

Abstract
Increasing evidence supports the concept that cancer stem cells (CSCs) are responsible for cancer progression and metastasis, therapy resistance and relapse. In addition to conventional therapies for colon cancer, the development of immunotherapies targeting cancer stem cells appears to be a promising strategy to suppress tumor recurrence and metastasis. In the present study, dendritic cells (DCs) were pulsed with whole-tumor cell lysates or total RNA of CD44+ colon cancer stem cells (CCSCs) isolated from mouse colon adenocarcinoma CT-26 cell cultures and investigated for their antitumor immunity against CCSCs in vivo and in vitro. In a model of colon adenocarcinoma using BALB/c mice, a sequential reduction in tumor volume and weight was associated with an extended survival in tumor-bearing mice vaccinated with DCs pulsed with RNA or CCSC lysate. In addition, a lactate dehydrogenase assay indicated that cytotoxic T-cells derived from the treated mice exhibited strong cytotoxic activity. Additionally, an enzyme-linked immunosorbent assay revealed that the cytotoxic T-cells of the treated mice released higher levels of interferon-γ against CCSCs compared with those of the control group. In all experiments, the antitumor efficacy of the lysate-pulsed DC-treated and RNA-pulsed DC-treated groups were significantly higher compared with that of the DC-treated and control groups. The results of the present study indicated the potential use of DCs pulsed with cancer stem cell lysates as a potent therapeutic antigen to target CSCs in colon cancer. Additionally, the results provided a rationale for using lysate-pulsed DCs in vivo to eliminate residual tumor deposits in post-operative patients.
Keywords: immunotherapy, cancer stem cell, cytotoxic T lymphocyte, colon cancer
Introduction
Colorectal cancer is the second most common cancer, with an estimated 145,600 newly diagnosed cases and 51,020 cases of mortality in the United States in 2019 (1). Traditional treatment modalities, including surgery, chemotherapy and radiation therapy have prolonged survival and improved the outcome for patients with colorectal cancer; however, recurrent disease after initial treatment and metastasis occurs in ~50% of cases (2). This is partially caused by the ability of colon cancer cells to evade the host immune-surveillance by suppressing cell-mediated immunity (3). The intra-tumor infiltration of colon cancer by lymphocytes suggests that the immune system can induce an immune response against the tumor, but this is not effective enough to prevent tumor growth (4–6). Based on this assumption, treating colon cancer with immunotherapy, especially therapies using dendritic cells (DCs), which are the most effective antigen-presenting cells of the adaptive immune system, has been extensively investigated over the past two decades (7,8). DCs capture, process and present antigens to T lymphocytes in association with major histocompatibility complex (MHC) class molecules (9). MHC class II molecules present antigens on their surface, stimulating the maturation of helper T cells/CD4+ cells (10). MHC class I molecules cross-present antigens to CD8+ cytotoxic T lymphocytes (CTLs) to elicit an antigen-specific immune response (11). Accordingly, immunotherapies using ex vivo-generated DC vaccines loaded with tumor-derived antigens can induce effective antigen-specific humoral and cell-mediated immune responses in several cancer models, including colon cancer (12–14).
Another major cause of cancer therapy failure is the development of acquired resistance to chemotherapy and radiation therapy by tumor cell subpopulations referred to as cancer stem cells (CSCs). CSCs display distinctive immunophenotypes and exhibit the capacity for unlimited self-renewal and heterogeneous-lineage differentiation (15–17). The inability to recognize, target and eliminate CSC populations results in tumor recurrence and metastasis (18). Currently available DC vaccines are loaded with synthesized tumor-associated peptide antigens, such as carcinoembryonic antigen (19), melanoma-associated antigen 2 (20) and human epidermal growth factor receptor 2 (21). However, since CSCs do not express these differentiated tumor antigens, using these antigens for vaccination may increase the risk of immune escape due to antigen loss. Previous studies have demonstrated that DC vaccines loaded with CSC extracts exerted promising anticancer immunity against various malignancies (14,22–27). Selection of the tumor antigen and transfection method to pulse DCs are crucial components of an effective cancer vaccination strategy (28). Several forms of antigens can be loaded onto DCs; antigens can be added exogenously as whole-cell lysates (29), RNA (30), peptides (31), whole proteins (19), apoptotic debris (32) or antibody complexes (33). Antigens can also be synthesized endogenously by transfection of mRNA or cDNA encoding the antigen (30,34,35). The advantage of using whole-cell lysates is that they may contain peptides that can be effectively presented by the majority of MHC molecules (28). In addition, the delivery of RNA to target cells can be achieved through transfection with naked RNA (35,36), RNA/liposome complexes (37), RNA/DOTAP complexes (30), electroporation (38) and gene gun (biolistic) (39).
To the best of our knowledge, the potential antitumor activities of DCs pulsed with colon CSC (CCSC)-derived materials have not been previously investigated using a colon cancer model. The aim of the present study was to evaluate the antitumor effects of DC vaccines prepared by pulsing DCs with CCSC-derived lysate (Pro-DC) or RNA (RNA-DC) isolated from CD44+ mouse colon adenocarcinoma CT-26 cells using a BALB/c murine model of colon adenocarcinoma.
Materials and methods
Animals
A total of 140 female BALB/c mice (6–8 weeks old; 16–18 g) were purchased from Beijing HFK Bioscience Co., Ltd. and housed in the Laboratory Animal Center at Jilin University (Changchun, China) under sterile conditions (room temperature, 25±2°C; humidity, 55±5%) on a 12-h light/dark cycle (lights on at 6:30 a.m.). Animals had ad libitum access to water and mouse chow diet. An acclimation period of at least 1 week was implemented for all mice prior to use in experiments. The experiments were approved by the Animal Care and Protection Committee of the Laboratory Animal Center at Jilin University. Mice remained on study until the tumor diameter exceeded 2.0 cm in any dimension unless ulceration, necrosis, or other complications was observed. Mice were evaluated for clinical signs including cachexia (weight loss exceeding 20% of the body weight), anorexia, dehydration, dyspenia, neurological impairment, hunched posture, body condition scoring system score 2 or less, or tumor burden greater than 15% of body weight.
Cell line
The mouse CT-26 cell line was purchased from The Cell Bank of Type Culture Collection of the Chinese Academy of Sciences, and cultured in RPMI 1640 medium (Gibco; Thermo Fisher Scientific, Inc.) containing 10% FBS (Zhejiang Tianhang Biotechnology, Co., Ltd.) and 100 U/ml penicillin/streptomycin (Beyotime Institute of Biotechnology) at 37°C with 5% CO2.
Generation of bone marrow-derived DCs
DCs were generated as previously described by Lutz et al (40). Briefly, the bone marrow was flushed from femurs and tibias obtained from 60 female BALB/c mice (6–8 weeks old, 16–18 g). Mice were euthanized by carbon dioxide asphyxiation for approximately 6 min (air displacement rate: 20%/min; carbon dioxide flow rate: 1.7 l/min; the mortality was ensured by cervical dislocation). Cells (1×106 cells/well) were washed twice with PBS and seeded in each well of a 6-well plate in 2 ml RPMI 1640 medium supplemented with 10 ng/ml recombinant murine granulocyte-macrophage colony stimulating factor (rmGM-CSF), 20 ng/ml recombinant murine interleukin (rmIL)-4 (both from PeproTech, Inc.) and 10% FBS at 37°C with 5% CO2 for 8 days. The morphology of DCs was observed and images were captured using an inverted light microscope (Olympus Corporation) at a magnification of ×200.
Magnetic-activated cell sorting (MACS)
CT-26 cells were harvested and incubated with an anti-CD44 monoclonal antibody conjugated with biotin (cat. no. 130-110-082; Miltenyi Biotec, Inc.) for 20 min at 4°C, followed by fractionation using a CELLection Biotin Binder kit (Invitrogen; Thermo Fisher Scientific, Inc.) according to the manufacturer's instructions. Briefly, microbeads were added to the CD44 antibody-labeled cells, which were incubated at 4°C for 20 min with gentle tilting and rotation and separated using a magnet. Subsequently, 10 µl Releasing buffer (DNase I) was added to the cell suspension and incubated for 15 min at room temperature with gentle tilting and rotation to release the cells. CD44+ CT-26 cells were separated using a magnet. The sorted CD44+ CT-26 cells were cultured in DMEM/F12 (Gibco; Thermo Fisher Scientific, Inc.), supplemented with 20 ng/ml basic fibroblast growth factor (bFGF) and 20 ng/ml epidermal growth factor (EGF; both from PeproTech, Inc.), 2% B27 (Gibco; Thermo Fisher Scientific, Inc.), 100 U/ml penicillin/streptomycin (Beyotime Institute of Biotechnology) and 8 mM HEPES (HyClone; GE Healthcare Life Sciences) at 37°C with 5% CO2.
On day 8 of culture, bone marrow-derived cells were harvested and incubated with CD11c MicroBeads UltraPure (Miltenyi Biotec, Inc.) for 20 min at 4°C according to the manufacturer's instructions. The negative flow-through fraction was discarded, and the positive fraction containing CD11c+ cells was analyzed by flow cytometry as described below.
Tumorsphere formation assay
Sorted CD44+ CT-26 cells were cultured in serum-free DMEM/F12 supplemented with 20 ng/ml bFGF, 20 ng/ml EGF, 2% B27, 100 U/ml penicillin/streptomycin and 8 mM HEPES for 7 days at 37°C with 5% CO2. Cells were seeded into uncoated 6-well culture plates (Corning Inc.) at a density of 1×104 cells/well with fresh medium added every 3 days. Tumorsphere formation was observed and images are representative of at least five random fields and were captured using an inverted light microscope (Olympus Corporation) at a magnification of ×100.
Serum-induced differentiation
A total of 5×105 CD44+ CT-26 cells were resuspended and incubated for 3 days in RPMI-1640 medium supplemented with 10% FBS at 37°C with 5% CO2. Images of cells were acquired using an inverted light microscope (Olympus Corporation) at a magnification of ×400.
Flow cytometric detection of cell surface markers
CT-26 cells and CD44+ CT-26 cells (termed CCSCs) were dissociated into single cells, and CCSCs were prepared at a concentration of 2×105 cells in 0.1 ml PBS. A FITC-conjugated anti-CD44 antibody (1:200; cat. no. 11-0441-82; eBioscience; Thermo Fisher Scientific, Inc.) was added to the cell suspension, which was subsequently incubated in the dark for 10 min at 4°C. A FITC-conjugated rat-anti mouse IgG2bκ isotype was used as isotype control (1:100; cat. no. 11-4031-81; eBioscience; Thermo Fisher Scientific, Inc.). Following two washes with PBS, cells were collected and analyzed using Beckman Coulter FC500 Flow Cytometer with the CellQuest Pro software (version 6.0; BD Biosciences) to determine the number of CD44+ cells.
Bone marrow-derived cells from an 8-day culture and DCs isolated following MACS were harvested, washed once with precooled PBS containing 2% FBS and adjusted to a concentration of 1×106 cells in 0.1 ml PBS. A FITC-conjugated CD11c Monoclonal Antibody (1:400; cat. no. 11-0114-82; eBioscience; Thermo Fisher Scientific, Inc.) was added to the cell suspension, which was subsequently incubated in the dark for 10 min at 4°C. After two washes in PBS, cells were acquired and analyzed using Beckman Coulter FC500 Flow Cytometer with the FlowJo software (version 10.0; FlowJo) to determine the number of CD11c+ cells.
Preparation of CCSC lysate and total RNA
CCSC lysates were prepared as previously described by Schnurr et al (41). Briefly, CCSCs were trypsinized using TrypLE (Gibco; Thermo Fisher Scientific, Inc.), resuspended in serum-free DMEM/F12 medium supplemented with 20 ng/ml bFGF, 20 ng/ml EGF, 2% B27, 100 U/ml penicillin/streptomycin and 8 mM HEPES at a concentration of 3×106 cells/ml and lysed by four freeze-thaw cycles. The lysed cells were centrifuged at 500 × g for 30 min at 4°C. The supernatants were collected and used as CCSC-associated antigens. Total RNA from CCSCs was extracted using TRIzol® reagent (Invitrogen; Thermo Fisher Scientific, Inc.) according to the manufacturer's instructions.
Pulsing of DCs with CCSC extracts
RNA and lysates were loaded to DCs by coincubation of DCs without any transfection reagents. Pulsing of DCs with RNA was performed as previously described by Nair et al (35) and Pan et al (36). DCs (5×105 cells) obtained following MACS were incubated with total RNA (40 µg) or lysate from 1.5×106 CCSCs in RPMI-1640 medium supplemented with 10 ng/ml rmGM-CSF and 20 ng/ml rmIL-4 for 4 h at 37°C. To induce DC maturation, tumor necrosis factor-α (5 ng/ml) was added to the medium and incubated with antigen-loaded DCs for 48 h at 37°C with 5% CO2. Subsequently, RNA-DCs and Pro-DCs were obtained and suspended in PBS for the following animal studies.
DC-based immunotherapy
The colon cancer model was established by subcutaneously injecting 1×106 CCSCs into the right hind limb of BALB/c mice on day 0. When tumors became visible on day 7, BALB/c mice were randomized into three treatment groups and one control group (n=20 mice/group). Mice in the treatment groups received a dose of 1×106 DCs, RNA-DCs or Pro-DCs in the left hind limb, whereas mice in the control group received 0.1 ml PBS. Mice were injected once per week for 3 weeks. The two perpendicular dimensions of each tumor were measured with a Vernier caliper every 3 days to calculate the tumor volume as follows: V (mm3)=0.5 × a × b2, where a is the maximum length of the tumor, and b is the maximum transverse diameter. On day 35, ten mice in each group were sacrificed by carbon dioxide asphyxiation (air displacement rate: 20%/min; carbon dioxide flow rate: 1.7 l/min) for approximately 6 min followed by cervical dislocation. Tumors were excised and weighed, and spleens were collected for further analysis. For the survival tests, the time of death was recorded for the remaining ten mice in each group to calculate the survival rate.
CTL induction
Spleens were recovered from mice on day 35. A single cell preparation of splenocytes was obtained by pressing the spleen through a stainless steel wire mesh (200 meshes), suspending the material in PBS and collecting it by centrifugation (350 × g for 5 min at room temperature. Red blood cells were removed by lysis in 0.83% ammonium chloride. Lymphocytes were further separated from the splenocytes by density gradient centrifugation using Lymphocyte Separation Medium (Beyotime Institute of Biotechnology, Co., Ltd.) according to the manufacturer's instructions. Briefly, splenocytes collected were mixed with an equal volume of PBS and subsequently added on top of lymphocyte separation medium (same volume of sample). The mixture was centrifuged at 400 × g for 30 min at room temperature. The cells in the interface were collected, mixed with RPMI-1640 medium, and centrifuged at 300 × g for 10 min at room temperature. The supernatant was discarded and the cell pellets were washed twice in RPMI-1640 medium. A single suspension of lymphocytes was harvested by centrifugation (400 × g for 5 min at room temperature) and seeded in each well of a 96-well plate at a density of 2×106 cells/ml in 0.1 ml RPMI 1640 medium supplemented with 10% FBS and 20 ng/ml rmIL-2 for 7 days at 37°C with 5% CO2.
Cytotoxicity assay
The CTLs from each group were examined for cytotoxic activity toward target CCSCs by performing a lactate dehydrogenase (LDH) release assay using the CytoTox96® Non-Radioactive Cytotoxicity Assay kit (Promega Corporation) according to the manufacturer's instructions. Briefly, CTLs and CCSCs were mixed at effector-to-target (E/T) ratios of 10:1, 20:1 and 40:1 in RPMI-1640 medium supplemented with 10% FBS with a final volume of 100 µl/well in a 96-well plate (Corning Inc.). The percent specific cytotoxicity was calculated as [(experimental value)-(effector cell spontaneous LDH release control)/[(target cell maximum LDH release control)-(target cells spontaneous LDH release control)] ×100. All assays were performed in triplicate.
ELISA
CTLs and CCSCs were mixed at an E/T ratio of 20:1 in a 96-well plate and incubated for 18 h at 37°C with 5% CO2. Interferon-γ (IFN-γ) secreted from CTLs was measured using the Mouse IFN-γ ELISA kit (R&D Systems, Inc.) according to the manufacturer's instructions. All experiments were performed in triplicate.
Statistical analysis
Data are presented as the mean ± standard deviation. The results of the in vitro experiments were obtained from three independent experiments. One-way ANOVA followed by a Tukey-Kramer multiple comparisons test was used to compare the data using SPSS software (v.17.0; SPSS, Inc.). Kaplan-Meier analysis was used for survival estimations and a log-rank (Mantel-Cox) test was used to evaluate the differences between the survival curves. P<0.05 was considered to indicate a statistically significant difference.
Results
Isolation and characterization of CD44+CT-26 cells
Single-cell suspensions of CCSCs isolated from murine cell line CT-26 by MACS were cultured in serum-free DMEM/F12 medium. These cells grew as suspended individual cells and gradually formed cell aggregates on day 3. On day 7, tumorsphere clones appeared and became visible to the naked eye (Fig. 1A). When these tumorsphere were cultured in serum-supplemented medium, cells began to detach from the tumorspheres and differentiated into adherent monolayers of CT-26 cells on day 3 (Fig. 1B). Moreover, the percentages of CD44+ cells in unsorted CT-26 cells and CCSCs were 6.98 and 95.26%, respectively, as analyzed by flow cytometry (Fig. 2).
Figure 1.

Isolation and characterization of CCSCs. (A) Optical micrographs demonstrating the morphology of CCSCs obtained by magnetic-activated cell sorting over 7 days. Scale bar, 200 µm. (B) Serum-induced differentiation of CCSCs into adherent cells. Scale bar, 50 µm. CCSCs, colon cancer stem cells.
Figure 2.

Flow cytometric analysis of CD44+ cell marker expression in CT-26 cells. Colon cancer stem cell percentage in CT-26 cells (A) before and (B) after magnetic-activated cell sorting evaluated by flow cytometry. White, isotype control; grey, CD44+ expression.
Characteristics of bone marrow-derived DCs in culture
Bone marrow-derived DCs were cultured and induced in RPMI 1640 medium supplemented with rmGM-CSF and rmIL-4. On day 2, bone marrow-derived cells floating as suspensions in culture medium exhibited spherical morphology (Fig. 3A). Cells became larger and started to form multicellular clusters on day 5 (Fig. 3B). On day 8, DC colonies became large and exhibited protrusions with a branched and extended morphology on the cell membrane (Fig. 3C). Semi-adherent and suspended cells were collected and considered to be immature DCs.
Figure 3.

Isolation and characterization of bone marrow-derived DCs. (A-C) Optical micrographs presenting the morphology of DCs. (A) On day 2, bone marrow-derived cells floated as suspensions in culture and exhibited a spherical morphology. (B) Cells grew larger and started to form multicellular clusters on day 5. (C) On day 8, DC colonies became large and exhibited protrusions with a branched and extended morphology on the cell membrane. Scale bars, 100 µm. (D and E) The percentage of CD11c+ cells (D) before and (E) after magnetic-activated cell sorting evaluated by flow cytometry. DCs, dendritic cells.
Bone marrow-derived cells and DCs were incubated with FITC-conjugated CD11c monoclonal antibody and analyzed by flow cytometry to determine the CD11c+ cell yield. The percentages of CD11c+ cells before and after MACS were 73.1 and 94.8%, respectively (Fig. 3D and E). This result indicated that relatively pure bone marrow-derived DCs could be obtained through a two-step extraction procedure.
Pro-DC and RNA-DC treatment of tumor-bearing mice induces antitumor activity
CCSC-derived tumors grew to a palpable size of ~5 mm in diameter prior to treatment initiation with unpulsed or pulsed DCs. On day 35, the average tumor volume in control mice was 2,617±55 mm3, whereas that in mice treated with unpulsed DCs, RNA-DCs, or Pro-DCs was 1,162±62, 1,063±71 and 381±93 mm3, respectively (Fig. 4A). Therefore, DC treatment suppressed tumor growth in mice. The suppressive effect of Pro-DCs was the strongest among all treatments; the effect elicited by RNA-DCs was similar to that of unpulsed DCs. The tumor weight in mice treated with Pro-DCs was significantly lower compared with that in mice treated with unpulsed DCs, RNA-DCs and PBS (P<0.05 for all comparisons; Fig. 4B). Tumor volume and weight in the pulsed and unpulsed DC groups were significantly different compared with those of the PBS group (P<0.05 for all comparisons).
Figure 4.

Therapeutic effects of unpulsed DCs, Pro-DCs and RNA-DCs against CCSCs in tumor-bearing mice. (A) Average tumor volume was measured every 3 days when tumors became visible on day 11 following CCSC implantation. (B) On day 35, the average tumor weight was determined. Data are presented as the mean ± standard deviation. *P<0.05 vs. PBS; #P<0.05 vs. unpulsed DCs; †P<0.05 vs. RNA-DCs. n=10 mice per group. DCs, dendritic cells; Pro-DC, tumor lysate protein-pulsed dendritic cells; RNA-DC, tumor total RNA-pulsed dendritic cells; CCSCs, colon cancer stem cells.
For survival studies, 10 tumor-bearing mice in each group were observed for their survival duration (Fig. 5). The results revealed that tumor-bearing mice vaccinated with unpulsed DCs, RNA-DCs or Pro-DCs survived longer compared with mice treated with PBS (P<0.05 for all comparisons); tumor-bearing mice treated with Pro-DCs survived the longest among all groups (P<0.05 for all comparisons), and tumor-bearing mice treated with RNA-DCs survived longer compared with mice treated with unpulsed DCs (P<0.05). On day 60, survival rates in mice treated with Pro-DCs and RNA-DCs were 60 and 10%, respectively.
Figure 5.
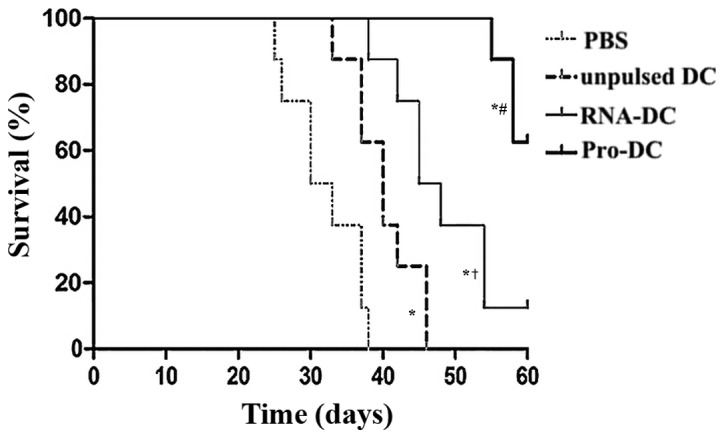
Pro-DCs enhance the survival of tumor-bearing mice. Tumor- bearing BALB/c mice treated with PBS, unpulsed DCs, RNA-DCs and Pro-DCs once per week for 3 weeks were observed for survival duration until day 60. A Kaplan-Meier curve of mouse survival was plotted using survival data. #P<0.05 vs. PBS, unpulsed DCs, or RNA-DCs; *P<0.05 vs. PBS; †P<0.05 vs. unpulsed DCs. Statistical analysis was performed using a log-rank test. n=10 mice per group. DC, dendritic cell; Pro-DC, tumor lysate protein-pulsed dendritic cell; RNA-DC, tumor total RNA-pulsed dendritic cell.
Cytotoxic response and tumor-specific immunity mediated by CTLs from unpulsed and pulsed DC-treated tumor-bearing mice
Lymphocytes from DC-treated mice were tested for cytotoxicity using an LDH assay. Unpulsed and pulsed DC treatments induced potent antitumor activity and tumor-specific cytotoxicity in vitro. As demonstrated in Fig. 6A, the immunization of tumor-bearing mice with Pro-DCs induced significantly stronger cytotoxic activity against CCSCs at E/T ratios of 10:1, 20:1 and 40:1. The percent cell lysis in Pro-DC samples at an E/T ratio of 40:1 was 63.9±3.31%, whereas those in the RNA-DC, unpulsed DC and PBS control groups were 31.42±3.64, 27.76±1.78 and 0.4±1.13%, respectively (P<0.05 for all comparisons).
Figure 6.

Induction of CTL response in tumor-bearing mice via immunization with unpulsed DCs, Pro-DCs or RNA-DCs. (A) CTL-stimulated lysis of CCSCs by pulsed or unpulsed DCs at E/T ratios of 10:1, 20:1 and 40:1. (B) Levels of IFN-γ secreted by CTLs derived from pulsed or unpulsed DCs against CCSCs at an E/T ratio of 20:1. The results represent three independent experiments. Data are presented as the mean ± standard deviation. *P<0.05 vs. PBS; #P<0.05 vs. unpulsed DCs; †P<0.05 vs. RNA-DCs. CTL, cytotoxic T lymphocyte; DC, dendritic cell; Pro-DC, tumor lysate protein-pulsed dendritic cell; RNA-DC, tumor total RNA-pulsed dendritic cell; CCSCs, colon cancer stem cells; E/T, effector-to-target; IFN-γ, interferon-γ.
Additionally, lymphocytes from Pro-DC-treated tumor-bearing mice released high levels of IFN-γ when stimulated with CCSCs. When CTLs were mixed with CCSCs at an E/T ratio of 20:1, the IFN-γ levels produced by splenic CTLs from the Pro-DC (745.53±62.28 ng/l) and RNA-DC (521.39±34.22 ng/l) groups were significantly higher compared with those from the unpulsed DC (403.09±46.71 ng/l) and control (131.89±21.87 ng/l) groups (P<0.05; Fig. 6B).
Discussion
CD44, a prominent transmembrane glycoprotein, is a unique cell adhesion molecule that serves an important role in cancer cell migration and matrix adhesion. CD44 is overexpressed in colorectal cancer and is associated with enhanced tumorigenesis and tumorsphere formation in vitro, as well as the initiation of xenograft tumors in vivo (42–44). In the present study, CD44 was used as a marker to isolate subsets enriched in CSCs from the mouse colon adenocarcinoma cell line CT-26 by MACS; the results demonstrated that CD44+ CT-26 CSCs accounted for 6.98% of all CT-26 cells. Highly-purified CCSCs (>95%) were obtained and their differentiation potential, tumorsphere formation capacity and surface marker expression were confirmed. Using a technique already used in clinical immunotherapy trials (45–48), DCs were pulsed with tumor cell lysate or total RNA to transfer a wide range of CSC antigens to DCs, increasing targets for specific T-cell clones and providing antigenic epitopes for MHC class I and II processing pathways.
The results of the present study demonstrated that DCs loaded with CCSC lysate or total RNA induced effective antitumor responses in tumor-bearing mice, which was demonstrated by the diminished tumor volume and weight, as well as an increased lifespan, compared with those in control animals. In addition, an induced antigen-specific CTL response against CCSCs was identified in murine therapeutic models, as well as specific CTL activity in vitro based on high levels of IFN-γ secretion. Similarly, DCs pulsed with total RNA from CD133+ hepatocellular carcinoma cells have been previously demonstrated to induce a specific CTL response to kill hepatocellular carcinoma CSCs via antigen-specific T-cell proliferation and the stimulation of IFN-γ secretion (26). In the majority of previous studies, CSCs were subjected to immunologic recognition and elimination by CD8+ CTLs in an antigen-specific manner, resulting in favorable outcomes in animal models, including delayed tumor growth, tumor regression and extended survival times (13,22–26). In the present study, however, a significantly more potent antitumor immune response was elicited by lysate-loaded DCs against CCSCs compared with RNA-DCs or unpulsed DCs. These results suggested that CSC lysates may be a more promising antigen for DC loading compared with total RNA; this was in agreement with the results of a previous immunotherapy study, which reported that DCs pulsed with whole-cell lysates are highly effective against breast CSCs compared with DCs loaded with tumor total RNA (22). Similarly, the level of IFN-γ expression in the CTL stimulated by lysate-loaded DCs is greater compared with that in T cells stimulated by RNA-loaded DCs in B cell chronic lymphocytic leukemia (49). By contrast, other studies have demonstrated enhanced CTL responses induced by total RNA or mRNA compared with tumor lysate (36,50). In addition, DCs pulsed with a tumor lysate derived from the mouse colon adenocarcinoma cell line CT-26 are able to induce CTL activity against target cancer cells, but not B16 melanoma cells, suggesting the selective targeting of the original tumor cells (12). In the present study, RNA-DCs elicited only a slightly stronger antitumor effect compared with unpulsed DCs, although the difference was statistically significant. This antitumor immune response may be enhanced by transfecting cells with tumor mRNA through electroporation or with lipofection instead of conventional RNA/DC co-culture (51). Vaccination with mRNA-transfected DCs stimulates robust CTL responses and antitumor immunity in mice and induced tumor antigen-specific CD8+ T cell responses in patients with prostate and renal cancer (34). However, Nair et al (35) have demonstrated efficient transfection of DCs with naked RNA when the DCs are immature, a stage at which they take up exogenous materials avidly, and revealed that DCs transfected with naked RNA are comparable to those transfected with RNA/lipid complexes in the stimulation of antigen-specific CTL responses. Using RNA as an antigen has an advantage since it requires only a small amount of tumor tissue to produce sufficient antigens for DC sensitization, which is crucial for postoperative patients or patients with a low tumor burden (46). Although the majority of tumors exclusively express MHC class I and the adoptive transfer of activated CTLs has been successful (33), a long-term memory-based immune response needs to be verified based on its association with cancer patient survival.
In conclusion, the results of the present study demonstrated that DCs pulsed with lysates of CCSCs isolated from CD44+ CT-26 mouse colon adenocarcinoma cells exhibited potent anticancer efficacy in tumor-bearing mice. The results revealed that DCs pulsed with tumor lysate decreased the tumor volume, extended survival time, induced anti-tumor immunity in vivo and generated tumor-specific CTL responses. In addition, Pro-DC vaccination resulted in significantly stronger anticancer effects compared with other methods of DC vaccination. Post-operative vaccination with DCs pulsed with CCSC lysates may decrease the recurrence of colon cancer and prolong tumor-free survival time for patients, suggesting a potentially effective immunotherapeutic strategy for the selectively targeting of CCSCs in established tumors.
Acknowledgements
Not applicable.
Glossary
Abbreviations
- bFGF
basic fibroblast growth factor
- CTL
cytotoxic T lymphocyte
- CCSC
colon cancer stem cell
- CSC
cancer stem cell
- DC
dendritic cell
- EGF
epidermal growth factor
- E/T
effector-to-target
- rmGM-CSF
recombinant murine granulocyte-macrophage colony stimulating factor
- IFN-γ
interferon-γ
- IL
interleukin
- LDH
lactate dehydrogenase
- MACS
magnetic-activated cell sorting
- MHC
major histocompatibility complex
- Pro-DC
tumor lysate protein-pulsed dendritic cell
- RNA-DC
tumor total RNA-pulsed dendritic cell
Funding
The present study was supported by the Jilin Province Science Foundation (grant no. 20160204036YY) and the Jilin Province Health Technology Innovation Project (grant no. 2017J062).
Availability of data and materials
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.
Authors' contributions
CF and NZ performed the experiments. CF drafted the manuscript and supervised the experiments. CF, YZ, JD, and HX performed data analysis and interpretation. CF and YW conceived and designed the experiments. YW verified the results of the experiments, helped with statistical analysis, and revised the manuscript critically for intellectual content. All authors read and approved the final version of the manuscript.
Ethics approval and consent to participate
The study was approved by the Animal Care and Protection Committee of Laboratory Animal Center, School of Pharmaceutical Sciences, Jilin University (approval no. 20180005).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.American Cancer Society, corp-author. Cancer Facts and Figures 2019. Atlanta: American Cancer Society;2019. https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2019/cancer-facts-and-figures-2019.pdf. [Feb 20;2019 ];
- 2.Van Cutsem E, Oliveira J. ESMO Guidelines Working Group: Advanced colorectal cancer: ESMO clinical recommendations for diagnosis, treatment and follow-up. Ann Oncol. 2009;20(Suppl 4):S61–S63. doi: 10.1093/annonc/mdp130. [DOI] [PubMed] [Google Scholar]
- 3.Wang T, Niu G, Kortylewski M, Burdelya L, Shain K, Zhang S, Bhattacharya R, Gabrilovich D, Heller R, Coppola D, et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat Med. 2004;10:48–54. doi: 10.1038/nm976. [DOI] [PubMed] [Google Scholar]
- 4.Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res. 2015;21:687–692. doi: 10.1158/1078-0432.CCR-14-1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Camus M, Tosolini M, Mlecnik B, Pagès F, Kirilovsky A, Berger A, Costes A, Bindea G, Charoentong P, Bruneval P, et al. Coordination of intratumoral immune reaction and human colorectal cancer recurrence. Cancer Res. 2009;69:2685–2693. doi: 10.1158/0008-5472.CAN-08-2654. [DOI] [PubMed] [Google Scholar]
- 6.Baier PK, Wimmenauer S, Hirsch T, von Specht BU, von Kleist S, Keller H, Farthmann EH. Analysis of the T cell receptor variability of tumor-infiltrating lymphocytes in colorectal carcinomas. Tumour Biol. 1998;19:205–212. doi: 10.1159/000030008. [DOI] [PubMed] [Google Scholar]
- 7.Hsu FJ, Benike C, Fagnoni F, Liles TM, Czerwinski D, Taidi B, Engleman EG, Levy R. Vaccination of patients with B-cell lymphoma using autologous antigen-pulsed dendritic cells. Nat Med. 1996;2:52–58. doi: 10.1038/nm0196-52. [DOI] [PubMed] [Google Scholar]
- 8.Rains N, Cannan RJ, Chen W, Stubbs RS. Development of a dendritic cell (DC)-based vaccine for patients with advanced colorectal cancer. Hepatogastroenterology. 2001;48:347–351. [PubMed] [Google Scholar]
- 9.Trombetta ES, Mellman I. Cell biology of antigen processing in vitro and in vivo. Annu Rev Immunol. 2005;23:975–1028. doi: 10.1146/annurev.immunol.22.012703.104538. [DOI] [PubMed] [Google Scholar]
- 10.Datta J, Terhune JH, Lowenfeld L, Cintolo JA, Xu S, Roses RE, Czerniecki BJ. Optimizing dendritic cell-based approaches for cancer immunotherapy. Yale J Biol Med. 2014;87:491–518. [PMC free article] [PubMed] [Google Scholar]
- 11.Anguille S, Smits EL, Lion E, van Tendeloo VF, Berneman ZN. Clinical use of dendritic cells for cancer therapy. Lancet Oncol. 2014;15:e257–e267. doi: 10.1016/S1470-2045(13)70585-0. [DOI] [PubMed] [Google Scholar]
- 12.Wu YG, Wu GZ, Wang L, Zhang YY, Li Z, Li DC. Tumor cell lysate-pulsed dendritic cells induce a T cell response against colon cancer in vitro and in vivo. Med Oncol. 2010;27:736–742. doi: 10.1007/s12032-009-9277-x. [DOI] [PubMed] [Google Scholar]
- 13.Brown CE, Starr R, Martinez C, Aguilar B, D'Apuzzo M, Todorov I, Shih CC, Badie B, Hudecek M, Riddell SR, Jensen MC. Recognition and killing of brain tumor stem-like initiating cells by CD8+ cytolytic T cells. Cancer Res. 2009;69:8886–8893. doi: 10.1158/0008-5472.CAN-09-2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang S, Darrow TL, Vervaert CE, Seigler HF. Immunotherapeutic potential of tumor antigen-pulsed and unpulsed dendritic cells generated from murine bone marrow. Cell Immunol. 1997;179:84–95. doi: 10.1006/cimm.1997.1151. [DOI] [PubMed] [Google Scholar]
- 15.Vlashi E, Pajonk F. Cancer stem cells, cancer cell plasticity and radiation therapy. Semin Cancer Biol. 2015;31:28–35. doi: 10.1016/j.semcancer.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dawood S, Austin L, Cristofanilli M. Cancer stem cells: Implications for cancer therapy. Oncology (Williston Park) 2014;28:1101–1107, 1110. [PubMed] [Google Scholar]
- 17.Wang T, Shigdar S, Gantier MP, Hou Y, Wang L, Li Y, Shamaileh HA, Yin W, Zhou SF, Zhao X, Duan W. Cancer stem cell targeted therapy: Progress amid controversies. Oncotarget. 2015;6:44191–44206. doi: 10.18632/oncotarget.6176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Snyder V, Reed-Newman TC, Arnold L, Thomas SM, Anant S. Cancer stem cell metabolism and potential therapeutic targets. Front Oncol. 2018;8:203. doi: 10.3389/fonc.2018.00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu KJ, Wang CC, Chen LT, Cheng AL, Lin DT, Wu YC, Yu WL, Hung YM, Yang HY, Juang SH, et al. Generation of carcinoembryonic antigen (CEA)-specific T-cell responses in HLA-A*0201 and HLA-A*2402 late-stage colorectal cancer patients after vaccination with dendritic cells loaded with CEA peptides. Clin Cancer Res. 2004;10:2645–2651. doi: 10.1158/1078-0432.CCR-03-0430. [DOI] [PubMed] [Google Scholar]
- 20.Hasegawa H, Mori M, Haraguchi M, Ueo H, Sugimachi K, Akiyoshi T. Expression spectrum of melanoma antigen-encoding gene family members in colorectal carcinoma. Arch Pathol Lab Med. 1998;122:551–554. [PubMed] [Google Scholar]
- 21.Park DI, Kang MS, Oh SJ, Kim HJ, Cho YK, Sohn CI, Jeon WK, Kim BI, Han WK, Kim H, et al. HER-2/neu overexpression is an independent prognostic factor in colorectal cancer. Int J Colorectal Dis. 2007;22:491–497. doi: 10.1007/s00384-006-0192-8. [DOI] [PubMed] [Google Scholar]
- 22.Chen Q, Cui XX, Liang PF, Dou JX, Liu ZY, Sun WW. Immunotherapy with dendritic cells and cytokine-induced killer cells for MDA-MB-231 breast cancer stem cells in nude mice. Am J Transl Res. 2016;8:2947–2955. [PMC free article] [PubMed] [Google Scholar]
- 23.Dashti A, Ebrahimi M, Hadjati J, Memarnejadian A, Moazzeni SM. Dendritic cell based immunotherapy using tumor stem cells mediates potent antitumor immune responses. Cancer Lett. 2016;374:175–185. doi: 10.1016/j.canlet.2016.01.021. [DOI] [PubMed] [Google Scholar]
- 24.Jachetti E, Mazzoleni S, Grioni M, Ricupito A, Brambillasca C, Generoso L, Calcinotto A, Freschi M, Mondino A, Galli R, Bellone M. Prostate cancer stem cells are targets of both innate and adaptive immunity and elicit tumor-specific immune responses. Oncoimmunology. 2013;2:e24520. doi: 10.4161/onci.24520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang XF, Weng DS, Pan K, Zhou ZQ, Pan QZ, Zhao JJ, Tang Y, Jiang SS, Chen CL, Li YQ, et al. Dendritic-cell-based immunotherapy evokes potent anti-tumor immune responses in CD105+ human renal cancer stem cells. Mol Carcinog. 2017;56:2499–2511. doi: 10.1002/mc.22697. [DOI] [PubMed] [Google Scholar]
- 26.Sun JC, Pan K, Chen MS, Wang QJ, Wang H, Ma HQ, Li YQ, Liang XT, Li JJ, Zhao JJ, et al. Dendritic cells-mediated CTLs targeting hepatocellular carcinoma stem cells. Cancer Biol Ther. 2010;10:368–375. doi: 10.4161/cbt.10.4.12440. [DOI] [PubMed] [Google Scholar]
- 27.Li Q, Lu L, Tao H, Xue C, Teitz-Tennenbaum S, Owen JH, Moyer JS, Prince ME, Chang AE, Wicha MS. Generation of a novel dendritic-cell vaccine using melanoma and squamous cancer stem cells. J Vis Exp. 2014:e50561. doi: 10.3791/50561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gilboa E. DC-based cancer vaccines. J Clin Invest. 2007;117:1195–1203. doi: 10.1172/JCI31205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fields RC, Shimizu K, Mulé JJ. Murine dendritic cells pulsed with whole tumor lysates mediate potent antitumor immune responses in vitro and in vivo. Proc Natl Acad Sci USA. 1998;95:9482–9487. doi: 10.1073/pnas.95.16.9482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boczkowski D, Nair SK, Snyder D, Gilboa E. Dendritic cells pulsed with RNA are potent antigen-presenting cells in vitro and in vivo. J Exp Med. 1996;184:465–472. doi: 10.1084/jem.184.2.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nestle FO, Alijagic S, Gilliet M, Sun Y, Grabbe S, Dummer R, Burg G, Schadendorf D. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat Med. 1998;4:328–332. doi: 10.1038/nm0398-328. [DOI] [PubMed] [Google Scholar]
- 32.Ruben JM, van den Ancker W, Bontkes HJ, Westers TM, Hooijberg E, Ossenkoppele GJ, de Gruijl TD, van de Loosdrecht AA. Apoptotic blebs from leukemic cells as a preferred source of tumor-associated antigen for dendritic cell-based vaccines. Cancer Immunol Immunother. 2014;63:335–345. doi: 10.1007/s00262-013-1515-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosenberg SA. Cancer vaccines based on the identification of genes encoding cancer regression antigens. Immunol Today. 1997;18:175–182. doi: 10.1016/S0167-5699(97)84664-6. [DOI] [PubMed] [Google Scholar]
- 34.Kyte JA, Gaudernack G. Immuno-gene therapy of cancer with tumor-mRNA transfected dendritic cells. Cancer Immunol Immunother. 2006;55:1432–1442. doi: 10.1007/s00262-006-0161-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nair SK, Boczkowski D, Morse M, Cumming RI, Lyerly HK, Gilboa E. Induction of primary carcinoembryonic antigen (CEA)-specific cytotoxic T lymphocytes in vitro using human dendritic cells transfected with RNA. Nat Biotechnol. 1998;16:364–369. doi: 10.1038/nbt0498-364. [DOI] [PubMed] [Google Scholar]
- 36.Pan K, Zhao JJ, Wang H, Li JJ, Liang XT, Sun JC, Chen YB, Ma HQ, Liu Q, Xia JC. Comparative analysis of cytotoxic T lymphocyte response induced by dendritic cells loaded with hepatocellular carcinoma-derived RNA or cell lysate. Int J Biol Sci. 2010;6:639–648. doi: 10.7150/ijbs.6.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Song Z, Guo C, Li Y, Tan B, Fan L, Xiao J. Enhanced antitumor effects of a dendritic cell vaccine transfected with gastric cancer cell total RNA carrying the 4-1BBL gene in vitro. Exp Ther Med. 2012;3:319–323. doi: 10.3892/etm.2011.394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van Tendeloo VF, Ponsaerts P, Lardon F, Nijs G, Lenjou M, Van Broeckhoven C, Van Bockstaele DR, Berneman ZN. Highly efficient gene delivery by mRNA electroporation in human hematopoietic cells: Superiority to lipofection and passive pulsing of mRNA and to electroporation of plasmid cDNA for tumor antigen loading of dendritic cells. Blood. 2001;98:49–56. doi: 10.1182/blood.V98.1.49. [DOI] [PubMed] [Google Scholar]
- 39.Qui P, Ziegelhoffer P, Sun J, Yang NS. Gene gun delivery of mRNA in situ results in efficient transgene expression and genetic immunization. Gene Ther. 1996;3:262–268. [PubMed] [Google Scholar]
- 40.Lutz MB, Kukutsch N, Ogilvie AL, Rössner S, Koch F, Romani N, Schuler G. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods. 1999;223:77–92. doi: 10.1016/S0022-1759(98)00204-X. [DOI] [PubMed] [Google Scholar]
- 41.Schnurr M, Galambos P, Scholz C, Then F, Dauer M, Endres S, Eigler A. Tumor cell lysate-pulsed human dendritic cells induce a T-cell response against pancreatic carcinoma cells: An in vitro model for the assessment of tumor vaccines. Cancer Res. 2001;61:6445–6450. [PubMed] [Google Scholar]
- 42.Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12:1167–1174. doi: 10.1038/nm1483. [DOI] [PubMed] [Google Scholar]
- 43.Dalerba P, Dylla SJ, Park IK, Liu R, Wang X, Cho RW, Hoey T, Gurney A, Huang EH, Simeone DM, et al. Phenotypic characterization of human colorectal cancer stem cells. Proc Natl Acad Sci USA. 2007;104:10158–10163. doi: 10.1073/pnas.0703478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Du L, Wang H, He L, Zhang J, Ni B, Wang X, Jin H, Cahuzac N, Mehrpour M, Lu Y, Chen Q. CD44 is of functional importance for colorectal cancer stem cells. Clin Cancer Res. 2008;14:6751–6760. doi: 10.1158/1078-0432.CCR-08-1034. [DOI] [PubMed] [Google Scholar]
- 45.Janikashvili N, Larmonier N, Katsanis E. Personalized dendritic cell-based tumor immunotherapy. Immunotherapy. 2010;2:57–68. doi: 10.2217/imt.09.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aarntzen EH, Figdor CG, Adema GJ, Punt CJ, de Vries IJ. Dendritic cell vaccination and immune monitoring. Cancer Immunol Immunother. 2008;57:1559–1568. doi: 10.1007/s00262-008-0553-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nestle FO, Farkas A, Conrad C. Dendritic-cell-based therapeutic vaccination against cancer. Curr Opin Immunol. 2005;17:163–169. doi: 10.1016/j.coi.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 48.Tuyaerts S, Aerts JL, Corthals J, Neyns B, Heirman C, Breckpot K, Thielemans K, Bonehill A. Current approaches in dendritic cell generation and future implications for cancer immunotherapy. Cancer Immunol Immunother. 2007;56:1513–1537. doi: 10.1007/s00262-007-0334-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kokhaei P, Choudhury A, Mahdian R, Lundin J, Moshfegh A, Osterborg A, Mellstedt H. Apoptotic tumor cells are superior to tumor cell lysate, and tumor cell RNA in induction of autologous T cell response in B-CLL. Leukemia. 2004;18:1810–1815. doi: 10.1038/sj.leu.2403517. [DOI] [PubMed] [Google Scholar]
- 50.Junking M, Grainok J, Thepmalee C, Wongkham S, Yenchitsomanus PT. Enhanced cytotoxic activity of effector T-cells against cholangiocarcinoma by dendritic cells pulsed with pooled mRNA. Tumour Biol. 2017;39:1010428317733367. doi: 10.1177/1010428317733367. [DOI] [PubMed] [Google Scholar]
- 51.Tyagi RK, Mangal S, Garg N, Sharma PK. RNA-based immunotherapy of cancer: Role and therapeutic implications of dendritic cells. Expert Rev Anticancer Ther. 2009;9:97–114. doi: 10.1586/14737140.9.1.97. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.