

ABSTRACT
Growth Hormone-Releasing Hormone (GHRH) regulates the release of growth hormone from the anterior pituitary gland. GHRH also acts as a growth and inflammatory factor in a variety of experimental models in oncology. In the current study, we used bovine pulmonary arterial cells in order to investigate the effects of GHRH and its antagonistic and agonistic analogs in key intracellular pathways that regulate endothelial permeability. GHRH antagonists suppressed the activation of MLC2, ERK1/2, JAK2/STAT3 pathway and increased the intracellular P53 and pAMPK levels. In contrast, both GHRH and GHRH agonist MR409 exerted the opposite effects. Furthermore, GHRH antagonists supported the integrity of endothelial barrier, while GHRH and GHRH agonists had the contrary effects, as reflected in measurements of transendothelial resistance. Our observations support the evidence for the anti – inflammatory role of GHRH antagonists in the vasculature. Moreover, our results suggest that GHRH antagonists should be considered as promising therapeutic agents for treating severe respiratory abnormalities, such as the lethal Acute Respiratory Distress Syndrome (ARDS).
KEYWORDS: Inflammation, endothelial function, P53
Introduction
Growth Hormone – Releasing Hormone [GHRH (1–44) NH2] is a hypothalamic hormone which consists of 44 amino acids. The biological activity of GHRH is retained in the first 29 amino acids [GHRH (1–29) NH2].1,2 Antagonists of GHRH exert powerful anticancer and anti – inflammatory activities.3 Those compounds could provide therapeutic approaches toward malignancies, since they “target” tumor cells and reduce their growth under experimental conditions “in vivo” and “in vitro”.4
GHRH antagonists suppress the most common and lethal human malignancies, such as prostate, breast, ovarian, renal, and lung cancers.3 Those protective effects are in part due to the inhibition of major inflammatory pathways, such as the ERK1/25 and JAK2/STAT3 signaling cascades.6 Moreover, it has been previously shown that GHRH antagonists induce the expression of the anti-inflammatory transcription factor P53.7 The P53, “Guardian of the Genome”, which is a negative regulator of the inflammatory transcription factor NFKB, has been shown to support the endothelium against inflammation.7 Thus, the anti-inflammatory activity of GHRH antagonists; is partially exerted due to their capacity to increase the levels of P53 in the intracellular niche. Indeed, the protective effects of GHRH antagonists are not limited to cancers. Those compounds have been shown to counteract the development of Benign Prostate Hyperplasia (BPH) through the suppression PCNA, as well as by opposing the stimulatory activity of IGF I and II, and FGF-II in the BPH I cell line.8
A severe consequence of pulmonary inflammation is the development of ARDS, which represents an important threat to human health. Described over 50 years ago, ARDS remains a manifestation of the corrupted lung homeostasis, destined to cause non-hydrostatic pulmonary edema, respiratory abnormalities and death.9 ARDS appears in 10 to 86 cases per 100,000 patients, with the highest rates reported in Australia and the United States. The development of ARDS is due to direct lung injury (pneumonia and gastric aspiration); or indirect injury (sepsis and pancreatitis), which in turn result in inflammation and hypoxemia. The disruption of the alveolar-capillary membrane produces an influx of protein-rich edema fluid, pulmonary dysfunction and endothelial hyperpermeability.10 ARDS outcomes are similar to those found in survivors of critical illnesses and largely impair the physical, social, emotional and neurocognitive functions of those unfortunate individuals.11
It has been recently demonstrated that P53 is an integral component of the lung endothelium, and that this protein supports the function of the lung microvasculature. P53 has been demonstrated to protect the endothelium against the inflammatory activities of LPS, through the suppression of the inflammatory RhoA/MLC2 pathway.12 Moreover, P53 induced the activation of the Rac1 GTPase, which in turn blocks the actin-severing activity of cofilin.13 Furthermore, latest observations suggest that P53 partially mediates the anti – inflammatory effects of Hsp90 inhibitors,14 and supports the endothelial function by suppressing the expression of the Apurinic/apyrimidinic endonuclease 1 (APE1)/Redox effector factor 1 (Ref1).15
APE1/Ref1 is a multifunctional enzyme involved in the base excision repair (BER) pathway. That pathway repairs oxidative base damage inflicted by environmental agents.16 APE1/Ref1 acts as a regulator of many signaling cascades, and affects cellular homeostasis via the activation of several transcription factors (activator protein-1, nuclear factor kappa B, hypoxia-inducible factor 1α, signal transducer activator of transcription 3). Those transcription factors regulate a variety of crucial physiological processes, such as apoptosis, inflammation, and angiogenesis.17 APE1/Ref1 is also involved in the regulation of redox balancing agents (thioredoxin, catalase and superoxide dismutase); which are enzymes in charge of the maintenance of the reactive oxygen and nitrogen species..17
An earlier GHRH antagonist JMR-132 has been previously shown to upregulate P53.3 In the present study we investigate the possibility that the latest generation of GHRH antagonists (MIA series) support the endothelial barrier function of Bovine Pulmonary Aortic Endothelial Cells (BPAECs). First, we demonstrated the GHRH-R receptor in the BPAECs. Then, we treated the cells with GHRH (1–29) NH2, GHRH antagonist MIA-602, as well as the GHRH agonist MR-409. Our results demonstrate that MIA-602 induces the expression of P53 and pAMPK, inhibits the actin-severing activity of cofilin, and suppresses the activation of the inflammatory pathways JAK2/STAT3, ERK1/2. Moreover, it suppressed the formation of F- actin, as reflected by the downregulation of the pMLC2. On the other hand, GHRH and GHRH agonist MR-409 exerted the opposite effects, suggesting that both compounds (GHRH (1–29) NH2, MR-409) trigger inflammatory responses in the endothelium.
To further substantiate our findings, we proceeded with Real – Time measurements of endothelial permeability, by employing the Electric Cell Substrate Impedance Sensing assay (ECIS). Our observations reveal that MIA-602 supports the endothelial barrier function of the bovine lung endothelial cells, since it enhances the transendothelial resistance (TEER) of the BPAEC monolayers. Moreover, this GHRH antagonist protects against the LPS-induced hyperpermeability, by suppressing the LPS-induced MLC2 and cofilin activation. In bold contract, GHRH and MR-409 increase the endothelial permeability of those cells, as reflected in the reduced TEER values. Collectively, our studies suggest that GHRH antagonists may represent an exciting new approach for the treatment of ALI/ARDS. Those compounds induce endothelial barrier enhancers (P53, pAMPK),10 suppress major inflammatory pathways (ERK1/2, JAK2/STAT3), and support the barrier function of BPAECs.
Results
Bovine pulmonary arterial endothelial cells (BPAECs) express GHRH receptor
BPAECs, HeLa cervical cancer cells, MCF7 breast cancer cells, and NIH/3T3 mouse embryonic fibroblast cells were subjected to Western blotting. The results shown in Figure 1(a) demonstrate the expression of GHRH receptor in BPAECs. HeLa, MCF7 and 3T3 cell lines were used as negative controls.5,18,19
Figure 1.
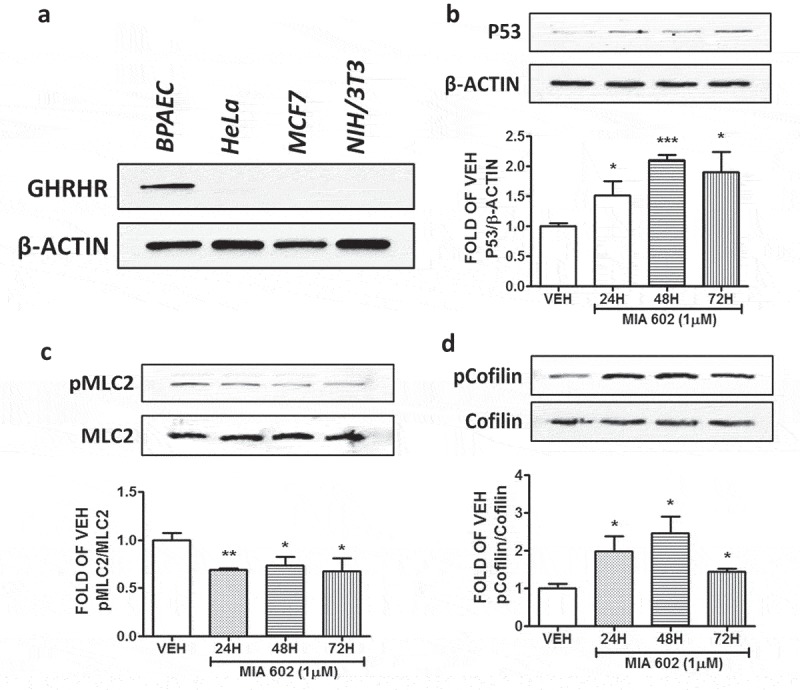
Detection of the GHRH receptor, and the effects of MIA 602 in the expression levels of P53, pMLC2, and pCofilin in BPAEC.
(a) Western Blot analysis of GHRH receptors and β-actin in BPAEC, HeLa cervical cancer cells, MCF7 breast cancer cells, and NIH/3T3 mouse embryonic fibroblast cells. B-actin was used as a loading control. Western Blot analysis of (b) P53 and β-actin, (c) phosphorylated MLC2 (pMLC2) and MLC2, (d) phosphorylated Cofilin (pCofilin) and Cofilin after treatment of BPAEC with either vehicle (0.1% DMSO) or GHRH antagonist MIA 602 (1μM) for 24, 48 and 72 hours. The blots shown are representative of 3 independent experiments. The signal intensity of the protein bands was analyzed by densitometry. Protein levels of P53, pMLC2 and pCofilin were normalized to β-actin, MLC2 and Cofilin respectively. *P < .05, **P < .01, ***P < .001 vs. vehicle. Means ± SEM.
MIA 602 induces P53 and suppresses the activation of MLC2 in BPAEC
BPAEC were treated with either vehicle (0.1% DMSO) or 1 μM MIA 602 for 24, 48 and 72 hours. This GHRH antagonist significantly induced p53 (Figure 1(b)); and suppressed pMLC2 expression levels (Figure 1(c)) in all treatments.
MIA 602 induces cofilin deactivation and suppresses JAK2 activation in BPAEC
BPAEC were treated with either vehicle (0.1% DMSO) or 1 μM MIA 602 for 24, 48 and 72 hours. Figure 1(d) shows that this compound significantly induced pCofilin expression levels in all treatments. Moreover, MIA 602 significantly reduced the phosphorylation of JAK2 in all experiments (Figure 2(a)).
Figure 2.
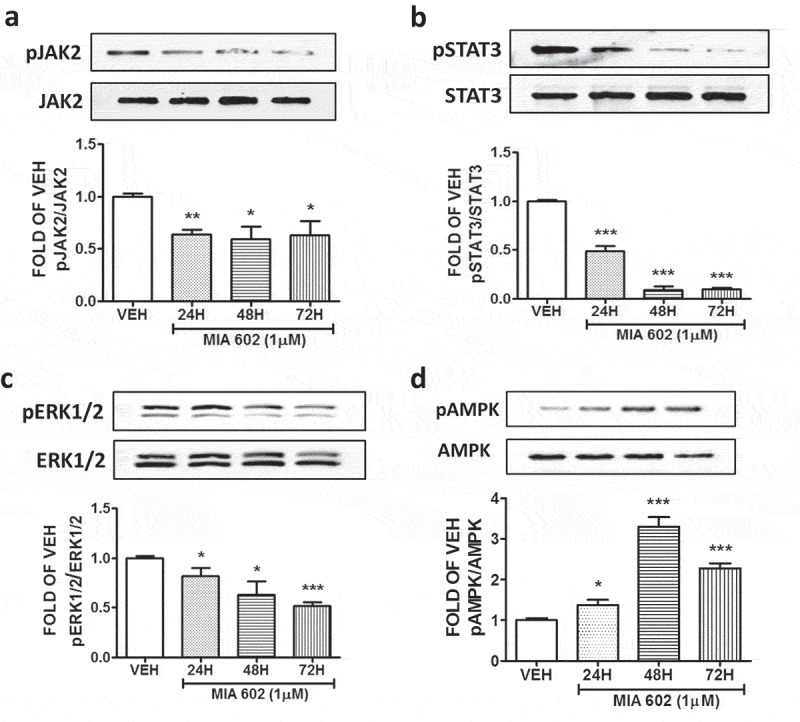
GHRH antagonist suppresses the activation of JAK2, STAT3, ERK1/2, and induces the activation of AMPK in BPAECs.
Western Blot analysis of (a) Phosphorylated JAK2 (pJAK2) and JAK2, (b) Phosphorylated STAT3 (pSTAT3) and STAT3, (c) Phosphorylated ERK1/2 (pERK1/2) and ERK1/2, (d) Phosphorylated AMPK and AMPK after treatment of BPAEC with either vehicle (0.1% DMSO) or GHRH antagonist MIA 602 (1μM) for 24, 48 and 72 hours. The blots shown are representative of 3 independent experiments. The signal intensity of the protein bands was analyzed by densitometry. Protein levels of pJAK2, pSTAT3, pERK1/2 and pAMPK were normalized to JAK2, STAT3, ERK1/2 and AMPK respectively. *P < .05, **P < .01, ***P < .001 vs vehicle. Means ± SEM.
MIA 602 suppresses the activation of STAT3 and ERK1/2, and induces AMPK phosphorylation
BPAEC were treated with vehicle (0.1% DMSO) or the GHRH antagonist MIA 602 for 24, 48 and 72 hours. This compound significantly suppressed the expression levels of pSTAT3 in all tests. The greatest suppression occurred after exposure to MIA 602 for 48 and 72 hours (Figure 2(b)). MIA 602 suppressed the phosphorylation of ERK1/2, with the greatest inhibition occurred after 72 hours of treatment (Figure 2(c)). Furthermore, this antagonist induced the phosphorylated form of AMPK (Figure 2(d)).
GHRH suppresses the expression levels of P53
BPAEC were treated with vehicle (0.1% DMSO), or 1 μΜ GHRH for 24, 48 and 72 hours. The results depicted in Figure 3(a) show that exposure of those lung cells to GHRH results in the reduction of the P53 expression levels.
Figure 3.
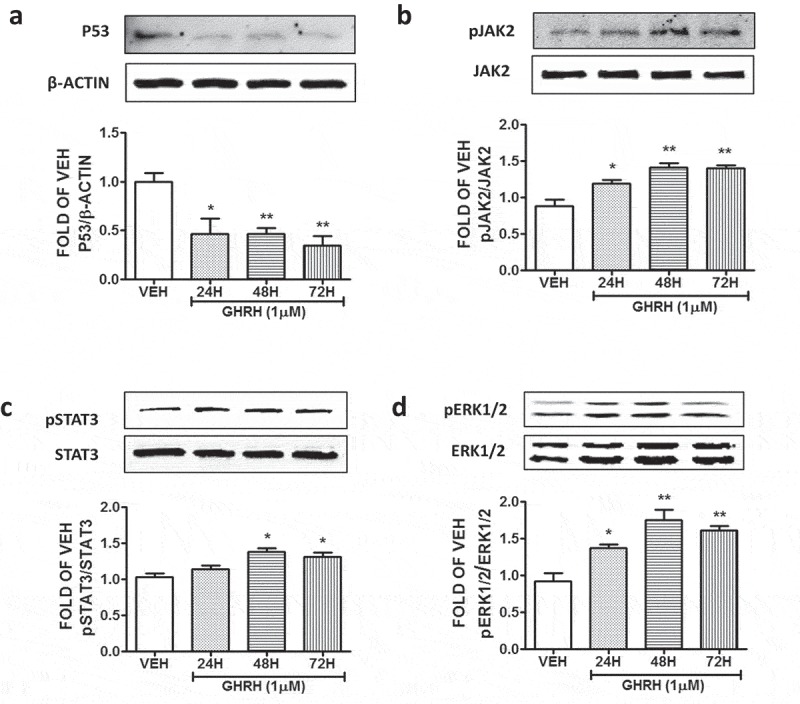
Effects of GHRH in the P53, pJAK2, pSTAT3, pERK1/2 expression levels of BPAEC.
Western Blot analysis of (a) P53 and β-actin, (b) Phosphorylated JAK2 (pJAK2) and JAK2, (c) Phosphorylated STAT3 (pSTAT3) and STAT3, (d) Phosphorylated ERK1/2 (pERK1/2) and ERK1/2, after treatment of BPAEC with either vehicle (0.1% DMSO) or GHRH (1μM) for 24, 48 and 72 hours. The blots shown are representative of 3 independent experiments. The signal intensity of the protein bands was analyzed by densitometry. Protein levels of P53, pJAK2, pSTAT3, and pERK1/2 were normalized to β-actin, JAK2, STAT3 and ERK1/2 respectively. *P < .05, **P < .01 vs. vehicle. Means ± SEM.
GHRH induces the activation of the JAK2, STAT3 and ERK1/2 pathways
The endothelial cells were treated with 1 μΜ GHRH for 24, 48 and 72 hours. The expression of pJAK2 and JAK2 was measured by Western Blot. This neuropeptide was able to activate this inflammatory pathway (Figure 3(b)). Moreover, GHRH induced the pSTAT3 (Figure 3(c)), as well as the pERK1/2 expression levels (Figure 3(d)). Our observations suggest that this growth factor (GHRH) activates all those three major inflammatory pathways.
MR 409 suppresses P53 in BPAEC
BPAEC were treated with either vehicle (0.1% DMSO) or 1 μM of the GHRH agonist MR 409 for 24, 48 and 72 hours. This GHRH agonist significantly suppressed p53 expression levels after 48 and 72 hours of treatment (Figure 4(a)).
Figure 4.
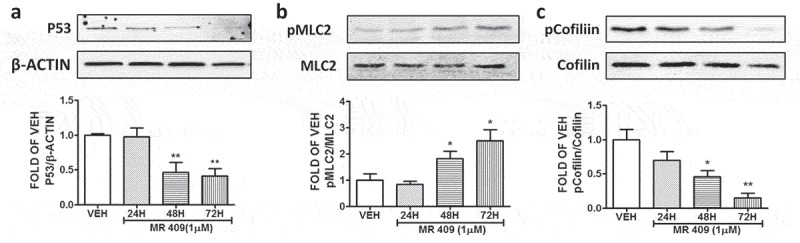
Effects of GHRH agonist MR 409 in the P53, pMLC2, and pCofilin expression levels of BPAEC.
Western Blot analysis of (a) P53 and β-actin, (b) Phosphorylated MLC2 (pMLC2) and MLC2, (c) Phosphorylated Cofilin (pCofilin) and Cofilin after treatment of BPAEC with either vehicle (0.1% DMSO) or GHRH agonist MR 409 (1μM) for 24, 48 and 72 hours. The blots shown are representative of 3 independent experiments. The signal intensity of the protein bands was analyzed by densitometry. Protein levels of P53, pMLC2 and pCofilin were normalized to β-actin, MLC2 and Cofilin respectively. *P < .05, **P < .01 vs. vehicle. Means ± SEM.
MR 409 induces MLC2 activation and activates cofilin in BPAEC
The endothelial cells were treated with MR 409 or vehicle (0.1% DMSO) for 24, 48 and 72 hours. That peptide induced pMLC2 (Figure 4(b)); and decreased pCofilin expression in BPAEC after 48 and 72 hours of treatment (Figure 4(c)).
MR 409 induces the activation of JAK2, STAT3 and ERK1/2
The endothelial cells were subjected to treatment with either vehicle (0.1% DMSO), or 1 μΜ MR 409 for 24, 48 and 72 hours. Our observations indicated MR 409 significantly increased the phosphorylation of JAK2 (Figure 5(a)) and STAT3 (Figure 5(b)) after 48 and 72 hours of exposure. Furthermore, treatment of those cells with DMSO or the GHRH agonist MR 409 (1 μΜ) for 24, 48 and 72 hours, resulted to an induction of the phosphorylated ERK1/2 (Figure 5(c)).
Figure 5.
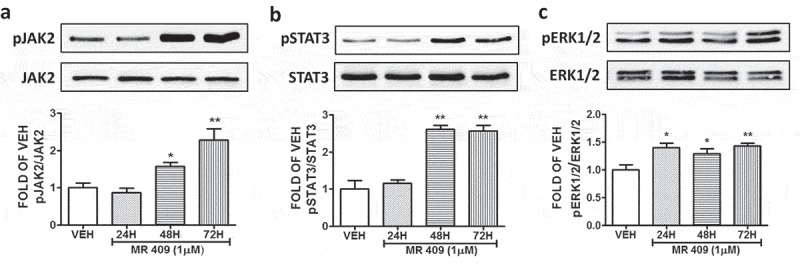
Effects of GHRH agonist MR 409 in the expression levels of pJAK2, pSTAT3, pERK1/2 in BPAEC.
Western Blot analysis of (a) Phosphorylated JAK2 (pJAK2) and JAK2, (b) phosphorylated STAT3 (pSTAT3) and STAT3, (c) Phosphorylated ERK1/2 (pERK1/2) and ERK1/2 after treatment of BPAEC with either vehicle (0.1% DMSO) or the GHRH agonist MR 409 (1μM) for 24, 48 and 72 hours. The blots shown are representative of 3 independent experiments. The signal intensity of the protein bands was analyzed by densitometry. Protein levels of pJAK2, pSTAT3, and pERK1/2 were normalized to JAK2, STAT3 and ERK1/2 respectively. *P < .05, **P < .01 vs vehicle. Means ± SEM.
Effects of GHRH, GHRH agonist MR 409 and GHRH antagonist MIA 602 on the endothelial permeability of BPAEC
Confluent monolayers of BPAEC were treated with either vehicle (0.1% DMSO), 1 μΜ GHRH or 1 μΜ GHRH agonist MR 409 for 36 hours. Both GHRH and MR 409 increased the permeability (lower TEER values) of the endothelial monolayers after the aforementioned period (Figure 6(a)). Furthermore, confluent monolayers of BPAECs were treated with either vehicle (DMSO), or 1 μΜ of the GHRH antagonist MIA 602. Cells that were exposed to MIA 602 exhibited a significant induction in TEER values (decreased permeability), compared to the DMSO – treated cells (Figure 6(b)). Moreover, this GHRH antagonist protected the cells against the LPS-triggered hyperpermeability. MIA 602 suppressed the LPS-induced MLC2 phosphorylation (Figure 6(c)), as well as the LPS-triggered cofilin dephosphorylation (activation) (Figure 6(d)). In all cases DMSO did not affect the endothelial permeability of BPAEC.
Figure 6.
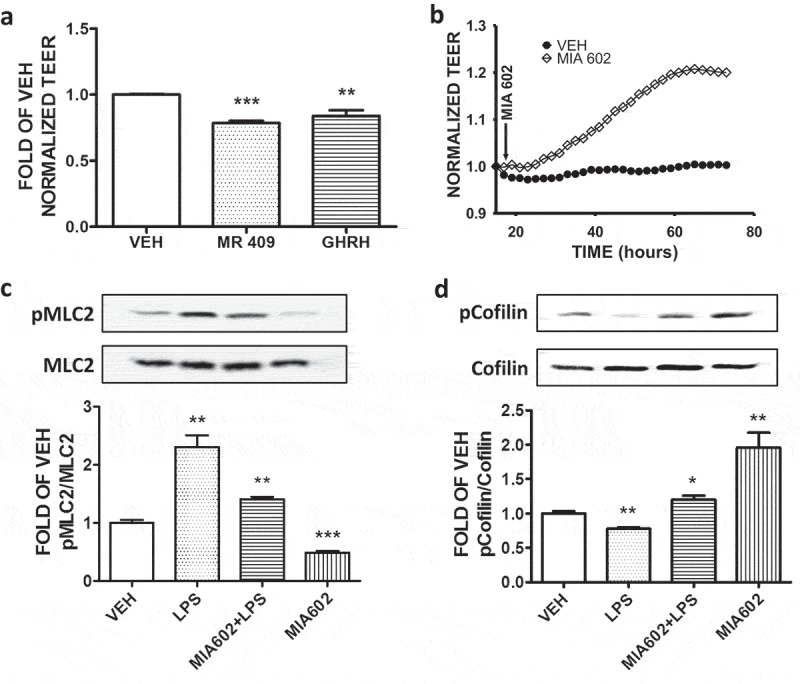
Effects of GHRH antagonist MIA 602, GHRH and GHRH agonist MR 409 in the endothelial permeability of BPAEC.
(a) BPAEC were treated with either vehicle (0.1% DMSO), GHRH (1μΜ) or 1 μΜ of GHRH agonist MR 409 for 36 hours. Both GHRH- and MR 409 – treated groups exerted increased permeability (decreased TEER values) compared to the vehicle – treated group. n = 3 per group, **P < .01, ***P < .001 vs vehicle. Means ± SEM. (b) Confluent monolayers of BPAEC were treated with either vehicle (DMSO) or the GHRH antagonist MIA 602 (1µM). A gradual increase of the TEER values (decreased permeability) was observed in that group that was treated with MIA 602 compared to the vehicle – treated group. n = 3 per group. Means ± S.E. The arrow indicate the addition of MIA 602 and DMSO in the confluent monolayers. Western Blot analysis of (c) phosphorylated MLC2 (pMLC2) and MLC2, (D) phosphorylated Cofilin (pCofilin) and Cofilin. BPAEC were treated for 48 hours with either vehicle (DMSO) or GHRH antagonist MIA 602 (1μM) prior to vehicle (PBS) or LPS (10μg/ml) exposure. The blots shown are representative of 3 independent experiments. The signal intensity of the protein bands was analyzed by densitometry. Protein levels of pMLC2 and pCofilin were normalized to MLC2 and Cofilin respectively. *P < .05, **P < .01, ***P < .001 vs. vehicle. Means ± SEM.
Discussion
Growth Hormone-Releasing Hormone (GHRH) is secreted by the hypothalamus, and regulates the release of Growth Hormone (GH) from the anterior pituitary gland. In addition to its endocrine role, this peptide has been shown to act as a growth factor in cancers. Remarkably, the inhibition of GHRH production by siRNA means suppressed the proliferation rate of MDA-MB-435s, MDA-MB-468, T47D, NCI H838, MCF7 and LNCAP cancer cells.20 Moreover, It has been previously shown that the splice variant 1 (SV1) of the pituitary type GHRH receptor, exerts ligand-independent activities in in vitro models of experimental human cancers.19,21
An emerging body of evidence suggests that GHRH regulates several important physiological processes, including inflammation. GHRH-R mediates cytokine production in ciliary and iris epithelial cells during LPS-induced ocular inflammation. GHRH receptor is upregulated in the ciliary and iris epithelial cells, as well as in the aqueous humor in a rat model of acute anterior uveitis. In explant cultures of rat ciliary body and iris, LPS caused a substantial increase in levels of GHRH-R in 24 h. Further investigations revealed an elevated expression of IL-6 and IL-1β in ciliary and iris epithelial cells after LPS treatment. That toxin also elevates the levels of IL-1β, IL-6, IL-1β, IL-6, and iNOS from the explants. MIA 602 suppresses the elevated expression of IL-1β and IL-6, and reduces the release of IL-6.22 It has been also previously reported that GHRH induces iNOS expression in vitro.23
GHRH antagonists can similarly suppress inflammation – induced prostatic enlargement. This activity was consistent with a decrease in prostatic GHRH, COX-2, IGF-1, and TGF-β1 protein levels. In other studies, GHRH stimulated multiple pathways involved in inflammation and growth in both BPH-1 and PrEp cells including NFκB p65, AKT, ERK1/2, EGFR, and STAT3.24
The current study investigated the effects of GHRH antagonist MIA 602, GHRH and GHRH agonist MR 409 in the regulation of inflammatory processes; which strongly affect the endothelial permeability of BPAECs. We decided to focus on the effects of those compounds toward the expression of P53, since P53 has been recently shown to be a major regulator of the endothelium permeability.7 Our results indicate that GHRH antagonist induces P53 expression levels, while GHRH and GHRH agonist exert the opposite effects. Similar observations were previously obtained in LNCAP prostate cancer cells.25
Furthermore, the GHRH antagonist MIA 602 can deactivate cofilin. Cofilin is an actin-binding protein that influence actin dynamics to regulate the initiation and shape of cell protrusions.26 Cofilin is deactivated by phosphorylation, and its severing activity generates free filament ends that are accessible to G-actin. Thus, deactivation of cofilin supports the endothelial integrity.13 Moreover, MIA 602 suppresses the inflammatory activity of the RhoA/MLC2 pathway. This pathway is responsible for the formation of the F actin fibers; and the consequent endothelial hyperpermeability responses.10 In contrast, both GHRH and GHRH agonist counteracted those effects, indicating that the targeted action of those compounds is through the GHRH receptors.
The involvement of the MAP kinases in lung endothelial permeability, has been previously described. It was reported that MAPK signaling plays a critical role in the barrier dysfunction mediated by thrombin, pertussis toxin, TNF-α, transforming growth factor-β1, hydrogen peroxide, and VEGF. Furthermore, the protein kinase MEK1/2 mediates vascular endothelial growth factor- and histamine-induced hyperpermeability in porcine coronary venules.27 It was also suggested that there is a direct link between p38 MAPK activation and EC barrier regulation.28 It was recently described that the JAK2/STAT3/SOCS axis contributes to the development of macrovascular complications by mediating inflammation associated with vascular endothelial cells and/or monocytes.29
Treatment of BPAEC with the MIA 602 suppressed the JAK2/STAT3, and ERK1/2 pathways. In line with previous observations, the GHRH antagonist induced the phosphorylation of AMPK.30 Such modification have been previously shown to support endothelial integrity; and represents a promising strategy for the protection of the vascular endothelial function.10,31 Moreover, it protected the endothelium against the LPS-induced MLC2 and cofilin activation. On the other hand, both GHRH and GHRH agonist MR 409 activated the JAK2/STAT3 and ERK1/2 pathways, indicating their strong potential to trigger inflammatory processes. It has been recently shown that GHRH agonists may suppress cancer through downregulation of GHRH receptors.32 The latter effect may be due to mechanisms associated with the direct action of the agonists on the cancer cells that operate both in vivo and in vitro, as well as the systemic (endocrine) action of GHRH that operates only in vivo and modulates tumor growth indirectly.33,34 Thus, a potential beneficial effect of GHRH agonists toward the suppression of inflammation cannot be excluded.
The effects of the earlier GHRH antagonist JMR-132 have been previously associated with the ER stress marker CHOP.35 CHOP indicates the activation of the Unfolded Protein Response (UPR) element. Moreover, the function of P53 is closely associated to UPR.36 It was recently shown that UPR activation regulates P53 levels in pulmonary endothelium.37 Hence, future studies will delineate the involvement of UPR activation in the MIA 602 – triggered endothelial barrier enhancement. A robust UPR induction, causes lethal ER stress.38 However, a moderate induction of that pathway has been associated with protective responses in the endothelium.39 In particular, the transfection of human pulmonary artery endothelial cells with siRNA for BiP (the ER Hsp70) promoted the filamentous actin formation and abrogated endothelial permeability.40 The LDL–induced inflammatory responses in human mesangial cells were significantly reduced after knocking down of the IRE1alpha (UPR component). Pretreatment of those cells with Tunicamycin significantly attenuated the induction of the LDL – induced pro – inflammatory cytokines.41
The severe manifestation of lung inflammation results in ARDS, which is a respiratory syndrome associated with unacceptably high mortality rates. Currently, there are no efficient therapies for those ARDS patients. Our study demonstrates that the GHRH antagonist MIA 602 suppresses major inflammatory pathways (i.e. pJAK2/STAT3, ERK1/2) induces the “Endothelial Defender” P53, thus it protects against hyperpermeability responses. Moreover, MIA 602 supports the integrity of the vascular barrier, as reflected in measurements of transendothelial resistance. Hence, we suggest that GHRH antagonists may be of therapeutic value for the treatment of ALI/ARDS.
Materials and methods
Reagents
RIPA buffer (AAJ63306-AP), anti-mouse (95017–554) and anti-rabbit (95017–556) IgG HRP-linked antibodies, nitrocellulose membranes (10063–173) and GHRH (103663–156) were obtained from VWR (Radnor, PA). The P53 (9282S), Phospho-MLC2 (3674S), MLC2 (3672), Phospho-cofilin (3313S), Cofilin (3318S), Phospho-p44/42 MAPK (Erk1/2) (9101S), p44/42 MAPK (Erk1/2) (9102S), Phospho-JAK2 (3776S), JAK2 (3230S), Phospho-STAT3 (9145S), STAT3 (4904S), Phospho-AMPKa (2535S) and AMPKa (2793S) antibodies were obtained from Cell Signaling Technology (Danvers, MA). The GHRH-R antibody (ab28692) was purchased from Abcam (Cambridge, MA). The β-actin antibody (A5441) was purchased from Sigma-Aldrich (St Louis, MO). The MIA – 602 and MR- 409 were synthesized in the laboratory of one of us (AVS).42
Cell Culture
Bovine Pulmonary Arterial Endothelial Cells (BPAEC) were purchased from Genlantis (San Diego, CA). HeLa, MCF7, and NIH/3T3 cells were purchased from ATCC (Manassas, VA). Those cells were cultured in DMEM (cat. no. VWRL0101-0500) medium supplemented with 10% fetal bovine serum and 1X penicillin/streptomycin. Cultures were maintained at 37°C in a humidified atmosphere of 5% CO2 – 95% air. All the reagents were purchased from VWR (Radnor, PA).
Protein isolation and Western Blot Analysis
Proteins were isolated from cells using RIPA buffer. Protein-matched samples were separated by electrophoresis through on 12% sodium dodecyl sulfate (SDS–PAGE) Tris-HCl gels. Wet transfer was used to transfer the proteins onto nitrocellulose membranes. The membranes were incubated for 1 h at room temperature in 5% nonfat dry milk in Tris-buffered saline (TBS) – 0.1% (v/v) Tween 20. The blots were then incubated at 4°C overnight with the appropriate primary antibody (1:1000). The signal for the immunoreactive proteins was developed by using the corresponding secondary antibody (1:2000) and visualized in a ChemiDocTM Touch Imaging System from Bio-Rad (Hercules, CA). The β-Actin antibody (1:5000) was used as a loading control.
Measurement of endothelial barrier function
The barrier function of endothelial cell monolayers was estimated by electric cell‐substrate impedance sensing (ECIS), utilizing ECIS model ΖΘ (Applied Biophysics, Troy, NY, USA). All the experiments were conducted on confluent cells which had reached a steady‐state resistance of at least 800 Ω.12
Densitometry and Statistical Analysis
Image J software (NIH) was used to perform densitometry of immunoblots. All data are expressed as mean values ± SEM. A value of P < .05 was considered significant. GraphPad Prism 5.01 from GraphPad (CA, USA) was used for data analysis. The letter n represents the number of experimental repeats.
Funding Statement
Dr. Barabutis’ research is supported by 1) R&D, Research Competitiveness Subprogram (RCS), Louisiana Board of Regents through the Board of Regents Support Fund (LEQSF(2019-22)-RD-A-26) (PI: N.B) 2) Faculty Research Support Program from Dean’s Office, College of Pharmacy, ULM (PI: N.B). 3) National Institute of General Medical Sciences of the National Institutes of Health (5 P20 GM103424-15, 3 P20 GM103424-15S1) 4) Malcolm Feist PAC Seed Program, LSU Health Shreveport. Dr. Schally’s research is supported by the Medical Research Service of the Department of Veterans Affairs and by the University of Miami Miller School of Medicine.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Villanova T, Gesmundo I, Audrito V, Vitale N, Silvagno F, Musuraca C, Righi L, Libener R, Riganti C, Bironzo P, et al. Antagonists of growth hormone-releasing hormone (GHRH) inhibit the growth of human malignant pleural mesothelioma. Proc Natl Acad Sci U S A. 2019;116(6):1–11. doi: 10.1073/pnas.1818865116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bodart G, Farhat K, Renard-Charlet C, Becker G, Plenevaux A, Salvatori R, Geenen V, Martens H.. The severe deficiency of the somatotrope GH-releasing hormone/growth hormone/insulin-like growth factor 1 axis of Ghrh(-/-) Mice is associated with an important splenic atrophy and relative B lymphopenia. Front Endocrinol (Lausanne). 2018;9:296. doi: 10.3389/fendo.2018.00420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barabutis N, Schally AV, Siejka A. P53, GHRH, inflammation and cancer. EBioMedicine. 2018;37:557–562. doi: 10.1016/j.ebiom.2018.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cui T, Schally AV. Growth hormone-releasing hormone (GHRH) and its agonists inhibit hepatic and tumoral secretion of IGF-1. Oncotarget. 2018;9(47):28745–28756. doi: 10.18632/oncotarget.25676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barabutis N, Siejka A, Schally AV, Block NL, Cai R, Varga JL. Activation of mitogen-activated protein kinases by a splice variant of GHRH receptor. J Mol Endocrinol. 2010;44(2):127–134. doi: 10.1677/JME-09-0121. [DOI] [PubMed] [Google Scholar]
- 6.Siejka A, Schally AV, Barabutis N. Activation of Janus kinase/signal transducer and activator of transcription 3 pathway by growth hormone-releasing hormone. Cell Mol Life Sci. 2010;67(6):959–964. doi: 10.1007/s00018-009-0224-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Uddin MA, Barabutis N. P53: the endothelium defender. J Cell Biochem. 2019. doi: 10.1002/jcb.28511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Siejka A, Schally AV, Barabutis N. The effect of LHRH antagonist cetrorelix in crossover conditioned media from epithelial (BPH-1) and stromal (WPMY-1) prostate cells. Horm Metab Res. 2014;46(1):21–26. doi: 10.1055/s-0033-1349127. [DOI] [PubMed] [Google Scholar]
- 9.Nanchal RS, Truwit JD. Recent advances in understanding and treating acute respiratory distress syndrome. F1000Res. 2018;7. doi: 10.12688/f1000research.15493.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barabutis N, Verin A, Catravas JD. Regulation of pulmonary endothelial barrier function by kinases. Am J Physiol Lung Cell Mol Physiol. 2016;311(5):L832–L845. doi: 10.1152/ajplung.00233.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herridge MS, Moss M, Hough CL, Hopkins RO, Rice TW, Bienvenu OJ, Azoulay E. Recovery and outcomes after the acute respiratory distress syndrome (ARDS) in patients and their family caregivers. Intensive Care Med. 2016;42(5):725–738. doi: 10.1007/s00134-016-4321-8. [DOI] [PubMed] [Google Scholar]
- 12.Barabutis N, Dimitropoulou C, Birmpas C, Joshi A, Thangjam G, Catravas JD. p53 protects against LPS-induced lung endothelial barrier dysfunction. Am J Physiol Lung Cell Mol Physiol. 2015;308(8):L776–87. doi: 10.1152/ajplung.00334.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barabutis N, Dimitropoulou C, Gregory B, Catravas JD. Wild-type p53 enhances endothelial barrier function by mediating RAC1 signalling and RhoA inhibition. J Cell Mol Med. 2018;22(3):1792–1804. doi: 10.1111/jcmm.13460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barabutis N, Uddin MA, Catravas JD. Hsp90 inhibitors suppress P53 phosphorylation in LPS - induced endothelial inflammation. Cytokine. 2019;113:427–432. doi: 10.1016/j.cyto.2018.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Uddin MA, Akhter MS, Siejka A, Catravas JD, Barabutis N. P53 supports endothelial barrier function via APE1/Ref1 suppression. Immunobiology. 2019;224(4):532–538. doi: 10.1016/j.imbio.2019.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kelley MR, Georgiadis MM, Fishel ML. APE1/Ref-1 role in redox signaling: translational applications of targeting the redox function of the DNA repair/redox protein APE1/Ref-1. Curr Mol Pharmacol. 2012;5:36–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choi S, Joo HK, Jeon BH. Dynamic regulation of APE1/Ref-1 as a therapeutic target protein. Chonnam Med J. 2016;52(2):75–80. doi: 10.4068/cmj.2016.52.2.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kiaris H, Schally AV, Busto R, Halmos G, Artavanis-Tsakonas S, Varga JL. Expression of a splice variant of the receptor for GHRH in 3T3 fibroblasts activates cell proliferation responses to GHRH analogs. Proc Natl Acad Sci U S A. 2002;99(1):196–200. doi: 10.1073/pnas.012590999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barabutis N, Tsellou E, Schally AV, Kouloheri S, Kalofoutis A, Kiaris H. Stimulation of proliferation of MCF-7 breast cancer cells by a transfected splice variant of growth hormone-releasing hormone receptor. Proc Natl Acad Sci U S A. 2007;104(13):5575–5579. doi: 10.1073/pnas.0700407104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barabutis N, Schally AV. Knocking down gene expression for growth hormone-releasing hormone inhibits proliferation of human cancer cell lines. Br J Cancer. 2008;98(11):1790–1796. doi: 10.1038/sj.bjc.6604386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kiaris H, Chatzistamou I, Schally AV, Halmos G, Varga JL, Koutselini H, Kalofoutis A. Ligand-dependent and -independent effects of splice variant 1 of growth hormone-releasing hormone receptor. Proc Natl Acad Sci U S A. 2003;100(16):9512–9517. doi: 10.1073/pnas.1533185100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ren JL, Yu QX, Ma D, Liang WC, Leung PY, Ng TK, Chu WK, Schally AV, Pang CP, Chan SO. Growth hormone-releasing hormone receptor mediates cytokine production in ciliary and iris epithelial cells during LPS-induced ocular inflammation. Exp Eye Res. 2019;181:277–284. doi: 10.1016/j.exer.2019.02.021. [DOI] [PubMed] [Google Scholar]
- 23.Barabutis N, Siejka A, Schally AV. Growth hormone releasing hormone induces the expression of nitric oxide synthase. J Cell Mol Med. 2011;15(5):1148–1155. doi: 10.1111/j.1582-4934.2010.01096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Popovics P, Cai R, Sha W, Rick FG, Schally AV. Growth hormone-releasing hormone antagonists reduce prostatic enlargement and inflammation in carrageenan-induced chronic prostatitis. Prostate. 2018;78(13):970–980. doi: 10.1002/pros.23655. [DOI] [PubMed] [Google Scholar]
- 25.Barabutis N, Schally AV. Growth hormone-releasing hormone: extrapituitary effects in physiology and pathology. Cell Cycle. 2010;9(20):4110–4116. doi: 10.4161/cc.9.20.13787. [DOI] [PubMed] [Google Scholar]
- 26.Bravo-Cordero JJ, Magalhaes MA, Eddy RJ, Hodgson L, Condeelis J. Functions of cofilin in cell locomotion and invasion. Nat Rev Mol Cell Biol. 2013;14(7):405–415. doi: 10.1038/nrm3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu MH, Yuan SY, Granger HJ. The protein kinase MEK1/2 mediate vascular endothelial growth factor- and histamine-induced hyperpermeability in porcine coronary venules. J Physiol. 2005;563(Pt 1):95–104. doi: 10.1113/jphysiol.2004.076075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Birukova AA, Birukov KG, Gorshkov B, Liu F, Garcia JG, Verin AD. MAP kinases in lung endothelial permeability induced by microtubule disassembly. Am J Physiol Lung Cell Mol Physiol. 2005;289(1):L75–84. doi: 10.1152/ajplung.00447.2004. [DOI] [PubMed] [Google Scholar]
- 29.Shan PF, Li Q, Khamaisi M, Qiang GF. Type 2 diabetes mellitus and macrovascular complications. Int J Endocrinol. 2017;2017:4301461. doi: 10.1155/2017/4301461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Siejka A, Barabutis N, Schally AV. GHRH antagonist MZ-5-156 increases the expression of AMPK in A549 lung cancer cells. Cell Cycle. 2011;10(21):3714–3718. doi: 10.4161/cc.10.21.17904. [DOI] [PubMed] [Google Scholar]
- 31.Zou MH, Wu Y. AMP-activated protein kinase activation as a strategy for protecting vascular endothelial function. Clin Exp Pharmacol Physiol. 2008;35(5–6):535–545. doi: 10.1111/j.1440-1681.2007.04851.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schally AV, Wang H, He J, Cai R, Sha W, Popovics P, Perez R, Vidaurre I, Zhang X. Agonists of growth hormone-releasing hormone (GHRH) inhibit human experimental cancers in vivo by down-regulating receptors for GHRH. Proc Natl Acad Sci U S A. 2018;115(47):12028–12033. doi: 10.1073/pnas.1813375115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kiaris H, Chatzistamou I. Inhibition of tumor growth by agonists of growth hormone-releasing hormone. Proc Natl Acad Sci U S A. 2018;115(47):11876–11878. doi: 10.1073/pnas.1817342115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schally AV, Zhang X, Cai R, Hare JM, Granata R, Bartoli M. Actions and potential therapeutic applications of growth hormone-releasing hormone agonists. Endocrinology. 2019;160:1600–1612. doi: 10.1210/en.2019-00111. [DOI] [PubMed] [Google Scholar]
- 35.Liu AX, Zhang D, Zhu YM, Gao HJ, Jiang JY, Hu XL, Lv PP, Leung PC, Huang HF. Impact of axis of GHRH and GHRH receptor on cell viability and apoptosis of the placental choriocarcinoma cell line. Curr Mol Med. 2016;16:299–311. [DOI] [PubMed] [Google Scholar]
- 36.Lopez I, Tournillon AS, Prado Martins R, Karakostis K, Malbert-Colas L, Nylander K, Fahraeus R. p53-mediated suppression of BiP triggers BIK-induced apoptosis during prolonged endoplasmic reticulum stress. Cell Death Differ. 2017;24(10):1717–1729. doi: 10.1038/cdd.2017.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Akhter MS, Uddin MA, Barabutis N. Unfolded protein response regulates P53 expression in the pulmonary endothelium. J Biochem Mol Toxicol. 2019;e22380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hsu SK, Chiu CC, Dahms HU, Chou CK, Cheng CM, Chang WT, Cheng KC, Wang HD, Lin IL. Unfolded protein response (UPR) in survival, dormancy, immunosuppression, metastasis, and treatments of cancer cells. Int J Mol Sci. 2019;20(10). doi: 10.3390/ijms20102518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barabutis N. Unfolded protein response supports endothelial barrier function. Biochimie. 2019;165:206–209. doi: 10.1016/j.biochi.2019.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leonard A, Paton AW, El-Quadi M, Paton JC, Fazal F. Preconditioning with endoplasmic reticulum stress ameliorates endothelial cell inflammation. PLoS One. 2014;9(10):e110949. doi: 10.1371/journal.pone.0110949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu Y, Zhang L, Liu Q, Tang L, Sun H, Guo H. Endoplasmic reticulum stress preconditioning antagonizes low-density lipoprotein-induced inflammation in human mesangial cells through upregulation of XBP1 and suppression of the IRE1alpha/IKK/NF-kappaB pathway. Mol Med Rep. 2015;11(3):2048–2054. doi: 10.3892/mmr.2014.2960. [DOI] [PubMed] [Google Scholar]
- 42.Zarandi M, Cai R, Kovacs M, Popovics P, Szalontay L, Cui T, Sha W, Jaszberenyi M, Varga J, Zhang X, et al. Synthesis and structure-activity studies on novel analogs of human growth hormone releasing hormone (GHRH) with enhanced inhibitory activities on tumor growth. Peptides. 2017;89:60–70. doi: 10.1016/j.peptides.2017.01.009. [DOI] [PubMed] [Google Scholar]