b | 9.1 | |||
| Pentamidine | 4.8 | |||
| AmB | 0.3 | |||
|
L. amazonensis [IC50 (µM)] |
L. donovani [IC50 (µM)] |
[99] | |
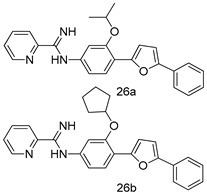
| 26a | 0.13 | 0.31 | ||
| 26b | 0.14 | 0.17 | ||
|
L. major [IC50 (µM)] |
L. donovani [IC50 (µM)] |
[100] | |
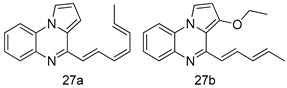
| 27a | 1.2 ± 0.7 | 10.5 ± 0.6 | ||
| 27b | 1.5 ± 1.1 | 4.0 ± 0.6 | ||
| Pentamidine | 4.3 ± 0.8 | 5.5 ± 0.8 | ||
|
L. aethiopica [IC50 (µg/mL)] | [101] | ||
| Promastigotes | Amastigotes | |||
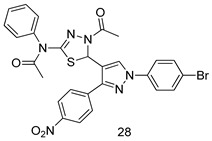
| 28 | 0.0142 ± 0.004 | 0.13 ± 0.02 | ||
| Miltefosine | 3.19 ± 14 | 0.3 ± 0.04 | ||
| AmB | 0.0472 ± 0.002 | 0.2 ± 0.02 | ||
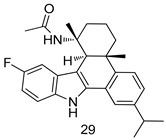
|
L. infantum [IC50 (µM)] | [102] | ||
| 29 | 1.5 ± 0.1 | |||
| Miltefosine | 3.4 ± 0.6 | |||
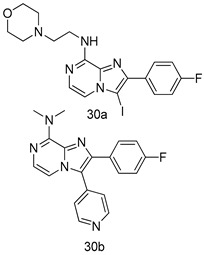
|
L. major [IC50 (µM)] | [103] | ||
| Promastigotes | Amastigotes | |||
| 30a | 2.8 ± 0.4 | 0.2 ± 0.1 | ||
| 30b | 6.4 ± 0.2 | 0.8 | ||
| Pentamidine | 4.6 ± 1.1 | 3 ± 1 | ||
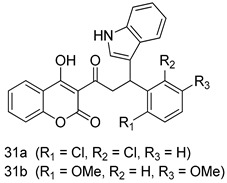
|
L. major [IC50 (µg/mL)] | [104] | ||
| 31a | 95.00 | |||
| 31b | 99.00 | |||
| SSG | 490.00 | |||
| Pentamidine | 5.50 | |||
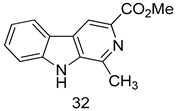
|
L. donovani (Promastigotes) [IC50 (µM)] |
[105] | ||
| 32 | 9.0 ± 2.80 | |||
| Miltefosine | 11.9 ± 2.70 | |||
|
L. donovani (Antiamastigotes) [IC50 (µM)] |
[106] | ||
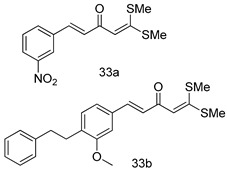
| 33a | 5.12 | |||
| 33b | 3.56 | |||
| Miltefosine | 12.50 | |||
|
L. major [IC50 (µM)] | [107] | ||
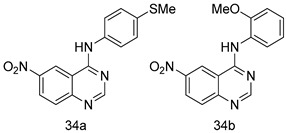
| 34a | 1.87 ± 0.31 | |||
| 34b | 4.37 ± 0.02 | |||
| Pentamidine | 5.09 ± 0.09 | |||
|
L. donovani [IC50 (µM)] | [108] | ||
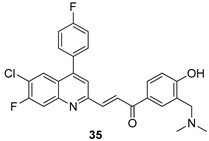
| 35 | 0.84 ± 0.12 | |||
| Miltefosine | 8.10 ± 0.60 | |||
|
L. infantum [IC50 (µM)] | [109] | ||
| 36a | 1.40 ± 0.09 | |||
| 36b | 1.47 ± 0.07 | |||
| 36c | 0.83 ± 0.04 | |||
| Miltefosine | 2.84 ± 0.10 | |||
| Edelfosine | 0.82 ± 0.13 | |||
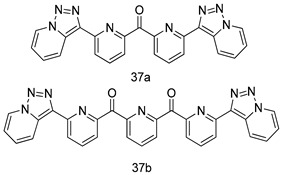
|
L. infantum [IC50(µM)] |
L. amazonesis [IC50(µM)] |
[110] | |
| 37a | 19.5 | 114.6 | ||
| 37b | 38.0 | inactive | ||
| Miltefosine | 23.7 | 20.9 | ||
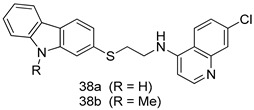
|
L. donovani [IC50 (µM)] | [111] | ||
| Promastogote | Amastogote | |||
| 38a | 8.57 ± 1.58 | 1.11 ± 0.19 | ||
| 38b | 6.27 ± 0.65 | 0.36 ± 0.10 | ||
| Miltefosine | 1.10 ± 0.26 | 8.10 ± 1.41 | ||
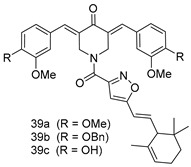
|
L. donovani [IC50 (µM)] | [112] | ||
| 39a | 3.75 ± 0.31 | |||
| 39b | 5.02 ± 0.49 | |||
| 39c | 1.83 ± 0.21 | |||
| Miltefosine | 8.10 ± 0.51 | |||
| SSG | 53.12 ± 4.56 | |||
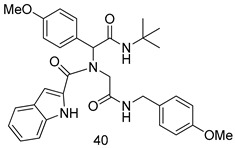
|
L. donovani Amastigote [IC50 (µM)] |
[113] | ||
| 40 | 0.6 ± 0.2 | |||
| Miltefosine | 8.4 ± 1.2 | |||
| SSG | 56.1 ± 3.2 | |||
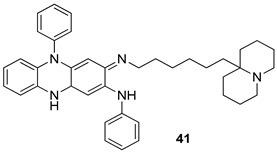
|
L. infantum [IC50 (µM)] |
L. tropica [IC50 (µM)] |
[114] | |
| 41 | 0.23 ± 0.05 | 0.12 ± 0.03 | ||
| Clofazimine | 4.48 ± 1.06 | 2.96 ± 1.23 | ||
| AmB | 0.08 ± 0.02 | 0.09 ± 0.04 | ||
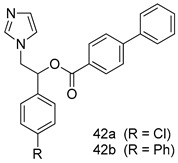
|
L. infantum [IC50 (µM)] | [115] | ||
| 42a | 12.7 | |||
| 42b | 8.0 | |||
| Miltefosine | 10.4 | |||
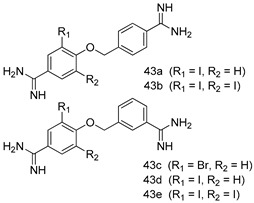
|
L. donovani [IC50 (µM)] | [116] | ||
| 43a | 1.94 | |||
| 43b | 1.6 | |||
| 43c | 1.86 | |||
| 43d | 1.27 | |||
| 43e | 1.39 | |||
| Pentamidine | 1.84 | |||
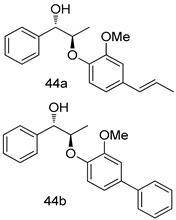
|
L. donovani [IC50 (µg/mL)] | [117] | ||
| 44a | 2.29 | |||
| 44b | 2.48 | |||
|
L. donovani [IC50 (µg/mL)] | [118] | ||
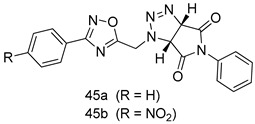
| 45a | 1.6 | |||
| 45b | 2.0 | |||
| Miltefosine | 0.171 | |||

|
L. donovani (Amastigotes) [IC50 (µM)] |
[119] | ||
| 46a | 5.4 | |||
| 46b | 1.1 | |||
| 46c | 2.4 | |||
| AmB | 0.10 | |||
|
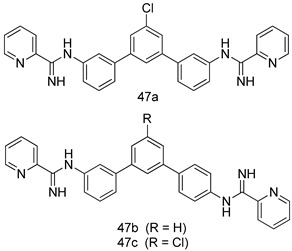
L. amazonensis [IC50 (µM)] | [120] | ||
| 47a | 0.095 | |||
| 47b | 0.123 | |||
| 46c | 0.211 | |||
| AmB | 0.124 | |||
|
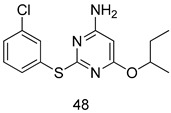
L. infantum [IC50 (µM)] | [121] | ||
| 48 | 29.43 ± 1.34 | |||
| AmB | 0.52 ± 1.34 | |||
|
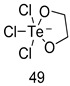
L. donovani [IC50 (µM)] | [122] | ||
| Promastigotes | Amastigotes | |||
| 49 | 26.9 | 10.6 | ||
| Miltefosine | 6.1 | 8.2 | ||
|
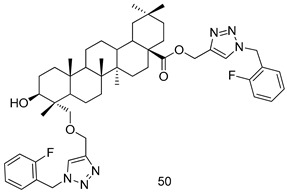
L. infantum [IC50 (µM)] | [123] | ||
| 50 | 5.6 ± 0.14 | |||
|
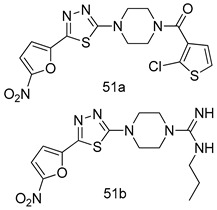
L. major [IC50 (µM)] | [124] | ||
| Promastigotes | Amastigotes | |||
| 51a | 9.35 | 2.7 | ||
| 51b | 0.08 | - | ||
Table 3.
Selected data of reported antimalarial agents.
| Compounds | Activity | Ref. | ||
|---|---|---|---|---|
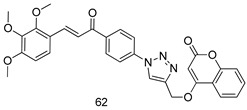
|
P. falciparum [IC50 (µM)] | [143] | ||
| 62 | 1.60 | |||
| Dihydro-artemisinin | 0.0011 | |||
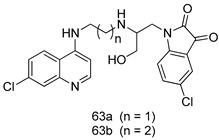
|
P. falciparum [IC50 (nM)] | [144] | ||
| 63a | 11.7 | |||
| 63b | 13.5 | |||
| Chloroquine | 36.37 | |||
| Artemisinin | 4.37 | |||
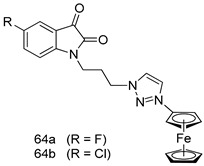
|
P. falciparum [IC50 (µM)] | [145] | ||
| 3D7 strain | W2 strain | |||
| 64a | 3.76 | 5.97 | ||
| 64b | 8.49 | 4.58 | ||
| Chloroquine | 0.021 | 0.49 | ||
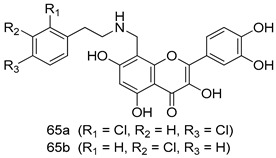
|
P. falciparum [IC50 (µM)] | [146] | ||
| W2 strain | D6 strain | |||
| 65a | 0.071 | 0.065 | ||
| 65b | 0.079 | 0.069 | ||
| Artemisinin | 0.007 | 0.009 | ||
| Chloroquine | 0.63 | 0.014 | ||
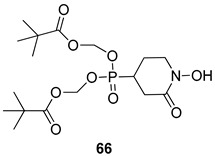
|
P. falciparum [IC50 (nM)] | [147] | ||
| 66 | 27.4 | |||
| Chloroquine | 300 | |||
| Mefloquine | 92 | |||

|
P. falciparum [IC50 (µM)] | [148] | ||
| D6 strain | W2 strain | |||
| 67 | 2.56 | 3.41 | ||
| Artemisinin | <0.09 | <0.09 | ||
| Chloroquine | <0.08 | 0.72 | ||
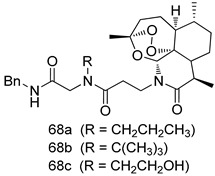
|
P. falciparum [IC50 (nM)] | [149] | ||
| 68a | 0.3 | |||
| 68b | 0.7 | |||
| 68c | 1.5 | |||
| Artemisinin | 19 | |||
|
P. falciparum [IC50 (nM)] | [150] | ||
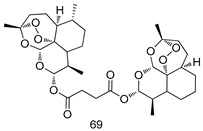
| 69 | 2.6 ± 0.4 | |||
| Chloroquine | 9.8 ± 2.8 | |||
| Dihydro-Artemisinin | 1.1 ± 0.5 | |||

|
P. falciparum [IC50 (µM)] | [151] | ||
| 3D7 | K1 | |||
| 70a | 0.62 | 1.78 | ||
| 70b | 0.29 | 0.496 | ||
| Chloroquine | 0.005 | 0.254 | ||
|
% Dead asexual parasites (P. falciparum) |
[152] | ||
| 3D7 | RKL-2 | |||
| 71a | 25.7 | 5.0 | ||
| 71b | 20.0 | 1.0 | ||
|
P. falciparum [IC50 (µM)] 3D7 strain |
[153] | ||
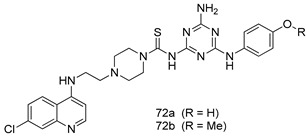
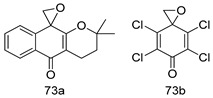
| 72a | 3.71 | |||
| 72b | 3.95 | |||
| Chloroquine | 0.18 | |||
|
P. falciparum [IC50 (µM)] | [154] | ||
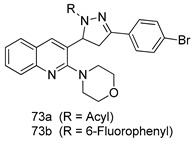
| 73a | 0.018 | |||
| 73b | 0.015 | |||
| Chloroquine | 0.062 | |||
|
P. falciparum [IC50 (µM)] | [155] | ||
| 3D7 | K1 | |||
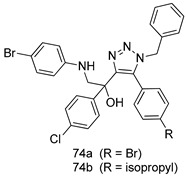
| 74a | 0.87 | 2.04 | ||
| 74b | 0.3 | 2.11 | ||
| Chloroquine | 0.011 | 1.2 | ||
|
P. falciparum [IC50 (µM)] 3D7 strain |
[156] | ||
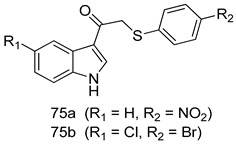
| 75a | 0.24 | |||
| 75b | 0.09 | |||
| Chloroquine | 0.028 | |||
|
P. falciparum [IC50 (µM)] 3D7 strain |
[157] | ||
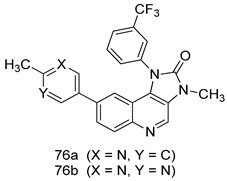
| 76a | 0.007 | |||
| 76b | 0.017 | |||

|
P. falciparum [IC50 (µM)] K1 strain |
[158] | ||
| 77a | 0.28 | |||
| 77b | 0.095 | |||
| Chloroquine | 0.12 | |||
|
P. falciparum [IC50 (µM)] K1 strain |
[115] | ||
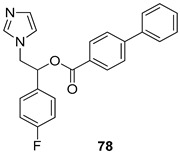
| 78 | 0.6 | |||
| Chloroquine | 0.14 | |||
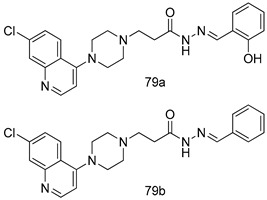
|
P. falciparum [(IC50 µM)] W2 strain |
[159] | ||
| 79a | 0.33 ± 0.095 | |||
| 79b | 0.2 ± 0.05 | |||
| Mefloquine | 0.04 ± 0.01 | |||
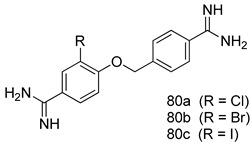
|
P. falciparum [IC50 (µM)] | [116] | ||
| 80a | 0.006 | |||
| 80b | 0.004 | |||
| 80c | 0.006 | |||
| Chloroquine | 0.201 | |||
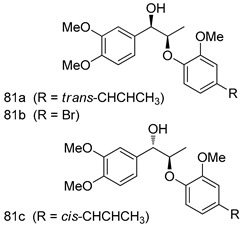
|
P. falciparum [IC50 (µg/mL)] | [117] | ||
| 81a | 0.608 | |||
| 81b | 2.50 | |||
| 81c | 1.71 | |||
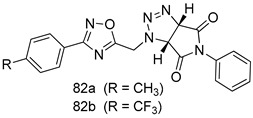
|
P. falciparum [IC50 (µg/mL)] | [118] | ||
| 82a | 13.2 | |||
| 82b | 14.7 | |||
| Chloroquine | 0.073 | |||
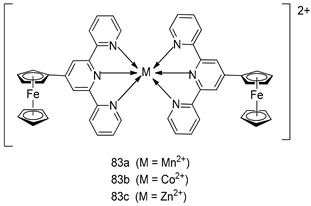
|
P. falciparum [IC50 (µM)] | [160] | ||
| 83a | 1.1 ± 1.1 | |||
| 83b | 1.9 ± 0.9 | |||
| 83c | 1.5 ± 0.14 | |||
| Mefloquine | 0.004 ± 0.02 | |||
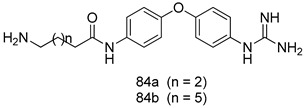
|
P. falciparum [IC50 (µM)] | [161] | ||
| 84a | 0.106 | |||
| 84b | 0.149 | |||
| Chloroquine | 0.0039 | |||
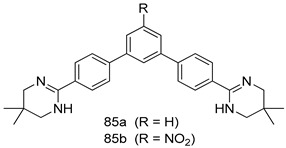
|
P. falciparum [IC50 (µM)] | [120] | ||
| 85a | 0.002 | |||
| 85b | 0.003 | |||
| Chloroquine | 0.125 | |||
| Artemisinin | 0.004 | |||
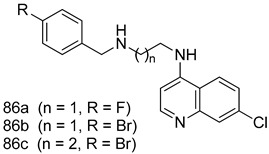
|
P. falciparum [IC50 (nM)] | [162] | ||
| W2 | D6 | |||
| 86a | 45.78 | 26.38 | ||
| 86b | 9.47 | 4.10 | ||
| 86c | 5.93 | 3.95 | ||
| Artemisinin | 6.7 | 9.00 | ||
|
P. falciparum [IC50 (nM)] | [163] | ||
| D6 | Dd2 | |||
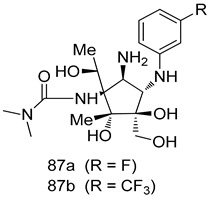
| 87a | 6.5 | 7.4 | ||
| 87b | 6.7 | 3.5 | ||
|
P. falciparum [IC50 (µM)] | [164] | ||
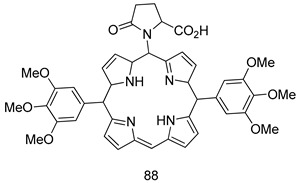
| 88 | 0.02 | |||
| Chloroquine | 15~25 | |||
|
P. falciparum [IC50 (µM)] | [165] | ||
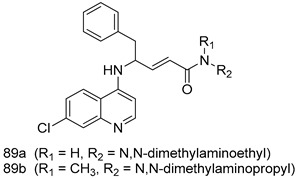
| 89a | 0.17 ± 0.01 | |||
| 89b | 0.23 ± 0.01 | |||

|
P. falciparum [IC50 (µM)] | [166] | ||
| 90a | 0.43 ± 0.02 | |||
| 90b | 0.53 ± 0.04 | |||
| 90c | 0.37 ± 0.03 | |||
| Chloroquine | 0.06 | |||
Table 4.
Selected data of reported antitrichomonal agents.
| Compound | Activity | Ref. | ||
|---|---|---|---|---|
|
T. vaginalis [IC50 (µM)] | [63] | ||
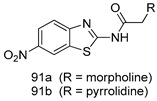
| 91a | 0.331 | |||
| 91b | 0.221 | |||
| MTZ | 0.290 | |||
|
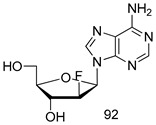
T. vaginalis [IC50 (µM)] | [191] | ||
| 92 | 0.09 | |||
| MTZ | 0.72 | |||
|
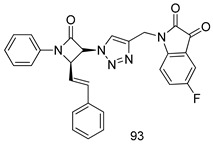
T. vaginalis [IC50 (µM)] | [192] | ||
| 93 | 7.06 | |||
| MTZ | 0.72 | |||
|
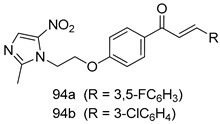
T. vaginalis [IC50 (µg/mL)] | [193] | ||
| MTZ-susceptible | MTZ-resistant | |||
| 94a | 1.56 | 3.125 | ||
| 94b | 1.56 | 3.125 | ||
| MTZ | 1.56 | 12.5 | ||
|
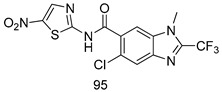
T. vaginalis [IC50 (µM)] | [66] | ||
| 95 | 0.023± 0.005 | |||
| Nitazoxanide | 0.107 ± 0.009 | |||
| MTZ | 0.213 ± 0.004 | |||
|
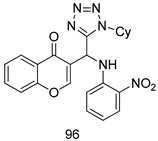
T. vaginalis [IC50 (µg/mL)] | [62] | ||
| 96 | 83.9 | |||
| MTZ | 0.037 | |||
|
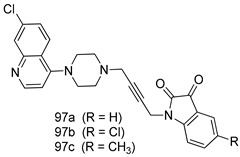
T.foetus [IC50 (µM)] | [194] | ||
| 97a | 22.2 | |||
| 97b | 11.3 | |||
| 97c | 24.5 | |||
| MTZ | 0.72 | |||

|
T. vaginalis [IC50 (µM)] | [195] | ||
| 98a | 9.86 | |||
| 98b | 9.79 | |||
| MTZ | 0.72 | |||
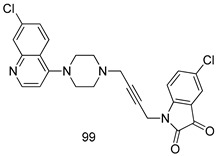
|
T. vaginalis [IC50 (µM)] | [196] | ||
| 99 | 23 | |||
| MTZ | 0.72 | |||
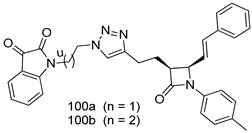
|
T. vaginalis [IC50 (µM)] | [197] | ||
| 100a | 10.49 ± 1.05 | |||
| 100b | 25.60 ± 1.08 | |||
| MTZ | 0.72 | |||
|
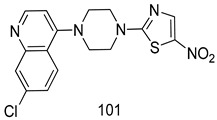
T. vaginalis [IC50 (µg/mL)] | [198] | ||
| 101 | 4.3 ± 1.2 | |||
| MTZ | 8.5 ± 0.9 | |||
|
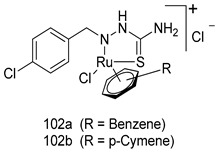
T. vaginalis [IC50 (µM)] | [199] | ||
| 102a | 5.47 | |||
| 102b | 7.56 | |||
| MTZ | 0.72 | |||
|
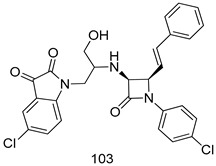
T. vaginalis [IC50 (µM)] | [200] | ||
| 103 | 9.73 ± 1.13 | |||
| MTZ | 0.72 | |||
|
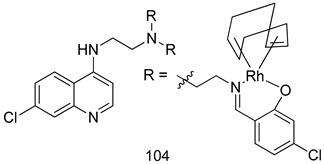
T. vaginalis [IC50 (µM)] | [201] | ||
| 104 | 4.80 ± 0.54 | |||
| MTZ | 0.72 | |||
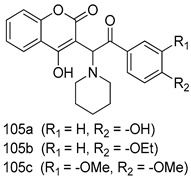
|
T. vaginalis [IC50 (µg/mL)] | [202] | ||
| MTZ-susceptible | MTZ-resistant | |||
| 105a | 0.0294 | 0.2363 | ||
| 105b | 0.0140 | 0.1122 | ||
| 105c | 0.0136 | 0.1091 | ||
| MTZ | 0.0182 | 0.3655 | ||
Table 5.
Selected data of reported antitrypanosomal agents.
| Compound | Activity | Ref. | ||
|---|---|---|---|---|
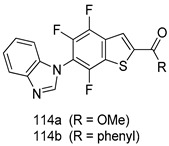
|
T. b. rhodesiense [IC50 (µM)] | [217] | ||
| 114a | 0.60 | |||
| 114b | 0.53 | |||
| Melarsoprol | 0.051 | |||
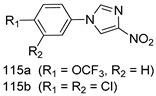
|
T. b. rhodesiense [IC50 (µM)] | [218] | ||
| 115a | 0.16 | |||
| 115b | 0.10 | |||
| Melarsoprol | 0.009 | |||
| Eflornithine | 3.80 | |||
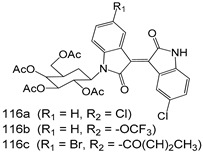
|
T. brucei [IC50 (µM)] | [219] | ||
| 116a | 0.84 | |||
| 116b | 0.60 | |||
| 116c | 0.51 | |||
| Melarsoprol | 0.013 | |||
|
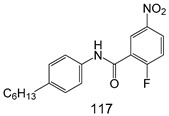
T. brucei [IC50 (µM)] | [220] | ||
| 117 | 1.5 ± 0.4 | |||
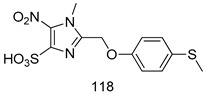
|
T. brucei [IC50 (µM)] | [221] | ||
| 118a | 0.25 ± 0.1 | |||
| 118b | 0.67 ± 10.1 | |||
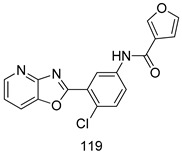
|
T. b. rhodesiense [IC50 (µM)] | [222] | ||
| 119 | 0.091 | |||
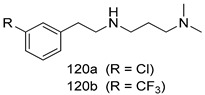
|
T. b. brucei [IC50 (µM)] | [223] | ||
| 120a | 0.7 | |||
| 120b | 0.5 | |||
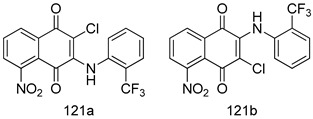
|
T. brucei [IC50 (µM)] | [224] | ||
| 121a | 0.07 ± 0.01 | |||
| 121b | 0.05 ± 0.01 | |||
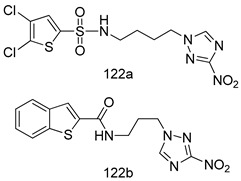
|
T. b. rhodesiense [IC50 (µM)] |
T. cruzi [IC50 (µM)] |
[225] | |
| 122a | 0.218 | 0.373 | ||
| 122b | 0.187 | 0.239 | ||
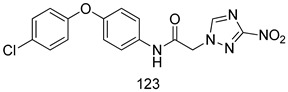
|
T. cruzi [IC50 (µM)] | [226] | ||
| 123 | 0.008 | |||
| Benznidazole | 2.153 ± 0.176 | |||
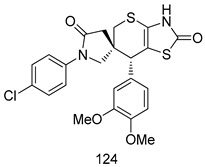
|
T. b. brucei [IC50 (µM)] | [227] | ||
| 124 | 0.2624 ± 0.0139 | |||
| pentamidine | 0.0032 ± 0.0004 | |||
|
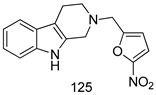
T. brucei [IC50 (µM)] | [98] | ||
| 125 | 1.01 | |||
| Pentamidine | 0.0041 | |||

|
T. cruzi [IC50 (µM)] | [228] | ||
| 126 | 8.5 | |||
| Nifurtimox | 7.7 | |||
|
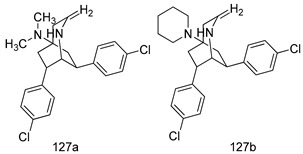
T. b. rhodesiense [IC50 (µM)] | [158] | ||
| 127a | 0.061 | |||
| 127b | 0.065 | |||
| Melarsoprol | 0.0039 | |||
| Suramin | 0.0075 | |||

|
T. b. rhodesiense [IC50 (µM)] |
T. cruzi [IC50 (µM)] |
[229] | |
| 128a | 1.81 | 0.25 | ||
|
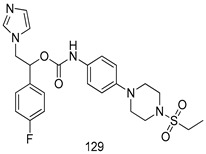
T. cruzi [IC50 (µM)] | [115] | ||
| 129 | 0.04 | |||
| Benznidazole | 1.95 | |||
|
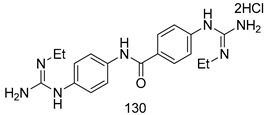
T. b. rhodesiense [IC50 (µM)] | [230] | ||
| 130 | 0.009 | |||
| Melarsoprol | 0.008 | |||

|
T. b. rhodesiense [IC50 (µM)] | [116] | ||
| 131 | 0.003 | |||
| Pentamidine | 0.003 | |||
| Melarsoprol | 0.006 | |||
|
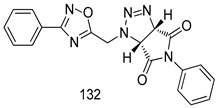
T. b. rhodesiense [IC50(µg/mL)] | [118] | ||
| 132 | 7.0 | |||
| Melarsoprol | 0.005 | |||
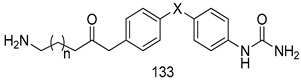
|
T. b. rhodesiense [IC50(µM)] | [161] | ||
| 133a | 13.1 | |||
| 133b | 20.2 | |||
| Pentamidine | 0.005 | |||
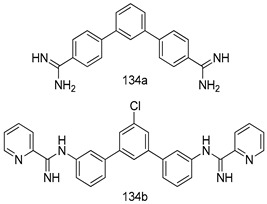
|
T. b. rhodesiense [IC50 (µM)] |
T. cruzi [IC50 (µM)] |
[120] | |
| 134a | 0.004 | 71.6 | ||
| 134b | 0.019 | 0.053 | ||
| Pentamidine | 0.003 | - | ||
| Melarsoprol | 0.004 | - | ||
| Benznidazole | - | 1.30 | ||
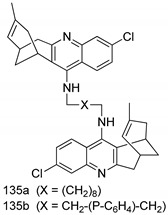
|
T. brucei [IC50 (µM)] | [231] | ||
| 135a | 0.52 ± 0.01 | |||
| 135b | 0.57 ± 0.02 | |||
2. Recent Progress of Antiprotozoan Agents
2.1. Anti-Giardiasis
Giardiasis is caused by the protozoan parasite G. intestinalis (also known as G. lamblia or G. duodenalis). It is also known by the common name “beaver fever,” because this infection was reported in campers who drank contaminated water that was inhabited by beavers. Giardiasis is the most common protozoan infection in human beings, and it occurs in both developing and industrialized countries [48]. Its global incidence is believed to range between 20%–60% [49], with 2%–7% incidence in industrialized nations [50]. Giardia intestinalis was first described in 1681 after the Dutch microscopist Antonie van Leeuwenhoek observed the protozoan in his own diarrheic stools. This disease is often prevalent in poor countries and communities that have untreated water, inadequate sanitation, and poor dietary status [51]. The infection is caused by fecal–oral transmission and initiated by the ingestion of infectious cysts from contaminated water or through person-to-person contact. After excystation, flagellated trophozoites colonize the upper small intestine, where they attach to the epithelial lining but do not invade the mucosa. The duration of Giardia infection is variable; however, chronic infection and reinfection commonly occur [52]. Approximately 50% of the symptoms classically associated with giardiasis are asymptomatic including diarrhea, abdominal pain, nausea, vomiting, and anorexia. However, infected individuals can also develop extraintestinal and postinfectious complications [53,54]. Chronic extraintestinal sequelae may affect the joints, skin, eyes, and central nervous system, but the underlying mechanisms are unknown [53,54]. According to research data, Giardia has eight distinct genetic assemblages labelled as assemblage “A” through “H” [55,56], and assemblages “A” and “B” are responsible for infection in humans.
Three classes of drugs are currently used for the treatment of giardiasis: metronidazole, mepacrine analogs, and nitrofurans, such as furazolidone (Figure 1). Metronidazole is the most widely used drug for the treatment of giardiasis globally and it is generally effective and well-tolerated. However, the United States Food and Drug Administration (FDA) has not yet approved this drug for the treatment of Giardia infection because of its toxicity and major side effects, such as seizures, ataxia, peripheral neuropathy, transient myopia, gastric mucosal irritation, sperm damage, and hematuria [14,15,16,17,18]. Tinidazole is an N1-position modified 5-nitro imidazole, which has been approved by the FDA for the treatment of giardiasis. Nitazoxanide (NTZ) belongs to an emerging class of 5-nitrothiazole compounds with potential antigiardial activity [57]. However, although NTZ is generally well tolerated, some adverse effects such as abdominal pain, diarrhea, and nausea limit the safe use for human beings. Mepacrine is no longer available in the United States, and it is being replaced in most of its applications with safer and more specific drugs. Furazolidone also has serious side effects, such as gastrointestinal disturbances, hemolytic anemia, disulfiram-like reactions to alcohol, and hypersensitivity reactions, as well as evidence of tumorigenicity in rodent studies. Furthermore, G. lamblia resistance to this drug has also been reported [58,59,60]. Therefore, research focusing on the development of novel, alternative drugs for the treatment of giardiasis is highly desirable. In view of these considerations, researchers have designed and synthesized some novel molecules for the treatment of giardiasis, their results are summarized in Table 1.
Figure 1.

Currently available antigiardial drugs.
Important Highlights of Table 1 Compounds
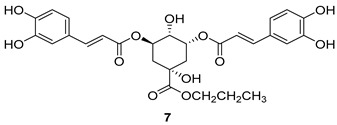
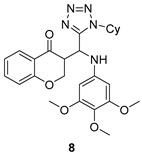
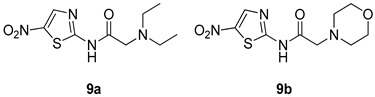
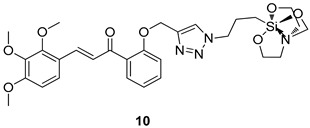
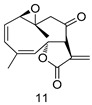
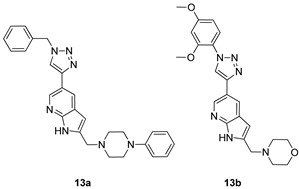
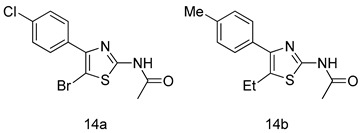
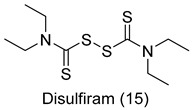
All of the novel compounds designed for giardiasis treatment are listed in Table 1 and their efficacies were compared with those of the standard drugs metronidazole (MTZ), formononetin, aminitrozole, nitazoxanide, tizoxanide, nitazoxanide and mebendazole. In brief, Zhang et al. isolated 3,5-dicaffeoylquinic acid from Artemisia argyi, and from it, developed a series of ester derivatives as potential antigiardial agents. Amongst the synthesized compounds, Compound 7 was reported as the most potent inhibitor against G. lamblia (IC50 = 4.62 µg/mL) [61]. A series of 3-tetrazolylmethyl-4H-chromen-4-ones were synthesized by Cano et al. via an Ugi-azide multicomponent reaction and evaluated for antigiardial activity and, compound 8 was found to be the most potential antigiardial agent (IC50 = 84.2 µg/mL) in the series [62]. Navarrete-Vázquez et al. [63] synthesized a series of four 5-nitrothiazole compounds and reported them as novel antigiardial agents. Among them, compounds 9a (IC50 = 0.122 µM) and 9b (IC50 = 0.151 µM) exhibited potential inhibitory activity against G. intestinalis. Singh et al. [64] designed and developed a series of chalconyl blended triazole allied silatranes; these compounds are hybrids of three pharmacologic scaffolds, namely chalcone, triazole and metal complex (silatranes). All the derivatives were evaluated for the antigiardial activity; among them compound 10 showed excellent activity against G. lamblia (IC50 = 19.58 µM). Compound 11 is a derivative of naturally occurring sesquiterpene lactone, which was isolated from Decachaeta incompta by Bautista et al. It showed greater antigiardial activity (IC50 = 30.6 µg/mL) than its parent compound [65]. Novel nitazoxanide–N-methylbenzimidazole hybrids were designed and synthesized by Soria-Arteche et al. [66], and evaluated for their in vitro biological activity. Compounds 12a–d expressed good antigiardial activity (IC50 = 0.021–0.027 µM) by inhibiting an G. intestinalis culture. Using a novel methodology based on a double Sonogashira coupling reaction in 2-amino-3,5-diiodopyridine, Leboho et al. synthesized a series of 2,3,5-trisubstituted 7-azaindoles, as well as 2,5-disubstituted 7- azaindoles. These synthesized series were evaluated against a G. duodenalis strain, and the results showed that compounds 13a (IC50 = 14.3 µg/mL) and 13b (IC50 = 8.2 µg/mL) were the most potent agents [67]. Another series of 2-amino-4-arylthiazole derivatives were prepared and evaluated by Mocelo-Castell et al. [68] as potential anti-giardial agents. The results revealed that compounds 14a (IC50 = 0.87 µM) and 14b (IC50 = 0.39 µM) were the most potent inhibitors of G. intestinalis. Disulfiram (compound 15) was proposed to inactivate G. lamblia kinase, and Castillo-Villanueva et al. hypothesized that it acts on enzymes of G. lamblia. Accordingly, compound 15 (IC50 = 6.6 µM) was efficient inactivator of immunoreceptor tyrosine-based inhibition motif (ITIM). Therefore, it is feasible that compound 15 could lead to new pharmacotherapies against G. lamblia [69]
In summary, the reported antigiardial agents could be categorized as the natural compounds and their analogues (e.g., 3,5-dicaffeoylquinic acid derivatives and 8-acyl and 8-alkyl incomptine A derivatives) and hybrids compounds with the known acitve drug such as nitazoxanide-based and benzimidazole-based hybrids. It is noteworthy that the hybrid of nitazoxanide and N-alkylbenzimidazole tethered by amide linker exhibited good activity profiles compared with nitazoxanide or albendazole, which suggests that hybridization of active compounds could provide good option for antigiardial drug discovery.
2.2. Anti-Leishmaniasis
Leishmaniasis, a parasitic disease spread by the bite of an infected female phlebotomine sand fly, has been known for many years it was first clinically described in 1756 by Alexander Russell, who named it Aleppo boil [70]. Many names correlate to this group of diseases such as kala-azar, Dum-dum fever, white leprosy, espundia, and pian bois. A vector borne disease, leishmaniasis is caused by an obligate intramacrophage protozoan, and it is characterized by its diversity and complexity [71,72]. Approximately 21 Leishmania species have been identified to be pathogenic to humans. Leishmania is one of several genera within the family Trypanosomatidae, and its species are characterized by the possession of a kinetoplast, a unique form of mitochondrial DNA. Based on species type host immune system responses, leishmaniasis has three basic clinical forms: Cutaneous (with skin ulcers), mucocutaneous (with skin, mouth, and nose ulcers), and visceral (with liver, and spleen enlargement, as well as bone marrow dysfunctions) [73,74]. Cutaneous leishmaniasis, the most common form of the disease, is caused by L. braziliensis, L. major, L. mexicana, L. tropica, and several other species [75]. It can be eventually defeated by the immune system; however, in most cases it progresses and is converted in to the mucocutaneous form, in which the parasites metastasize to the mucosal tissues. Mucosal leishmaniasis is usually caused by L. braziliensis, and it is associated with damage to the palate, nasal septum, and mucous membranes [76,77]. This form is usually refractory to therapy and can be fatal. The most dangerous form of the disease is visceral leishmaniasis, commonly called kala-azar. It can become fatal if it is not rapidly diagnosed and treated, it is responsible for most leishmaniasis-related deaths [78,79]. In visceral leishmaniasis, which is caused by L. donovani and L. infantum (synonym of L. chagasi), the parasite mainly infects the liver, spleen, and bone marrow. The infected host shows symptoms such as fever, weight loss, and anemia [80]. This disease has been recognized as an increasing health problem worldwide by the World Health Organization (WHO) [81], with high morbidity and mortality rates in Africa, Asia, and America. Among all tropical diseases, leishmaniasis is ranked fourth in morbidity and second in mortality rates [82]. Leishmaniasis is widespread, having been reported in 88 countries across all continents, with the exception of Antarctica [71,83]. It has an annual death rate of approximately 80,000 people [84], and there are two million new cases occurring annually, with 12 million people currently infected globally [83,85].
The life cycle of Leishmania sp. begins when the invertebrate host (sand fly) feeds on infected mammalian blood, thereby imbibing the amastigotes present within the macrophages. In the intestine of the insect vector, the amastigote transforms into procyclic promastigotes, and later into metacyclic promastigotes. When the insect bites a mammalian host again, the inoculated virulent promastigotes enter the blood stream and are internalized by the macrophages, where they differentiate again into amastigotes, completing the cycle [86]. No effective vaccine is available against leishmaniasis; chemotherapy is the only effective way to treat all forms of the disease [87,88,89]. However, some drugs are available for the treatment of this disease. The first-choice treatment for leishmaniasis involves the use of the pentavalent antimonial derivatives, sodium stibogluconate, which is highly toxic with serious side effects, and requires a prolonged treatment regimen [90,91]. Alternatives include paromomycin, pentamidine, miltefosine and amphotericin-B (Figure 2) However, these drugs have not found extensive use owing to their severe toxicities and difficulties associated with parenteral administration and drug resistance [19,20,21,22]. Generally, the drugs currently used for the treatment of human cutaneous and visceral leishmaniasis are toxic, and can cause severe adverse reactions such as pancreatitis, pancytopenia, reversible peripheral neuropathy, nephrotoxicity, cardiotoxicity, bone pain, and myalgia [92,93]. Therefore, the development of novel, effective, and safe antileishmanial agents with reduced side effects is a major priority for health researchers. In view of these considerations, some researchers have designed and synthesized novel molecules for the treatment of leishmaniasis; their results are summarized in Table 2.
Figure 2.

Currently available antileishmanial drugs.
Important Highlights of Table 2 Compounds
Compounds of different heterocycles classes are included in Table 2 as potent antileishmanial agents and the results of their analysis were compared with those of the standard drugs pentamidine, sodium stibogluconate (SSG), miltefosine, amphotericin-B (AmB), edelfosine and clofazimine (CFM).
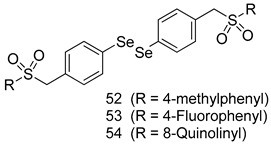
In brief, a series of thiosemicarbazide derivatives were prepared and evaluated for thier antileishmanial activities. Compounds 21a and 21b were reported as the most active candidates, with IC50 values in the range of 16.4 µM to 22.0 µM, after screening the whole series against L. amazonensis cultures using promastigotes and amastigote assays [94]. Among the compounds developed by Rashid et al., compound 22 was found to be a highly active antileishmanial agent. Basically, compound 22 is a designed hybrid compound in which two biological scaffolds, pyrazoline and pyrimidine are linked with each other. It showed excellent biological activity the highest among all theh hybrids in its series against L. major and L. donovani with IC50 values of 0.47 ± 0.02 and 1.5 ± 0.17 µg/mL, respectively [95]. Sangshetti et al. [96] synthesized 4,5,6,7-tetrahydrothieno[3,2-c]pyridine-based hydrazone derivatives and determined their antileishmanial inhibitory activities. Among the derivatives in this series, compounds 23a and 23b showed significant biological activities against L. donovani promastigotes, IC50 values of 98.75 and 93.75 µg/mL, respectively compared to that of the strandard drug SSG (IC50 = 490 µg/mL). A series of chalcones are also included in Table 2, and their activity against L. donovani cultures clearly showed that among them, the compounds containing chromane, pyridine, and a substituted 4-hydroxy phenyl ring (compounds 24a and 24b) exhibited excellent antileishmanial activities (IC50 values of 2.8 and 2.0 µM, respectively) [97]. Among a series of carboline derivatives reported by Manda et al. [98], compounds 25a (IC50 = 12.7 µM) and 25b (IC50 = 9.1 µM), which are derivatives of the commercially available tetrahydro-β-carboline prepared in a single procedure were the best candidates against L. donovani (promastigotes). Zhu et al. [99] reported compounds 26a and 26b, which are derived from arylamidamide using the reaction between amino diarylfurans and 2-pyridyl thioimidate analogs, as the most active antileishmanial agents against both intracellular L. donovani and L. amazonensis amastigotes, with IC50 values ranging from 0.13 to 0.31 µM. A series of 4-alkapolyenylpyrrolo[1,2-a]quinoxaline derivatives, including compounds 27a and 27b, which exhibited remarkable inhibitory potential against two Leishmania spp. strains namely L. major and L. donovani (IC50 values between 1.2 and 10.5 µM), were prepared and reported by Ronga et al. [100]. Of all the compounds synthesized by Bekhit et al. [101], compound 28, whose hybrid analog baasically resulted from hybridization with five-membered heterocyclici moieties, including 1,3,4-thiadiazoles and pyrazolines, exhibited the greatest potential in terms of antileishmanial activity. It was the best candidate among the series against L. aethiopica promastigotes (IC50 = 0.0142 µM) and amastigotes (IC50 = 0.13 µM). Compound 29, an acetamide derivative of (+) -dehydroabietylamine derivative reported by Dae-ayuela et al [102], was found to be the most potent leishmanicidal agent among the series (IC50 = 1.5 µM). It was even more active than the reference compound, miltefosine (IC50 = 3.4 µM). A series of 2-phenyl-3-(pyridin-4-yl)imidazo[1,2-a]pyrazine derivatives were synthesized by Marchand et al. [103], and their antileishmanial activities were evaluated. Among the synthesized molecules, compounds 30a and 30b were found to be the most potent analogs against L major promastigotea and amastigotes with IC50 values between 0.2 and 6.4 µM. Sangshetti et al. [105] synthesized a series of indolyl–coumarin hybrids, and after evaluating their antileishmanial potential in vitro reported that compounds 31a and 31b were excellent antileishmanial agents (IC50 values between 95 and 99 µg/mL) compared to the standard drug SSG (IC50 = 490 µg/mL) [104]. The sntileishmanial potentials of 1,3,6-trisubstituted β-carboline derivatives, synthesized by Lunagariya et al. were evaluated, and compound 32 (IC50 = 9.0 µM) was found to show an comparable antileishmanial activity comparable to that of the standard drug, miltefosine (IC50 = 11.9 µM). Kumar et al. [106] reported the synthesis of a new series of aryl substituted ketene dithioacetals, which were evaluated in vitro for their activity against L. donovani. Based on their results, compounds 33a and 33b were reported as the most potent antileishmanial agents, with IC50 values of 5.12 and 3.56 µM respectively. Among the 4-arylamino-6-nitroquinazolines synthesized by Saad et al. [107], compounds 34a and 34b were found to be the most potent inhibitors of L. major promastigotes (IC50 values of 1.87 and 4.37 µM, respectively) compared to the standard drug, pentamidine (IC50 = 5.09 µM). Gopinath et al. [108] developed a series of substituted quinoline analogs and assessed their antileishmanial activities. They found compound 35 to be the most active (IC50 = 0.84 µM). Among the diselenide and sulfonamide derivatives developed by Baquedano et al. [109] compounds 36a–c were found to be potent antileishmanial agents, with IC50 values of 1.40, 1.47 and 0.83 µM, respectively, against L. infantum intracellular amastigotes. An assessment of the antileishmanial potential of triazolopyridyl pyridyl ketone derivatives developed by Adam et al. [110] revealed that compounds 37a and 37b elicited potent growth inhibition against cultured Leishmania spp. promastigotes and amastiogotes with IC50 values ranging between 19.5 and 114.6 µM. Sharma et al. [111] synthesized Triazino indole–quinoline hybrid as antileishmanial agents targeting L. donovani. Their results showed that compounds 38a and 38b significantly inhibited L. donovani extracellular promastigotes and intracellular amastigotes with IC50 values ranging between 0.36 and 8.57 µM. Among the heteroretinoid-bisbenzylidine ketone hybrids developed by Tiwari et al. [112], compounds 39a–c were identified as the most potent agents against L. donovani intramacrophagic amastigotes, with IC50 values between 1.83 and 5.02 µM. Pandey et al. [113] synthesized indole-2-carboxamide derivatives using utilizing the isocyanide based multicomponent reaction and evaluated them against L. donovani. Their results showed that among them, compound 40 (IC50 = 0.6 µM) exhibited a more promising antileishmanial activity than those of standard drugs, including SSG (IC50 = 56.1 µM) and miltefosine (IC50 = 8.4 µM). Several Clofazimine analogs were synthesized by Barteselli et al. [114] and their antileishmanial activities were screened using an in vitro evaluation, which demonstrated that compound 41 was the most potent antileishmanial agent against L. infantum, and L. tropica. A series of 8 imidazole derivatives were developed by Vita et al. [115], and an in vitro analysis of their antileishmanial activities showed that out of the 8 compounds, compounds 42a and 42b were the most potent antileishmanial molecules with IC50 values of 12.7 and 8.0 µM against L. infantum respectively. Reddy et al. [116] synthesized a large series of benzyl phenyl ether derivatives and evaluated their biological activities. Their results showed that compounds 43a–e were potent antileishmanial agents and compounds 43b (IC50 = 1.6 µM), 43d (IC50 = 1.27 µM) and 43e (IC50 = 1.39 µM) showed the greater antileishmanial activity against L. donovani compared to that of the standard drug, pentamidine (IC50 = 1.84 µM). Zhang et al. [117] prepared a novel series of oxyneolignans virolin, surinamensin, and analogs using an asymmetric synthetic method. Thereafter, their ability to inhibit the growth of different protozoal strains was tested. Their results showed that compounds 44a and 44b exerted the maximum antileishmanial activities with IC50 of 2.29 µg/mL and 2.48 µg/mL, respectively. The antiprotozoal activities of novel oxadiazolyl pyrrolo triazole diones derivatives synthesized by Dürüst et al. [118] was investigated. The results showed that compounds 45a (IC50 = 1.6 µg/mL) and 45b (IC50 = 2.0 µg/mL) were the most active antileishmanial agents against L. donovani. Pierson et al. [119] synthesized a series of novel 4-arylcoumarin derivatives, and the determination of their antiprotozoal activities against various biological strains revealed that compounds 46a–c, with different substitutions on phenyl rings, displayed the most potent activity against L. donovani amastigotes (IC50 = 1.1–5.4 µM). Patric et al. [120] developed a series of bis-pyridylimidamide derivatives, and an assessment of their antileishmanial activities showed that among them, compound 47a was the most active, and inhibited the L. amazonensis strain with an IC50 value of 0.095 µM, while the others, including 47b (IC50 = 0.123 µM) and 47c (IC50 = 0.211 µM), exhibited slightly more potent activity compared with amphotericin B (IC50 = 0.124 µM). Diaryl sulfide inhibits L. infantum promastigotes, and an evaluation of the inhibitory activity of its deriatives by Saccoliti et al. [121] showed that compound 48 inhibited L. infantum promastigotes by a dose-dependent amount with an IC50 value of 29.43 µM. Preeti et al. [122] developed tellurium derivate, immunomodulatory, and demonstrated that compound 49 could eliminate L. donovani promastigotes, and an evaluation by in vitro assay showed that it had a significant growth inhibitory effect on L. donovani promastigotes with an IC50 value of 26.9 µM. Rodríguez-Hernandez et al. [123] converted hederagenin into 1,2,3-trizolyl derivatives aiming to obtain antileishmanial and cytotoxic compounds. Of the synthesized compounds, compound 50 was found to be the most potent antileishmanial molecule, with an IC50 value of 5.6 µM. The thiadazole scaffold is a prevalent heterocyclic ring with antiparasitic activity. Tahghighi et al. [124] developed its derivatives, among which compounds 51a and 51b (IC50 between 0.08=9.35 µM) were found to be the most potent antileishmanial agents inhibiting extracellular promastigotes and amastigotes.
In summary, antileishmanial agents of diverse scaffolds such as chalcone, arylamidine, thiohydrazone, and polyheteroaromatics, were reported. Som of the compounds exhibited comparable activity with pentamidine or miltefosine. In particular, 1,5-diphenylpenta-1,4-dien-3-one derivatives (39) and Ether-tether phenylamidine (43) displayed good activity profiles.
2.3. Anti-Malaria
Malaria is a deadly mosquito-borne disease that mainly affects humans. It is an infectious disease caused by protozoan parasites belonging to the genus Plasmodium. Five different species of Plasmodium, P. falciparum, P. ovale, P. malariae, P. vivax, and P. knowlesi are responsible for the spread of malaria; P. falciparum is considered the most dangerous and virulent form. The disease is transmitted via the sucking of human blood by infected female Anopheles mosquitoes [125], and symptoms include fever, fatigue, vomiting, and headache; in severe cases, it can cause yellow skin, seizures, coma, and death [126]. The symptoms usually begin 10 to 15 days after infection, and disease recurrence may be observed months later if not properly treated. In those who have recently survived an infection, reinfection usually causes milder symptoms. This partial resistance disappears over months to years if the person has no continuing exposure to malaria [126]. According to the 2014 world malaria report by the WHO, a total of 198 million cases of malaria and nearly 584,000 malaria deaths occurred in 2013 [127]. In 2013, 3.4 billion people were at risk of malaria, of whom 1.2 billion were at a higher risk with more than one case per 1000 people, especially in over 97 countries in tropical areas with the ongoing transmission of malaria. Ninety percent of the above-mentioned deaths occurred in sub-Saharan Africa, of which 77% were children under the age of five. In 2010, an estimated 660,000 malaria deaths were reported worldwide [128]. Additional reports suggest that malarial infection and mortality are more widespread than previously estimated by the report of Murray et al., with up to 200 million clinical cases and 1.2 million deaths reported in 2010 alone [129]. Because of its devastating effects on the human population, the WHO rates malaria as one of the top three infectious diseases worldwide [130]. In brief, malaria has become one of the major causes of illness in humans, with approximately 250 million clinical cases reported around the world annually, particularly in poor or developing countries [127]. P. falciparum causes most of the severe cases and the most deaths, and nearly 80% of all reported cases and as mentioned above 90% of malaria-attributed deaths occur in Africa [131]. Moreover, P. vivax is the predominant species, in Asia, the Middle East, Central and South America, and the Western Pacific [132]. Malarial parasites exhibit a complex life cycle involving an insect vector (mosquito) and a vertebrate host (human), and all species exhibit a similar life cycle with only minor variations. The infection is initiated when sporozoites are injected into the human body through the saliva of a feeding Anopheles mosquito. When sporozoites enter the human body and travel through the bloodstream to the liver they transform into exoerythrocytic forms (EEFs) [133]. Based on the Plasmodium species, these forms are converted into mature exoerythrocytic-stage schizonts, or enter a dormant phase in which they are called hypnozoites, which only two species of Plasmodiums, P. vivax and P. ovale make. These hypnozoites reactivate several weeks to months (or years) after the primary infection and are responsible for malaria relapses weeks, months, or even years after the initial infection [134]. Fully developed exoerythrocytic-stage merozoites eventually exit the liver and re-enter the bloodstream [133]. They enter the red blood cells (RBCs) and replicate asexually causing RBC destruction, which leads to the characteristic symptoms associated with malaria such as anemia, fever, and chills [135]. A small percentage of these asexual blood-stage parasites then differentiate into sexual erythrocytic stages (female and male gametocytes) whose transmission back to the mosquito vector during a subsequent blood meal completes the life cycle [136]. Important antimalarial agents are presented in Figure 3. In the past, malaria was treated with the bark of cinchona (Cinchona rubra [Rubiaceae]); however, at this time, it was not known that cinchona bark contains quinine, which was later isolated and shown to have antimalarial properties [137,138]. Some medicinal chemists developed simpler synthetic analogs of quinine such as chloroquine, amodiaquine, primaquine, and piperaquine, which all had a quinine pharmacophore, but did not have multiple stereogenic centers. Among them, chloroquine was found to be the most efficient drug, and it has served humanity for over five decades [42,139]. However, the spread of chloroquine resistance prompted medicinal chemists to re-investigate the chemistry and pharmacology of alternative 4-aminoquinoline antimalarials such as amodiaquine [132], which has proven to be effective against chloroquine-resistant parasite strains [23,24,25,26,27]. Amodiaquine is effective against many chloroquine-resistant strains of P. falciparum [126]. However, its clinical use has been severely restricted because of its associations with hepatotoxicity and agranulocytosis [28,29]. Although in recent years a natural endoperoxide artemisinin and its semisynthetic derivatives artemether, artesunate, and dihydroartemisinin have been employed for the treatment of malaria owing to chloroquine resistance in parasites, but the global deployment of artemisinin-based combination therapy is limited by its relatively high cost of treatment, safety concerins during pregnancy, and early signs of resistance in Southeast Asia [140,141,142]. Despite the market availability of large numbers of antimalarial drugs, no perfect drug is known because individual drugs and drug combinations have their own limitations including poor compliance, side effects, toxicity, and resistance. Therefore, owing to the aforementioned conditions, some researchers have designed and synthesized novel molecules for antimalarial purposes. In this review article, we have compiled the latest data on antimalarial agents designed from 2012–2017. The details of these antimalarial agents are summarized in Table 3.
Figure 3.

Currently available antimalarial drugs.
Important Highlights of Table 3 Compounds
Compounds of different heterocycle classes are included in Table 3 as potent antimalarial agents and the results of their assessment were compared to those of the standard drugs dihydroartemisinine, chloroquine, artemisinin, mefloquine, puromycin, artesunate, artemether, artesunic acid, artemisinin derived alcohol, di-artemisinin, proguanil, and quinone.
Pingaew et al. [143] synthesized 11 chalcone coumarin hybrids linked by the 1,2,3-triazole ring. All the derivatives were screened, and the evaluation of their potent antimalarial activity showed that among them, compound 62 significantly inhibited a P. falciparum culture with an IC 50 value of 1.60 µM. Nisha et al. [144] developed β-amino-alcohol tethered 4-aminoquinoline-isatin conjugates, and evaluation of their antimalarial activities revealed that compounds 63a and 63b were the most potent antimalarial agents, with IC50 values of 11.7 and 13.5 nM respectively. Kumar et al. [145] synthesized various triazole tethered isatin-ferrocene derivatives and evaluated their antimalarial activities against chloroquine-susceptible and chloroquine-resistant P. falciparum strains. The results showed that compounds 64a and 64b were potent antimalarial agents. Several aminoalkylated quercetin derivatives were synthesized by Helgren et al. [146] by using the mannich reaction were screened for antimalarial activity using an in vitro assay. The results demonstrated THAT compounds 65a and 65b were the most potent antimalarial agents against three drug-resistant malarial strains (D6 and W2), with IC50 values between 0.065 and 0.079 µM. A series of fosmidomycin analogs were developed by Phillips et al. [147] and analyzed in vitro. The results showed that of all the synthesized compounds, compound 66 was the most potent antimalarial molecule with an IC50 values of 27.4 nM against chloroquine- and mefloquine-resistant Dd2 strains of P. falciparum. Yadav et al. [148] synthesized a series of marine-derived indole alkaloid derivatives, and an evaluation of their biological activities showed that compound 67 was the most potent antimalarial agent. Le et al. [149] designed a novel series of 11-aza-artemisinin analogus and tested them to determine their ability to inhibit the growth of FcB1 strains of P. falciparum. The results showed that compounds 68a–c exerted the greatest antimalarial activities with IC50 values of 0.3, 0.7, and 1.5 µM respectively. Two dimers and two trimers of artemisinin hybrids were synthesized by Reiter et al. [150] and their antimalarial activities were investigated using an antimalarial assay against P. falciparum 3D7 strains. Of all the compounds, compound 69 was reported to be the most active antimalarial agents, with an IC50 value of 2.6 ± 0.4 nM. Parthiban et al. [151] synthesized a series of chloroquinoline-4H-chromene conjugates with piperazine and azpane rings as tethers, and the determination of their antimalarial activities against 3D7 and K1 strains of P. falciparum showed that compounds 70a and 70b displayed the most potent antimalarial activity with IC50 values between 0.29 and 1.78 µM. Bhat et al. [152] developed a series of hybrid 4-aminoquinoline 1,3,5-triazine derivatives, and an assessment their antimalarial activities showed that compounds 71a and 71b were the most active inhibiting chloroquine-sensitive (3D7) and chloroquine-resistant (RKL-2) P. falciparum strains with IC50 values between 1 and 25 µM. The results of the docking studies, revealed that the most active compounds bound well with P. falciparum dihydrofolate reductase thymidylate synthase (pf-DHFR-TS). Different oxirane compounds were synthesized by Carneiro et al. [153] and subjected to antimalarial screening against chloroquine-sensitive 3D7 strains of P. falciparum after evaluation, compounds 72a and 72b were reported to be the most active species among 18 synthesized compounds. Karad et al. [154] synthesized various analogs of morpholinoquinoline-based conjugates with an incorporated pyrazoline ring, and the evaluation of their antimalarial activities in vitro using the biological assay revealed that compounds 73a and 73b were the most active antimalarial agents with IC50 values between 0.015 and 0.018 µM. Devender et al. [155] synthesized a new series of triazoles and in vitro antimalarial activity evaluation found that among the different compounds synthesized, compounds 74a and 74b displayed excellent biological activity against 3D7 and K1 P. falciparum strains, with IC50 value between 0.3 and 2.11 µM. Svogie et al. [156] prepared a series of indolyl-3-ethanone α-thioethers, and an evaluation of their antimalarial activities revealed that compounds 75a (IC50 = 0.24 µM) and 75b (IC50 = 0.09 µM) were the most active species against 3D7 P. falciparum strains. The development of new antimalarial agents, led to the synthesis of a new series of imidazo[4,5-c]quinolin-2-one derivatives by Patel et al. [157] using a four-step synthetic route. The evaluation of an antimalarial activities of derivatives using an allamar-Blue gametocytocidal assay showed that compound 76a was the most potent candidate among the series. Seebacher et al. [158] prepared a series of several azabicyclic compounds, and an evaluation of their antimalarial activities showed that compounds 77a and 77b, with IC50 values of 0.28 and 0.095 µM, respectively, showed a remarkable antimalarial activity against 3D7 P. falciparum strains. Further analysis proved that compound 77b was the most potent P. falciparum inhibitor of the active species. A new imidazole-based series of substituted ester and carbamate derivatives were synthesized by Vita et al. [115], and an in vitro investigation revealed that compound 78 showed the highest antimalarial effect against K1 P. falciparum strains (IC50 = 0.6 µM). Inam et al. [159] designed and synthesized several acylhydrazine derivatives, attached to chloroqunoline nuclei with piperazine rings. An evaluation of their antiprotozoal activities revealed that among the compounds, compounds 79a (IC50 = 0.33) and 79b (IC50 = 0.2 µM) were moderately active against w2 strains of P. falciparum. Patrick et al. [116] developed a large series of cationic benzyl phenyl ether derivatives for application in antiprotozoal drug development. Their results showed that compounds 80a–c were extremely potent P. falciparum inhibitors, with IC50 values of 0.006, 0.004, and 0.006 µM, respectively. The screening and evaluation of the antiprotozoal activities of fifteen 8,4-oxyneolignans analogs prepared by Rye et al. [117], using asymmetric synthesis revealed that compounds 81a–c showed IC50 values between 0.608 and 2.50 µM against P. falciparum. Dürüst et al. [118] developed several derivatives of triazoles coupled with 1,2,4-oxadiazole moieties, and an investigation of their antiprotozoal activities revealed that compounds 82a and 82b were the most active P. falciparum inhibitors, with an IC50 values of 13.2 and 14.7 µg/mL, respectively. A new series of metal complexes designed and prepared by Juneja et al. [160] were evaluated for their antimalarial activity against w2 P. falciparum strains. The in vitro study revealed that compounds 83a–c were the best candidates with IC50 values between 1.1 and 1.9 µM. McKeever et al. [161] developed aminoalkyl derivatives form guanidine diaromatic minor groove binder, and evaluated their antiprotozoal activities. The results showed that compounds 84a (IC50 = 0.106 µM) and 84b (IC50 = 0.149 µM) elicited a significant P. falciparum culture inhibitory. Patrick et al. [120] synthesized thirty-six 4,4″-Diamidino-m-terphenyl analogs, which were tested for their antiprotozoal drug development. Among these 36 compounds, compounds 85a and 85b, which bear a dimethyltetrahydropyrimidinyl ring at the para-position, exhibited significant antimalarial activity with IC50 values of 0.002 and 0.003 µM, respectively, which were lower than that of their parent drug, chloroquine. From the study, it was evident that a substituent attached at position 4 of a phenyl ring would lead to the exhibition of excellent inhibitory action. Opsenica et al. [162] synthesized a novel series of aminochloroquinoline derivatives, and assessed their antimalarial activities in vivo using different P. falciparum strains. The results revealed that compounds 86a–c were notable antimalarial agents with IC50 values between 3.95 and 45.78 nM. Hanessian et al. [163] designed a novel series of pactamycin analogs, and an evaluation of their biological activities found that compounds 87a (IC50 = 3.5 nM) and 87b (IC50 = 6.7 nM) were the most potent molecules against D6 and Dd2 strains of P. falciparum. Abada et al. [164] evaluated 24 porphyrin precursors and derivatives against different protozoal strains. Their results showed that compound 88 was the most active member of the series. Its IC50 value 0.02 µM was 100 to 200 times lower than that of standard drug chloroquine (15–25 µM). Yeo S J et al. [165] synthesized chloroquine derivatives with phenylmethyl groups and unsaturated amides, which have anti-malarial activity. Among them, compounds 89a and 89b showed greater antimalarial activity against 3D7 P. falciparum strains with IC50 values of 0.17 and 0.23 µM, respectively. Singh et al. [166] synthesized 4-aminoquinolin-ferrocenyl-chalcone derivatives, and the evaluation of their pharmacological properties revealed that compounds 90a–c were the most potent compounds against W2 strain of P. falciparum; their IC50 values ranged from 0.37 to 0.53 µM.
In summary, various chemical scaffolds and its analogues such as triazole-tethered chalcone-coumarin hybrids, chloroquinoline-isatin hybrids, flavonones, indolesulfonamides, artemisinin derivatives, N,N′-diaryl substituted piperizines, porphyrin derivatives, and polyaryls present significant antimalarial activities. In particular, artemisinin derivatives (68) in which lactone was transformed to lactam, then various hydrophilic substituents were introduced, showed good inhibitor activity. As novel scaffolds, symmetric terphenyl cyclic amidines (85) exhibited slightly better antimalarial activity than chloroquine and artemisinin.
2.4. Anti-Trichomoniasis
Trichomoniasis is a protozoan infection caused by the flagellate protozoan T. vaginalis. It is one of the most prevalent nonviral sexually transmitted diseases worldwide [167]. The protozoan T. vaginalis affects both men and women. In women, the symptoms of this infection worsen during menstruation whereas in men the infection is largely asymptomatic; these asymptomatic men are considered carriers [168]. Trichomoniasis in men and women is associated with birth outcomes [169], infertility [170], cervical and prostate cancers [171], and pelvic inflammatory disease [172]. In men, this disease is characterized by irritation inside the penis, mild discharge, or slight burning after urination or ejaculation and in women, it is associated with yellow-green vaginal discharge with a strong odor. The infection may also cause discomfort during sex and urination, as well as irritation and itching of the female genital area. In rare cases, lower abdominal pain can occur. The symptoms usually appear in women within 5–28 days of exposure. Generally, the prevalence of T. vaginalis infection is found to be higher among women than men. A recent report on trichomoniasis revealed that the prevalence of trichomoniasis in non-human immunodeficiency virus (HIV)-infected persons was 10.1% among women vs. 2.0% among men [173], when compared to HIV-infected women (10%–20% prevalence of trichomoniasis) [174]. Data regarding the prevalence of T. vaginalis in men who have sex with men (MSM) are scarce. T. vaginalis infection damages the vaginal epithelium, which increases the risk of women being infected by HIV, and thereby considerably increasing the chances of infected women transmitting HIV to her sexual partner(s) [175,176]. In short, T. vaginalis is a co-factor in HIV transmission and acquisition [177,178]. According to a WHO report, approximately 248 million new cases of trichomoniasis are reported worldwide annually. It is believed that two to three million symptomatic infections occur annually among sexually active women in the United States [179]. In the Republic of Korea, 10.4% of women complaining of vaginal symptoms and signs were found to be infected with T. vaginalis [180].
According to the literature the pathogenicity of T. vaginalis is due to cysteine peptidases (CP) enzymes [181,182] which may present on their cell surfaces as secretion products of the parasite [183,184,185,186,187]. These enzymes play a critical role in pathogenicity, as well as in the biological processes of this protozoan. Although, MTZ, which is approved by the FDA is the drug of choice for trichomoniasis, [188] recent studies have shown that this drug has several toxic effects [14,15,16,17,18], and the clinical resistance of many microbes has reduced its efficiency [189,190]. In view of these major drawbacks, there is an urgent need for the development of new and efficient scaffolds against trichomoniasis. Therefore, researchers have designed and synthesized some novel agents as inhibitors of T. vaginalis growth. In this review article, we have compiled the latest data (from 2012–2017) on the development of novel antitrichomonial agents. The results are summarized in Table 4.
Important Highlights of Table 4 Compounds
Compounds of different class heterocycles are included in Table 4 as potent agents against trichomoniasis and the results of their assessments were compared with those of the standard drugs nitazoxanide, tizoxanide, MTZ, albendazole, and nonoxynol.
In brief, Navarrete-Vázquez et al. [63] synthesized a novel series eight of nitrothiazole and benzothiazole derivatives and among them, compounds 91a and 91b showed effective T. vaginalis cell growth inhibition, with IC50 values of 0.331 and 0.221 µM, respectively. During structure-activity relationship studies of adenosine and uridine analogs against T. vaginalis, Shokar et al. [191] reported that compound 92 (IC50 = 0.09 µM) was the most potent candidate for trichomoniasis treatment. Interestingly, it has also been approved by the US FDA as a potential drug candidate for trichomoniasis treatment. In a one-step reaction of N-substituted β-lactams with a free azide group and 5- substituted isatins containing a terminal alkyne, Raj et al. synthesized a series of β-lactam-isatin-triazole conjugates, and one of them, compounds 93, was found to be capable of selectively inhibiting T. vaginalis growth with an IC50 value of 7.06 µM [192]. A novel series of metronidazole-chalcone conjugates were designed and developed by Anthwal et al. [193], and their antitrichomonal activities against a T. vaginalis culture were evaluated for in vitro. The results revealed that two of the compounds in the series, compounds 94a and 94b were the most effective candidates against both MTZ-susceptible and MTZ-resistant parasite strains. Soria-Arteche et al. [66] synthesized a series of hybrid compounds bearing nitazoxanide and N-methylbenzimidazole moieties using different reagents, and the evaluation of their antiprotozoal activities revealed that among the 13 synthesized molecules, compound 95 (IC50 = 0.023 µM) displayed greater antitrichomonal activity against T. vaginalis compared to those of the other 12 compounds. Several hybrid analogs bearing tetrazole and chromane as bioactive scaffolds were synthesized by Cano et al. [62] using the one pot Ugi-azide multicomponent reaction in the presence of InCl3 as catalyst. Thereafter, they were screened, and their antiprotozoal activities were evaluated and among them, compound 96 was identified as the most potent antitrichomonal agent against T. vaginalis with an IC50 value of 83.9 µM. Nisha et al. [194] prepared a series of N-propargylated-isatin Mannich derivatives as potential antitrichomonal agents, using the one-pot CuCl-catalyzed Mannich-type reaction. An evaluation of their activities against T. foetus revealed that three of the synthesized compounds 97a–c elicited promising inhibitory activity on T. foteus culture growth, with IC50 values between 11.3 and 24.5 µM. In order to develop novel and effective antitrichomonal agents, a series of mono- and bis-uracil-isatin conjugates were prepared using single-step synthetic procedure. After they were evaluated against a T. vaginalis culture. Kumar et.al [195] found that compounds 98a (IC50 = 9.86 µM) and 98b (IC50 = 9.79 µM) were the best candidate. Using a single-step CuCl-catalyzed Mannich-type reaction, Nisha et al. [196] synthesized a large series of Mannish-based compounds in which isatin and 4-aminoquinoline ring are linked to the piperazine nucleus. An evaluation of their antitrichomonal activities revealed that compound 99 was the most potent agent against T. vaginalis with an IC50 value of 23 µM. A series of hybrid conjugates with incorporated β-lactone, triazole, and isatin nuclei were designed and prepared by Raj et al. [197] as as novel T. vaginalis inhibitors. The evaluation of the antitrichomonal activities of these compounds in-vitro demonstrated that compounds 100a and 100b were the most active agents inhibiting the growth of a T. vaginalis culture. Saleh et al. [198] synthesized a new series of hybrid compounds bearing 5-nitrothiazole moiety, and an in vitro assessment of their antiprotozoal activities, and consequently compound 101 displayed the most promising biological activity against T. vaginalis strains, with an IC50 value of 4.3 µg/mL. In a study aimed at developing of antiparasitic agents, Adams et al. [199] prepared thiosemicarbazone-derived ruthenium metal complexes, and after evaluating their inhibitory properties against the in vitro growth of a G3 T. vaginalis strain, they found that the compounds 102a (IC50 = 5.47 µM) and 102b (IC50 = 7.56 µM) displayed the most potent antitrichomonal activities. A novel combinatorial library of β-amino alcohol-based β-lactam–isatin chimeras were designed and developed by Nisha et al. [201], and an evaluation of their potential against T. vaginalis in vitro demonstrated that among the synthesized compounds, compound 103 possessed significant antitrichomonal activity, with an IC50 value of 9.73 µM. Stringer et al. [201] prepared a series of rhodium metal complexes and analyzed them for their antiprotozoal activity. The results showed that compound 104 displayed an IC50 value of 4.80 µM against a T. vaginalis culture. A novel series of coumarin-glyoxal hybrid compounds were synthesized by Gupta et al. [202], and their inhibitory activities against T. vaginalis, as well as their spermicidal activities were evaluated. The results led to the conclusion that compounds 105a–c displayed the most potent trichomonacidal activity.
2.5. Anti-Trypanosomiasis
Parasitic infections caused by trypanosomatids constitute a major health problem in countries where poor sanitary conditions are prevalent. Diseases acquired by such infections are considered ‘neglected’ because they receive limited funding for the research and development of new treatments. Amongst the neglected tropical diseases (NTD), human African trypanosomiasis (HAT) is endemic throughout sub-Saharan Africa, while American trypanosomiasis (Chagas disease) affects populations in South and Central America.
2.5.1. HAT/African Sleeping Sickness
HAT, also known as African sleeping sickness, is one of the most neglected diseases in regions of sub-Saharan Africa; it affects 70 million people in 36 countries. HAT is a vector-borne disease caused by the protozoa parasite Trypanosoma brucei [203,204,205]. It is transmitted through the bite of an infected tsetse fly or passed from an infected mother to her child through the placenta. This disease has two stages: the first stage involves parasite-included seizures of the hemolymphatic system, and the second involves the transmission of the parasites into the central nervous system (CNS) across the blood–brain barrier [206]. Infection of the CNS leads to a number of symptoms including mental impairment, severe headaches, fever, chronic encephalopathy, and eventual death. The development of effective vaccines would be an option for preventing this deadly disease; however, trypanosomes can evade the host immune’s system because of the high degree of antigenic variation in glycoproteins forming their surface coat [207,208,209,210,211,212]. Therefore, chemotherapy remains the only viable strategy for the treatment and control of infection. The current chemotherapy for HAT comprises only four drugs. Three of these drugs, suramin, pentamidine, and melarsoprol (Figure 4), were developed over 60 years ago, and exhibit severe side effects. Melarsoprol is an arsenical derivative used for the treatment of HAT in the neurological stage, and they have many undesirable or fatal (3%–10%) side effects in addition to the development of drug resistance, exhibiting a drug failure rate of up to 30%. Pentamidine and suramin are used for treatment in the early stage of the disease, before the involvement of the CNS. Nevertheless, they have many severe side effects such as low blood pressure, decreased level of consciousness, kidney problems, low blood cell levels, and wheezing [30]. Moreover, eflornithine, which is less toxic, is only effective against T. brucei gambiense subspecies [213]. Treatment with a combination of nifurtimox and eflornithine is less toxic, but ineffective against the T. brucei rhodesiense subspecies.
Figure 4.

Currently available antitrypanosomal drugs (use in medical practice).
2.5.2. Chagas Disease
American trypanosomiasis (Chagas disease) is caused by the flagellate parasitic protozoan T. cruzi. This infection was first discovered by the Brazilian physician Carlos Chagas (1879−1934) in 1909. While Trypanosoma cruzi is transmitted to animals and humans via insect vectors of the Triatominae, a subfamily of the Reduviidae family of insects, commonly known as “kissing bugs” [214], outbreaks of chagas disease are usually caused by foodborne T. cruzi since it produces a very aggressive acute form which is often missed by clinicians. This infection is one of the most threatening diseases in Central and South America. A large number of people, approximately 18 million annually, are infected with this parasite. Among them, 50,000 died owing to heart failure. This disease has become a major public health problem in 22 developing countries in Latin America, where more than eight million people suffer from this infection annually [215]. As depicted in its life cycle, the infection is transmitted by two predominant modes: The first is vectorial, through the infected feces/urine of triatomine bugs, and the second is by blood transfusion: the metacyclic trypomastigotes are released in the feces of the insect vector as it takes a blood meal, and they enter the bloodstream via a bite wound or mucosal membrane. Once inside the host, the metacyclic trypomastigotes invade nearby cells and differentiate into their intracellular amastigote form, which multiplies by binary fission. The transformation into trypomastigotes occurs prior to the release from the cells back into the bloodstream, proliferating the infection cycle. Other forms of transmission include congenital, blood transfusion, and contaminated food, to a lesser extent. This disease is endemic to Latin America, affecting people from Mexico to Argentina [216]. The origin and development of this disease is divided into three phases: The (i) acute (short), (ii) latent (long lasting), and (iii) chronic (very serious condition) phases. In the chronic phase, symptoms such as cardiomyopathy and malformation of the intestines (e.g., megaesophagus and megacolon) have been reported. The administration of the antiparasitic drugs benznidazole and nifurtimox is 100% effective in the short acute phase, and both drugs act through the generation of free radicals that kill the parasite. If no treatment occurs at this stage, then this infection silently progresses into the chronic phase in which internal organs such as the heart, peripheral nervous system, esophagus, and colon are irreversibly affected. Benznidazole and nifurtimox are no longer efficient in the chronic phase, and the health of the patients deteriorates rapidly leading to death, usually due to heart failure. Therefore, the development of new antichagasic drugs is of the utmost importance and urgency [216]. In Table 5, we compiled the latest data (2012–2017) on drug development against trypanosomiasis.
Important Highlights of Table 5 Compounds
Compounds of different class heterocycles are included in Table 5 as potent agents against Trypanosoma species and the results of their assessments were compared to those of standard drugs Tryparsamide, melarsoprol, difluoromethylornitithine, pentamidine, diminazene, suramin, benznidazole and nifurtimox. Among the substituted benzothiophene derivatives developed by Bhambra et al. [217], compound 14a and 114b were found to be the most active candidates for HAT treatment with IC50 values of 0.60 and 0.53 µM, respectively. In another series of imidazole compounds synthesized by Trunz et al. [218], compounds 115a and 115b demonstrated good potency against T.b.rhodesiense and their reported IC50 values were 0.16 and 0.10 µM, respectively. Bouchikhi et al. [219] designed a series of compounds based on glycosyl-isoindigo conjugates, and an evaluation of their antitrypanosomal activities revealed that among the compounds synthesized, compounds 116a–c expressed significant biological activity against T. b. brucei strains with IC50 values between 0.51 and 0.84 µM. Among the several halonitrobenzamides derivatives synthesized by Hwang et al. [220], and evaluated against a T. b. brucei culture compounds, compound 117 was found to be the most potent inhibitor (IC50 = 1.5 µM). An assessment of the antiprotozoal activities (antitrypanosomal) of a series of nitroimidazole analogs developed by Samant et al. [221] revealed that among the synthesized molecules, compound 118 was the most active antitrypanosomal agent (IC50 = 0.25 µM). Ferrins et al. [222] synthesized various anilides and the evaluation of their antitrypanosomal activity revealed that among the compounds synthesized, compound 119 (IC50 = 0.091 µM) significantly inhibited a T. b. rhodesiense culture. A large series of convolutamine analogs were prepared by Pham et al. [223]. Basically, convolutamine, which is a natural product, is a highly effective antitrypanosomal. All the synthesized convolutamine derivatives were screened against T. b. brucei culture, and compounds 120a and 120b were reported as the most potent inhibitors with IC50 values of 0.7 µM and 0.5 µM, respectively. Samant et al. [224] synthesized a big library of naphthoquinone derivatives, and the evaluation of their antiprypanosomal activities in vitro showed that compounds 121a (IC50 = 0.07 µM) and 121b (IC50 = 0.05 µM) were the most potent compounds in the series. Papadopoulou et al. [225] synthesized a series of azole-based compounds. An assessment of their antitrypanosomal potentials revealed that compounds 122a and 122b were the most active agents with IC50 values against T. b. rhodesiense and T. cruzi vetween 0.187 and 0.373 µM. Papadopoulou et al. [226] also designed and developed nitrotriazole-based amide derivatives; an investigation of their antitrypanosomal activities against T. cruzi revealed that among them compound 123 was the most potent agent against T. cruzi (IC50 = 0.008 µM). A series of heterocyclic spiro compounds synthesized by Zelisko et al. [227] were evaluated for their antitrypanosomal activities. The results showed that compound 124 was the most active molecule (IC50 = 0.2624 µM) against T. b. rhodesiense. An in vitro assessment of the inhibitory effect of a novel series of β-carboline analogs prepared by Manda et al. [98] against T. b. brucei revealed that compound 125 was the most potent inhibitory agent among the compounds (IC50 = 1.01 µM). Alves et al. [228] developed several semicarbazone derivatives, and evaluated their antiprotozoal activities in vitro by testing their potential to inhibit biological strains. Compound 126 (IC50 = 8.5 µM) appeared to be the most potent T. cruzi culture inhibitor, with an IC50 values of 8.5 µM. Several azabicyclo nonane type derivatives synthesized by Seebacher et al. [158] were screened for their anti-protozoal activity. The results showed that among them, compounds 127a (IC50 = 0.061 µM) and 127b (IC50 = 0.065 µM) exhibited the greatest activity profiles. Upadhayaya et al. [229] developed a series of quinolone- and indenoquinoline-based heterocycles, and an evaluation of their antiprotozoal potentials revealed that compound 128 was a potential candidate against T. cruzi and T. b. rhodesiense with IC50 values of 0.25 and 1.81 µM, respectively. Vita et al. [115] synthesized series of potent and effective imidazole incorporated phenylethanol derivatives, of which compound 129 exhibited the highest antitrypanosomal activity (IC50 = 0.04 µM), and showed more potency against T. cruzi strains. Martínez et al. [230] reported the synthesis of several bisguanidine T. b. rhodesiense strain inhibitors among which compound 130 (IC50 = 0.009 µM) was identified as the most active agent for trypanosomiasis treatment. Patrick et al. [116] developed a new series of benzyl phenyl ether diamidine derivatives, and investigated their potential against a T. b. rhodesiense culture. Compound 131, which exhibited a good therapeutic potential (IC50 = 0.003 µM), was identified as the best candidate. Dürüst et al. [118] synthesized a new series of 1,2,4-oxadiazole-linked triazole derivatives using the 1,3-dipolar cycloaddition reaction, and their antiprotozoal activities were analyzed. Of these compounds, compound 132 was identified as the most potent antitrypanosomal agents (IC50 = 7.0 µM). As already reported, Mckeever et al. [161] developed a series ofguanidine diaromatic minor grove binder aminoalkyl derivatives. They did not only evaluate the antimalarial activities of these compounds, they also evaluated their antitrypnosomal properties and found that compounds 133a (IC50 = 13.1 µM) and 133b (IC50 = 20.2 µM) were the most potent candidates against T. b. rhodesiense strains. A series of 1,3-dipyridylbenzene derivatives were evaluated for their antitrypanosomal activity. The results showed that compounds 134a and 134b were the best candidates against T. b. rhodesiense and T. cruzi strains [120]. Sola et al. [231] synthesized a novel series of huprine Y dimer derivatives, and assessment of their antiprotozoal activities using a bioassay showed that compounds 135a (IC50 = 0.52 µM) and 135b (IC50 = 0.57 µM) were the most significant T. brucei culture inhibitors.
3. Conclusions
Infections caused by protozoan parasites such as giardiasis, leishmaniasis, malaria, trichomoniasis, and trypanosomiasis are responsible for considerable morbidity and mortality worldwide, with devastating social and economic consequences. The currently available drugs for the treatment of and protection against protozoan parasites were discovered over 50 years ago, and a number of factors limit their utility, such as high cost, poor compliance, drug resistance, low efficacy, and safety concerns. Therefore, the development of new and more effective drugs with fewer side effects presents a crucial challenge. Currently, research focused on the developing new drugs to protect against and treat protozoans are increasing steadily. In this review article, we have presented some of the developments in this field, with the aim of showing the recent significant advances in the discovery of new antiprotozoal drugs.
Abbreviations
| HAT | Human African trypanosomiasis |
| MTZ | Metronidazole |
| ML | Mucosal leishmaniasis |
| SSG | Sodium gluconate |
| AmB | Amphotericin-B |
| CFM | Clofazimine |
| EEF | Exoerythrocytic form |
| RBC | Red blood cell |
| pf-DHFR-TS | P.falciparum dihydrofolate reductase thymidylate synthase |
| CP | Cysteine peptidase |
| NTD | Neglected tropical disease |
| CNS | Central nervous system |
Author Contributions
Conceptualization, F.H., D.S., Literature search, S.-M.L., M.-S.K., F.H., Writing-original draft preparation, S.-M.L., M.-S.K., F.H., Writing-Review & Editing, D.S.
Funding
This work was supported by grants from the Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (Grant No. NRF-2015R1D1A1A01056620, NRF-2014M3C1A3054139, and NRF-2018M3A9C8024360).
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Nash T.E. Parasitic Diseases that Cause Seizures. Epilepsy Curr. 2014;14(Suppl. 1):29–34. doi: 10.5698/1535-7511-14.s2.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Renslo A.R., McKerrow J.H. Drug discovery and development for neglected parasitic diseases. Nat. Chem. Biol. 2006;2:701–710. doi: 10.1038/nchembio837. [DOI] [PubMed] [Google Scholar]
- 3.Pozio E. World distribution of Trichinella spp. Infections in animals and humans. Vet. Parasitol. 2007;149:3–21. doi: 10.1016/j.vetpar.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 4.Garcia H.H., Gonzalez A.E., Gilman R.H. Diagnosis, treatment and control of Taenia solium cysticercosis. Curr. Opin. Infect. Dis. 2003;16:411–419. doi: 10.1097/00001432-200310000-00007. [DOI] [PubMed] [Google Scholar]
- 5.Fennell B. Microtubules as antiparasitic drug targets. Expert Opin. Drug Discov. 2008;3:501–518. doi: 10.1517/17460441.3.5.501. [DOI] [PubMed] [Google Scholar]
- 6.Pink R. Opportunities and challenges in antiparasitic drug discovery. Nat. Rev. Drug Discov. 2005;4:727–740. doi: 10.1038/nrd1824. [DOI] [PubMed] [Google Scholar]
- 7.Tekwani B.L., Walker L.A. 8-Aminoquinolines: Future role as antiprotozoal drugs. Curr. Opin. Infect. Dis. 2006;19:623–631. doi: 10.1097/QCO.0b013e328010b848. [DOI] [PubMed] [Google Scholar]
- 8.Watkins B.M. Drugs for the control of parasitic diseases: Current status and development. Trends Parasitol. 2003;19:477–478. doi: 10.1016/j.pt.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 9.Woodhall D. Neglected parasitic infections: What every family physician needs to know. Am. Fam. Physician. 2014;89:803–811. [PubMed] [Google Scholar]
- 10.Mathers C. The Global Burden of Disease: 2004 Update. World Health Organization; Geneva, Switzerland: 2008. [Google Scholar]
- 11.Hotez P.J. Control of neglected tropical diseases. N. Engl. J. Med. 2007;357:1018–1027. doi: 10.1056/NEJMra064142. [DOI] [PubMed] [Google Scholar]
- 12.Utzinger J. Neglected tropical diseases: Diagnosis, clinical management, treatment and control. Swiss Med. Wkly. 2012;142:w13727. doi: 10.4414/smw.2012.13727. [DOI] [PubMed] [Google Scholar]
- 13.Secor W.E. Neglected parasitic infections in the United States: Trichomoniasis. Am. J. Trop. Med. Hyg. 2014;90:800–804. doi: 10.4269/ajtmh.13-0723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agarwal A.S. Targeting Amoebiasis: Status and Developments. Curr. Bioact. Compd. 2007;3:121–133. [Google Scholar]
- 15.Ordaz-Pichardo C., Shibayama M., Villa-Treviño S., Arriaga-Alba M., Angeles E., de la Garza M. Antiamoebic and toxicity studies of a carbamic acid derivative and its therapeutic effect in a hamster model of hepatic amoebiasis. Antimicrob. Agents Chemother. 2005;49:1160–1168. doi: 10.1128/AAC.49.3.1160-1168.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.El-Nahas A.F., el-Ashmawy I.M. Reproductive and cytogenetic toxicity of metronidazole in male mice. Basic Clin. Pharmacol. Toxicol. 2004;94:226–231. doi: 10.1111/j.1742-7843.2004.pto940505.x. [DOI] [PubMed] [Google Scholar]
- 17.Purohit V., Basu A.K. Mutagenicity of nitroaromatic compounds. Chem. Res. Toxicol. 2000;13:673–692. doi: 10.1021/tx000002x. [DOI] [PubMed] [Google Scholar]
- 18.Evans J., Levesque D., Knowles K., Longshore R., Plummer S. Diazepam as a treatment for metronidazole toxicosis in dogs: A retrospective study of 21 cases. J. Vet. Intern. Med. 2003;17:304–310. doi: 10.1111/j.1939-1676.2003.tb02452.x. [DOI] [PubMed] [Google Scholar]
- 19.Jha T.K. Drug unresponsiveness & combination therapy for kala-azar. Indian J. Med. Res. 2006;123:389–398. [PubMed] [Google Scholar]
- 20.Rudrapaul P., Sarma I.S., Das N., De U.C., Bhattacharjee S., Dinda B. New flavonol methyl ether from the leaves of Vitex peduncularis exhibitspotential inhibitory activity against Leishmania donovani through activation of iNOS expression. Eur. J. Med. Chem. 2014;87:328–335. doi: 10.1016/j.ejmech.2014.09.076. [DOI] [PubMed] [Google Scholar]
- 21.Guimarães T.T., Pinto Mdo C., Lanza J.S., Melo M.N., do Monte-Neto R.L., de Melo I.M., Diogo E.B., Ferreira V.F., Camara C.A., Valença W.O., et al. Potent naphthoquinones against antimony-sensitive and -resistant Leishmania parasites: Synthesis of novel α-and nor-α-lapachone-based 1,2,3-triazoles by copper-catalyzed azide–alkyne cycloaddition. Eur. J. Med. Chem. 2013;63:523–530. doi: 10.1016/j.ejmech.2013.02.038. [DOI] [PubMed] [Google Scholar]
- 22.Oh S., Kim S., Kong S., Yang G., Lee N., Han D., Goo J., Siqueira-Neto J.L., Freitas-Junior L.H., Song R. Synthesis and biological evaluation of 2,3-dihydroimidazo[1,2-a]benzimidazole derivatives against Leishmania donovani and Trypanosoma cruzi. Eur. J. Med. Chem. 2014;84:395–403. doi: 10.1016/j.ejmech.2014.07.038. [DOI] [PubMed] [Google Scholar]
- 23.World Health Organization The World Health Report 1997—Conquering suffering, enriching humanity. World Health Forum. 1997;18:248–260. [PubMed] [Google Scholar]
- 24.Watkins W.M., Sixsmith D.G., Spencer H.C., Boriga D.A., Kariuki D.M., Kipingor T., Koech D.K. Effectiveness of amodiaquine as treatment for chloroquine-resistant Plasmodium falciparum infections in Kenya. Lancet. 1984;1:357–359. doi: 10.1016/S0140-6736(84)90410-0. [DOI] [PubMed] [Google Scholar]
- 25.White N.J. Can amodiaquine be resurrected? Lancet. 1996;348:1184–1185. doi: 10.1016/S0140-6736(05)65475-X. [DOI] [PubMed] [Google Scholar]
- 26.Olliaro P., Nevill C., LeBras J., Ringwald P., Mussano P., Garner P., Brasseur P. Systematic review of amodiaquine treatment in uncomplicated malaria. Lancet. 1996;348:1196–1201. doi: 10.1016/S0140-6736(96)06217-4. [DOI] [PubMed] [Google Scholar]
- 27.O’Neill P.M., Mukhtar A., Stocks P.A., Randle L.E., Hindley S., Ward S.A., Storr R.C., Bickley J.F., O’Neil I.A., Maggs J.L., et al. Isoquine and related amodiaquine analogues: A new generation of improved 4-aminoquinoline antimalarials. J. Med. Chem. 2003;46:4933–4945. doi: 10.1021/jm030796n. [DOI] [PubMed] [Google Scholar]
- 28.Neftel K.A., Woodtly W., Schmid M., Frick P.G., Fehr J. Amodiaquine induced agranulocytosis and liver damage. Br. Med. J. (Clin. Res. Ed.) 1986;292:721–723. doi: 10.1136/bmj.292.6522.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lind D.E., Levi J.A., Vincent P.C. Amodiaquine-induced agranulocytosis: Toxic effect of amodiaquine in bone marrow cultures in vitro. Br. Med. J. 1973;1:458–460. doi: 10.1136/bmj.1.5851.458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zuckerman J. Principles and Practice of Travel Medicine. J. Travel Med. 2003;10:313–314. doi: 10.2310/7060.2003.2585. [DOI] [Google Scholar]
- 31.Gelb M.H., Hol W.G. Drugs to combat tropical protozoan parasites. Science. 2002;297:343–344. doi: 10.1126/science.1073126. [DOI] [PubMed] [Google Scholar]
- 32.Njogu P.M., Guantai E.M., Pavadai E., Chibale K. Computer-Aided Drug Discovery Approaches against the Tropical Infectious Diseases Malaria, Tuberculosis, Trypanosomiasis, and Leishmaniasis. ACS Infect. Dis. 2016;2:8–31. doi: 10.1021/acsinfecdis.5b00093. [DOI] [PubMed] [Google Scholar]
- 33.Torresa F.A.E., Passalacquaa T.G., Velásqueza A.M.A., de Souzab R.A., Colepicolod P., Graminha M.A.S. New drugs with antiprotozoal activity from marine algae: A review. Rev. Bras. Farmacogn. 2014;24:265–276. doi: 10.1016/j.bjp.2014.07.001. [DOI] [Google Scholar]
- 34.Andrews K.T., Fisher G., Skinner-Adams T.S. Drug repurposing and human parasitic protozoan diseases. Int. J. Parasitol. Drugs Drug Resist. 2014;4:95–111. doi: 10.1016/j.ijpddr.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sangshetti J.N., Kalam Khan F.A., Kulkarni A.A., Arote R., Patil R.H. Antileishmanial Drug Discovery: Comprehensive Review of the Last 10 Years. RSC Adv. 2015;5:32376–32415. doi: 10.1039/C5RA02669E. [DOI] [Google Scholar]
- 36.Nagle A.S., Khare S., Kumar A.B., Supek F., Buchynskyy A., Mathison C.J., Chennamaneni N.K., Pendem N., Buckner F.S., Gelb M.H., et al. Recent Developments in Drug Discovery for Leishmaniasis and Human African Trypanosomiasis. Chem. Rev. 2014;114:11305–11347. doi: 10.1021/cr500365f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Biamonte M.A., Wanner J., Le Roch K.G. Recent advances in malaria drug discovery. Bioorg. Med. Chem. Lett. 2013;23:2829–2843. doi: 10.1016/j.bmcl.2013.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen C. Development of antimalarial drugs and their application in China: A historical review. Infect. Dis. Poverty. 2014;3:9. doi: 10.1186/2049-9957-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lawal B., Shittu O.K., Kabiru A.Y., Jigam A.A., Umar M.B., Berinyuy E.B., Alozieuwa B.U. Potential antimalarials from African natural products: A reviw. J. Intercult. Ethnopharmacol. 2015;4:318–343. doi: 10.5455/jice.20150928102856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barnett D.S., Guy R.K. Antimalarials in Development in 2014. Chem. Rev. 2014;114:11221–11241. doi: 10.1021/cr500543f. [DOI] [PubMed] [Google Scholar]
- 41.Sinha S., Singh A., Medhi B., Sehgal R. Systematic Review: Insight into Antimalarial Peptide. Int. J. Pept. Res. Ther. 2016;2:325–340. doi: 10.1007/s10989-016-9512-1. [DOI] [Google Scholar]
- 42.Mushtaque M. Reemergence of chloroquine (CQ) analogs as multi-targeting antimalarial agents: A review. Eur. J. Med. Chem. 2015;90:280–295. doi: 10.1016/j.ejmech.2014.11.022. [DOI] [PubMed] [Google Scholar]
- 43.Vandekerckhove S., D’Hooghe M. Quinoline-based antimalarial hybrid compounds. Bioorg. Med. Chem. 2015;23:5098–5119. doi: 10.1016/j.bmc.2014.12.018. [DOI] [PubMed] [Google Scholar]
- 44.Kumar A., Paliwal D., Saini D., Thakur A., Aggarwal S., Kaushik D. A comprehensive review on synthetic approach for antimalarial agents. Eur. J. Med. Chem. 2014;85:147–178. doi: 10.1016/j.ejmech.2014.07.084. [DOI] [PubMed] [Google Scholar]
- 45.Mishra M., Mishra V.K., Kashaw V., Iyer A.K., Kashaw S.K. Comprehensive review on various strategies for antimalarial drug discovery. Eur. J. Med. Chem. 2017;125:1300–1320. doi: 10.1016/j.ejmech.2016.11.025. [DOI] [PubMed] [Google Scholar]
- 46.Jones A.J., Grkovic T., Sykes M.L., Avery V.M. Trypanocidal activity of marine natural products. Mar. Drugs. 2013;11:4058–4082. doi: 10.3390/md11104058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.de Brum Vieira P., Giordani R.B., Macedo A.J., Tasca T. Natural and synthetic compound anti-Trichomonas vaginalis: An update review. Parasitol. Res. 2015;114:1249–1261. doi: 10.1007/s00436-015-4340-3. [DOI] [PubMed] [Google Scholar]
- 48.Eckmann L., Gillin F.D. Microbes and microbial toxins: Paradigms for microbial-mucosal interactions I. Pathophysiological aspects of enteric infections with the lumen-dwelling protozoan pathogen Giardia lamblia. Am. J. Physiol. Gastrointest. Liver Physiol. 2001;280:G1–G6. doi: 10.1152/ajpgi.2001.280.1.G1. [DOI] [PubMed] [Google Scholar]
- 49.Tripathi D.M., Gupta N., Lakshmi V., Saxena K.C., Agrawal A.K. Antigiardial and immunostimulatory effect of Piper longum on giardiasis due to Giardia lamblia. Phytother. Res. 1999;13:561–565. doi: 10.1002/(SICI)1099-1573(199911)13:7<561::AID-PTR479>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 50.Upcroft P., Upcroft J.A. Drug targets and mechanisms of resistance in the anaerobic protozoa. Clin. Microbiol. Rev. 2001;14:150–164. doi: 10.1128/CMR.14.1.150-164.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kfir R., Hilner C., du Preez M., Bateman B. Studies on the prevalence of Giardia cysts and Cryptosporidium oocysts in South African water. Water Sci. Technol. 1995;31:435–438. doi: 10.2166/wst.1995.0654. [DOI] [Google Scholar]
- 52.Nash T.E. Surface antigenic variation in Giardia lamblia. Mol. Microbiol. 2002;45:585–590. doi: 10.1046/j.1365-2958.2002.03029.x. [DOI] [PubMed] [Google Scholar]
- 53.Halliez M.C., Buret A.G. Extra-intestinal and long term consequences of Giardia duodenalis infections. World J. Gastroenterol. 2013;19:8974–8985. doi: 10.3748/wjg.v19.i47.8974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wensaas K.A., Langeland N., Hanevik K., Morch K., Eide G.E., Rortveit G. Irritable bowel syndrome and chronic fatigue 3 years after acute giardiasis: Historic cohort study. Gut. 2012;61:214–219. doi: 10.1136/gutjnl-2011-300220. [DOI] [PubMed] [Google Scholar]
- 55.Monis P.T., Caccio S.M., Thompson R.C. Variation in Giardia: Towards a taxonomic revision of the genus. Trends Parasitol. 2009;25:93–100. doi: 10.1016/j.pt.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 56.Lasek-Nesselquist E., Welch D.M., Sogin M.L. The identification of a new Giardia duodenalis assemblage in marine vertebrates and a preliminary analysis of G. duodenalis population biology in marine systems. Int. J. Parasitol. 2010;40:1063–1074. doi: 10.1016/j.ijpara.2010.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Anderson V.R., Curran M.P. Nitazoxanide: A review of its use in the treatment of gastrointestinal infections. Drugs. 2007;67:1947–1967. doi: 10.2165/00003495-200767130-00015. [DOI] [PubMed] [Google Scholar]
- 58.Chen C.Z., Kulakova L., Southall N., Marugan J.J., Galkin A., Austin C.P., Herzberg O., Zheng W. High-throughput Giardia lamblia viability assay using bioluminescent ATP content measurements. Antimicrob. Agents Chemother. 2011;55:667–675. doi: 10.1128/AAC.00618-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Upcroft J.A., Upcroft P., Boreham P.F. Drug resistance in Giardia intestinalis. Int. J. Parasitol. 1990;20:489–496. doi: 10.1016/0020-7519(90)90196-T. [DOI] [PubMed] [Google Scholar]
- 60.Wright J.M. Efficacy of antigiardial drugs. Expert Opin. Drug Saf. 2003;2:529–541. doi: 10.1517/14740338.2.6.529. [DOI] [PubMed] [Google Scholar]
- 61.Zhang Y.H., Xue M.Q., Bai Y.C., Yuan H.H., Zhao H.L., Lan M.B. 3,5-Dicaffeoylquinic acid isolated from Artemisia argyi and its ester derivatives exert anti-leucyl-tRNA synthetase of Giardia lamblia (GlLeuRS) and potential anti-giardial effects. Fitoterapia. 2012;83:1281–1285. doi: 10.1016/j.fitote.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 62.Cano P.A., Islas-Jacome A., Gonzalez-Marrero J., Yepez-Mulia L., Calzada F., Gamez-Montano R. Synthesis of 3-tetrazolylmethyl-4H-chromen-4-ones via Ugi-azide and biological evaluation against Entamoeba histolytica, Giardia lamblia and Trichomona vaginalis. Bioorg. Med. Chem. 2014;22:1370–1376. doi: 10.1016/j.bmc.2013.12.069. [DOI] [PubMed] [Google Scholar]
- 63.Navarrete-Vazquez G., Chavez-Silva F., Colin-Lozano B., Estrada-Soto S., Hidalgo-Figueroa S., Guerrero-Alvarez J., Mendez S.T., Reyes-Vivas H., Oria-Hernandez J., Canul-Canche J., et al. Synthesis of nitro(benzo)thiazole acetamides and in vitro antiprotozoal effect against amitochondriate parasites Giardia intestinalis and Trichomonas vaginalis. Bioorg. Med. Chem. 2015;23:2204–2210. doi: 10.1016/j.bmc.2015.02.059. [DOI] [PubMed] [Google Scholar]
- 64.Singh G., Arora A., Mangat S.S., Rani S., Kaur H., Goyal K., Sehgal R., Maurya I.K., Tewari R., Choquesillo-Lazarte D., et al. Design, synthesis and biological evaluation of chalconyl blended triazole allied organosilatranes as giardicidal and trichomonacidal agents. Eur. J. Med. Chem. 2016;108:287–300. doi: 10.1016/j.ejmech.2015.11.029. [DOI] [PubMed] [Google Scholar]
- 65.Bautista E., Calzada F., Lopez-Huerta F.A., Yepez-Mulia L., Ortega A. Antiprotozoal activity of 8-acyl and 8-alkyl incomptine A analogs. Bioorg. Med. Chem. Lett. 2014;24:3260–3262. doi: 10.1016/j.bmcl.2014.06.017. [DOI] [PubMed] [Google Scholar]
- 66.Soria-Arteche O., Hernandez-Campos A., Yepez-Mulia L., Trejo-Soto P.J., Hernandez-Luis F., Gres-Molina J., Maldonado L.A., Castillo R. Synthesis and antiprotozoal activity of nitazoxanide-N-methylbenzimidazole hybrids. Bioorg. Med. Chem. Lett. 2013;23:6838–6841. doi: 10.1016/j.bmcl.2013.10.011. [DOI] [PubMed] [Google Scholar]
- 67.Leboho T.C., Giri S., Popova I., Cock I., Michael J.P., de Koning C.B. Double Sonogashira reactions on dihalogenated aminopyridines for the assembly of an array of 7-azaindoles bearing triazole and quinoxaline substituents at C-5: Inhibitory bioactivity against Giardia duodenalis trophozoites. Bioorg. Med. Chem. 2015;23:4943–4951. doi: 10.1016/j.bmc.2015.05.024. [DOI] [PubMed] [Google Scholar]
- 68.Mocelo-Castell R., Villanueva-Novelo C., Cáceres-Castillo D., Carballo R.M., Quijano-Quiñones R.F., Quesadas-Rojas M., Cantillo-Ciau Z., Cedillo-Rivera R., Moo-Puc R.E., Moujir L.M., et al. 2-Amino-4-arylthiazole Derivatives as Anti-giardial Agents: Synthesis, Biological Evaluation and QSAR Studies. Open Chem. 2015;13:1127–1136. doi: 10.1515/chem-2015-0127. [DOI] [Google Scholar]
- 69.Castillo-Villanueva A., Rufino-Gonzalez Y., Mendez S.T., Torres-Arroyo A., Ponce-Macotela M., Martinez-Gordillo M.N., Reyes-Vivas H., Oria-Hernandez J. Disulfiram as a novel inactivator of Giardia lamblia triosephosphate isomerase with antigiardial potential. Int. J. Parasitol. Drugs Drug Resist. 2017;7:425–432. doi: 10.1016/j.ijpddr.2017.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hayani K., Dandashli A., Weisshaar E. Cutaneous leishmaniasis in Syria: Clinical features, current status and the effects of war. Acta Derm. Venereol. 2015;95:62–66. doi: 10.2340/00015555-1988. [DOI] [PubMed] [Google Scholar]
- 71.Herwaldt B.L. Leishmaniasis. Lancet. 1999;354:1191–1199. doi: 10.1016/S0140-6736(98)10178-2. [DOI] [PubMed] [Google Scholar]
- 72.Desjeux P. The increase in risk factors for leishmaniasis worldwide. Trans. R. Soc. Trop. Med. Hyg. 2001;95:239–243. doi: 10.1016/S0035-9203(01)90223-8. [DOI] [PubMed] [Google Scholar]
- 73.Chappuis F., Sundar S., Hailu A., Ghalib H., Rijal S., Peeling R.W., Alvar J., Boelaert M. Visceral leishmaniasis: What are the needs for diagnosis, treatment and control? Nat. Rev. Microbiol. 2007;5:873–882. doi: 10.1038/nrmicro1748. [DOI] [PubMed] [Google Scholar]
- 74.Murray H.W., Berman J.D., Davies C.R., Saravia N.G. Advances in leishmaniasis. Lancet. 2005;366:1561–1577. doi: 10.1016/S0140-6736(05)67629-5. [DOI] [PubMed] [Google Scholar]
- 75.Reithinger R., Dujardin J.C., Louzir H., Pirmez C., Alexander B., Brooker S. Cutaneous leishmaniasis. Lancet Infect. Dis. 2007;7:581–596. doi: 10.1016/S1473-3099(07)70209-8. [DOI] [PubMed] [Google Scholar]
- 76.Scope A., Trau H., Bakon M., Yarom N., Nasereddin A., Schwartz E. Imported mucosal leishmaniasis in a traveler. Clin. Infect. Dis. 2003;37:e83–e87. doi: 10.1086/377045. [DOI] [PubMed] [Google Scholar]
- 77.Marsden P.D. Mucosal leishmaniasis (“espundia” Escomel, 1911) Trans. R. Soc. Trop. Med. Hyg. 1986;80:859–876. doi: 10.1016/0035-9203(86)90243-9. [DOI] [PubMed] [Google Scholar]
- 78.Desjeux P. Leishmaniasis: Public health aspects and control. Clin. Dermatol. 1996;14:417–423. doi: 10.1016/0738-081X(96)00057-0. [DOI] [PubMed] [Google Scholar]
- 79.Habib Z.H., Yusuf M.A., Ahmed I., Jhora S.T. Clinical Burden of Kala-azar in Bangladesh: A Review Update. J. Sci. Found. 2012;10:70–79. doi: 10.3329/jsf.v10i2.17960. [DOI] [Google Scholar]
- 80.Sharlow E.R., Grogl M., Johnson J., Lazo J.S. Anti-leishmanial drug discovery: Rising to the challenges of a highly neglected disease. Mol. Interv. 2010;10:72–75. doi: 10.1124/mi.10.2.4. [DOI] [PubMed] [Google Scholar]
- 81.WHO Expert Committee on the Control of the Leishmaniases & World Health Organizatio . Conotrol of the Leishmaniases: Report of a Meeting of the WHO Expert Commitee on the Control of Leishmaniases. WHO Expert Committee on the Control of the Leishmaniases & World Health Organizatio; Geneva, Switzerland: 2010. [Google Scholar]
- 82.Mathers C.D., Ezzati M., Lopez A.D. Measuring the burden of neglected tropical diseases: The global burden of disease framework. PLoS Negl. Trop. Dis. 2007;1:e114. doi: 10.1371/journal.pntd.0000114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.den Boer M., Argaw D., Jannin J., Alvar J. Leishmaniasis impact and treatment access. Clin. Microbiol. Infect. 2011;17:1471–1477. doi: 10.1111/j.1469-0691.2011.03635.x. [DOI] [PubMed] [Google Scholar]
- 84.Fuertes M.A., Nguewa P.A., Castilla J., Alonso C., Perez J.M. Anticancer compounds as leishmanicidal drugs: Challenges in chemotherapy and future perspectives. Curr. Med. Chem. 2008;15:433–439. doi: 10.2174/092986708783503221. [DOI] [PubMed] [Google Scholar]
- 85.Bern C., Maguire J.H., Alvar J. Complexities of assessing the disease burden attributable to leishmaniasis. PLoS Negl. Trop. Dis. 2008;2:e313. doi: 10.1371/journal.pntd.0000313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vannier-Santos M.A., Martiny A., de Souza W. Cell biology of Leishmania spp.: Invading and evading. Curr. Pharm. Des. 2002;8:297–318. doi: 10.2174/1381612023396230. [DOI] [PubMed] [Google Scholar]
- 87.Croft S.L., Coombs G.H. Leishmaniasis--current chemotherapy and recent advances in the search for novel drugs. Trends Parasitol. 2003;19:502–508. doi: 10.1016/j.pt.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 88.Hussain H., Al-Harrasi A., Al-Rawahi A., Green I.R., Gibbons S. Fruitful decade for antileishmanial compounds from 2002 to late 2011. Chem. Rev. 2014;114:10369–10428. doi: 10.1021/cr400552x. [DOI] [PubMed] [Google Scholar]
- 89.Alvar J., Croft S., Olliaro P. Chemotherapy in the Treatment and Control of Leishmaniasis. Adv. Parasitol. 2006;61:223–274. doi: 10.1016/S0065-308X(05)61006-8. [DOI] [PubMed] [Google Scholar]
- 90.Plano D., Baquedano Y., Moreno-Mateos D., Font M., Jimenez-Ruiz A., Palop J.A., Sanmartin C. Selenocyanates and diselenides: A new class of potent antileishmanial agents. Eur. J. Med. Chem. 2011;46:3315–3323. doi: 10.1016/j.ejmech.2011.04.054. [DOI] [PubMed] [Google Scholar]
- 91.Rodrigues R.F., Castro-Pinto D., Echevarria A., dos Reis C.M., Del Cistia C.N., Sant’Anna C.M., Teixeira F., Castro H., Canto-Cavalheiro M., Leon L.L., et al. Investigation of trypanothione reductase inhibitory activity by 1,3,4-thiadiazolium-2-aminide derivatives and molecular docking studies. Bioorg. Med. Chem. 2012;20:1760–1766. doi: 10.1016/j.bmc.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 92.Maarouf M., Adeline M.T., Solignac M., Vautrin D., Robert-Gero M. Development and characterization of paromomycin-resistant Leishmania donovani promastigotes. Parasite. 1998;5:167–173. doi: 10.1051/parasite/1998052167. [DOI] [PubMed] [Google Scholar]
- 93.Croft S.L., Sundar S., Fairlamb A.H. Drug resistance in leishmaniasis. Clin. Microbiol. Rev. 2006;19:111–126. doi: 10.1128/CMR.19.1.111-126.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.de Melos J.L.R., Torres-Santos E.C., Faioes V.D.S., Del Cistia C.D.N., Sant’Anna C.M.R., Rodrigues-Santos C.E., Echevarria A. Novel 3,4-methylenedioxyde-6-X-benzaldehyde-thiosemicarbazones: Synthesis and antileishmanial effects against Leishmania amazonensis. Eur. J. Med. Chem. 2015;103:409–417. doi: 10.1016/j.ejmech.2015.09.009. [DOI] [PubMed] [Google Scholar]
- 95.Rashid U., Sultana R., Shaheen N., Hassan S.F., Yaqoob F., Ahmad M.J., Iftikhar F., Sultana N., Asghar S., Yasinzai M., et al. Structure based medicinal chemistry-driven strategy to design substituted dihydropyrimidines as potential antileishmanial agents. Eur. J. Med. Chem. 2016;115:230–244. doi: 10.1016/j.ejmech.2016.03.022. [DOI] [PubMed] [Google Scholar]
- 96.Sangshetti J.N., Shaikh R.I., Khan F.A., Patil R.H., Marathe S.D., Gade W.N., Shinde D.B. Synthesis, antileishmanial activity and docking study of N′-substitutedbenzylidene-2-(6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl)acetohydraz ides. Bioorg. Med. Chem. Lett. 2014;24:1605–1610. doi: 10.1016/j.bmcl.2014.01.035. [DOI] [PubMed] [Google Scholar]
- 97.Gupta S., Shivahare R., Korthikunta V., Singh R., Gupta S., Tadigoppula N. Synthesis and biological evaluation of chalcones as potential antileishmanial agents. Eur. J. Med. Chem. 2014;81:359–366. doi: 10.1016/j.ejmech.2014.05.034. [DOI] [PubMed] [Google Scholar]
- 98.Manda S., Khan S.I., Jain S.K., Mohammed S., Tekwani B.L., Khan I.A., Vishwakarma R.A., Bharate S.B. Synthesis, antileishmanial and antitrypanosomal activities of N-substituted tetrahydro-beta–carbolines. Bioorg. Med. Chem. Lett. 2014;24:3247–3250. doi: 10.1016/j.bmcl.2014.06.030. [DOI] [PubMed] [Google Scholar]
- 99.Zhu X., Farahat A.A., Mattamana M., Joice A., Pandharkar T., Holt E., Banerjee M., Gragg J.L., Hu L., Kumar A., et al. Synthesis and pharmacological evaluation of mono-arylimidamides as antileishmanial agents. Bioorg. Med. Chem. Lett. 2016;26:2551–2556. doi: 10.1016/j.bmcl.2016.03.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ronga L., Del Favero M., Cohen A., Soum C., Le Pape P., Savrimoutou S., Pinaud N., Mullie C., Daulouede S., Vincendeau P., et al. Design, synthesis and biological evaluation of novel 4-alkapolyenylpyrrolo[1,2-a]quinoxalines as antileishmanial agents—Part III. Eur. J. Med. Chem. 2014;81:378–393. doi: 10.1016/j.ejmech.2014.05.037. [DOI] [PubMed] [Google Scholar]
- 101.Bekhit A.A., Hassan A.M., Abd El Razik H.A., El-Miligy M.M., El-Agroudy E.J., Bekhit Ael D. New heterocyclic hybrids of pyrazole and its bioisosteres: Design, synthesis and biological evaluation as dual acting antimalarial-antileishmanial agents. Eur. J. Med. Chem. 2015;94:30–44. doi: 10.1016/j.ejmech.2015.02.038. [DOI] [PubMed] [Google Scholar]
- 102.Dea-Ayuela M.A., Bilbao-Ramos P., Bolas-Fernandez F., Gonzalez-Cardenete M.A. Synthesis and antileishmanial activity of C7-and C12-functionalized dehydroabietylamine derivatives. Eur. J. Med. Chem. 2016;121:445–450. doi: 10.1016/j.ejmech.2016.06.004. [DOI] [PubMed] [Google Scholar]
- 103.Marchand P., Bazin M.-A., Pagniez F., Rivière G., Bodero L., Marhadour S., Nourrisson M.-R., Picot C., Ruchaud S., Bach S., et al. Synthesis, antileishmanial activity and cytotoxicity of 2,3-diaryl- and 2,3,8-trisubstituted imidazo[1,2-a]pyrazines. Eur. J. Med. Chem. 2015;103:381–395. doi: 10.1016/j.ejmech.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 104.Sangshetti J.N., Kalam Khan F.A., Kulkarni A.A., Patil R.H., Pachpinde A.M., Lohar K.S., Shinde D.B. Antileishmanial activity of novel indolyl-coumarin hybrids: Design, synthesis, biological evaluation, molecular docking study and in silico ADME prediction. Bioorg. Med. Chem. Lett. 2016;26:829–835. doi: 10.1016/j.bmcl.2015.12.085. [DOI] [PubMed] [Google Scholar]
- 105.Lunagariya N.A., Gohil V.M., Kushwah V., Neelagiri S., Jain S., Singh S., Bhutani K.K. Design, synthesis and biological evaluation of 1,3,6-trisubstituted beta–carboline derivatives for cytotoxic and anti-leishmanial potential. Bioorg. Med. Chem. Lett. 2016;26:789–794. doi: 10.1016/j.bmcl.2015.12.095. [DOI] [PubMed] [Google Scholar]
- 106.Kumar S., Tiwari A., Suryawanshi S.N., Mittal M., Vishwakarma P., Gupta S. Chemotherapy of leishmaniasis. Part IX: Synthesis and bioevaluation of aryl substituted ketene dithioacetals as antileishmanial agents. Bioorg. Med. Chem. Lett. 2012;22:6728–6730. doi: 10.1016/j.bmcl.2012.08.096. [DOI] [PubMed] [Google Scholar]
- 107.Saad S.M., Ghouri N., Perveen S., Khan K.M., Choudhary M.I. 4-Arylamino-6-nitroquinazolines: Synthesis and their activities against neglected disease leishmaniasis. Eur. J. Med. Chem. 2016;108:13–20. doi: 10.1016/j.ejmech.2015.11.016. [DOI] [PubMed] [Google Scholar]
- 108.Gopinath V.S., Rao M., Shivahare R., Vishwakarma P., Ghose S., Pradhan A., Hindupur R., Sarma K.D., Gupta S., Puri S.K., et al. Design, synthesis, ADME characterization and antileishmanial evaluation of novel substituted quinoline analogs. Bioorg. Med. Chem. Lett. 2014;24:2046–2052. doi: 10.1016/j.bmcl.2014.03.065. [DOI] [PubMed] [Google Scholar]
- 109.Baquedano Y., Moreno E., Espuelas S., Nguewa P., Font M., Gutierrez K.J., Jimenez-Ruiz A., Palop J.A., Sanmartin C. Novel hybrid selenosulfonamides as potent antileishmanial agents. Eur. J. Med. Chem. 2014;74:116–123. doi: 10.1016/j.ejmech.2013.12.030. [DOI] [PubMed] [Google Scholar]
- 110.Adam R., Bilbao-Ramos P., Lopez-Molina S., Abarca B., Ballesteros R., Gonzalez-Rosende M.E., Dea-Ayuela M.A., Alzuet-Pina G. Triazolopyridyl ketones as a novel class of antileishmanial agents. DNA binding and BSA interaction. Bioorg. Med. Chem. 2014;22:4018–4027. doi: 10.1016/j.bmc.2014.05.069. [DOI] [PubMed] [Google Scholar]
- 111.Sharma R., Pandey A.K., Shivahare R., Srivastava K., Gupta S., Chauhan P.M. Triazino indole-quinoline hybrid: A novel approach to antileishmanial agents. Bioorg. Med. Chem. Lett. 2014;24:298–301. doi: 10.1016/j.bmcl.2013.11.018. [DOI] [PubMed] [Google Scholar]
- 112.Tiwari A., Kumar S., Shivahare R., Kant P., Gupta S., Suryawanshi S.N. Chemotherapy of leishmaniasis part XIII: Design and synthesis of novel heteroretinoid-bisbenzylidine ketone hybrids as antileishmanial agents. Bioorg. Med. Chem. Lett. 2015;25:410–413. doi: 10.1016/j.bmcl.2014.09.078. [DOI] [PubMed] [Google Scholar]
- 113.Pandey S., Chauhan S.S., Shivahare R., Sharma A., Jaiswal S., Gupta S., Lal J., Chauhan P.M. Identification of a diverse indole-2-carboxamides as a potent antileishmanial chemotypes. Eur. J. Med. Chem. 2016;110:237–245. doi: 10.1016/j.ejmech.2016.01.028. [DOI] [PubMed] [Google Scholar]
- 114.Barteselli A., Casagrande M., Basilico N., Parapini S., Rusconi C.M., Tonelli M., Boido V., Taramelli D., Sparatore F., Sparatore A. Clofazimine analogs with antileishmanial and antiplasmodial activity. Bioorg. Med. Chem. 2015;23:55–65. doi: 10.1016/j.bmc.2014.11.028. [DOI] [PubMed] [Google Scholar]
- 115.De Vita D., Moraca F., Zamperini C., Pandolfi F., Di Santo R., Matheeussen A., Maes L., Tortorella S., Scipione L. In vitro screening of 2-(1H-imidazol-1-yl)-1-phenylethanol derivatives as antiprotozoal agents and docking studies on Trypanosoma cruzi CYP51. Eur. J. Med. Chem. 2016;113:28–33. doi: 10.1016/j.ejmech.2016.02.028. [DOI] [PubMed] [Google Scholar]
- 116.Patrick D.A., Bakunov S.A., Bakunova S.M., Jones S.K., Wenzler T., Barszcz T., Kumar A., Boykin D.W., Werbovetz K.A., Brun R., et al. Synthesis and antiprotozoal activities of benzyl phenyl ether diamidine derivatives. Eur. J. Med. Chem. 2013;67:310–324. doi: 10.1016/j.ejmech.2013.06.033. [DOI] [PubMed] [Google Scholar]
- 117.Rye C.E., Barker D. Asymmetric synthesis and anti-protozoal activity of the 8,4′-oxyneolignans virolin, surinamensin and analogues. Eur. J. Med. Chem. 2013;60:240–248. doi: 10.1016/j.ejmech.2012.12.013. [DOI] [PubMed] [Google Scholar]
- 118.Durust Y., Karakus H., Kaiser M., Tasdemir D. Synthesis and anti-protozoal activity of novel dihydropyrrolo[3,4-d][1,2,3]triazoles. Eur. J. Med. Chem. 2012;48:296–304. doi: 10.1016/j.ejmech.2011.12.028. [DOI] [PubMed] [Google Scholar]
- 119.Pierson J.T., Dumetre A., Hutter S., Delmas F., Laget M., Finet J.P., Azas N., Combes S. Synthesis and antiprotozoal activity of 4-arylcoumarins. Eur. J. Med. Chem. 2010;45:864–869. doi: 10.1016/j.ejmech.2009.10.022. [DOI] [PubMed] [Google Scholar]
- 120.Patrick D.A., Ismail M.A., Arafa R.K., Wenzler T., Zhu X., Pandharkar T., Jones S.K., Werbovetz K.A., Brun R., Boykin D.W., et al. Synthesis and antiprotozoal activity of dicationic m-terphenyl and 1,3-dipyridylbenzene derivatives. J. Med. Chem. 2013;56:5473–5494. doi: 10.1021/jm400508e. [DOI] [PubMed] [Google Scholar]
- 121.Saccolitia F., Angiullib G., Pupoa G., Pescatoria L., Madiaa V.N., Messorea A., Colottib G., Fiorillob A., Scipionea L., Santoa R.D., et al. Inhibition of Leishmania infantum trypanothione reductase by diaryl sulfide derivatives. J. Enzyme Inhit. Med. Chem. 2017;32:304–310. doi: 10.1080/14756366.2016.1250755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Vishwakarma P., Parmar N., Chandrakar P., Sharma T., Kathuria M., Agnihotri P.K., Siddiqi M.I., Mitra K., Kar S. Ammonium trichloro [1,2-ethanediolato-O,O′]-tellurate cures experimental visceral leishmaniasis by redox modulation of Leishmania donovani trypanothione reductase and inhibiting host integrin linked PI3K/Akt pathway. Cell. Mol Life Sci. 2018;75:563–588. doi: 10.1007/s00018-017-2653-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rodríguez-Hernández D., Barbosa L.C.A., Demuner A.J., Nain-Perez A., Ferreira S.R., Fujiwara R.T., de Almeida R.M., Heller L., Csuk R. Leishmanicidal and cytotoxic activity of hederagenin-bistriazolyl derivatives. Eur. J. Med. Chem. 2017;140:624–635. doi: 10.1016/j.ejmech.2017.09.045. [DOI] [PubMed] [Google Scholar]
- 124.Tahghighi A., Babalouei F. Thiadiazoles: The appropriate pharmacological scaffolds with leishmanicidal and antimalarial activities: A review. Iran. J. Basic Med. Sci. 2017;20:613–622. doi: 10.22038/IJBMS.2017.8828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Njoroge M., Njuguna N.M., Mutai P., Ongarora D.S., Smith P.W., Chibale K. Recent Approaches to Chemical Discovery and Development against Malaria and the Neglected Tropical Diseases Human African Trypanosomiasis and Schistosomiasis. Chem. Rev. 2014;114:11138–11163. doi: 10.1021/cr500098f. [DOI] [PubMed] [Google Scholar]
- 126.Caraballo H., King K. Emergency department management of mosquito-borne illness: Malaria, dengue, and West Nile virus. Emerg. Med. Pract. 2014;16:1–23. [PubMed] [Google Scholar]
- 127.World Health Organization . World Malaria Report 2014. World Health Organization; Geneva, Switzerland: 2014. [Google Scholar]
- 128.World Health Organization . World Malaria Report 2012. World Health Organization; Geneva, Switzerland: 2012. [Google Scholar]
- 129.Murray C.J., Rosenfeld L.C., Lim S.S., Andrews K.G., Foreman K.J., Haring D., Fullman N., Naghavi M., Lozano R., Lopez A.D. Global malaria mortality between 1980 and 2010: A systematic analysis. Lancet. 2012;379:413–431. doi: 10.1016/S0140-6736(12)60034-8. [DOI] [PubMed] [Google Scholar]
- 130.Sachs J., Malaney P. The economic and social burden of malaria. Nature. 2002;415:680–685. doi: 10.1038/415680a. [DOI] [PubMed] [Google Scholar]
- 131.Brieger W. The World Malaria Report 2015: Prospects for Malaria Elimination. Africa Health. 2016:14–16. [Google Scholar]
- 132.Baird J.K. Neglect of Plasmodium vivax malaria. Trends Parasitol. 2007;23:533–539. doi: 10.1016/j.pt.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 133.Prudencio M., Rodriguez A., Mota M.M. The silent path to thousands of merozoites: The Plasmodium liver stage. Nat. Rev. Microbiol. 2006;4:849–856. doi: 10.1038/nrmicro1529. [DOI] [PubMed] [Google Scholar]
- 134.Wells T.N., Burrows J.N., Baird J.K. Targeting the hypnozoite reservoir of Plasmodium vivax: The hidden obstacle to malaria elimination. Trends Parasitol. 2010;26:145–151. doi: 10.1016/j.pt.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 135.Clark I.A., al Yaman F.M., Jacobson L.S. The biological basis of malarial disease. Int. J. Parasitol. 1997;27:1237–1249. doi: 10.1016/S0020-7519(97)00121-5. [DOI] [PubMed] [Google Scholar]
- 136.Baker D.A. Malaria gametocytogenesis. Mol. Biochem. Parasitol. 2010;172:57–65. doi: 10.1016/j.molbiopara.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Smith D.C. Quinine and fever: The development of the effective dosage. J. Hist. Med. Allied Sci. 1976;31:343–367. doi: 10.1093/jhmas/XXXI.3.343. [DOI] [PubMed] [Google Scholar]
- 138.Croft S. Antimalarial Chemotherapy: Mechanisms of Action, Resistance and New Directions in Drug Discovery. Drug Discov. Today. 2001;6:1151. doi: 10.1016/S1359-6446(01)02035-9. [DOI] [PubMed] [Google Scholar]
- 139.Salas P.F., Herrmann C., Orvig C. Metalloantimalarials. Chem. Rev. 2013;113:3450–3492. doi: 10.1021/cr3001252. [DOI] [PubMed] [Google Scholar]
- 140.Bloland P.B., Ettling M., Meek S. Combination therapy for malaria in Africa: Hype or hope? Bull. World Health Organ. 2000;78:1378–1388. [PMC free article] [PubMed] [Google Scholar]
- 141.Mutabingwa T.K. Artemisinin-based combination therapies (ACTs): Best hope for malaria treatment but inaccessible to the needy! Acta Trop. 2005;95:305–315. doi: 10.1016/j.actatropica.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 142.Jambou R. Resistance of Plasmodium falciparum field isolates to in-vitro artemether and point mutations of the SERCA-type PfATPase6. Lancet. 2005;366:1960–1963. doi: 10.1016/S0140-6736(05)67787-2. [DOI] [PubMed] [Google Scholar]
- 143.Pingaew R., Saekee A., Mandi P., Nantasenamat C., Prachayasittikul S., Ruchirawat S., Prachayasittikul V. Synthesis, biological evaluation and molecular docking of novel chalcone-coumarin hybrids as anticancer and antimalarial agents. Eur. J. Med. Chem. 2014;85:65–76. doi: 10.1016/j.ejmech.2014.07.087. [DOI] [PubMed] [Google Scholar]
- 144.Gut J., Rosenthal P.J., Kumar V. β-beta-amino-alcohol tethered 4-aminoquinoline-isatin conjugates: Synthesis and antimalarial evaluation. Eur. J. Med. Chem. 2014;84:566–573. doi: 10.1016/j.ejmech.2014.07.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kumar K., Pradines B., Madamet M., Amalvict R., Benoit N., Kumar V. 1H-1,2,3-triazole tethered isatin-ferrocene conjugates: Synthesis and in vitro antimalarial evaluation. Eur. J. Med. Chem. 2014;87:801–804. doi: 10.1016/j.ejmech.2014.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Helgren T.R., Sciotti R.J., Lee P., Duffy S., Avery V.M., Igbinoba O., Akoto M., Hagen T.J. The synthesis, antimalarial activity and CoMFA analysis of novel aminoalkylated quercetin analogs. Bioorg. Med. Chem. Lett. 2015;25:327–332. doi: 10.1016/j.bmcl.2014.11.039. [DOI] [PubMed] [Google Scholar]
- 147.Faisca Phillips A.M., Nogueira F., Murtinheira F. Synthesis and antimalarial evaluation of prodrugs of novel fosmidomycin analogues. Bioorg. Med. Chem. Lett. 2015;25:2112–2116. doi: 10.1016/j.bmcl.2015.03.077. [DOI] [PubMed] [Google Scholar]
- 148.Yadav R.R., Khan S.I., Singh S., Khan I.A., Vishwakarma R.A., Bharate S.B. Synthesis, antimalarial and antitubercular activities of meridianin derivatives. Eur. J. Med. Chem. 2015;98:160–169. doi: 10.1016/j.ejmech.2015.05.020. [DOI] [PubMed] [Google Scholar]
- 149.Nguyen Le T., De Borggraeve W.M., Grellier P., Pham V.C., Dehaen W., Nguyen V.H. Synthesis of 11-aza-artemisinin derivatives using the Ugi reaction and an evaluation of their antimalarial activity. Tetrahedron Lett. 2014;55:4892–4894. doi: 10.1016/j.tetlet.2014.07.027. [DOI] [Google Scholar]
- 150.Reiter C., Frohlich T., Gruber L., Hutterer C., Marschall M., Voigtlander C., Friedrich O., Kappes B., Efferth T., Tsogoeva S.B. Highly potent artemisinin-derived dimers and trimers: Synthesis and evaluation of their antimalarial, antileukemia and antiviral activities. Bioorg. Med. Chem. 2015;23:5452–5458. doi: 10.1016/j.bmc.2015.07.048. [DOI] [PubMed] [Google Scholar]
- 151.Parthiban A., Muthukumaran J., Manhas A., Srivastava K., Krishna R., Rao H.S. Synthesis, in vitro and in silico antimalarial activity of 7-chloroquinoline and 4H-chromene conjugates. Bioorg. Med. Chem. Lett. 2015;25:4657–4663. doi: 10.1016/j.bmcl.2015.08.030. [DOI] [PubMed] [Google Scholar]
- 152.Bhat H.R., Singh U.P., Thakur A., Kumar Ghosh S., Gogoi K., Prakash A., Singh R.K. Synthesis, antimalarial activity and molecular docking of hybrid 4-aminoquinoline-1,3,5-triazine derivatives. Exp. Parasitol. 2015;157:59–67. doi: 10.1016/j.exppara.2015.06.016. [DOI] [PubMed] [Google Scholar]
- 153.Carneiro P.F., Pinto M.C.R.F., Marra R.K.F., da Silva F.C., Resende J.A.L.C., Rocha E., Silva L.F., Alves H.G., Barbosa G.S., de Vasconcellos M.C., et al. Synthesis and antimalarial activity of quinones and structurally-related oxirane derivatives. Eur. J. Med. Chem. 2016;108:134–140. doi: 10.1016/j.ejmech.2015.11.020. [DOI] [PubMed] [Google Scholar]
- 154.Karad S.C., Purohit V.B., Thakor P., Thakkar V.R., Raval D.K. Novel morpholinoquinoline nucleus clubbed with pyrazoline scaffolds: Synthesis, antibacterial, antitubercular and antimalarial activities. Eur. J. Med. Chem. 2016;112:270–279. doi: 10.1016/j.ejmech.2016.02.016. [DOI] [PubMed] [Google Scholar]
- 155.Devender N., Gunjan S., Chhabra S., Singh K., Pasam V.R., Shukla S.K., Sharma A., Jaiswal S., Singh S.K., Kumar Y., et al. Identification of beta-Amino alcohol grafted 1,4,5 trisubstituted 1,2,3-triazoles as potent antimalarial agents. Eur. J. Med. Chem. 2016;109:187–198. doi: 10.1016/j.ejmech.2015.12.038. [DOI] [PubMed] [Google Scholar]
- 156.Svogie A.L., Isaacs M., Hoppe H.C., Khanye S.D., Veale C.G. Indolyl-3-ethanone-alpha-thioethers: A promising new class of non-toxic antimalarial agents. Eur. J. Med. Chem. 2016;114:79–88. doi: 10.1016/j.ejmech.2016.02.056. [DOI] [PubMed] [Google Scholar]
- 157.Patel P.R., Sun W., Kim M., Huang X., Sanderson P.E., Tanaka T.Q., McKew J.C., Simeonov A., Williamson K.C., Zheng W., et al. In vitro evaluation of imidazo[4,5-c]quinolin-2-ones as gametocytocidal antimalarial agents. Bioorg. Med. Chem. Lett. 2016;26:2907–2911. doi: 10.1016/j.bmcl.2016.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Seebacher W., Wolkinger V., Faist J., Kaiser M., Brun R., Saf R., Bucar F., Groblacher B., Brantner A., Merino V., et al. Synthesis of 3-azabicyclo[3.2.2]nonanes and their antiprotozoal activities. Bioorg. Med. Chem. Lett. 2015;25:1390–1393. doi: 10.1016/j.bmcl.2015.02.044. [DOI] [PubMed] [Google Scholar]
- 159.Inam A., Siddiqui S.M., Macedo T.S., Moreira D.R., Leite A.C., Soares M.B., Azam A. Design, synthesis and biological evaluation of 3-[4-(7-chloro-quinolin-4-yl)-piperazin-1-yl]-propionic acid hydrazones as antiprotozoal agents. Eur. J. Med. Chem. 2014;75:67–76. doi: 10.1016/j.ejmech.2014.01.023. [DOI] [PubMed] [Google Scholar]
- 160.Juneja A., Macedo T.S., Magalhaes Moreira D.R., Pereira Soares M.B., Lima Leite A.C., Kelle de Andrade Lemoine Neves J., Alves Pereira V.R., Avecilla F., Azam A. Synthesis of 4′-(2-ferrocenyl)-2,2′:6′2″-terpyridine: Characterization and antiprotozoal activity of Mn(II), Co(II), Ni(II), Cu(II) and Zn(II) complexes. Eur. J. Med. Chem. 2014;75:203–210. doi: 10.1016/j.ejmech.2014.01.051. [DOI] [PubMed] [Google Scholar]
- 161.McKeever C., Kaiser M., Rozas I. Aminoalkyl derivatives of guanidine diaromatic minor groove binders with antiprotozoal activity. J. Med. Chem. 2013;56:700–711. doi: 10.1021/jm301614w. [DOI] [PubMed] [Google Scholar]
- 162.Opsenica I.M., Tot M., Gomba L., Nuss J.E., Sciotti R.J., Bavari S., Burnett J.C., Solaja B.A. 4-Amino-7-chloroquinolines: Probing ligand efficiency provides botulinum neurotoxin serotype A light chain inhibitors with significant antiprotozoal activity. J. Med. Chem. 2013;56:5860–5871. doi: 10.1021/jm4006077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Hanessian S., Vakiti R.R., Chattopadhyay A.K., Dorich S., Lavallee C. Probing functional diversity in pactamycin toward antibiotic, antitumor, and antiprotozoal activity. Bioorg. Med. Chem. 2013;21:1775–1786. doi: 10.1016/j.bmc.2013.01.037. [DOI] [PubMed] [Google Scholar]
- 164.Abada Z., Cojean S., Pomel S., Ferrie L., Akagah B., Lormier A.T., Loiseau P.M., Figadere B. Synthesis and antiprotozoal activity of original porphyrin precursors and derivatives. Eur. J. Med. Chem. 2013;67:158–165. doi: 10.1016/j.ejmech.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 165.Yeo S.J., Liu D.X., Kim H.S., Park H. Anti-malarial effect of novel chloroquine derivatives as agents for the treatment of malaria. Malar. J. 2017;16:80. doi: 10.1186/s12936-017-1725-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Singh A., Gut J., Rosenthal P.J., Kumar V. 4-Aminoquinoline-ferrocenyl-chalcone conjugates: Synthesis and anti-plasmodial evaluation. Eur. J. Med. Chem. 2017;125:269–277. doi: 10.1016/j.ejmech.2016.09.044. [DOI] [PubMed] [Google Scholar]
- 167.World Health Organization, Department of Reproductive Health and Research . Global Incidence and Prevalence of Selected Curable Sexually Transmitted Infections. World Health Organization; Geneva, Switzerland: 2008. [Google Scholar]
- 168.Petrin D., Delgaty K., Bhatt R., Garber G. Clinical and microbiological aspects of Trichomonas vaginalis. Clin. Microbiol. Rev. 1998;11:300–317. doi: 10.1128/CMR.11.2.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Klebanoff M.A., Carey J.C., Hauth J.C., Hillier S.L., Nugent R.P., Thom E.A., Ernest J.M., Heine R.P., Wapner R.J., Trout W., et al. Failure of metronidazole to prevent preterm delivery among pregnant women with asymptomatic Trichomonas vaginalis infection. N. Engl. J. Med. 2001;345:487–493. doi: 10.1056/NEJMoa003329. [DOI] [PubMed] [Google Scholar]
- 170.Goldman M.B., Cramer D.W. Relation of tubal infertility to history of sexually transmitted diseases. Am. J. Epidemiol. 1993;137:577–584. doi: 10.1093/oxfordjournals.aje.a116711. [DOI] [PubMed] [Google Scholar]
- 171.Viikki M., Pukkala E., Nieminen P., Hakama M. Gynaecological infections as risk determinants of subsequent cervical neoplasia. Acta Oncol. 2000;39:71–75. doi: 10.1080/028418600431003. [DOI] [PubMed] [Google Scholar]
- 172.Cherpes T.L., Wiesenfeld H.C., Melan M.A., Kant J.A., Cosentino L.A., Meyn L.A., Hillier S.L. The associations between pelvic inflammatory disease, Trichomonas vaginalis infection, and positive herpes simplex virus type 2 serology. Sex. Transm. Dis. 2006;33:747–752. doi: 10.1097/01.olq.0000218869.52753.c7. [DOI] [PubMed] [Google Scholar]
- 173.Laga M., Manoka A., Kivuvu M., Malele B., Tuliza M., Nzila N., Goeman J., Behets F., Batter V., Alary M., et al. Non-ulcerative sexually transmitted diseases as risk factors for HIV-1 transmission in women: Results from a cohort study. AIDS. 1993;7:95–102. doi: 10.1097/00002030-199301000-00015. [DOI] [PubMed] [Google Scholar]
- 174.Cu-Uvin S., Ko H., Jamieson D.J., Hogan J.W., Schuman P., Anderson J., Klein R.S. Prevalence, incidence, and persistence or recurrence of trichomoniasis among human immunodeficiency virus (HIV)-positive women and among HIV-negative women at high risk for HIV infection. Clin. Infect. Dis. 2002;34:1406–1411. doi: 10.1086/340264. [DOI] [PubMed] [Google Scholar]
- 175.Shethwala N.D., Mulla S.A., Kosambiya J.K., Desai V.K. Sexually transmitted infections and reproductive tract infections in female sex workers. Indian J. Pathol. Microbiol. 2009;52:198–199. doi: 10.4103/0377-4929.48916. [DOI] [PubMed] [Google Scholar]
- 176.Mavedzenge S.N., Pol B.V., Cheng H., Montgomery E.T., Blanchard K., de Bruyn G., Ramjee G., Straten A.V. Epidemiological synergy of Trichomonas vaginalis and HIV in Zimbabwean and South African women. Sex. Transm. Dis. 2010;37:460–466. doi: 10.1097/OLQ.0b013e3181cfcc4b. [DOI] [PubMed] [Google Scholar]
- 177.Sorvillo F., Smith L., Kerndt P., Ash L. Trichomonas vaginalis, HIV, and African-Americans. Emerg. Infect. Dis. 2001;7:927–932. doi: 10.3201/eid0706.010603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.van Der Pol B., Kwok C., Pierre-Louis B., Rinaldi A., Salata R.A., Chen P.L., van de Wijgert J., Mmiro F., Mugerwa R., Chipato T., et al. Trichomonas vaginalis infection and human immunodeficiency virus acquisition in African women. J. Infect. Dis. 2008;197:548–554. doi: 10.1086/526496. [DOI] [PubMed] [Google Scholar]
- 179.Wolner-Hanssen P., Krieger J.N., Stevens C.E., Kiviat N.B., Koutsky L., Critchlow C., DeRouen T., Hillier S., Holmes K.K. Clinical manifestations of vaginal trichomoniasis. JAMA. 1989;261:571–576. doi: 10.1001/jama.1989.03420040109029. [DOI] [PubMed] [Google Scholar]
- 180.Ryu J.S., Chung H.L., Min D.Y., Cho Y.H., Ro Y.S., Kim S.R. Diagnosis of trichomoniasis by polymerase chain reaction. Yonsei Med. J. 1999;40:56–60. doi: 10.3349/ymj.1999.40.1.56. [DOI] [PubMed] [Google Scholar]
- 181.Coombs G.H., North M.J. An analysis of the proteinases of Trichomonas vaginalis by polyacrylamide gel electrophoresis. Pt 1Parasitology. 1983;86:1–6. doi: 10.1017/S0031182000057103. [DOI] [PubMed] [Google Scholar]
- 182.Lockwood B.C., North M.J., Scott K.I., Bremner A.F., Coombs G.H. The use of a highly sensitive electrophoretic method to compare the proteinases of trichomonads. Mol. Biochem. Parasitol. 1987;24:89–95. doi: 10.1016/0166-6851(87)90119-8. [DOI] [PubMed] [Google Scholar]
- 183.Arroyo R., Alderete J.F. Trichomonas vaginalis surface proteinase activity is necessary for parasite adherence to epithelial cells. Infect. Immun. 1989;57:2991–2997. doi: 10.1128/iai.57.10.2991-2997.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Neale K.A., Alderete J.F. Analysis of the proteinases of representative Trichomonas vaginalis isolates. Infect. Immun. 1990;58:157–162. doi: 10.1128/iai.58.1.157-162.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Mallinson D.J., Lockwood B.C., Coombs G.H., North M.J. Identification and molecular cloning of four cysteine proteinase genes from the pathogenic protozoon Trichomonas vaginalis. Pt 10Microbiology. 1994;140:2725–2735. doi: 10.1099/00221287-140-10-2725. [DOI] [PubMed] [Google Scholar]
- 186.Garber G.E., Lemchuk-Favel L.T. Analysis of the extracellular proteases of Trichomonas vaginalis. Parasitol. Res. 1994;80:361–365. doi: 10.1007/BF00932372. [DOI] [PubMed] [Google Scholar]
- 187.Scott D.A., North M.J., Coombs G.H. The pathway of secretion of proteinases in Trichomonas vaginalis. Int. J. Parasitol. 1995;25:657–666. doi: 10.1016/0020-7519(95)99370-X. [DOI] [PubMed] [Google Scholar]
- 188.Wendel K.A., Workowski K.A. Trichomoniasis: Challenges to appropriate management. Clin. Infect. Dis. 2007;44(Suppl. 3):S123–S129. doi: 10.1086/511425. [DOI] [PubMed] [Google Scholar]
- 189.Wright J.M., Webb R.I., O’Donoghue P., Upcroft P., Upcroft J.A. Hydrogenosomes of laboratory-induced metronidazole-resistant Trichomonas vaginalis lines are downsized while those from clinically metronidazole-resistant isolates are not. J. Eukaryot. Microbiol. 2010;57:171–176. doi: 10.1111/j.1550-7408.2009.00455.x. [DOI] [PubMed] [Google Scholar]
- 190.Upcroft J.A., Dunn L.A., Wal T., Tabrizi S., Delgadillo-Correa M.G., Johnson P.J., Garland S., Siba P., Upcroft P. Metronidazole resistance in Trichomonas vaginalis from highland women in Papua New Guinea. Sex. Health. 2009;6:334–338. doi: 10.1071/SH09011. [DOI] [PubMed] [Google Scholar]
- 191.Shokar A., Au A., An S.H., Tong E., Garza G., Zayas J., Wnuk S.F., Land K.M. S-Adenosylhomocysteine hydrolase of the protozoan parasite Trichomonas vaginalis: Potent inhibitory activity of 9-(2-deoxy-2-fluoro-beta,D-arabinofuranosyl)adenine. Bioorg. Med. Chem. Lett. 2012;22:4203–4205. doi: 10.1016/j.bmcl.2012.03.087. [DOI] [PubMed] [Google Scholar]
- 192.Raj R., Singh P., Haberkern N.T., Faucher R.M., Patel N., Land K.M., Kumar V. Synthesis of 1H-1,2,3-triazole linked beta-lactam-isatin bi-functional hybrids and preliminary analysis of in vitro activity against the protozoal parasite Trichomonas vaginalis. Eur. J. Med. Chem. 2013;63:897–906. doi: 10.1016/j.ejmech.2013.03.019. [DOI] [PubMed] [Google Scholar]
- 193.Anthwal A., Rajesh U.C., Rawat M.S., Kushwaha B., Maikhuri J.P., Sharma V.L., Gupta G., Rawat D.S. Novel metronidazole-chalcone conjugates with potential to counter drug resistance in Trichomonas vaginalis. Eur. J. Med. Chem. 2014;79:89–94. doi: 10.1016/j.ejmech.2014.03.076. [DOI] [PubMed] [Google Scholar]
- 194.Kumar K., Bhargava G., Land K.M., Chang K.H., Arora R., Sen S., Kumar V. N-Propargylated isatin-Mannich mono-and bis-adducts: Synthesis and preliminary analysis of in vitro activity against Tritrichomonas foetus. Eur. J. Med. Chem. 2014;74:657–663. doi: 10.1016/j.ejmech.2014.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Kumar K., Liu N., Yang D., Na D., Thompson J., Wrischnik L.A., Land K.M., Kumar V. Synthesis and antiprotozoal activity of mono-and bis-uracil isatin conjugates against the human pathogen Trichomonas vaginalis. Bioorg. Med. Chem. 2015;23:5190–5197. doi: 10.1016/j.bmc.2015.04.075. [DOI] [PubMed] [Google Scholar]
- 196.Nisha N., Tran R., Yang D., Hall D., Hopper M.J., Wrischnik L.A., Land K.M., Kumar V. Cu(I)Cl-promoted synthesis of novel N-alkylated isatin analogs with an extension toward isatin-4-aminoquinoline conjugates: In vitro analysis against Trichomonas vaginalis. Med. Chem. Res. 2014;23:4570–4578. doi: 10.1007/s00044-014-1024-y. [DOI] [Google Scholar]
- 197.Raj R., Sharma V., Hopper M.J., Patel N., Hall D., Wrischnik L.A., Land K.M., Kumar V. Synthesis and preliminary in vitro activity of mono- And bis-1H-1,2,3-triazole-tethered-βlactam-isatin conjugates against the human protozoal pathogen Trichomonas vaginalis. Med. Chem. Res. 2014;23:3671–3680. doi: 10.1007/s00044-014-0956-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Saleh Y.R.H., Saadeh H.A., Kaur H., Goyal K., Sehgal R., Mubarak M.S. The synthesis of novel hybrid compounds containing 5-nitrothiazole moiety as potential antiparasitic agents. Monatshefte für Chemie. 2015;146:2087–2095. doi: 10.1007/s00706-015-1511-y. [DOI] [Google Scholar]
- 199.Adams M., Li Y., Khot H., De Kock C., Smith P.J., Land K., Chibale K., Smith G.S. The synthesis and antiparasitic activity of aryl-and ferrocenyl-derived thiosemicarbazone ruthenium(II)-arene complexes. Dalton Trans. 2013;42:4677–4685. doi: 10.1039/c3dt32740j. [DOI] [PubMed] [Google Scholar]
- 200.Nisha, Mehra V., Hopper M., Patel N., Hall D., Wrischnik L.A., Land K.M., Kumar V. Design and synthesis of β-amino alcohol based β-lactam-isatin chimeras and preliminary analysis of in vitro activity against the protozoal pathogen Trichomonas vaginalis. MedChemComm. 2013;4:1018–1024. doi: 10.1039/c3md00057e. [DOI] [Google Scholar]
- 201.Stringer T., Taylor D., Guzgay H., Shokar A., Au A., Smith P.J., Hendricks D.T., Land K.M., Egan T.J., Smith G.S. Polyamine quinoline rhodium complexes: Synthesis and pharmacological evaluation as antiparasitic agents against Plasmodium falciparum and Trichomonas vaginalis. Dalton Trans. 2015;44:14906–14917. doi: 10.1039/C5DT02378E. [DOI] [PubMed] [Google Scholar]
- 202.Gupta S., Kushwaha B., Srivastava A., Maikhuri J.P., Sankhwar S.N., Gupta G., Dwivedi A.K. Design and synthesis of coumarin-glyoxal hybrids for spermicidal and antimicrobial actions: A dual approach to contraception. RSC Adv. 2016;6:76288–76297. doi: 10.1039/C6RA12156J. [DOI] [Google Scholar]
- 203.Deborggraeve S., Koffi M., Jamonneau V., Bonsu F.A., Queyson R., Simarro P.P., Herdewijn P., Buscher P. Molecular analysis of archived blood slides reveals an atypical human Trypanosoma infection. Diagn. Microbiol. Infect. Dis. 2008;61:428–433. doi: 10.1016/j.diagmicrobio.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 204.Truc P., Jamonneau V., N’Guessan P., N’Dri L., Diallo P.B., Cuny G. Trypanosoma brucei ssp. and T congolense: Mixed human infection in Cote d’Ivoire. Trans. R. Soc. Trop. Med. Hyg. 1998;92:537–538. doi: 10.1016/S0035-9203(98)90904-X. [DOI] [PubMed] [Google Scholar]
- 205.Joshi P.P., Shegokar V.R., Powar R.M., Herder S., Katti R., Salkar H.R., Dani V.S., Bhargava A., Jannin J., Truc P. Human trypanosomiasis caused by Trypanosoma evansi in India: The first case report. Am. J. Trop. Med. Hyg. 2005;73:491–495. doi: 10.4269/ajtmh.2005.73.491. [DOI] [PubMed] [Google Scholar]
- 206.Brun R., Blum J., Chappuis F., Burri C. Human African trypanosomiasis. Lancet. 2010;375:148–159. doi: 10.1016/S0140-6736(09)60829-1. [DOI] [PubMed] [Google Scholar]
- 207.Ferguson M.A.J., Homans S.W., Dwek R.A., Rademacher T.W. Glycosyl-phosphatidylinositol moiety that anchors Trypanosoma brucei variant surface glycoprotein to the membrane. Science. 1988;239:753–759. doi: 10.1126/science.3340856. [DOI] [PubMed] [Google Scholar]
- 208.Mehlert A., Richardson J.M., Ferguson M.A.J. Structure of the glycosylphosphatidylinositol membrane anchor glycan of a class-2 variant surface glycoprotein from Trypanosoma brucei 11Edited by I. B. Holland. J. Mol. Biol. 1998;277:379–392. doi: 10.1006/jmbi.1997.1600. [DOI] [PubMed] [Google Scholar]
- 209.Mehlert A., Sullivan L., Ferguson M.A. Glycotyping of Trypanosoma brucei variant surface glycoprotein MITat1.8. Mol. Biochem. Parasitol. 2010;174:74–77. doi: 10.1016/j.molbiopara.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Mehlert A., Zitzmann N., Richardson J.M., Treumann A., Ferguson M.A. The glycosylation of the variant surface glycoproteins and procyclic acidic repetitive proteins of Trypanosoma brucei. Mol. Biochem. Parasitol. 1998;91:145–152. doi: 10.1016/S0166-6851(97)00187-4. [DOI] [PubMed] [Google Scholar]
- 211.Zamze S.E., Ashford D.A., Wooten E.W., Rademacher T.W., Dwek R.A. Structural characterization of the asparagine-linked oligosaccharides from Trypanosoma brucei type II and type III variant surface glycoproteins. J. Biol. Chem. 1991;266:20244–20261. [PubMed] [Google Scholar]
- 212.Zamze S.E., Wooten E.W., Ashford D.A., Ferguson M.A., Dwek R.A., Rademacher T.W. Characterisation of the asparagine-linked oligosaccharides from Trypanosoma brucei type-I variant surface glycoproteins. Eur. J. Biochem. 1990;187:657–663. doi: 10.1111/j.1432-1033.1990.tb15350.x. [DOI] [PubMed] [Google Scholar]
- 213.Alirol E., Schrumpf D., Amici Heradi J., Riedel A., de Patoul C., Quere M., Chappuis F. Nifurtimox-eflornithine combination therapy for second-stage gambiense human African trypanosomiasis: Medecins Sans Frontieres experience in the Democratic Republic of the Congo. Clin. Infect. Dis. 2013;56:195–203. doi: 10.1093/cid/cis886. [DOI] [PubMed] [Google Scholar]
- 214.Andrade L.O., Andrews N.W. The Trypanosoma cruzi-host-cell interplay: Location, invasion, retention. Nat. Rev. Microbiol. 2005;3:819–823. doi: 10.1038/nrmicro1249. [DOI] [PubMed] [Google Scholar]
- 215.Clayton J. Chagas disease: Pushing through the pipeline. Nature. 2010;465:S12–S15. doi: 10.1038/nature09224. [DOI] [PubMed] [Google Scholar]
- 216.Rassi A., Jr., Dias J.C., Marin-Neto J.A., Rassi A. Challenges and opportunities for primary, secondary, and tertiary prevention of Chagas’ disease. Heart. 2009;95:524–534. doi: 10.1136/hrt.2008.159624. [DOI] [PubMed] [Google Scholar]
- 217.Bhambra A.S., Edgar M., Elsegood M.R.J., Li Y., Weaver G.W., Arroo R.R.J., Yardley V., Burrell-Saward H., Krystof V. Design, synthesis and antitrypanosomal activities of 2,6-disubstituted-4,5,7-trifluorobenzothiophenes. Eur. J. Med. Chem. 2016;108:347–353. doi: 10.1016/j.ejmech.2015.11.043. [DOI] [PubMed] [Google Scholar]
- 218.Trunz B.B., Jedrysiak R., Tweats D., Brun R., Kaiser M., Suwinski J., Torreele E. 1-Aryl-4-nitro-1H-imidazoles, a new promising series for the treatment of human African trypanosomiasis. Eur. J. Med. Chem. 2011;46:1524–1535. doi: 10.1016/j.ejmech.2011.01.071. [DOI] [PubMed] [Google Scholar]
- 219.Bouchikhi F., Anizon F., Brun R., Moreau P. Biological evaluation of glycosyl-isoindigo derivatives against the pathogenic agents of tropical diseases (malaria, Chagas disease, leishmaniasis and human African trypanosomiasis) Bioorg. Med. Chem. Lett. 2011;21:6319–6321. doi: 10.1016/j.bmcl.2011.08.116. [DOI] [PubMed] [Google Scholar]
- 220.Hwang J.Y., Smithson D., Connelly M., Maier J., Zhu F., Guy K.R. Discovery of halo-nitrobenzamides with potential application against human African trypanosomiasis. Bioorg. Med. Chem. Lett. 2010;20:149–152. doi: 10.1016/j.bmcl.2009.11.022. [DOI] [PubMed] [Google Scholar]
- 221.Samant B.S., Sukhthankar M.G. Compounds containing 2-substituted imidazole ring for treatment against human African trypanosomiasis. Bioorg. Med. Chem. Lett. 2011;21:1015–1018. doi: 10.1016/j.bmcl.2010.12.040. [DOI] [PubMed] [Google Scholar]
- 222.Ferrins L., Rahmani R., Sykes M.L., Jones A.J., Avery V.M., Teston E., Almohaywi B., Yin J., Smith J., Hyland C., et al. 3-(Oxazolo[4,5-b]pyridin-2-yl)anilides as a novel class of potent inhibitors for the kinetoplastid Trypanosoma brucei, the causative agent for human African trypanosomiasis. Eur. J. Med. Chem. 2013;66:450–465. doi: 10.1016/j.ejmech.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 223.Pham N.B., Deydier S., Labaied M., Monnerat S., Stuart K., Quinn R.J. N1,N1-Dimethyl-N3-(3-(trifluoromethyl)phenethyl)propane-1,3-diamine, a new lead for the treatment of human African trypanosomiasis. Eur. J. Med. Chem. 2014;74:541–551. doi: 10.1016/j.ejmech.2013.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Samant B.S., Chakaingesu C. Novel naphthoquinone derivatives: Synthesis and activity against human African trypanosomiasis. Bioorg. Med. Chem. Lett. 2013;23:1420–1423. doi: 10.1016/j.bmcl.2012.12.075. [DOI] [PubMed] [Google Scholar]
- 225.Papadopoulou M.V., Bloomer W.D., Rosenzweig H.S., Wilkinson S.R., Kaiser M. Novel nitro(triazole/imidazole)-based heteroarylamides/sulfonamides as potential antitrypanosomal agents. Eur. J. Med. Chem. 2014;87:79–88. doi: 10.1016/j.ejmech.2014.09.045. [DOI] [PubMed] [Google Scholar]
- 226.Papadopoulou M.V., Bloomer W.D., Rosenzweig H.S., O’Shea I.P., Wilkinson S.R., Kaiser M., Chatelain E., Ioset J.R. Discovery of potent nitrotriazole-based antitrypanosomal agents: In vitro and in vivo evaluation. Bioorg. Med. Chem. 2015;23:6467–6476. doi: 10.1016/j.bmc.2015.08.014. [DOI] [PubMed] [Google Scholar]
- 227.Zelisko N., Atamanyuk D., Vasylenko O., Grellier P., Lesyk R. Synthesis and antitrypanosomal activity of new 6,6,7-trisubstituted thiopyrano[2,3-d][1,3]thiazoles. Bioorg. Med. Chem. Lett. 2012;22:7071–7074. doi: 10.1016/j.bmcl.2012.09.091. [DOI] [PubMed] [Google Scholar]
- 228.Alves M.A., de Queiroz A.C., Alexandre-Moreira M.S., Varela J., Cerecetto H., González M., Doriguetto A.C., Landre I.M., Barreiro E.J., Lima L.M. Design, synthesis and in vitro trypanocidal and leishmanicidal activities of novel semicarbazone derivatives. Eur. J. Med. Chem. 2015;100:24–33. doi: 10.1016/j.ejmech.2015.05.046. [DOI] [PubMed] [Google Scholar]
- 229.Upadhayaya R.S., Dixit S.S., Foldesi A., Chattopadhyaya J. New antiprotozoal agents: Their synthesis and biological evaluations. Bioorg. Med. Chem. Lett. 2013;23:2750–2758. doi: 10.1016/j.bmcl.2013.02.054. [DOI] [PubMed] [Google Scholar]
- 230.Rios Martinez C.H., Lagartera L., Kaiser M., Dardonville C. Antiprotozoal activity and DNA binding of N-substituted N-phenylbenzamide and 1,3-diphenylurea bisguanidines. Eur. J. Med. Chem. 2014;81:481–491. doi: 10.1016/j.ejmech.2014.04.083. [DOI] [PubMed] [Google Scholar]
- 231.Sola I., Artigas A., Taylor M.C., Gbedema S.Y., Pérez B., Clos M.V., Wright C.W., Kelly J.M., Muñoz-Torrero D. Synthesis and antiprotozoal activity of oligomethylene-and p-phenylene-bis(methylene)-linked bis(+)-huprines. Bioorg. Med. Chem. Lett. 2014;24:5435–5438. doi: 10.1016/j.bmcl.2014.10.025. [DOI] [PubMed] [Google Scholar]