

Abstract
Background
Carotenoids are a class of terpenoid pigments that contribute to the color and nutritional value of many fruits. Their biosynthetic pathways have been well established in a number of plant species; however, many details of the regulatory mechanism controlling carotenoid metabolism remain to be elucidated. Apricot is one of the most carotenoid-rich fruits, making it a valuable system for investigating carotenoid metabolism. The purpose of this study was to identify key genes and regulators associated with carotenoid metabolism in apricot fruit based on transcriptome sequencing.
Results
During fruit ripening in the apricot cultivar ‘Luntaixiaobaixing’ (LT), the total carotenoid content of the fruit decreased significantly, as did the levels of the carotenoids β-carotene, lutein and violaxanthin (p < 0.01). RNA sequencing (RNA-Seq) analysis of the fruit resulted in the identification of 44,754 unigenes and 6916 differentially expressed genes (DEGs) during ripening. Among these genes, 33,498 unigenes were annotated using public protein databases. Weighted gene coexpression network analysis (WGCNA) showed that two of the 13 identified modules (‘blue’ and ‘turquoise’) were highly correlated with carotenoid metabolism, and 33 structural genes from the carotenoid biosynthetic pathway were identified. Network visualization revealed 35 intramodular hub genes that putatively control carotenoid metabolism. The expression levels of these candidate genes were determined by quantitative real-time PCR analysis, which showed ripening-associated carotenoid accumulation. This analysis revealed that a range of genes (NCED1, CCD1/4, PIF3/4, HY5, ERF003/5/12, RAP2–12, AP2, AP2-like, BZR1, MADS14, NAC2/25, MYB1R1/44, GLK1/2 and WRKY6/31/69) potentially affect apricot carotenoid metabolism during ripening. Based on deciphering the molecular mechanism involved in ripening, a network model of carotenoid metabolism in apricot fruit was proposed.
Conclusions
Overall, our work provides new insights into the carotenoid metabolism of apricot and other species, which will facilitate future apricot functional studies and quality breeding through molecular design.
Keywords: Carotenoids, Apricot, Color, WGCNA, Prunus armeniaca L
Background
Carotenoids are widely distributed secondary metabolites that play important roles in plant physiology and are beneficial to human health as dietary components [1]. They confer flowers and fruits with yellow, orange and red colors, thereby helping to attract insects or animals to disperse seeds and pollen, and they are involved in photosynthesis and photoprotection. Carotenoids also scavenge free radicals in plants and help resist biotic and abiotic stresses [2]. In humans, dietary carotenoids provide vitamin A precursors, scavenge free radicals, strengthen immunity and prevent various diseases, such as certain cancers and cardiovascular diseases [3].
The carotenoid biosynthesis pathway has been well characterized in many plants [4]. The process starts with geranylgeranyl diphosphate (GGPP) synthesis, which involves a condensation reaction between isopentenyl diphosphate isomerase (IPP) and dimethylallyl diphosphate (DMAPP). GGPP is then converted into phytoene by phytoene synthase (PSY), which is in turn converted into lycopene via a series of desaturation and isomerization steps. Lycopene is cyclized by ε-cyclase (LCYE) and β-cyclase (LCYB) or by LCYB to produce α-carotene or β-carotene, respectively. These carotenes are further hydroxylated to produce zeaxanthin. Carotenoids can be cleaved into apocarotenoids (e.g., β-ionone, strigolactones, β-citraurin and abscisic acid) by carotenoid cleavage dioxygenase (CCD) and 9-cis-epoxycarotenoid dioxygenase (NCED) [5–7]. The associated structural genes in this biosynthetic pathway have been studied in detail in a number of plant species [1]. In tomato, the increased production of lycopene during the ripening-related fruit color change from green to red is caused by increased transcription of genes from early ‘upstream’ steps in the biosynthetic pathway, including PSY, phytoene desaturase (PDS), carotenoid isomerase (CRTISO) and deoxy-d-xylulose-5-phosphate synthase (DXS), and the downregulation of the ‘downstream genes’ LCYB, LCYE, and β-carotene hydroxylase (CHYB) [8–11]. Additionally, several transcription factors associated with carotenoid accumulation have been identified. For example, ethylene response factors (ERFs) and phytochrome interacting factors (PIFs) have been shown to reduce carotenoid accumulation by binding specifically to the AtPSY and AtPDS promoter in Arabidopsis thaliana [12, 13]. In tomato fruit tissues, the MADS box transcription factor ripening inhibitor (RIN) has been reported to regulate carotenoid accumulation by interacting with the SlPSY1 promoter [14, 15]. However, many details of the regulatory network controlling carotenoid metabolism have yet to be identified.
Apricot (Prunus armeniaca L.) is an important fruit crop that is grown in temperate climates. Fresh apricots are an excellent source of diverse nutrients, including carotenoids, polyphenols, ascorbic acid and various microelements [16]. Processed apricot fruit byproducts have also been suggested to be valuable for human nutrition and the treatment of different diseases [16]. The ‘Luntaixiaobaixing’ (LT) apricot cultivar from China is commercially popular and valued by consumers due to its attractive flavor, shape and color. During ripening, its color changes from green to yellow and then to light yellow, and the fruit exhibits a reduction in carotenoid levels [17], which involves a range of carotenoid metabolic reactions, including synthesis, accumulation and cleavage. Thus, the ‘LT’ cultivar represents an excellent model for studying the regulatory mechanism of carotenoid metabolism.
RNA sequencing (RNA-Seq) is a powerful technology for studying gene expression, identifying genes and performing functional analysis [18], due to its high throughput, low cost, high accuracy and high sensitivity [19]. In recent years, RNA-Seq has been applied to the study of many fruit species to characterize the molecular processes underlying ripening and secondary metabolism [20–23]. It is a particularly effective approach for species for which whole-genome sequence data are not yet available, such as apricot. At present, the public resources of apricot transcriptome data are still very limited [24, 25].
In this study, we integrated weighted gene coexpression network analysis (WGCNA), gene-trait correlations and differential expression analysis in apricot fruit to identify key genes and regulators related to carotenoid metabolism during fruit ripening. Based on this analysis, a network model of carotenoid metabolism is proposed. These data will help advance the functional genomic analysis of carotenoid metabolism in apricot and other species.
Results
Changes in basic quality parameters and carotenoid content during apricot fruit ripening
During the three apricot ripening stages (T, turning, 57 DPA (days post anthesis); CM, commercial maturation, 65 DPA; FR, fully ripe, 74 DPA), ‘LT’ fruit color changed from green to yellow and then from yellow to light yellow (Fig. 1a). Over the same period, fruit firmness dramatically decreased from 25.55 N to 2.77 N compression (p < 0.01), but fruit weight sharply increased from an average of 8.7 g to 19.0 g (Fig. 1b). Similarly, the content of fruit total soluble solids (TSS) significantly increased from 10.1 oBrix to 19.9 oBrix, while titratable acidity (TA) decreased substantially from 2.43 mg/mL to 0.4 mg/mL (p < 0.01) (Fig. 1b). We also observed that the citrus color index (CCI) values of the fruit significantly increased from − 6.77 to − 0.64 during ripening (p < 0.01) (Fig. 1c). Notably, the levels of lutein, violaxanthin and β-carotene significantly decreased during ripening (Fig. 1c), and the total carotenoid content markedly decreased from 51.4 μg/g fresh weight (FW) to 21.3 μg/g FW (p < 0.01). Conversely, the contents of neoxanthin and phytoene increased during the process (Fig. 1c). These results indicated that the ‘LT’ fruit underwent normal ripening based on a range of physiological and biochemical characteristics and that the cultivar is a ‘carotenoid loss’ type of cultivar.
Fig. 1.

Characterization of fruit quality traits during ripening. a Color changes of apricot fruit. b Fruit firmness, fruit weight, total soluble solid (TSS) and titratable acid (TA) contents. c Changes in the citrus color index (CCI) and carotenoid content in apricot fruit. LSD, least significant difference (p < 0.01). Error bars represent ± SD of the means of three biological replicates
Transcriptome analysis of fruit ripening
Nine mRNA libraries were generated from the T, CM and FR stages of ‘LT’ fruit, with three biological replicates per stage. Average clean read numbers of 26.3, 26.0 and 27.2 million were derived from the T, CM and FR stages, respectively, corresponding to approximately 2.37, 2.34 and 2.45 gigabase pairs of nucleotides (nt), with GC percentages of 45.88, 45.58 and 46.16% (Additional file 1). Principal component analysis (PCA) confirmed the reproducibility of the sequence data for each stage (Fig. 2).
Fig. 2.
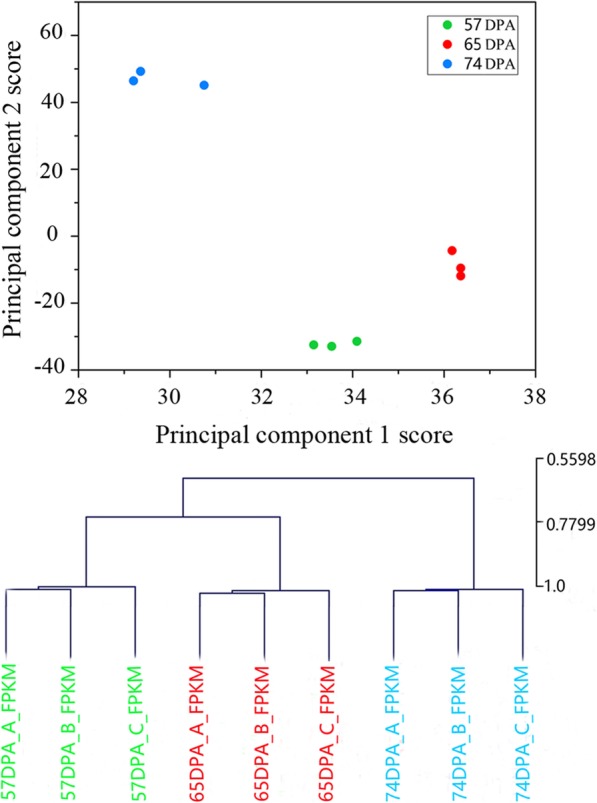
Principal component analysis (PCA) of transcriptome data. Three biological replicates per sample were analyzed for each ripening stage. The percentages on the axes indicate the values explained by each PCA. The green, red, and sky blue dots represent the transcriptomes of the samples obtained at 57 DPA (days post anthesis), 65 DPA and 74 DPA, respectively
Next, a total of 44,754 unigenes with a mean length of 1196 bp and an N50 length of 1860 nt and a total of 71,243 contigs with a mean length of 529 bp and an N50 of 1303 nt, were assembled using Trinity software (Additional file 2). A total of 23,784 unigenes were assigned gene ontology (GO) terms and classified into 55 subcategories within the three standard categories of ‘biological process’, ‘cellular component’ and ‘molecular function’. The subcategories ‘metabolic process’ and ‘cellular process’ were the most highly enriched in the ‘biological process’ domain, while ‘cell’ and ‘cell part’ were the most highly enriched subcategories in the ‘cellular component’ domain, and ‘catalytic activity’ and ‘binding’ were the top ranked terms in the ‘molecular function’ domain (Additional file 3).
Identification of differentially expressed genes (DEGs)
On the basis of the mapped reads, a total of 6916 DEGs were identified using fragments per kilobase of transcript per million mapped reads (FPKM) values, with 2436, 4801 and 5728 DEGs present in the T, CM and FR stages, respectively (Fig. 3a). The numbers of unique DEGs in the different stages were 630, 1288, and 1561 at 57 vs. 65 DPA, 65 vs .74 DPA and 57 vs. 74 DPA, respectively, and a total of 448 genes were expressed at all three stages. The identified DEGs were selected for further analysis (Fig. 3b and Additional file 4).
Fig. 3.
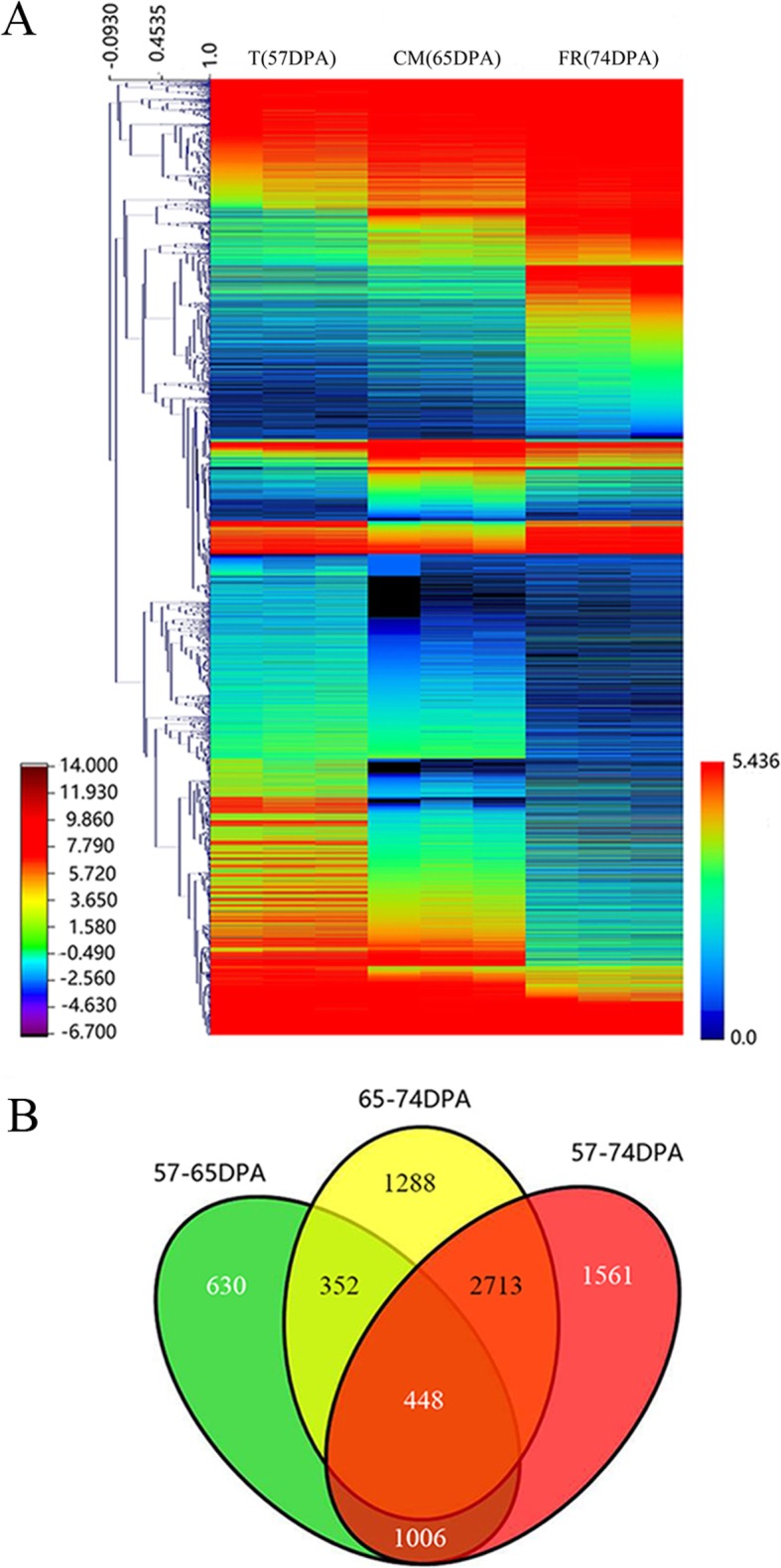
Expression profiles of differentially expressed genes (DEGs) during apricot fruit ripening. a DEG expression dynamics during fruit ripening. b Venn diagram indicates the number of DEGs in each of the three ripening stages
GO enrichment and Kyoto encyclopedia of genes and genomes (KEGG) pathway analysis
To help identify the biological functions of the DEGs, the GO and KEGG databases were used to identify enriched terms/pathways at the three ripening stages for the identified DEGs. ‘Cellular process’, ‘metabolic process’ and ‘single organism process’ were the main categories in the ‘biological process’ domain, while the ‘cell’, ‘cell part’ and ‘organelle’ categories were enriched in the ‘cellular component’ domain. Finally, molecular functions such as ‘binding’, ‘catalytic activity’ and ‘transporter activity’ were the most highly enriched during the three ripening stages (Additional file 5).
KEGG pathway enrichment analysis was carried out to assess the biological significance of the DEGs during the three ripening stages. A total of 1773, 4719, 3215 unigenes from the T, CM and FR stages, respectively, were mapped to 20 KEGG pathways. ‘Metabolic pathways’, ‘plant-pathogen interaction’ and ‘plant hormone signal transduction’ were the most significantly enriched metabolic pathways. We identified 19, 33 and 16 DEGs from the carotenoid biosynthetic pathway enriched in the T, CM and FR stages, respectively. This analysis suggested that the three enriched metabolic pathways may play a role in carotenoid metabolism in apricot fruit (Additional file 6).
Weighted gene coexpression network analysis (WGCNA)
WGCNA is a method for describing the correlations in patterns of gene expression and for revealing clusters, or modules, of genes whose expression is highly correlated. This method allows the identification of module eigengenes and intramodular hub genes and the association of modules with one another and with sample traits [26]. Here, 13 coexpression modules were identified by WGCNA, among which the ‘blue’ and ‘turquoise’ modules were significantly associated with carotenoid metabolism during ripening (Fig. 4). Analysis of the module-trait relationships suggested that the ‘blue’ module, containing 4981 genes, was highly positively correlated with levels of violaxanthin (r = 0.99, P = 8e-07), lutein (r = 0.84, P = 0.005) and β-carotene (r = 0.94, P = 2e-04) and negatively correlated with neoxanthin (r = − 0.47, P = 0.2) and phytoene (r = − 0.75, P = 0.02) contents during ripening. The abundance of phytoene was highly positively correlated with gene expression in the ‘turquoise’ module, which contained 5258 genes, with a coefficient of 0.95 (P = 7e-05), but the levels of violaxanthin and lutein were significantly negatively correlated with gene expression, with coefficients of − 0.87 (P = 0.002) and − 0.97 (P = 2e-05), respectively (Fig. 4b). These results suggested that the genes in these two modules were associated with carotenoid accumulation.
Fig. 4.

Weighted gene coexpression network analysis (WGCNA) of apricot fruit during ripening. a Hierarchical clustering showing 13 modules of coexpressed genes. Each leaf in the tree represents one gene. b Module-carotenoid correlations and corresponding p-values. The left panel shows the 13 modules and the number of genes in each module. The right panel shows a color scale for module/trait correlations from − 1 to 1
DEGs involved in carotenoid biosynthesis
In the carotenoid biosynthesis pathway (Fig. 5a), we identified 33 DEGs that were annotated as functioning in carotenoid metabolism; their expression levels were represented by both FPKM and qPCR values (Fig. 5b, Additional file 7), and good linear positive correlations were observed between them (Fig. 5c). During ripening, the expression levels of two IPI genes (U7591 and U10599) were downregulated and upregulated, respectively. In addition, the expression of the geranylgeranyl diphosphate synthase (GGPPS) gene was significantly upregulated, and the expression levels of two PSY (U14684 and U3487) genes were upregulated and up/downregulated, respectively. The expression levels of four PDS1 genes gradually increased during ripening, with PDS1 (U17285) representing the exception. Three ζ-carotene desaturase (ZDS) genes (CL1821.1, CL3007.2 and U15347) displayed similar up/downregulated expression patterns, and other ZDS genes showed significant upregulation. CRTISO expression markedly decreased during ripening, and the expression of three LCYB genes showed similarly upregulated expression, with LYCB (CL4657.1) representing the exception, but the expression of two CHYB genes showed the opposite expression pattern. Five zeaxanthin epoxidase (ZEP) genes exhibited different levels of expression. Two violaxanthinde-epoxidase 1 (VDE1) genes were downregulated, while the expression of neoxanthin synthase (NXS) was upregulated at the FR stage. The expression of the NCED1 gene as well as CCD1 and CCD4 gradually increased during ripening. Thus, numerous DEGs that were involved in the ripening-related synthesis and accumulation of carotenoids were identified.
Fig. 5.

Expression of genes related to carotenoid biosynthesis in apricot fruit. a Carotenoid biosynthesis pathway in apricot fruit. IPI, isopentenyl diphosphate isomerase; GGPPS, geranylgeranyl diphosphate synthase; PSY, phytoene synthase; PDS, phytoene desaturase; ZDS, ζ-carotene desaturase; CRTISO, carotenoid isomerase; LCYE, lycopene ε-cyclase; LCYB, lycopene β-cyclase; CHYB, β-carotene hydroxylase; ZEP, zeaxanthin epoxidase; VDE, violaxanthin de-epoxidase; NXS, neoxanthin synthase; CCD, carotenoid cleavage dioxygenase; NCED, 9-cis-epoxycarotenoid dioxygenase. b Heatmap of the expression of genes related to carotenoid biosynthesis during fruit ripening. Columns and rows represent samples and gene names in the heatmap, respectively. Red, blue and white indicate high expression, low expression and the absence (or undetectable levels) of detectable transcripts at the corresponding stage, respectively. c The linear fitting equation and R squared values between the FPKM and qRT-PCR values
Visualization of gene networks
A total of 78 genes in the ‘blue’ module and 15 genes in the ‘turquoise’ module were analyzed using Cytoscape version 2.8. Both modules exhibited a significant association with carotenoid contents and gene expression of > 0.4 (Fig. 6 and Additional file 8). In the two networks, 15 ERF, 9 bHLH and 24 WRKY family members were identified as intramodular hub genes and were therefore putatively associated with the regulation of carotenoid metabolism. The carotenoid biosynthesis pathway genes PDS1, CCD1, CCD4, VDE1 and ZEP were identified as key control points for carotenoid metabolism (Fig. 6a and b). In addition to these structural genes, we identified transcription factors in the two modules, including U7919, C4195.1 and U6922, which are homologs of the Arabidopsis thaliana MYB1R1, MYB21 and MYB44 proteins, respectively, involved in the regulation of carotenoid metabolism. Homologs of the A. thaliana transcription factors MYC2, GLK1, GLK2, NAC25, HY5, PIF3, MADS14, WRKY6, WRKY31, WRKY69, ERF003, RAP2–12 (ERF), NAC and bHLH68 were also identified.
Fig. 6.

Coexpression networks of transcription factors and structural genes involved in carotenoid metabolism. The network includes transcription factors and structural genes from the ‘blue’ (a) and ‘turquoise’ (b) modules. Dot sizes and colors represent the numbers of connections between genes
Expression of candidate genes and regulators during fruit ripening
To verify the reliability of the RNA-Seq results and ascertain the relationship between candidate genes and carotenoid metabolism, we also measured the expression levels of the 12 candidate structural genes and 35 transcription factors by qRT-PCR during fruit ripening (Fig. 7a). Overall, all upstream genes related to carotenoid biosynthesis, such as PDS1 (CL1571.2), ZEP (U21300 and U10800), CCD1/4 and capsanthin/capsorubin synthase (CCS), were upregulated in the ‘LT’ fruit, while the expression levels of 15-cis-zeta-carotene isomerase (Z-ISO), PDS1 (U17285), ZEP (U20649) and VDE1 were downregulated. ERF003, AP2-like, RAP2–12, NAC (CL3330.2), WRKY6/31 and BZR1 were significantly upregulated during ripening, but other genes showed opposite expression patterns. Linear regression analysis demonstrated that the FPKM values were highly correlated with the qRT-PCR results (Fig. 7b).
Fig. 7.

The expression of genes and regulators related to carotenoid metabolism in the visualized networks for the ‘blue’ and ‘turquoise’ modules. a Heatmap of the expression of structural genes related to carotenoid biosynthesis during fruit ripening. Columns and rows in the heat map represent the developmental stage and gene name, respectively. The black and blue genes represent structural genes and transcription factors, respectively. Red, green and white indicate high expression, low expression and the absence (or undetectable levels) of detectable transcripts at the corresponding stage, respectively. b The linear fitting equation and R squared values between the FPKM and qRT-PCR values
Discussion
An increasing number of studies highlight the complexity and diversity of carotenoid metabolism in different plant species [1, 4]. Here, we used the apricot cultivar ‘LT’, which exhibits a specific carotenoid accumulation pattern that includes biosynthesis, sequestration and degradation during fruit ripening, to identify key candidate genes and regulators related to carotenoid metabolism by WGCNA. Based on our results, we conclude that carotenoid metabolism in apricot fruit is cocontrolled by light signals, phytohormones, and developmental factors, and a network regulatory model was proposed (Fig. 8).
Fig. 8.
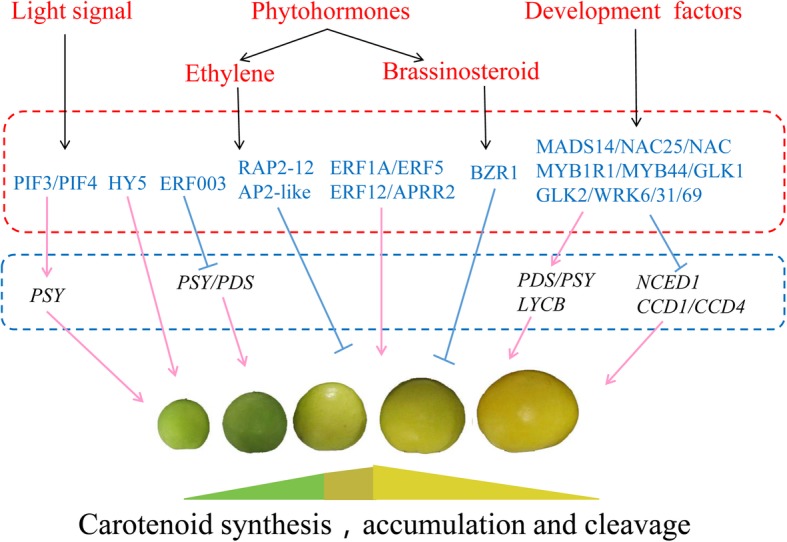
The proposed model of carotenoid metabolism in apricot during fruit ripening. Pink arrows represent positive regulation; blue lines represent negative regulation; black and skyblue genes represent structural genes and transcription factors, respectively. PIF, phytochrome interacting factor; HY5, LONG HYPOCOTYL5; RAP2–12, Ethylene-responsive transcription factor RAP2–12; ERF, Ethylene response factor; BZR1, BRASSINAZOLE RESISTANT1; MADS14, MADS-box transcription factor 14; NAC, NAC transcription factor; MYB, MYB transcription factor; GLK, GOLDEN 2-LIKE transcription factor
Light is a key factor in plant growth and development, since it provides energy for photosynthesis and regulates photomorphogenesis as a signal [4, 5]. PIF is a member of the bHLH transcription factor family and can integrate multiple environmental and intercellular signals to control growth and development, stress responses and secondary metabolism. A light signal causes a rapid transfer of active phytochrome to the nucleus, where it interacts with PIFs [27, 28]. In the present study, we observed that the expression of two PIF3 genes and two PIF4 genes was positively correlated with carotenoid accumulation during ripening (Fig. 1c and Fig. 7a). Our results are in agreement with the observation that PIF5 functions as a positive regulator of the MEP pathway and increases the accumulation of chlorophylls and carotenoids in cultured cells [29], but they contrast with the finding that PIF1 proteins bind directly to the G-box element in the PSY promoter to inhibit its expression, resulting in reduced carotenoid accumulation [13], which suggests that PIF gene members may play a different role in carotenoid metabolism. Two light signaling transcription factors, LONG HYPOCOTYL5 (HY5) and COP1-like, were previously identified in tomato fruit. In contrast to LeCOP1LIKE, repression of LeHY5 results in a decrease in carotenoid content [30]. In this study, we observed that the expression level of HY5 gradually decreased during ripening, in parallel with the decline in carotenoid levels in apricot fruit. In Arabidopsis, HY5 and PIFs form a dynamic activation-suppression transcriptional module that is responsive to light and temperature cues and regulates carotenoid and chlorophyll accumulation by direct binding to the same G-box cis element of a target gene [31]. However, more work is required to confirm whether this fine-tuning module exists in fruit. Taken together, these results suggest that light signaling-associated transcription factors are involved in regulating carotenoid accumulation in ripening apricot fruit.
Previous studies have shown that the synthesis and accumulation of carotenoids are regulated by various phytohormones. Tomato fruit ripening is triggered by an increase in ethylene biosynthesis and is accompanied by a large increase in β-carotene and lycopene content as well as the expression of SIPSY1 and SIPDS, which is ethylene dependent [32]. Similarly, ethylene induces the expression of PSY1 and PDS and promotes the accumulation of the corresponding carotenoid product in apricot [32]. This is consistent with our observation that the upregulation of the expression of PDS1 (CL1571.2) and the ethylene response factor ERF003 was correlated with increased phytoene and neoxanthin levels during ripening. We propose that PDS1 may also be regulated by ethylene in apricots to promote carotenoid accumulation (Fig. 1c and Fig. 7). In tomato, the suppression of SIAP2a mediated by RNAi results in lower levels of carotenoids, and reduced expression of SIERF6 enhances carotenoid accumulation, suggesting that these genes play positive and negative roles, respectively, in carotenoid accumulation during ripening [33, 34]. In our study, ERF genes showed two distinct expression patterns, indicating that ethylene signaling member has different functions in the regulation of carotenoid accumulation in apricot fruit (Fig. 7a). In A. thaliana, the ERF gene RAP2.2 directly binds to the AtPSY and AtPDS promoters to reduce carotenoid accumulation [12]. Here, we also observed that the increased expression levels of RAP2–12 showed a negative correlation with the levels of carotenoid accumulation, while ERF1A, ERF5/12, and APRR2 were positively correlated with carotenoid accumulation (Fig. 7a).
In other studies, the application of the brassinosteroid (BR) 2,4-epibrassinolide (EBR) to tomato has led to increased levels of lycopene and a reduced chlorophyll content in the fruit [35]. Additionally, the transcription factor BRASSINAZOLE RESISTANT1 (BZR1) has been shown to play a role in carotenoid accumulation in tomato fruit [36]. In this study, we observed increased expression of BZR1 during ripening, suggesting that the BR signaling pathway may also be associated with carotenoid accumulation (Fig. 7b). Over time, our results are consistent with the regulation of carotenoid accumulation during apricot fruit ripening by both ethylene and BR.
Numerous developmental regulatory factors have been shown to affect the synthesis and accumulation of carotenoids. These include the MADS-box genes AGAMOUS-LIKE 1 and FRUITFUL1, which modulate carotenoid accumulation during tomato fruit ripening [37], and the MADS-box transcription factors RIN and FUL1/TDR4, which directly bind to the SlPSY1 and SIPDS1 promoters to regulate carotenoid synthesis [38, 39]. Recently, a sweet orange (Citrus sinensis) transcription factor, CsMADS6, was found to promote carotenoid accumulation by coordinately modulating the expression of LCYB1, PSY, PDS and CCD1 [40]. Here, we observed that the downregulation of MADS14 expression was accompanied by upregulated expression of CCD1/4 and downregulated expression of PDS1 (U17285), consistent with their involvement in carotenoid accumulation during ripening (Fig. 1c and Fig. 7a). NAC transcription factors are plant specific and play important roles in the development of various organs and fruit [41]. Overexpression of SlNAC1 and SlNAC4 results in reduced carotenoid levels and inhibition of fruit ripening by altering the carotenoid pathway flux and decreasing the levels of ethylene biosynthesis genes belonging to system-2, but a newly identified tomato NAC (N AM/A TAF1/2/C UC2), SlNAC4, functions as a positive regulator of fruit ripening and carotenoid accumulation [41, 42]. In this study, we observed that three NAC (NAC25, NAC_U12188, NAC_CL3330.2) genes with different expression patterns were involved in regulating carotenoid accumulation (Fig. 7a).
MYB TFs regulate diverse plant developmental processes. The R2R3-MYB TF RCP1 is a master regulator that controls the entire CBP during flower development and positively regulates carotenoid biosynthesis during Mimulus lewisii flower development [43]. MYB7 can activate the AdLCYB promoter, thereby modulating carotenoid pigment accumulation in kiwifruit (Actinidia deliciosa) [44]. Similarly, the significantly reduced expression of MYB1R1 and MYB44 during ripening suggested their involvement in carotenoid accumulation in apricots (Fig. 7a). In contrast, CrMYB68 suppresses the expression of CrBCH2 and CrNCED5 in the citrus carotenoid pathway [45]. Another MYB transcription factor, GOLDEN2-LIKE (GLK2), has been shown to increase lycopene levels in tomato fruit at the red ripe stage [46–48]. Here, the expression levels of GLK1 and GLK2 decreased during ripening, suggesting that their expression may limit carotenoid biosynthesis during ripening in apricots (Fig. 7a). Overexpression of SlPRE2, an atypical bHLH transcription factor, negatively affects plant morphology and fruit pigment accumulation in tomato by downregulating the expression of the chlorophyll-related genes GLK2 and RbcS and the carotenoid biosynthesis-related genes PSY1A and ZDS in ripening tomato [49], suggesting that bHLH TFs may regulate carotenoids upstream of GLKs. In the present study, we identified three bHLHs with downregulated expression patterns, but their regulatory roles are still unknown. We also examined the expression patterns of the WRKY6/31/69 transcription factors (Fig. 7b), whose homolog OfWRKY3 has been implicated as a positive regulator of the OfCCD4 gene in Osmanthus fragrans [50]. Collectively, these results indicated that many developmental factors take part in regulating carotenoid biosynthesis and accumulation in apricots.
We noted that higher expression levels of PDS1, ZEP and VDE1 were detected in ‘LT’ apricot fruit. In addition, the two genes responsible for carotenoid cleavage (CCD1 and CCD4) exhibited remarkable upregulated expression patterns in the ‘carotenoid loss’ type of cultivar during fruit ripening (Fig. 7a). Similarly, studies in strawberry showed that changes in lutein content were closely related to the expression level of CCD1 during ripening [51]. CCD4 has also been shown to control peach flesh pigmentation, with high CCD4 transcript abundance being observed in white flesh peach, which is associated with the emission of carotenoid-derived volatiles [52]. In this study, the upregulated expression of two ZEP genes (U10800 and U21300) was accompanied by a decrease in violaxanthin during ripening. Loss of function of ZEP in the aba1 mutant of A. thaliana and the aba2 mutant of tobacco causes the accumulation of high zeaxanthin levels in leaves [53, 54]. These observations suggest that ZEP, CCD1 and CCD4 play central roles in determining carotenoid levels in apricots and that degradation is the key control point for carotenoid accumulation.
Conclusions
In this study, WGCNA analysis revealed that two modules (‘blue’ and ‘turquoise’) are highly correlated with carotenoid metabolism. Structural genes (CCD1, CCD4, VDE1, ZEP and PDS1) are associated with the carotenoid biosynthesis pathway, and transcription factors related to light signaling (PIF3/4 and HY5), phytohormones (ERF4/5/12, AP2, AP2-like and BZR1) and development factors (MADS14, NAC2/25, MYB1R1/44, GLK1/2, and WRKY6/31/69) may play important regulatory roles in carotenoid metabolism, but the specific regulatory mechanism still needs to be elucidated. Our findings not only provide new insights into the mechanisms of carotenoid metabolism, but these datasets also provide a useful platform for further functional studies of candidate genes.
Methods
Plant materials
From May to July 2017, fruits from the ‘LT’ cultivar were harvested at different ripening stages (turning, T, 57 DPA (days post anthesis); commercial maturation, CM, 65 DPA and fully ripe, FR, 74 DPA) from the National Fruit Tree Germplasm Repository, Academy of Xinjiang Agricultural Sciences, Luntai, Xinjiang, China (Fig. 1a). All samples were transported to the laboratory on the day of harvest. A set of fruit of uniform size and with no obvious mechanical damage was selected for the subsequent experiments. Fifty fruits were used as one replicate, and three biological replicates were used for each sample. For each replicate, 20 fruit were used to measure a range of physiological indices. Other fruit were cut into small cubes and then immediately frozen in liquid nitrogen and stored at − 80 °C for measurement of carotenoid contents and for RNA-Seq and gene expression analysis.
Determination of basic quality parameters
Flesh firmness was measured using a firmness tester (Model: HL-300, Xianlin Non Detection Device Co., Ltd., Nanjing, China) with an 8 mm probe. To determine TSS values, the juice was squeezed from three fruits per replicate, pooled and analyzed using a handheld digital refractometer (B32T Brix Meter, Guangzhou Ruiqi Trading Company, Guangdong, China). TA values were determined after the juice sample was diluted 100 times with pure water. The fruit color parameters were measured using a Hunter’s Mini Scanning Colorimeter (Hunter Associates Laboratory, Inc., Reston, VA, USA). The CCI was calculated with the following formula: CCI = 1000 × a*/(L* × b*), using three fruits as a single replicate and three biological replicates.
Extraction and identification of carotenoids
Carotenoids were extracted as previously described with some modifications [55]. Eight grams of fruit peel was placed in a screw-top tube, and 50 mL of extraction solvent (hexane/acetone/ethanol, 50:25:25, v/v) was added. After standing for 30 min, the samples were centrifuged for 5 min at 6500 rpm. The colored top layer of hexane was recovered and transferred to a volumetric flask, where it was dried under nitrogen, and dissolved in 2 mL of methyl tert-butyl ether (MTBE) before being combined with 2 mL of 10% methanol/potassium hydroxide. The mixture was allowed to stand for 1 h and separated in a separating funnel and was rinsed twice with water and once with 0.1% butylhydroxytoluene (BHT)/MTBE. The rinsed solution was transferred to a brown bottle and dried under nitrogen before 2 mL methanol/acetone (2,1) was added, and the solution was filtered through a 0.22 m filter membrane.
Carotenoids were identified by high-pressure liquid chromatography (HPLC, Waters, Milford, MA, USA) with a C30 chromatography column (250 mm × 4.6 mm, 5 μm, YMC, Wilmington, NC, USA). The mobile phase flow rate was 1 mL/min, and the column temperature was 25 °C, with a detection wavelength of 450 nm and an injection volume of 20 μL. The mobile phase composition was as follows: methanol, MTBE and water. Carotenoids were identified by comparison to standard retention times and UV-visible spectral peaks. The quantification of carotenoids was performed using a standard curve and expressed as μg/g FW. Three biological replicates for each sample were used.
RNA extraction, library construction and data analysis
Total RNA was isolated and extracted from 1 g of fruit peel for each sample using a Tiangen reagent kit (Tiangen, Beijing, China). RNA purity, concentration and integrity were determined using a Nanodrop 2000 (NanoDrop 2000, Wilmington, DC, USA) and denaturing agarose gel electrophoresis. ‘LT’ RNA-Seq libraries of T-, CM- and FR-stage fruit were constructed using an Illumina TruSeq RNA Library Prep Kit v2 following standard procedures, and three biological replicates were used for each stage. The libraries were sequenced using an Illumina HiSeq™ 2000 system at the Beijing Genomics Institute (BGI), China. Clean reads were obtained by removing the linker, repetitive, redundant and low-quality sequences from the raw reads. The sequence data from this study have been deposited in the National Center for Biotechnology Information (NCBI) under Sequence Read Archive (SRA) accession number PRJNA530709.
De novo assembly and functional annotation
De novo transcriptome assembly was carried out using the short read assembly program Trinity (http://trinityrnaseq.sourceforge.net/) [56]. The assembled unigenes were first filtered to remove redundant sequences and further spliced using the TGICL (https://sourceforge.net/projects/tgicl/files/tgicl%20v2.1/) version 2.1 software package [57]. The sequences were then clustered into homologous transcripts to obtain the final unigenes. Subsequently, the unigenes were divided into two classes: one with several unigenes with > 70% similarity, with the prefix CL followed by the cluster ID, and the other consisting of singletons, with the prefix unigene. Finally, BLASTx alignment (e-value < 0.00001) was performed between the unigenes and the NR, Swiss-Prot, KEGG and COG protein databases, and the best alignments were used to determine the sequence direction of the unigenes. The GO annotations of unigenes were obtained using Blast2GO Version 2.3.4 software (https://www.blast2go.com) [58]. WEGO Version 2.0 (Web Gene Ontology Annotation Plot) software (http://wego.genomics.org.cn) was used to provide the GO functional classifications of all the unigenes [59].
Principal component analysis (PCA) and identification of DEGs
PCA was performed to verify the reproducibility of the sequence data using a previously described method [60]. DEGs between each sample were identified on the basis of FPKM values [61] and pairwise comparisons, with a false discovery rate (FDR) threshold of < 0.001 and an absolute value of the log2 Ratio ≥ 1 using the DESeq R package (v1.10.1) [62]. The DEGs were also analyzed by heatmap clustering using Origin Pro 2018 (Origin Lab, Northampton, MA, USA).
GO and KEGG pathway enrichment analysis
The DEGs were mapped to each term in the GO database (http://www.geneontology.org/) [63], and the number of genes associated with each term was determined. Based on the gene numbers for each GO term, a hypergeometric test was applied to determine the GO terms that were significantly enriched for DEGs compared with the genome background. A corrected p-value < 0.05 was used as the threshold, and the GO terms meeting this condition were defined as significantly enriched for DEGs. A KEGG pathway enrichment analysis was performed using the KEGG database (http://www.genome.jp/kegg/) [64]. After multiple tests and corrections, we determined that pathways with a Q-value 0.05 were significantly enriched in DEGs.
WGCNA and visualization of gene networks
A total of 16,773 unigenes with FPKM values > 1 were used to perform WGCNA by using the WGCNA package in R [26]. The modules were obtained using the automatic network construction function. Modules were identified using default settings, except that the soft power was 10, the min module size was 30, and the merge cut height was 0.25. Finally, Cytoscape version 2.8 (http://www.cytoscape.org/) was used to visualize the carotenoid metabolism regulatory network based on the WGCNA modules [65].
Real-time quantitative PCR
The expression levels of candidate structural genes and transcription factors were measured in ‘LT’ fruit at three ripening stages by qRT-PCR analysis. Ribosomal RNA and actin gene expression was used as a normalization reference as previously described [24]. Specific primers were designed using Primer5 (Additional file 9). Gene expression levels were detected using an iQ5 instrument (Bio-Rad Laboratories, Inc. America) with the SYBR® Premix Ex TaqTM II Kit (TaKaRa Biotechnology (Dalian) Co, Ltd., China). The amplification program was as follows: 95 °C for 1 min, followed by 40 cycles at 95 °C for 20 s, 58 °C for 20 s and 72 °C for 30 s. Each qRT-PCR analysis was performed in triplicate, and the mean value was used for the qRT-PCR analysis. The relative expression of the genes was calculated according to the 2 –ΔΔCT method [66], and OriginPro 2018 (OriginLab, Northampton, MA, USA) was used to analyze the data.
Supplementary information
Additional file 1. Throughput and quality of RNA-Seq data in apricot fruit.
Additional file 2. Statistics of assembly quality.
Additional file 3. Gene ontology (GO) classification map. The horizontal axis represents the type of GO function; the vertical axis on the right represents the number of unigenes annotated to the corresponding GO function; and the vertical axis on the left represents the percentage of the number of unigenes within the total.
Additional file 4. Differentially expressed genes (DEGs) shown in Fig. 3b.
Additional file 5. Gene ontology (GO) annotation of differentially expressed genes (DEGs). (A) T vs CM, (B) T vs FR, (C) CM vs FR. The unigenes were annotated in three categories: biological process, cellular component and molecular functions. The right y-axis indicates the number of genes in a category, and the left y-axis indicates the percentage of a specific gene.
Additional file 6. Significantly enriched differentially expressed genes (DEGs) in Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways during different ripening stages.
Additional file 7. The expression and functional annotation of genes shown in Fig. 5b.
Additional file 8. The expression and functional annotation of genes shown in Fig. 6.
Additional file 9. Primer sequences used for real-time quantitative PCR.
Acknowledgements
We thank Junhui Zhou (Department of Cell Biology and Molecular Genetics, University of Maryland) for comments on an earlier version of this manuscript.
Abbreviations
- AP2
Ethylene-responsive transcription factor AP2
- bHLH
Basic helix-loop-helix protein
- BHT
Butylhydroxytoluene
- BR
Brassinosteroid
- BZR1
BRASSINAZOLE RESISTANT1
- CCD1/4
Carotenoid cleavage dioxygenase 1/4
- CCI
Citrus color index
- CCS
Capsanthin/capsorubin synthase
- CHYB
β-carotene hydroxylase
- CM
Commercial maturation
- COP1-like
CONSTITUTIVE PHOTOMORPHOGENIC 1-like
- CRTISO
Carotenoid isomerase
- DEGs
Differentially expressed genes
- DMAPP
Dimethylallyl diphosphate
- DXS
Deoxy-d-xylulose-5-phosphate synthase
- EBR
2,4-epibrassinolide
- ERF003/5/12
Ethylene-responsive transcription factor 003/5/12
- FDR
False discovery rate
- FPKM
Fragments per kilobase of transcript per million mapped reads
- FR
Fully ripe
- FW
Fresh weight
- GGPP
Geranylgeranyl diphosphate
- GGPPS
Geranylgeranyl diphosphate synthase
- GLK1/2
GOLDEN 2-LIKE transcription factor
- GO
Gene ontology
- HPLC
High-pressure liquid chromatography
- IPP
Isopentenyl diphosphate isomerase
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- LCYB
β-cyclase
- LCYE
ε-cyclase
- LT
Luntaixiaobaixing
- MADS14
MADS-box transcription factor 14
- MEP
Methyl-D-erythritol 4-phosphate
- MTBE
Methyl tert-butyl ether
- MYB1R1/44
Transcription factor MYB1R1/44
- MYC2
Transcription factor MYC2
- NAC2/25
Transcription factor NAC2/25
- NCED1
9-cis-epoxycarotenoid dioxygenase 1
- NXS
Neoxanthin synthase
- PCA
Principal component analysis
- PDS1
Phytoene desaturase 1
- PIF3/4
Phytochrome interacting factor 3/4
- PSY
Phytoene synthase
- qRT-PCR
Quantitative real time reverse transcriptase PCR
- RAP2–12
Ethylene-responsive transcription factor RAP2–12
- RIN
Ripening inhibitor
- RNA-Seq
RNA sequencing
- SRA
Sequence Read Archive
- T
Turning
- TA
Titratable acidity
- TSS
Total soluble solids
- VDE1
Violaxanthinde-epoxidase 1
- WGCNA
Weighted gene coexpression network analysis
- WRKY6/31/69
Transcription factor WRKY6/31/69
- ZDS
ζ-carotene desaturase
- ZEP
Zeaxanthin epoxidase
- Z-ISO
15-cis-zeta-carotene isomerase.
Authors’ contributions
WX designed the project. LZ measured the carotenoids and prepared the manuscript. QZ analyzed the data. WL participated in collecting the materials. SZ participated in assaying the physiological parameters. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (No. 31872046). The funding body played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Availability of data and materials
The datasets supporting the results of this article are included within the article and its additional files. All of the obtained sequences from apricot were deposited in the NCBI Sequence Read Archive (SRA) repository under accession number PRJNA530709 and were released on April 3, 2019.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Lina Zhang and Qiuyun Zhang contributed equally to this work.
Contributor Information
Lina Zhang, Email: Zhangln0923@163.com.
Qiuyun Zhang, Email: swuzqy@163.com.
Wenhui Li, Email: lwh-2003@163.com.
Shikui Zhang, Email: zsk2366287@163.com.
Wanpeng Xi, Phone: +86 23 68250483, Email: xwp1999@zju.edu.cn.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12864-019-6261-5.
References
- 1.Yuan H, Zhang J, Nageswaran D, Li L. Carotenoid metabolism and regulation in horticultural crops. Hortic Res. 2015;2:15036. doi: 10.1038/hortres.2015.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakabayashi R, Saito K. Integrated metabolomics for abiotic stress responses in plants. Curr Opin Plant Biol. 2015;24:10–16. doi: 10.1016/j.pbi.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 3.Rao AV, Rao LG. Carotenoids and human health. Pharmacol Res. 2007;55(3):207–216. doi: 10.1016/j.phrs.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 4.Nisar N, Li L, Lu S, Khin NC, Pogson BJ. Carotenoid metabolism in plants. Mol Plant. 2015;8(1):68–82. doi: 10.1016/j.molp.2014.12.007. [DOI] [PubMed] [Google Scholar]
- 5.Sun T, Yuan H, Cao H, Yazdani M, Tadmor Y, Li L. Carotenoid metabolism in plants: the role of plastids. Mol Plant. 2018;11(1):58–74. doi: 10.1016/j.molp.2017.09.010. [DOI] [PubMed] [Google Scholar]
- 6.Giuliano G, Aquilani R, Dharmapuri S. Metabolic engineering of plant carotenoids. Trends Plant Sci. 2000;5(10):406–409. doi: 10.1016/S1360-1385(00)01749-0. [DOI] [PubMed] [Google Scholar]
- 7.Hirschberg J. Carotenoid biosynthesis in flowering plants. Curr Opin Plant Biol. 2001;4(3):210–218. doi: 10.1016/S1369-5266(00)00163-1. [DOI] [PubMed] [Google Scholar]
- 8.Tal I, Gil R, Dani Z, Joseph H. Cloning of tangerine from tomato reveals a carotenoid isomerase essential for the production of beta-carotene and xanthophylls in plants. Plant Cell. 2002;14(2):333–342. doi: 10.1105/tpc.010303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ronen G, Cohen M, Zamir D, Hirschberg J. Regulation of carotenoid biosynthesis during tomato fruit development: expression of the gene for lycopene epsilon-cyclase is down-regulated during ripening and is elevated in the mutant Delta. Plant J. 1999;17(4):341–351. doi: 10.1046/j.1365-313X.1999.00381.x. [DOI] [PubMed] [Google Scholar]
- 10.Fraser PD, Truesdale MR, Bird CR, Schuch W, Bramley PM. Carotenoid biosynthesis during tomato fruit development (evidence for tissue-specific gene expression) Plant Physiol. 1994;105(1):405–413. doi: 10.1104/pp.105.1.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lois LM, Rodríguez-Concepción M, Gallego F, Campos N, Boronat A. Carotenoid biosynthesis during tomato fruit development: regulatory role of 1-deoxy-D-xylulose 5-phosphate synthase. Plant J. 2000;22(6):503–513. doi: 10.1046/j.1365-313x.2000.00764.x. [DOI] [PubMed] [Google Scholar]
- 12.Welsch R, Maass D, Voegel T, DellaPenna D, Beyer P. Transcription factor RAP2. 2 and its interacting partner SINAT2: stable elements in the carotenogenesis of Arabidopsis leaves. Plant Physiol. 2007;145(3):1073–1085. doi: 10.1104/pp.107.104828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toledo-Ortiz G, Huq E, Rodríguez-Concepción M. Direct regulation of phytoene synthase gene expression and carotenoid biosynthesis by phytochrome-interacting factors. Proc Natl Acad Sci U S A. 2010;107(25):11626–11631. doi: 10.1073/pnas.0914428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Catherine M, Julia V, Petra T, Giovannoni JJ. The tomato MADS-box transcription factor RIPENING INHIBITOR interacts with promoters involved in numerous ripening processes in a COLORLESS NONRIPENING-dependent manner. Plant Physiol. 2011;157(3):1568–1579. doi: 10.1104/pp.111.181107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruizsola MÁ, Rodríguezconcepción M. Carotenoid biosynthesis in Arabidopsis: a colorful pathway. In: Bak S, Beisson F, Bishop G, Hamberger B, Höfer R, Paquette S, Werck-Reichhart D, editors. The Arabidopsis book. Rockville: MD American Society of Plant Biologists; 2012. pp. 1–28. [Google Scholar]
- 16.Fratianni F, Ombra MN, d’Acierno A, Cipriano L, Nazzaro F. Apricots: biochemistry and functional properties. Curr Opin Food Sci. 2018;19:23–29. doi: 10.1016/j.cofs.2017.12.006. [DOI] [Google Scholar]
- 17.Xi W, Zheng H, Zhang Q, Li W. Profiling taste and aroma compound metabolism during apricot fruit development and ripening. Int J Mol Sci. 2016;17:998. doi: 10.3390/ijms17070998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dillies MA, Rau A, Aubert J, Hennequet-Antier C, Jeanmougin M, Servant N, et al. A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Brief Bioinform. 2013;14(6):671–683. doi: 10.1093/bib/bbs046. [DOI] [PubMed] [Google Scholar]
- 19.Malone JH, Oliver B. Microarrays, deep sequencing and the true measure of the transcriptome. BMC Biol. 2011;9:34. doi: 10.1186/1741-7007-9-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li G, Ma J, Tan M, Mao J, An N, Sha G, et al. Transcriptome analysis reveals the effects of sugar metabolism and auxin and cytokinin signaling pathways on root growth and development of grafted apple. BMC Genomics. 2016;17(1):150. doi: 10.1186/s12864-016-2484-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garcia-Seco D, Zhang Y, Gutierrez-Mañero FJ, Martin C, Ramos-Solano B. RNA-Seq analysis and transcriptome assembly for blackberry (Rubus sp. Var. Lochness) fruit. BMC Genomics. 2015;16(1):5. doi: 10.1186/s12864-014-1198-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu K, Xu Q, Da X, Guo F, Ding Y, Deng X. Transcriptome changes during fruit development and ripening of sweet orange (Citrus sinensis) BMC Genomics. 2012;13(1):10. doi: 10.1186/1471-2164-13-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sánchez-Sevilla JF, Cruz-Rus E, Valpuesta V, Botella MA, Amaya I. Deciphering gamma-decalactone biosynthesis in strawberry fruit using a combination of genetic mapping, RNA-Seq and eQTL analyses. BMC Genomics. 2014;15(1):218. doi: 10.1186/1471-2164-15-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Q, Feng C, Li W, Qu Z, Zeng M, Xi W. Transcriptional regulatory networks controlling taste and aroma quality of apricot (Prunus armeniaca L.) fruit during ripening. BMC Genomics. 2019;20(1):45. doi: 10.1186/s12864-019-5424-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manganaris GA, Rasori A, Bassi D, Geuna F, Ramina A, Tonutti P, et al. Comparative transcript profiling of apricot (Prunus armeniaca L.) fruit development and on-tree ripening. Tree Genet Genomes. 2011;7(3):609–616. doi: 10.1007/s11295-010-0360-4. [DOI] [Google Scholar]
- 26.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Llorente B, Martinez-Garcia JF, Stange C, Rodriguez-Concepcion M. Illuminating colors: regulation of carotenoid biosynthesis and accumulation by light. Curr Opin Plant Biol. 2017;37:49–55. doi: 10.1016/j.pbi.2017.03.011. [DOI] [PubMed] [Google Scholar]
- 28.Leivar P, Monte E. PIFs: systems integrators in plant development. Plant Cell. 2014;26(1):56–78. doi: 10.1105/tpc.113.120857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mannen K, Matsumoto T, Takahashi S, Yamaguchi Y, Tsukagoshi M, Sano R, et al. Coordinated transcriptional regulation of isopentenyl diphosphate biosynthetic pathway enzymes in plastids by phytochrome-interacting factor 5. Biochem Biophys Res Commun. 2014;443(2):768–774. doi: 10.1016/j.bbrc.2013.12.040. [DOI] [PubMed] [Google Scholar]
- 30.Yongsheng L, Sherry R, Zhibiao Y, Cornelius B, Ageeth VT, Julia V, et al. Manipulation of light signal transduction as a means of modifying fruit nutritional quality in tomato. Proc Natl Acad Sci U S A. 2004;101(26):9897–9902. doi: 10.1073/pnas.0400935101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Toledo-Ortiz G, Johansson H, Lee KP, Bou-Torrent J, Stewart K, Steel G, et al. The HY5-PIF regulatory module coordinates light and temperature control of photosynthetic gene transcription. PLoS Genet. 2014;10(6):e1004416. doi: 10.1371/journal.pgen.1004416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marty I, Bureau S, Sarkissian G, Gouble B, Audergon JM, Albagnac G. Ethylene regulation of carotenoid accumulation and carotenogenic gene expression in colour-contrasted apricot varieties (Prunus armeniaca) J Exp Bot. 2005;56(417):1877–1886. doi: 10.1093/jxb/eri177. [DOI] [PubMed] [Google Scholar]
- 33.Chung M-Y, Vrebalov J, Alba R, Lee J, McQuinn R, Chung J-D, et al. A tomato (Solanum lycopersicum) APETALA2/ERF gene, SlAP2a, is a negative regulator of fruit ripening. Plant J. 2010;64(6):936–947. doi: 10.1111/j.1365-313X.2010.04384.x. [DOI] [PubMed] [Google Scholar]
- 34.Lee JM, Joung JG, Mcquinn R, Chung MY, Fei Z, Tieman D, et al. Combined transcriptome, genetic diversity and metabolite profiling in tomato fruit reveals that the ethylene response factor SlERF6 plays an important role in ripening and carotenoid accumulation. Plant J. 2012;70(2):191–204. doi: 10.1111/j.1365-313X.2011.04863.x. [DOI] [PubMed] [Google Scholar]
- 35.Vardhini B. Vidya, Rao SSR. Acceleration of ripening of tomato pericarp discs by brassinosteroids. Phytochemistry. 2002;61(7):843–847. doi: 10.1016/S0031-9422(02)00223-6. [DOI] [PubMed] [Google Scholar]
- 36.Liu L, Jia C, Zhang M, Chen D, Chen S, Guo R, et al. Ectopic expression of a BZR1-1D transcription factor in brassinosteroid signalling enhances carotenoid accumulation and fruit quality attributes in tomato. Plant Biotechnol. 2014;12(1):105–115. doi: 10.1111/pbi.12121. [DOI] [PubMed] [Google Scholar]
- 37.Julia V, Pan IL, Arroyo AJM, Ryan MQ, Miyoung C, Mervin P, et al. Fleshy fruit expansion and ripening are regulated by the tomato SHATTERPROOF gene TAGL1. Plant Cell. 2009;21(10):3041–3062. doi: 10.1105/tpc.109.066936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Masaki F, Toshitsugu N, Yoko S, Yasuhiro I. A large-scale identification of direct targets of the tomato MADS box transcription factor RIPENING INHIBITOR reveals the regulation of fruit ripening. Plant Cell. 2013;25(2):371–386. doi: 10.1105/tpc.112.108118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Masaki F, Yoko S, Hiroyuki N, Mamiko K, Junji K, Toshitsugu N, et al. Transcriptional regulation of fruit ripening by tomato FRUITFULL homologs and associated MADS box proteins. Plant Cell. 2014;26(1):89–101. doi: 10.1105/tpc.113.119453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu S, Zhang Y, Zhu K, Yang W, Ye J, Chai L, et al. The citrus transcription factor CsMADS6 modulates carotenoid metabolism by directly regulating carotenogenic genes. Plant Physiol. 2018;176(4):2657–2676. doi: 10.1104/pp.17.01830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ma N, Feng H, Meng X, Li D, Yang D, Wu C, et al. Overexpression of tomato SlNAC1 transcription factor alters fruit pigmentation and softening. BMC Plant Biol. 2014;14(1):351. doi: 10.1186/s12870-014-0351-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu M, Chen G, Zhou S, Tu Y, Wang Y, Dong T, et al. A new tomato NAC (NAM/ATAF1/2/CUC2) transcription factor, SlNAC4, functions as a positive regulator of fruit ripening and carotenoid accumulation. Plant Cell Physiol. 2014;55(1):119–135. doi: 10.1093/pcp/pct162. [DOI] [PubMed] [Google Scholar]
- 43.Sagawa JM, Stanley LE, LaFountain AM, Frank HA, Liu C, Yuan Y-W. An R2R3-MYB transcription factor regulates carotenoid pigmentation in Mimulus lewisii flowers. New Phytol. 2016;209(3):1049–1057. doi: 10.1111/nph.13647. [DOI] [PubMed] [Google Scholar]
- 44.Ampomah-Dwamena C, Thrimawithana AH, Dejnoprat S, Lewis D, Espley RV, Allan AC. A kiwifruit (Actinidia deliciosa) R2R3-MYB transcription factor modulates chlorophyll and carotenoid accumulation. New Phytol. 2019;221(1):309–325. doi: 10.1111/nph.15362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu F, Luo T, Liu C, Wang Y, Yang H, Yang W, et al. An R2R3-MYB transcription factor represses the transformation of α- and β-branch carotenoids by negatively regulating expression of CrBCH2 and CrNCED5 in flavedo of Citrus reticulate. New Phytol. 2017;216:178–192. doi: 10.1111/nph.14684. [DOI] [PubMed] [Google Scholar]
- 46.Waters MT, Moylan EC, Langdale JA. GLK transcription factors regulate chloroplast development in a cell-autonomous manner. Plant J. 2010;56(3):432–444. doi: 10.1111/j.1365-313X.2008.03616.x. [DOI] [PubMed] [Google Scholar]
- 47.Waters MT, Peng W, Muris K, Capper RG, Saunders NJ, Langdale JA. GLK transcription factors coordinate expression of the photosynthetic apparatus in Arabidopsis. Plant Cell. 2009;21(4):1109–1128. doi: 10.1105/tpc.108.065250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Powell ALT, Bennett AB. Uniform ripening encodes a Golden 2-like transcription factor regulating tomato fruit chloroplast development. Science. 2012;336(6089):1711–1715. doi: 10.1126/science.1222218. [DOI] [PubMed] [Google Scholar]
- 49.Liang M-H, Jiang J-G. Analysis of carotenogenic genes promoters and WRKY transcription factors in response to salt stress in Dunaliella bardawil. Sci Rep. 2017;7(1):37025. doi: 10.1038/srep37025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Han Y, Wu M, Cao L, Yuan W, Dong M, Wang X, et al. Characterization of OfWRKY3, a transcription factor that positively regulates the carotenoid cleavage dioxygenase gene OfCCD4 in Osmanthus fragrans. Plant Mol Biol. 2016;91(4):485–496. doi: 10.1007/s11103-016-0483-6. [DOI] [PubMed] [Google Scholar]
- 51.García-Limones C, Schnäbele K, Blanco-Portales R, Luz Bellido M, Caballero JL, Schwab W, et al. Functional characterization of FaCCD1: a carotenoid cleavage dioxygenase from strawberry involved in lutein degradation during fruit ripening. J Agric Food Chem. 2008;56(19):9277–9285. doi: 10.1021/jf801096t. [DOI] [PubMed] [Google Scholar]
- 52.Brandi F, Bar E, Mourgues F, Horváth G, Turcsi E, Giuliano G, et al. Study of'Redhaven'peach and its white-fleshed mutant suggests a key role of CCD4 carotenoid dioxygenase in carotenoid and norisoprenoid volatile metabolism. BMC Plant Biol. 2011;11(1):24. doi: 10.1186/1471-2229-11-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rock CD, Zeevaart J. The ABA mutant of Arabidopsis thaliana is impaired in epoxy-carotenoid biosynthesis. Proc Natl Acad Sci U S A. 1991;88(17):7496–7499. doi: 10.1073/pnas.88.17.7496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marin E, Nussaume L, Quesada A, Gonneau M, Sotta B, Hugueney P, et al. Molecular identification of zeaxanthin epoxidase of Nicotiana plumbaginifolia, a gene involved in abscisic acid biosynthesis and corresponding to the ABA locus of Arabidopsis thaliana. EMBO J. 1996;15(10):2331–2342. doi: 10.1002/j.1460-2075.1996.tb00589.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee HS, Castle WS. Seasonal changes of carotenoid pigments and color in Hamlin, Earlygold, and budd blood orange juices. J Agric Food Chem. 2001;49(2):877–882. doi: 10.1021/jf000654r. [DOI] [PubMed] [Google Scholar]
- 56.Grabherr MG, Haas BJ, Moran Y, Levin JZ, Thompson DA, Ido A, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pertea G, Huang X, Liang F, Antonescu V, Sultana R, Karamycheva S, et al. TIGR gene indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics. 2003;19(5):651–652. doi: 10.1093/bioinformatics/btg034. [DOI] [PubMed] [Google Scholar]
- 58.Ana C, Stefan GT, Juan Miguel GG, Javier T, Manuel T, Montserrat R. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 59.Jia Y, Lin F, Hongkun Z, Yong Z, Jie C, Zengjin Z, et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 2006;34:293–297. doi: 10.1093/nar/gkl031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yeung KY, Ruzzo WL. Principal component analysis for clustering gene expression data. Bioinformatics. 2001;17(9):763–774. doi: 10.1093/bioinformatics/17.9.763. [DOI] [PubMed] [Google Scholar]
- 61.Mortazavi A, Williams B, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 62.Wang L, Feng Z, Wang X, Wang X, Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2009;26(1):136–138. doi: 10.1093/bioinformatics/btp612. [DOI] [PubMed] [Google Scholar]
- 63.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 64.Minoru K, Michihiro A, Susumu G, Masahiro H, Mika H, Masumi I, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36(S1):D480–D484. doi: 10.1093/nar/gkm882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shannon P, Markiel A, Ozier O. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Throughput and quality of RNA-Seq data in apricot fruit.
Additional file 2. Statistics of assembly quality.
Additional file 3. Gene ontology (GO) classification map. The horizontal axis represents the type of GO function; the vertical axis on the right represents the number of unigenes annotated to the corresponding GO function; and the vertical axis on the left represents the percentage of the number of unigenes within the total.
Additional file 4. Differentially expressed genes (DEGs) shown in Fig. 3b.
Additional file 5. Gene ontology (GO) annotation of differentially expressed genes (DEGs). (A) T vs CM, (B) T vs FR, (C) CM vs FR. The unigenes were annotated in three categories: biological process, cellular component and molecular functions. The right y-axis indicates the number of genes in a category, and the left y-axis indicates the percentage of a specific gene.
Additional file 6. Significantly enriched differentially expressed genes (DEGs) in Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways during different ripening stages.
Additional file 7. The expression and functional annotation of genes shown in Fig. 5b.
Additional file 8. The expression and functional annotation of genes shown in Fig. 6.
Additional file 9. Primer sequences used for real-time quantitative PCR.
Data Availability Statement
The datasets supporting the results of this article are included within the article and its additional files. All of the obtained sequences from apricot were deposited in the NCBI Sequence Read Archive (SRA) repository under accession number PRJNA530709 and were released on April 3, 2019.