

ABSTRACT
There is increasing interest in defining the location, content, and role of extracellular matrix (ECM) components in brain structure and function during development, aging, injury, and neurodegeneration. Studies in vivo confirm brain ECM has a dynamic composition with constitutive and induced alterations that impact subsequent cell-cell and cell-matrix interactions. Moreover, it is clear that for any given ECM component, the brain region, and cell type within that location, determines the direction, magnitude, and composition of those changes. This review will examine the ECM at the neurovascular unit (NVU) and the blood–brain barrier (BBB) within the NVU. The discussion will begin at the glycocalyx ECM on the luminal surface of the vasculature, and progress to the abluminal side with a focus on changes in basement membrane ECM during aging and neurodegeneration.
KEYWORDS: Glycocalyx, basement membrane, aged, neurodegeneration
Introduction
The vascular blood–brain barrier (BBB) is a dynamic and complex interface designed to control the exchange of substances between blood and the brain’s interstitial fluid.1,2 The BBB can be thought of as the anatomically central component of the neurovascular unit (NVU), a structure that includes astrocytes, neurons, pericytes, and microglia. The cellular components of the BBB are largely comprised of a monolayer of brain endothelial cells (BEC) that are in contact with each other, as well as pericytes and astrocytic endfeet, via a network of regulatory proteins. The cells of the BBB, in conjunction with the endothelial glycocalyx and the basement membrane (BM), interact with perivascular neurons to create a highly regulatory environment that forms a barrier, restricting the passage of molecules between blood and brain. This barrier also serves to limit the access of cells, such as leukocytes, that have increased access into the brain parenchyma during inflammation, to optimize the neuronal microenvironment and subsequent function. This review will focus on the origins, components and functional roles of the extracellular matrix (ECM) of the glycocalyx and the BBB. A discussion regarding the junctional components of the BBB, such as gap junctions, tight junctions, and the numerous receptors of the BBB, is beyond the scope of this article and has been recently reviewed by others.3,4 When possible, the effect of normal aging and neurodegeneration (with a focus on Alzheimer’s disease) on the ECM in humans will be discussed, with animal studies presented when relevant. To avoid confounding, we will avoid studies in the context of significant vascular disease or trauma, which creates additional stresses on the vasculature that could affect the expression of ECM in the BBB.
Brain endothelial cells (ECs) form a polarized intact monolayer that lines the cerebral blood vessels and have unique characteristics including: lack of fenestrae; circumferential tight junctions between adjacent endothelial cells; presence of solute carriers that regulate ion and small molecule transport; expression of efflux transporters including P-glycoprotein (P-gp), Breast Cancer Resistance Protein, and Multidrug Resistance Proteins; low levels of macropinocytosis; carrier-proteins that can be ubiquitous or specific for transport of macromolecules and other substances.2,5,6 Brain ECs of the BBB have a greater density of mitochondria than other vasculature, which generates a higher potential for oxidative stress and other mediators that influence the inflammatory response.5,7
Pericytes are multifunctional cells that invest and support the endothelial layer throughout the vasculature. Pericytes share some common origins with smooth muscle cells and electron microscopy studies have shown that brain pericytes can contain microfilaments that express alpha-smooth muscle actin, a contractile protein that could confer the ability to regulate blood flow.8 The ratio of pericytes to endothelial cells in the central nervous system (CNS) tends to be higher than any other organ in the body indicating additional regulatory functions.9,10 Known roles include: supporting structural elements of the BBB; regulating vascular stability; and even angiogenesis.11 Pericytes and endothelial cells share and are typically separated by a common BM with attachment points to ECM components mediated by integrins and less ubiquitous cell-ECM receptors such as dystroglycan.12,13 Direct contact between the cells can also occur at gap junctions, adherens junctions, and peg-and-socket structures that lack BM.14 At adherens junctions, the cytoskeletons of pericytes and endothelial cells are connected by cadherins and catenins that provide support and anchor the cells to each other. N-cadherin is one of the many adherens junction proteins at the pericyte-endothelium interface, whereas connexin-43 hemichannels actually function as gap junctions that allow for direct intercellular connections between endothelial cells and pericytes.15,14 Pericyte communication with other cells in the BBB occurs at multiple levels, including direct contact as well as autocrine and paracrine signaling pathways that optimize BBB function.10 In the healthy brain, these highly dynamic and regulated properties allow for rapid changes in blood flow that meets the demand for oxygen and nutrients in the setting of increased neuronal activity.
Astrocytes are the most common sub-type of glial cell, a group defined as non-neuronal cells that provide structural and metabolic support to neurons.16 Glial cells also include oligodendrocytes, microglia, and ependymal cells, which do not have established roles in the normal physiology of the BBB. Astrocytes perform myriad tasks essential for BBB function including: neurovascular coupling; release of chemokines and glutamate; ionic buffering; control of brain pH; metabolization of dopamine and other substrates by monoamine oxidases; uptake of glutamate and γ-aminobutyric acid by specific transporters; and production of antioxidant compounds like glutathione and enzymes such as superoxide dismutases.17,18
Endothelial cells, pericytes, and to a lesser extent astrocytes (via their endfeet), come together to establish the microvasculature of the brain. Studies of changes in the microvasculature with aging or neurodegeneration are often confounded by the concurrent presence of vascular disease, such as strokes and microinfarcts. Despite these concerns, it is generally accepted that brain vessel density (vessels per area) decreases throughout the brain with normal aging.19,20 In most organs, aging also results in thickening of the vasculature that reflects changes in BM.20 BM thickening is due, in part, to carbonyl stress induced by methylglyoxal followed by the formation of advanced glycation endproducts that alter brain ECs. Subsequent changes to receptor for advanced glycation end products (RAGE), occludin and ZO-1 also promote the expression of pro-inflammatory interleukins, such as IL-6 and IL-8.21 The functional implications of less microvascular density, concurrent with vessel thickening in the aging brain, include less perfusion to deep structures in watershed regions and slower vascular reactivity to flow and pressure changes.22 Less is known about specific changes in BBB physiology during normal aging. It is probable that healthy aged brains retain BBB function, although changes in transporters have been found.20,22–24 In contrast to normal aging, it is controversial whether vessel density or morphology changes in Alzheimer’s disease (AD) relative to age-matched controls.25,26 AD brains demonstrate decreases in the function of certain BBB proteins such as GLUT-1, a glucose receptor that is a key transporter.1,27 It is possible that AD neuropathologic progression, which occurs on a regional basis throughout the brain with plaques of amyloid beta (Abeta) and neurofibrillary tangles of tau, influences the microvasculature in an analogous manner that precludes consistent measures of vessel density, morphology and physiology across brain regions. Brain EC, pericytes and astrocytes all contribute to deposition of BBB ECM throughout the vasculature.13 The contribution of neurons to this ECM is variable, as individual neurons display unique biosynthetic signatures depending on the region of the brain and the specific cell layer.28,29
Glycocalyx and perivascular parenchymal ECM
The endothelial glycocalyx serves as the initial gatekeeper for barrier functions at the luminal side of the BBB.30,31 The thickness and structure of the glycocalyx layer varies from approximately 0.2 µm in capillaries to over 5.0 μm in large arteries.32 Although the luminal surface of brain capillaries has a significantly greater area of glycocalyx coverage than other organs, there is no consensus as to whether brain glycocalyx differs with respect to depth or consistency of thickness relative to the vasculature of other parts of the body.30,33,34 The glycocalyx is comprised of proteoglycans, glycosaminoglycans (GAGs), glycoproteins and associated plasma proteins (Figure 1). These components vary in their contribution to glycocalyx structure and permeability throughout the microvasculature.32,35 Proteoglycans are core proteins, often anchored to the apical membrane of endothelial cells, and to which 1 or more GAG chains are covalently attached. As such, they are generally considered to function as the most important “backbone” molecules of the glycocalyx. There is a notable variation among the proteoglycan core proteins with regard to their size, number of attached GAG chains, and whether or not they are bound to the cell membrane. Core groups of proteoglycans comprised of syndecans and glypicans have a firm connection to the endothelial membrane via membrane spanning domains or a glycosylphosphatidylinositol anchor, respectively. Other proteoglycans, such as biglycan and versican, are secreted after assembly and GAG modification.30,31 These proteoglycans are less tethered to the endothelium and can even diffuse into the bloodstream.
Figure 1.
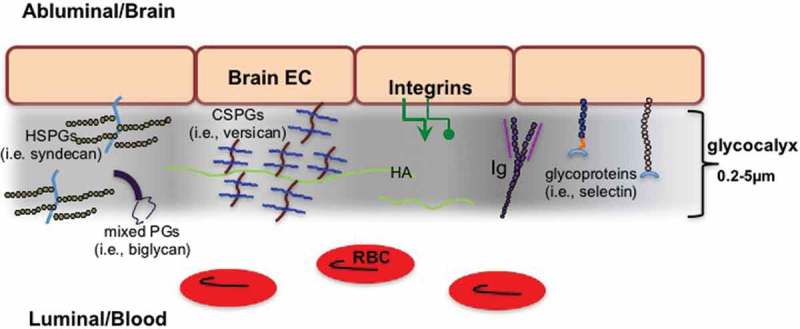
Glycocalyx of the brain vasculature. To focus on ECM, note that gap junctions, tight junctions, and regulatory molecules are not shown. Heparan sulfate proteoglycans (HSPGs), Proteoglycans (PGs), Chondroitin sulfate proteoglycans (CSPGs), Endothelial cell (EC), Hyaluronan (HA), Immunoglobulins (Ig), Red blood cells (RBC).
GAG chains are ubiquitous in the glycocalyx and confer additional difficulty in characterizing the exact components of ECM in the BBB.31 The GAG polymers of disaccharides (composed of a uronic acid and a hexosamine) have variable lengths that are modified by sulfation and/or (de)acetylation. Classification of the glycosaminoglycans depends on which uronic acid or hexosamine is incorporated and on the pattern of sulfation. There are at least five types of GAGs: heparan sulfates (HS), chondroitin sulfates (CS), dermatan sulfates (DS), keratin sulfates (KS) and the non-sulfated GAG, hyaluronan (HA). In the vasculature, heparan sulfate proteoglycans (HSPGs), i.e., syndecans, glypicans, constitute a high proportion of the proteoglycans present in the glycocalyx.35 The second most common GAG in the endothelial glycocalyx is chondroitin sulfate/dermatan sulfate (the latter can be regarded as a separate class of a type B chondroitin sulfate), i.e., versican and biglycan, with reportedly a 4:1 ratio of HS to CS in the vasculature.36,37 Less well studied, especially in brain glycocalyx, is the expression of keratan sulfate GAGs (i.e., phosphacan), although it should be noted that some CS/DS proteoglycans (i.e., aggrecan) can have a KS GAG chain. Regardless of the GAG, modifications such as sulfation alter subsequent cell-matrix and matrix-matrix interactions that determine functional effects. Moreover, proteoglycans can bind to a variety of GAG chains and the proportion of the GAG chains they bind changes in a dynamic fashion during injury and inflammation.37 This results in inconsistent nomenclature. As an example, syndecan-1 proteoglycan is often referred to as an HSPG, due to binding of heparan sulfate GAGs, when it actually contains as many chondroitin sulfate GAG chains as heparan sulfate GAG chains.38 As a further example of the complexity, it is worth noting that dozens of sulfation patterns may exist per disaccharide resulting in theoretically thousands of different combinations with unique functions and biological activity. Moreover, aging and neurodegeneration alter sulfation in a dynamic fashion that is unpredictable.
It is difficult to know the exact composition of GAGs in the glycocalyx as the presence of the non-sulfated GAG, hyaluronan (HA), was initially underappreciated due to losses of this carbohydrate during tissue processing. HA, a disaccharide polymer of glucuronic acid/N-acetyl glucosamine, is the only GAG with no core protein and can range from 2 to 25,000 disaccharides with a size span from 1->2x104 kDa.39,40 HA is a significant component of the vasculature throughout the body and serves a key role in regulating the permeability of the glycocalyx.41 In the brain, HA is assembled at the cytosolic side of the cell membrane and is extruded from the surfaces of cells by the action of HA synthases 1–3 (HAS) in endothelial cells and astrocytes. Whether specific HA synthases have effects on the size of HA produced is a matter of debate, but there is some evidence that the three isoforms of mammalian hyaluronan synthases have distinct enzymatic properties. Comparisons of hyaluronan secreted into the culture media by stable HAS transfectants showed that HAS1 and HAS3 generated hyaluronan with broad size distributions (molecular masses of 2 × 105 to approximately 2 × 106 Da), whereas HAS2 generated hyaluronan with a broad, but extremely large size (average molecular mass of >2 x 106 Da). Newly synthesized HA is constantly and rapidly processed in the extracellular milieu into fragments ranging from 2 to 25,000 disaccharides by several mechanisms, including the activity of hyaluronidases. There are six hyaluronidase-associated genes in humans, but the primary hyaluronidases in the brain are hyaluronidases 1–3. Hyaluronidase activity and HA degradation are accelerated and upregulated by inflammation and ischemia.42,43 HA’s association with cells is mediated by binding to its cellular receptors, such as CD44.44,45 HA size determines receptor binding with lower molecular weight (MW) forms favoring TLR2 over CD44 and RHAMM. Activation of the TLRs incites inflammation, thereby perpetuating a cycle of cellular injury.46–50
HA is highly hydrophilic and functions as a scaffold for assembly and organization of multiple other ECM components. This interaction confers stability and modulates subsequent cell-ECM functions, but HA itself is a simple repeating disaccharide that is not linked to a core protein.39 Proteins with specific HA binding domains are called hyaladherins and are present throughout tissues and plasma. Hyaladherins are also found inside cells indicating a role for HA within intracellular compartments.51 Hyaladherins in the glycocalyx include: tumor necrosis factor-inducible gene 6 protein (TSG-6); the protein complex Inter-alpha-inhibitor (IαI), and the chondroitin sulfate proteoglycan (CSPG) versican, which are present in quantities that remain to be established. The association of HA with TSG-6 is of particular interest, as this secreted protein is thought to mitigate inflammation and provide tissue protection in many organs including the brain after traumatic injury.52–54 TSG-6 is constitutively expressed in the brains of adult rodents and likely organizes HA during glial scar formation in the CNS.54 Moreover, TSG-6 alters the structure of HA via its direct crosslinking of HA chains, which enhances HA-receptor interactions, and perhaps contributes to the anti-inflammatory effects of HA.55–57 Recent studies of hyaluronidase treatment indicate that eliminating HA has subsequent functional effects on the BBB, with substantial increases in passage of larger molecules such as 150 kDa dextran, based on real-time images of single blood vessels using 2 photon microscopy.58
The lecticans are specific brain CSPGs characterized by structural similarities: a central core protein that binds CS GAG side chains; an N-terminal globular G1 domain with a link domain that binds HA, and a C-terminal G3 domain with a lectin-type sequence. The lecticans include: the large aggrecan that contains an additional G2 domain, three forms of versican (V0, V1, and V2), neurocan, and brevican.59 Individual lectican molecules differ in the number of CS-GAG chains attached to their core proteins, with over one hundred GAG side chains being found in aggrecan, which can be over 400 kDa, and as little as zero to five GAG chains being found in brevican and neurocan. Lecticans are produced by several cell types in the CNS, including neurons and astrocytes, and are well studied for their contribution to perineural nets (PNNs) in the brain parenchyma. It is worth noting that brevican, aggrecan, neurocan and versican differ in their relevance to the glycocalyx in the adult brain. Neurocan is present primarily during development, brevican has low levels of expression in adult brain and aggrecan is dominant in the PNNs, leaving versican as the primary CSPG in the adult brain vasculature and glycocalyx.59,60
Additional components of the glycocalyx include well-studied ECM components and families of endothelial cell adhesion molecules, such as selectins, integrins, and immunoglobulins. Many of the ECM molecules in this group are small glycoproteins (for example, the selectins) with short sugar residues and branched carbohydrate chains that work in conjunction with transmembrane proteoglycans, such as syndecans and glypicans, to provide support by connecting the glycocalyx to the EC membrane.34
Sulfated GAGs and HA sequester water, stabilize the gel-like structure of the glycocalyx, and along with glycoproteins, interact with cell-adhesion molecules and soluble constituents of the glycocalyx.39 Plasma and endothelium-derived soluble components include albumin, circulating HA, proteoglycans (e.g., thrombomodulin), and clotting factors, such as fibrinogen and antithrombin III. The resulting interface serves to maintain a balance between plasma and endothelium that functions to optimize vascular homeostasis. Subsequent small chain modifications and size changes can alter the interaction of GAGs and HA with these constituents, resulting in significant differences in vascular permeability and function over time and under different stresses.34,61,62 Consequently, there is great interest in how GAG sulfation and other ECM modifications affect cell-cell and cell-ECM interactions that have effects on microvascular physiology during aging and neurodegeneration.
Glycocalyx and associated ECM changes in aging
The glycocalyx is a delicate structure that is easily lost during tissue retrieval or fixation. As such, changes in the glycocalyx ECM with age are difficult to quantify and are often measured as part of the content that includes the abluminal side of the BBB. Although it is likely that changes in the glycocalyx are reflected in the relevant ECM molecules present in the brain parenchyma, precise measures of isolated glycocalyx would advance understanding of changes in its barrier contribution with age. To that end, a recent study of microvessel glycocalyx in easily accessible tissues, such as skin, found thickness decreased with aging and correlated with measures of reduced barrier function (as measured by intravital microscopy) in both mice and humans.63 Other studies provide only indirect evidence of potential changes in glycocalyx ECM with aging. As an early example, a moderate increase in HA concentration was found in the brain tissue of 30 month-old rats compared to tissues from younger rats by electrophoretic separation of GAGs.64 More recently, HA content, as measured by a highly sensitive assay, has been reported to increase with normal aging, especially in general brain gray matter.65 However, others report that HA expression might decrease in non-gray matter regions of the brain.66 Studies of brain regions in normal aging indicate HA accumulation was higher in aged brain relative to young brain in the cerebral cortex and cerebellum, but not in other regions examined. In contrast, CSPGs did not change with aging in any of the brain regions examined. HA and CSPGs colocalized with a subset of neuronal cell bodies and astrocytes, and at the microvasculature.67 In specific studies of the vasculature, which should primarily reflect the content on the luminal side, brain microvasculature isolated from the gray matter cortex from aged mice showed significantly greater HA content and synthesis than young mice.68 The increase in HA in aging reflected increased activity of HAS2, the primary source of HA in the brain.
HA and CSPG synthesis and accumulation are associated with glial scar formation after injury in the CNS.69 These changes are likely mimicked in the aging brain, which incurs scarring over time, but direct studies are lacking. Microscopically, HA and the lecticans appear in densely packed areas of parenchymal ECM called perineural nets, specialized structures that control plasticity in development and aging. A recent study in aged mice has found that Bral2, a link protein that stabilizes HA and CSPG interactions in the brain, is reduced in aged mice with resulting disruption of PNNs.70 PNN function also depends on the GAG chains of CSPGs. It is generally accepted that C4 sulfation of GAGs is inhibitory at the same time C6 sulfation promotes neural plasticity. IHC studies of aged rats indicate a progressive increase in C4 to C6 sulfation ratio in PNNs with aging.71 Whether changes in this ratio are reflected in the aged brain glycocalyx is unknown. Proteomic analysis of the aging brain of mice identified multiple changes, including decreases in enzymes that balance metabolic and oxidative stress, but yielded little information on changes in ECM components.72,73
Glycocalyx and associated ECM changes in neurodegeneration
Similar to studies in aging, examination of the glycocalyx of AD brains is difficult and is often extrapolated from measures of the general brain parenchyma. Increased HA is found in many areas of inflammation or ischemia where it inhibits oligodendrocyte precursor cell maturation and modulates signals that can potentiate additional neuronal injury.43,47,74,75 Deposition of HA is found in areas of brain ischemia that appear as white matter hyperintensities on MRI.47 Whether the appearance of HA and its associated ECM is ultimately beneficial or detrimental to neural plasticity and repair in AD is a matter of ongoing debate.76 The sizes of HA that mediate specific functions in the CNS are also an area of active investigation. Injection of HA of distinct sizes and modulation of HA size (via hyaluronidase) can affect the response to injury and ongoing inflammatory processes, but whether HA of lower MW is beneficial or detrimental varies with the experimental model.77,78,50,79 Similar to experiments performed in muscle microvasculature by Henry and Duling in 1999, degradation of HA by hyaluronidase has been recently shown to enhance large molecule passage through the BBB.41,58
In addition to difficulties in localization, the timeline for changes in expression of ECM in human neurodegeneration is difficult as studies occur post-mortem. HA content was recently measured using carefully classified autopsy samples from research subjects with no AD (CERAD = 0, Braak = 0–II, n = 12–21), intermediate AD (CERAD = 2, Braak = III–IV, n = 13–18) and high AD (CERAD = 3, Braak = V–VI, n = 32–40) neuropathology. By histochemistry, HA was associated with deposits of amyloid and tau, and was also found diffusely in brain parenchyma, with overall HA quantity (measured by enzyme-linked sorbent assays) significantly greater in brains with high AD neuropathology. Although mean HA MW was similar among the samples, HAS2 mRNA was higher in high AD relative to no AD samples. TSG6 mRNA and protein were also significantly increased in AD brains.80
It is generally accepted that CSPG accumulation is increased during neurodegeneration.59 Similar to what has been reported during CNS scar formation, the characteristic lesions of Abeta and neurofibrillary tangles NFT are surrounded by reactive gliosis as measured by GFAP positive astrocytes.81 This reactive gliotic reaction contains CSPGs, HSPGs, DSPGs, and HA. CSPG accumulation might be a general reaction to neurodegeneration as indicated by higher levels in Huntington’s Disease and Parkinson’s Disease.59 This increase is mediated, in part by an upregulation of bone morphogenic protein (BMP), which stimulates astrocytes to increase the synthesis of CSPGs surrounding neurologic lesions.82 Much of the increase in CSPGs is accompanied by increases in the C4:C6 sulfation ratio, similar to that noted during normal aging. Studies in human cortex with AD showed increased HA, but no overall change in HSPGs and CSPGs, relative to controls. However, analysis of cell layers suggests differences in localized expression of ECM, especially as it relates to AD neuropathology.29 A connection between changes in HSPGs and CSPGs with alterations in the glycocalyx during neurodegeneration remains to be established.
Proteomics analysis of brain samples affected by AD has provided additional information regarding ECM that might be reflected in the glycocalyx.83,84 Studies in human cortex and hippocampus found broad differences between AD and age-matched controls that include changes in ECM, such as down-regulation of tenascin-R, in the hippocampus.85 There is great interest in making correlations between proteomic analysis of CSF, which is available in vivo, with brain tissues studies, but no specific localization to the glycocalyx was performed.86
Basement membrane ECM
The BM is a unique construct of ECM at the abluminal surface of the endothelial cell (Figure 2). In the brain, BM is generally viewed as having endothelial and parenchymal compartments that are separated by pericytes.6,10,87 The endothelial and parenchymal compartments are typically indistinguishable and in the absence of pericytes appear as one. Although often thought of as a supportive anatomic boundary between ECs and the brain, the BM is also a dynamic interface that provides signals for cell-ECM communication. The thickness of the BM is approximately 40–100 nm and consists of at least five major forms of ECM proteins: Laminin, Collagen IV, Nidogen, HSPGs (perlecan and agrin), and Fibronectin. Organization begins when a member of the laminin family interacts with cells (primarily via integrins) to polymerize and then binds to nidogen, perlecan, and agrin. Subsequent assembly is mediated by interactions with type IV collagen to stabilize the network.
Figure 2.
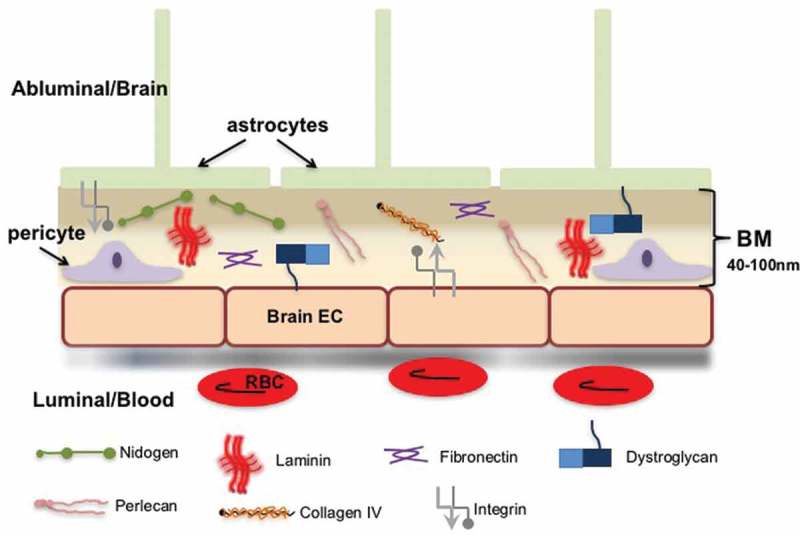
Extracellular matrix (ECM) in the vascular and parenchymal basement membrane (BM). To focus on ECM, note that gap junctions, tight junctions, and regulatory molecules are not shown. Endothelial cell (EC), Red blood cells (RBC).
It is accepted that BM thickening occurs in response to stress such as sheer forces. The impact of longstanding vascular flow on the BM has led to consensus that normal aging results in eventual BM thickening.20,88 Whether these changes confer functional alteration is less clear. It is also noted that specific brain regions demonstrate differential changes in BM ECM with aging in both mouse and humans.89–91 Changes with AD are likely similar to that of aging, with a general increase in BM thickening, but accurate assessment is again complicated by the regional changes that are a foundation of AD progression and classification.92 AD patients with cerebral amyloid angiopathy (CAA) provide additional difficulty in the study.46 Although CAA primarily affects arterioles, this pathology induces alterations in BM that exceed, or at the minimum differ from, that found in microvasculature without significant Abeta deposition.90,93 It should also be noted that in most human samples, autofluorescence and the presence of residual blood contents in brain specimens can make the differentiation of changes in ECM content difficult to discern, and can reflect regional bias that affects analysis within and among brain specimens.94 Measures of BM ECM are further complicated by whether the quality is considered in the evaluation of immunostaining (for example, linear integrity of the BM) or discrete bands on electrophoresis.
Laminin
The foundation for BM formation is self-assembly of laminin, a complicated hetero-trimer consisting of an alpha chain, a beta chain, and a gamma chain. Laminin in the brain is primarily deposited by astrocytes and is identified by three Arabic numerals based on the number of the specific chain that contributes to the composition of the trimer. Five alpha, four beta, and three gamma chains exist and can combine to form over 16 different laminin isoforms. Only B1 and G1 are typically found in the brain microvasculature; therefore, alpha chains 1, 2, 4, or 5 determine unique isoforms. Whereas laminin 411 is secreted by EC throughout the body, laminin 111 and 211 are secreted by astrocytes and regulated by pericytes and thereby deemed specific for brain microvasculature.13 There are exceptions to the above; the gamma 3 chain demonstrated staining in BM zones of multiple adult tissues, including the brain, which was determined by subsequent ultrastructural immunogold staining to localize specifically to the basement membrane tissues.95 In addition, it has recently been found that laminin 421 is deposited in cerebral vasculature in a murine model of the BBB in vitro.96
Laminin changes in normal aging
Studies of changes in brain ECM with aging are complicated by regional differences in expression. For example, studies in normal aged mice have found decreases in laminin in the cortex and striatum, but not other regions.91 In related functional studies, older (22 months) mice demonstrated impaired BBB function, which was associated with age-dependent changes in capillary density, relative to young mice (3 and 7 months of age). Decreases in the levels of laminin in vascular basement membranes were also noted in aged mice relative to young mice.97
Laminin changes in neurodegeneration
Few studies have specifically examined laminin in the BBB with neurodegeneration. Studies in transgenic APP mice that have cerebral amyloid angiopathy indicate a decrease in laminin in vasculature.91 Early work in human brain parenchyma identified punctate deposits of increased alpha 1 and gamma 1 chains in plaques of Abeta and in astrocytes of both the gray and white matter. These data suggest that these laminins and their specific domains may have distinct functions in the pathophysiology of AD.98
In immunohistochemical studies of Braak stage III-VI AD patients relative to age-matched controls, laminin exhibited patterns of BM fragmentation in AD and colocalized with beta-amyloid in senile plaques.99 Fragmentation, which is often visualized by immunofluorescence-based histologic analyses, underscores the difficulty in quantification that can also affect western blotting and other measures of ECM. The implications of fragmentation, whether defined by histologic appearance or degradation on immunoblotting, for ECM quality is another consideration when discussing changes that occur in AD.
Collagen IV
Collagen type IV (Col IV) is the most abundant fibrous protein within the interstitial ECM and is secreted by ECs, astrocytes, and pericytes in brain vasculature.100 In other tissues, Col IV is essential for regulating cell adhesion and directing tissue development through its role in chemotaxis and cell migration, while also providing strength to the ECM.101 Col IV consists of polypeptide chains derived from six different α chains, which can combine and form three different collagen IV isoforms. The Col IV isoform predominantly expressed in the brain consists of two α1 and one α2 chains that fuse to form the collagen IV [a 1(IV)]2a 2(IV) isoforms.87 Inside cells, the three alpha-chains assemble forming triple helical molecules, termed protomers, which oligomerize outside the cells into a supramolecular network.102 The self-assembly of the Col IV protomers is associated with both lateral (side-by-side) as well as C- and N-terminal non-covalent bindings of the Col IV monomers.102 Col IV stabilizes BM by retaining laminin and other ECM proteins, such as nidogen, and perlecan. Modest mutations in the Col4α1 gene are associated with fragile vessels that predispose to stress-induced hemorrhage and adult-onset stroke in humans.103,104 Null mutations of Col IV in mice are embryonic lethal due, in part, to impaired BM stability.105
Col IV changes in aging
Col IV is presumed to contribute to the BM thickening that occurs throughout the body with normal aging. Hawkes originally reported no change in Col IV in the brain vasculature with aging.97 However, in studies of old mice with significant thickening of capillary basement membranes in cerebral cortex, hippocampus, and thalamus, western blotting and immunocytochemistry experiments actually showed a significant reduction in Col IV in the cortex and hippocampus with age.91 In contrast, the brains of aged humans (mean age of 74) demonstrated increased Col IV accumulation in the BM of cerebral microvessels compared to those of controls (mean age of 39).89 These data underscore the difficulties in making quantitative assessments of ECM changes in the BM.
Col IV changes in neurodegeneration
Although it is controversial whether AD neuropathology results in changes in vessel density, it is likely that there are some alterations in microvascular morphology, such as decreased vessel diameter, which could have clinical correlations with cognition.106,107 One would predict that neurodegeneration should be accompanied by increased deposition of Col IV reflecting stress on the vasculature that enhances the deposition of BM proteins.89 Early studies in transgenic models of AD reported BM thickening with increased collagen content.108,109 Similarly, the brains of 11‐month‐old triple (APP × PS1 × Tau) transgenic mice showed decreased cerebrovascular volume, higher levels of Col IV and increased basement membrane thickness.110 In contrast, studies in transgenic APP mice that have cerebral amyloid angiopathy, which can confound studies of the microvascular BM, indicate a significant reduction in Col IV in vasculature.97
Early studies in humans found a 55% increase in Col IV content in cerebral microvessels in AD patients compared to controls.33 Additional investigations in AD patients also reported a thickening of the basement membrane together with an increase in collagen content.111–114 A recent study notes an increase in Col IV in the microvessels of subjects with AD, with both subclinical AD and AD groups similarly demonstrating significantly higher levels of Col IV staining than the control group in frontal and temporal cortex.115 In contrast, Christov reports that Col I and III increased, but Col IV decreased, in AD microvessels relative to controls.116 Studies in vitro indicate collagen deposition influences Abeta fibril formation, highlighting the possibility that Col IV levels change, but the direction of the change could be determined by location in the brain as well as proximity to the neuropathologic changes of AD.117 Although studies in humans with CAA are confounded by the influence of amyloid deposition on the BM, changes in collagen IV were not found.90
Nidogen
The laminin and Col IV networks are held together by nidogens, sulfated monomeric glycoproteins also called entactins. In the brain, nidogen has been shown to come from ECs as well as astrocytes and pericytes.13,93 Two isoforms of nidogen, nidogen-1 and −2 (entactin-1 and −2), have been identified and consist of three globes (G1, G2, G3) connected by segments of variable lengths. In addition to laminin and Col IV, nidogen also binds fibulin and perlecan. Antisense treatment of astrocytes in culture that normally expresses nidogen results in astrocyte detachment,118 which can compromise the BBB. Studies have found stronger nidogen expression in immature than in adult brain, and a dramatic increase of vascular staining in kainate-injured hippocampus, suggesting a contribution of nidogen to both development and reactive angiogenesis.119 Nidogen-1 is important for connecting and stabilizing the self-assembled layers of laminin and Col IV, and mutation of nidogen-1 reduces brain capillary BM.120 Mutations in nidogen-2 do not have a brain BM phenotype, but studies of nidogen-1 knockout mice indicate that nidogen-2 compensates for the loss of nidogen-1.120,121
Nidogen changes in aging
Nidogens function in neural plasticity and one would predict levels of these glycoproteins are reduced in normal aging, which is characterized by decreased plasticity in most organs.122 Indeed, results from Western blotting and immunocytochemistry experiments showed a significant reduction in nidogen 2 in the cortex and striatum during normal aging in mice.91 In contrast, studies in humans showed an increase in nidogen 2 with age that might reflect differences in techniques, regions and concurrent presence of CAA.90
Nidogen changes in neurodegeneration
Although studies in transgenic APP mice that have cerebral amyloid angiopathy and humans with CAA indicate a decrease in nidogen 2 in vasculature, these studies are confounded by the influence of amyloid deposition on the BM.90,91 Changes in nidogen have not been specifically studied in AD without CAA, but it has been shown nidogen binds Abeta plaque-associated BM components in vitro.123
Perlecan and agrin (HSPGs)
The HSPGs found in BM are exemplified by perlecan and agrin, which promote barrier function. In the brain, HSPGs come from BEC, astrocytes and likely pericytes.93 Perlecan, also called HSPG2, is a large multidomain proteoglycan that binds to and cross-links other components of ECM, thereby stabilizing BM and ensuring maintenance of the endothelial barrier function.124 Less is known about agrin in aging and neurodegeneration. Agrin is also a large HSPG that has barrier functions and is found extensively in the basal lamina of the brain microvasculature.125 Agrin’s connection to the BM has been shown to be mediated by binding of its amino terminus with laminin.126
Perlecan changes in aging
Age-related increases in the levels of perlecan in small vessel BM have been noted in wild-type mice.97 In this same study, capillary density was decreased and drainage of small molecular weight dextran along BM was impaired in the hippocampal vasculature of aged mice compared to young mice, but the role of perlecan in the vessel changes was not defined.
Perlecan changes in neurodegeneration
In early investigations, no significant difference was seen in perlecan mRNA levels in the hippocampus of AD patients when compared to age‐matched controls.127 However, in studies of cerebral microvessels, total uronic acids (surrogates for perlecan content) were found to be increased in AD.33 Histologic examination of AD brains showed fragmentation of BM-associated agrin with immunoreactivity also found within Abeta plaques and neurofibrillary tangles. Soluble agrin levels were increased approximately 30% in Braak stage III-VI AD patients relative to age-matched controls.99 More recent studies support the finding that HSPGs are ubiquitously present in the BM, are increased in AD, and significantly correlated with staining for Abeta.115,128
Fibronectin
Fibronectin is a protein dimer comprised of 2 monomers linked by disulfide bonding, and is secreted by brain ECs, astrocytes and pericytes. Cellular fibronectin starts out soluble, but incorporates with other ECM to become part of the insoluble BM matrix.129 Fibronectin is directly involved in mediating cell attachment (via integrins) and functions to facilitate the organization of the interstitial ECM. Fibronectin directly affects the barrier properties of brain ECs as measured by TEER, and stimulates the proliferation and survival of brain ECs in vitro.130,131 Like Col IV, null mutations of fibronectin are embryonic lethal, partly due to impaired BM stability.132
Fibronectin changes in aging
Early studies reported a substantial decrease of fibronectin content in ECM that was thought to contribute to reduced size of brain ECs, changes in BBB function, and subsequent detrimental effects on memory.133 Later measures of fibronectin with aging showed no changes in the large 250 kDa monomer, at the same time there were decreases in the 60 kDa monomer, in the brain cortex microvasculature of aged mice relative to young mice. As with other ECM findings, the relationship between reduced small monomer fibronectin and decreased brain vessel density and function in aged mice was a correlative finding.97
Fibronectin changes in neurodegeneration
Fibronectin exists in plasma and is, therefore, a logical target as a biomarker for examining changes in brain BM with AD. Although an early study showed no differences between AD and controls, more recent reports found that high molecular fibronectin forms appear more frequently, and at higher amounts, in the plasma of AD patients compared to age‐matched healthy controls and those with mild cognitive impairment.134–136
Immunohistochemistry studies demonstrate cellular fibronectin staining correlates highly with Abeta in the temporal cortex, but not the frontal cortex, of AD patients. However, subsequent quantitative imaging showed high variability in fibronectin content, which demonstrated an increase in frontal, but not temporal, cortex in AD brains relative to age-matched controls.115 Although studies in humans with CAA are confounded by the influence of amyloid deposition on the BM, changes in fibronectin were not found in the human samples.90
Summary
The ECM is a dynamic component of the NVU and the BBB within the NVU. The glycocalyx is difficult to study in isolation and is comprised of ECM molecules that are intricately linked. Glycocalyx ECM components are exposed to environmental stresses that result in constant modifications in structure, which elude precise quantification and labeling with aging or neurodegeneration. BM ECM undergoes less modifications, but tends to show alterations with age and neurodegeneration that might reflect responses to longstanding vascular stress. Measures of any ECM component are complicated by regional variations within the brain and differing techniques to analyze ECM quantity versus quality, all of which contributes to variable findings with aging and neurodegeneration.
Funding Statement
This work was supported by the NIH grants [R21AG056883 to M.J.R., R01AG046619 to W.A.B.]
Disclosure statement
The authors report no conflict of interest.
References
- 1.Sweeney MD, Kisler K, Montagne A, Toga AW, Zlokovic BV.. The role of brain vasculature in neurodegenerative disorders. Nat Neurosci. 2018;21(10):1–17. doi: 10.1038/s41593-018-0234-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Erickson MA, Banks WA. Neuroimmune axes of the blood-brain barriers and blood-brain interfaces: bases for physiological regulation, disease states, and pharmacological interventions. Pharmacol Rev. 2018;70(2):278–314. doi: 10.1124/pr.117.014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stamatovic SM, Johnson AM, Keep RF, Andjelkovic AV. Junctional proteins of the blood-brain barrier: new insights into function and dysfunction. Tissue Barriers. 2016;4(1):e1154641. doi: 10.1080/21688370.2016.1154641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Erickson MA, Banks WA. Age-associated changes in the immune system and blood(-)brain barrier functions. Int J Mol Sci. 2019;20(7):1632. doi: 10.3390/ijms20071632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nag S. Morphology and properties of brain endothelial cells. Methods Mol Biol. 2011;686:3–47. doi: 10.1007/978-1-60761-938-3_1. [DOI] [PubMed] [Google Scholar]
- 6.Gastfriend BD, Palecek SP, Shusta EV. Modeling the blood-brain barrier: beyond the endothelial cells. Curr Opin Biomed Eng. 2018;5:6–12. doi: 10.1016/j.cobme.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oldendorf WH, Cornford ME, Brown WJ. The large apparent work capability of the blood-brain barrier: a study of the mitochondrial content of capillary endothelial cells in brain and other tissues of the rat. Ann Neurol. 1977;1(5):409–417. doi: 10.1002/ana.410010502. [DOI] [PubMed] [Google Scholar]
- 8.Bandopadhyay R, Orte C, Lawrenson JG, Reid AR, De Silva S, Allt G. Contractile proteins in pericytes at the blood-brain and blood-retinal barriers. J Neurocytol. 2001;30:35–44. [DOI] [PubMed] [Google Scholar]
- 9.Sweeney MD, Ayyadurai S, Zlokovic BV. Pericytes of the neurovascular unit: key functions and signaling pathways. Nat Neurosci. 2016;19(6):771–783. doi: 10.1038/nn.4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu S, Agalliu D, Yu C, Fisher M. The role of pericytes in blood-brain barrier function and stroke. Curr Pharm Des. 2012;18(25):3653–3662. doi: 10.2174/138161212802002706. [DOI] [PubMed] [Google Scholar]
- 11.Daneman R. The blood-brain barrier in health and disease. Ann Neurol. 2012;72(5):648–672. doi: 10.1002/ana.23648. [DOI] [PubMed] [Google Scholar]
- 12.Diaz-Flores L, Gutierrez R, Madrid JF, Varela H, Valladares F, Acosta E, Martin-Vasallo P, Diaz-Flores L Jr.. Pericytes. Morphofunction, interactions and pathology in a quiescent and activated mesenchymal cell niche. Histol Histopathol. 2009;24(7):909–969. doi: 10.14670/HH-24.909. [DOI] [PubMed] [Google Scholar]
- 13.Baeten KM, Akassoglou K. Extracellular matrix and matrix receptors in blood-brain barrier formation and stroke. Dev Neurobiol. 2011;71(11):1018–1039. doi: 10.1002/dneu.20954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Winkler EA, Bell RD, Zlokovic BV. Central nervous system pericytes in health and disease. Nat Neurosci. 2011;14(11):1398–1405. doi: 10.1038/nn.2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bobbie MW, Roy S, Trudeau K, Munger SJ, Simon AM, Roy S. Reduced connexin 43 expression and its effect on the development of vascular lesions in retinas of diabetic mice. Invest Ophthalmol Vis Sci. 2010;51(7):3758–3763. doi: 10.1167/iovs.09-4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cabezas R, Avila M, Gonzalez J, El-Bacha RS, Baez E, Garcia-Segura LM, Jurado Coronel JC, Capani F, Cardona-Gomez GP, Barreto GE. Astrocytic modulation of blood brain barrier: perspectives on Parkinson’s disease. Front Cell Neurosci. 2014;8:211. doi: 10.3389/fncel.2014.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kimelberg HK, Nedergaard M. Functions of astrocytes and their potential as therapeutic targets. Neurotherapeutics. 2010;7(4):338–353. doi: 10.1016/j.nurt.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hamby ME, Sofroniew MV. Reactive astrocytes as therapeutic targets for CNS disorders. Neurotherapeutics. 2010;7(4):494–506. doi: 10.1016/j.nurt.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deak F, Freeman WM, Ungvari Z, Csiszar A, Sonntag WE. Recent developments in understanding brain aging: implications for Alzheimer’s disease and vascular cognitive impairment. J Gerontol A Biol Sci Med Sci. 2016;71(1):13–20. doi: 10.1093/gerona/glv206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scioli MG, Bielli A, Arcuri G, Ferlosio A, Orlandi A. Ageing and microvasculature. Vasc Cell. 2014;6:19. doi: 10.1186/2045-824X-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hussain M, Bork K, Gnanapragassam VS, Bennmann D, Jacobs K, Navarette-Santos A, Hofmann B, Simm A, Danker K, Horstkorte R. Novel insights in the dysfunction of human blood-brain barrier after glycation. Mech Ageing Dev. 2016;155:48–54. doi: 10.1016/j.mad.2016.03.004. [DOI] [PubMed] [Google Scholar]
- 22.Brown WR, Thore CR. Review: cerebral microvascular pathology in ageing and neurodegeneration. Neuropathol Appl Neurobiol. 2011;37(1):56–74. doi: 10.1111/j.1365-2990.2010.01139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murugesan N, Demarest TG, Madri JA, Pachter JS. Brain regional angiogenic potential at the neurovascular unit during normal aging. Neurobiol Aging. 2012;33(5):1004.e1–1004.e16. doi: 10.1016/j.neurobiolaging.2011.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ungvari Z, Kaley G, de Cabo R, Sonntag WE, Csiszar A. Mechanisms of vascular aging: new perspectives. J Gerontol A Biol Sci Med Sci. 2010;65(10):1028–1041. doi: 10.1093/gerona/glq113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Biron KE, Dickstein DL, Gopaul R, Jefferies WA. Amyloid triggers extensive cerebral angiogenesis causing blood brain barrier permeability and hypervascularity in Alzheimer’s disease. PLoS One. 2011;6(8):e23789. doi: 10.1371/journal.pone.0023789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burke MJ, Nelson L, Slade JY, Oakley AE, Khundakar AA, Kalaria RN. Morphometry of the hippocampal microvasculature in post-stroke and age-related dementias. Neuropathol Appl Neurobiol. 2014;40(3):284–295. doi: 10.1111/nan.12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nelson AR, Sweeney MD, Sagare AP, Zlokovic BV. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim Biophys Acta. 2016;1862(5):887–900. doi: 10.1016/j.bbadis.2015.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu C, Li Q, Efimova O, He L, Tatsumoto S, Stepanova V, Oishi T, Udono T, Yamaguchi K, Shigenobu S, et al. Human-specific features of spatial gene expression and regulation in eight brain regions. Genome Res. 2018;28(8):1097–1110. doi: 10.1101/gr.231357.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morawski M, Bruckner G, Jager C, Seeger G, Matthews RT, Arendt T. Involvement of perineuronal and perisynaptic extracellular matrix in Alzheimer’s disease neuropathology. Brain Pathol. 2012;22(4):547–561. doi: 10.1111/j.1750-3639.2011.00557.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ando Y, Okada H, Takemura G, Suzuki K, Takada C, Tomita H, Zaikokuji R, Hotta Y, Miyazaki N, Yano H, et al. Brain-specific ultrastructure of capillary endothelial glycocalyx and its possible contribution for blood brain barrier. Sci Rep. 2018;8(1):17523. doi: 10.1038/s41598-018-35976-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Uchimido R, Schmidt EP, Shapiro NI. The glycocalyx: a novel diagnostic and therapeutic target in sepsis. Crit Care. 2019;23(1):16. doi: 10.1186/s13054-018-2292-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reitsma S, Slaaf DW, Vink H, van Zandvoort MA, Oude Egbrink MG. The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch. 2007;454(3):345–359. doi: 10.1007/s00424-007-0212-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kalaria RN, Pax AB. Increased collagen content of cerebral microvessels in Alzheimer’s disease. Brain Res. 1995;705(1–2):349–352. doi: 10.1016/0006-8993(95)01250-8. [DOI] [PubMed] [Google Scholar]
- 34.Kolarova H, Ambruzova B, Svihalkova Sindlerova L, Klinke A, Kubala L. Modulation of endothelial glycocalyx structure under inflammatory conditions. Mediators Inflamm. 2014;2014:1–17. doi: 10.1155/2014/694312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao L, Lipowsky HH. Composition of the endothelial glycocalyx and its relation to its thickness and diffusion of small solutes. Microvasc Res. 2010;80(3):394–401. doi: 10.1016/j.mvr.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mulivor AW, Lipowsky HH. Inflammation- and ischemia-induced shedding of venular glycocalyx. Am J Physiol Heart Circ Physiol. 2004;286(5):H1672–H1680. doi: 10.1152/ajpheart.00832.2003. [DOI] [PubMed] [Google Scholar]
- 37.Rapraeger A. Transforming growth factor (type beta) promotes the addition of chondroitin sulfate chains to the cell surface proteoglycan (syndecan) of mouse mammary epithelia. J Cell Biol. 1989;109(5):2509–2518. doi: 10.1083/jcb.109.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Voyvodic PL, Min D, Liu R, Williams E, Chitalia V, Dunn AK, Baker AB. Loss of syndecan-1 induces a pro-inflammatory phenotype in endothelial cells with a dysregulated response to atheroprotective flow. J Biol Chem. 2014;289(14):9547–9559. doi: 10.1074/jbc.M113.541573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garantziotis S, Savani RC. Hyaluronan biology: A complex balancing act of structure, function, location and context. Matrix Biol. 2019;78–79:1–10. doi: 10.1016/j.matbio.2019.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chappell D, Jacob M, Paul O, Rehm M, Welsch U, Stoeckelhuber M, Conzen P, Becker BF. The glycocalyx of the human umbilical vein endothelial cell: an impressive structure ex vivo but not in culture. Circ Res. 2009;104(11):1313–1317. doi: 10.1161/CIRCRESAHA.108.187831. [DOI] [PubMed] [Google Scholar]
- 41.Henry CB, Duling BR. Permeation of the luminal capillary glycocalyx is determined by hyaluronan. Am J Physiol. 1999;277(2):H508–H514. doi: 10.1152/ajpheart.1999.277.2.H508. [DOI] [PubMed] [Google Scholar]
- 42.Stern R, Jedrzejas MJ. Hyaluronidases: their genomics, structures, and mechanisms of action. Chem Rev. 2006;106(3):818–839. doi: 10.1021/cr050247k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Al’Qteishat A, Gaffney J, Krupinski J, Rubio F, West D, Kumar S, Kumar P, Mitsios N, Slevin M. Changes in hyaluronan production and metabolism following ischaemic stroke in man. Brain. 2006;129(Pt 8):2158–2176. doi: 10.1093/brain/awl139. [DOI] [PubMed] [Google Scholar]
- 44.Evanko SP, Tammi MI, Tammi RH, Wight TN. Hyaluronan-dependent pericellular matrix. Adv Drug Deliv Rev. 2007;59(13):1351–1365. doi: 10.1016/j.addr.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Misra S, Hascall VC, Markwald RR, Ghatak S. Interactions between Hyaluronan and Its Receptors (CD44, RHAMM) Regulate the Activities of Inflammation and Cancer. Front Immunol. 2015;6:201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Al-Ahmad AJ, Patel R, Palecek SP, Shusta EV. Hyaluronan impairs the barrier integrity of brain microvascular endothelial cells through a CD44-dependent pathway. J Cerebr Blood Flow Metab. 2019;39(9):1759–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Back SA, Tuohy TMF, Chen H, Wallingford N, Craig A, Struve J, Luo NL, Banine F, Liu Y, Chang A, et al. Hyaluronan accumulates in demyelinated lesions and inhibits oligodendrocyte progenitor maturation. Nat Med. 2005;11(9):966–972. doi: 10.1038/nm1279. [DOI] [PubMed] [Google Scholar]
- 48.Jiang D, Liang J, Noble PW. Hyaluronan as an immune regulator in human diseases. Physiol Rev. 2011;91(1):221–264. doi: 10.1152/physrev.00052.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Struve J, Maher PC, Li YQ, Kinney S, Fehlings MG, Kuntz CT, Sherman LS. Disruption of the hyaluronan-based extracellular matrix in spinal cord promotes astrocyte proliferation. Glia. 2005;52(1):16–24. doi: 10.1002/glia.20215. [DOI] [PubMed] [Google Scholar]
- 50.Winkler CW, Foster SC, Itakura A, Matsumoto SG, Asari A, McCarty OJ, Sherman LS. Hyaluronan oligosaccharides perturb lymphocyte slow rolling on brain vascular endothelial cells: implications for inflammatory demyelinating disease. Matrix Biol. 2013;32(3–4):160–168. doi: 10.1016/j.matbio.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dicker KT, Gurski LA, Pradhan-Bhatt S, Witt RL, Farach-Carson MC, Jia X. Hyaluronan: a simple polysaccharide with diverse biological functions. Acta Biomater. 2014;10(4):1558–1570. doi: 10.1016/j.actbio.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Watanabe J, Shetty AK, Hattiangady B, Kim DK, Foraker JE, Nishida H, Prockop DJ. Administration of TSG-6 improves memory after traumatic brain injury in mice. Neurobiol Dis. 2013;59:86–99. doi: 10.1016/j.nbd.2013.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Day AJ, Milner CM. TSG-6: A multifunctional protein with anti-inflammatory and tissue-protective properties. Matrix Biol. 2019; 78-79:60–83. [DOI] [PubMed] [Google Scholar]
- 54.Coulson-Thomas VJ, Lauer ME, Soleman S, Zhao C, Hascall VC, Day AJ, Fawcett JW. Tumor necrosis factor-stimulated gene-6 (TSG-6) Is Constitutively Expressed in Adult Central Nervous System (CNS) and associated with astrocyte-mediated Glial scar formation following spinal cord injury. J Biol Chem. 2016;291(38):19939–19952. doi: 10.1074/jbc.M115.710673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baranova NS, Nileback E, Haller FM, Briggs DC, Svedhem S, Day AJ, Richter RP. The inflammation-associated protein TSG-6 cross-links hyaluronan via hyaluronan-induced TSG-6 oligomers. J Biol Chem. 2011;286(29):25675–25686. doi: 10.1074/jbc.M111.247395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lesley J, Gal I, Mahoney DJ, Cordell MR, Rugg MS, Hyman R, Day AJ, Mikecz K. TSG-6 modulates the interaction between hyaluronan and cell surface CD44. J Biol Chem. 2004;279(24):25745–25754. doi: 10.1074/jbc.M313319200. [DOI] [PubMed] [Google Scholar]
- 57.Lawrance W, Banerji S, Day AJ, Bhattacharjee S, Jackson DG. Binding of hyaluronan to the native lymphatic vessel endothelial receptor LYVE-1 dependent on receptor clustering is critically and hyaluronan organization. J Biol Chem. 2016;291(15):8014–8030. doi: 10.1074/jbc.M115.708305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kutuzov N, Flyvbjerg H, Lauritzen M. Contributions of the glycocalyx, endothelium, and extravascular compartment to the blood-brain barrier. Proc Natl Acad Sci U S A. 2018;115(40):E9429–E9438. doi: 10.1073/pnas.1802155115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Siebert JR, Conta Steencken A, Osterhout DJ. Chondroitin sulfate proteoglycans in the nervous system: inhibitors to repair. Biomed Res Int. 2014;2014:1–15. doi: 10.1155/2014/845323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Avram S, Shaposhnikov S, Buiu C, Mernea M. Chondroitin sulfate proteoglycans: structure-function relationship with implication in neural development and brain disorders. Biomed Res Int. 2014;2014:1–11. doi: 10.1155/2014/642798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vogl-Willis CA, Edwards IJ. High-glucose-induced structural changes in the heparan sulfate proteoglycan, perlecan, of cultured human aortic endothelial cells. Biochim Biophys Acta. 2004;1672(1):36–45. doi: 10.1016/j.bbagen.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 62.Ballinger ML, Nigro J, Frontanilla KV, Dart AM, Little PJ. Regulation of glycosaminoglycan structure and atherogenesis. Cell Mol Life Sci. 2004;61(11):1296–1306. doi: 10.1007/s00018-004-3389-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Machin DR, Bloom SI, Campbell RA, Phuong TTT, Gates PE, Lesniewski LA, Rondina MT, Donato AJ. Advanced age results in a diminished endothelial glycocalyx. Am J Physiol Heart Circ Physiol. 2018;315(3):H531–H539. doi: 10.1152/ajpheart.00104.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jenkins HG, Bachelard HS. Developmental and age-related changes in rat brain glycosaminoglycans. J Neurochem. 1988;51(5):1634–1640. doi: 10.1111/j.1471-4159.1988.tb01134.x. [DOI] [PubMed] [Google Scholar]
- 65.Cargill R, Kohama SG, Struve J, Su W, Banine F, Witkowski E, Back SA, Sherman LS. Astrocytes in aged nonhuman primate brain gray matter synthesize excess hyaluronan. Neurobiol Aging. 2012;33(4):830.e13–830.e24. doi: 10.1016/j.neurobiolaging.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lindwall C, Olsson M, Osman AM, Kuhn HG, Curtis MA. Selective expression of hyaluronan and receptor for hyaluronan mediated motility (Rhamm) in the adult mouse subventricular zone and rostral migratory stream and in ischemic cortex. Brain Res. 2013;1503:62–77. doi: 10.1016/j.brainres.2013.01.045. [DOI] [PubMed] [Google Scholar]
- 67.Reed MJ, Damodarasamy M, Pathan JL, Erickson MA, Banks WA, Vernon RB. The effects of of hyaluronan and chondroitin normal aging on regional accumulation sulfate proteoglycans in the mouse brain. J Histochem Cytochem. 2018;66(10):697–707. doi: 10.1369/0022155418774779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Reed MJ, Vernon RB, Damodarasamy M, Chan CK, Wight TN, Bentov I, Banks WA. Microvasculature of the mouse cerebral cortex exhibits increased accumulation and synthesis of hyaluronan with aging. J Gerontol A Biol Sci Med Sci. 2017;72(6):740–746. doi: 10.1093/gerona/glw213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Howell MD, Gottschall PE. Lectican proteoglycans, their cleaving metalloproteinases, and plasticity in the central nervous system extracellular microenvironment. Neuroscience. 2012;217:6–18. doi: 10.1016/j.neuroscience.2012.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cicanic M, Edamatsu M, Bekku Y, Vorisek I, Oohashi T, Vargova L. A deficiency of the link protein Bral2 affects the size of the extracellular space in the thalamus of aged mice. J Neurosci Res. 2018;96(2):313–327. doi: 10.1002/jnr.24136. [DOI] [PubMed] [Google Scholar]
- 71.Foscarin S, Raha-Chowdhury R, Fawcett JW, Kwok JCF. Brain ageing changes proteoglycan sulfation, rendering perineuronal nets more inhibitory. Aging. 2017;9(6):1607–1622. doi: 10.18632/aging.101256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang S, Liu T, Li S, Zhang X, Ding Q, Que H, Yan X, Wei K, Liu S. Comparative proteomic analysis of brains of naturally aging mice. Neuroscience. 2008;154(3):1107–1120. doi: 10.1016/j.neuroscience.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 73.Stauch KL, Purnell PR, Villeneuve LM, Fox HS. Proteomic analysis and functional characterization of mouse brain mitochondria during aging reveal alterations in energy metabolism. Proteomics. 2015;15(9):1574–1586. doi: 10.1002/pmic.201400277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fernandez-Lopez D, Faustino J, Daneman R, Zhou L, Lee SY, Derugin N, Wendland MF, Vexler ZS. Blood-brain barrier permeability is increased after acute adult stroke but not neonatal stroke in the rat. J Neurosci. 2012;32(28):9588–9600. doi: 10.1523/JNEUROSCI.5977-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sloane JA, Batt C, Ma Y, Harris ZM, Trapp B, Vartanian T. Hyaluronan blocks oligodendrocyte progenitor maturation and remyelination through TLR2. Proc Natl Acad Sci U S A. 2010;107(25):11555–11560. doi: 10.1073/pnas.1006496107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Frischknecht R, Gundelfinger ED. The brain’s extracellular matrix and its role in synaptic plasticity. Adv Exp Med Biol. 2012;970:153–171. doi: 10.1007/978-3-7091-0932-8_7. [DOI] [PubMed] [Google Scholar]
- 77.Sunabori T, Koike M, Asari A, Oonuki Y, Uchiyama Y. Suppression of Ischemia-Induced Hippocampal pyramidal neuron death by hyaluronan tetrasaccharide through inhibition of toll-like receptor 2 signaling pathway. Am J Pathol. 2016;186(8):2143–2151. doi: 10.1016/j.ajpath.2016.03.016. [DOI] [PubMed] [Google Scholar]
- 78.Vinukonda G, Dohare P, Arshad A, Zia MT, Panda S, Korumilli R, Kayton R, Hascall VC, Lauer ME, Ballabh P. Hyaluronidase and hyaluronan oligosaccharides promote neurological recovery after intraventricular Hemorrhage. J Neurosci. 2016;36(3):872–889. doi: 10.1523/JNEUROSCI.3297-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Winkler CW, Foster SC, Matsumoto SG, Preston MA, Xing R, Bebo BF, Banine F, Berny-Lang MA, Itakura A, McCarty OJT, et al. Hyaluronan anchored to activated CD44 on central nervous system vascular endothelial cells promotes lymphocyte extravasation in experimental autoimmune encephalomyelitis. J Biol Chem. 2012;287(40):33237–33251. doi: 10.1074/jbc.M112.356287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Reed MJ, Damodarasamy M, Pathan JL, Chan CK, Spiekerman C, Wight TN, Banks WA, Day AJ, Vernon RB, Keene CD. Increased hyaluronan and TSG-6 in association with neuropathologic changes of Alzheimer’s disease. J Alzheimer’s Dis. 2019;67(1):91–102. doi: 10.3233/JAD-180797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Frost GR, Li YM. The role of astrocytes in amyloid production and Alzheimer’s disease. Open Biol. 2017;7(12):170228. doi: 10.1098/rsob.170228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fuller ML, DeChant AK, Rothstein B, Caprariello A, Wang R, Hall AK, Miller RH. Bone morphogenetic proteins promote gliosis in demyelinating spinal cord lesions. Ann Neurol. 2007;62(3):288–300. doi: 10.1002/ana.21179. [DOI] [PubMed] [Google Scholar]
- 83.Hondius DC, van Nierop P, Li KW, Hoozemans JJ, van der Schors RC, van Haastert ES, van der Vies SM, Rozemuller AJ, Smit AB. Profiling the human hippocampal proteome at all pathologic stages of Alzheimer’s disease. Alzheimer’s Dement. 2016;12(6):654–668. doi: 10.1016/j.jalz.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 84.Zhang Q, Ma C, Gearing M, Wang PG, Chin LS, Li L. Integrated proteomics and network analysis identifies protein hubs and network alterations in Alzheimer’s disease. Acta Neuropathol Commun. 2018;6(1):19. doi: 10.1186/s40478-018-0524-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Manavalan A, Mishra M, Feng L, Sze SK, Akatsu H, Heese K. Brain site-specific proteome changes in aging-related dementia. Exp Mol Med. 2013;45:e39. doi: 10.1038/emm.2013.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Begcevic I, Kosanam H, Martinez-Morillo E, Dimitromanolakis A, Diamandis P, Kuzmanov U, Hazrati LN, Diamandis EP. Semiquantitative proteomic analysis of human hippocampal tissues from Alzheimer’s disease and age-matched control brains. Clin Proteomics. 2013;10(1):5. doi: 10.1186/1559-0275-10-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Xu L, Nirwane A, Yao Y. Basement membrane and blood–brain barrier. Stroke Vasc Neurol. 2018;4:78–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kurtz A, Oh SJ. Age related changes of the extracellular matrix and stem cell maintenance. Prev Med. 2012;54(Suppl):S50–S56. doi: 10.1016/j.ypmed.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 89.Uspenskaia O, Liebetrau M, Herms J, Danek A, Hamann GF. Aging is associated with increased collagen type IV accumulation in the basal lamina of human cerebral microvessels. BMC Neurosci. 2004;5:37. doi: 10.1186/1471-2202-5-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Keable A, Fenna K, Yuen HM, Johnston DA, Smyth NR, Smith C, Al-Shahi Salman R, Samarasekera N, Nicoll JAR, Attems J, et al. Deposition of amyloid beta in the walls of human leptomeningeal arteries in relation to perivascular drainage pathways in cerebral amyloid angiopathy. Biochim Biophys Acta. 2016;1862(5):1037–1046. doi: 10.1016/j.bbadis.2015.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hawkes CA, Gatherer M, Sharp MM, Dorr A, Yuen HM, Kalaria R, Weller RO, Carare RO. Regional differences in the morphological and functional effects of aging on cerebral basement membranes and perivascular drainage of amyloid-beta from the mouse brain. Aging Cell. 2013;12(2):224–236. doi: 10.1111/acel.12045. [DOI] [PubMed] [Google Scholar]
- 92.Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, et al. National institute on aging-Alzheimer’s association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 2012;123(1):1–11. doi: 10.1007/s00401-011-0910-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Thomsen MS, Routhe LJ, Moos T. The vascular basement membrane in the healthy and pathological brain. J Cerebr Blood Flow Metab. 2017a;37(10):3300–3317. doi: 10.1177/0271678X17722436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tian J, Shi J, Smallman R, Iwatsubo T, Mann DM. Relationships in Alzheimer’s disease between the extent of Abeta deposition in cerebral blood vessel walls, as cerebral amyloid angiopathy, and the amount of cerebrovascular smooth muscle cells and collagen. Neuropathol Appl Neurobiol. 2006;32(3):332–340. doi: 10.1111/j.1365-2990.2006.00732.x. [DOI] [PubMed] [Google Scholar]
- 95.Gersdorff N, Kohfeldt E, Sasaki T, Timpl R, Miosge N. Laminin gamma3 chain binds to nidogen and is located in murine basement membranes. J Biol Chem. 2005;280(23):22146–22153. doi: 10.1074/jbc.M501875200. [DOI] [PubMed] [Google Scholar]
- 96.Thomsen MS, Birkelund S, Burkhart A, Stensballe A, Moos T. Synthesis and deposition of basement membrane proteins by primary brain capillary endothelial cells in a murine model of the blood-brain barrier. J Neurochem. 2017b;140(5):741–754. doi: 10.1111/jnc.13747. [DOI] [PubMed] [Google Scholar]
- 97.Hawkes CA, Hartig W, Kacza J, Schliebs R, Weller RO, Nicoll JA, Carare RO. Perivascular drainage of solutes is impaired in the ageing mouse brain and in the presence of cerebral amyloid angiopathy. Acta Neuropathol. 2011;121(4):431–443. doi: 10.1007/s00401-011-0801-7. [DOI] [PubMed] [Google Scholar]
- 98.Palu E, Liesi P. Differential distribution of laminins in Alzheimer disease and normal human brain tissue. J Neurosci Res. 2002;69(2):243–256. doi: 10.1002/jnr.10292. [DOI] [PubMed] [Google Scholar]
- 99.Berzin TM, Zipser BD, Rafii MS, Kuo-Leblanc V, Yancopoulos GD, Glass DJ, Fallon JR, Stopa EG. Agrin and microvascular damage in Alzheimer’s disease. Neurobiol Aging. 2000;21:349–355. [DOI] [PubMed] [Google Scholar]
- 100.Marchand M, Monnot C, Muller L, Germain S. Extracellular matrix scaffolding in angiogenesis and capillary homeostasis. Semin Cell Dev Biol. 2018; 89:147–156. [DOI] [PubMed] [Google Scholar]
- 101.Rozario T, DeSimone DW. The extracellular matrix in development and morphogenesis: a dynamic view. Dev Biol. 2010;341(1):126–140. doi: 10.1016/j.ydbio.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cummings CF, Pedchenko V, Brown KL, Colon S, Rafi M, Jones-Paris C, Pokydeshava E, Liu M, Pastor-Pareja JC, Stothers C, et al. Extracellular chloride signals collagen IV network assembly during basement membrane formation. J Cell Biol. 2016;213(4):479–494. doi: 10.1083/jcb.201510065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Alamowitch S, Plaisier E, Favrole P, Prost C, Chen Z, Van Agtmael T, Marro B, Ronco P. Cerebrovascular disease related to COL4A1 mutations in HANAC syndrome. Neurology. 2009;73(22):1873–1882. doi: 10.1212/WNL.0b013e3181c3fd12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gould DB, Phalan FC, van Mil SE, Sundberg JP, Vahedi K, Massin P, Bousser MG, Heutink P, Miner JH, Tournier-Lasserve E, et al. Role of COL4A1 in small-vessel disease and hemorrhagic stroke. N Engl J Med. 2006;354(14):1489–1496. doi: 10.1056/NEJMoa053727. [DOI] [PubMed] [Google Scholar]
- 105.Poschl E, Schlotzer-Schrehardt U, Brachvogel B, Saito K, Ninomiya Y, Mayer U. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development. 2004;131(7):1619–1628. doi: 10.1242/dev.01037. [DOI] [PubMed] [Google Scholar]
- 106.Bouras C, Kovari E, Herrmann FR, Rivara CB, Bailey TL, von Gunten A, Hof PR, Giannakopoulos P. Stereologic analysis of microvascular morphology in the elderly: Alzheimer disease pathology and cognitive status. J Neuropathol Exp Neurol. 2006;65(3):235–244. doi: 10.1097/01.jnen.0000203077.53080.2c. [DOI] [PubMed] [Google Scholar]
- 107.Bennett RE, Robbins AB, Hu M, Cao X, Betensky RA, Clark T, Das S, Hyman BT. Tau induces blood vessel abnormalities and angiogenesis-related gene expression in P301L transgenic mice and human Alzheimer’s disease. Proc Natl Acad Sci U S A. 2018;115(6):E1289–E1298. doi: 10.1073/pnas.1710329115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Thal DR, Capetillo-Zarate E, Larionov S, Staufenbiel M, Zurbruegg S, Beckmann N. Capillary cerebral amyloid angiopathy is associated with vessel occlusion and cerebral blood flow disturbances. Neurobiol Aging. 2009;30(12):1936–1948. doi: 10.1016/j.neurobiolaging.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 109.Rockenstein E, Adame A, Mante M, Larrea G, Crews L, Windisch M, Moessler H, Masliah E. Amelioration of the cerebrovascular amyloidosis in a transgenic model of Alzheimer’s disease with the neurotrophic compound cerebrolysin. J Neural Transm. 2005;112(2):269–282. doi: 10.1007/s00702-004-0181-4. [DOI] [PubMed] [Google Scholar]
- 110.Bourasset F, Ouellet M, Tremblay C, Julien C, Do TM, Oddo S, LaFerla F, Calon F. Reduction of the cerebrovascular volume in a transgenic mouse model of Alzheimer’s disease. Neuropharmacology. 2009;56(4):808–813. doi: 10.1016/j.neuropharm.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 111.Tong XK, Nicolakakis N, Kocharyan A, Hamel E. Vascular remodeling versus amyloid beta-induced oxidative stress in the cerebrovascular dysfunctions associated with Alzheimer’s disease. J Neurosci. 2005;25(48):11165–11174. doi: 10.1523/JNEUROSCI.4031-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Challa VR, Thore CR, Moody DM, Anstrom JA, Brown WR. Increase of white matter string vessels in Alzheimer’s disease. J Alzheimer’s Dis. 2004;6(4):379–383; discussion 443-379. [DOI] [PubMed] [Google Scholar]
- 113.Farkas E, De Jong GI, de Vos RA, Jansen Steur EN, Luiten PG. Pathological features of cerebral cortical capillaries are doubled in Alzheimer’s disease and Parkinson’s disease. Acta Neuropathol. 2000;100(4):395–402. doi: 10.1007/s004010000195. [DOI] [PubMed] [Google Scholar]
- 114.Zarow C, Barron E, Chui HC, Perlmutter LS. Vascular basement membrane pathology and Alzheimer’s disease. Ann N Y Acad Sci. 1997;826:147–160. doi: 10.1111/j.1749-6632.1997.tb48467.x. [DOI] [PubMed] [Google Scholar]
- 115.Lepelletier FX, Mann DM, Robinson AC, Pinteaux E, Boutin H. Early changes in extracellular matrix in Alzheimer’s disease. Neuropathol Appl Neurobiol. 2017;43(2):167–182. doi: 10.1111/nan.12295. [DOI] [PubMed] [Google Scholar]
- 116.Christov A, Ottman J, Hamdheydari L, Grammas P. Structural changes in Alzheimer’s disease brain microvessels. Curr Alzheimer Res. 2008;5:392–395. [DOI] [PubMed] [Google Scholar]
- 117.Kiuchi Y, Isobe Y, Fukushima K. Type IV collagen prevents amyloid beta-protein fibril formation. Life Sci. 2002;70(13):1555–1564. doi: 10.1016/s0024-3205(01)01528-4. [DOI] [PubMed] [Google Scholar]
- 118.Grimpe B, Probst JC, Hager G. Suppression of nidogen-1 translation by antisense targeting affects the adhesive properties of cultured astrocytes. Glia. 1999;28:138–149. [DOI] [PubMed] [Google Scholar]
- 119.Niquet J, Represa A. Entactin immunoreactivity in immature and adult rat brain. Brain Res Dev Brain Res. 1996;95:227–233. [DOI] [PubMed] [Google Scholar]
- 120.Dong L, Chen Y, Lewis M, Hsieh J-C, Reing J, Chaillet JR, Howell CY, Melhem M, Inoue S, Kuszak JR, et al. Neurologic defects and selective disruption of basement membranes in mice lacking entactin-1/nidogen-1. Lab Invest. 2002;82(12):1617–1630. doi: 10.1097/01.LAB.0000042240.52093.0F. [DOI] [PubMed] [Google Scholar]
- 121.Schymeinsky J, Nedbal S, Miosge N, Poschl E, Rao C, Beier DR, Skarnes WC, Timpl R, Bader BL. Gene structure and functional analysis of the mouse nidogen-2 gene: nidogen-2 is not essential for basement membrane formation in mice. Mol Cell Biol. 2002;22(19):6820–6830. doi: 10.1128/mcb.22.19.6820-6830.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kohling R, Nischt R, Vasudevan A, Ho M, Weiergraber M, Schneider T, Smyth N. Nidogen and nidogen-associated basement membrane proteins and neuronal plasticity. Neurodegener Dis. 2006;3(1–2):56–61. doi: 10.1159/000092094. [DOI] [PubMed] [Google Scholar]
- 123.Kiuchi Y, Isobe Y, Fukushima K. Entactin-induced inhibition of human amyloid beta-protein fibril formation in vitro. Neurosci Lett. 2001;305(2):119–122. doi: 10.1016/s0304-3940(01)01831-6. [DOI] [PubMed] [Google Scholar]
- 124.Farach-Carson MC, Warren CR, Harrington DA, Carson DD. Border patrol: insights into the unique role of perlecan/heparan sulfate proteoglycan 2 at cell and tissue borders. Matrix Biol. 2014;34:64–79. doi: 10.1016/j.matbio.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Barber AJ, Lieth E. Agrin accumulates in the brain microvascular basal lamina during development of the blood-brain barrier. Dev Dyn. 1997;208(1):62–74. doi:. [DOI] [PubMed] [Google Scholar]
- 126.Denzer AJ, Brandenberger R, Gesemann M, Chiquet M, Ruegg MA. Agrin binds to the nerve-muscle basal lamina via laminin. J Cell Biol. 1997;137(3):671–683. doi: 10.1083/jcb.137.3.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Maresh GA, Erezyilmaz D, Murry CE, Nochlin D, Snow AD. Detection and quantitation of perlecan mRNA levels in Alzheimer’s disease and normal aged hippocampus by competitive reverse transcription-polymerase chain reaction. J Neurochem. 1996;67(3):1132–1144. doi: 10.1046/j.1471-4159.1996.67031132.x. [DOI] [PubMed] [Google Scholar]
- 128.Zhang GL, Zhang X, Wang XM, Li JP. Towards understanding the roles of heparan sulfate proteoglycans in Alzheimer’s disease. Biomed Res Int. 2014;2014:516028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Singh P, Carraher C, Schwarzbauer JE. Assembly of fibronectin extracellular matrix. Annu Rev Cell Dev Biol. 2010;26:397–419. doi: 10.1146/annurev-cellbio-100109-104020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Tilling T, Korte D, Hoheisel D, Galla HJ. Basement membrane proteins influence brain capillary endothelial barrier function in vitro. J Neurochem. 1998;71(3):1151–1157. doi: 10.1046/j.1471-4159.1998.71031151.x. [DOI] [PubMed] [Google Scholar]
- 131.Wang J, Milner R. Fibronectin promotes brain capillary endothelial cell survival and proliferation through alpha5beta1 and alphavbeta3 integrins via MAP kinase signalling. J Neurochem. 2006;96(1):148–159. doi: 10.1111/j.1471-4159.2005.03521.x. [DOI] [PubMed] [Google Scholar]
- 132.George EL, Georges-Labouesse EN, Patel-King RS, Rayburn H, Hynes RO. Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development. 1993;119:1079–1091. [DOI] [PubMed] [Google Scholar]
- 133.Sykova E, Mazel T, Simonova Z. Diffusion constraints and neuron-glia interaction during aging. Exp Gerontol. 1998;33:837–851. [DOI] [PubMed] [Google Scholar]
- 134.Muenchhoff J, Poljak A, Song F, Raftery M, Brodaty H, Duncan M, McEvoy M, Attia J, Schofield PW, Sachdev PS. Plasma protein profiling of mild cognitive impairment and Alzheimer’s disease across two independent cohorts. J Alzheimer’s Dis. 2015;43(4):1355–1373. doi: 10.3233/JAD-141266. [DOI] [PubMed] [Google Scholar]
- 135.Lemanska-Perek A, Leszek J, Krzyzanowska-Golab D, Radzik J, Katnik-Prastowska I. Molecular status of plasma fibronectin as an additional biomarker for assessment of Alzheimer’s dementia risk. Dement Geriatr Cogn Disord. 2009;28(4):338–342. doi: 10.1159/000252764. [DOI] [PubMed] [Google Scholar]
- 136.Rattan SI, Rasmussen LM, Bjerring P, Bhatia P, Clark BF. Unchanged concentrations of plasma fibronectin in Alzheimer’s disease. J Clin Pathol. 1988;41(4):476. doi: 10.1136/jcp.41.4.476. [DOI] [PMC free article] [PubMed] [Google Scholar]