

Alemtuzumab is an efficacious therapy for active relapsing-remitting MS (RRMS), but its use is complicated by the potential development of secondary autoimmunity.1 Recent data from phase 3 extension studies confirm thyroid autoimmunity as the most abundant entity of secondary autoimmunity found in up to 30%–40% of treated patients, with most events mild or moderate in severity. Data also show low rates of previously known autoimmune phenomena, such as immune thrombocytopenia and nephropathy.2 However, further entities including sarcoidosis, vitiligo, and hemophagocytic lymphohistiocytosis (HLH) have been described in real-world cohorts.3 Here, we present a case of another lymphoproliferative syndrome, namely idiopathic multicentric Castleman disease (iMCD), after alemtuzumab treatment.
Case report
A 30-year-old Caucasian man was first diagnosed with RRMS in 2003. After initial interferon-β1a and natalizumab (61 infusions) treatment, he received 2 courses of alemtuzumab (December 2014 and December 2015) because of persistent disease activity. Infusions were administered without complications, and disease activity stabilized over time. The patient developed Graves' disease 24 months after alemtuzumab initiation.
In April 2018, he presented to his local hospital with fatigue, fever, dyspnea, and thoracic and abdominal pain. Clinical examination revealed generalized lymphadenopathy, hepatosplenomegaly, and edema. Sonography indicated pleural and pericardial effusion and ascites suggestive of polyserositis. He developed pancytopenia and showed elevated serum C-reactive protein (CRP, figure, A and B) and hypoalbuminemia (2.24 g/dL). A mesenteric lymph node (LN) biopsy was performed and was deemed inconclusive.
Figure. Diagnostic work-up and treatment regimes in an alemtuzumab-treated patient with RRMS developing idiopathic multicentric Castleman disease.

(A–E) Charts show the course of CRP, hemoglobin, platelets, white blood cell count, and blood urea nitrogen since the initial presentation during hospitalization. Time points and length of different treatment regimens are outlined in (A) (♦: IV methylprednisolone 250 mg, cumulative dose 2,750 mg; •: immunoadsorption; *: IVIG single dose, 1 mg/kg). Arrows represent platelet (B) and erythrocyte concentrate (C) transfusions and hemodialysis (E). (F) Abdominal CT scan outlining extensive abdominal (red arrows) and inguinal (yellow arrows) lymphadenopathy. (G–H) Axial thoracic and abdominal CT scan indicating polyserositis with pleural (G: yellow arrows) and pericardial (G: red arrows) effusions and ascites (F: yellow arrows) as well as hepatosplenomegaly (F: red arrows). (I) H&E staining of bone marrow puncture displaying megakaryocytosis. (J–K) Mediastinal lymph node biopsy consistent with iMCD plasma cell type. (J) Haematoxylin and eosin stain displaying regressive germinal centers (blue arrows), with small vessels reaching into germinal centers (“lollipop vessels,” black arrows). (K) Giemsa staining displaying interfollicular proliferation of plasma cells (black arrows).
He was referred to our clinic. Repeated serologic testing for autoimmune, paraneoplastic, and infectious causes remained inconspicuous, especially regarding antinuclear antibodies and herpesvirus (including human herpesvirus (HHV)-8 and HIV PCR in peripheral blood). Whole-body 18F-FDG PET/CT, bronchoscopy, and gastro-/colonoscopy provided no further explanations. Persistent disseminated lymphadenopathy, polyserositis, and organomegaly were detected through CT (figure, F–H). An axillary LN biopsy was performed 3 weeks after the initial presentation, but changes were nonspecific despite extensive immunohistochemistry and clonality analysis. Bone marrow puncture revealed increased megakaryocytosis (figure, I).
After immunosuppressive therapy with high-dose IV methylprednisolone (IVMPS, figure, A♦) and immunoadsorption (IA, figure, A•), inflammatory activity decreased and blood cell counts rose again. Pleural and ascites punctures had to be performed less frequently.
However, 28 days after symptom onset, the patient rapidly redeteriorated with increasing serum CRP-levels and re-emerging cytopenia, especially thrombocytopenia refractory to IV immunoglobulins (figure, A and B*). Because of intrathoracic bleeding, he required multiple erythrocyte and platelet transfusions and an emergency thoracotomy. Thereafter, the patient was referred to the intensive care unit because of renal failure (predominantly caused by systemic inflammation (procalcitonin 18 ng/mL) and third spacing) and underwent continuous hemodialysis under which blood cell counts, effusions, edema, and retention parameters slowly recovered (figure, A–E). A LN extracted during thoracotomy revealed findings typical for Castleman disease, including hyperplasia, germinal center regression, plasma cell expansion, and hyalinized vessels (figure, J and K). HHV-8 immunohistochemical staining was negative. Ascites interleukin (IL)-6 levels were strongly increased (4,030 pg/mL, reference: <3 pg/mL, otherwise published value: 4,440 pg/mL4). Consequently, a diagnosis of iMCD with TAFRO (Thrombocytopenia, Anasarca, MyeloFibrosis, Renal failure, Organomegaly) subtype (typical histopathologic findings, HHV-8 negative immunohistochemistry, thrombocytopenia (figure, B), fever, organomegaly, absence of hypergammaglobulinemia (0.8 g/dL), lymphadenopathy, and hyperplasia of megakaryocytes) was established.5
Repeated IVMPS and IA treatment (figure, A♦,•) induced ongoing remission. The patient fully recovered and continues to receive a small dose of oral prednisolone (5 mg/d). He is regularly screened (every 3 months) for re-emerging disease activity and development of secondary malignancies.6
Discussion
Development of secondary autoimmunity after alemtuzumab treatment is a well-known phenomenon and has been addressed by a strict risk management program screening especially for thrombocytopenia, thyroid, and kidney autoimmunity. Although the incidence of these disorders might have been overestimated according to postmarketing studies,2 a variety of other rare entities have been described in the past 2 years. These reports include both organ-restricted diseases and systemic conditions.3 Two additional iMCD cases have been described, with disease onset at 31 and 41 months after alemtuzumab initiation.6,7
One feature shared by HLH and iMCD is their association with different malignancies which can occur before, concurrent with, or after iMCD diagnosis (with an incidence of up to 19%). These require careful consideration when adjusting the patient's risk management plan, especially beyond 48 months after the last alemtuzumab infusion.5 Furthermore, differentiation of HLH and iMCD is crucial because the latter can be treated with the monoclonal anti-IL6 antibody siltuximab.5
In conclusion, the current case highlights iMCD as another rare but potentially life-threatening secondary autoimmune event after alemtuzumab treatment. Until risk management plans have been adjusted to include these new disorders, clinicians should perform a thorough clinical follow-up (e.g., screening of B symptoms, examination of LNs) and, in suspicious cases, seek early interdisciplinary support.
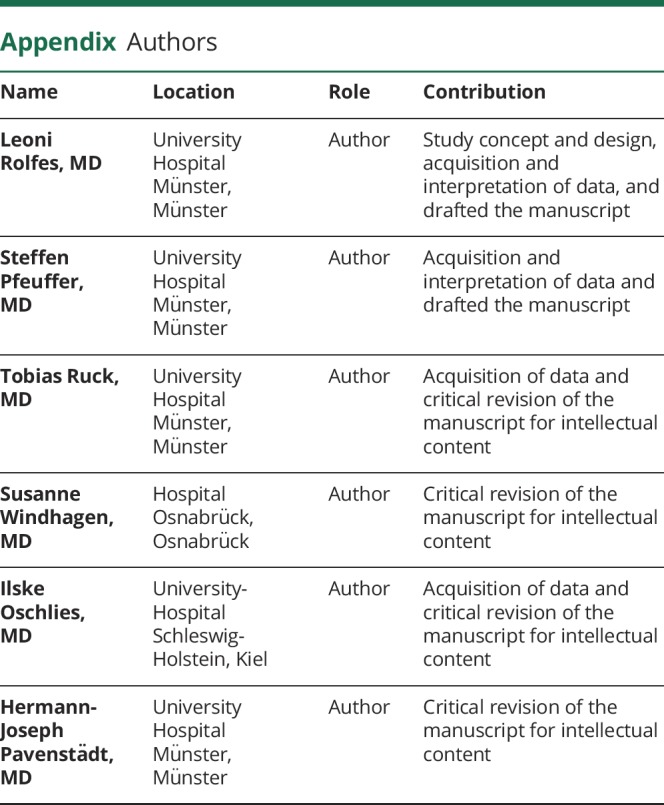
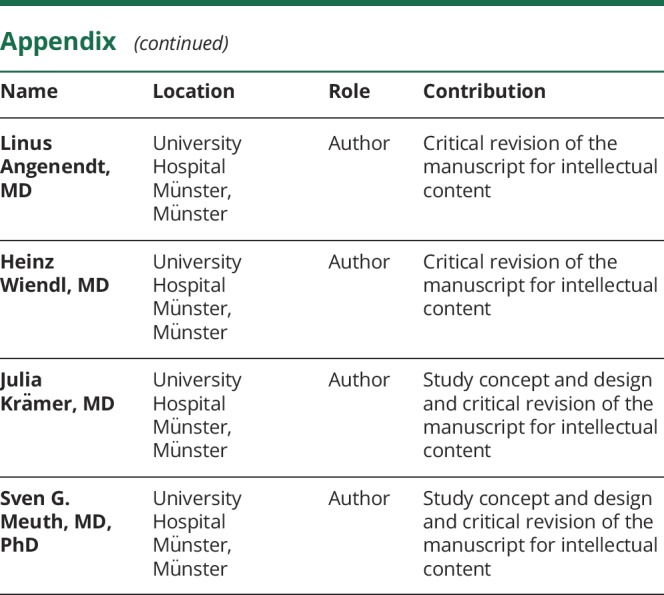
Appendix. Authors
Study funding
This study was financially supported by the Competence Network Multiple Sclerosis (01GI1603D, PROGRAMMS to T. Ruck, H. Wiendl and S.G. Meuth). This study was also supported by Genzyme Therapeutics Ltd., United Kingdom (“Alemtuzumab in autoimmune inflammatory neurodegeneration: mechanisms of action and neuroprotective potential” [ALAIN01] to H. Wiendl, S.G.Meuth, and T. Ruck).
Disclosure
L. Rolfes received travel reimbursements from Merck Serono and Sanofi Genzyme. S. Pfeuffer received travel reimbursements from Sanofi Genzyme and Merck Serono; received honoraria for lecturing from Sanofi Genzyme, Biogen, and Mylan Healthcare; and received research support from Merck Serono, DiaMed, and the German Multiple Sclerosis Society. T. Ruck received travel expenses and financial research support from Genzyme and Novartis and received honoraria for lecturing from Roche, Merck, Genzyme, Biogen, and Teva. S. Windhagen, I. Oschlies, H.-J. Pavenstädt, and L. Angenendt report no disclosures. H. Wiendl received compensation for serving on Scientific Advisory Boards/Steering Committees for Bayer HealthCare, Biogen Idec, Sanofi Genzyme, Merck Serono, and Novartis; received speaker honoraria and travel support from Bayer Vital GmbH, Bayer Schering AG, Biogen, CSL Behring, EMD Serono, Fresenius Medical Care, Genzyme, Merck Serono, OmniaMed, Novartis, and Sanofi-Aventis; received compensation as a consultant from Biogen Idec, Merck Serono, Novartis, Roche, and Sanofi Genzyme; and also received research support from Bayer HealthCare, Bayer Vital, Biogen Idec, Merck Serono, Novartis, Sanofi Genzyme, Sanofi US, and Teva. J. Krämer received honoraria for lecturing from Biogen, Novartis, Mylan, and Teva and financial research support from Sanofi Genzyme. S.G. Meuth received honoraria for lecturing and travel expenses for attending meetings from Almirall, Amicus Therapeutics Germany, Bayer HealthCare, Biogen, Celgene, DiaMed, Genzyme, MedDay Pharmaceuticals, Merck Serono, Novartis, Novo Nordisk, Ono Pharma, Roche, Sanofi-Aventis, Chugai Pharma, QuintilesIMS, and Teva and his research is funded by the German Ministry for Education and Research (BMBF), Deutsche Forschungsgemeinschaft (DFG), Else Kröner Fresenius Foundation, German Academic Exchange Service, Hertie Foundation, Interdisciplinary Center for Clinical Studies (IZKF) Muenster, German Foundation Neurology, Almirall, Amicus Therapeutics Germany, Biogen, DiaMed, Fresenius Medical Care, Genzyme, Merck Serono, Novartis, Ono Pharma, Roche, and Teva. Go to Neurology.org/NN for full disclosures.
References
- 1.Coles AJ, Twyman CL, Arnold DL, et al. . Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet 2012;380:1829–1839. [DOI] [PubMed] [Google Scholar]
- 2.Coles AJ, Cohen JA, Fox EJ, et al. . Alemtuzumab CARE-MS II 5-year follow-up: efficacy and safety findings. Neurology 2017;89:1117–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Saarela M, Senthil K, Jones J, et al. . Hemophagocytic lymphohistiocytosis in 2 patients with multiple sclerosis treated with alemtuzumab. Neurology 2018;90:849–851. [DOI] [PubMed] [Google Scholar]
- 4.Kubokawa I, Yachie A, Hayakawa A, et al. . The first report of adolescent TAFRO syndrome, a unique clinicopathologic variant of multicentric Castleman's disease. BMC Pediatr 2014;14:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu AY, Nabel CS, Finkelman BS, et al. . Idiopathic multicentric Castleman's disease: a systematic literature review. Lancet Haematol 2016;3:e163–e175. [DOI] [PubMed] [Google Scholar]
- 6.Somerfield J, Hill-Cawthorne GA, Lin A, et al. . A novel strategy to reduce immunogenicity of biological therapies. J Immunol 2010;185:763–768. [DOI] [PubMed] [Google Scholar]
- 7.Boddu PC, Hassan A, Mauer A, Singh DA, Weisenberg ES. Rare case of multicentric castleman disease secondary to alemtuzumab therapy. Int J Tumor Ther 2015;4:1–4. [Google Scholar]