

Abstract
Background
For infectious diseases, the probability that an animal gets infected depends on its own susceptibility, and on the number of infectious herd mates and their infectivity. Together with the duration of the infectious period, susceptibility and infectivity determine the basic reproduction ratio of the disease (). is the average number of secondary cases caused by a typical infectious individual in an otherwise uninfected population. An infectious disease dies out when is lower than 1. Thus, breeding strategies that aim at reducing disease prevalence should focus on reducing , preferably to a value lower than 1. In animal breeding, however, has received little attention. Here, we estimate the additive genetic variance in host susceptibility, host infectivity, and for the endemic claw disease digital dermatitis (DD) in Holstein Friesian dairy cattle, and estimate genomic breeding values (GEBV) for these traits. We recorded DD disease status of both hind claws of 1513 cows from 12 Dutch dairy farms, every 2 weeks, 11 times. The genotype data consisted of 75,904 single nucleotide polymorphisms (SNPs) for 1401 of the cows. We modelled the probability that a cow got infected between recordings, and compared four generalized linear mixed models. All models included a genetic effect for susceptibility; Models 2 and 4 also included a genetic effect for infectivity, while Models 1 and 2 included a farm*period interaction. We corrected for variation in exposure to infectious herd mates via an offset.
Results
GEBV for from the model that included genetic effects for susceptibility only had an accuracy of ~ 0.39 based on cross-validation between farms, which is very high given the limited amount of data and the complexity of the trait. Models with a genetic effect for infectivity showed a larger bias, but also a slightly higher accuracy of GEBV. Additive genetic standard deviation for was large, i.e. ~ 1.17, while the mean was 2.36.
Conclusions
GEBV for showed substantial variation. The mean was only about one genetic standard deviation greater than 1. These results suggest that lowering DD prevalence by selective breeding is promising.
Background
Infectious disease transmission in a host population is a dynamic process. The probability that an animal is infected depends on its own susceptibility, on the number of infected contact animals (“group mates”), and on the infectivity of those group mates. The number and composition of the infectious contact animals in a population varies over time, since some animals get infected while others recover. Whereas state-of-the-art epidemiological models take this variation into account, it should probably also be taken into account to make optimal genetic inference. However, most genetic studies use linear mixed models that ignore the dynamics of the transmission process in the population, such as the variation in exposure of susceptible individuals to infectious contact individuals. Moreover, studies on host genetic variability commonly connect an individual’s disease status to its own pedigree or genotype only, and therefore capture genetic effects related to susceptibility (or resistance) only [1, 2]. However, individuals probably also differ in infectivity, as suggested for example by the observation of superspreaders, which are animals that infect substantially more contact individuals compared to a typical infectious animal [3]. Recently, studies on selection against disease transmission have started that include the infectivity of the herd mates [4–8].
Genetic inference on infectious diseases can probably be improved by using quantitative genetic models that are founded on epidemiological theory. Moreover, such models would give estimates of genetic variation and breeding values for fundamental epidemiological parameters, such as the basic reproduction number (see also below). Such knowledge would also facilitate the prediction of response to selection while accounting for the non-linear nature of infectious diseases, including phenomena such as positive feedback and the eradication of a disease when falls below 1 [9].
An animals’ infectivity affects the disease status of other animals, rather than its own disease status. Thus, if the observed variation in infectivity has a genetic component, then infectivity is a so-called indirect genetic effect (IGE). IGE can have a considerable effect on the rate and direction of evolution by natural selection, and on response to selective breeding [10–12]. Hence, IGE can and should be used for the genetic improvement of populations whenever they play a role.
Together, susceptibility, infectivity, and the duration of the infectious period determine the basic reproduction ratio (), which is the average number of secondary cases caused by a typical infectious individual in a fully susceptible population [13]. contains information on the ability of an infection to transmit and establish itself in the population [13], and has a threshold value of 1; if is lower than 1, an infectious animal will infect, on average, less than one susceptible animal and the disease will die out with certainty. If is higher than 1, an epidemic disease can affect a substantial proportion of the population, while an endemic disease may persist in the population. For endemic diseases, the prevalence at the equilibrium state depends on , and is (ignoring differences in susceptibility between individuals) equal to when is higher than 1. Because determines the prevalence of a disease in a group of animals (e.g., a herd), breeding strategies that aim at reducing the prevalence should focus on reducing , ideally to a value lower than 1 [4].
In this study, we focused on the endemic infectious disease digital dermatitis (DD). DD is a claw disorder that affects (mainly) the hind feet of (dairy) cattle [14, 15]. Typically, affected cattle have a round lesion above the interdigital space next to the heel bulbs [16]. These lesions can be painful and prone to bleed, and can develop filiform papillae or be surrounded by hyperkeratotic skin with hairs longer than normal [15]. Severely affected cows show signs of lameness; they bear their weight on the toes of the affected foot, shake the foot as if in pain, and show reluctance to move [15, 17, 18]. The herd prevalence of DD in 383 dairy herds in the Netherlands was estimated at 21.2% in 2003, and within these herds cow level prevalence estimates ranged from 0 to 83.0% [19]. Thus, DD has an impact on the welfare of cows and, furthermore, causes economic losses for the farmer [20, 21].
The prevalence of DD is affected by many factors, such as herd management, lactation stage, flooring system, climate, and breed [19]. Optimizing management strategies are one way to reduce the DD prevalence on a dairy farm [22]. An additional strategy could be to improve claw health through genetic selection [23–25]. As we will argue, this is best done by selecting for lower .
The objective of our work was to quantify the genetic variation in host susceptibility, host infectivity, and for DD in Holstein Friesian dairy cattle, and to investigate our ability to estimate genomic breeding values (GEBV) for these traits. In addition, for model validation, models with and without genetic variation for infectivity will be compared for their ability to predict whether susceptible (infection-free) claws of an animal get infected.
Methods
Phenotype data
Phenotypes for DD were collected on 12 dairy cattle farms in the Netherlands, between November 2014 and April 2015. Two observers (author FB being one of them) visited these farms 11 times with a 2-week interval between visits [26]. On each farm, one of the observers rinsed and scored the hind feet using the method of Relun et al. [27], while the other observer recorded cow ID and DD status. Six distinct classes were scored: skin on which lesions were macroscopically absent (M0), displayed a small lesion of 0 to 2 cm (M1), a lesion of more than 2 cm (M2), a lesion covered by a scab (M3), altered skin with dyskeratotis or surface proliferation (M4), and a small lesion in addition to altered skin (M4.1) [28–30]. These classes were divided into susceptible (M0) and infected (M1, M2, M3, M4, and M4.1). Farmers were not informed on the DD status of the cows, but were allowed to identify and treat affected cows using their normal routine. Phenotypes were collected on 1513 cows, of which 1401 were genotyped (see below). On average, a cow was scored 8.7 times. Table 1 shows several characteristics of the farms enrolled in the study.
Table 1.
Characteristics of the farms enrolled in the study
| Farm | Number of cows examineda | Number of cows genotypeda | Number of observationsb | Average (days)c | Number of foot bathsd | Prevalence (SD)e | |
|---|---|---|---|---|---|---|---|
| Cow level | Foot level | ||||||
| A | 134 | 116 | 11 | 14 | 7 | 78.0 (5.4) | 69.6 (6.6) |
| B | 105 | 101 | 11 | 14 | 0 | 56.3 (7.5) | 46.9 (7.9) |
| C | 159 | 152 | 11 | 14 | 5 | 49.7 (2.8) | 40.2 (1.9) |
| D | 118 | 116 | 11 | 14 | 7 | 57.8 (5.0) | 49.2 (5.1) |
| E | 102 | 90 | 11 | 13.55 | 9 | 62.8 (5.0) | 54.6 (5.4) |
| F | 133 | 112 | 10 | 15.56 | 10 | 59.2 (10.0) | 48.7 (10.4) |
| G | 100 | 98 | 11 | 14 | 3 | 65.6 (8.1) | 58.2 (7.6) |
| H | 189 | 180 | 11 | 14 | 7 | 64.9 (6.2) | 56.7 (5.8) |
| I | 104 | 75 | 11 | 14 | 0 | 56.4 (5.1) | 45.6 (4.9) |
| J | 88 | 88 | 11 | 14 | 0 | 65.8 (10.8) | 58.1 (10.9) |
| K | 130 | 116 | 9 | 14 | 13 | 63.6 (9.6) | 52.5 (8.5) |
| L | 151 | 147 | 11 | 13.90 | 3 | 70.9 (7.2) | 62.0 (7.7) |
| Total | 1513 | 1401 | 129 | 14.07 | 64 | 62.6 (7.5) | 53.9 (11.0) |
aTotal number of different cows on a farm
bTotal number farm visits
cAverage number of days between two scorings ()
dTotal number of footbaths given during the study period
eAverage percentage of animals and feet scored as infected, standard deviation (SD) in brackets
Genotype data
The cows were genotyped with the Eurogenomics 10 K chip. A first round of quality control was performed before imputation, following the standard procedure of the breeding company CRV. A marker (single nucleotide polymorphism, SNP) was included only when its call rate was higher than 0.85, the deviation of the observed frequency from the expected Hardy Weinberg equilibrium frequency was less than 0.15, and the minor allele frequency was higher than 0.025. Furthermore, inconsistent genotypes between parents and offspring were set to missing. SNPs that passed the quality control were imputed to a set of 76,438 SNPs based on the Illumina BovineSNP50 chip and a custom chip from the breeding company CRV, with a reference population of more than 1000 animals with genotypes on both chips and the combination of Beagle [31] and PHASEBOOK [32] software. A second round of quality control was performed on the imputed data. A SNP was included only when there was no strong deviation from Hardy Weinberg equilibrium (p > 10−15), the missing rate was lower than 0.05, and the minor allele frequency was higher than 0.02. In total, 75,904 SNPs passed the quality control and were included in the analysis.
Models
In this section, we develop a generalized linear mixed model (GLMM) founded on epidemiological principles, to estimate genetic parameters for susceptibility, infectivity and . To develop the GLMM, we need to find the probability that a susceptible cow gets infected between two observations. We present an epidemiological model, derive the infection probability from this model, and present the resulting GLMM, building on work of Velthuis et al. [5], Lipschutz-Powell et al. [6], Anacleto et al. [7], Anche et al. [4], and Biemans et al. [8].
DD is an endemic infectious disease with infections reoccurring in the same animals. DD is transmitted via the environment [33], where the environment is defined as any possible pathogen reservoir through which transmission can occur. Thus, we used a stochastic compartmental susceptible-infected-susceptible-model (SIS-model) with an environmental route (E) see e.g. [34] to model disease transmission (Fig. 1). An SIS-model has two categories of individuals: (1) non-infected individuals who can become infected; these are referred to as susceptible; and (2) infected individuals that are also infectious, and who can recover; these are referred to as infected. Hence, the term “susceptible” merely denotes a non-infected individual that can, in principle, become infected. It does not indicate a high degree of susceptibility. In the SIS-model, the infection of a susceptible claw occurs randomly with a probability per observation interval, depending on the parameters of the model, the number of infected claws in the group, and the infection pressure coming from the environment.
Fig. 1.
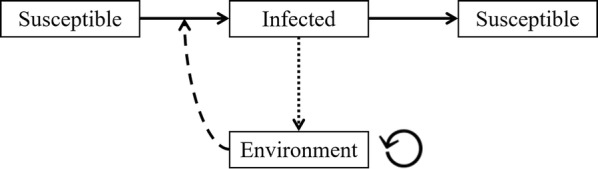
Susceptible-infected-susceptible model with environmental route
In the SIS-model with an environmental route (), the expected rate with which susceptible claws get infected is , where is the number of currently infectious claws, the number of susceptible claws, and the total number of claws in a group (twice the number of cows). Here, it is assumed that infected cows are immediately infectious to their herd mates. is the infection pressure coming from the environmental reservoir, expressed as the equivalent number of currently infectious claws (i.e., and are on the same scale). is the transmission rate parameter that contains information on the contact rate and transmission probability between individuals [35].
Because our interest is in genetic variation in susceptibility and infectivity among cows, we consider the pairwise between a susceptible cow and her infected herd mate. This pairwise depends on the susceptibility of the cow and the infectivity of the herd mate. Thus, the transmission rate parameter from an infected claw of herd mate , with infectivity , to a susceptible claw of cow , with susceptibility , is:
| 1 |
In Eq. 1, the overall contact rate serves as a scaling parameter, so that mean susceptibility and infectivity are approximately 1, , and mean logarithms are 0, The latter is relevant for the inclusion of random effects in mixed models, which are commonly assumed to have a mean of 0.
Thus, the expected (i.e., average) rate with which a single susceptible claw of cow gets infected when exposed to all infected claws in the group depends on the susceptibility of cow , on the number of infected claws in the group, and on their average infectivity:
| 2 |
where is the pairwise transmission rate parameter averaged over the genotyped infected herd mates of susceptible cow , is the number of infected claws of herd mates that had genotype records, and is the total number of infected claws at the start of the observation interval. We distinguished between and because some of the cows were not genotyped. While we estimated infectivity for the genotyped cows only, the non-genotyped infected cows also contributed to transmission. To account for all infected claws, we assumed that the claws of the non-genotyped cows ( = 112) had the same infectivity as their infected genotyped herd mates ().
In the term (Eq. 2), we should ideally average the infectivity over all claws that contribute to the current infection pressure. This also includes the infection pressure via the environment of the claws that were infected at an earlier time. However, in the statistical software we did not manage to keep track of all the weighted genotypes of claws that were infected at earlier times. Therefore, we only included claws that were infected at the start of the observation interval in the term. In contrast, the -term included infection pressure of the full number of previously infected claws, but these were not weighted according to their infectivity. Thus, our estimates of genetic variation in infectivity use only part of the variation in the infection pressure. This issue is further addressed in the “Discussion” section.
The probability that a susceptible claw becomes infected in an observation interval varies among observation intervals, because it depends on the number of infected herd mates, the infectivity of those herd mates, and on the infection pressure coming from the environment. To estimate this probability, we assumed that the transmission rate (probability per unit of time) is constant within the interval, which is the default assumption in disease transmission models. With this assumption, transmission follows a so-called Poisson process, where the number of transmissions within the interval has a Poisson distribution with a mean equal to the product of the rate and the length of the interval; . A claw becomes a case when it is infected at least once within the interval. Hence, the probability of becoming a case is the complement of the probability of no infection, , where is the probability of a zero outcome from a Poisson distribution. Using Eq. 2:
| 3 |
where, is the probability that a susceptible claw of is a case (becomes infected) in interval , given the number of infected herd mates, the infectivity of those herd mates, and on the infection pressure coming from the environment at the start of the interval.
The number of cases for each interval (counting process in discrete time) follows a binomial distribution with a probability that follows from a Poisson process within the interval (counting process in continuous time). Therefore, the complementary log–log is the appropriate link function to connect the explanatory variables to the expected value of the observed variable [36]. Thus, the GLMM follows from applying the complementary log–log transformation to Eq. 3:
| 4 |
where is an intercept, is the logarithm of susceptibility of the focal individual, the log of the mean infectivity of infected (genotyped) herd mates, and is an offset, i.e., a known “explanatory variable”. The offset accounts for the infectious pressure coming from the infected cows (both genotyped and non-genotyped) at time : ( and from the environment at : (), and for the length of the interval (). Note that the dependent variable for Eq. 4 is the number of cases for a cow with at least one susceptible claw. For cows with one susceptible claw, the binomial total equals 1 and the number of cases takes values = 0 or 1. For cows with two susceptible claws, the binomial total equals 2 and the number of cases takes values = 0, 1 or 2.
Equation 4 is linear in the logarithm of susceptibility, but not in the logarithm of infectivity. To allow the fitting of a linear model, we linearized the model term for infectivity following [37] in the models in which the infectivity of the group mates was included (Models 2 and 4, see below):
| 5 |
Equation 5 follows from approximating the , which is an arithmetic mean, by the corresponding geometric mean (see [37] for details). The errors caused by this approximation are less than 5% for infectivity effects up to a factor of 3 (i.e., between 0.33 and 3.0). However, this error estimation is based on a model with SNP effects rather than polygenic breeding values. The size of the error is not known for a trait determined by many genes each with a small effect. The method will anyway take the real differences in exposure for different observation periods on the different farms into account. We acknowledge that this is a relevant issue that requires further investigation.
We calculated the infectious pressure coming from the environment () as described in detail in Biemans et al. [26]. In short, claws that were infected at an earlier time could still contribute partly to the environmental reservoir at the moment of observation. Their contribution was assumed to decrease each interval by a factor , which may be interpreted as a survival rate of the pathogen. The estimate for this survival rate is 0.9 [26]. Thus, the number of pathogens in the environment coming from a claw that was infected at , is a fraction 0.9 at , a fraction 0.92 = 0.81 at , a fraction 0.93 = 0.729 at , etc. Therefore, the values for were calculated as:
| 6 |
where is the total number of infected claws at , and is the infectious pressure coming from the environmental reservoir at .
Because we did not observe the number of infected claws in the period before the first observation (), we estimated this number with a linear model. For each farm, we fitted the model to the number of infected claws over the observation period. The intercept of the model, , was used as the average number of infected claws on that particular farm in the periods before observations started (). Thereafter, the value for the environmental reservoir at the first observation was estimated as, , and was used in Eq. 6.
Implementation of the model
With the GLMM, we modelled the expectation of the number of cases over the number of susceptible feet (claws) of cow within the interval , . Only the hind feet of the cows were scored, so a susceptible cow could have one or two susceptible feet () at the start of an interval, which were zero, one, or two cases by the end of the interval. Thus, the number of cases (0, 1 or 2) for each susceptible cow followed a binomial distribution with binomial total (1 or 2).
We tested four models (Table 2). Model 1 included a genetic effect for susceptibility only:
where is the intercept. The fixed effects were farm ( with = A to L), period ( with = 1 to 10), parity ( with = 1, 2, or > 2), and months in milk (, a continuous covariate with = 1 to 12). Random effects included an interaction between farm and period ( with = A to L and = 1 to 10), a non-genetic permanent animal effect for the susceptible animal () to account for repeated observations on the same animal in different periods, and an additive genetic effect for susceptibility of animal (, where is the genomic relationships matrix among animals).
Table 2.
Overview of the fixed and random effects included in the four models
| Model | Random effects | ||
|---|---|---|---|
| 1 | Genetic susceptibility focal individual | – | Farm*period |
| 2 | Genetic susceptibility focal individual | Genetic infectivity herd mates | Farm*period |
| 3 | Genetic susceptibility focal individual | – | – |
| 4 | Genetic susceptibility focal individual | Genetic infectivity herd mates | – |
All models contained fixed effects for farm, period, parity, and months in milk; and a non-genetic random animal effect for the susceptible animal
Model 2 included genetic effects for both susceptibility and infectivity:
where are the random genetic effects for infectivity of the infected group mates of animal , with ().
We expected that the interaction between farm and period could be partly confounded with the genetic effect for infectivity, because previous IGE studies showed that omitting group-effects may substantially inflate estimated genetic parameters for infectivity [38]. To investigate this issue, we dropped the farm by period interaction from Models 1 and 2, resulting in Models 3 and 4, respectively.
Data analyses
The -matrix was computed using method 1 of VanRaden [39], with the calc_grm software [40]. We fitted the four models with ASReml v4.1.0 [41]. Model fit was assessed with Akaike information criterion (AIC). The susceptibility and infectivity estimates from ASReml were on the log scale because of the complementary log–log link function (Eq. 5). These random effects are zero on average. By taking the exponent of the estimates, we obtained the genomic estimated breeding values (GEBV) for susceptibility and infectivity, relative to a typical (average) individual that has a GEBV of 1 (thus , therefore ).
Cross-validation
We validated the GEBV to investigate their bias and accuracy with a 12-fold cross-validation on all four models. In each analysis, all the cases () of one of the 12 farms were censored from the dataset. For each susceptible claw of cow at interval at the censored farm, we predicted the dependent variable of the GLMM, , based on the information of the other 11 farms. In the following, we refer to this prediction as the predicted probability .
However, as the fixed effects were nonlinear on the normal scale because they were estimated with a complementary log–log link function, correction of the observed records for fixed effects was not straightforward. To solve this issue, we translated both the predicted probabilities and the observed records to a standard (i.e., average) farm. Subsequently, we validated the models using a weighted correlation and a regression of observed records on predicted probabilities (see “Appendix” for detailed methods). To calculate the correlation and regression coefficients, we used observations and predictions averaged over the number of times an animal was susceptible at the start of an interval, and this number was used as the weight.
Genetic variance and breeding values for the basic reproduction ratio
For the best fitting model (lowest bias, highest accuracy), we calculated the genetic variance and GEBV () for the basic reproduction ratio.
The additive genetic variance of was calculated as [4]:
where the approximation follows from
The GEBV for the basic reproduction ratio is the product of the GEBV for susceptibility (), the GEBV for infectivity (, the contact rate (), and the average duration of the infectious period () [4]:
| 7 |
With a previously estimated for DD of 2.36 on these farms [26] and the average product of the estimated relative susceptibility and relative infectivity, the value for was calculated as . The breeding value for in Eq. 7 is on the absolute scale, and thus has an average equal to , rather than zero. This is convenient, because absolute breeding values for can be interpreted relative to a value of 1, which is the threshold for eradication of a disease. Note that, in Models 1 and 3, variation in infectivity is not estimated. Hence, for these models we used for all individuals.
GEBV are prone to overestimation, as illustrated by regression coefficients of validation phenotypes on predicted GEBV, which are often less than 1 e.g. [42]. To avoid overestimation of the variation in breeding values for , we shrunk the estimates for the transmission rate parameter
where is the regression coefficient of the observed on the predicted probability, averaged over cross-validation sets. The corrected individual breeding values for the basic reproduction ratio were:
After this correction, the regression coefficient of the observed on the predicted probability in cross-validation was 1, implying that the GEBV have the correct variance. Moreover, when the regression coefficient of phenotypes on predicted breeding values equals 1, and observations and predictions follow an approximate normal distribution, then the accuracy of EBV equals the ratio of the standard deviations, [43]. We used this result to obtain the approximate accuracy of the GEBV for .
Results
Fixed effects
The farm effect was significant (P < 0.05) in Models 1 and 3, but not in Models 2 and 4. For all models, there was a significant effect for period, parity, and months in milk. The probability of becoming infected during an interval increased during the first six periods and stabilized thereafter. The transmission rate parameter increased with increasing parity; it was 21% higher for parity 2 compared to parity 1, and 69% higher for parities greater than 2 compared to parity 1. For months in milk, the transmission rate parameter decreased by 4% with every month in milk. A table of fixed effect estimates and standard errors on the log scale from Model 1 is presented in Additional file 1.
Estimated variance components
Table 3 shows the estimated variance components and their standard errors (SE) on the log scale. The estimated variance components are similar on the normal scale, because around ( = 1) [44]. For Models 1, 2, and 4, the estimated genetic variance for susceptibility was about 0.55, and was strongly significant. These models had a genetic effect for infectivity and/or the interaction term between farm and period. For Model 3, the estimated genetic variance for susceptibility was smaller (0.49); this model had neither a genetic effect for infectivity, nor a term for the interaction between farm and period. Similarly, the variance of the non-genetic random animal effect was about 0.95 for Models 1, 2, and 4; and was smaller, ~ 0.92, for Model 3. The estimated variance for infectivity was large, and not significant for Model 2. The interpretation of the magnitude of these variance components is discussed below, in the section on .
Table 3.
Estimated variance components and their standard errors (SE) for the genetic effect of susceptibility and infectivity, the interaction between farm and period, and the animal effect for the four models
| Model | Estimated variance (SE) of the random terms | |||
|---|---|---|---|---|
| Susceptibility | Infectivity | Farm*period | Animal | |
| 1 | 0.555 (0.142) | – | 0.262 (0.050) | 0.949 (0.130) |
| 2 | 0.558 (0.142) | 25.43 (16.41) | 0.144 (0.067) | 0.952 (0.130) |
| 3 | 0.490 (0.131) | – | – | 0.922 (0.123) |
| 4 | 0.556 (0.142) | 68.27 (14.13) | – | 0.952 (0.130) |
Figure 2 shows the infectivity estimates from Model 4 plotted against the infectivity estimates from Model 2. The estimates varied from − 3.20 to 3.80 for Model 2 and from − 8.76 to 9.04 for Model 4. Thus, the GEBV for infectivity from Model 2 showed less variation compared to the GEBV from Model 4; part of the variation that is attributed to the interaction between farm and period in Model 2, is attributed to the genetic infectivity effect in Model 4. This suggests that the GEBV for infectivity from Model 4 include both a genetic and a non-genetic component, and may therefore be inflated.
Fig. 2.
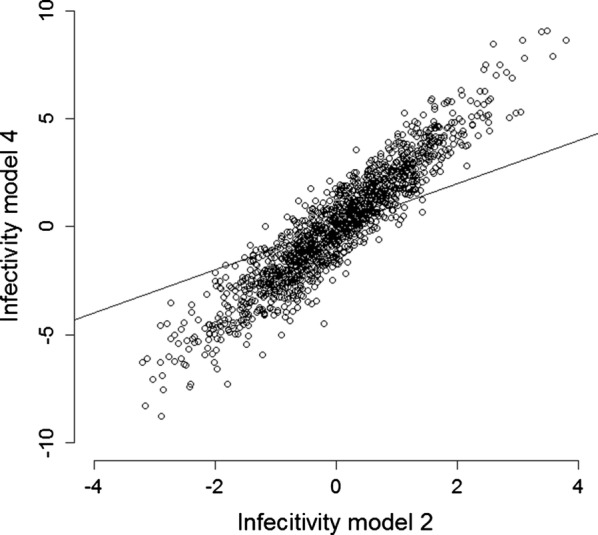
Estimated infectivity effect from Model 2 versus Model 4. Each point represents one cow. The line shows y = x
Cross-validation
Figure 3 shows the weighted linear regression of the average observed number of cases over the number of susceptible feet () on the average predicted probability, and their correlation. Bias was smallest for models without the infectivity effect (Models 1 and 3), since regression coefficients of these models were closest to 1. The weighted correlations coefficients were higher for models with the infectivity effect (Models 2 and 4). Thus, models without an infectivity effect showed significantly lower bias, while models with an infectivity effect had a slightly higher accuracy. The higher bias for Model 4 compared to Model 2 agrees with results in Fig. 2, and confirms that omitting the random farm-by-period interaction inflates the variation in GEBV for infectivity.
Fig. 3.

Weighted linear regression and correlation coefficients between the average observed number of cases over the number of susceptible feet () and the average predicted probability for the observations. Regression coefficients smaller than 1 indicate over prediction
Basic reproduction ratio
The genomic estimated breeding values (GEBV) for susceptibility corrected for bias were approximately the same for Models 1, 2 and 4, and ranged from 0.49 to 2.81; the GEBV from Model 3 ranged from 0.50 to 2.53. A cow with a susceptibility GEBV of 0.50 is genetically about two times less susceptible than an average cow, whereas a cow with a susceptibility GEBV of 2.50 is genetically about 2.5 times more susceptible than an average cow. The GEBV for infectivity showed substantially more variation than the GEBV for susceptibility.
We chose to illustrate the variation in GEBV for using results from Model 3, which is the most conservative model as judged by the estimated genetic variance in susceptibility (smallest value) and the bias. For this model, we calculated individual breeding values for the basic reproduction ratio () corrected for the bias that we found from the cross-validation. The average GEBV for susceptibility was 1.081, which is slightly higher than 1 because of the transformation from the log scale to the normal scale. In Model 3, there was no infectivity effect, thus infectivity was 1.00 for all individuals. With an average for DD of 2.36 on our farms [26], was estimated at 2.18. GEBV for ranged from 1.089 to 5.515 (Fig. 4) (the GEBV not corrected for bias ranged from 0.62 to 6.68). This result indicates very substantial genetic variation in . The expected prevalence in a population of individuals similar to the genetically best individual equals approximately [45], while the corresponding value for the genetically worst individual equals 81.9%.
Fig. 4.
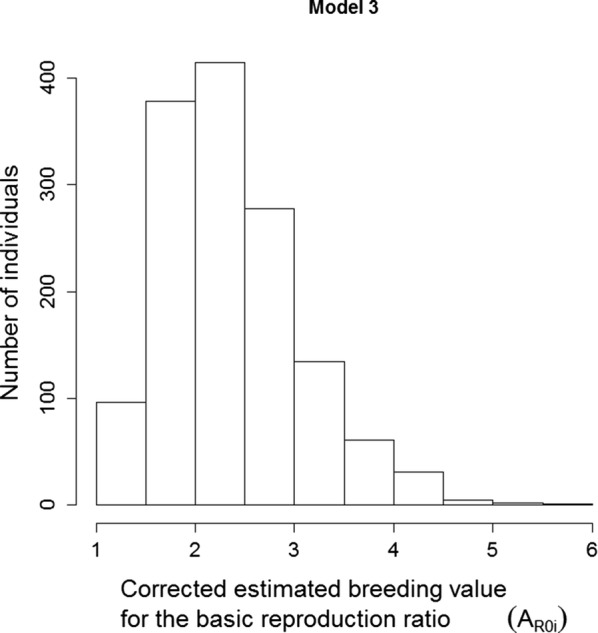
Histogram of the individual GEBV for the basic reproduction ratio corrected for bias for all genotyped cows, based on results from Model 3
The estimated additive genetic standard deviation for was 1.17 (this value is corrected for bias). This result shows that the current of 2.36 [26] is only about one genetic standard deviation greater than 1. Hence, this suggests that a genetic improvement of by a bit more than one genetic standard deviation would be sufficient to eradicate DD.
The approximate accuracy of follows from the ratio of the standard deviation of GEBV for over the additive genetic standard deviation of the true breeding values for ; . This accuracy equals ~ 39%. To be conservative, we did not correct (i.e., shrink) the in the denominator of this expression, while we did correct the numerator for the bias. Note that this accuracy is based on validation within a generation, rather than on validation forward in time. The accuracy based on validation forward in time is relevant for response to selection, and may be somewhat lower because genetic relationships may be a little weaker.
Discussion
We estimated the additive genetic variation in host susceptibility, infectivity, and the basic reproduction ratio () for DD in dairy cattle. Furthermore, we calculated GEBV for susceptibility, infectivity, and for each animal. Four models were compared for their ability to predict whether a susceptible animal becomes infected. All four models included a genetic effect for susceptibility; Models 2 and 4 also included a genetic effect for infectivity, while Models 1 and 2 included an interaction term between farm and period. In all models, the estimates are corrected for the variation in exposure of the susceptible individuals to infected group mates via the offset of the model. The estimated additive genetic standard deviation for was large, ~ 1.17, and the mean (2.36) was only about one genetic standard deviation greater than the important threshold value of 1. Furthermore, GEBV for (corrected for bias) showed large variation, ranging from 1.089 to 5.515, and the approximate accuracy of GEBV for was ~ 0.39. These results show that genetic selection against DD is very promising; there is substantial heritable variation and a meaningful accuracy can be obtained from a limited amount of data.
Farm, parity, period, and months in milk of the focal cow were included in the models as fixed effects. The transmission rate parameter was 21% higher for parity 2 compared to parity 1, and 69% higher for parity greater than 2 compared to parity 1. The prevalence also increased with parity. This is in contrast with most previous studies where DD was most prevalent in first and second parity cows [15, 46]. For months in milk, the transmission rate parameter decreased by 4% per month in milk. This is in agreement with Argáez-Rodríguez et al. [46] who found that cows had the highest risk of getting DD in the first and third months of lactation, after which the risk decreased. The potential effect of parity and months in milk on the infectivity of a cow was not considered, because incorporating these factors in the summed effects of the infected group mates of a focal cow was methodologically too challenging.
We managed, only partly, to account for genetic effects on infectivity. For technical reasons, we included only the genetic effects of claws that were infected at the start of the observation interval () in the statistical model. However, the majority of the infection pressure originated from earlier infected claws that still contribute to transmission via the environment. We estimated a survival rate (λ) of the pathogen of 0.9 [26], meaning that 90% of the total infectious pressure originates from infectivity present in the environment from claws that were infected before the start of the observation interval. This suggests that we missed a large part of the potential heritable variation in infectivity. Hence, the relevance of genetic variation in infectivity for DD may be larger than suggested by the estimates presented here (Table 3). Unfortunately, in the statistical software we did not manage to keep track of all the weighted genotypes (i.e., weighing the genotypes of the infected individuals for their contribution to the environment). Nevertheless, the accuracy of predicting the phenotype from the cross-validation for the model with genetic effects of infectivity (Model 2) was 9% higher than the accuracy for the corresponding model without these effects (Model 1). Thus, even including a small proportion of the apparent variation in infectivity in the model appears to increase the accuracy of GEBV.
Estimates of infectivity show relatively larger standard errors because variation in infectivity must be estimated indirectly, unlike variation in susceptibility. Infectivity estimates are based on the number of susceptible group mates of an infectious individual that become infected, and on differences in genotype among the infected group mates at different time points. When there are multiple infected group mates, the accuracy of the infectivity estimates decreases [7]. Especially in large groups, as in this study (~ 100 cows), more records and groups are needed to estimate genetic variation in infectivity accurately [47]. This issue is very similar to the estimation of indirect genetic effects from large groups. In addition, we observed DD transmission for a relatively short period of time. More accurate estimation of genetic variation in infectivity requires data that are recorded over longer time periods (i.e., to be able to observe the entire infectious period for the majority of the cows), more groups (herds), a better statistical model (i.e., inclusion of the genetic effects from earlier infected cows of which the pathogens are still present in the environment), and the inclusion of infectivity effects of cows without genotypes. The latter could be done using single-step GBLUP [48].
In the first two models, we included an interaction between farm and period to account for non-genetic effects of infectivity. This interaction serves to avoid overestimation of the genetic variance in infectivity [49], similar to the inclusion of a random group effect in the analysis of indirect genetic effects [38, 50]. The genetic effect for infectivity and the interaction term were partly confounded, because both terms have an effect on the number of susceptible animals that become a case within a certain period on a certain farm. However, confounding is not complete because of genetic relationships between the infected animals across farms and periods. Our results also suggest that inclusion of a random farm-by-period effect is essential to avoid overestimation of the genetic variation due to infectivity.
The estimated variances for susceptibility (genetic and non-genetic) were lower for Model 3 that included neither a genetic effect for infectivity nor an interaction between farm and period. Anacleto et al. [7] showed that estimates for susceptibility are less accurate when genetic variation in infectivity is not accounted for. Indeed, we also found a slightly higher correlation in the cross-validation when infectivity was included in the model, although this was accompanied by an inflation of the GEBV, as shown by the regression coefficients in Fig. 3. However, inflation of GEBV can be remedied by shrinking them based on the results of cross-validation, whereas a reduction in correlation cannot. Therefore, even when genetic variation in infectivity is small, it might be beneficial to include infectivity in the model to more accurately estimate susceptibility GEBV [7].
In the cross-validation, we estimated a weighted correlation of about 0.2 between the observed and predicted number of cases over the number of susceptible feet. This value can be used to approximate the accuracy of the GEBV () [51],
| 8 |
where, is the correlation between the observations and the predictions, and is the heritability of the trait. Heritability estimates for digital dermatitis from previous studies range from 0.05 to 0.29, depending on the model used [52]. Assuming a heritability of 0.28 for DD [53], the accuracy of the predicted number of cases is 0.37.
In general, studies on genetic variability of infectious diseases commonly focus on individual differences in susceptibility only, and those differences are estimated with linear models that ignore variation in exposure between individuals. In this study, we used a GLMM founded on epidemiological theory to estimate genetic variability in susceptibility and infectivity. An important advantage of models founded on epidemiological theory is that they provide estimates for epidemiological parameters, such as , that are interpretable in the context of infectious disease dynamics. Our results, for example, show that is only slightly more than one genetic standard deviation away from the threshold value of 1, which suggests that eradication of DD by genetic improvement is in principle feasible. Further work is needed to quantify the benefits of GLMM based on epidemiological theory over simpler linear models, to better account for the potential genetic variation in infectivity via the environment, and to include genetic variation in the duration of the infectious period.
Conclusions
Genetic variance components for susceptibility and infectivity for digital dermatitis were estimated with four generalized linear mixed models. We managed, only partly, to account for genetic effects on infectivity. We estimated GEBV for the basic reproduction ratio from a (conservative) model including genetic effects for susceptibility only. GEBV for were corrected for bias, and showed substantial variation, ranging from 1.089 to 5.515. The mean (2.36) was only about one genetic standard deviation greater than 1. Based on cross-validation between farms, the approximate accuracy of GEBV for was 0.39, in spite of the relatively small dataset of only 12 genotyped herds. These results suggest that lowering prevalence of DD by selective breeding is very promising.
Supplementary information
Additional file 1. Fixed effect estimates. Table of fixed effect estimates and standard errors on the log scale from Model 1.
Acknowledgements
We thank Natasja Boots for her help with the phenotype collection, the farmers for their participation, and the breeding company CRV for providing the genotype data.
Appendix
Cross-validation
In the 12-fold cross-validation, we censored the number of cases in each period and predicted for each susceptible animal the number of cases over the number of susceptible feet () based on information of the 11 other farms. In general, the predicted probability for the number of cases over the number of susceptible feet can be calculated with the estimated effects. Because of the complementary log–log link function, these effects need to be back-calculated to the original scale:
| 9 |
when the genetic effect for infectivity was not included (Models 1 and 3), and
| 10 |
when the genetic effect for infectivity was included (Models 2 and 4).
In Eqs. (9) and (10), is the predicted probability for the number of cases over the number of susceptible feet for animal in the time from to , is the sum of the estimates for the fixed effects, i.e., the estimates for the intercept, farm , period , parity , and months in milk. The is the estimated genetic effect for susceptibility on the log scale and is the sum of the estimated genetic effects for infectivity of the infected group mates of the susceptible individual on the log scale. The last term in Eqs. (9) and (10) is the offset. In the 12-fold cross-validation, the cases () of one entire farm were censored from the dataset, therefore, the random effects for the interaction between farm and period, and non-genetic animal effect for animal could not be estimated for this farm. Thus, these random effects did not contribute to the predicted probabilities, and they are not included in Eqs. (9) and (10).
To validate the estimated genetic effects, we wanted to estimate the probability that an animal would be a case during an interval independent of the fixed effects in the model. We achieved this independence by standardizing both and . We obtained the regression coefficients for the fixed effects for each of the four models that were applied to the full dataset where no data was censored. With these regression coefficients, we calculated the average value of summed fixed effects . The can be interpreted as a standard/average farm and a standard/average cow. This was used in Eqs. (9) and (10) instead of the estimated fixed effects:
| 11 |
for Models 1 and 3, and
| 12 |
for Models 2 and 4.
Here, is the predicted probability for the number of cases over the number of susceptible feet for animal in a period, as if it were an average cow with an average parity and months in milk, during a standard period, on a standard farm. Note that the genetic susceptibility and infectivity did differ between cows, and thus had an effect on the predictions.
Similarly, we wanted to standardize the observed cases over the number of susceptible claws , so that they would be independent of the fixed effects that contributed to that observation. The observations were transformed to the complementary log–log scale so that they were linear in the effects:
| 13 |
for Models 1 and 3, and,
| 14 |
for Models 2 and 4.
Next, the summed fixed effects in Eqs. (13) and (14) were replaced with the average value of the summed fixed effects , and back-calculated to the original scale to obtain the observed number of cases over the number of susceptible feet independent of the fixed effects , for all models:
| 15 |
Here, is the observed number of cases over the number of susceptible feet for animal in a period, as if observed on an average cow with an average parity and months in milk, on a standard farm. Again, the genetic susceptibility and infectivity did differ between cows (see Eqs. (13) and (14)), and affected the dependent variable.
We calculated weighted correlation coefficients between the “corrected” observed number of cases over the number of susceptible feet () and the “corrected” predicted probabilities (), averaged over the number of times an animal was susceptible at the start of an interval, and this number was used as the weight.
Authors’ contributions
FB collected the phenotype data and conducted the study. FB, MdJ, and PB designed the statistical methods. FB, MdJ, and PB wrote the manuscript. All authors read and approved the final manuscript.
Funding
This work is part of the research programme “Meer met Minder” with project number 847.13.004, which is financed by the Netherlands Organisation for Scientific Research (NWO).
Availability of data and materials
Datasets used and analysed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
The committee on animal ethics approved the design of the field trail (phenotype collection) under reference 2014089.b.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Floor Biemans, Email: floor.biemans@wur.nl.
Mart C. M. de Jong, Email: mart.dejong@wur.nl
Piter Bijma, Email: piter.bijma@wur.nl.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12711-019-0505-3.
References
- 1.Springbett AJ, MacKenzie K, Woolliams JA, Bishop SC. The contribution of genetic diversity to the spread of infectious diseases in livestock populations. Genetics. 2003;165:1465–1474. doi: 10.1093/genetics/165.3.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Woolhouse MEJ, Stringer SM, Matthews L, Hunter N, Anderson RM. Epidemiology and control of scrapie within a sheep flock. Proc Biol Sci. 1998;265:1205–1210. doi: 10.1098/rspb.1998.0421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stein RA. Super-spreaders in infectious diseases. Int J Infect Dis. 2011;15:e510–e513. doi: 10.1016/j.ijid.2010.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anche MT, de Jong MCM, Bijma P. On the definition and utilization of heritable variation among hosts in reproduction ratio R0 for infectious diseases. Heredity (Edinb). 2014;113:364–374. doi: 10.1038/hdy.2014.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Velthuis AGJ, de Jong MCM, Kamp EM, Stockhofe N, Verheijden JHM. Design and analysis of an Actinobacillus pleuropneumoniae transmission experiment. Prev Vet Med. 2003;60:53–68. doi: 10.1016/S0167-5877(03)00082-5. [DOI] [PubMed] [Google Scholar]
- 6.Lipschutz-Powell D, Woolliams JA, Doeschl-Wilson AB. A unifying theory for genetic epidemiological analysis of binary disease data. Genet Sel Evol. 2014;46:15. doi: 10.1186/1297-9686-46-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anacleto O, Garcia-Cortés LA, Lipschutz-Powell D, Woolliams JA, Doeschl-Wilson AB. A novel statistical model to estimate host genetic effects affecting disease transmission. Genetics. 2015;201:871–884. doi: 10.1534/genetics.115.179853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Biemans F, de Jong MCM, Bijma P. A model to estimate effects of SNPs on host susceptibility and infectivity for an endemic infectious disease. Genet Sel Evol. 2017;49:53. doi: 10.1186/s12711-017-0327-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bishop SC, MacKenzie KM. Genetic management strategies for controlling infectious diseases in livestock populations. Genet Sel Evol. 2003;35:S3–S17. doi: 10.1186/1297-9686-35-S1-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Griffing B. Selection in reference to biological groups I. Individual and group selection applied to populations of unordered groups. Aust J Biol Sci. 1967;20:127–140. doi: 10.1071/BI9670127. [DOI] [PubMed] [Google Scholar]
- 11.Bijma P, Wade MJ. The joint effects of kin, multilevel selection and indirect genetic effects on response to genetic selection. J Evol Biol. 2008;21:1175–1188. doi: 10.1111/j.1420-9101.2008.01550.x. [DOI] [PubMed] [Google Scholar]
- 12.Bijma P, Muir WM, van Arendonk JAM. Multilevel selection 1: quantitative genetics of inheritance and response to selection. Genetics. 2007;175:277–288. doi: 10.1534/genetics.106.062711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.May RM, Anderson RM. Transmission dynamics of HIV infection. Nature. 1987;326:137–142. doi: 10.1038/326137a0. [DOI] [PubMed] [Google Scholar]
- 14.Sogstad ÅM, Fjeldaas T, Østerås O, Forshell KP. Prevalence of claw lesions in Norwegian dairy cattle housed in tie stalls and free stalls. Prev Vet Med. 2005;70:191–209. doi: 10.1016/j.prevetmed.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 15.Read DH, Walker RL. Papillomatous digital dermatitis (Footwarts) in California dairy cattle: clinical and gross pathologic findings. J Vet Diagn Investig. 1998;10:67–76. doi: 10.1177/104063879801000112. [DOI] [PubMed] [Google Scholar]
- 16.Walker RL, Read DH, Loretz KJ. Spirochetes isolated from dairy cattle with papillomatous digital dermatitis and interdigital dermatitis. Vet Microbiol. 1995;47:342–355. doi: 10.1016/0378-1135(95)00114-X. [DOI] [PubMed] [Google Scholar]
- 17.Collighan RJ, Woodward MJ. Spirochaetes and other bacterial species associated with bovine digital dermatitis. FEMS Microbiol Lett. 1997;156:37–41. doi: 10.1016/S0378-1097(97)00399-6. [DOI] [PubMed] [Google Scholar]
- 18.Bassett HF, Monaghan ML, Lenhan P, Doherty ML, Carter ME. Bovine digital dermatitis. Vet Rec. 1990;126:164–165. [PubMed] [Google Scholar]
- 19.Holzhauer M, Hardenberg C, Bartels CJM, Frankena K. Herd- and cow-level prevalence of digital dermatitis in The Netherlands and associated risk factors. J Dairy Sci. 2006;89:580–588. doi: 10.3168/jds.S0022-0302(06)72121-X. [DOI] [PubMed] [Google Scholar]
- 20.Bruijnis MRN, Beerda B, Hogeveen H, Stassen EN. Assessing the welfare impact of foot disorders in dairy cattle by a modeling approach. Animal. 2012;6:962–970. doi: 10.1017/S1751731111002606. [DOI] [PubMed] [Google Scholar]
- 21.Bruijnis MRN, Beerda B, Hogeveen H, Stassen EN. Foot disorders in dairy cattle: impact on cow and dairy farmer. Anim Welf. 2012;21:33–40. doi: 10.7120/096272812X13345905673601. [DOI] [PubMed] [Google Scholar]
- 22.Wells SJ, Garber LP, Wagner BA. Papillomatous digital dermatitis and associated risk factors in US dairy herds. Prev Vet Med. 1999;38:11–24. doi: 10.1016/S0167-5877(98)00132-9. [DOI] [PubMed] [Google Scholar]
- 23.van der Waaij EH, Holzhauer M, Ellen E, Kamphuis C, de Jong G. Genetic parameters for claw disorders in Dutch dairy cattle and correlations with conformation traits. J Dairy Sci. 2005;88:3672–3678. doi: 10.3168/jds.S0022-0302(05)73053-8. [DOI] [PubMed] [Google Scholar]
- 24.van der Spek D, van Arendonk JAM, Vallée AAA, Bovenhuis H. Genetic parameters for claw disorders and the effect of preselecting cows for trimming. J Dairy Sci. 2013;96:6070–6078. doi: 10.3168/jds.2013-6833. [DOI] [PubMed] [Google Scholar]
- 25.van der Linde C, de Jong G, Koenen EPC, Eding H. Claw health index for Dutch dairy cattle based on claw trimming and conformation data. J Dairy Sci. 2010;93:4883–4891. doi: 10.3168/jds.2010-3183. [DOI] [PubMed] [Google Scholar]
- 26.Biemans F, Bijma P, Boots NM, de Jong MCM. Digital dermatitis in dairy cattle: the contribution of different disease classes to transmission. Epidemics. 2018;23:76–84. doi: 10.1016/j.epidem.2017.12.007. [DOI] [PubMed] [Google Scholar]
- 27.Relun A, Guatteo R, Roussel P, Bareille N. A simple method to score digital dermatitis in dairy cows in the milking parlor. J Dairy Sci. 2011;94:5424–5434. doi: 10.3168/jds.2010-4054. [DOI] [PubMed] [Google Scholar]
- 28.Döpfer D, ter Huurne AAHM, Cornelisse JL, van Asten AJAM, Koopmans A, Meijer FA, et al. Histological and bacteriological evaluation of digital dermatitis in cattle, with special reference to spirochaetes and Campylobacter faecalis. Vet Rec. 1997;140:620–623. doi: 10.1136/vr.140.24.620. [DOI] [PubMed] [Google Scholar]
- 29.Döpfer D. The dynamics of digital dermatitis in dairy cattle and the manageable state of disease. In: Proceedings of the CanWest Veterinary Conference: 17–20 October 2009; Banff; 2009.
- 30.Berry SL, Read DH, Famula TR, Mongini A, Döpfer D. Long-term observations on the dynamics of bovine digital dermatitis lesions on a California dairy after topical treatment with lincomycin HCl. Vet J. 2012;193:654–658. doi: 10.1016/j.tvjl.2012.06.048. [DOI] [PubMed] [Google Scholar]
- 31.Browning SR, Browning BL. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet. 2007;81:1084–1097. doi: 10.1086/521987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Druet T, Georges M. A hidden markov model combining linkage and linkage disequilibrium information for haplotype reconstruction and quantitative trait locus fine mapping. Genetics. 2010;184:789–798. doi: 10.1534/genetics.109.108431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laven R. Control of digital dermatitis in cattle. In Pract. 2001;23:336–341. doi: 10.1136/inpract.23.6.336. [DOI] [Google Scholar]
- 34.Bravo de Rueda C, de Jong MCM, Eblé PL, Dekker A. Quantification of transmission of foot-and-mouth disease virus caused by an environment contaminated with secretions and excretions from infected calves. Vet Res. 2015;46:43. doi: 10.1186/s13567-015-0156-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roberts M, Heesterbeek H. Bluff your way in epidemic models. Trends Microbiol. 1993;1:343–348. doi: 10.1016/0966-842X(93)90075-3. [DOI] [PubMed] [Google Scholar]
- 36.McCullagh P. Generalized linear models. Eur J Oper Res. 1984;16:285–292. doi: 10.1016/0377-2217(84)90282-0. [DOI] [Google Scholar]
- 37.Anche MT, Bijma P, de Jong MCM. Genetic analysis of infectious diseases: estimating gene effects for susceptibility and infectivity. Genet Sel Evol. 2015;47:85. doi: 10.1186/s12711-015-0163-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bergsma R, Kanis E, Knol EF, Bijma P. The contribution of social effects to heritable variation in finishing traits of domestic pigs (Sus scrofa) Genetics. 2008;178:1559–1570. doi: 10.1534/genetics.107.084236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.VanRaden PM. Efficient methods to compute genomic predictions. J Dairy Sci. 2008;91:4414–4423. doi: 10.3168/jds.2007-0980. [DOI] [PubMed] [Google Scholar]
- 40.Calus MPL, Vandenplas J. Calc_grm—a program to compute pedigree, genomic, and combined relationship matrices. Wageningen: Animal Breeding and Genomics Centre; 2016. [Google Scholar]
- 41.Gilmour AR, Gogel BJ, Cullis BR, Welham SJ, Thompson R. ASReml user guide release 4.1 structural specification. Hemel Hempstead: VSN International Ltd; 2015. [Google Scholar]
- 42.Vitezica ZG, Aguilar I, Misztal I, Legarra A. Bias in genomic predictions for populations under selection. Genet Res (Camb). 2011;93:357–366. doi: 10.1017/S001667231100022X. [DOI] [PubMed] [Google Scholar]
- 43.Mrode RA. Linear models for the prediction of animal breeding values. 3. Wallingford: CABI; 2014. [Google Scholar]
- 44.Hosmer DW, Lemeshow S, May S. Applied survival analysis : regression modeling of time-to-event data, appendix 1: the delta method. 2. Hoboken: Wiley-Interscience; 2008. [Google Scholar]
- 45.Fine PEM. Herd immunity: history, theory, practice. Epidemiol Rev. 1993;15:265–302. doi: 10.1093/oxfordjournals.epirev.a036121. [DOI] [PubMed] [Google Scholar]
- 46.Argáez-Rodríguez FJ, Hird DW, de Anda JH, Read DH, Rodríguez-Lainz A. Papillomatous digital dermatitis on a commercial dairy farm in Mexicali, Mexico: incidence and effect on reproduction and milk production. Prev Vet Med. 1997;32:275–286. doi: 10.1016/S0167-5877(97)00031-7. [DOI] [PubMed] [Google Scholar]
- 47.Bijma P. Estimating indirect genetic effects: precision of estimates and optimum designs. Genetics. 2010;186:1013–1028. doi: 10.1534/genetics.110.120493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Legarra A, Christensen OF, Aguilar I, Misztal I. Single step, a general approach for genomic selection. Livest Sci. 2014;166:54–65. doi: 10.1016/j.livsci.2014.04.029. [DOI] [Google Scholar]
- 49.Anche MT. Estimating host genetic effects on susceptibility and infectivity to infectious diseases and their contribution to response to selection. PhD thesis, Wageningen University. 2016.
- 50.Bijma P, Muir WM, Ellen ED, Wolf JB, van Arendonk JAM. Multilevel selection 2: estimating the genetic parameters determining inheritance and response to selection. Genetics. 2007;175:289–299. doi: 10.1534/genetics.106.062729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Calo LL, McDowell RE, VanVleck LD, Miller PD. Genetic aspects of beef production among Holstein–Friesians pedigree selected for milk production. J Anim Sci. 1973;37:676–682. doi: 10.2527/jas1973.373676x. [DOI] [Google Scholar]
- 52.Schöpke K, Gomez A, Dunbar KA, Swalve HH, Döpfer D. Investigating the genetic background of bovine digital dermatitis using improved definitions of clinical status. J Dairy Sci. 2015;98:8164–8174. doi: 10.3168/jds.2015-9485. [DOI] [PubMed] [Google Scholar]
- 53.Biemans F, de Jong MCM, Bijma P. A genome-wide association study for susceptibility and infectivity of dairy cattle to Digital Dermatitis. J Dairy Sci. 2019;102:6248_62. doi: 10.3168/jds.2018-15876. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Fixed effect estimates. Table of fixed effect estimates and standard errors on the log scale from Model 1.
Data Availability Statement
Datasets used and analysed during the current study are available from the corresponding author on reasonable request.