

Summary
Target identification for biologically active small molecules remains a major barrier for drug discovery. Cancer cells exhibiting defective DNA mismatch repair (dMMR) have been used as a forward genetics system to uncover compound targets. However, this approach has been limited by the dearth of cancer cell lines that harbor naturally arising dMMR. Here, we establish a platform for forward genetic screening using CRISPR-Cas9 to engineer dMMR into mammalian cells. We demonstrate the utility of this approach to identify mechanisms of drug action in mouse and human cancer cell lines using in vitro selections against three cellular toxins. In each screen, compound-resistant alleles emerged in drug-resistant clones, supporting the notion that engineered dMMR enables forward genetic screening in mammalian cells.
Keywords: Forward genetics, phenotypic screening, target identification, mismatch repair deficiency, hypermutation, mismatch repair
Graphical Abstract
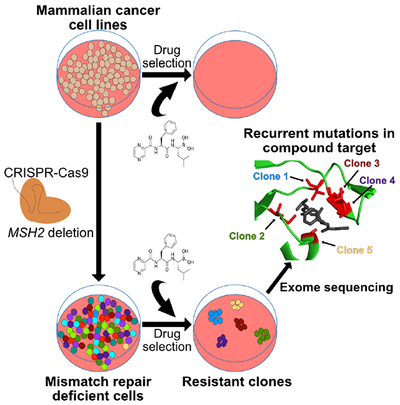
eTOC
Target identification of small molecules remains a barrier for drug discovery. Here, Povedano et al. establish a platform for forward genetic screening using CRISPR-Cas9 to engineer DNA mismatch repair deficiency in cancer cell lines and demonstrate the feasibility of this approach to identify the known targets of three anti-cancer toxins.
Introduction
The identification of therapeutic targets in cancer can be divided into two complementary approaches. Target-based approaches use tumor sequencing or laboratory-based genetic studies to identify cancer driver genes followed by screening for small molecules that impair the protein products of cancer driver genes. Phenotypic high-throughput small molecule screens (HTS) first identify drug-like chemicals that selectively impair the growth of cancer cells. The latter approach has been limited by the technical challenge of identifying the direct protein targets of small molecules exhibiting anti-cancer effects. One strategy to identify chemical targets is through the identification of compound resistant alleles that impair compound-target interaction. Cancer cells with dMMR exhibit mutation rates increased as much as 50-750-fold compared to cells with intact MMR (Glaab and Tindall, 1997). As a result, these cells are predisposed to develop resistance through the acquisition of compound resistant alleles. Following selection, these compound resistant alleles can be identified by transcriptome or whole exome sequencing of multiple independent drug-resistant clones (Han et al., 2017, Han et al., 2016, Wacker et al., 2012). However, it is not clear whether this approach can be applied to other cancer cell lines, particularly those established from cancers without MMR deficiency or those harboring low mutation frequencies, such as pediatric malignancies. To date, a single cancer cell line, HCT116, derived from a human colorectal cancer harboring a naturally-arising mutation in the MMR protein MLH1, has been used successfully for these studies. Here, we sought to determine if somatic deletion of the MMR protein MSH2 using CRISPR-Cas9 could be used to expand the repertoire of dMMR cancer cell lines for use in forward genetic screens.
Results
MSH2 deletion in cancer cell lines induces MSI and hypermutation
We sought to develop cancer cell lines with engineered loss of MSH2 in order to establish forward genetics screening in other cancer types, including Ewing sarcoma (EWS) and small cell lung cancer (SCLC). Ewing sarcoma is a pediatric malignancy without approved targeted therapies, and the overall mutational burden in these tumors is extremely low, similar to other pediatric malignancies (Pishas and Lessnick, 2016, Yu et al., 2017, Tirode et al., 2014, Brohl et al., 2014, Crompton et al., 2014). We used CRISPR-Cas9 to generate MSH2-null Ewing sarcoma A673 cells (Figure 1A, Methods). We examined multiple microsatellite regions by PCR and capillary electrophoresis for evidence of microsatellite instability (MSI), a hallmark of dMMR cells, in two independently generated MSH2-null A673 lines (A673-M1, A673-M8). MSI was observed in three out of five loci analyzed in both A673-M1 and A673-M8 clones compared to the parental, MMR-proficient, A673 cell line (Fig. 1B,C)(Boland et al., 1998).
Figure 1. CRISPR-mediated MMR loss in Ewing sarcoma cells induces MSI and hypermutation.
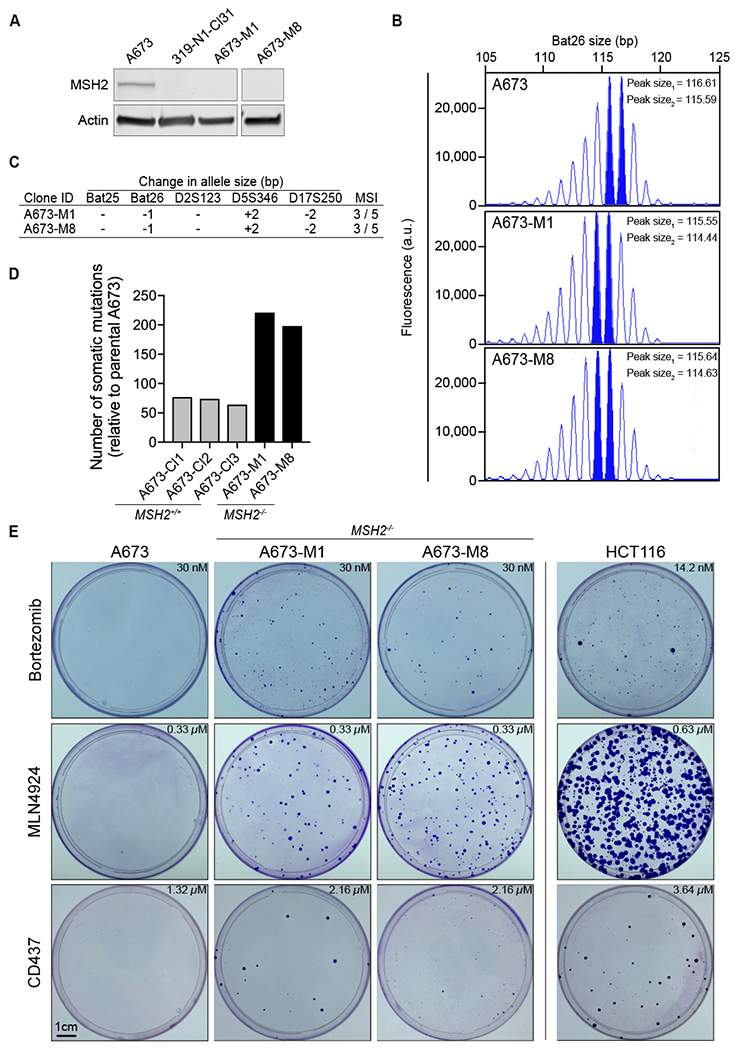
(A) MSH2 and Actin protein levels showed for A673 (MSH2-wild-type), 319-N1-CI31 (Msh2-edited mouse small cell lung cancer cell line (see Figure S1)), and MSH2-edited A673-M1 and A673-M8 (right panel for A673-M8 cells performed in an independent blot, as stated by the break between the first three lanes and A673-M8). (B) Capillary electrophoresis histogram of microsatellite Bat26 locus for A673, A673-M1 and A673-M8. The most prominent peaks in the histogram determine the allele size is shown (solid blue). (C) Table of microsatellites assessed in A673 cells. Difference in the dominant allele size between the parental A673 and each MMR deficient cell line is indicated. Number of microsatellites affected in relation to the number of microsatellites studied displayed in the last column. (D) Number of somatic mutations present in A673-M1, and A673-M8 identified by exome sequencing. Parental A673 cells were used as a normal reference. Mutations with an allelic fraction (AF) higher or equal to 0.15 were reported. (E) Crystal violet staining of A673 (MSH2-wild-type), A673-M1 (MSH2-null), A673-M8 (MSH2-null), and HCT116 (MLH1-null) cells following compound selection. Concentrations used for selection shown in each panel. See also Figure S1 and S2.
We next tested whether MMR deletion would facilitate MSI in tumor cell lines derived from genetically-engineered mouse cancer models (GEMMs), which also exhibit very low mutation frequencies compared to many human malignancies (McFadden et al., 2014, McFadden et al., 2016). Msh2 was deleted in a cell line generated from a small cell lung cancer (SCLC) GEMM initiated by loss of the p53, retinoblastoma (Rb), and p130 tumor suppressors (319-N1 cell line) (Figure S1A)(Schaffer et al., 2010). Msh2-null murine SCLC (mSCLC) cells also exhibited evidence of MSI, with two of three microsatellite loci exhibiting instability (Figure S1B, C)(Bacher et al., 2005).
Following observation of MSI in MMR-edited human and murine cells, we sought direct evidence of an increased mutation frequency using whole exome sequencing (WES). Two MSH2-null A673 clones (M1 and M8) and three independent MSH2-wild-type parental A673 clones (CL1, CL2, and CL3) were subjected to WES. Because a reference germline genome was not available for A673 cells, we used the parental A673 cell line as the germline reference (Methods). We observed an increased frequency of somatic mutations in M1 (n=221) and M8 (n=198) clones, compared to MSH2-wild-type parental clones (n= 77, 74, 64) (Table S1).
We performed WES on multiple mSCLC tumor cell lines generated from independent tumors isolated from the same mouse in order to accurately compare mutation frequencies between MMR-proficient and dMMR cell lines (Figure S2A). Common variants between two independent primary tumor cell lines (319-T1, T2) represented germline variants, whereas variants unique to individual cell lines represented somatic mutations. The Msh2-wild type cell line 319-N1 exhibited 25 high-confidence somatic mutations (Table S2). In contrast, 319-N1-Cl31 cells with engineered Msh2 loss exhibited 352 somatic mutations. Therefore, elevated somatic mutation frequencies were observed following MMR editing in both human and murine cancer cell lines. (Figure 1D, Figure S2B,C).
Engineered dMMR enables forward genetic screening in mammalian cancer cells
After establishing the dMMR phenotype of CRISPR-edited MSH2-null cell lines, we determined whether CRISPR-mediated dMMR facilitated the emergence of compound-resistant clones. We performed drug selections using MSH2-wild type A673 cells (parental A673), MSH2-null A673 cells (A673-M1 and A673-M8), and HCT116 cells. Selections were performed against three cellular toxins: bortezomib (an inhibitor of the subunit β5 of the proteasome, PSMB5), MLN4924 (a NEDD8-activating enzyme (NAE) inhibitor), and CD437 (a DNA polymerase alpha (POLA1) inhibitor)(Soucy et al., 2009, Han et al., 2016, Lu and Wang, 2013, Chen et al., 2011). Following compound selection at lethal doses, resistant colonies growing in the presence of compound were visualized by crystal violet staining. Consistent with the notion that CRISPR-mediated MSH2 loss enabled acquisition and emergence of compound resistant clones, colonies were observed following selection only on the MSH2-null A673 and HCT116 plates (Figure 1E). No colonies were observed on the parental MSH2-wild type A673 plates.
We next performed additional selections using bortezomib, CD437, and MLN4924. We selected MSH2-null A673 cells and Msh2-null mSCLC cells using bortezomib at three concentrations close to the lethal dose, as determined by one week of compound exposure (EC1001wk) for bortezomib (see methods). Following 2 weeks of selection, compound-resistant colonies emerged and were expanded from both A673-M1 (4 clones) and Msh2-null mSCLC cells (11 clones).
Bortezomib-resistant alleles have been reported within exon 2 of PSMB5, which encodes a binding pocket for the drug (Lu and Wang, 2013, Chen et al., 2011). We therefore amplified and sequenced exon 2 in bortezomib-resistant MSH2-null A673 and Msh2-null mSCLC clones. These regions were also amplified and sequenced from the parental A673 and mSCLC cell lines to ensure these mutations did not exist prior to MMR impairment. We identified PSMB5 mutations in MMR-deficient A673 (4 out of 4 clones harbored mutations) and mSCLC cell lines (11 out of 11 clones harbored PSMB5 exon 2 mutations), including mutations previously reported to mediate bortezomib resistance (Fig. 2B, D, E) (Lu and Wang, 2013, Wacker et al., 2012). All mutations identified in PSMB5 mapped to the bortezomib binding pocket (Huber et al., 2016) (Fig. 2F). We confirmed in vitro resistance to bortezomib in all 4 clones harboring putative compound-resistant alleles from A673-M1 by cell viability assay (CellTiter Glo, Promega) following 72 hours of drug exposure (Figure 2A, B). We also tested 7 out of the 11 mSCLC clones that harbored Psmb5 mutations and confirmed in vitro resistance to bortezomib (Figure 2C, D). We confirmed that all clones harboring PSMB5 mutations exhibited bortezomib resistance (2.36 to 13.84-fold increase in EC50), whereas resistance to etoposide was not observed (Figure 2A–D; Figure S3A, B).
Figure 2. Emergence and validation of PSMB5-resistant alleles following bortezomib selection.
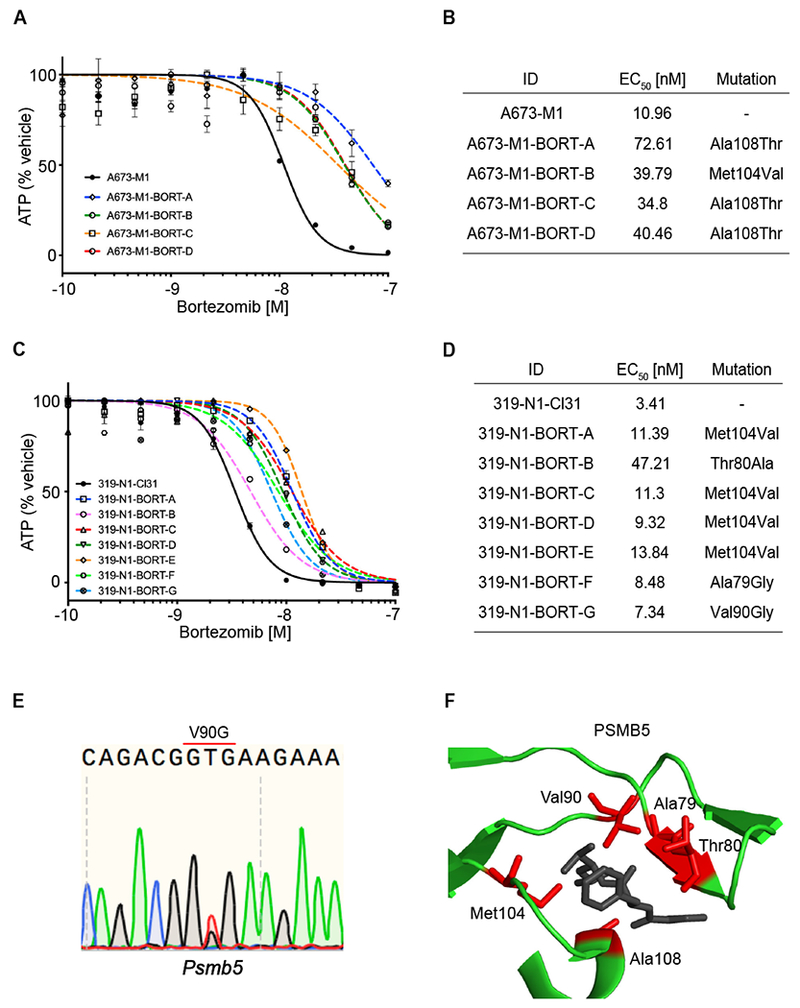
(A) Cell Titer Glo assay of bortezomib 10-point dose response curve (DRC) for parental A673 cells and bortezomib-resistant clones A, B, C, and D. Data are represented as mean ± SD. (B) Table showing EC50 for bortezomib from parental A673-M1 and each bortezomib-resistant clone. Mutations detected in exon 2 of PSMB5 reported for each resistant clone. (C) Cell Titer Glo assay of bortezomib in 10-point dose response curve (DRC) for parental 319-N1 cells and bortezomib-resistant clones A, B, C, D, E, F and G. Data are represented as mean ± SD. (D) Table showing EC50 for bortezomib from parental 319-N1-CI31 and each bortezomib-resistant clone. Mutations found in exon 2 of Psmb5 showed for each resistant clone. (E) Sequencing trace for V90G mutation found in Psmb5 from mSCLC clone G. (F) Crystal structure of PSMB5 bound to bortezomib (Protein Data Bank accession 5L5Z). Depicted all mutated codons found in bortezomib-resistant clones from Ewing sarcoma and mSCLC clones (red). See also Figure S3 A–B.
To further confirm that CRISPR-dMMR cells exhibited the capacity to acquire compound resistant alleles to different classes of cellular toxins, we performed additional selections using MSH2-null A673 cells and Msh2-null mSCLC cells against the DNA polymerase-alpha inhibitor CD437. After 2 weeks of selection, compound-resistant colonies emerged and were expanded from both A673-M1 (4 clones) and Msh2-null mSCLC cells (17 clones). For CD437-selected clones, cDNA flanking exons 19 to 25 was amplified and sequenced after clonal expansion. We identified POLA1 mutations in the MMR-deficient A673 (4 out of 4 clones harbored mutations) and mSCLC lines (11 out of 17 clones harbored mutations) (Figure 3B, D, E) (Han et al., 2016). None of these mutations were detected in the parental, MMR proficient, cell lines. Mutations impacted five amino acids that clustered within the POLA1 structure (Coloma et al., 2016) (Figure 3F).
Figure 3. Emergence and validation of POLA1-resistant alleles following CD437 selection.
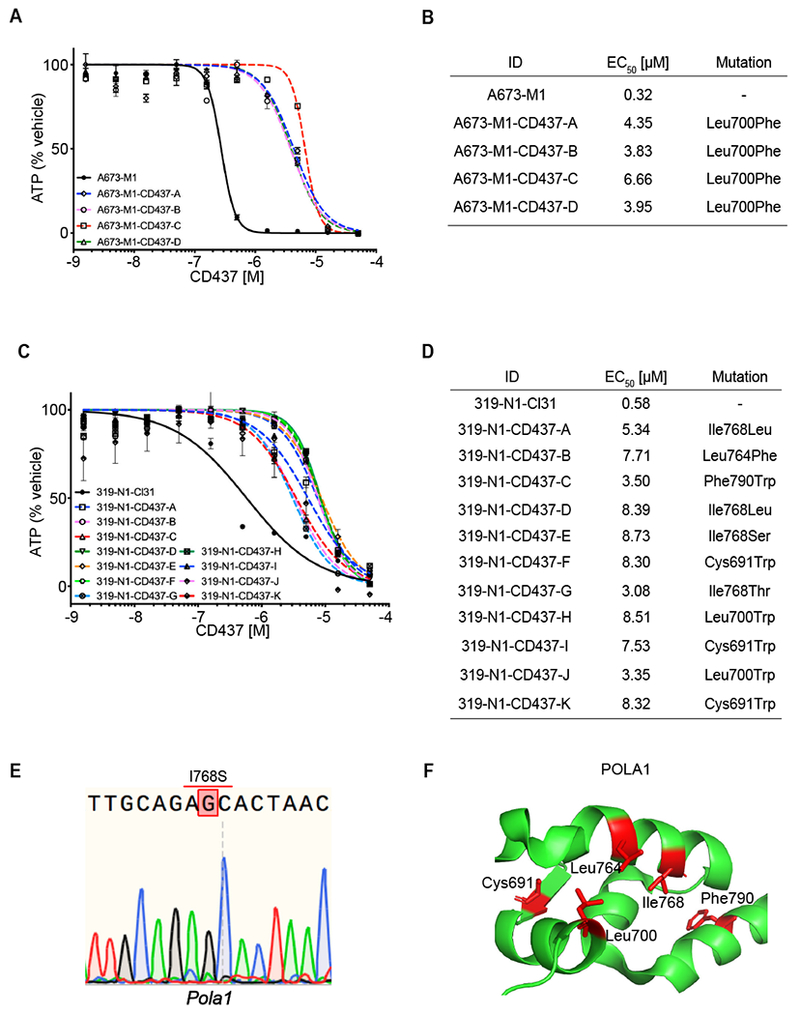
(A) Cell Titer Glo assay of CD437 10-point DRC for parental cell line and CD437-resistant clones A, B, C, and D. Data are represented as mean ± SD. (B) Table showing EC50 for CD437 from parental A673-M1 and each CD437-resistant clone. POLA1 mutations reported for each resistant clone. (C) Cell Titer Glo assay of CD437 10-point DRC for parental cell line and clones A, B, C, D, E, F, G, H, I and J. Data are represented as mean ± SD. (D) Table showing EC50 for CD437 from parental 319-N1-Cl31 and each CD437-resistant clone. Polal mutations detected in each resistant clone. (E) Sequencing trace for I768S mutation found in Pola1 from mSCLC clone A. (F) Crystal structure of POLA1 (Protein Data Bank accession 5IUD). Mutated codons found in CD437-resistant clones from Ewing sarcoma and mSCLC (red). See also Figure S3 C–D.
We confirmed CD437-resistance in all 4 clones from A673-M1 cell line and 11 out of 17 mSCLC clones harboring putative compound-resistant alleles to CD437 (Figure 3A, B). The six mSCLC resistant clones that did not harbor POLA1 mutations exhibited resistance to both CD437 and etoposide, which suggested a generalized mechanism of acquired resistance drove expansion of these clones during compound selection (Figure S3C, D). The ten mSCLC clones harboring POLA1 mutations all exhibited CD437 resistance (5.31 to 15.05-fold increase in EC50), whereas no difference in sensitivity to etoposide was observed (Figure 3C, D; Figure S3E, F).
The selections performed with bortezomib and CD437 demonstrate that engineered MSH2 loss in enables the emergence of compound resistant alleles during drug selections in A673 and mSCLC cells. We finally tested whether prospective identification of compound targets could be accomplished using exome sequencing of compound resistant clones. Therefore, we performed selections using MLN4924 in the MSH2-null clone, A673-M1, at three concentrations for MLN4924 (see Methods). Following two weeks of selection, six colonies emerged and were expanded. To confirm in vitro resistance, we determined the EC50 for both parental A673-M1 cells and six resistant clones (Figure 4A,B). To identify potential clones exhibiting general resistance, including increased expression of drug efflux pumps, we assessed sensitivity to the topoisomerase II-inhibitor, etoposide (Figure S3F). We validated that all MLN clones exhibited resistance to MLN4924 (21.75 to 135.03-fold increase in EC50), while no difference in etoposide resistance was observed (Figure 4A,B; Figure S3F).
Figure 4. WES of MLN4924 resistant clones uncovers recurrent compound-resistant alleles in UBA3.
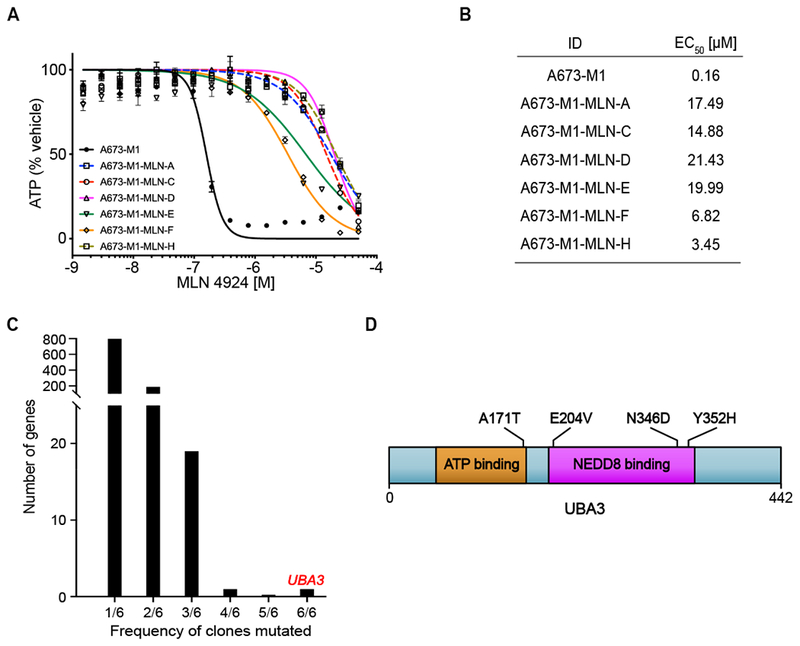
(A) Cell Titer Glo assay of MLN4924 10-point DRC for parental A673-M1 cells and MLN4924-resistant clones A, C, D, E, F, and H. Data are represented as mean ± SD. (B) Table showing EC50 for MLN4924 from parental A673-M1 and each resistant clone. (C) UBA3 is the only gene mutated in all six clones. (D) UBA3 mutations identified in MLN4924-resistant clones. DRC, dose response curve.
We next determined if the target of MLN4924, NAE subunit encoded by UBA3, could be identified by WES of MLN4924-resistant clones. Indeed, UBA3 was identified as the single gene mutated in 6/6 MSH2-null clones (Figure 4C). This gene encodes the NAE subunit targeted by MLN4924, and three of the mutations observed, A171T, E204K, and Y352H, were previously reported as MLN4924-resistant alleles (Figure 4D) (Michael et al., 2012, Xu et al., 2014). We compared somatic mutations in MLN4924-resistant A673 clones to establish whether the MLN4924-resistant clones arose independently (see Methods). A673-MLN-D and A673-MLN-H shared a majority of mutations (783/965), establishing that these two clones arose from the same founder cell. However, no other clones shared more than 6 somatic mutations, suggesting that the other clones arose independently, including A673-MLN-A and A673-MLN-C that shared the A171T mutation in UBA3.
Discussion
We establish that induced MMR deficiency using CRISPR-Cas9 methods is sufficient to induce MSI, hypermutation, and facilitate the emergence of compound resistant alleles in established human and murine cancer cell lines derived from diverse cancer lineages. This approach offers the potential to significantly expand the use of forward genetics to identify the mechanisms of action of compounds with anticancer activity. In particular, we demonstrate that this strategy can be employed in cancers with low mutation rates such as pediatric malignancies. Therefore, cell lines with engineered MMR deficiency represent an experimental tool to facilitate the identification of mechanisms of action of selective cancer toxins identified by HTS campaigns, and to model genetic mechanisms of acquired resistance to anti-cancer therapies in current use. However, we recognize limitations of the current study. First, following identification of candidate compound-resistant alleles (i.e., recurrent mutations in compound-resistant clones), additional biochemical studies are necessary to establish the direct molecular target. Second, the forward genetics approach requires that the small molecule target a protein essential for viability of the cancer cell. In addition, we cannot from our current data establish a frequency of mutation necessary to facilitate the emergence of compound-resistant clones. Hypermutation due to endogenous defects in DNA repair might also be more broadly applied to other phenotypic genetic screens, including in vivo screens in GEMMs. Therefore, cancer cell lines with induced MMR deficiency and hypermutation represent a tool with wide potential application in cancer genetics and drug discovery.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagent should directed to and will be fulfilled by the Lead Contact, David McFadden (david.mcfadden@utsouthwestern.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Ewing sarcoma A673 cell line were cultured at 37°C and 5% CO2 in RPMI (R8758, Sigma-Aldrich) supplemented with 10% FBS (#35-150-CV, Corning), 2 mM L-glutamine (G7513, Sigma-Aldrich), and penicillin streptomycin (P0781, Sigma-Aldrich). Cells were expanded using Trypsin (T4049, Sigma-Aldrich) every 3-4 days. A673 cell lines are derived from a female subject and were authenticated by STR profiling.
The Trp53fl/fl; Rb1fl/fl; Rbl2fl/fl; RosaLSL-Tomato/+ mouse model of small cell lung cancer mice has been previously described (Schaffer et al., 2010). 319-T1 and 319-T2 were established from primary tumors in the lung, and 319-N1 was established from a lymph node metastasis, all developed in a PRP female mouse. 319-N1 as well as clones derived from those cell lines were cultured with DMEM (D6429, Sigma-Aldrich) supplemented with 5% FBS (#35-150-CV, Corning), 2 mM L-glutamine (G7513, Sigma-Aldrich), and penicillin streptomycin (P0781, Sigma-Aldrich). Cells were expanded using Trypsin (T4049, Sigma-Aldrich) diluted in PBS (2:1 ratio) every 3-4 days.
All animal experiments were approved by the UTSW IACUC 2018-102383 (D.G.M., P.I.).
METHOD DETAILS
sgRNAs, surveyor, CRISPR KO of MSH2, MLH1.
Single-guide RNA (sgRNA) targeting human MSH2 and murine Msh2 were designed by “sgRNA Designer: CRISPR KO” (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design) from the Broad Institute(Doench et al., 2016). sgRNAs were cloned into LentiCRISPR V2 plasmid (Addgene plasmid #52961) as previously described(Ran et al., 2013), and validated by T7 endonuclease assay (T7 endonuclease I from NEB, Cat. #M0302). sgRNA sequences used were as follows: human MSH2 5’-TGAGAGGCTGCTTAATCCAC-3’; murine Msh2 5’-GGTTAATACCCT GATACAGT-3’. For the generation of lentiviral vectors, 293T/17 cell were transfected with LentiCRISPR V2, psPAX2 (Addgene plasmid #12260), and pMD2.G (Addgene plasmid #12259) in a ratio (4:3:1) using TransIT®-LT1Transfection reagent (MIR 2304, Mirus Bio) as described by manufacturer. Mouse SCLC and Ewing sarcoma cell lines were plated at 106 cells in a 10 cm dish and cultured overnight. The next day, cells were transduced with lentiviruses (MOI<0.5 determined by visual assessment) using 8ug/mL of polybrene transfection agent (TR-1003-G, EMD Millipore). Cells were selected with 2 mg/ml of Puromycin (P8833, Sigma-Aldrich) for 72 hours. Then surviving clones were picked and expanded for validation by western-blotting.
Western-blotting
Western-blotting for Ewing sarcoma and mouse SCLC protein samples was performed using standard methods. Odyssey Nitrocellulose membrane (LICOR, #926-31092) were used for protein transference and then blocked using Odyssey® Blocking Buffer in PBS (LICOR, #927-40000) for 1h at RT. Primary antibodies were incubated for 1h at RT diluted 1:1,000 in Odyssey® Blocking Buffer : PBS-Tween (0.1%). Antibodies used were anti-Msh2 [D24B5] XP rabbit mAb (#2017, Cell Signaling Technology), and anti-β-actin (8H10D10) mouse mAb (#3700, Cell Signaling Technologies). Membrane was washed with PBS-Tween (0.1%) three times for 5 minutes each wash. Secondary antibodies were incubated for 30 min at RT using IRDye 800CW donkey anti-rabbit IgG (H+L) (#926-32213, LI-COR), and IRDye 680RD donkey anti-mouse IgG (H+L) (#926-68072, LI-COR), at dilution 1:10,000. Visualization was performed with Odyssey CLx Imaging System (LI-COR).
Compounds
Bortezomib was purchased from Selleck Chemicals (#S1013). Etoposide was purchased from Sigma-Aldrich (#E1383-100MG). CD437 was purchased from Sigma-Aldrich (#C5865). MLN4924 was purchased from ApexBio (#B1036). Compounds were diluted using DMSO (Sigma-Aldrich, D650-100ML) and aliquoted at 10 mM and aliquots were exposed to a maximum of three freeze-thaw cycles.
Dose response curves
Mouse SCLC cell lines were plated in 96-well plates, 6,600 cells per well in 200 μL of media. Ewing sarcoma cell lines were plated in 96-well plates, 10,000 cells per well in 200 μL of media. After overnight incubation, compounds were dispensed using a D300e Digtal Dispenser (TECAN). Cell viability assay was assessed after 72 hours using CellTiter-Glo luminescent cell viability assay (Promega, #G7571). The CellTiter-Glo reagent was diluted by adding PBS-Triton-X (1%) (1:1 ratio). EC1001wk determination for bortezomib was performed in a 12-well plate seeding 25,000 cells per well. After 24h, Bortezomib was dispensed using TECAN D300e setting up a minimum concentration of EC5072h and a maximum concentration of EC10072h. Media was changed every 3-4 days to refresh compounds. Cell viability was determined visually following seven days.
Selection of resistant clones.
10 cm plates for each MMR deficient cell lines (1 million cells per plate) were treated with bortezomib or CD437 at EC1001wk ÷ 1.5, EC1001wk, and EC1001wk × 1.5 concentrations. Media with bortezomib or CD437 was replenished every 3 – 4 days over the course of 2 weeks. Surviving clones were expanded. Exon 2 of PSMB5 from both human and mouse cell lines was amplified and sequenced using primers specified in Table S3.
Amplification and sequence of exon 19 to 25 of POLA1 from both human and mouse cell lines was amplified and sequenced using primers specified in Table S3.
Crystal Violet experiment
10 cm plates for each MMR deficient cell lines (3 million cells per plate) were treated with botezomib, MLN4924, and CD437 at EC1001wk ÷ 1.5, EC1001wk ÷ 1.25, EC1001wk, and EC1001wk × 1.25, EC1001wk × 1.5 concentrations. Media with either bortezomib, MLN4924, or CD437 was replenished every 3 – 4 days over the course of 2 weeks followed by growth in media without compound for 1 week.
Staining solution was prepared with 1% (weight/volume ratio) crystal violet from Sigma-Aldrich (#C6158-50G) in 10% ethanol.
Whole Exome Sequencing Analysis
Whole-exome sequencing of cell line samples was performed by BGI Genomics using SureSelect Human All Exon V5 (Ewing sarcoma samples) and SureSelect Mouse All Exon V1 (mSCLC) and BGISEQ-500. The analysis workflow was based on Genome Analysis Toolkit (GATK, v3.8-0) best practices(McKenna et al., 2010, DePristo et al., 2011). The qualities of sequencing reads were evaluated using NGS QC Toolkit (v2.3.3)(Patel and Jain, 2012) and the extracted high-quality reads were mapped to human and mouse reference genome (UCSC hg19 and Ensembl 91) using Burrows-Wheeler Aligner (BWA, v0.7.15a)(Li and Durbin, 2009). Picard (v2.12.0) (https://broadinstitute.github.io/picard.) was used to remove PCR duplicates and GATK was used to recalibrate base qualities. For murine SCLC cell lines, calling variants and joint genotyping together were performed using HaplotypeCaller and the variant calls were filtered by applying the following criteria: QD (Variant Confidence/Quality by Depth) < 2, FS (Phred-scaled p-value using Fisher’s exact test to detect strand bias) > 60, MQ (RMS Mapping Quality) < 40, DP (Approximate read depth) < 10, GQ (Genotype Quality) < 20, maximum VAF (variant allele fraction) < 0.2. For each murine SCLC cell lines, the somatic mutations in 319-N1 were defined by the VAF > 0.15 and VAF < 0.05 for 319-T1 and 319-T2. Somatic mutations in 319-N1-Cl31 were defined by the VAF > 0.15 and VAF < 0.05 for the other cell lines. For human cell lines, MuTect2(Cibulskis et al., 2013) was used to identify somatic mutations in clones A673-M1 and A673-M8 comparing to the A673 parental cell line. Somatic mutations for each clones (A673-M1 and A673-M8) were defined by VAF > 0.15 and VAF < 0.05 for the other human cell lines.
Identification of recurrently mutated genes in MLN4924-resistant clones
We defined acquired somatic mutations for each A673-M1 MLN-resistant clone by VAF > 0.01 and VAF < 0.01 for the parental A673-M1 cell line. Non-coding mutations were excluded.
QUANTIFICATION ANS STATISTICAL ANALYSIS
Data were analyzed using Prism 8.0 by GraphPad. Dose response curves were fitting in Figure 2A, 2C, 3A, 3C and 4A, to calculate IC50. Hill coefficients and standard error were done using Log [inhibitor] vs Normalized response. Quantitative data are presented as mean.
DATA AND CODE AVAILABILITY
The variants were annotated using a custom Perl script (https://github.com/jiwoongbio/Annomen) with mouse transcripts, proteins, and variations (Ensembl 91 for mouse, RefSeq and dbSNP build 150 for human). The variant allele frequencies were calculated using a custom Perl script and SAMtools (v1.4) (Goncearenco et al., 2017, Li et al., 2009) (all analysis scripts are available at https://github.com/jiwoongbio/Annomen).
The sequencing datasets exposed in this study have been deposited in SRA under accession code PRJNA543281.
Supplementary Material
Table S1. WES_A673 Ewing_Related to Figures 1 and 4. This table shows whole exome sequencing data for the A673 cell lines described in this manuscript.
Tab 1. Raw data. Whole exome sequencing data for A673 cell line, A673 MMR-proficient and MMR-deficient cell lines, and MLN4924-resistant clones.
Tab 2. Somatic mutations in A673-Cl1 (A673 VAF < 0.05; A673-Cl1 > 0.15).
Tab 3. Somatic mutations in A673-Cl2 (A673 VAF < 0.05; A673-Cl2 > 0.15).
Tab 4. Somatic mutations in A673-Cl4 (A673 VAF < 0.05; A673-Cl4 > 0.15).
Tab 5. Somatic mutations in A673-M1 (A673 VAF < 0.05; A673-M1 > 0.15).
Tab 6. Somatic mutations in A673-M8 (A673 VAF < 0.05; A673-M8 > 0.15).
Table S2. WES_319 mSCLC_Related to Figures 1, S1 and S2. This table shows whole exome sequencing data for the murine small cell lung cancer cell lines described in this manuscript.
Tab 1. Raw data. Whole exome sequencing data for 319-T1, 319-T2, 319-N1, and MMR-deficient 319-N1-Cl31 cell line.
Tab 2. Somatic mutations in 319-N1 (319-T1 VAF < 0.05; 319-T2 VAF < 0.05; 319-N1 > 0.15).
Tab 3. Somatic mutations in 319-N1-Cl31 (319-T1 VAF < 0.05; 319-T2 VAF < 0.05; 319-N1-Cl31 > 0.15).
Table S3. Oligos PCR and sequencing_Related to STAR Methods. This table shows the oligos used for amplification and sequencing of exon 2 from PSMB5 and exons 19 to 25 from POLA1.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Msh2 [D24B5] XP rabbit mAb | Cell Signaling Technology | Cat#2017 |
| Anti-β-actin (8H10D10) mouse mAb | Cell Signaling Technologies | Cat#3700 |
| IRDye 800CW donkey anti-rabbit IgG (H+L) | LI-COR | Cat#926-32213 |
| IRDye 680RD donkey anti-mouse IgG (H+L) | LI-COR | Cat#926-68072 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Bortezomib | Selleck Chemicals | Cat#S1013; CAS ID: 179324-69-7 |
| Etoposide | Sigma-Aldrich | Cat#E1383; CAS ID: 33419-42-0 |
| CD437 | Sigma-Aldrich | Cat#C5865; CAS ID: 125316-60-1 |
| MLN4924 | ApexBio | Cat#B1036; CAS ID: 905579-51-3 |
| Critical Commercial Assays | ||
| CellTiter-Glo luminescent cell viability assay | Promega | Cat#G7571 |
| Deposited Data | ||
| WES from human and mouse cell lines | This paper | SRA ID: PRJNA543281 |
| Experimental Models: Cell Lines | ||
| Ewing sarcoma A673 cell lines | ATCC | ATCC® CRL-1598™ |
| Trp53fl/fl; Rb1fl/fl; Rbl2fl/fl; RosaLSL-Tomato/+ murine SCLC cell lines | This paper | N/A |
| 293T/17 | ATCC | ATCC® CRL-11268™ |
| Experimental Models: Organisms/Strains | ||
| Mouse: Trp53fl/fl Rbfl/fl P130fl/fl | Schaffer et al., 2010 | Mouse: Trp53fl/fl JAX: 008462 Mouse: Rbfl/fl JAX: 008186 Mouse: P130fl/fl JAX: 008177 |
| Oligonucleotides | ||
| Primers for PCR and sequencing of PSMB5 and POLA1, see Table S3 | This paper | N/A |
| Recombinant DNA | ||
| LentiCRISPR V2 plasmid | Addgene | Cat#52961 |
| psPAX2 plasmid | Addgene | Cat#12260 |
| pMD2.G plasmid | Addgene | Cat#12259 |
| Software and Algorithms | ||
| Genome Analysis Toolkit (GATK, v3.8-0) | DePristo et al., 2011; McKenna et al., 2010 | N/A |
| NGS QC Toolkit (v2.3.3) | Patel and Jain, 2012 | N/A |
| Burrows-Wheeler Aligner (BWA, v0.7.15a) | Li and Durbin, 2009 | N/A |
| Picard (v2.12.0) | N/A | https://broadinstitute.github.io/picard |
| HaplotypeCaller | N/A | N/A |
| MuTect2 | Cibulskis et al., 2013 | N/A |
| Annomen | N/A | https://github.com/jiwoongbio/Annomen |
| SAMtools | (Li et al., 2009) | N/A |
| Other | ||
HIGHLIGHTS.
MSH2 deletion induces hypermutation in mammalian cancer cell lines.
Compound-resistant clones emerged in DNA mismatch repair deficient cell lines.
This approach expands forward genetic screening in mammalian cancer cell lines.
Significance.
Phenotypic small molecule screens can identify chemicals that selectively kill subsets of cancers. In theory, these molecules target a protein uniquely required for viability by the cancer cells. This selective toxicity is the hallmark feature of molecular targeted therapies in cancer. However, identifying the mechanism of action of chemicals identified in phenotypic screens has remained a major barrier for drug discovery and development. Cancer cells harboring defective DNA mismatch repair have been used to identify targets of selective cancer toxins. However, naturally-arising DNA mismatch repair defects are infrequent in many types of cancer. Here, we demonstrate that CRISPR-Cas9-mediated mutation of MSH2 is sufficient to induce hypermutation and enable forward genetic screening for drug targets in mammalian cells. We propose that this approach offers the potential to expand the repertoire of cell lines and small molecules suitable for forward genetics screening.
Acknowledgements
The authors thank Florentina Moreno for technical support and Anderson Frank, James McGinnis and Alberto Bremauntz Enriquez for reviewing the manuscript. LentiCRISPR v2 was a gift from Feng Zhang (Addgene plasmid #52961). psPAX2 and pMD2.G were a gift from Didier Trono (Addgene plasmids #12260 and #12259, respectively). J.K. and Y.X. were supported by the Cancer Prevention and Research Institute of Texas (RP150596). D.G.M. was supported by a St. Baldrick’s Foundation Scholar Award (524523), the Cancer Prevention and Research Institute of Texas (RR140084) and a Disease-Oriented Scholar Award from UT Southwestern Medical Center. D.N. is supported by Welch Foundation I-1879-20180324, NIH R37CA226771-02, NIH RO1CA217333-02.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest
The authors declare no competing interests.
REFERENCES
- BACHER JW, ABDEL MEGID WM, KENT-FIRST MG & HALBERG RB 2005. Use of mononucleotide repeat markers for detection of microsatellite instability in mouse tumors. Mol Carcinog, 44, 285–92. [DOI] [PubMed] [Google Scholar]
- BOLAND CR, THIBODEAU SN, HAMILTON SR, SIDRANSKY D, ESHLEMAN JR, BURT RW, MELTZER SJ, RODRIGUEZ-BIGAS MA, FODDE R, RANZANI GN & SRIVASTAVA S 1998. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res, 58, 5248–57. [PubMed] [Google Scholar]
- BROHL AS, SOLOMON DA, CHANG W, WANG J, SONG Y, SINDIRI S, PATIDAR R, HURD L, CHEN L, SHERN JF, LIAO H, WEN X, GERARD J, KIM JS, LOPEZ GUERRERO JA, MACHADO I, WAI DH, PICCI P, TRICHE T, HORVAI AE, MIETTINEN M, WEI JS, CATCHPOOL D, LLOMBART-BOSCH A, WALDMAN T & KHAN J 2014. The genomic landscape of the Ewing Sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet, 10, e1004475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN D, FREZZA M, SCHMITT S, KANWAR J & DOU QP 2011. Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr Cancer Drug Targets, 11, 239–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CIBULSKIS K, LAWRENCE MS, CARTER SL, SIVACHENKO A, JAFFE D, SOUGNEZ C, GABRIEL S, MEYERSON M, LANDER ES & GETZ G 2013. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol, 31, 213–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COLOMA J, JOHNSON RE, PRAKASH L, PRAKASH S & AGGARWAL AK 2016. Human DNA polymerase alpha in binary complex with a DNA:DNA template-primer. Sci Rep, 6, 23784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CROMPTON BD, STEWART C, TAYLOR-WEINER A, ALEXE G, KUREK KC, CALICCHIO ML, KIEZUN A, CARTER SL, SHUKLA SA, MEHTA SS, THORNER AR, DE TORRES C, LAVARINO C, SUNOL M, MCKENNA A, SIVACHENKO A, CIBULSKIS K, LAWRENCE MS, STOJANOV P, ROSENBERG M, AMBROGIO L, AUCLAIR D, SEEPO S, BLUMENSTIEL B, DEFELICE M, IMAZ-ROSSHANDLER I, SCHWARZ-CRUZ YCA, RIVERA MN, RODRIGUEZ-GALINDO C, FLEMING MD, GOLUB TR, GETZ G, MORA J & STEGMAIER K 2014. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov, 4, 1326–41. [DOI] [PubMed] [Google Scholar]
- DEPRISTO MA, BANKS E, POPLIN R, GARIMELLA KV, MAGUIRE JR, HARTL C, PHILIPPAKIS AA, DEL ANGEL G, RIVAS MA, HANNA M, MCKENNA A, FENNELL TJ, KERNYTSKY AM, SIVACHENKO AY, CIBULSKIS K, GABRIEL SB, ALTSHULER D & DALY MJ 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet, 43, 491–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOENCH JG, FUSI N, SULLENDER M, HEGDE M, VAIMBERG EW, DONOVAN KF, SMITH I, TOTHOVA Z, WILEN C, ORCHARD R, VIRGIN HW, LISTGARTEN J & ROOT DE 2016. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol, 34, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GLAAB WE & TINDALL KR 1997. Mutation rate at the hprt locus in human cancer cell lines with specific mismatch repair-gene defects. Carcinogenesis, 18, 1–8. [DOI] [PubMed] [Google Scholar]
- GONCEARENCO A, RAGER SL, LI M, SANG QX, ROGOZIN IB & PANCHENKO AR 2017. Exploring background mutational processes to decipher cancer genetic heterogeneity. Nucleic Acids Res, 45, W514–W522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAN T, GORALSKI M, CAPOTA E, PADRICK SB, KIM J, XIE Y & NIJHAWAN D 2016. The antitumor toxin CD437 is a direct inhibitor of DNA polymerase alpha. Nat Chem Biol, 12, 511–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAN T, GORALSKI M, GASKILL N, CAPOTA E, KIM J, TING TC, XIE Y, WILLIAMS NS & NIJHAWAN D 2017. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science, 356. [DOI] [PubMed] [Google Scholar]
- HUBER EM, HEINEMEYER W, DE BRUIN G, OVERKLEEFT HS & GROLL M 2016. A humanized yeast proteasome identifies unique binding modes of inhibitors for the immunosubunit beta5i. EMBO J, 35, 2602–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI H & DURBIN R 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 25, 1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI H, HANDSAKER B, WYSOKER A, FENNELL T, RUAN J, HOMER N, MARTH G, ABECASIS G, DURBIN R & GENOME PROJECT DATA PROCESSING S 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25, 2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LU S & WANG J 2013. The resistance mechanisms of proteasome inhibitor bortezomib. Biomark Res, 1, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCFADDEN DG, PAPAGIANNAKOPOULOS T, TAYLOR-WEINER A, STEWART C, CARTER SL, CIBULSKIS K, BHUTKAR A, MCKENNA A, DOOLEY A, VERNON A, SOUGNEZ C, MALSTROM S, HEIMANN M, PARK J, CHEN F, FARAGO AF, DAYTON T, SHEFLER E, GABRIEL S, GETZ G & JACKS T 2014. Genetic and clonal dissection of murine small cell lung carcinoma progression by genome sequencing. Cell, 156, 1298–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCFADDEN DG, POLITI K, BHUTKAR A, CHEN FK, SONG X, PIRUN M, SANTIAGO PM, KIM-KISELAK C, PLATT JT, LEE E, HODGES E, ROSEBROCK AP, BRONSON RT, SOCCI ND, HANNON GJ, JACKS T & VARMUS H 2016. Mutational landscape of EGFR-, MYC-, and Kras-driven genetically engineered mouse models of lung adenocarcinoma. Proc Natl Acad Sci U S A, 113, E6409–E6417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCKENNA A, HANNA M, BANKS E, SIVACHENKO A, CIBULSKIS K, KERNYTSKY A, GARIMELLA K, ALTSHULER D, GABRIEL S, DALY M & DEPRISTO MA 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res, 20, 1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MICHAEL, MICHAEL, NARAYANAN U, TRAORE T, RICEBERG J, BENJAMIN, NEIL, JOSEPH, BROWNELL J, LAWRENCE, LOKE H-K, ALICE, MA J, MARK, TODD, MIKE, YANG X, XU Q, ERIK, JAMES & PETER 2012. Treatment-Emergent Mutations in NAEP Confer Resistance to the NEDD8-Activating Enzyme Inhibitor MLN4924. 21, 388–401. [DOI] [PubMed] [Google Scholar]
- PATEL RK & JAIN M 2012. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One, 7, e30619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PISHAS KI & LESSNICK SL 2016. Recent advances in targeted therapy for Ewing sarcoma. F1000Res, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAN FA, HSU PD, WRIGHT J, AGARWALA V, SCOTT DA & ZHANG F 2013. Genome engineering using the CRISPR-Cas9 system. Nat Protoc, 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHAFFER BE, PARK KS, YIU G, CONKLIN JF, LIN C, BURKHART DL, KARNEZIS AN, SWEET-CORDERO EA & SAGE J 2010. Loss of p130 accelerates tumor development in a mouse model for human small-cell lung carcinoma. Cancer Res, 70, 3877–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SOUCY TA, SMITH PG, MILHOLLEN MA, BERGER AJ, GAVIN JM, ADHIKARI S, BROWNELL JE, BURKE KE, CARDIN DP, CRITCHLEY S, CULLIS CA, DOUCETTE A, GARNSEY JJ, GAULIN JL, GERSHMAN RE, LUBLINSKY AR, MCDONALD A, MIZUTANI H, NARAYANAN U, OLHAVA EJ, PELUSO S, REZAEI M, SINTCHAK MD, TALREJA T, THOMAS MP, TRAORE T, VYSKOCIL S, WEATHERHEAD GS, YU J, ZHANG J, DICK LR, CLAIBORNE CF, ROLFE M, BOLEN JB & LANGSTON SP 2009. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature, 458, 732–6. [DOI] [PubMed] [Google Scholar]
- TIRODE F, SURDEZ D, MA X, PARKER M, LE DELEY MC, BAHRAMI A, ZHANG Z, LAPOUBLE E, GROSSETETE-LALAMI S, RUSCH M, REYNAUD S, RIO-FRIO T, HEDLUND E, WU G, CHEN X, PIERRON G, OBERLIN O, ZAIDI S, LEMMON G, GUPTA P, VADODARIA B, EASTON J, GUT M, DING L, MARDIS ER, WILSON RK, SHURTLEFF S, LAURENCE V, MICHON J, MAREC-BERARD P, GUT I, DOWNING J, DYER M, ZHANG J, DELATTRE O, ST. JUDE CHILDREN’S RESEARCH HOSPITAL-WASHINGTON UNIVERSITY PEDIATRIC CANCER GENOME, P. & THE INTERNATIONAL CANCER GENOME, C. 2014. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov, 4, 1342–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WACKER SA, HOUGHTALING BR, ELEMENTO O & KAPOOR TM 2012. Using transcriptome sequencing to identify mechanisms of drug action and resistance. Nat Chem Biol, 8, 235–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XU GW, TOTH JI, DA SILVA SR, PAIVA S-L, LUKKARILA JL, HURREN R, MACLEAN N, SUKHAI MA, BHATTACHARJEE RN, GOARD CA, GUNNING PT, DHE-PAGANON S, PETROSKI MD & SCHIMMER AD 2014. Mutations in UBA3 Confer Resistance to the NEDD8-Activating Enzyme Inhibitor MLN4924 in Human Leukemic Cells. 9, e93530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YU H, GE Y, GUO L & HUANG L 2017. Potential approaches to the treatment of Ewing’s sarcoma. Oncotarget, 8, 5523–5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. WES_A673 Ewing_Related to Figures 1 and 4. This table shows whole exome sequencing data for the A673 cell lines described in this manuscript.
Tab 1. Raw data. Whole exome sequencing data for A673 cell line, A673 MMR-proficient and MMR-deficient cell lines, and MLN4924-resistant clones.
Tab 2. Somatic mutations in A673-Cl1 (A673 VAF < 0.05; A673-Cl1 > 0.15).
Tab 3. Somatic mutations in A673-Cl2 (A673 VAF < 0.05; A673-Cl2 > 0.15).
Tab 4. Somatic mutations in A673-Cl4 (A673 VAF < 0.05; A673-Cl4 > 0.15).
Tab 5. Somatic mutations in A673-M1 (A673 VAF < 0.05; A673-M1 > 0.15).
Tab 6. Somatic mutations in A673-M8 (A673 VAF < 0.05; A673-M8 > 0.15).
Table S2. WES_319 mSCLC_Related to Figures 1, S1 and S2. This table shows whole exome sequencing data for the murine small cell lung cancer cell lines described in this manuscript.
Tab 1. Raw data. Whole exome sequencing data for 319-T1, 319-T2, 319-N1, and MMR-deficient 319-N1-Cl31 cell line.
Tab 2. Somatic mutations in 319-N1 (319-T1 VAF < 0.05; 319-T2 VAF < 0.05; 319-N1 > 0.15).
Tab 3. Somatic mutations in 319-N1-Cl31 (319-T1 VAF < 0.05; 319-T2 VAF < 0.05; 319-N1-Cl31 > 0.15).
Table S3. Oligos PCR and sequencing_Related to STAR Methods. This table shows the oligos used for amplification and sequencing of exon 2 from PSMB5 and exons 19 to 25 from POLA1.
Data Availability Statement
The variants were annotated using a custom Perl script (https://github.com/jiwoongbio/Annomen) with mouse transcripts, proteins, and variations (Ensembl 91 for mouse, RefSeq and dbSNP build 150 for human). The variant allele frequencies were calculated using a custom Perl script and SAMtools (v1.4) (Goncearenco et al., 2017, Li et al., 2009) (all analysis scripts are available at https://github.com/jiwoongbio/Annomen).
The sequencing datasets exposed in this study have been deposited in SRA under accession code PRJNA543281.