

Abstract
Objective
To test the hypothesis that patients with amyotrophic lateral sclerosis (ALS) inheriting the common interleukin 6 receptor (IL6R) coding variant (Asp358Ala, rs2228145, C allele) have associated increases in interleukin 6 (IL6) and IL6R levels in serum and CSF and faster disease progression than noncarriers.
Methods
An observational, case-control study of paired serum and CSF of 47 patients with ALS, 46 healthy, and 23 neurologic disease controls from the Northeastern ALS Consortium Biofluid Repository (cohort 1) was performed to determine serum levels of IL6, sIL6R, and soluble glycoprotein 130 and compared across groups and IL6R genotype. Clinical data regarding disease progression from a separate cohort of 35 patients with ALS from the Wake Forest ALS Center (cohort 2) were used to determine change in ALSFRS-R scores by genotype.
Results
Patients with ALS had increased CSF IL6 levels compared with healthy (p < 0.001) and neurologic (p = 0.021) controls. Patients with ALS also had increased serum IL6 compared with healthy (p = 0.040) but not neurologic controls. Additive allelic increases in serum IL6R were observed in all groups (average increase of 52% with the presence of the IL6R C allele; p < 0.001). However, only subjects with ALS had significantly increased CSF sIL6R levels compared with controls (p < 0.001). When compared across genotypes, only patients with ALS inheriting the IL6R C allele exhibit increased CSF IL6. ALSFRS-R scores decreased more in patients with ALS with the IL6R C allele than in those without (p = 0.019).
Conclusions
Theses results suggest that for individuals inheriting the IL6R C allele, the cytokine exerts a disease- and location-specific role in ALS. Follow-up, prospective studies are necessary, as this subgroup of patients may be identified as ideally responsive to IL6 receptor–blocking therapies.
Amyotrophic lateral sclerosis (ALS) is a uniformly fatal disease of upper and lower motoneurons (MNs). It remains unclear whether the body's inflammatory responses, either autoimmune or in response to stress and injury, have a role in initiating ALS, although data clearly show that these responses involved in disease progression (for review, see references 1 and 2). Although several inflammatory biomarkers are elevated in the serum of patients with ALS,3–5 interleukin 6 (IL6) has been considered potential therapeutic target in ALS (NCT02469896).5,6 IL6, a multifunctional cytokine, influences diverse cellular mechanisms including cell growth, metabolism, differentiation, death, and inflammatory and anti-inflammatory processes with critical influences in the CNS, peripherally at the neuromuscular junction (NMJ), and within muscle itself.7–9
IL6 differentially regulates these mechanisms through 2 signaling paradigms: classic IL6 signaling and IL6 trans-signaling (for review, see reference 8). Although classic IL6 signaling occurs through membrane-bound IL6 receptors, IL6 trans-signaling is driven by systemic and localized increases in extracellular soluble IL6 receptor (sIL6R) generated by proteolytic cleavage, termed “shedding,” of the receptor from cell surfaces. These soluble receptors can be activated by IL6 and activate IL6 signaling cascades through constitutively expressed gp130 coreceptor. Thus, IL6 trans-signaling allows IL6 signaling pathways to be activated in cells that do not express IL6R. Of relevance to ALS, IL6 trans-signaling may mediate trophic activity for neurons, diffuse proinflammatory actions, and glial activation.3,7,9
In humans, there is an IL6R coding change (Asp358Ala, A/C; rs2228145), where the A allele (Asp358) is the major allele, and the C allele (Ala358) is the variant allele. IL6R shedding is enhanced by the commonly inherited C allele, with over 50% of the variability in increased sIL6R plasma levels accounted for by the C allele.10–12 In individuals inheriting the IL6R C allele, increased shedding of the receptor enhances both localized and systemic IL6 trans-signaling in the presence of IL6. Given the role of IL6 signaling in mediating multiple cell functions, IL6 trans-signaling may be an important modifier for diseases associated with IL6 signaling.13–17
To determine whether IL6 trans-signaling could have a potential role in ALS, IL6 and sIL6R levels were measured in paired samples of serum and CSF collected at a single time point in a cohort of patients with ALS and compared with healthy (HCs) and disease controls (DCs) and across IL6R variant groups. Our results suggest that the IL6R C allele influences IL6 signaling in the CNS of patients with ALS. In a second cohort of subjects with ALS with more defined clinical data, the presence of the IL6R C allele was associated with faster disease progression. These results suggest that identifying patients who possess the IL6R C allele may provide insight for predicting disease progression and identifying those who might benefit most from IL6R-blocking therapies. Follow-up studies are warranted.
Methods
Subjects
All subjects were unrelated persons of self-reported European ancestry. Genetic status regarding disease causative mutations (e.g., SOD1) was unknown. Cohort 1 consisted of paired samples of serum and CSF collected at a single time point from patients with ALS, DCs, and healthy subjects. All samples for cohort 1 were obtained from the Northeastern ALS Consortium (NEALS) multicenter biorepository. Subjects with ALS were diagnosed as probable or definite by the El Escorial criteria; diseased controls were diagnosed with a degenerative or inflammatory neurologic disorder that may mimic ALS such as MS, myasthenia gravis, and hereditary spastic paraplegia.18 CSF and blood samples were collected by lumbar and venipuncture, respectively. Cohort 2 included male and female patients, 18 years and older with a probable or definite diagnosis of ALS18 without other potentially confounding neurologic diseases as determined by 2 independent neuromuscular disease specialists. Clinical data for these subjects were collected at the Wake Forest ALS Center during routine clinical practice, deidentified, and entered into the Wake Forest ALS biorepository database.
Standard protocol approvals, registrations, and patient consents
All subjects provided informed consent as required by the respective institutional review boards.
Serum and CSF samples
The NEALS biorepository provided deidentified samples with a separate document containing clinical information and subject IDs. Before assays, thawed aliquots were cleared by 10-minute centrifugation at 18,000g at 4°C. Protein concentrations for serum and CSF were determined using bicinchoninic acid assay total protein assay (Thermo Scientific, 23,225) to assure that sample protein content fell within the normal range for adults (serum 6.4–8.3 g/dL; CSF15–45 mg/dL) and did not differ between groups (data not shown).
ELISA assays
IL6, sIL6R, and sgp130 concentrations were measured with commercial kits (R&D Systems, Q6000B, DR600, and DGP00, respectively) according to the manufacturer's instructions. ELISA plates were read on a Wallac plate reader. Detection limits of assays are reported by the manufacturer to be 0.16 pg/mL for IL6, 6.5 pg/mL for sIL6R, and 0.25 ng/mL for sgp130. Intra- and inter-assay precisions were determined experimentally using a single consistent sample across assays to be <3.3% and <2.1% for sIL6R, <5.8% and <6.8% for IL6, and <1.9% and <5.8% for sgp130, respectively.
Genotyping of IL6R Ala358 variant rs2228145
DNA from the NEALS cohort was unavailable; therefore, DNA was extracted from residual serum samples assuming (1) cell-free DNA was present, or (2) small amounts of peripheral mononuclear blood cells could still be present. Genomic DNA was isolated from 100–500 μL of serum using the ChargeSwitch gDNA Serum kit (Invitrogen, CS11040) resulting in 3–25 ng/μL of DNA per sample. DNA fragments were cleaned using DNA clean-up spin columns (Zymo Research, D4014) to obtain a 260/280 ratio >1.7. For cohort 2, DNA was purified from whole blood using the Qiagen AutoPure LS using standard Purgene chemistry. DNA was genotyped using a validated TaqMan assay for the IL6R variant rs2228145 according to the manufacturer's instructions (Applied Biosystems, assay ID: C_16170664_10; IL6R Ala358 variant) and read using the allelic discrimination protocol on an rtPCR system (Applied Biosystems, model 7500). Positive controls for each genotype and 3 no template controls (blanks) were run for each assay set.
Measures of disease progression
ALS outcome is significantly related to the decline in various measures of disease progression, especially percent predicted forced vital capacity and the revised ALS functional rating scale (ALSFRS-R) score.19 The ALSFRS-R is a patient-reported survey of symptoms and motor ability commonly used in patient care and clinical trials to follow functional decline. The progression rate of ALSFRS-R, often referred to as the ΔFS, calculated as differential of the total score from 1 time point to another, divided by disease duration (time), correlates closely with prognosis and has been found to be a significant predictor of survival and overall disease progression.20,21 Here, we calculate ΔFS as a “preslope,” or the loss in function experienced before diagnosis, for patients who presented to the Wake Forest ALS Center within 1 year of symptom onset.21 Patients qualified as definite or probable ALS by the El Escorial diagnostic criteria, confirmed by 2 independent neuromuscular clinicians. Time of symptom onset was reported by the patient during their first visit to the clinic as when they first noticed any symptom of motor dysfunction.
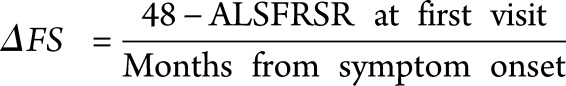 |
Statistical analyses
Serum and CSF IL6 levels were winsorized to within 2 SDs of the mean of the cohort as a whole to limit the influence of outlying values (4 CSF values and 5 serum values winsorized; table e-1, links.lww.com/NXI/A152). IL6 raw values were natural log transformed to normalize the distribution before any parametric comparisons; sIL6R and sgp130 were well approximated by the normal distribution and did not require transformation.
Significant age differences had previously measured between ALS and control groups' serum IL6 levels (p < 0.001, ρ = 0.12, Spearman correlation). Therefore, age was a covariate in all serum IL6 analyses involving generalized linear models (GLM) to test for associations (table e-2, links.lww.com/NXI/A152). The data's fit to the model's distributional assumptions of conditional normality and homogeneity of variance were examined. Estimated least square means and standard errors were computed for summarizing data and individual comparisons. All comparisons were determined before data analysis to be as follows: ALS vs HC, ALS vs DC (whole group, within AA genotype, or within *C genotypes), and AA vs *C (within ALS, HC, or DC groups; see table e-3, links.lww.com/NXI/A152). As these were planned comparisons, tests for pairwise differences were performed using the Fisher protected least significant difference multiple comparison procedure. The Student 2-way t test was used on the ΔFS values. Corresponding effect sizes were estimated via Cohen's D statistic.
Data availability
Data not provided in the article because of space limitations will be shared upon request of other investigators for purposes of replicating procedures and results.
Results
Subject characteristics
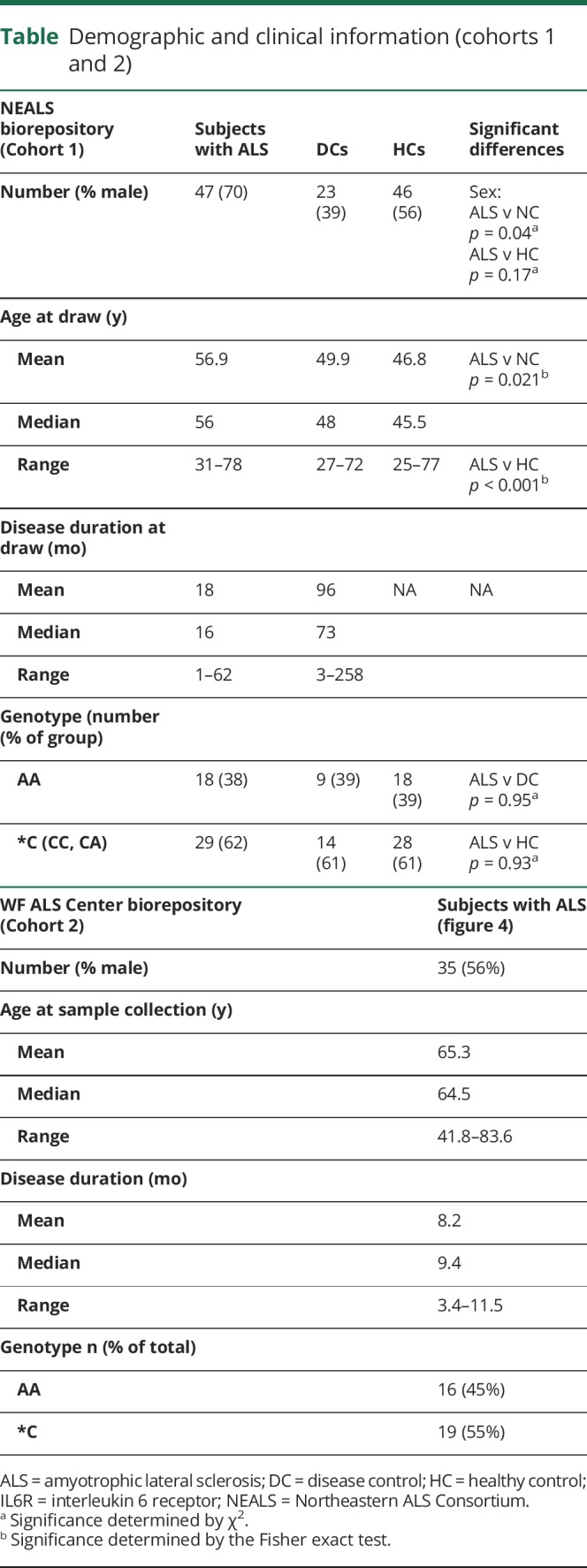
Cohort 1 consisted of serum and CSF samples provided by the NEALS repository and comprised 47 subjects with ALS, 46 HCs, and 23 subjects with neurologic diseases that mimic ALS. Demographics for each sample included age, sex, race, and disease duration (ALS and DCs; table). Two subjects with ALS and 11 neurologic DCs did not have disease duration information. Significant differences in age existed between the ALS and control groups; therefore, this factor was controlled for in subsequent analyses when appropriate, as described in Methods. Cohort 2 consisted of 35 subjects with ALS, all of whom had contributed DNA samples along with detailed demographic and clinical data from their initial visit to the Wake Forest ALS Clinic (table).
Table.
Demographic and clinical information (cohorts 1 and 2)
Serum IL6 levels are elevated in subjects with ALS and DCs compared with HCs, but only subjects with ALS exhibit increased CSF levels.
Both ALS and DC individuals exhibited elevated serum IL6 compared with HCs (figure 1A). CSF IL6 levels were significantly higher in subjects with ALS compared with both HCs and DCs (figure 1B). There were no observed differences between CSF and serum sIL6R levels between groups (Figure 1, C and D). Together, these data indicate a possible CNS-specific role for IL6 in ALS. Although the concentrations of IL6 were comparable between serum and CSF, serum levels of sIL6R and its naturally occurring, soluble gp130 (sgp130) were consistently ten-fold higher than the CSF levels in all groups. We found no significant differences in sgp130 in either serum or CSF between groups (figure 1, E and F). These results suggest that in ALS, sgp130 does not compensate for the observed increases in IL6.
Figure 1. Subjects with amyotrophic lateral sclerosis (ALS) exhibit increased levels of interleukin 6 (IL6) in serum and CSF.

(A) Serum IL6 is significantly elevated in subjects with ALS compared with healthy controls (HCs) (p = 0.040, Cohen's D = 0.62) but not to disease controls (DCs) (p = 0.94) controls. (B) CSF IL6 is elevated in subjects with ALS compared with both HCs (p < 0.001, D = 0.61) and DCs (p = 0.021, D = 0.28). (C) Neither serum (ALS vs HC p = 0.30, vs DC p = 0.21) nor (D) CSF (ALS vs HC p = 0.32, vs DC p = 0.85) sIL6R varies with disease status. (E) Serum (ALS vs HC p = 0.28, vs DC p = 0.09) and (F) CSF sgp130 (ALS vs HC p = 0.18, vs DC p = 0.07) do not vary with disease status. (A) = p < 0.05, b = p < 0.001. For sgp130 and CSF IL6, all were planned comparisons where significance was determined by the Student t test. For serum IL6, significance was determined with generalized linear models, followed by Fisher protected least significant difference for planned individual comparisons of ALS vs HCs and ALS vs DCs; graphed values for IL6 are least squared means.
IL6R C allele accounts for serum sIL6R levels in all groups
The IL6C allele occurs at a frequency of ∼10% in African, ∼40% in European, ∼30% in East Asian, and ∼50% in Native American ancestries (reference 13, NCBI dbSNP). To investigate the role of the IL6R C allele on sIL6R levels and the potential influence of trans-signaling in ALS, we evaluated the distribution of the IL6R C allele in all subjects (table; those with AC or CC genotype will be referred to as *C). When contrasting subjects with ALS with control sets in cohort 1, the allele frequencies were comparable (ALS minor allele frequency (MAF) = 0.394 and p = 0.91, healthy MAF = 0.380 and p = 0.98, DC MAF = 0.386 and p = 0.81). Similar results were also observed in cohort 2 (table). Importantly, both within each cohort across groups (table e-1, links.lww.com/NXI/A152) and across the 2 cohorts, the IL6R C allele frequencies (MAF) were comparable and consistent with previous reports (0.387 and 0.417 for cohorts 1 and 2, respectively).13 Both cohorts were consistent with Hardy-Weinberg equilibrium proportions (p = 0.72 and p = 0.61 for cohorts 1 and 2, respectively). Univariate analysis found no association between the IL6R C allele and sex (p = 0.77), age at onset (p = 0.55), or disease duration (p = 0.19). These results suggest that the presence of the C allele does not predispose individuals to ALS. As expected for an additive genetic model, in all groups, the IL6R C allele was also associated with increased (52%) serum sIL6R (figure 2A). Only in subjects with ALS were CSF sIL6R levels significantly increased (23%) with the presence of the C allele (figure 2B), suggesting potential for CSF trans-signaling to be correlated in a genotypic manner with occurrence of ALS-specific pathologic processes in *C individuals.
Figure 2. Interleukin 6 receptor (IL6R) C allele associates with serum levels of IL6R in all subjects, but only in patients with amyotrophic lateral sclerosis (ALS), does it account for CSF IL6R and IL6 increases.

(A) Concentrations of the sIL6R are significantly higher in subjects with the C allele compared with those without (p < 0.001, all groups). (B) CSF sIL6R are significantly increased only in subjects with ALS with the C allele compared with those without (CSF p = 0.021). (C) In serum, there is no significant difference in IL6 levels between subjects with ALS with and without the C allele; subjects with ALS with the C allele have more IL6 than healthy patients with the C allele (p = 0.045, D = 0.71). (D) Subjects with ALS with the C allele have more CSF IL6 than subjects with ALS without the C allele and than subjects with the C allele in the healthy control (HC) or disease control (DC) groups (p = 0.021, D = 0.56 compared with AA ALS; p < 0.001, D = 0.84, compared with *C HCs; p = 0.039, D = 0.18 compared with *C DCs). For serum and CSF sIL6R, and CSF IL6 all analyses were planned comparisons where significance was determined by the 2-sided Student t test. For serum IL6, significance was determined with generalized linear models, followed by Fisher protected least significant difference for planned individual comparisons of AA vs *C.
Subjects with ALS with the IL6R C allele have increased IL6 in the CSF
Univariate analysis was used to check for potential correlations between CSF or serum IL6, sIL6R, sgp130, and demographics. Sex and duration of symptoms were unrelated to any measure; however, serum IL6 was positively correlated with age (IL6 p < 0.001, ρ = 0.12, Spearman correlation). We next investigated whether the presence of the IL6R C allele contributed to differences in IL6 levels in subjects with ALS in cohort 1. There was no significant difference in IL6 levels between subjects with ALS with and without the C allele (figure 2C). Serum IL6 was higher in subjects with *C ALS compared with *C HCs (figure 2C).
*C ALS subjects had significantly higher levels of CSF IL6 compared with AA ALS subjects or controls with or without the C allele (figure 2D). In contrast, levels of CSF IL6 were nearly equivalent between AA ALS subjects (2.33 pg/mL) and AA HCs (2.25 pg/mL). We and others have reported that IL6 is increased in the CSF of subjects with ALS compared with control populations.5 Our results here suggest that this observed increase is not present in every subject but rather is driven by *C subjects, thus indicating the potential that IL6 trans-signaling could be active in ALS CSF and dependent on inheriting the IL6R C allele. In both serum and CSF, there were no significant differences in sgp130 between affected and unaffected subjects (data not shown).
Subjects with ALS who inherit the IL6R C allele exhibit faster disease progression
We next questioned whether the presence of the C allele is associated with accelerated ALS disease progression. Because there were no disease progression data available for cohort 1, and to investigate whether the presence of the C allele resulted in faster disease progression compared with patients without the variant, we turned to an independent sample (cohort 2) that included clinical outcomes, although paired CSF and serum samples were not available. Subjects in cohort 2 had reliable information regarding symptom onset with diagnosis and clinic visits within 1 year of onset. We limited our progression comparison to those subjects who were diagnosed within 1 year of their first symptom to most accurately obtain disease onset time. Thus, we were able to assess early stages of disease as secondary pathologic outcomes where IL6 may be a contributing factor (e.g., lung inflammatory responses and advanced muscle atrophy), have either not yet occurred, or would be less significantly progressed. Functional decline (ALSFRS-R progression rate) as measured by the ΔFS of patients diagnosed within the first year after symptom onset revealed that patients with the IL6R C allele have greater loss of function than those without (figure 3). There was no difference in the number of patients with bulbar or spinal onset between the 2 compared groups (p = 0.68).
Figure 3. Subjects with amyotrophic lateral sclerosis (ALS) with the interleukin 6 receptor (IL6R) C allele have faster disease progression.
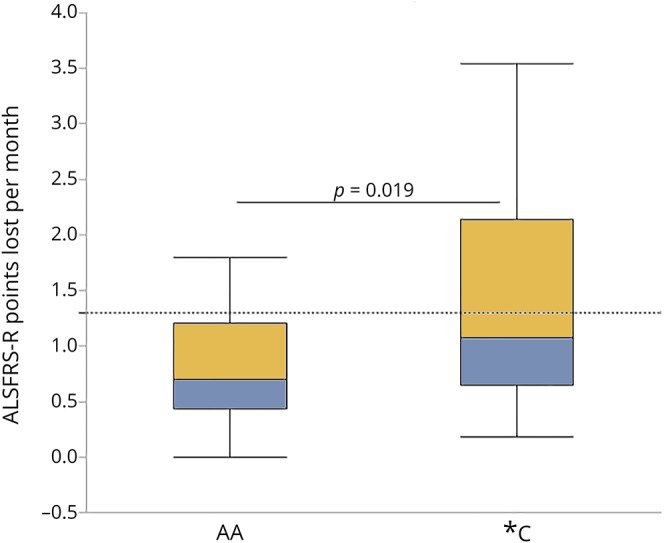
Subjects with ALS who are diagnosed within 12 months of symptom onset (table) who have the C allele lose more points on the ALSFRS-R between symptom onset and their first ALS clinic visit than those without the C allele. Results express as mean +SD; AA: n = 16; *C: n = 19; a = p = 0.019, D = 0.70; significance by Wilcoxon rank-sum.
Discussion
ALS is a fatal disease with a short survival time after diagnosis, an extensive clinical diagnostic process, and few treatment options. To further our understanding of which factors influence disease progression and severity, we completed a retrospective study of 2 cohorts of subjects with ALS and controls. Our results from cohort 1 show that only subjects with ALS with the IL6R C allele have increased IL6 and IL6R in CSF compared with HCs and neurologic DCs. The additive nature of the allele appears to be magnified only in CSF of patients with ALS and not in controls. From cohort 2, we found that individuals inheriting the IL6R C allele exhibit faster disease progression. The IL6R C allele regulates IL6 signaling mechanisms reported to influence several disease pathologies.13,14 Knowledge that a specific subgroup of patients with ALS, such as those carrying the IL6R C allele, will experience faster decline can contribute to earlier, informed diagnosis and application of patient-specific treatments and interventions (e.g., noninvasive ventilation) to improve quality of life. These results of the current study may have clinical significance and serve as a foundation for future, prospective studies to determine whether IL6R genotype and CNS signaling modify disease progression and are targets of therapeutic intervention with IL6R-blocking agents.
Serum levels of IL6 can easily fluctuate within 12 pg/mL range due to environmental factors unrelated to disease, such as age, adiposity, recent exercise, tobacco consumption, or comorbid inflammatory conditions.22–25 Variability in serum IL6 levels must also be interpreted in context of a disease, especially one like ALS where secondary disease processes may occur coincident with decreased functionality.3,26,27 Patients inheriting the IL6R C allele present with elevated serum IL6 at disease onset compared with AA patients because sIL6R extends the half-life of circulating IL6.28 Early in disease, MN degeneration and muscle atrophy stimulate production of IL6.29,30 However, throughout the disease course, other peripheral mechanisms can produce IL6, such as aspiration pneumonia or hypoxia resulting from decreased bulbar muscle and lung function3,13,14,31,32 limiting the diagnostic or prognostic value of the measure. Although IL6 levels may be partially dependent on IL6R genotype and subject age, extensive individual clinical characterization accounting for the aforementioned IL6-stimulating factors is necessary for appropriate interpretation.
Unlike in the periphery, levels of CSF IL6 likely reflect CNS-specific processes and are less influenced by acute peripheral undulations. Although there may be a specific transport mechanism for IL6 across the blood-brain barrier (BBB) that has only been minimally investigated in animals,33 we found no correlation between serum and CSF IL6 levels (figure e-1, links.lww.com/NXI/A152) underlining that CSF IL6 levels are not simply a reflection of serum IL6 at any given time. In addition, measurements of the albumin quotient (CSF: serum albumin) indicate that there is no BBB disruption in subjects with ALS as a whole or within IL6R variant groups (data not shown).
To determine whether CSF IL6 is potentially a diagnostic biomarker, we estimated by the area under the corresponding receiver operating characteristic curve for cohort 1 (data not shown). When age is included as a covariate, CSF IL6 was an informative biomarker, with an area under curve (AUC) of 0.75, sensitivity of 76.6%, and specificity of 70.6% (data not shown). Given the less variable nature of CSF IL6, a normal IL6 CSF level could be a good biomarker to exclude an ALS diagnosis and is at least comparable to other recently reported CSF diagnostic biomarkers neurofilament (NF) light (AUC = 0.78, 76% sensitivity, and 73% specificity) or phosphorylated NF heavy (AUC = 0.87, 84% sensitivity, and 83% specificity).34 Considering that patients with ALS with the C allele exhibited the highest levels of CSF IL6, we further estimated the ability of CSF IL6 to discriminate between ALS and HCs or DCs using a separate receiver operating characteristic only including subjects with the C allele, with age as a covariate. In this case, CSF IL6 as a diagnostic tool has superior sensitivity (AUC = 0.82, 90% sensitivity, and 69% specificity).
Because cohort 1 did not include clinical data, we were not able to evaluate whether CSF IL6 levels associate with disease progression as reported to chitotriosidase-1 (CHIT-1), a biomarker associated with microglial activation in ALS.35 A prospective study evaluating IL6R genotype, CSF levels of IL6, CHIT-1, and NF could provide insight into how biomarkers of glial activation and neuronal responses correlate with disease symptomology and progression.
As CSF IL6 levels are highest in *C patients, elevated IL6 trans-signaling would be expected. The source of CSF IL6 may be activated microglia and astrocytes.9 Although as shown in animal models, activation/IL6 levels may vary throughout disease,36,37 in the presence of circulating CSF IL6R observed in *C ALS patients, sIL6R/IL6 complexes could enhance activation via trans-signaling mechanisms to further promote MN dysfunction and degeneration.9,38,39 Indeed, as high levels of CSF CHIT-1, a biomarker of microglial activation are associated with faster disease progression,35 we hypothesize that *C patients who experience elevated CNS IL6 trans-signaling may be associated with increased glial activation and experience a more rapid disease progression. A change in the ALSFRS-R score of ≥3 points is an indicator of a substantial decline in function.40 In our data set, only 3 of 16 (19%) AA patients met this threshold (1.3 points lost per month; dotted line, figure 3) as opposed to 8 of 19 (42%) *C patients. Future studies that include assessment of IL6R genotype, CSF IL6R and IL6, and clinical progression will provide more insights into this hypothesis.
Although the limitations of this study prevent mechanistic evaluation, the potential role of IL6 signaling in ALS should be considered to provide perspective for current results and design of future studies. IL6 signaling has distinct, localized roles (e.g., peripheral muscle/NMJ, diaphragm and lung, and spinal cord/brain) throughout ALS disease progression, with some potentially being beneficial and others exacerbating disease progression (figure 4). To discover and use effective treatments, it is crucial to better understand physiologic causes of variability in ALS disease progression. The possibility that *C patients experience faster progression, possibly mediated by CNS IL6 trans-signaling, merits further investigation and consideration in future therapeutic trials of IL6R-blocking strategies.
Figure 4. In amyotrophic lateral sclerosis (ALS), interleukin 6 (IL6) trans-signaling effects on motor neurons may shift the balance from trophic activity to promoting a toxic environment.

In conditions of axonal injury, muscle injury, muscle stress (e.g., exercise), and atrophy (e.g., sarcopenia), increases in muscle-specific expression of IL6 are correlated with increased levels of circulating IL6.9,38,39 Of relevance to MNs, IL6 is a member of the CNTF/CT/LIF gp130 family of trophic factors that are critical in MN development and survival following injury and disease.7 In the local environment, in the presence of sIL6R, the myokine-receptor complex can bind to gp130 on MNs acting as a trophic factor. Accordingly, IL6 signaling may be beneficial in maintaining NMJ innervation, promoting MN survival, and rebuilding muscle (lower panel). Despite the potential prosurvival effect of IL6 on neurons in the periphery, it may be that in *C patients, CNS IL6 trans-signaling effects on glial cells promote damaging effects in the CNS.9,38,39 In the CNS, microglia become activated in response to NMJ denervation, and both microglial and astrocytes become active in response to disease-associated MN pathology. These and other CNS cells have been reported to be sources of IL6 (upper panel). In this central environment, IL6 can bind sIL6R and, as in the periphery, bind gp130 on MNs promoting trophic activity. However, the IL6-sIL6R complex can also bind to gp130 on glial cells to further activate them. In ALS, activated astrocytes have been shown to create a toxic environment for MNs.38,39 We hypothesize that this toxic activity overpowers any potential trophic activity, shifting the balance for MNs toward dysfunction and degeneration.
Study limitations
As an observational and correlative study, there are several limitations. First, the 2 cohorts were not collected under identical recruitment schemes, thus availability of samples and phenotypic data was not consistent. As a result, it was not possible to perform the same assays in each cohort because cohort 1 did not include clinical or outcome data. Although containing clinical data, cohort 2 did not include paired serum and CSF samples. The difference in cohort design did not allow comparative, longitudinal data to be collected to make direct comparisons for disease progression in the same cohort. For this reason, we caution the reader not to conflate results presented from each cohort. In addition, ALS is a highly heterogeneous in demographics and clinical phenotypes. As such, it is imperative to replicate the above findings in a larger cohort of patients where longitudinal clinical indicators of disease and multiple biological samples are collected simultaneously.
Glossary
- ALS
amyotrophic lateral sclerosis
- BBB
blood-brain barrier
- CHIT-1
chitotriosidase-1
- DC
disease control
- HC
healthy control
- IL6
interleukin 6
- IL6R
IL6 receptor
- MAF
minor allele frequency
- MN
motoneuron
- NEALS
Northeastern ALS Consortium
- NMJ
neuromuscular junction
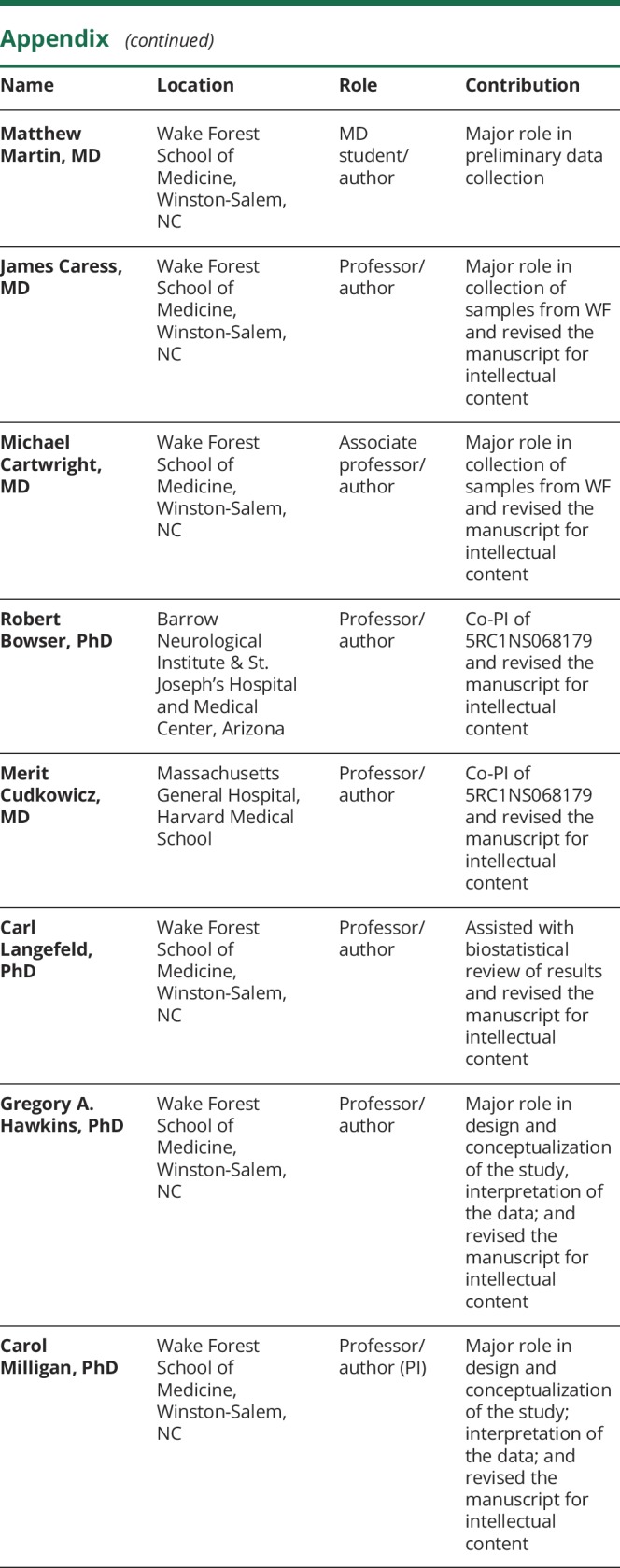
Appendix. Authors
Study funding
This work was supported by Hope for Tomorrow, a gift in the memory of Murray Sherman, the WFHS Brian White Fund, and the Tab A. Williams Funds at Wake Forest School of Medicine. Matched CSF and serum samples for the first cohort in this study were provided by the Northeastern ALS Biofluid Repository and were collected as part of the Multi-Center Validation of Biomarkers for Motor Neuron Disease Project (5RC1NS068179, Bowser and Cudkowicz). MWK is supported by the Argenta Scholarship at WFSM.
Disclosure
Drs. Milligan, Hawkins, Langefeld, Caress, Cartwright, Cudkowicz, Bowser, Martin, Robinson, Mr. Arounleut, Ms. Strupe, and Ms. Wosiski-Kuhn report no disclosures. Go to Neurology.org/NN for full disclosures.
References
- 1.Lyon MS, Wosiski-Kuhn M, Gillespie R, Caress J, Milligan C. Inflammation, immunity, and amyotrophic lateral sclerosis: I. Etiology and pathology. Muscle Nerve 2019;59:10–22. [DOI] [PubMed] [Google Scholar]
- 2.Wosiski-Kuhn M, Lyon MS, Caress J, Milligan C. Inflammation, immunity, and amyotrophic lateral sclerosis: II. Immune-modulating therapies. Muscle Nerve 2019;59:23–33. [DOI] [PubMed] [Google Scholar]
- 3.Moreau C, Devos D, Brunaud-Danel V, et al. Elevated IL-6 and TNF-alpha levels in patients with ALS: inflammation or hypoxia? Neurology 2005;65:1958–1960. [DOI] [PubMed] [Google Scholar]
- 4.Rentzos M, Rombos A, Nikolaou C, et al. Interleukin-17 and interleukin-23 are elevated in serum and cerebrospinal fluid of patients with ALS: a reflection of Th17 cells activation? Acta Neurol Scand 2010;122:425–429. [DOI] [PubMed] [Google Scholar]
- 5.Mizwicki MT, Fiala M, Magpantay L, et al. Tocilizumab attenuates inflammation in ALS patients through inhibition of IL6 receptor signaling. Am J Neurodegen Dis 2012;1:305–315. [PMC free article] [PubMed] [Google Scholar]
- 6.Sekizawa T, Openshaw H, Ohbo K, Sugamura K, Itoyama Y, Niland JC. Cerebrospinal fluid interleukin 6 in amyotrophic lateral sclerosis: immunological parameter and comparison with inflammatory and non-inflammatory central nervous system diseases. J Neurol Sci 1998;154:194–199. [DOI] [PubMed] [Google Scholar]
- 7.Zigmond RE. gp130 cytokines are positive signals triggering changes in gene expression and axon outgrowth in peripheral neurons following injury. Front Mol Neurosci 2011;4:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schaper F, Rose-John S. Interleukin-6: biology, signaling and strategies of blockade. Cytokine Growth Factor Rev 2015;26:475–487. [DOI] [PubMed] [Google Scholar]
- 9.Campbell IL, Erta M, Lim ML, et al. Trans-signaling is a dominant mechanism for the pathogenic actions of interleukin-6 in the brain. J Neurosci 2014;34:2503–2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garbers C, Monhasery N, Aparicio-Siegmund S, et al. The interleukin-6 receptor Asp358AL single nucleotide polymorphism rs2228145 confers increased proteolytic conversion rates by ADAM proteases. Biochemica Biophys Acta 2014;1842:1485–1494. [DOI] [PubMed] [Google Scholar]
- 11.van Dongen J, Jansen R, Smit D, et al. The contribution of the functional IL6R polymorphism rs2228145, eQTLs and other genome-wide SNPs to the heritability of plasma sIL-6R levels. Behav Genet 2014;44:368–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bagli M, Papassoritopoulos A, Hampel H, et al. Polymorphisms of the gene encoding the inflammatory cytokine interleukin-6 determine the magnitude of the increase in soluble interleukin-6 receptor levels in Alzheimer's disease. Eur Arch Psychiatry Clin Neurosci 2003;253:44–48. [DOI] [PubMed] [Google Scholar]
- 13.Hawkins GA, Robinson MB, Hastie AT, et al. The IL6R variation Asp(358)Ala is a potential modifier of lung function in subjects with asthma. J Allergy Clin Immunol 2012;130:510–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peters MS, McGrath KW, Hawkins GA, et al. Plasma interleukin-6 concentrations, metabolic, dysfunction, and asthma severity: a cross-sectional analysis of two cohorts. Lancet Respir Med 2016;4:574–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mitsuyama K, Toyonaga A, Sasaki E, et al. Soluble interleukin-6 receptors in inflammatory bowel disease: relation to circulating interleukin-6. Gut 1995;36:45–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stephens OW, Zhang Q, Pingping Q, et al. An intermediate-risk multiple myeloma subgroup is defined by sIL-6r: levels synergistically increase with incidence of SNP rs2228145 and 1q21 amplification. Blood 2016;119:503–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sasayama D, Wakabayashi C, Hori H, et al. Association of plasma IL-6 and soluble IL-6 receptor levels with the Asp358AL polymorphism of the IL-6 receptor gene in schizophrenic patients. J Psychiatr Res 2011;45:1439–1444. [DOI] [PubMed] [Google Scholar]
- 18.Ludolph A, Drory V, Hardiman O, et al. A revision of the El Escodial criteria. Amyotroph Lateral Scler Frontotemporal Degener 2015;16:291–292. [DOI] [PubMed] [Google Scholar]
- 19.Chiò A, Logroscino G, Hardiman O, et al. Prognostic factors in ALS: a critical review. Amyotroph Lateral Scler 1009;10:310–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Labra J, Menon P, Byth K, Morrison S, Vucic S. Rate of disease progression: a prognostic biomarker in ALS. J Neurol Neurosurg Psychiatry 2016;87;628–632. [DOI] [PubMed] [Google Scholar]
- 21.Thakore NJ, Lapin BR, Pioro EP. Trajectories of impairment in amyotrophic lateral sclerosis: insights from the pooled resource open-access ALS clinical trials cohort. Muscle Nerve 2018;57:937–945. [DOI] [PubMed] [Google Scholar]
- 22.Cullen T, Thomas AW, Webb R, Hughes MG. The relationship between interleukin-6 in saliva, venous and capillary plasma, at rest and in response to exercise. Cytokine 2015;71:397–400. [DOI] [PubMed] [Google Scholar]
- 23.Park HS, Park JY, Yu R. Relationship of obesity and visceral adiposity with serum concentrations of CRP, TNF-α and IL-6. Diabetes Res Clin Pract 2005;69:29–35. [DOI] [PubMed] [Google Scholar]
- 24.Ishihara K, Hirano T. IL-6 in autoimmune disease and chronic inflammatory proliferative disease. Cytokine Growth Factor Rev 2002;13:357–368. [DOI] [PubMed] [Google Scholar]
- 25.Kastelein T, Duffield R, Marino F. Human in situ cytokine and leukocyte responses to acute smoking. J Immunotoxicol 2017;14:109–115. [DOI] [PubMed] [Google Scholar]
- 26.Lee CW, Chen HJ, Liang JA, Kao CH. Risk of sepsis in patients with amyotrophic lateral sclerosis: a population-based retrospective cohort study in Taiwan. BMJ Open 2017;7:e013761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lechtzin N, Wiener CM, Clawson L, Chaudhry V, Diette GB. Hospitalization in amyotrophic lateral sclerosis: causes, costs, and outcomes. Neurology 2001;56:753–757. [DOI] [PubMed] [Google Scholar]
- 28.Schöbitz B, Pezeshki G, Pohl T, et al. Soluble interleukin-6 receptor augments central effects of IL-6 in vivo. FASEB J 1995;9:659–664. [DOI] [PubMed] [Google Scholar]
- 29.Munoz-Canoves P, Scheele C, Pedersen BK, Serrano AL. Interleukin-6 myokine signaling in skeletal muscle: a double edged sword? FEBS J 2013;280:4131–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gadient RA, Otten UH. Interleukin-6 (IL-6)—a molecule with both beneficial and destructive potentials. Prog Neurobiol 1997;52:379–390. [DOI] [PubMed] [Google Scholar]
- 31.Glynn P, Coakley R, Kilgallen I, Murphy N, O'Neill S. Circulating interleukin 6 and interleukin 10 in community acquired pneumonia. Thorax 1999;54:51–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hedlund J. Community-acquired pneumonia requiring hospitalisation. Factors of importance for the short-and long term prognosis. Scand J Infect Dis Suppl 1995;97:1–60. [PubMed] [Google Scholar]
- 33.Banks WA, Kastin AJ, Gutierrez EG. Penetration of interleukin-6 across the murine blood-brain barrier. Neurosci Lett 1994;179:53–56. [DOI] [PubMed] [Google Scholar]
- 34.Rossi D, Volanti P, Brambilla L, et al. CSF neurofilament proteins as diagnostic and prognostic biomarkers for amyotrophic lateral sclerosis. J Neurol 2018;265:510–521. [DOI] [PubMed] [Google Scholar]
- 35.Gille B, De Schaepdryver M, Dedeene L, et al. Inflammatory markers in cerebrospinal fluid: independent prognostic biomarkers in amytrophic lateral sclerosis? J Neurol Neurosurg Psychiatry 2019;Epub 2019 June 7. [DOI] [PubMed] [Google Scholar]
- 36.Weydt P, Yuen EC, Ransom BR, Möller T. Increased cytotoxic potential of microglia from ALS-transgenic mice. Glia 2004;48:179–182. [DOI] [PubMed] [Google Scholar]
- 37.Gifondorwa DJ, Jimenz-Moreno R, Hayes CD, et al. Administration of recombinant heat shock protein 70 delays pripheral muscle denervation in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Neurol Res Int 2012;2012:170426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barbeito LH, Pehar M, Cassina P, et al. A role for astrocytes in motor neuron loss in amyotrophic lateral sclerosis. Brain Res Brain Res Rev 2004:47:263–274. [DOI] [PubMed] [Google Scholar]
- 39.Liddelow SA, Guttenplan KA, Clarke LE, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017;541:481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Castrillo-Viguera C, Grasso DL, Simpson E, Shefner J, Cudkowicz ME. Clinical significance in the change of decline in ALSFRS-R. Amyotroph Lateral Scler 2010;11:178–180. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data not provided in the article because of space limitations will be shared upon request of other investigators for purposes of replicating procedures and results.