

Abstract
Liver cancer is one of the most common cancer types globally. However, the acquisition of drug resistance limits the effectiveness of chemotherapy and commonly results in metastasis. Therefore, an effective therapeutic approach to target chemoresistance-associated cellular molecules is imperative. Claudin-1 (CLDN1) has previously been reported to be associated with the development of drug resistance. The present study investigated the effect of CLDN1 on the sensitivity of 5-fluorouracil (5-FU)-resistant liver cancer cells. Firstly, a 5-FU-resistant HepG2 liver cancer cell line (Hep/5FU) was developed by continuous 5-FU treatment. MTT proliferation, Transwell and Matrigel assays indicated that Hep/5FU cells were significantly resistant to 5-FU, and demonstrated increased migration and invasion abilities, compared with parental HepG2 cells. Furthermore, reverse transcription-quantitative polymerase chain reaction and western blot analysis indicated that mRNA and protein expression levels of CLDN1 were significantly increased in Hep/5FU cells, compared with HepG2 cells. CLDN1 was knocked down by transfection with small interference RNA. MTT and Annexin V-fluorescein isothiocyanate/propidium iodide assays demonstrated that CLDN1 silencing significantly inhibits proliferation and enhances apoptosis induced by 5-FU treatment in Hep/5FU cells, compared with non-silenced Hep/5FU cells. Additionally, CLDN1 silencing attenuated the migration and invasion capabilities of Hep/5FU cells. In addition, it was identified that CLDN1 silencing decreased drug resistance by inhibiting autophagy, which was associated with a decrease in the ratio of microtubule-associated protein 1A/1B-light chain 3 (LC3)-II/LC3-I and upregulation of P62. A cell proliferation assay revealed that the addition of autophagy inhibitor 3-methyladenine decreased drug resistance of Hep/5FU cells. By contrast, incubation with the autophagy agonist Rapamycin elevated drug resistance of CLDN1-silenced Hep/5FU cells. In summary, these data indicate that CLDN1 may be a potential target for resensitizing resistant liver cancer HepG2 cells to 5-FU by regulating cell autophagy.
Keywords: claudin-1, 5-fluorouracil, proliferation, migration, invasion, autophagy
Introduction
Liver cancer is one of the most common malignant tumor types, exhibiting the highest rate of cancer-associated mortality globally in 2014 due to uncontrolled cell growth in the liver and high metastatic ability (1). Anticancer drugs are frequently applied to liver cancer treatment combined with surgical therapy and/or radiation therapy (2). However, the majority of patients are diagnosed at an advanced stage, when liver cancer is highly resistant to the existing therapeutics, which results in poor prognosis (3,4). 5-Fluorouracil (5-FU) is an anticancer drug that is used as one of the standard chemotherapies for liver cancer treatment (5). However, 5-FU therapy has been demonstrated to frequently be inefficient due to the acquisition of drug resistance and cytotoxicity at high concentrations (6). Therefore, investigating the mechanism underlying 5-FU resistance and investigating therapeutic strategies, to reverse drug resistance and resensitize cancer cells to 5-FU, is vital.
Tight junctions are a type of cell-to-cell adhesion in epithelial or endothelial cell sheets, forming continuous seals around cells and functioning as a physical barrier to prevent solutes and water from passing freely through the paracellular space (7). Tight junctions maintain cell polarity by preventing the lateral diffusion of integral membrane proteins between the lateral/basal and apical surfaces, allowing the specialized functions of each surface to be preserved (8). It has also been reported that tight junctions are associated with cellular survival, proliferation, migration and differentiation (9). Claudin-1 (CLDN1), a protein that is encoded by the CLDN1 gene in humans, belongs to the group of CLDNs and serves a crucial role in tight junctions (10,11). Abnormal expression of CLDN1 has been demonstrated to destroy the epithelial permeability barrier and disrupt cellular polarity, which results in decreased cell adhesion (12). Additionally, abnormal expression of CLDN1 has been revealed to be associated with mechanisms of tumor progression and development, including proliferation, migration, invasion and chemotherapy resistance (13–16). CLDN1 has been identified to be expressed in multiple tumor tissue types and is involved in tumor growth, metastasis and prognosis (15,16). However, the function of CLDN1 is distinct in different types of tumor (17). To the best of our knowledge, the role of CLDN1 in the development of 5-FU resistance in liver cancer remains unclear (18,19). The present study developed a 5-FU-resistant liver cancer HepG2 cell line and investigated the effect of CLDN1 and the underlying mechanism in 5-FU resistance of HepG2 cells. Additionally, CLDN1 was investigated as a potential therapeutic target for enhancing the sensitivity of HepG2 cells to 5-FU.
Materials and methods
Cell culture
The human liver cancer cell line HepG2 was purchased from the Cell Bank of Type Culture Collection the Chinese Academy of Sciences (Shanghai, China). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS; Lonza Group, Ltd., Basel, Switzerland), 5 mM L-glutamine, 5 mM non-essential amino acids, and 100 U/ml penicillin and streptomycin (Invitrogen; Thermo Fisher Scientific, Inc.), in a humidified 5% CO2 incubator at 37°C.
Cultivation of a 5-FU-resistant cell line
5-FU-resistant HepG2 cells were developed by exposing HepG2 cells to increasing concentrations of 5-FU ranging from 10 to 50 mg/l in complete medium (Gibco; Thermo Fisher Scientific, Inc.), as described previously (20). Briefly, HepG2 cells (2×106 cells/plate) were seeded in 60 mm culture plates and allowed to grow. Following incubation for 24 h at 37°C, 10 mg/l 5-FU was added for a further 48 h at 37°C. Subsequently, the medium was removed and fresh drug-free medium (cat. no. C11995500BT; Gibco; Thermo Fisher Scientific, Inc.) was added. The cells were incubated at 37°C. When 90% confluence was reached, cells were trypsinized, replated at a density of 2×106 cells/plate and re-exposed to 20 mg/l 5-FU as previously described. This process was repeated with increasing doses (40 and 80 mg/l) until clones developed resistance to 50 mg/l 5-FU. Following exposure to 5-FU for ≥3 months, living cells were collected, termed drug-resistant cells (Hep/5FU) and used for subsequent experiments.
Proliferation assay
Cell proliferation was evaluated with an MTT assay, for which MTT was obtained from Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). A total of 1×104 Hep/5FU cells and HepG2 cells with 100 µl Dulbecco's Modified Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific, Inc.) were plated in each well of a 96-well plate and incubated for 24 h at 37°C. The cells were then treated with 0, 10, 25, 50, 100, 200 or 400 mg/l 5-FU, 5 mM 3-methyladenine (3-MA; Sigma-Aldrich; Merck KGaA) or 10 nM Rapamycin (Selleck Chemicals, Houston, TX, USA) for 48 h at 37°C in a 5% CO2 incubator. Subsequently, cells were incubated with 20 µl 5 mg/ml MTT for 4 h and lysed for 10 min at room temperature by addition of 200 µl dimethyl sulfoxide (OriGene Technologies, Inc., Rockville, MD, USA). Absorbance was measured at 490 nm using a Rainbow microplate reader (Tecan Group, Ltd., Mannedorf, Switzerland). Cell proliferation was expressed as a percentage of the untreated control cells.
Migration and invasion assays
Cell migration was assessed with a Transwell assay using 6.5 mm chambers (Corning Incorporated, Corning, NY, USA) with 8-µm pore membranes (17). A total of 600 µl DMEM with 40 mg/l 5-FU was added to the lower chamber. A suspension of 5×104 cells (Hep/5FU and HepG2 cells) in 100 µl DMEM with 1% FBS was plated into the upper chamber followed by addition of 5-FU. Cells on the undersurface of the polycarbonate membranes were stained with crystal violet (Amresco, LCC, Solon, OH, USA) for 10 min at room temperature. Stained cells were observed using a light microscope (magnification, ×100) and six fields were selected at random to measure the mean cell coverage using Image J software version 1.8.0 (National Institutes of Health, Bethesda, MD, USA). The migration of Hep/5FU cells was calculated relative to the migration of control HepG2 cells. Invasion was assayed using the same protocol as the migration assay, with the exception that 70 µl 1 mg/ml Matrigel (BD Biosciences, Franklin Lakes, NJ, USA) was added into the upper face of the membrane.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA in Hep/5FU cells and HepG2 cells was extracted using the Total RNA isolation kit (A&A Biotechnology, Gdynia, Poland). cDNA was obtained by RT using a RevertAidi cDNA synthesis kit (Fermentas; Thermo Fisher Scientific, Inc.) and was amplified using a TaqMan® Gene Expression assay (Applied Biosystems; Thermo Fisher Scientific, Inc.). The reverse transcription reaction parameters were 37°C for 60 min, followed by 85°C for 5 min. The PCR parameters were as follows: 95°C for 10 min, followed by 40 cycles of 95°C for 10 sec, 60°C for 20 sec and 72°C for 3 sec. The primer sequences used for RT-qPCR were as follows: CLDN1 forward, 5′-CAGAAGATGAGGATGGCTGTCATT-3′ and reverse, 5′-AAGGGGGGCACAGCCTCTATTA-3′; GAPDH forward, 5′-TCCCTGAGCTGAACGGGAAG-3′ and reverse, 5′-GGAGGAGTGGGTGTCGCTGT-3′. RT-qPCR was performed using an ABI PRISM 7700 Sequence Detector (PerkinElmer, Inc., Waltham, MA, USA). mRNA expression levels were calculated using the 2−ΔΔCq method and were normalized to the expression level of GAPDH (21). The expression level of mRNA in cells was made relative to the expression level of mRNA in control cells.
Western blot analysis
Hep/5FU cells and HepG2 cells were seeded in a culture dish (2×106 cells/dish) with DMEM, cultured overnight and then incubated at room temperature with 50 mg/l 5-FU for 48 h. Cells were harvested and washed twice with cold PBS, and then lysed in cell lysis buffer (catalog no. P0013; Beyotime Institute of Biotechnology, Haimen, China) containing a protease inhibitor cocktail (Roche Diagnostics, Basel, Switzerland) at 4°C for 15 min. Following centrifugation at 12,000 × g for 10 min at room temperature, the supernatant was collected and quantified using a bicinchoninic acid quantification kit (Beyotime Institute of Biotechnology). The proteins (50 µg/lane) were separated by 12% SDS-PAGE (Beijing Solarbio Science & Technology Co., Ltd., Beijing, China) and transferred to polyvinylidene fluoride membranes (EMD Millipore, Billerica, MA, USA). The membranes were blocked with 5% non-fat dried milk in TBS and Tween-20 for 1 h at room temperature. The membranes were then incubated overnight at 4°C with the following specific primary antibodies: Anti-CLDN1 (1:1,000; catalog no. ab15098; Abcam, Cambridge, UK), anti-LC3 I/II (1:1,000; catalog no. 4108S; Cell Signaling Technology, Inc., Danvers, MA, USA), anti-P62 (1:1,000; catalog no. 8025S; Cell Signaling Technology) and rabbit polyclonal anti-GAPDH (1:2,000; catalog no. sc-25778; Santa Cruz Biotechnology, Inc., Dallas, TX, USA). Following the primary incubation, membranes were incubated for 2 h at room temperature with either of the following horseradish peroxidase-conjugated secondary antibodies: Goat anti-mouse (1:2,000; catalog no. sc-2005; Santa Cruz Biotechnology, Inc.) and goat anti-rabbit IgG (1:2,000; catalog no. sc-2004; Santa Cruz Biotechnology, Inc.). Protein bands were visualized using ECL-detecting reagent (GE Healthcare, Chicago, IL, USA) and quantified by densitometry using Quantity One software (version 4.6.6; Bio-Rad Laboratories, Inc., Hercules, CA, USA). Protein amounts were expressed relative to the internal reference GAPDH.
RNA interference of CLDN1
Small interference (si)RNAs against CLDN1 (siCLDN1) and the negative control (siNC) were designed and chemically synthesized by Shanghai GenePharma Co., Ltd. (Shanghai, China). Cell transfections were conducted using Lipofectamine® 2000 (Invitrogen; Thermo Fisher Scientific, Inc.), according to the manufacturer's protocol. Briefly, 1.5 l 25 pmol/l siCLDN1 or siNC and 1 µl Lipofectamine® 2000 were added into vials containing 50 µl Opti Memi medium (Thermo Fisher Scientific, Inc.) at room temperature. After 5 min, the liquids in the vials were mixed before standing for 20 min at room temperature, and then the mixture was added to the cells and incubated for 6 h. Fresh DMEM supplemented with 10% FBS was provided and cells were grown for 48 h prior to subsequent experiments.
Furthermore, Hep/5FU cells transfected with negative control siRNA or siRNA against CLDN1 were treated with 50 mg/l 5-FU for 48 h. Non-silenced Hep/5FU cells were treated with or without 5 mM 3-MA, an autophagy inhibitor, for 48 h. CLDN1-silenced Hep/5FU cells were treated with or without 10 nM Rapamycin, an autophagy agonist, for 48 h.
Apoptosis assay
Hep/5FU cells and HepG2 cells were grown to 80% confluence at room temperature and treated with 50 mg/l 5-FU for 48 h at room temperature. Cells were collected, washed three times using TBS + 0.1% Tween-20 for 5 min and resuspended in PBS at room temperature. Apoptosis was analyzed using Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) assay (eBioscience, USA), according to the manufacturer's protocol. Briefly, the cells were washed three times with PBS + 0.1% Tween-20 and subsequently incubated for 15 min at room temperature in the dark with 100 µl 1X Annexin binding buffer containing 5 µl Annexin V-FITC and 2 µl PI. This detected the amount of phosphatidylserine on the outer surface of the plasma membrane, a biochemical alteration unique to membranes of apoptotic cells, and the amount of PI, a dye that enters dead cells or cells in the late stages of apoptosis and binds to DNA but does not bind to the plasma membrane of viable cells. Fluorescence was detected using a flow cytometer and data were analyzed using CellQuest software version 5.1 (BD Biosciences). Cells with phosphatidylserine on their surface were considered to be apoptotic, including early apoptotic cells and late apoptotic cells.
Transmission electron microscopy (TEM)
Cells were trypsinized and fixed in 0.1 M cacodylate buffer (pH 7.4) and 2.5% glutaraldehyde for 2 h at room temperature and were washed twice using precooled PBS. Subsequently, cells were post-fixed in 0.1 M cacodylate buffer (pH 7.4) and 1% osmium tetroxide. The fixed cells were rinsed with PBS, dehydrated with various doses of ethanol and embedded in epoxy resin. The ultrastructures of autophagic cells were observed and images were obtained using an electron microscope (magnification, ×10,000 or ×20,000; JEM-1200; JEOL, Ltd., Tokyo, Japan) at 80 kV.
Statistical analysis
Data were obtained from a minimum of three experiments. Statistical analysis was performed using SPSS software (version 13.0; SPSS, Inc., Chicago, IL, USA). Data are presented as the mean ± standard error of the mean. One-way analysis of variance was used to assess differences between groups. Duncan's multiple range test was employed for pairwise comparison, followed by Bonferroni correction. P<0.05 was considered to indicate a statistically significant difference.
Results
Development of 5-FU-resistant HepG2 cells
To examine the mechanism underlying 5-FU resistance in liver cancer, a 5-FU resistant Hep/5FU cell model was first developed. Cell growth was assessed using an MTT assay and growth curves of Hep/5FU and HepG2 cells are depicted in Fig. 1A. The data demonstrated that Hep/5FU cells grew faster, compared with HepG2 cells, at the same time interval. Additionally, the maximal half inhibitory concentration (IC50) values of 5-FU were determined by exposing Hep/5FU and HepG2 cells to different concentrations of 5-FU for 48 h. IC50 values of 5-FU in Hep/5FU and HepG2 cells were calculated to be 260 and 41 mg/l, respectively (Fig. 1B). Additionally, migration and invasion capabilities of Hep/5FU cells were identified to be significantly increased, compared with HepG2 cells (Fig. 1C and D). These data indicate that a 5-FU-resistant HepG2 cell line was successfully constructed, which demonstrated an increased migration and invasion ability, compared with HepG2 cells.
Figure 1.

Characterization of Hep/5FU and HepG2 cells. (A) Cells were maintained in 96-well plates and cell proliferation was assessed with an MTT assay at the indicated time intervals. Growth curves of the two cell lines were generated. (B) Cells were incubated in 100 µl DMEM for 24 h and treated with 0, 10, 25, 50, 100, 200 or 400 mg/l 5-FU for 48 h. Cell proliferation was evaluated with an MTT assay. Maximal half inhibitory concentration values of 5-FU for the two cell lines were calculated. (C) Hep/5FU and HepG2 cells were seeded into the upper chamber for migration assays. (D) For invasion assays the upper face of the membrane of chamber was covered with 70 µl Matrigel (1 mg/ml) prior to Hep/5FU and HepG2 cells being added. Following incubation, cell migration and invasion were evaluated. Migration and invasion were calculated relative to that of HepG2 cells. *P<0.05 vs. HepG2 cells. Data are presented as the mean ± standard error of the mean of three independent experiments. Hep/5FU, 5-fluorouracil-resistant HepG2.
CLDN1 silencing resensitizes resistant HepG2 cells to 5-FU
To detect the role of CLDN1 in the generation of 5-FU resistance in HepG2 cells, the expression level of CLDN1 was examined. RT-qPCR demonstrated that the mRNA expression level of CLDN1 was significantly increased in 5-FU-resistant HepG2 cells, compared with HepG2 cells (Fig. 2A). Additionally, western blot analysis demonstrated that the protein expression level of CLDN1 was significantly increased in 5-FU-resistant HepG2 cells, compared with HepG2 cells (Fig. 2B). Furthermore, the expression of CLDN1 was silenced by transfection siRNA and western blot analysis revealed a significant decrease of CLDN1 expression in CLDN1-silenced Hep/5FU cells, compared with non-silenced control cells (Fig. 2C). Additionally, Hep/5FU cells were transfected with negative control or siRNA against CLDN1 and cultured in the presence or absence of 50 mg/l 5-FU for 48 h. Cell proliferation was analyzed with an MTT assay. A slight decrease was observed in the proliferation of CLDN1-silenced Hep/5FU cells, compared with non-silenced Hep/5FU control cells, under identical experimental conditions without 5-FU treatment. Additionally, cell proliferation of CLDN1-silenced Hep/5FU cells was significantly decreased following 5-FU treatment, compared with 5-FU-treated non-silenced Hep/5FU control cells, and the growth of non-silenced Hep/5FU control cells was not significantly affected by 5-FU treatment, compared with untreated control cells (Fig. 2D).
Figure 2.

Role of CLDN1 in proliferation and apoptosis of Hep/5FU cells. (A) Cells were collected and total RNA was extracted for reverse transcription-quantitative polymerase chain reaction to evaluate the mRNA expression level of CLDN1 in Hep/5FU and HepG2 cells. **P<0.01 vs. HepG2 cells. (B) Cells were collected and lysed, and western blot analysis with an anti-CLDN1 antibody was used to examine the protein level of CLDN1. GAPDH expression level was detected as a reference. The protein bands were quantified by densitometry and the expression levels were expressed as a ratio relative to the expression level in HepG2 cells. Data are presented as the mean ± SEM of three independent experiments. **P<0.01 vs. HepG2 cells. (C) Hep/5FU cells were transfected with siCLDN1 or negative control for 48 h. Subsequently, western blot analysis was used to detect CLDN1 protein expression using an anti-CLDN1 antibody. GAPDH expression level was detected as a reference. The protein bands were quantified by densitometry and the expression levels were expressed as a ratio relative to the expression level in HepG2 cells. Data are presented as the mean ± SEM of three independent experiments. **P<0.01 vs. control cells. Hep/5FU cells were transfected with negative control or siCLDN1 and cultured in the presence or absence of 50 mg/l 5-FU for 48 h. (D) Cell proliferation was analyzed with an MTT assay and (E) cell apoptosis was evaluated by Annexin V-fluorescein isothiocyanate/propidium iodide assay. Data are presented as the mean ± SEM of three independent experiments. **P<0.01. NS, no significance; CLDN1, claudin-1; Hep/5FU, 5-fluorouracil-resistant HepG2; SEM, standard error of the mean; siCLDN1, small interfering RNA targeting claudin-1.
Subsequently, Hep/5FU cells were transfected with negative control or siRNAs against CLDN1 and cultured in the presence or absence of 50 mg/l 5-FU for 48 h. Cell apoptosis was evaluated with an Annexin V-FITC/PI assay. These data indicate that CLDN1 silencing significantly increased apoptosis following treatment with 5-FU in Hep/5FU cells, compared with non-silenced Hep/5FU cells (Fig. 2E). However, 5-FU treatment did not significantly induce apoptosis in non-silenced Hep/5FU cells, compared with untreated control cells (Fig. 2E). These results indicate that CLDN1 is associated with the resistance of HepG2 cells to 5-FU and CLDN1 silencing may increase the sensitivity of resistant Hep/5FU cells to 5-FU by enhancing the apoptosis triggered by 5-FU treatment.
CLDN1 silencing decreases migration and invasion of Hep/5FU cells
To further investigate the effect of CLDN1 on 5-FU resistance of Hep/5FU cells, the expression of CLDN1 was downregulated using siRNA, and cells were cultured in the presence or absence of 50 mg/l 5-FU for 48 h. Cell migration and invasion were then evaluated. Transwell and Matrigel assays demonstrated the migration and invasion abilities, respectively. Following treatment with 5-FU, the migration and invasion abilities of CLDN1-silenced Hep/5FU cells were significantly decreased, compared with CLDN1 non-silenced Hep/5FU cells (Fig. 3). These data indicate that regulation of cell motility by CLDN1 may be involved in the development of drug resistance in Hep/5FU cells.
Figure 3.
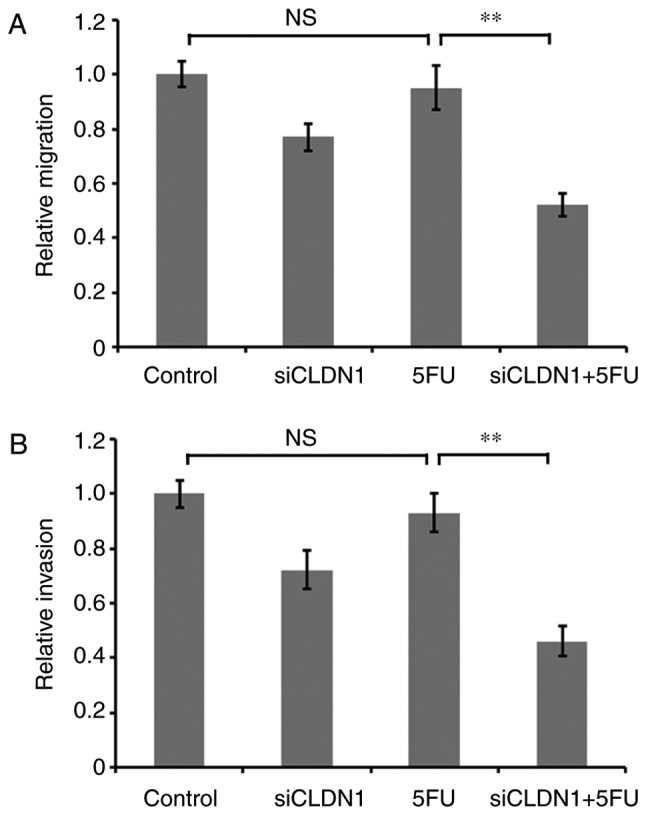
Effect of CLDN1 silencing on the motility of Hep/5FU cells. Hep/5FU cells were transfected with negative control or siCLDN1 and cultured in the presence or absence of 50 mg/l 5-FU for 48 h. (A) Migration and (B) invasion were evaluated. Data are presented as the mean ± SEM of three independent experiments. **P<0.01. NS, no significance; CLDN1, claudin-1; Hep/5FU, 5-fluorouracil-resistant HepG2; siCLDN1, small interfering RNA targeting claudin-1.
CLDN1 silencing reduces drug resistance of Hep/5FU cells by inhibiting autophagy
To investigate whether CLDN1 silencing enhances drug sensitivity of Hep/5FU cells by affecting autophagy, the ultrastructures of cells were observed and imaged using TEM. The results demonstrate that an increased number of autophagosomes were observed in drug-resistant Hep/5FU cells, compared with HepG2 cells (Fig. 4A). By contrast, CLDN1 silencing reversed the phenomenon and inhibited cell autophagy of Hep/5FU cells, compared with non-silenced Hep/5FU cells (Fig. 4B). Additionally, it was revealed that the ratio of LC3B II/I was significantly increased in Hep/5FU cells, compared with HepG2 cells, while CLDN1 silencing significantly decreased the ratio of LC3B II/I in Hep/5FU cells, compared with non-silenced Hep/5FU cells (Fig. 4C). Additionally, a significantly reduced expression of P62 was observed in Hep/5FU cells, compared with HepG2 cells, while CLDN1 silencing significantly increased the expression of P62 in Hep/5FU cells, compared with non-silenced Hep/5FU cells (Fig. 4C).
Figure 4.

Association between autophagy and drug resistance of Hep/5FU cells. (A) HepG2 and Hep/5FU cells were cultured to 80% confluence and autophagy was detected. The ultrastructures of cells undergoing autophagy were observed and imaged using TEM. Magnification, ×10,000. Black arrows indicate autophagosomes. (B) Autophagy of Hep/5FU cells transfected with siNC or siCLDN1 was observed using TEM. Magnification, ×20,000. Black arrows indicate autophagosomes. (C) Protein expression levels of LC3 I/II and P62 in HepG2 and Hep/5FU cells, as well as Hep/5FU cells transfected with siNC or siCLDN1 were assessed by western blot analysis using the corresponding antibodies. Protein bands were quantified by densitometry and the expression levels were expressed as a ratio relative to the expression level in HepG2 or siNC-transfected cells. **P<0.01 vs. HepG2 cells or siNC-transfected cells. (D) Hep/5FU cells were treated with or without siCLDN1, 5 mM 3-MA or 10 nM Rapa combined with 50 mg/l 5-FU for 48 h, and cell proliferation was detected with an MTT assay. **P<0.01. CLDN1, claudin-1; Hep/5FU, 5-fluorouracil-resistant HepG2; siCLDN1, small interfering RNA targeting claudin-1; TEM, transmission electron microscopy; LC3, microtubule-associated protein 1A/1B-light chain 3; 3-MA, 3-methyladenine; Rapa, Rapamycin; NC, negative control.
Subsequently, cell proliferation was detected with an MTT assay. The results demonstrate that the addition of autophagy inhibitor 3-MA significantly decreased drug resistance of Hep/5FU cells, compared with control cells, while incubation with autophagy agonist Rapamycin significantly increased drug resistance of CLDN1-silenced Hep/5FU cells, compared with cells not treated with Rapamycin (Fig. 4D). These data indicate that CLDN1 silencing resensitizes resistant HepG2 cells to 5-FU, possibly by regulating cell autophagy.
Discussion
5-FU is one of the most common anticancer drugs and is used as one of the standard chemotherapies for liver cancer (22). However, cancer cells usually develop resistance to 5-FU, which is the main cause of treatment failure (7). Overcoming drug resistance may be significant to improve prognosis and survival rates of liver cancer. 5-FU has been demonstrated to irreversibly inhibit thymidylate synthase and act as an antimetabolite, resulting in defection of DNA and RNA synthesis, and thus inducing apoptosis and inhibiting cell growth (23). Therefore, it is important to investigate the mechanism of 5-FU resistance. The present study developed Hep/5FU cells and demonstrated that Hep/5FU cells exhibited enhanced migration, invasion and cell autophagy. Additionally, the expression of CLDN1 in Hep/5-FU cells was identified to be significantly increased, compared with 5-FU sensitive HepG2 cells, and was associated with tumor cell biological functions. In addition, it was revealed that downregulation of CLDN1 expression in Hep/5FU cells inhibited cell autophagy and resensitized drug-resistant Hep/5FU cells to 5-FU.
Tight junctions serve important roles in epithelial cells by maintaining cell polarity and the epithelial barrier (8,9). Abnormal expression and distribution of tight junctions is associated with characteristics of tumor progression and development, including growth, migration and invasion (10). CLDN1, a key protein in tight junctions, is associated with tumor metastasis and recurrence (14–16,18). It has been reported that CLDN1 is associated with the prognosis of colon cancer and enhances the invasion and migration of tumor cells (24). Additionally, it has been demonstrated that regulation of osteosarcoma cell motility by CLDN1 is associated with the intracellular localization of CLDN1 protein (25). It has been identified that the high expression of CLDN1 indicates a poor prognosis of patients with non-small-cell lung carcinoma and downregulation of CLDN1 expression hinders cellular migration in breast cancer (26). Furthermore, upregulation of CLDN1 was determined to stimulate cell proliferation and motility in liver cancer (27). It has been reported that CLDN1 expression is elevated in nasopharyngeal carcinoma cells following treatment with 5-FU and upregulation of CLDN1 expression confers resistance to 5-FU (23). However, to the best of our knowledge, the role of CLDN1 in drug resistance of liver cancer to 5-FU remains unclear.
In the present study, resistant Hep/5FU cells were constructed, which exhibited resistance to 50 mg/l 5-FU and demonstrated increased levels of cell proliferation, migration and invasion. Additionally, the expression level of CLDN1 was significantly increased in Hep/5FU cells, compared with HepG2 cells, which indicates that CLDN1 may be positively associated with drug resistance of HepG2 cells to 5-FU. The inhibitory effect of 5-FU on cell proliferation, migration and invasion increased following silencing of CLDN1 by transfection with siRNA. 5-FU induced cell apoptosis and therefore reduced drug resistance. These data indicate that CLDN1 expression increases drug resistance in Hep/5FU cells by attenuating the inhibition of 5-FU on proliferation and cell motility. It has previously been identified that the expression and anti-apoptotic activity of CLDN1 can be regulated by E-cadherin, which has been associated with the development of 5-FU resistance in nasopharyngeal carcinoma cells (28). Whether E-cadherin is involved in the modulation of CLDN1 and 5-FU resistance in HepG2 cells requires further investigated in future studies.
Autophagy is a homeostatic process that is responsible for degrading intracellular organelles and damaged proteins via the delivering of cytoplasmic cargo to the lysosome, which achieves cell metabolism and renews organelles (29). Autophagy serves an important role in the progression and development of liver cancer (30). However, the role of autophagy in drug resistance of liver cancer is not clear. In the present study, increased levels of autophagy were observed in Hep/5FU cells, compared with HepG2. Silencing of CLDN1 inhibited autophagy in Hep/5FU cells and was associated with a downregulation of the ratio of the LC3 II/I protein and the upregulation of P62. The inhibition of autophagy in Hep/5FU cells by 3-MA resulted in a decrease in drug resistance of the cells, while activation of autophagy by Rapamycin restored drug resistance of CLDN1-silenced Hep/5FU cells to 5-FU. It has been reported that unc-51 like autophagy activating kinase 1 (ULK1) serves a critical role in regulating autophagy of tumor cells and activates autophagy by phosphorylation, which can be mediated by adenosine monophosphate-activated protein kinase (AMPK) (30,31). Autophagy-related protein 13 (ATG13), a target of the target of rapamycin kinase signaling pathway, modulates autophagy via phosphorylation of ULK1 and ATG13 and the regulation of the ATG13/ULK1 complex (32,33). The ATG13/ULK1 complex regulates the kinase activity of cell proliferation (34). The present study indicated that CLDN1 modulates the resistance of HepG2 cells to 5-FU by cell autophagy. However, to the best of our knowledge, whether AMPK/ULK1/ATG13 signaling is involved in the modulation of autophagy by CLDN1 remains unknown. Therefore, the underlying mechanism of CLDN1 regulating autophagy and affecting drug resistance of HepG2 cells to 5-FU requires further investigation.
Acknowledgements
The authors would like to thank Dr. Xiaohui Liu (Ruijin Hospital, Shanghai) for his support and advice during this project.
Funding
The present study was supported by the Science and Technology Commission of Shanghai Municipality (grant no. 15411950404).
Availability of data and materials
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.
Authors' contributions
HT wrote the manuscript and conducted experiments. TL and WQ designed and helped conduct the experiments. ZZ designed the study and revised the manuscript. All the authors have reviewed the manuscript before submission and have approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Cancer Facts & Figures 2014. Atlanta: American Cancer Society; 2014. American Cancer Society. [Google Scholar]
- 2.Krishna R, Mayer LD. Multidrug resistance (MDR) in cancer: Mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. Eur J Pharm Sci. 2000;11:265–283. doi: 10.1016/S0928-0987(00)00114-7. [DOI] [PubMed] [Google Scholar]
- 3.Deng GL, Zeng S, Shen H. Chemotherapy and target therapy for hepatocellular carcinoma: New advances and challenges. World J Hepatol. 2015;7:787–798. doi: 10.4254/wjh.v7.i5.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Petraccia L, Onori P, Sferra R, Lucchetta MC, Liberati G, Grassi M, Gaudio E. MDR (multidrug resistance) in hepatocarcinoma clinical-therapeutic implications. Clin Ter. 2003;154:325–335. (In Italian) [PubMed] [Google Scholar]
- 5.Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 6.Ohtsu A. Chemotherapy for metastatic gastric cancer: Past, present, and future. J Gastroenterol. 2008;43:256–264. doi: 10.1007/s00535-008-2177-6. [DOI] [PubMed] [Google Scholar]
- 7.Obert E, Strauss R, Brandon C, Grek C, Ghatnekar G, Gourdie R, Rohrer B. Targeting the tight junction protein, zonula occludens-1, with the connexin43 mimetic peptide, aCT1, reduces VEGF-dependent RPE pathophysiology. J Mol Med (Berl) 2017;95:535–552. doi: 10.1007/s00109-017-1506-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matsuoka H, Shima A, Uda A, Ezaki H, Michihara A. The retinoic acid receptor-related orphan receptor a positively regulates tight junction protein claudin domain-containing 1 mRNA expression in human brain endothelial cells. J Biochem. 2017;161:441–450. doi: 10.1093/jb/mvw092. [DOI] [PubMed] [Google Scholar]
- 9.Liu W, Mi S, Ruan Z, Li J, Shu X, Yao K, Jiang M, Deng Z. Dietary tryptophan enhanced the expression of tight junction protein ZO-1 in intestine. J Food Sci. 2017;82:562–567. doi: 10.1111/1750-3841.13603. [DOI] [PubMed] [Google Scholar]
- 10.Morita K, Furuse M, Fujimoto K, Tsukita S. Claudin multigene family encoding four-transmembrane domain protein components of tight junction strands. Proc Natl Acad Sci USA. 1999;96:511–516. doi: 10.1073/pnas.96.2.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Furuse M, Fujita K, Hiiragi T, Fujimoto K, Tsukita S. Claudin-1 and-2: Novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J Cell Biol. 1998;141:1539–1550. doi: 10.1083/jcb.141.7.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim B, Breton S. The MAPK/ERK-signaling pathway regulates the expression and distribution of tight junction proteins in the mouse proximal epididymis. Biol Reprod. 2016;94:22. doi: 10.1095/biolreprod.115.134965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akizuki R, Shimobaba S, Matsunaga T, Endo S, Ikari A. Claudin-5, −7 and −18 suppress proliferation mediated by inhibition of phosphorylation of Akt in human lung squamous cell carcinoma. Biochim Biophys Acta. 2017;1864:293–302. doi: 10.1016/j.bbamcr.2016.11.018. [DOI] [PubMed] [Google Scholar]
- 14.Yang Y, Cheon S, Jung MK, Song SB, Kim D, Kim HJ, Park H, Bang SI, Cho D. Interleukin-18 enhances breast cancer cell migration via down-regulation of claudin-12 and induction of the p38 MAPK pathway. Biochem Biophys Res Commun. 2015;459:379–386. doi: 10.1016/j.bbrc.2015.02.108. [DOI] [PubMed] [Google Scholar]
- 15.Zhang WN, Li W, Wang XL, Hu Z, Zhu D, Ding WC, Liu D, Li KZ, Ma D, Wang H. CLDN1 expression in cervical cancer cells is related to tumor invasion and metastasis. Oncotarget. 2016;7:87449–87461. doi: 10.18632/oncotarget.13871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuo KT, Chen CL, Chou TY, Yeh CT, Lee WH, Wang LS. Nm23H1 mediates tumor invasion in esophageal squamous cell carcinoma by regulation of CLDN1 through the AKT signaling. Oncogenesis. 2016;5:e239. doi: 10.1038/oncsis.2016.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kominsky SL. Claudins: Emerging targets for cancer therapy. Expert Rev Mol Med. 2006;8:1–11. doi: 10.1017/S1462399406000056. [DOI] [PubMed] [Google Scholar]
- 18.Moldvay J, Fábián K, Jäckel M, Németh Z, Bogos K, Furák J, Tiszlavicz L, Fillinger J, Döme B, Schaff Z. Claudin-1 protein expression is a good prognostic factor in non-small cell lung cancer, but only in squamous cell carcinoma cases. Pathol Oncol Res. 2017;23:151–56. doi: 10.1007/s12253-016-0115-0. [DOI] [PubMed] [Google Scholar]
- 19.Sun BS, Yao YQ, Pei BX, Zhang ZF, Wang CL. Claudin-1 correlates with poor prognosis in lung adenocarcinoma. Thorac Cancer. 2016;7:556–563. doi: 10.1111/1759-7714.12368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meena AS, Sharma A, Kumari R, Mohammad N, Singh SV, Bhat MK. Inherent and acquired resistance to paclitaxel in hepatocellular carcinoma: Molecular events involved. PLoS One. 2013;8:e61524. doi: 10.1371/journal.pone.0061524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 22.Vu NB, Nguyen TT, Tran LC, Do CD, Nguyen BH, Phan NK, Pham PV. Doxorubicin and 5-fluorouracil resistant hepatic cancer cells demonstrate stem-like properties. Cytotechnology. 2013;65:491–503. doi: 10.1007/s10616-012-9511-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tolba MF, Abdel-Rahman SZ. Pterostilbine, an active component of blueberries, sensitizes colon cancer cells to 5-fluorouracil cytotoxicity. Sci Rep. 2015;5:15239. doi: 10.1038/srep15239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakagawa S, Miyoshi N, Ishii H, Mimori K, Tanaka F, Sekimoto M, Doki Y, Mori M. Expression of CLDN1 in colorectal cancer: A novel marker for prognosis. Int J Oncol. 2011;39:791–967. doi: 10.3892/ijo.2011.1102. [DOI] [PubMed] [Google Scholar]
- 25.Jian Y, Chen C, Li B, Tian X. Delocalized Claudin-1 promotes metastasis of human osteosarcoma cells. Biochem Biophys Res Commun. 2015;466:356–361. doi: 10.1016/j.bbrc.2015.09.028. [DOI] [PubMed] [Google Scholar]
- 26.Zhao X, Zou Y, Gu Q, Zhao G, Gray H, Pfeffer LM, Yue J. Lentiviral vector mediated Claudin1 silencing inhibits epithelial to mesenchymal transition in breast cancer cells. Viruses. 2015;7:2965–2979. doi: 10.3390/v7062755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fortier AM, Asselin E, Cadrin M. Keratin 8 and 18 loss in epithelial cancer cells increases collective cell migration and cisplatin sensitivity through claudin1 up-regulation. J Biol Chem. 2013;288:11555–7129. doi: 10.1074/jbc.M112.428920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee JW, Hsiao WT, Chen HY, Hsu LP, Chen PR, Lin MD, Chiu SJ, Shih WL, Hsu YC. Upregulated claudin-1 expression confers resistance to cell death of nasopharyngeal carcinoma cells. Int J Cancer. 2010;126:1353–1366. doi: 10.1002/ijc.24857. [DOI] [PubMed] [Google Scholar]
- 29.Kim H, Kim Y, Jeoung D. DDX53 promotes cancer stem cell-like properties and autophagy. Mol Cells. 2017;40:54–65. doi: 10.14348/molcells.2017.2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee YJ, Jang BK. The role of autophagy in hepatocellular carcinoma. Int J Mol Sci. 2015;16:26629–26643. doi: 10.3390/ijms161125984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jang JE, Eom JI, Jeung HK, Cheong JW, Lee JY, Kim JS, Min YH. Targeting AMPK-ULK1-mediated autophagy for combating BET inhibitor resistance in acute myeloid leukemia stem cells. Autophagy. 2017;13:761–762. doi: 10.1080/15548627.2016.1278328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nie T, Yang S, Ma H, Zhang L, Lu F, Tao K, Wang R, Yang R, Huang L, Mao Z, Yang Q. Regulation of ER stress-induced autophagy by GSK3b-TIP60-ULK1 pathway. Cell Death Dis. 2016;7:e2563. doi: 10.1038/cddis.2016.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mercer CA, Kaliappan A, Dennis PB. A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy. 2009;5:649–662. doi: 10.4161/auto.5.5.8249. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.