

Abstract
For decades, there has been debate regarding the origin of the catalytic power of enzymes. In this work, we use the approach of computational chemistry to study the enzyme catechol O-methyltransferase (COMT) and reveal that the two current views on the catalytic mechanism of enzymes, the rate-promoting vibrations and the electric field, may both be viewed as part of the chemical step catalyzed by COMT. However, we show that the rate-promoting vibrations cause the electrostatic effect. This work provides insight into the catalytic mechanism of COMT and resolves a longstanding controversy regarding this enzyme’s mechanism.
Keywords: methyltransferase, rate-promoting vibrations, electric field preorganization, transition path sampling, QM/MM simulation
Graphical Abstract
INTRODUCTION
Understanding the source of the catalytic power of enzymes is of vital importance in many practical fields including designing drugs that hinder specific reactions or artificially designing enzymes to catalyze reactions that do not exist in natural biological systems. Among many proposals aimed at explaining the catalytic power of enzymes, the effect of protein motions has been of significant interest. In addition to slow conformational motions that prepare the system to bind the substrate and bring it to a potentially reactive configuration, one type of protein motion directly coupled to barrier passage are rate-promoting vibrations (RPVs). They are fast, femtosecond to picosecond, protein motions that are coupled to the chemical step of enzymatic reactions. These motions have been found in several enzymes including alcohol dehydrogenase (ADH),1 lactate dehydrogenase (LDH),2,3 and purine nucleotide phosphorylase (PNP).4,5 Experimental work on LDH6 and PNP7 employing kinetic isotope effects (KIE) and “heavy enzymes” have further supported the importance of RPVs in reaching the transition state in these enzymes. For example, systems where the chemical step consists of a particle transfer, a rate-promoting vibration, may consist of residues located along the donor–acceptor axis. The concerted motion of these residues, behind the donor and the acceptor, reduces the donor–acceptor distance, and thus, lowers the free energy barrier to the reaction.
Another proposed explanation for the origin of the catalytic power of enzymes, which has also been widely studied, is the electrostatic preorganization effect. This proposal states that the active site of enzymes pre-organize before the transition state to stabilize nascent charge, thus lowering the barrier to the chemical step via electrostatic stabilization. Recent work has provided insight on this effect, including an experimental study by Fried et al. using the vibrational Stark effect as a probe of the strength of the electric field inside the active site of ketosteroid isomerase (KSI)8 and a computational study that calculates the strength of electric fields in KSI, LDH, and PNP.9 Seemingly distinct, the two proposals both involve motions of enzymes and may be inherently conveying the same piece of information from different perspectives.
In this study, we focus on the enzyme catechol O-methyltransferase (COMT), in which both a protein motion effect10 and electrostatic preorganization11 have been proposed to be the central cause of catalysis. The reaction catalyzed by COMT involves the transfer of a methyl group from S-adenosylmethionine (SAM) to a catechol molecule, shown schematically in Figure 1. The rate-limiting step of the COMT enzymatic process has been proposed to be the chemical step.12,13 COMT plays an important role in many physiological processes including the metabolism of catecholamine neuro-transmitters14 and the extracellular metabolism of dopamine.15 Insight on the source of the catalytic power of COMT can aid in the ongoing research in designing drugs targeting COMT to treat Parkinson’s disease,15,16 and COMT serves as a model system for comparing the rate-promoting vibration effect and the electrostatic preorganization effect in one enzyme.
Figure 1.
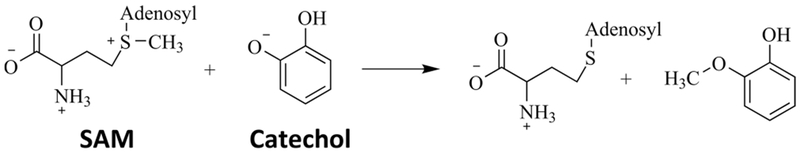
Reaction Mechanism of COMT.
The protein motion, proposed to be promoting the rate of the chemical step of COMT, takes the form of a compression that compacts the active site, specifically the SAM molecule. Earlier experimental studies studying the KIE of rat liver COMT discovered an inverse secondary deuterium KIE,12,13 suggesting the existence of a compressed transition state of the catalyzed reaction. This idea has been further supported by comparing the secondary deuterium KIE of wild-type COMT with several mutated COMT variants. Enzymes with mutations on Tyr68 have shown decreased catalytic efficiency with a larger inverse secondary deuterium KIE,10 suggesting that Tyr68, which resides right behind the donor molecule SAM, plays an important role in compacting the active site for the enzymatic reaction. Further theoretical studies on COMT have suggested that mutations on Tyr68 would increase both the donor–acceptor distance and the acceptor-methyl group C distance in the reactant state.17 The other proposal, which is the electrostatic preorganization in COMT, has also been studied extensively. As proposed by Lameira et al. in a computational investigation on COMT, the reduced free energy barrier of COMT is caused by the preorganization of the protein active site to stabilize the transition state of the reaction.11 Some recent computational studies have provided further insights into this proposal. Saez et al. stated that the major reduction of the activation barrier of wild-type COMT, calculated by the adaptive string method, takes place in the electronic rearranging, bond-breaking–bond-forming phase of the reaction.18 Swiderek et al.19 investigated the enzyme glycine N-methyltransferase (GNMT), which catalyzes a reaction similar to COMT, by mapping the free energy surface of the reaction process and calculating the evolution of charges and electrostatic potential generated by GNMT on the reaction-involved atoms along the reaction progress. This study has found that the wild-type GNMT creates a more negative electrostatic potential, which stabilizes the positively charged methyl group compared to other mutants.19 Besides the two proposals discussed above, there are other ideas aiming to explain the source of the catalytic power of COMT. Lau and Bruice proposed the idea of a near attack conformation, which can be viewed as a static approximation to a rate-promoting vibration.20 Recently, Czarnota et al.21 published a study that correlates the active site compaction effect and the electrostatic preorganization effect. This study stated that two “equatorial” residues, Asp141 and Met40, compact the active site and stabilizes the positive charge on the methyl group through hydrogen bonding between Met40 and the methyl group.21 Calculated electrostatic stabilization energies on molecular dynamic trajectories show a strong correlation with the Met40 Cα-Asp141 Cα distance, indicating that the compaction effect is potentially related to the electrostatic preorganization. As discussed in detail in the Results and Discussion section, we found this compaction motion in our study. Following on this work, we report further studies on the role of promoting vibrations in the catalytic process of COMT.
One major caveat regarding some recent computational studies on COMT is that mapping the progress of the enzymatic reaction onto a chosen donor–acceptor reaction coordinate does not necessarily give a complete description of the reaction pathway, since the reaction coordinate might be much more complicated. As a matter of fact, the reaction coordinate of the enzymatic reaction of COMT had not yet been rigorously found. Reaction pathways generated from an enhanced sampling method using simple sets of reaction coordinates may be heavily biased. In this study, we report an unbiased reaction coordinate through a combination of the transition path sampling (TPS) method and committor distribution calculations. The effects of protein motions and electrostatic field shaping have been examined on the nonbiased reactive trajectories generated by TPS. As will be described later, both effects have been found in the catalytic process of COMT, but protein motions are in fact causing the electric field shaping, the very concept of “preorganization”.
METHODS
System Setup.
We set up our system from the X-ray crystal structure of COMT co-crystalized with SAM, 3,5-dinitrocatechol and Mg2+ from Rutherford et al.22 (Protein Data Bank ID: 3BWM). System preparation follows the scheme of previous studies on enzymes.2,23 All molecular dynamic (MD) simulations are carried out with the CHARMM molecular dynamics program,24,25 with the CHARMM36 force field. CHARMM parameters of SAM were obtained from the work of Senn.26 Since a 3,5-dinitrocatechol molecule was co-crystalized with COMT in the crystal structure, we modified it back to a catechol molecule, which is the actual substrate of the methyl transfer reaction, and obtained CHARMM force field parameters with the CHARMM parameter preparation tool paramChem.27 The catechol molecule is singly protonated, as suggested by multiple theoretical studies.21,28,29 The catechol, the SAM molecule, and the magnesium ion coordinated with the catechol were treated quantum mechanically (QM) using the PM3 semi-empirical method, as shown in Figure 2a. The system was solvated with water molecules using TIP3P model in a nanodroplet, whose volume was set to be 15 A away from the surface of the protein, and then neutralized with potassium ions. A two-step minimization was then performed. The system was first minimized with 100 steps of a steepest decent method to avoid clashes between atoms, then with 1000 steps of an adopted basis Newton–Raphson (ABNR) method. Harmonic constraint forces (100 kcal mol−1 Å−2) were applied on the QM region during the steepest descent step then gradually relaxed during the ABNR process. The minimized system was then heated to 300 K and equilibrated for 1000 picoseconds. The coordination state of the Mg2+ is of vital importance for the active site geometry and needs to be treated carefully during heating and equilibration. Different coordination geometries of Mg2+ have been proposed.28,29 We followed the geometry proposed by Kulik et al. through large-scale density functional theory (DFT) calculations,29 in which Mg2+ is coordinated with the catechol, Asp141, Asp169, Asn170, and a water molecule. Also, two residues adjacent to the catechol, Lys144 and Glu199, are stabilizing the geometry of the catechol through hydrogen bonds.28 We applied harmonic constraint forces to put Asp141, Asp169, and Asn170 in their correct coordination geometry and constrained Lys144 and Glu199 to stay hydrogen-bonded to the catechol during the heating process. The relative positions of these residues are shown in Figure 2b. After the heating step, all these constraint forces are slowly relaxed during the equilibration process.
Figure 2.
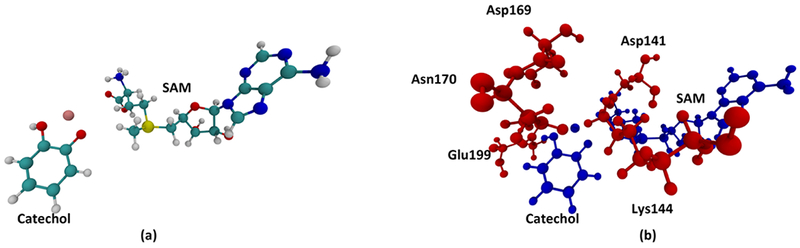
The QM region and adjacent residues. (a) QM region of the system. (b) Relative positions of residues constraint in the heating and equilibration step (red) to the QM region (blue).
Transition Path Sampling.
We used the Transition Path Sampling (TPS) method30–32 to generate MD trajectories that contain the chemical step of the reaction. TPS is a Monte Carlo method that samples the space of transition paths connecting two metastable states, without any prior knowledge of the reaction coordinate of the system. In the case of a chemical reaction, the two states will be the reactant and the product of the reaction. The first reactive trajectory is generated by applying constraint forces to bias the system toward the product state. To generate a new reactive trajectory, a slice is chosen randomly along the previous trajectory, and its momentum is perturbed randomly choosing from a Boltzmann distribution. One propagates the system by shooting in both backward and forward direction in time. If the new trajectory is reactive, we add it to the ensemble and use it as a seed for generating the next reactive trajectory. After generating a large number of reactive trajectories, we had the information needed to describe the chemical step of the methyl transfer reaction catalyzed by COMT. We used the donor-methyl group distance and the acceptor-methyl group distance as the order parameters that define reactants and products. The definitions for the states are: reactant state if the donor-methyl distance is smaller than 2.2 Å or product state if the acceptor-methyl distance is smaller than 1.6 Å. The length of both forward and backward shootings is 250 fs, making each reactive trajectory 500 fs long.
Committor Analysis.
We determined transition state structures from the trajectory ensemble of COMT through committor analysis.33,34 We selected several slices on a reactive trajectory and initiated fifty 250 fs-long trajectories from each slice with a randomly assigned momentum. From these 50 trajectories, we calculated the committer probability that the system ends at the product state. The time slice with 0.5 committor probability is the transition state of that trajectory.
The transition states obtained from the reactive trajectory ensemble of COMT form the stochastic separatrix of the ensemble. We determined the reaction coordinate of COMT through committor distribution analysis.34 From each transition state on the separatrix, a new 250 fs trajectory is initiated, with harmonic constraint forces on atoms and degrees of freedoms that are tested as part of the reaction coordinate. If we have chosen correctly all the degrees of freedom involved in the reaction coordinate, then these trajectories will stay on the stochastic separatrix. This is quantified by calculating committor values for slices along these constrained trajectories. If these trajectories did stay on the separatrix, then the calculated committor values would be centered at 0.5. If these trajectories deviate from the separatrix, new constrained trajectories must be initiated with new sets of constrained residues. This methodology has been used to successfully find reaction coordinates for enzymatic reactions in several previous studies.23,35
Electric Field Calculation.
Recent theoretical works have used different metrics to quantify the effect of electrostatic preorganization in COMT including calculating the contribution of the electrostatic terms in the total free energy barrier,11,21 calculating electrostatic potentials,19 and calculating charges on reaction-involved atoms.18 In this study, we use a methodology that has successfully reproduced experimentally measured electric field strength in the active site.9 We calculated Mulliken charges of atoms in the quantum region and calculated the electric field exerted onto the donor atom and the acceptor atom using Coulomb’s law, as shown in eq 1. In the earlier work, the field was projected onto a carbonyl dipole, which could be used as a probe for measuring the strength of the electric field in the active site.8 Since there is not a carbonyl group on both substrates, we directly projected the fields onto the donor–acceptor axis. Positive direction of the axis was defined as pointing from the donor atom to the acceptor atom. Half of the sum of the two projections would be the total projection of the electric field onto the donor–acceptor dipole.
| (1) |
RESULTS AND DISCUSSION
Transition Path Ensemble.
Using the transition path sampling (TPS) method, we have generated a trajectory ensemble containing 180 reactive trajectories. Six uncorrelated transition state structures have been selected for the trajectory ensemble using committor analysis. Some important characteristics of the transition state structures are shown in Table 1. These characteristics indicate that the transition state of the reaction lies roughly halfway on the methyl transfer reaction, as shown in Table 1 and in Figure S1 of the Supporting Information.. We noticed that the average donor–acceptor distance at the transition state of the reaction is significantly shorter than the donor–acceptor distance during the equilibration runs, which had an average of 4.94 Å. As a test of the semi-empirical methods we use, we compared to two high-level single-point calculations. Calculated transition state structures are slightly different from recent work by Czarnota et al.21 and Kulik et al.,29 specifically in that the transition state geometry we acquired possesses a slightly larger donor-methyl group distance and a smaller acceptor-methyl group distance, meaning that the transition state we calculated is slightly “earlier” along the course of the reaction. The differences though are modest and do not affect the overall quality of the TPS results.
Table 1.
Average Transition States Structures
Reaction Coordinate of COMT and the Compression Effect.
Committor distribution analysis was performed with different sets of constrained residues, starting from the calculated transition state structures. We started by applying constraints only to atoms in the quantum region of the QM/MM calculation. This yields a committor distribution significantly skewed toward products (Figure 3a). Amino acids adjacent to the quantum region were then incorporated into the constraint. We succeeded in finding an optimal committor distribution when 4 amino acids, Tyr68, Glu90, Leu198, and Glu199, in addition to the quantum region, are under constraint (Figure 3b). These four amino acids and the quantum region constitute the reaction coordinate of the methyl transfer reaction catalyzed by COMT (Figure 3c). We did carefully check the effect of two “equatorial” residues, Asp141 and Met40, which have been suggested to be involved in the electrostatic pre-organization effect of the active site.21 Through our methodology of committor distribution analysis, they were shown not to be involved in the reaction coordinate (Figure 4a) on the time scale of motion across the barrier. We did observe the compaction effect of Met40 and Asp141, as indicated by the Asp141 Cα-Met 40 Cα distance (labeled as the black dashed line in Figure 4b, plotted as the black solid line in Figure 4c) prior to transition state passage. This effect starts 2000 fs prior to the transition state of the reaction and finishes 500 fs prior to the transition state. The absence of these residues from the reaction coordinate is due to the limitation of committor distribution analysis method in studying motions taking place closer to the transition states, and one may view the residues as being an important “extended” reaction coordinate. We have previously found such motions long before transition state passage as being important in both purine nucleoside phosphorylase and lactate dehydrogenase.36–38
Figure 3.
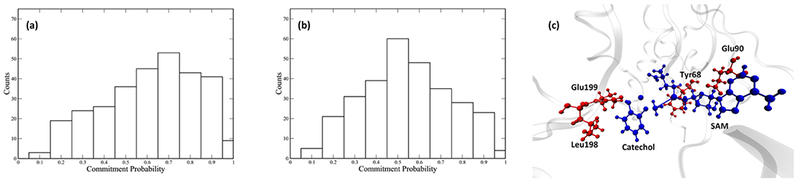
Committor distribution and reaction coordinate of the wild-type COMT. Panel a shows the committor distribution when only the QM region is under constraint, Panel b shows the committor distribution when the QM region and 4 residues. Leu198, Glu199, Tyr68, and Glu90 are under constraint. Panel c shows the relative positions of the 4 residues involved in the reaction coordinate (red) to the QM region (blue).
Figure 4.
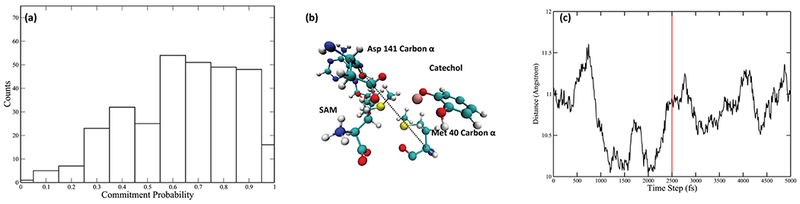
Examining the effect of the “equatorial” residues. (a) Commitor distribution with Met40, Asp41, and Asn170, together with the QM region, under constraint. (b) Relative position of Met40 and Asp141. Distance between Met40 Cα and Asp141 Cα is labeled by the dashed black line. (c) Distance mentioned above in a 5000 fs-long TPS trajectory (black solid line), with the transition state being labeled by the vertical red solid line. The reduction of this distance approximately 2000 fs prior to the transition state indicates the compaction effect of Met40 and Asp141.
Looking deeper into the role of Tyr68, Glu90, Leu198, and Glu199, we found three pushing motions, two from the catechol side and one from the SAM side, that are coupled to the progress of the reaction. Important time slices of these motions are taken from a representative trajectory and displayed in Figure 5. The first pushing motion is exerted by Leu198 Cγ onto the acceptor catechol molecule. Figure 5a,b describes the scheme of this motion: Leu198 moves toward the catechol molecule and pushes the catechol molecule toward the SAM molecule. We plotted the distance between Leu198 Cγ and the nearest carbon atom on the catechol molecule (catechol C61, labeled in Figure 5a,b) in the representative trajectory (solid purple line in Figure 5f). This motion reaches its maximum compression around 100 fs (the lowest point of the solid purple line). The second pushing motion is exerted by Glu199 onto the catechol, which also pushes the catechol molecule toward the SAM and takes place from the time slice of 150 fs to approximately 275 fs. Figure 5c shows the final geometry of the active site after the second pushing motion. The third pushing motion comes from the SAM side and is exerted by Tyr68 Cβ onto the donor atom. As shown in Figure 5d,e, Tyr68 Cβ moves toward the donor atom and pushes the donor atom toward the catechol molecule. The magnitude of the movement of Cβ is roughly 1 Å. This level of compaction is commensurate with previous promoting vibration studies, and as we have shown in alcohol dehydrogenase computations, is enough to significantly reduce free energy barriers in reactive trajectories. We also monitored the progress of this pushing motion by plotting the distance between Tyr68 Cβ and the donor atom, shown in Figure 5f as the solid green line. This pushing motion starts approximately at 200 fs and reaches its maximum compression at approximately 300 fs (the lowest point of the solid green line at around 300 fs). After this pushing, the donor–acceptor distance is significantly reduced from 5.13 Å, which is close to the average donor–acceptor distance during equilibration to 4.37 Å at the transition state. While the motion from Tyr68 clearly plays an important role in preparing the system for the chemical reaction by significantly reducing the donor–acceptor distance, the role of the first two motions from the catechol side merits further discussion. As reflected by the donor–acceptor distance in a 5000 fs TPS trajectory in Figure S2 of the Supporting Information, the active site of COMT is mobile and constantly fluctuating. The first two pushing motions from the catechol side play the role of preparing the geometry of the catechol molecule and thus preparing it for the chemical reaction.
Figure 5.
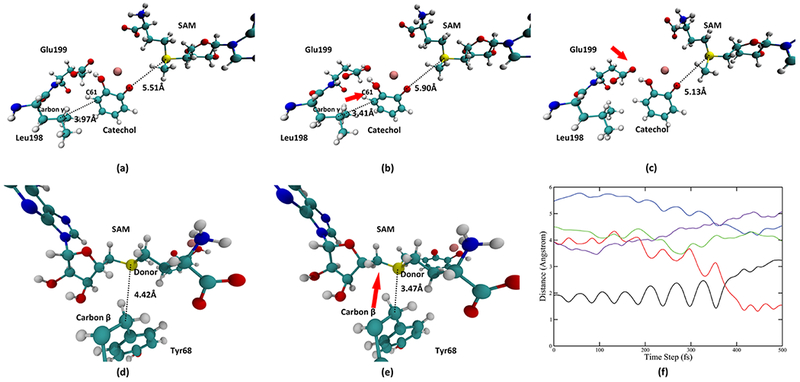
Representations of pushing motions in the wild-type COMT. (a, b) System before and at the maximum of the pushing motion from Leu198, respectively. Distance between Leu198 Cγ and the catechol C61 is reduced from 3.97 Å to 3.41 Å, as labeled in (a, b). Direction of this pushing is shown by the red arrow in (b). (c) System after the pushing from Glu199. Direction of this pushing is labeled by the red arrow in (c). (d, e) System before and at the maximum of the pushing motion from the Tyr68 Cβ to the donor atom, respectively. Distance between the Tyr68 Cβ and the donor atom is reduced from 4.42 Å to 3.47 Å. Direction of this pushing is indicated by the red arrow in (e). The first and the third pushing are further visualized in (f). In (f), the black solid line and the red solid line indicate the bond-breaking (donor-methyl group distance) and bond-forming (acceptor-methyl group distance) progress of the reaction. The blue solid line is the donor–acceptor distance, the green solid line is the distance between the Tyr68 Cβ and the donor atom. The purple solid line is the distance between the Leu198 Cγ and the catechol C61.
The fact that Tyr68 is involved in the reaction coordinate of COMT and is responsible for part of the protein motion is in accord with both experimental10 and theoretical17 studies stating the importance of Tyr68 in the catalytic process. To further test the role of Tyr68, we looked into the Y68A mutant, whose catalytic efficiency is significantly reduced relative to the wild-type.10 We prepared the mutant by replacing Tyr68 of the COMT wild-type crystal structure with an alanine in CHARMM. The same preparation scheme was followed, then we used the same methodologies to study the reactive trajectory ensemble generated by TPS. Transition state structures of Y68A show an extended donor–acceptor distance (Table S1 of the Supporting Information). The reaction coordinate of the Y68A mutant has been determined. Constraining only the QM region gave a committor distribution skewed toward the reactant side (Figure 6a). With Leu198, Glu199, and Met40 constrained during a walk that started from transition states, we were able to achieve a committor distribution centered at 0.5 (Figure 6b). The relative positions of these three residues and the QM region are shown in Figure 6c–e. Interestingly, both Ala68 and Glu90 were no longer involved in the reaction coordinate. Tracking the motions of Ala68 in the representative trajectory, we found that the distance between Ala68 Cβ and the donor atom (labeled as the dashed line in Figure 6e and green solid line in Figure 6f) is not decreasing as the system progresses toward the transition state, nor is it small enough to make van der Waals contact. Combined with the fact that Ala68 is not in the reaction coordinate of the Y68A mutant, it is clear that the pushing motion from the SAM side has been eliminated by the mutation. Mutating Tyr68 to an alanine, which has a smaller side chain, makes the SAM side of the active site more flexible and fails to preserve the correct geometry for a pushing motion. As for the catechol side, as shown in Figure 6d, the catechol molecule in Y68A mutant is located slightly further away from both Leu198 and Glu199, making both residues unable to exert a pushing motion on the catechol. The distance between Leu198 Cγ and C61 on the catechol molecule (solid purple line) is shown in Figure 6f, which behaves similarly to the Ala68 Cβ-donor distance mentioned above, meaning that the protein motions from the catechol side are also eliminated. Finally, Met40 shows little movement along the course of the reaction. It is hydrogen-bonded to the transferred methyl group through the backbone oxygen (Figure S3c of the Supporting Information), but this hydrogen bonding interaction is not altered during the reaction (Figure S3d of the Supporting Information). Therefore, we believe that the role Met40 plays in the reaction coordinate, similar to that of Leu198 and Glu199, is to fix the geometry of the catechol molecule. The fact that mutating Tyr68 to Ala68 eliminates protein motions from both sides is a strong indication that the protein motions in the wild-type COMT are sensitive to the active site geometry and can be influenced by a minor disruption of the active site. Also, in Y68A, a modest reduction in donor–acceptor distance (solid blue line in Figure 6e) still exists but is caused mainly by the change of the orientation of the catechol molecule itself (Figure S3a,b of the Supporting Information), not by any protein motions. Recalling that the catalytic efficiency of Y68A is significantly reduced compared to the wild-type COMT, the role of protein motions in COMT discussed above can be extended beyond compressing the donor–acceptor distance, to giving both the SAM and the catechol molecules sufficient momentum for crossing the barrier.
Figure 6.
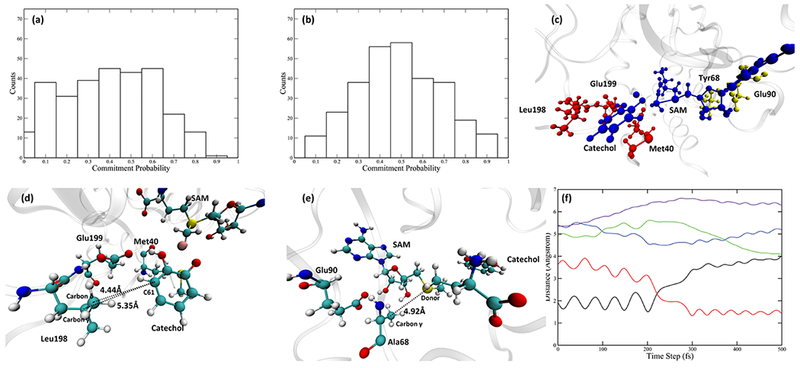
Committor distribution and the reaction coordinate of the Y68A mutant. (a) Committor distribution when only the QM region is under constraint. (b) Committor distribution when the QM region and 3 residues, Leu198, Glu199, and Met40, are under constraint. (c) Relative positions of the 3 residues involved in the reaction coordinate (red) to the QM region (blue). Ala68 and Glu90 are shown in yellow. (d) Another view of the QM region and residues in the reaction coordinate, focusing on the catechol molecule. Distance between Leu198 Cγ/Cδ and the catechol C61 are labeled by dashed lines. (e) Another view of the QM region, Ala68, and Glu90. Distance between Ala68 Cγ and the donor atom is labeled by the black dashed line. (f) Progress of the reaction: the black solid line and the red solid line indicate the bond breaking (donor-methyl group distance) and bond forming (acceptor-methyl group distance). The blue solid line is the donor–acceptor distance, the green solid line is the distance between the Ala68 Cγ and the donor atom. The purple solid line is the distance between the Leu198 Cγ and the catechol C61.
One test of how the overall protein structure changes during the reaction is to study the active site volume. We calculated the volume of the active site using POVME339 for both the wild-type COMT and the Y68A mutant. As shown in Figure S4a of the Supporting Information, in a representative reactive trajectory of the wild-type COMT, the active site volume remains at approximately 440 Å3 before the reaction and increases to 480Å3 along the progress of the reaction, while in the Y68A mutant, the size of the binding site remains at approximately 500Å3 along the reaction. The change of volume in the wild-type COMT does not correlate strongly with the rate-promoting vibrations. We believe that this is because the active site of COMT is complex, needing to accommodate both small and much larger substrate molecules. Compression motions along the donor–acceptor axis do not necessarily reduce the total active site volume to a large extent. However, it is obvious that the wild-type COMT has a more compact active site compared to the Y68A mutant. Interestingly, we did discover that the change of the active site volume in wild-type COMT is in good accordance with the compression of the “equatorial” compaction discovered by Czarnota et al,21 which has also been found in this study to take place 2000 fs prior to the reaction, as shown in Figure 5c and Figure S4b of the Supporting Information..
To further understand the contribution of rate-promoting vibrations to the energetics of the methyl transfer reaction, we calculated free energies barriers, as presented in Table 2, of the wild-type COMT ensemble, Y68A mutant ensemble, and a TPS ensemble starting from wild-type COMT, in which the 4 residues involved in the reaction coordinate is under constraint. Free energy barriers are calculated as the ensemble average of the work produced on the transferred methyl group, which has been used in several previous works.40,41 We acknowledge that our method of calculating free energy barriers underestimates the height of the barrier, as compared to some previous studies on the free energy barrier of COMT.29,42 This is because TPS creates only an ensemble of reactive trajectories. However, it is the difference of free energy barriers between different ensembles that is worthy of attention. As shown in Table 2, the wild-type COMT has a lower barrier (6 kcal/mol) compared to the Y68A mutant (10 kcal/mol). However, the constrained ensemble, even though possessing no mutation, has an average barrier height close to that of the Y68A mutant. By constraining the 4 residues involved in the reaction coordinate, the rate-promoting vibrations of COMT are eliminated. The comparison between the normal wild-type COMT ensemble and the ensemble without rate-promoting vibrations indicate that the rate-promoting vibrations of COMT contribute to lower the barrier height of the reaction by 4 kcal/mol. Finally, we note that the large error bars in the free energy are due to the modest size of the uncorrelated reactive trajectory ensembles.
Table 2.
Average Free Energy Barriers of Three Ensembles
| ensemble | wild-type COMT | Y68A mutant | constrained ensemble from wild-type COMT |
|---|---|---|---|
| free energy barrier height (kcal/mol) | 6.0 ± 5.0 | 10.0 ± 5.0 | 10.0 ± 5.0 |
Electric Field Calculations.
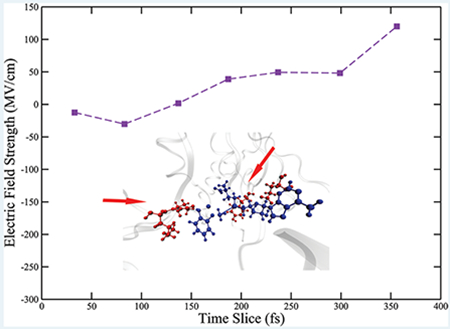
As described in the Methods section, we calculated the projection of the electric field onto the donor–acceptor axis. The positive direction of the projection is defined pointing from the donor to the acceptor. Intuitively, a larger positive projection of the electric field would push the positively charged donor atom and negatively charged acceptor atom closer and result in a shorter donor–acceptor distance. We observed a periodic fluctuation in the calculated fields (Figure S5 of the Supporting Information), that was caused by the bond vibration of the atoms involved in the reaction. We show here the maxima of the fluctuation envelopes before transition states and overlaid them with the progress of the reaction. As shown in Figure 7a, in the wild-type COMT, the field is slightly negative around the time slice of 100 fs. It then experiences a dramatic increase to around 50 MV/cm at 100 fs. The field plateaus for around 100 fs, until experiencing another increase to around 120 MV/cm right before the transition state. The magnitude of the field before the transition state is the same order of magnitude as calculations of fields in the active sites of KSI, LDH, PNP,9 and previous experimental measurement on KSI.8 Clearly, in wild-type COMT, the field projected onto the donor–acceptor axis significantly increases as the system prepares for the chemical reaction and stabilizes a compressed donor–acceptor distance. This observation would seem to support the proposal that the electrostatic preorganization of the active site stabilizes a closer donor–acceptor distance. However, as we will see, donor–acceptor compression caused by protein motions creates the increasing electric field, not the reverse.
Figure 7.

Electric fields and charges from representative trajectories of wild-type COMT and Y68A mutant. (a–d) Calculated electric fields or charges are overlaid with the progress of the reaction. In each of the four figures, the black solid line and the red solid line indicate the bond-breaking and bond-forming progress of the reaction. The blue solid line is the donor–acceptor distance, the green solid line is the distance between the Tyr68/Ala68 Cβ and the donor atom. The purple solid line is the distance between the Leu198 Cγ and the catechol C61. The red dashed line indicates the transition state of the corresponding trajectory. (a, c) Calculated electric field for wild-type COMT and Y68A mutant, respectively. In each figure, the dashed maroon line represents the electric field. (b, d) Charged evolution of the wild-type COMT and Y68A mutant, respectively. In each figure, the dashed maroon line represents the charge on the donor atom, while the dashed yellow line represents the charge on the acceptor atom.
Since the reaction coordinate of COMT has been identified, we can examine the influence of the four residues in the reaction coordinate on the electric field of the active site by overlaying the calculated field on their motions. Residues that are not part of the reaction coordinate are moving but do not influence the course of the reaction, either through physical contact or through modulating the electric field of the active site. Examining the two time periods when the field increases, we found that the first time period aligns with the time when the catechol molecule feels the first protein motion from Leu198 and continues throughout the pushing motion of Glu199, while the second time period aligns with the time the SAM molecule feels the protein motion from Tyr68. The plateau of the electric field starts approximately from when the pushing motion from Glu199 has been completed and lasts until the protein motion from Tyr68 reaches its maximum. The plateau of the electric field indicates that the moving of Tyr68 itself does not play the role of modulating the electrostatic environment of the donor atom. The field is only changing when either of the two substrate molecules “feel” the effect of the pushing motions from the protein, which causes further geometric changes on the substrate molecules. This observation is a very strong indication that the modification of the observed electric field is caused by the protein motions. We also calculated charges on the donor and the acceptor atom along the progress of the reaction, as shown in Figure 7b. Charges on both atoms are only fluctuating in a narrow range as the system proceeds to the transition state, meaning that the electrostatic environment of both atoms is not modified. The only possible explanation for the field modulation is that the active site geometry has been altered by the protein motions.
The electric field for the Y68A mutant was also calculated. As shown in Figure 7c, the field in Y68A remains little changed in the range from 120 MV/cm to 150 MV/cm as the system proceeds to the transition state. The field in Y68A remains at a high value throughout the progress of the reaction, which is caused by a minor change of the active geometry, specifically the Mg29+ ion (Figure S6 of the Supporting Information). Charges on the donor atom and the acceptor atom of the Y68A mutant were also calculated. As shown in Figure 7d, similar to the scenarios in the wild-type COMT and in the Y68A mutant, charges on both atoms are stable as the system progresses toward the transition state. We also calculated charges on other atoms in the QM region in both the Y68A mutant and the wild-type COMT. Charges on all atoms are virtually identical between the Y68A mutant and the wild-type COMT, meaning that the electrostatic environment of the two substrate molecules is little modified by mutating Tyr68 to an alanine. This allows us to make a direct link between the disappearance of protein motions in the Y68A mutant and the reduced field modification. This further supports the claim that protein motions are the cause of the modification in the electric field. In fact, the higher value of the field in the far less-proficient mutant further support the notion that the field along is not the cause of the catalytic effect.
CONCLUSIONS
In this study, we have determined that the reaction coordinate of COMT involves 4 residues in the protein: Tyr68, Glu90, Leu198, and Glu199. Three concerted protein motions that compress the catechol molecule and the donor atom have been identified in the wild-type COMT, which do not exist in the Y68A mutant. Calculation of the electric field on the wild-type COMT and Y68A mutant reveals that the electric field in wild-type COMT is modified to stabilize a compressed donor– acceptor distance; however, the modulation of the electric field is caused by the protein motions.
Supplementary Material
ACKNOWLEDGMENTS
All computer simulations were performed at the University of Arizona High Performance Computing Center on a Lenovo NeXtScale nx360 M5 supercomputer.
Funding
We acknowledge the support of the National Institute of Health Grant R01GM127594.
ABBREVIATIONS
- COMT
catechol O-methyltransferase
- QM/MM
quantum mechanics/molecular mechanics
- TPS
transition path sampling
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.9b02657.
The Supporting Information includes: illustration of the transition state of wild-type COMT, important characteristic of the transition state of Y68A mutant, important distances along the reaction progress of a 5000 fs long TPS trajectory, detailed reaction progress of the Y68A mutant, calculated active site volume of wild-type COMT and the Y68A mutant, calculated electric field of the wild-type COMT, and overlay of the QM region of the wild-type COMT and the Y68A mutant (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Antoniou D; Schwartz SD Internal enzyme motions as a source of catalytic activity: Rate-promoting vibrations and hydrogen tunneling. J. Phys. Chem. B 2001, 105, 5553–5558. [Google Scholar]
- (2).Masterson JE; Schwartz SD The enzymatic reaction catalyzed by lactate dehydrogenase exhibits one dominant reaction path. J. Chem. Phys 2014, 442, 132–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Masterson JE; Schwartz SD Changes in protein architecture and subpicosecond protein dynamics impact the reaction catalyzed by lactate dehydrogenase. J. Phys. Chem. A 2013, 117, 7107–7113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Núñez S; Antoniou D; Schramm VL; Schwartz SD Promoting vibrations in human purine nucleoside phosphorylase. A molecular dynamics and hybrid quantum mechanical/molecular mechanical study. J. Am. Chem. Soc 2004, 126, 15720–15729. [DOI] [PubMed] [Google Scholar]
- (5).Antoniou D; Ge X; Schramm VL; Schwartz SD Mass Modulation of Protein Dynamics Associated with Barrier Crossing in Purine Nucleoside Phosphorylase. J. Phys. Chem. Lett 2012, 3, 3538–3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Wang Z; Chang EP; Schramm VL Triple isotope effects support concerted hydride and proton transfer and promoting vibrations in human heart lactate dehydrogenase. J. Am. Chem. Soc 2016, 138, 15004–15010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Silva RG; Murkin AS; Schramm VL Femtosecond dynamics coupled to chemical barrier crossing in a Born-Oppenheimer enzyme. Proc. Natl. Acad. Sci. U. S. A 2011, 108, 18661–18665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Fried SD; Boxer SG Electric fields and enzyme catalysis. Annu. Rev. Biochem 2017, 86, 387–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Zoi I; Antoniou D; Schwartz SD Electric Fields and Fast Protein Dynamics in Enzymes. J. Phys. Chem. Lett 2017, 8, 6165–6170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Zhang J; Klinman JP Enzymatic methyl transfer: Role of an active site residue in generating active site compaction that correlates with catalytic efficiency. J. Am. Chem. Soc 2011, 133, 17134–17137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Lameira J; Bora RP; Chu ZT; Warshel A Methyltransferases do not work by compression, cratic, or desolvation effects, but by electrostatic preorganization. Proteins 2015, 83, 318–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Gray CH; Coward JK; Schowen KB; Schowen RL alpha.-Deuterium and carbon-13 isotope effects for a simple, intermolecular sulfur-to-oxygen methyl-transfer reaction. Transitionstate structures and isotope effects in transmethylation and transalkylation. J. Am. Chem. Soc 1979, 101, 4351–4358. [Google Scholar]
- (13).Hegazi MF; Borchardt RT; Schowen RL alpha.-Deuterium and carbon-13 isotope effects for methyl transfer catalyzed by catechol O-methyltransferase. SN2-like transition state. J. Am. Chem. Soc 1979, 101, 4359–4365. [DOI] [PubMed] [Google Scholar]
- (14).Roth JA Membrane-bound catechol-O-methyltransferase: a reevaluation of its role in the O-methylation of the catecholamine neurotransmitters. Rev. Physiol., Biochem. Pharmacol 1992, 120,1–29. [DOI] [PubMed] [Google Scholar]
- (15).Espinoza S; Manago F; Leo D; Sotnikova TD; Gainetdinov RR Role of catechol-O-methyltransferase (COMT)-dependent processes in Parkinsonfs disease and L-DOPA treatment. CNS Neurol. Disord.: Drug Targets 2012, 11, 251–263. [DOI] [PubMed] [Google Scholar]
- (16).Kiss LE; Soares-da-Silva P Medicinal chemistry of catechol O-methyltransferase (COMT) inhibitors and their therapeutic utility. J. Med. Chem 2014, 57, 8692–8717. [DOI] [PubMed] [Google Scholar]
- (17).Zhang J; Kulik HJ; Martinez TJ; Klinman JP Mediation of donor–acceptor distance in an enzymatic methyl transfer reaction. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 7954–7959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Saez DA; Zinovjev K; Tuñón I; Vöhringer-Martinez E Catalytic Reaction Mechanism in Native and Mutant Catechol-Omethyltransferase from the Adaptive String Method and Mean Reaction Force Analysis. J. Phys. Chem. B 2018, 122, 8861–8871. [DOI] [PubMed] [Google Scholar]
- (19).Świderek K; Tuñón I; Williams IH; Moliner V Insights on the Origin of Catalysis on Glycine N-Methyltransferase from Computational Modeling. J. Am. Chem. Soc 2018, 140, 4327–4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Lau EY; Bruice TC Importance of correlated motions in forming highly reactive near attack conformations in catechol O-methyltransferase. J. Am. Chem. Soc 1998, 120, 12387–12394. [Google Scholar]
- (21).Czarnota S; Johannissen LO; Baxter NJ; Rummel F; Wilson AL; Cliff MJ; Levy CW; Scrutton NS; Waltho JP; Hay S Equatorial Active Site Compaction and Electrostatic Reorganization in Catechol-O-methyltransferase. ACS Catal 2019, 9, 4394–4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Rutherford K; Trong IL; Stenkamp RE; Parson WW Crystal structures of human 108V and 108M catechol Omethyltransferase. J. Mol. Biol 2008, 380, 120–130. [DOI] [PubMed] [Google Scholar]
- (23).Varga MJ; Dzierlenga MW; Schwartz SD Structurally Linked Dynamics in Lactate Dehydrogenases of Evolutionarily Distinct Species. Biochemistry 2017, 56, 2488–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Brooks BR; Bruccoleri RE; Olafson BD; States DJ; Swaminathan S; Karplus M CHARMM: A program for macro-molecular energy, minimization, and dynamics calculations. J. Comput. Chem 1983, 4, 187–217. [Google Scholar]
- (25).Brooks BR; Brooks CL III; Mackerell AD Jr.; Nilsson L; Petrella RJ; Roux B; Won Y; Archontis G; Bartels C; Boresch S; Caflisch A; Caves L; Cui Q; Dinner AR; Feig M; Fischer S; Gao J; Hodoscek M; Im W; Kuczera K; Lazaridis T; Ma J; Ovchinnikov V; Paci E; Pastor RW; Post CB; Pu JZ; Schaefer M; Tidor B; Venable RM; Woodcock HL; Wu X; Yang W; York DM; Karplus M CHARMM: the biomolecular simulation program. J. Comput. Chem 2009, 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Senn HM; O’Hagan D; Thiel W Insight into Enzymatic C. F Bond Formation from Qm and Qm/Mm Calculations. J. Am. Chem. Soc 2005, 127, 13643–13655. [DOI] [PubMed] [Google Scholar]
- (27).Vanommeslaeghe K; Hatcher E; Acharya C; Kundu S; Zhong S; Shim J; Darian E; Guvench O; Lopes P; Vorobyov I; Mackerell AD Jr. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem 2009, 31, 671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Patra N; Ioannidis EI; Kulik HJ Computational Investigation of the Interplay of Substrate Positioning and Reactivity in Catechol O-Methyltransferase. PLoS One 2016, 11, No. e0161868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Kulik HJ; Zhang J; Klinman JP; Martínez TJ How large should the QM region be in QM/MM calculations? The case of catechol O-methyltransferase. J. Phys. Chem. B 2016, 120, 11381–11394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Dellago C; Bolhuis PG; Csajka FS; Chandler D Transition path sampling and the calculation of rate constants. J. Chem. Phys 1998, 108, 1964. [Google Scholar]
- (31).Dellago C; Bolhuis PG; Chandler D Efficient transition path sampling: Application to Lennard-Jones cluster rearrangements. J. Chem. Phys 1998, 108, 9236–9245. [Google Scholar]
- (32).Bolhuis PG; Dellago C; Chandler D Sampling ensembles of deterministic transition pathways. Faraday Discuss. 1998, 110, 421–436. [Google Scholar]
- (33).Antoniou D; Schwartz SD Protein Dynamics and Enzymatic Chemical Barrier Passage. J. Phys. Chem. B 2011, 115, 15147–15158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Bolhuis PG; Chandler D; Dellago C; Geissler PL TRANSITIONPATHSAMPLING: throwing ropes over rough mountain passes, in the dark. Annu. Rev. Phys. Chem 2002, 53, 291–318. [DOI] [PubMed] [Google Scholar]
- (35).Zoi I; Motley MW; Antoniou D; Schramm VL; Schwartz SD Enzyme homologues have distinct reaction paths through their transition states. J. Phys. Chem. B 2015, 119, 3662–3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Núñez S; Wing C; Antoniou D; Schramm VL; Schwartz SD Insight into catalytically relevant correlated motions in human purine nucleoside phosphorylase. J. Phys. Chem. A 2006, 110, 463–472. [DOI] [PubMed] [Google Scholar]
- (37).Ghanem M; Saen-oon S; Zhadin N; Wing C; Cahill SM; Schwartz SD; Callender R; Schramm VL Tryptophan-free human PNP reveals catalytic site interactions. Biochemistry 2008, 47, 3202–3215. [DOI] [PubMed] [Google Scholar]
- (38).Pineda JRET; Antoniou D; Schwartz SD Slow conformational motions that favor sub-picosecond motions important for catalysis. J. Phys. Chem. B 2010, 114, 15985–15990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Wagner JR; Sørensen J; Hensley N; Wong C; Zhu C; Perison T; Amaro RE POVME 3.0: software for mapping binding pocket flexibility. J. Chem. Theory Comput 2017, 13, 4584–4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Dzierlenga MW; Antoniou D; Schwartz SD Another look at the mechanisms of hydride transfer enzymes with quantum and classical transition path sampling. J. Phys. Chem. Lett 2015, 6, 1177–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Schafer JW; Zoi I; Antoniou D; Schwartz SD Optimization of the turnover in artificial enzymes via directed evolution results in the coupling of protein dynamics to chemistry. J. Am. Chem. Soc 2019, 141, 10431–10439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Kuhn B; Kollman PA QM-FE and molecular dynamics calculations on catechol O-methyltransferase: free energy of activation in the enzyme and in aqueous solution and regioselectivity of the enzyme-catalyzed reaction. J. Am. Chem. Soc 2000, 122, 2586–2596. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.