

Abstract
A decade of research and development in resting‐state functional MRI (RSfMRI) has opened new translational and clinical research frontiers. This review aims to bridge between technical and clinical researchers who seek reliable neuroimaging biomarkers for studying drug interactions with the brain. About 85 pharma‐RSfMRI studies using BOLD signal (75% of all) or arterial spin labeling (ASL) were surveyed to investigate the acute effects of psychoactive drugs. Experimental designs and objectives include drug fingerprinting dose‐response evaluation, biomarker validation and calibration, and translational studies. Common biomarkers in these studies include functional connectivity, graph metrics, cerebral blood flow and the amplitude and spectrum of BOLD fluctuations. Overall, RSfMRI‐derived biomarkers seem to be sensitive to spatiotemporal dynamics of drug interactions with the brain. However, drugs cause both central and peripheral effects, thus exacerbate difficulties related to biological confounds, structured noise from motion and physiological confounds, as well as modeling and inference testing. Currently, these issues are not well explored, and heterogeneities in experimental design, data acquisition and preprocessing make comparative or meta‐analysis of existing reports impossible. A unifying collaborative framework for data‐sharing and data‐mining is thus necessary for investigating the commonalities and differences in biomarker sensitivity and specificity, and establishing guidelines. Multimodal datasets including sham‐placebo or active control sessions and repeated measurements of various psychometric, physiological, metabolic and neuroimaging phenotypes are essential for pharmacokinetic/pharmacodynamic modeling and interpretation of the findings. We provide a list of basic minimum and advanced options that can be considered in design and analyses of future pharma‐RSfMRI studies. Hum Brain Mapp 38:2276–2325, 2017. © 2017 Wiley Periodicals, Inc.
Keywords: resting state fMRI, arterial spin labeling, drug, pharmacological neuroimaging, biomarkers, translational research, PK/PD modeling, functional connectivity, brain chemistry, pharma‐fMRI
INTRODUCTION
To study the neurochemical substrates of the brain function is one of the most important aspects of understanding inter‐individual variations in behavior or clinical outcomes of psychotherapeutic interventions. Direct evaluation of brain chemistry in vivo has been made possible by positron emission tomography (PET). In PET, the dynamics of the regional uptake of neurotransmitter‐specific radioligands serve as evidence of the local neurochemical modulation of the brain activity. Generally, PET provides a quantifiable measure of glucose metabolism through the use of 18FDG, a direct reflection of regional glutamate transmission. When receptor‐specific radioligands exist (e.g., 11C‐CARFENTANYL and 11C‐METHYLNALTRINDOLE for opioid; 11C‐RACLOPRIDE, 18F‐FALLYPRIDE for Dopamine R2; 11C‐SCH39166, 11C‐NNC‐112 for Dopamine R1; 11C‐WAY635 for serotonin 5HT1A; 11C‐AZ10419369 for 5HT1B; 18f‐SETOPERONE for 5HT2A; 18F‐FLUMAZENIL for GABA, etc.), PET serves as the gold standard for studying brain chemistry. The neural correlates of anesthetics [Alkire et al., 1995], analgesics [Hartvig et al., 1995; Wagner et al., 2001], drug‐induced psychosis [Lahti et al., 1995; Onoe et al., 1994; Vollenweider et al., 1997], depression [Fowler et al., 1987; Hartvig et al., 1995] or euphoric intoxication [Volkow et al., 1995, 1991] have long been investigated with this technology. However, PET is not an easy or cheap research tool. Appropriate ligands do not always exist, or if they do, facilities that can produce them are not accessible broadly. Even when the technology is accessible, experimental designs are constrained by the half‐life of both the drug and the tracer isotope. The time‐dependency of PET experiments can sometimes make them more difficult to conduct and analyze. Furthermore, the complex dynamics of drug actions on peripheral and autonomic autoregulation (which will be discussed in the following sections) increase the challenge of tracer kinetic modeling with respect to non‐specific uptake within a so‐called “reference region.” Importantly, dose‐radiation restrictions make PET unsuitable for repeated crossover study designs in humans. In fact, some Ethics Research Boards restrict PET scans in healthy individuals. These practical considerations have shifted the spotlight onto pharmacological fMRI (pharma‐fMRI) as an affordable exploratory option for accelerating bench‐to‐bed clinical research applications [Wise and Tracey, 2006].1
MRI methods are not without challenge either. Earlier reviews have thoroughly covered the practical and theoretical challenges and promises of task‐based pharma‐fMRI [Borsook et al., 2008, 2011; Iannetti and Wise, 2007] and resting‐state fMRI [Liu, 2013; Lu and Stein, 2014]. The most important limitation is that fMRI signals hinge on the assumption that neuronal metabolism and cerebral blood flow are proportionally related to electrophysiological activity of neurons—but these relations are far more complex than generally considered [Logothetis, 2008]. The question is whether existing RSfMRI studies of acute drug effects provide enough evidence to make this method a valid alternative to PET for studying the neurochemistry of the brain. We approached this question by surveying the state‐of‐the‐art resting‐state methods that have been used in pharmacological RSfMRI (Pharma‐RSfMRI) studies of acute drug effects on the brain. The survey is organized in a manner to bridge between neuroimaging experts–who like to become familiar with analytical complexities that arise from neurochemical modulation of the brain—as well as clinical researchers who need noninvasive neuroimaging biomarkers in pharmacological or diagnostic applications. We focused on identifying challenges and opportunities in pharma‐RSfMRI, and areas that need further development and testing.
This review includes (to the best of our knowledge) all pharma‐RSfMRI in any PubMed publications listed between Jan 2000 and Jan 2016. Our search was limited to studies in which acute effects of drugs on resting‐state brain function were investigated. For each of these 85 studies we have extracted information about experimental design, statistical modeling, acquisition parameters and subject demographics and clinical indications (Table 1). Reports in which the latent drug effects are studied after a few days were excluded. We summarize acquisition and analytical methods used in these studies by providing examples of application, interpretation and limitations (“Basics and Biomarkers”); describe various experimental approaches that demonstrate the potential of pharma‐RSfMRI in pharmacological, clinical and translational applications (“Experimental Objectives and Clinical Relevance”), and outline theoretical, practical and methodological factors that challenge current interpretations and demand further research (“Challenges and Limitations”). Finally, we propose a unifying framework that proposes standardizing design and multimodal data acquisition, as well as an integrative approach to data fusion and analytical approaches that will be important both for neuropharmacology, and basic and clinical research studies (“Opportunities and Future Directions”).
Table 1.
List of reviewed studies and their experimental design and data acquisition parameters
| Drug | Objective | Mode of delivery Dosage | PK | Placebo | Analytical approach | A Priori ROI | # scans per subject | Statistical Design | Patient (N male /N female) (age (mean/sd) | Healthy (N male /N female) (age range or mean/sd) | MRI parameters (Field, Sequence) | Physiological monitoring/Correction | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Morphine [Becerra et al., 2006] | DR | IV4 mg/70 kg morphine | X | ✓ | % BOLD signal (PhfMRI) | 2 | Mixed effect GLM (Morphine × time – Saline × time) | 8/0(28.3/2.86) | 1.5T, T2* EPI (TR = 6 cardiac cycles)N = 250 | ✓/✓ | ||
| 2 | Morphine [Khalili‐Mahani et al., 2012] | FP | IVBolus + pseudosteady14.5 mg/70 kg | ✓ | ✓ | NOI+DR | Beckmann's 8 RSN template | 21 | Mixed GLMDrugmor or alc(t1–6 – t0) –Placebo(t1–6 – t0) | 12/018–40 | 3T, T2* EPI (TR = 2.2 s) N = 220 | ✓/✓ | |
| 3 | Morphine [Khalili‐Mahani et al., 2011] | FP | IVBolus + pseudo‐steady14.5 mg/70 kg | ✓ | ✓ | Global CBF, rCBF | 6 | Mixed GLMDrugmor or alc(t2 – t0) –Placebo(t2 – t0) | 12/018–40 | 3T, PCASL, 30 pairs | ✓/✓ | ||
| 4 | Remifentanil [Leppa et al., 2006] | DR | IV 0.5 µg/kg 3 times 7 min each 0.5 µg/kg vs. 7 min saline | X | X | Whole brain % BOLD signal change (PhfMRI) | 4 | Fixed effect Mean0–420 s (BOLD1,BOLD2, BOLD3) – Mean0–420 s (BOLD salin) | 4/422–28 | 1.5T, T2* EPI (TR = 3 s) N = 600 | ✓/x | ||
| 5 | Heroin [Schmidt et al., 2015] | PR | IV (only in patients)Plasma Morphine levels (ng/mL at min 3, 10 and 60: 529 ± 726; 334 ± 156; and 224 ± 92) | X | ✓ | ICA+DR | Basal ganglia/limbic network | 2 | Placebo‐Heroine (patients)Patients – Controls (Placebo) | 12/8F (41.45 ± 6.70) | 14/6(40.24/10.91) | 3T, T2* EPI (TR = 2),N = 152 | X/X |
| 6 | Heroin [Denier et al., 2013] | FP | Regular morning dose per subject | X | ✓ | CBF | 4 | ANOVADrug (heroine – placebo) and Time (post – pre) | 9/6F (mean 40.9± 6.6) | 3T, PASL (FAIR, Q2TIPS + SS 3D GRASE), 28 pairs at TI 200–2,800 ms. | ✓/x | ||
| 7 | Remifentanil [MacIntosh et al., 2008] | DR | IV1.0 ng/mL | ✓ | X | AAT Cerebrovascular response = ΔCBF/ΔPetCo2 | 2 | Permutation‐based pairwise t‐test | 9/1(31/6) | 3T, PASL (FAIR, Q2TIPS, 3D GRASE), 10 pairs at TI = 500–2,500 ms | ✓/✓ | ||
| 8 | Buprenorphine [Upadhyay et al., 2012] | CV/FP/DR | IV (Cohort 1 0.1 mg/70 kg Cohort 2 0.2 mg/70 kg) andSublingual (2.0 mg) | ✓ | ✓ | Seed + PK/PD modeling | Putamen | 1 | Mixed Effect Paired comparison | 36/0(28.0/2.5) | 3T, T2* EPI (TR = 2.5)N = 360 | X/X | |
| 9 | Alcohol [Spagnolli et al., 2013] | FP | Oral 0.5 g/L | X | X | ICA+DR | DMN, VIS, FPN | 2 | Paired T‐test | 8/7(28/1.2) | 1.5T, T2* EPI (TR = 2)N = NA | X/X | |
| 10 | Alcohol [Esposito et al., 2010] | FP | Oral 0.7 mL/kg | X | X | ICA | VIS, DMN, FPN, SMN, AUD and the self‐referential (SRN) | 4 | 2‐factor ANOVA | 5/3(28/3.2) | 3T, T2* EPI (TR = 1.5 s) N = 240 | ✓/✓ | |
| 11 | Alcohol [Khalili‐Mahani et al., 2012] | FP | IV (clamping)0.6 g/L | ✓ | ✓ | NOI+DR | Beckmann's 8 RSN template | 21 | Mixed GLMDrugmor or alc(t1–6 – t0) – Placebo(t1–6 – t0) | 12/0(18–40) | 3T, T2* EPI (TR = 2.2 s) N = 220 | ✓/✓ | |
| 12 | Alcohol [Strang et al., 2015] | DR | IV0.6 g/LTwo doses 40% and 80% eBAC | X | ✓ | CBF | 6 | Mixed factorial ANOVAsexmale, female, drugplacebo alcohol, timeASL1, ASL2, ASL3 | 11/9(19.9/0.8)heavy drinkers | 3T, PCASL, 30 pairs | X/X | ||
| 13 | Alcohol [Marxen et al., 2014] | DR | Oral 0.6 g/kg | ✓ | ✓ | ΔCBF | 2 | Average ΔCBF (men and women) | 38/0(18–19) | 3T, PASL, 3D GRASE, 23 pairs, TI:300–2,600 | X/X | ||
| 14 | Alcohol [Rickenbacher et al., 2011] | FP | Oral F 0.55 g/kg M 0.6 g/kg | X | X | PWI, rCBF | Mixed factorial ANOVASex | 10/922–33 | 3T, PASL, FAIR, QUIPS2 | X/X | |||
| 15 | Alcohol [Khalili‐Mahani et al., 2011] | FP | IV (clamping)0.6 g/L | ✓ | ✓ | Global CBF, rCBF | 6 | Mixed GLMDrugmor or alc(t2 – t0) –Placebo(t2 – t0) | 12/018–40 | 3T, PCASL, 30 Pairs | ✓/✓ | ||
| 16 | Propofol (GABA‐A)[Jordan et al., 2013] | FP/CV | IVBolus 1.2 μg/mL + increments of 0.4 μg/mL until sedation scale 5–6 | ✓ | X | ICA + Seed | VIS, DMN, AUD, SMNThalamus | 1 | Full factorial ANOVA (Network, Conditionawake vs. unconcsious) | 15/0(25.8 + N/A). | 3T, T2* EPI (TR = 1.8 s) N = 300 | ✓/X | |
| 17 | Propofol [Gili et al., 2013) | FP | IV1.2 ± 0.2 μg/mL.pseudosteady | ✓ | X | Graph theory | Thalamus, Pons, Brainstem | 2 | Permutation‐based Paired t‐test Eigenvector centrality (sedation vs. awake) | 15/0(20–41) | 3T, T2* EPI (TR = 3 s)N = 160 | ✓/✓ | |
| 18 | Propofol [Guldenmund et al., 2013] | PR | IV1.71 μg/mL (sd 0.72), mild 3.02 μg/mL (sd 1.03), unconscious 0.59 μg/mL (sd 0.28) recovery | ✓ | X | ICASeed | DMN and AUDACC, Thalamus, brainstem and Hippocampus | 1 | Average t‐maps, Fingerprints and Connectivity graphs per state of consciousness (awake, mild, deep, recovery) | 4/13(21.9/1.9) | 3T, T2* EPI (TR = 2.5 s) N = 235 ± 74 | X/X | |
| 19 | Propofol [Boveroux et al., 2010] | PR | IV1.75 μg/mL (sd 0.67), mild 3.2 μg/mL (sd 0.99), unconscious 0.61 μg/mL (sd 0.22) recovery | ✓ | X | Seed | PCC (DMN), Middle Frontal gyrus (ECN) | 1 | ANOVA ( DMN or ECN connectivityWakeful, mild, deep, recovery) | 4/16(22.4/3.4) | 3T, T2* EPI (TR = 2.5 s) N = 196 | X/X | |
| 20 | Propofol [Schrouff et al., 2011] | CV | Same as [Boveroux et al., 2010] | Network hierarchy clustering | 1 | Same as [Boveroux et al., 2010] | Same as [Boveroux et al., 2010] | X/✓ CORSICA | |||||
| 21 | Propofol [Monti et al., 2013] | CV | Same as [Boveroux et al., 2010] | Graph theory, Pattern recognition | AAL‐based 196 ROIs | ANOVA (Wakeful, mild sedation, deep sedation, recovery) | 12/0 | 3T, T2* EPI (TR = 2.5 s) N = 196–350 | X/X | ||||
| 22 | Propofol [Schroter et al., 2012] | CV | IVBolus 1.2 μg/mL + increments of 0.4 μg/mL | ✓ | X | Graph theory + NOI | Graph properties, global and local network efficiency Functional connectivity of consciousness | 1 | Sedated – awake | 11/0(25.8/3.0) | 3T, T2* EPI (TR = 1.8 s) N = NA | X/X | |
| 23 | Propofol [Stamatakis et al., 2010] | CV | IV0.6 and 1.2 μg/mL | ✓ | X | Seed Power spectrum and BOLD amplitude | PCC (DMN) | 1 | T‐test (DMN connectivityAwake, mild, deep) | 16/0(19–52)(34.6/9) | 3T, T2* EPI (TR = 2 s)N = 150 | X/X | |
| 24 | Midazolam [Greicius et al., 2008] | CV | IV (4.1 ± 0.9) mg Tittered until Ramsay scale 3 | ✓ | X | ICA | DMN, SMN | 1 | Paired t‐test (rest, sedation) | 4/5(22–27) | 1.5T, T2* EPI (TR = 2 s)N = 82 | ✓/X | |
| 25 | Zolpidem [Rodriguez‐Rojas et al., 2013] | CL | Oral 10 mg | X | X | Hemodynamic modeling (PhfMRI) | Left frontal cortex | 4 | %BOLD (post – pre) | 0/1 brain injured (21) | Age matched control | 1.5T, T2* EPI (TR = 3 s)N = 60 | X/X |
| 26 | Zolpidem [Licata et al., 2013] | PR | Oral 0, 5, 10 or 20 mg | X | ✓ | ICA + DR | VIS, TEMP | One‐factor repeated measure ANOVA (0,5,10, 20 mg) | 6/624.2/2.3 | 3T, T2* EPI (TR = 3 s)N = 200 | ✓/X | ||
| 27 | Benzodiazepine (GABAA)[Flodin et al., 2012] | PR | Oral Oxazepam (20 mg) | X | ✓ | Seed,fALFF and REHO | DMN(PCC, vmPFC)PMN(M1, Put)Amyg, Primary visual and NAcc (aread affected by PD) | 1 | Two‐sample t‐test (Oxazepam – placebo) | 9/11(24.6/4.4) Oxazepam | 1.5T, T2* EPI (TR = 2.5 s) N = 192 | X/X | |
| 8/14(25.5/4.6)Placebo | |||||||||||||
| 28 | Dopamine [Flodin et al., 2012] | PR | Oral Levdopa (100 mg) | X | ✓ | 1 | Two‐sample t‐test (LDOPA – placebo) | 10/9(22.3/3.5) Ldopa | |||||
| 10/10F(22.8/4.5) placebo | |||||||||||||
| 29 | Dopamine [Cole et al., 2013] | PR | Oral Haloperidol (3 mg) | X | ✓ | ICA+DR | VIS, DMN, SMN, EXEC, AUD, LVDS, RVDS, | 1 | Two‐sample t‐test (Haloperidol – placebo) | 18/0(22.2/3.3) | 3T, T2* EPI (TR = 2.2 s) N = 220 | X/X | |
| Levodopa (100 mg) | X | ✓ | 1 | Two‐sample t‐test (Levdopa – placebo) | 16/0(23.4/5.3) | ||||||||
| Carbidopa (25 mg) | 1 | 15/0(21.5/3)Placebo | |||||||||||
| 30 | Dopamine [Esposito et al., 2013] | PR/CL | Oral Levodopa/carbodopa (250/25 mg) | X | ✓ | ICA and fALFF | SMN, DMN, Basal Ganglia | 2 PD1 HC | Two‐sample t‐tests and two‐way ANOVA interaction model | Drug‐naïve PD5/5 (60.8/2.7) | No treatment 10/859.2/1.52 | 3T, T2* EPI (TR = 1.5 s) N = 240 | ✓/✓ |
| Placebo | Placebo 5/5(66/1.7) | ||||||||||||
| 31 | Dopamine [Carbonell et al., 2014] | PR | Oralnutritionally balanced 100 g amino acid mixture (BAL), and tyrosine and phenylalanine deficient (APTD) | X | ✓ | Graph analysis Modularity | 2 | Mixed‐effects GLM (t‐test: BAL vs. APTD) + covariance of no interest (sex and age), | 11/6(23.6/4.4) | 3T, T2* EPI (TR = 2.04 s)N = 180 | X/X | ||
| 32 | Dopamine [Vytlacil et al., 2014] | PR | Oral 1.25 mg Bromocriptine (DA) | X | ✓ | Seed | Bilateral caudate, putamen and ventral striatum, brainstem | 2 | 2 × 2 ANOVA with factors of drug (bromocriptine vs placebo) and span (high vs low). | 8/0(18–22) | 3T, T2* EPI (TR = 2 s)N = NA | X/X | |
| 33 | Dopamine [Kelly et al., 2009] | PR | Oral Levodopa 100 mg (25 mg of benserazide) | X | ✓ | Seed (multiple regression) | inferior ventral striatum, superior ventral striatum, dorsal caudate and putamen | 4 | Repeated‐measures mixed‐effects (Drug vs placebo) | 12/7(26.2/NA) | 4T, T2* EPI (TR = 2.1 s) N = 200 | X/X | |
| 34 | Dopamine [Fernandez‐Seara et al., 2011] | DR | Oral 10 mg metoclopramide (DA) | X | ✓ | CBF, rCBF and seed | ‐ Haemodynamics in vertebral and internal carotid arteries with phase contrast MRI‐ putamen and insula | 4 | 2 × 2 repeated measures ANOVA factors treatment (metoclopramide, placebo) and session (baseline, post‐medication) | 8/10(23.9/4.5) | 3T, PCASL, 50 pairs | X/X | |
| 35 | Cocaine [Kufahl et al., 2005] | DR | IV(40 mg/70 kg) | ✓ | ✓ | %BOLD (PhfMRI) | ‐ Amygdala, PFC, Nacc | 2 | Single‐subject cocaine vs. saline Group: Correlation with craving scores | 8/7(N/a)Cocain abuser >6 years | 1.5T, EPI (MultiEcho Segmented EPI with z‐shimmed BAckground gradient Compensation (MESBAC)) | ✓/✓ | |
| 36 | Methylphenidate [Konova et al., 2013] | PR | Oral 20 mg | X | ✓ | Seed | VTA, Nacc, amygdala hippocampus, thalamus, rACC | 4 | t‐test: Post drug – baseline drug t‐test: baseline drug – baseline placebo t‐test: Placebo CD – Placebo HC | 16/2F(45/7.3)cocaine addicts | 4T, T2* EPI (TR = 1.6 s)N = 320 | X/X | |
| 37 | Methylphenidate [Ramaekers et al., 2013] | PR | Oral 40 mg | X | ✓ | Seed | NAcc and medial dorsal nucleus | 2 | Paired t‐test (Drug – Placebo) | 9/11(23–35) | 3T, T2* EPI (TR = 2 s)N = 192 | X/X | |
| 38 | Methylphenidate [An et al., 2013] | CL | Oral 10 mg | X | ✓ | REHO | 2 | Unpaired t‐test ADHD placebo – HC placeboPaired t‐test: ADHD drug – ADHD placebo | 23 M (12.5 ± 1.8) ADHD | 32/0(11.8/1.8) | 3T, T2* EPI (TR = 2 s)N = NA | X/X | |
| 39 | Methylphenidate [Konova et al., 2015] | CL | Oral 20 mg | Plasma measurement | ✓ | Global Connectivity | 2 × 2 repeated measure ANOVA factors illness (CUD vs. HC) and drug (MPH vs. placebo) | 17/2F46.2 ± 7.5Cocaine user | 15/0(39/7.4) | 4T, T2* EPI (TR = 1.6 s)N = | X/X | ||
| 40 | Methylphenidate [Mueller et al., 2014] | FP | Oral 40 mg | X | ✓ | ICA + DR | DMN, ECN, FPN, SMN, VIS, DAN | 2 | Paired t‐test drug vs. placebo | 54/0(23.65/2.97) | 3T, T2* EPI (TR = 3 s)N = 120 | X/X | |
| 41 | Methylphenidate [Sripada et al., 2013ab] | FP | Oral 40 mg | X | ✓ | SVM | 2 | Δ(Placebo,Drug) | 16/16(20.6/2.0) | 3T, T2* EPI (TR = 2 s)N = 180 | X/✓ | ||
| 42 | Modafinil [Schmaal et al., 2013] | CL | Oral 200 mg | X | ✓ | ICA | DMN, Salience, Executive | 2 | 2 × 2 repeated measure ANOVA factors illness (AD vs. HC) and drug (MOD vs. placebo) | 6/0 Alcohol (43/2.4) | 8/0(41 /1.8) | 3T, T2* EPI (TR = 2.3 s)N = 200 | X/X |
| 43 | Modafinil [Esposito et al., 2013b] | FP | Oral 100 mg | X | ✓ | ICA | DMN, ECN, FPN, SMN, VIS, ECN, DAN | 2 | Three way mixed design ANOVA (Group × time × treatment) | 13/0(23–35) | 3T, T2* EPI (TR = 1.67 s)N = 140 | X/X | |
| Placebo | 13/0(23–35) | ||||||||||||
| 44 | Cannabis [Klumpers et al., 2012] | FP | THC inhalation 2, 6 and 6 mg | ✓ | ✓ | NOI + DR | Beckmann's 8 RSN template | 16 | Mixed GLMDrugmor or alc(t1–7 – t0) –Placebo(t1–7 – t0) | 9/3(22/2.25) | ✓/✓ | ||
| 45 | Cannabis [Van Hell et al., 2011] | FP | THC inhalation 6 mg + 1 m updosage every 30 min | X | ✓ | temporal signal‐to‐noise ratio | 2 | Paired t‐test (THC – Placebo) (t3+ t4) | 20/0(21.1/2.1) | 3T, T2*, SENSE‐PRESTO (TR = 0.0225 s), N = 400 | ✓/✓ | ||
| 46 | Cannabis [Van Hell et al., 2011] | FP | Same as above | X | ✓ | Global CBF, rCBF | 2 | Paired t‐test (THC – Placebo) (t0–t1) | 20/0(21.1/2.1) | 3T, PCASL, 30 pairs | ✓/✓ | ||
| 47 | Insulin [Kullmann et al., 2013] | PR | Intranasal solution 40 IU insulin (400 IU/mL) × 4 times | X | ✓ | fALFF | 6 | Mixed GLMInsulin (post1 and 2 –pre) – placebo (post 1 and 2 –pre) | 0/17(24.5/2.2) | 3T, T2*, EPI (TR = 1.8 s)N = 320 | X/X | ||
| 48 | Sucrose [Kilpatrick et al., 2014] | PR | Oral (50 g truvia, low calorie) vs. (50 g sugar, high calorie) | X | ✓ | Seed + fALFF | Hypothalamus and brainstem | 2 | 3‐way ANOVA (Group, Treatment, Time) | 0/11 healthy obese (27/1.9) | 0/11(25/1.2) | 1.5T, T2* EPI (TR = 2 s)N = NA | X/X |
| 49 | Exenatide [Schlogl et al., 2013] | PR | IV0.12 pmol/kg/min target 0.1–0.2 ng/mL | ✓ | ✓ | Graph Theory (ECM) | Hypothalamus | 2 | Paired t‐test: drug – placebo | 24M (obese)(29/7) | 3T, T2* EPI (TR = 2 s)N = NA | X/X | |
| 50 | Ketamine [Deakin et al., 2008] | PR/DR/FP | Experiment 1: IV ketamine bolus 0.26 mg/kg plus 0.25 mg/kg/h | ✓ | ✓ | %BOLD signal (PhfMRI) | 2 | One‐way ANOVA on 8 blocks of %BOLD signal change (activation maps) | 12/0(22.2/3.85) | 1.5T, T2* EPI (TR = 5 s) N = NA | X/X | ||
| 51 | Ketamine and Lamorigine [Deakin et al., 2008] | Experiment 2:IV Ketamine: Bolus 0.26 mg/kg plus 0.25 mg/kg/h Oral Lamotrigine 300 mg | 19/0 (21.6/ 3.2) | ||||||||||
| 52 | Ketamine [Scheidegger et al., 2012] | FP | IV0.25 mg/kg | ✓ | ✓ | Seed | DLPFC (cognitive control network)PCC (DMN)sgACC; Affective network Amygdala | 4 | Paired‐t‐test (baseline – follow‐up) | 19/?(40.5/7.5) | 3T, T2* EPI (TR = 3 s)N = 200 | X/X | |
| 53 | Ketamine [De Simoni et al., 2013] | CV/DR | IVSame dose in two occasions 50 nmol/kg (5 subjects)and 75 nmol/kg (5 subjects) | ✓ | X | ICCGLMBOLD | Whole brain and ROI (ACC, PCC, Thalamus) | 2 | GLM with Gamma Variate model, shape capture, nuisance factors | 10/0(25.5 ±6.5) | 3T, T2* EPI (TR = 2 s)N = 400 | X/X | |
| 54 | Ketamine, Lamotrigine, Risperidone [Doyle et al., 2013] | PR/DR/CV | IV Ketamine 0.12 mh/kg bolus + 0.31 mk/kh per h Oral Lamotrigibe 300 mg Oral Risperidone 2 mg | ✓ | ✓ | %BOLD signal Gaussian process classification | 3 | Least‐square mean difference PLA‐KET, LAM‐KET, RIS‐KET, PLA‐SAL in “activated” regions | 16/0(25.8/5.7) | 3T, T2* EPI (TR = 2 s)N = 450 | X/X | ||
| 55 | Ketamine [Driesen et al., 2013] | PR | IV0.23 mg/kg bolus and 0.58 mg/kg/h | ✓ | X | Global brain connectivity (GBC) | 1 | SPM: regional correlation of GBC with symptoms (PANSS factor scores) | 14/8(22–45) | 3T, T2* EPI (TR = 1.5 s)N = 160 | ✓/X | ||
| 56 | Ketamine [Niesters et al., 2012] | FP/DR | IV (20 mg/70 kg/h) for 1 h, followed by (40 mg/70 kg/h) for another hour. | ✓ | ✓ | NOI/DR | Beckmann's 8 RSN template | 5 | Mixed GLMDrug (t1–4 – t0) – Placebo(t1–4 – t0) + Pain | 12/0(19–36) | 3T, T2* EPI (TR = 2.18 s)N = 220 | ✓/X | |
| 57 | Ketamine [Khalili‐Mahani et al., 2015] | PR | Same as [Niesters et al., 2012] | ✓ | ✓ | Seed | Hippocampus (head, body, tail) | 10 | Mixed GLMDrug (t1–4 – t0) – Placebo(t1–4 – t0) + cortisol | 12/0(19–36) | 3T, T2* EPI (TR = 2.18 s)N = 220 | ✓/X | |
| CBF, rCBF | 10 | Mixed GLMDrug (t1–4 – t0) – Placebo(t1–4 – t0) + cortisol | 12/0(19–36) | 3T, PCASL, 30 pairs | ✓/X | ||||||||
| 58 | Ketamine and scopolamine [Grimm et al., 2015] | PR | ICKetamine (0.5 mg/kg)Scopolamine (4 µg/kg) | ✓ | ✓ | Seed | Prelimbic cortex | 3 | T‐test Drug – Placebo | 12/12(25) | 3T, T2* EPI (TR = 1.79)N = 332 | X/X | |
| 59 | Psilocybin [Carhart‐Harris et al., 2012] | FP/PR | IV2 mg | X | ✓ | %BOLD signal (PhfMRI)Seed | MPFC | 2 | Mixed effect GLM drug related variations in % BOLD signal Mixed effect (drug – placebo) | 13/2(32/8.9) | 3T, T2* EPI (TR = 3 s)N = 240 | ✓/✓ | |
| FP | ✓ | Perfusion | 2 | Mixed effect GLM drug related variations in perfusion | 10/5(34.1 /8.2) | 3T, PASL, PICORE, QUIPSS2, 240 pairs | X/X | ||||||
| 60 | MDMA[Carhart‐Harris et al., 2015] | FP/PR | Oral 100 mg | X | ✓ | Seed | vmPFC, Amygdala, Hippocampus | 4 | Mixed effect least square (Drug – Placebo) | 18/7(34 /11) | 3T, T2* EPI (TR = 3 s)N = 240 | X/X | |
| AAT, CBF | 4 | 3T, pASL, Q2TIPS, TI = 750–1,650, 150 ms increment | X/X | ||||||||||
| 61 | Psilocybin and MDMA[Roseman et al., 2014] | FP | As [Carhart‐Harris et al., 2015] and [Carhart‐Harris et al., 2012] | X | ✓ | NOI+DR | 20 Smith's networks | Paired t‐test (drug – placebo) | As above | 3T, T2* EPI (TR = 3 s)N = 240 | X/X | ||
| 62 | Aripiprazole and Haloperidol [Handley et al., 2013] | FP | Oral Haloperidol 3 mg Aripiprazole 10 mg | X | ✓ | rCBF | Flexible Factorial,Random effect GLM3 pairs of T‐tests | 20/0(23/4.5) | 1.5T, pCASL, 3 pairs | ||||
| 63 | Sertraline [Klaassen et al., 2015] | FP | Oral 75 mg | ✓ | ✓ | NOI + DR | 10 Smith's 2009 networks | 10 | Mixed GLMDrugmor or alc(t1–4 – t0) –Placebo(t1–4 – t0) | 6/6(23/3) | 3T, t2* EPI (Tr = 2.2)N = 220 | ✓/✓ | |
| 64 | Nicotine [Stein et al, 1998] | DR | IV1.5 mg | ✓ | ✓ | %BOLD (PhfMRI) | 1 | PK model fitting, individual response | 15 (F/M?)(26) smokers | 1.5T, T2* EPI (TR = 6 s), TE = 40 | ✓/✓ | ||
| 65 | Nicotine [Wylie et al., 2012] | FP | Patch 7 mg | X | ✓ | Global/local efficiency Wavelet | Limbic and paralimbic regions | 4 | 2‐way repeated measures ANOVA (Drug, Time) | 9/6(29.4/7.5) | 3T, T2* EPI (TR = 2 s)N = NA | ✓/✓ | |
| 66 | Nicotine [Tanabe et al., 2011] | PR | Patch 7 mg | X | ✓ | ICASpectral analysis | DMN and CCN | 4 | 2‐way repeated measures ANOVA (Drug, Time) | 19/0(30/9) | 3T, T2* EPI (TR = 2 s)N = NA | ✓/✓ | |
| 67 | Acetazolamide [Bokkers et al., 2011] | CV | IVBolus 14 mg/kg (max dose 1,200 mg) | X | X | Global CBF, ΔCBF(post – pre) | 1 | Paired t‐test ΔCBF Patient ‐ ΔCBFHC | 12/4(56.3/ 13.8)ICA occlusion | 5/12(56.5/5.7) | 3T PCASL, 30 pairs | X/X | |
| 68 | Scopolamine [Suckling et al., 2008] | CV | Subcutaneous scopolamine hydrochloride 0.3 mg (0.75 mL) | X | ✓ | Estimation of the Hurst exponent (spectral power) | 2 | Mixed effect repeated measures ANOVA (age, drug) | 5/6 (20–25)5/6 (60–70) | 3T, T2* EPI (TR = 1.1 s)N = 450 | X/X | ||
| 69 | Caffeine [Rack‐Gomer et al., 2009] | PR/CV | Oral pill 200 mg | X | ✓ | CBF, rCBF | Motor Cortex (obtained from task‐activation) | 4 | 2‐way repeated measures ANOVA (Drug, time) | 5/4(23–41) | 3T, pASL, PICORE/QUIPSS2 | ✓/✓ | |
| Connectivity | 4 | 3T, T2* EPI (TR = 0.5 s)N = 450 | ✓/✓ | ||||||||||
| 70 | Caffeine [Rack‐Gomer and Liu 2012] | Same as [Rack‐Gomer et al., 2009] | Spectral power | ||||||||||
| 71 | Caffeine [Wong et al., 2012] | CV | Oral pill 200 mg | X | ✓ | CBF | 4 | 2‐way repeated measures ANOVA (Drug, time) | 4/6 (25/6) | 3T, PCASL (GE), Pairs NA | |||
| BOLD amplitude | DMN and TPN | 4 | 3T, T2* EPI (TR = 1.8 s)N = NA | ||||||||||
| 72 | Caffeine [Tal et al., 2013] | CV | [Wong et al., 2012] | X | ✓ | BOLD amplitude and MEG | |||||||
| 73 | Oxytocin [Sripada et al., 2013aa] | PR | Intranasal spray 40.32 mg (three puffs of 4 IU or 6.72 mg per nostril.) | X | ✓ | Seed | Amygdala | 2 | Paired t‐test drug – placebo | 15/0(30.7/10.2) | 3T, T2* EPI (TR = 2 s)N = 100 | X/X | |
| 74 | Oxytocin [Dodhia et al., 2014] | PR/CL | intranasal spray (three puffs of 4 IU or 6.72 mg per nostril)40.32 mg | X | ✓ | Seed | Amygdala | 2 | Repeated measures ANOVA (Group, treatment) | 18/0(29.4/ 9.0)GSAD | 18/0(29.9/10.2) | 3T, T2* EPI (TR = 2 s)N = NA | X/X |
| 75 | Oxytocin [Paloyelis et al., 2016] | FP | Intranasal spray One puff (4 IU) of IN‐OT (or placebo) every 30 s, alternating b/w nostrils 40 mg. | X | ✓ | Global CBF, rCBFPattern recognition | Flexible factorial model, ANCOVA (treatment × time) | 16/0(24/1.7)16/0(25.8/4.4) | 3T, PCASL (GE), pairs NA | X/X | |||
| *76 | Oxytocin [Frijling et al., 2015] | PR | intranasal spray Five puff (4 IU) of IN‐OT in each nostrile every 30 s (total 40 IU) | X | ✓ | Seed | Amygdala, MPFC, insula, dACC | 1 | Between group analysis Oxytocin vs Saline | Potential PTSD9/9 Placebo (32/11)9/10 Ox (28/11) | 3T, T2* EPI (TR = 2.3)N = NA | X/X | |
| *77 | Oxytocin [Koch et al., 2015] | PR/CL | Intranasal spray Five puff (4 IU) of IN‐OT in each nostrile every 30 s (total 40 IU) | X | ✓ | Seed | Amygdala, MPFC, insula, dACC | 2 | ANOVA(Drug × Sex × Group) | PTSD21/16 | 20/20 | 3T, T2* EPI (TR = 2)N = 238 | X/X |
| 78 | Chloral Hydrate [Wei et al., 2013] | PR | Oral 50 mg/kg Sedation score 5 (sleep) | X | X | Local/global network efficiency, between centrality | 1 | Paired t‐test (sedation – awake) | 20/8(10.3/2.6) | 1.5T, T2* EPI (TR = 2 s)N = 180 | X/X | ||
| 79 | Sevoflurane [Peltier et al., 2005] | PR | Intranasal 0%, 2.0% and 1.0% end‐tidal sevoflurane | ✓ | X | Seed | Primary motor M1 | 1 | # of connected voxels per state per subject | 6/0(22–24) | 3T, T2* EPI (TR = 0.75 s)N = 280 | ✓/✓ | |
| 80 | Sevoflurane [Qiu et al., 2008] | CV | 0.25 MAC (minimum alveolar concentration) | ✓ | ✓ | BOLD/rCBF coupling | Anterior cingulate, Posterior cingulate, intraparietal lobule | 1 | Paired t‐test:PerfusionAnesthesia (Blocks 2,4) – Perfusionno‐anesthesia (Blocks 1,3,5)BOLDAnesthesia (Blocks 2,4) – BOLDno‐anesthesia (Blocks 1,3,5) | 22(19–30) | 3T, pASL, STAR QUIPSS, interleaved BOLD (TR = 3 s)/ASL(TI = 700), fast suppression and phase correction | X/X | |
| 81 | Thiopental [Kiviniemi et al., 2000] | CV | IV3 mg/kg/h | ✓ | X | Spectral analysis Seed | Visual cortex seed | Subject‐based | 12 children various illness | 1.5 T, TR = 3N = NA | ✓/X | ||
| 82 | Thiopental [Kiviniemi et al., 2003] | CV | IV 6 mg/kg/h Midatzolam premedication (0.3 mg/kg) was given 2–3 h before IV | x | ✓ | Spectral, ICA | VIS, AUD, SMN and blood vessel related component | 1 | Individual | 8/7(2–9.5) | 1.5 T, TR = 2N = 84 | X/X | |
| 83 | Bupivacaine [Niesters et al., 2014] | FP/PR/DR | Spinal tap Lumbar 3–415 mg | x | ✓ | NOI, Seed | 8 Beckmann's NOIs Thalamic subsegments (Oxford)Insula | 6 | Mixed GLMDrug (t1–2 – t0) –Placebo(t1–2 – t0) + Pain | 12/0(23.7/3.4) | 3T, T2* EPI (TR = 2.18)N = 220 | ✓/X | |
| 84 | Hydrocortisone [Henckens et al., 2012] | PR | Oral 10 mg | X | ✓ | Seed | Amygdala | 1 | Independent t‐test (Cort – Placebo) | 23/0 CORT20/0PLACEBO(19–28) | 1.5T, T2* EPI (TR = 2 s)N = 265 | ✓/X | |
| 85 | Steroids [Sripada et al., 2014] | PR | Oral 400 mg pregnenolone 400 mg DHEAPlacebo | X | ✓ | Seed | Amygdala | 1 | Random effect 1‐sample and 2‐sample t‐test | 16/0pregn. | 3T, T2* EPI (TR = 2 s)N = 240 | X/X | |
| 14/0DHEA | |||||||||||||
| 15/0Placebo (22.6/3.6) |
FP, finger printing; DR, dose response; CL, clinical; CV, calibration validation; PR, probing; DMN, default mode network; ECN, executive control network; VIS, visual network; AUD, auditory network; CCN, cognitive control network; SMN, sensorimotor network; FPN, frontopariental network; DAN, dorsal attention network; TPN, temporoparietal network.
BASICS AND BIOMARKERS
Drugs modulate the neuronal and metabolic signals by acting on neurotransmitter receptors (directly or via indirect pathways). Figure 1A presents a schematic overview of the complex cascade of signaling pathways affected by different drugs. Drugs may act by directly targeting their receptors in the central or autonomic nervous systems. They can also act indirectly, by acting peripherally and causing sympathetic and parasympathetic responses related to adaptive feedback regulation. Together, this cascade of events translates to fast and slow alterations in mental and proprioceptive states, sending top‐down and bottom‐up signals between the central and autonomic nervous systems in order to adaptively regulate behavior and homeostasis. The fMRI technique measures changes in blood‐oxygen‐level dependent (BOLD) signal or blood flow and volume (CBF and CBV) in response to such cascade neuromodulation of the brain.
Figure 1.
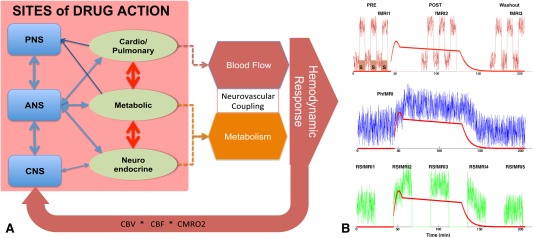
(A) Schematic diagram of the cascade of drug interactions that give rise to the hemodynamic response measured by fMRI (PNS, peripheral nervous system; ANS, autonomic nervous system, CNS, central nervous system). (B) Schematic diagram of possible pharmacological fMRI experiments. Top, task‐based or calibrated fMRI in which drugs modulate the magnitude of stimulus versus no‐stimulus signal; Middle, PhfMRI where the drugs cause a latent neurovascular response consistent with the pharmacokinetic profile of the drug; Below, repeated measurements of RSfMRI signal which will be used to derive connectivity or spectral metrics and study their dynamics over the pharmacodynamic profile. The red line shows the pharmacodynamic model of a drug (morphine). [Color figure can be viewed at http://wileyonlinelibrary.com]
Pharmacological fMRI (Pharma‐fMRI)
The overall goal of pharmacological fMRI is to identify the site of drug action fingerprinting to estimate the relation between drug‐dose, brain response and clinical outcome over time (pharmacokinetic/pharmacodynamic modeling); and to help make go–nogo decisions about the efficacy of drug‐treatment in clinical trials—aimed to accelerate the drug discovery process. Pharmacological probing experiments can also be important for basic neuroscience and validation studies, by allowing a controlled modulation of a specific pathway and studying its causal effect on other signals and systems. More on this is covered in “Experimental Objectives and Clinical Relevance.”
Initial pharma‐fMRI experiments involved collecting regular task‐fMRI data and either comparing the activation maps (i.e., % of the BOLD signal change in response to task) under drug (at one or more dose strengths) versus no‐drug conditions, or estimating differences in the hemodynamic response functions between drug and control conditions (Fig. 1B). Task‐based pharma‐fMRI is valuable for hypothesis testing, but difficult to conduct in translational or large‐scale clinical studies where standardization of tasks and normalization of performance metrics is virtually impossible (yet). Another important pharma‐fMRI approach is to evaluate the pharmacodynamics of the brain response by collecting fMRI data continuously over the course of drug infusion, and then evaluating the percentage of signal change from the pre‐drug baseline as a surrogate for drug‐induced neuronal activation [e.g., Becerra et al, 2006; Bloom et al, 1999; De Simoni et al, 2013]. This method is suitable for studying fast‐acting drugs or for anesthesiology experiments, where pharmacokinetic models help reach (pseudo)steady‐state plasma drug concentrations over time (Fig. 1C). We refer to this technique as phfMRI. A more recent pharma‐fMRI approach involves pharmacological “resting‐state” fMRI (pharma‐RSfMRI) in which spontaneous brain activity is recorded at several brief intervals over the course of drug administration, and the dynamics of change in spectral power or network properties of the brain are evaluated across different phases of the pharmacokinetic profile (Fig. 1D). This review excludes task pharma‐fMRI studies, and focuses on measuring BOLD and CBF signals under resting‐state.
More than 75% of studies surveyed here use T2*‐weighted echo planar imaging (EPI) which is sensitive to BOLD contrast. T2*‐weighted signals reflect NMR signal decay arising from local field inhomogeneities related to deoxygenation of hemoglobin—a proxy for neuronal activation resulting from increased oxidative metabolism. Oxygen metabolism is coupled to a hemodynamic response in order to deliver additional blood to the tissue, thus the BOLD signal summarizes the combined effects of vascular response, oxygen metabolism and cerebral perfusion. Around 14% of the studies that we have reviewed employed phfMRI and fit a model of the plasma drug concentration to the BOLD signal, and the rest used multiple metrics derived from RSfMRI measurements. The other 25% used different versions of the arterial spin labeling (ASL) techniques, which are sensitive to cerebral blood flow (CBF) (Fig. 2).
Figure 2.
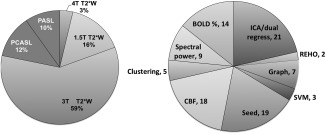
Distribution of RSfMRI data acquisition and analytical approaches.
Spontaneous BOLD Signal Fluctuations and Functional Networks
In RSfMRI, slow fluctuations in the BOLD signal (around 0.1 Hz frequency) serve as a proxy to regional spontaneous neuronal activity. Since the discovery of correlated patterns of BOLD signal fluctuations in the contralateral part of the un‐stimulated motor cortex [Biswal, et al., 2005], RSfMRI has been explored to evaluate networks that are active in the brain at “rest” [Beckmann et al., 2005; Damoiseaux et al., 2006; Smith et al., 2012] and to compare these resting‐state network maps or metrics to task‐related activity [Calhoun et al., 2008; Smith et al., 2009]. Similarities in the topography of RSfMRI networks with task‐activation networks [Calhoun et al., 2008; Smith et al., 2009], anatomical covariance networks [Bullmore and Sporns, 2009], white matter fiber‐tracked networks [Sui et al., 2014], and electrophysiological networks [Brookes et al., 2011] justify the use of their spatiotemporal dynamics as surrogate markers of the brain's response to drugs. Because they are task‐free, RSfMRI methods are practical in large cohort studies. Commonly, typical RSfMRI studies are conducted at spatiotemporal resolution of TR = 2–3 s, TE = 30–35 ms, 100–200 T2* frames, and 2 mm isotropic resolution explore this dataset and to spatial and temporal because such RSfMRI dataset offers a rich spatiotemporal representation of brain states, it lends itself to various modeling and signal processing approaches. Although the origin of the signal measured in a typical fMRI study remains the same, different analytical approaches can yield myriad biometrics that are more sensitive to one aspect of neurophysiology than other (Fig. 3). An ongoing and rewarding challenge in RSfMRI research is to use different mathematical formulations to explore different temporal and spectral characteristics of the same data in relation to neurobiological states. In this section we describe several of these methods that are used in pharma‐fMRI studies. We will discuss limitations in validity and interpretations of these methods in “Challenges and Limitations.”
Figure 3.
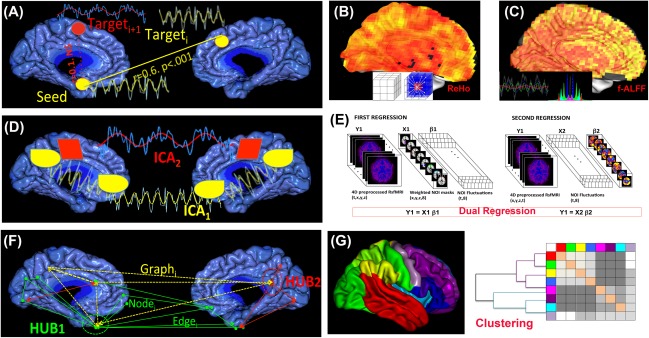
Schematic representation of analysis approaches that are used in pharma‐RSfMRI. (A) Seed‐based connectivity analysis; (B) REHO; (C) (f)ALFF; (D) ICA; (E) dual regression analysis; (F) Graph theory and (G) Hierarchical clustering.
Seed‐based analysis
Over 30% of pharmacological studies surveyed here have relied on region of interest (ROI) seed‐based analysis of resting‐state functional connectivity. Seed selection is based on a priori information about drug actions on local target receptors, hence this method provides a simple answer to the question of how local drug effects propagate across an associated network of regions that collectively subserve a behavior or a (somato)sensory function. For example, a seed in the thalamus can be used to investigate effects of a drug on the sensorimotor network. The seed‐based approach simplifies comparison and within‐subject reproducibility but, as will be discussed in “Challenges and Limitations,” anatomical variations and heterogeneity of brain regions can lead to “seed selection bias,” and confounds such as proximity to pulsating vessels, motion, susceptibility and registration artifacts can reduce signal to noise ratio. Cole et al. have illustrated that three non‐overlapping seeds, all taken from the posterior part of the default mode network (DMN), produce extensive non‐overlapping cortical connectivity maps [Cole et al., 2010]. On the plus side, these anatomical variations in network topography help explore the regional specificity of the response of specific brain regions, for example sub‐nuclei of the thalamus [Niesters et al., 2014], or subsections of the hippocampus [Khalili‐Mahani et al., 2015], to drugs that are expected to target these regions.
Seed‐based connectivity analysis is best suited to hypothesis‐driven research. In testing the neural circuitry of the dopamine system under the influence of dopaminergic drugs, significant results have been observed focusing on the functional connectivity of seeds from the ventral striatum or the nucleus accumbens [Flodin et al., 2012; Kelly et al., 2009; Konova et al., 2013]. Amygdala and the hippocampus have been used as seeds in studying effects of steroids on the brain [Henckens et al., 2012; Sripada et al., 2014]. Because of the consistent presence of the DMN across different species, its important role in metabolic and behavioral regulation [Raichle et al., 2001], and its importance in mentation during the resting‐state, often seeds in the precuneus or medial prefrontal areas are often used to define the strength of the DMN connectivity as a primary biomarker [Boveroux et al., 2010; Flodin et al., 2012; Guldenmund et al., 2013; Scheidegger et al., 2012; Stamatakis et al., 2010]. (See “Experimental Objectives and Clinical Relevance” for more detailed description of these studies). Seed‐based analysis lends itself to standardized protocols) and accommodates replication studies. However, by restricting the tests with a priori hypotheses, it does not provide a global view of the brain's response to a drug. Other limitations of this method in terms of data preparation and normalization are further discussed in “Challenges and Limitations.”
Regional homogeneity (REHO)
REHO is a voxel‐based measure of the coherence and synchrony of the BOLD signal fluctuations in adjacent voxels, based on the hypothesis that clusters of neighboring neurons drive intrinsic brain activity. Synchrony is computed using Kendall's coefficient of concordance (KCC) which measures the ranked correlations at each timepoint among the timeseries of adjacent voxels (usually 27) [Zang et al., 2004]. An alternative to KCC, which measures synchronization in the time domain, evaluates coherence and synchrony in frequency domain (Cohe‐REHO) instead of time domain, and thus eliminates sensitivity to phase variations [Liu et al., 2010]. One advantage of REHO over the seed‐based method is that it does not depend on an a priori definition of ROIs and can yield information about the local/regional consistency of activity throughout the brain. A major disadvantage is that the reliability and reproducibility of REHO is highly sensitive to motion and physiological noise, as well as the morphology, thus it requires careful pre‐processing and normalization [Zuo et al., 2013]. The functional interpretation of the REHO is still tentative, but there is preliminary evidence that suggests a correlation between neurovascular coupling and REHO variations [Yuan et al., 2013], and that regionally‐specific REHO effects under different resting‐state conditions, such as eye‐closed or eye‐open, are associated with differences in global BOLD signal fluctuation in these different conditions [Qing et al., 2015]. Application of REHO in pharma‐RSfMRI studies is still limited. REHO has been shown to be sensitive to the normalizing effect of a single dose of methylphenidate on the brains of ADHD children compared with healthy controls [An et al., 2013], however, a placebo‐controlled study of a single dose l‐dopa and benzodiazepine administration in 81 healthy individuals, showed that seed‐based connectivity analysis was statistically more powerful than Cohe‐REHO [Flodin et al., 2012]. Given that REHO is an entirely model‐free technique for data‐mining, further investigation of its viability as a biomarker for dissociating regional and global drug effects under different states is warranted.
Spectral analysis techniques
The spectral characteristics of spontaneous brain activity have long been studied in RSfMRI experiments [Kiviniemi et al., 2000, 2003; Suckling et al., 2008]. Two recent formulations, the amplitude of low frequency fluctuations (ALFF) and fractional ALFF (fALFF), use a Fourier decomposition of the RSfMRI BOLD signal, followed by integration of the amplitudes in a given spectral range, typically in 0.01–0.1 Hz band, to assess the strength of low frequency fluctuations in each voxel (ALFF). Normalization of ALFF by the power in the entire frequency range of the signal (fALFF) represents the relative contribution of a specific frequency band to the whole frequency range [Zou et al., 2008]. Seed‐ or REHO analyses assess coherence and correlation in temporal characteristics of signal fluctuations. In contrast, regional variations of (f)ALFF provide information about the spectral content of the spontaneous fluctuations in a given voxel independent of its neighboring, regional or network dependencies. Spectral analyses are usually complementary to other RSfMRI metrics such as regional homogeneity and functional connectivity. In a cross‐sectional randomized study, comparing effects of l‐dopa and benzodiazepine versus placebo [Flodin et al., 2012], fALFF detected moderate local differences between drug groups in the cerebellum which were not detectible with REHO—although fALFF was not as sensitive to detecting cortical effects compared to seed‐based functional connectivity [Flodin et al., 2012]. In another randomized placebo‐controlled study of intranasal insulin [Kullmann et al., 2013], fALFF revealed a significant reduction of hypothalamic and orbitofrontal power amplitude and a BMI‐correlated drug induced change in brain areas involved in cognitive appetitive control. ROI‐examination of fALFF in drug‐naive Parkinson's patients versus healthy controls [Esposito et al., 2013a] provided compelling evidence for a band‐specific modulation of the sensorimotor network with levodopa. As preprocessing schemes to improve the reliability of fALFF measurements in the presence of spurious noise become more established [Yan et al., 2013a], a re‐analysis of many connectivity‐based studies might shed a light on the sensitivity of this method to detecting drug‐specific neuromodulation.
Independent component analysis and dual regression
The limitations of seed‐based analysis are to some extent addressed by the data‐driven approach of independent component analysis (ICA). The majority of work reviewed here applies spatial ICA (sICA) [McKeown et al., 1998], which decomposes the 4D spatiotemporal fMRI dataset into a set of “hidden” signals characterized by a spatial map, and associated timecourses of BOLD signal fluctuations. These hidden signals may represent resting‐state networks, physiological and subject motion, or other artifacts that all give rise to the measured BOLD signals. In sICA, the decomposition is done in such a way as to maximize statistical independence in the spatial patterns, with no constraints on the timecourses. An alternative approach to sICA is temporal ICA (tICA), which decomposes the 4D spatiotemporal fMRI dataset into spatial maps and timecourses in such a way to maximize statistical independence in the temporal domain, with no constraints on the spatial maps [Biswal and Ulmer, 1999]. In other words, whereas sICA reveals different brain regions that define a dominant temporal behavior, tICA identifies different temporal behaviors that are dominant in different brain regions. Convergence or divergence of tICA and sICA results depend on assumptions about, for example, spatial versus temporal independence, and existence of spatially and temporally separable components in the data [Calhoun et al., 2012; Tian et al., 2013]. To date, because data in the spatial dimension has been much more abundant (e.g., BOLD signals are measured for many more voxels than timepoints), sICA has dominated the literature. However, fMRI data collected using recent fast acquisition techniques with TRs less than 0.25 s [Feinberg et al., 2010; Posse et al., 2013] have demonstrated the potential of tICA in doing more refined parcellation of brain networks [Smith et al., 2012], and clinical exploration of non‐stationarity in spontaneous fluctuations [Miller et al., 2014]. However, these new acquisition techniques are not yet widely available and to our knowledge, they have not been explored in pharmacological studies.
Application of ICA to pharma‐RSfMRI data was first done by [Kiviniemi et al., 2003], who used FastICA to analyze fMRI data collected in 15 anesthetized children. They were able to successfully delineate large vascular regions, as well as primary and sensory cortical areas in separate component maps. The discovery of consistent and functionally relevant ICA networks [Beckmann et al., 2005; Damoiseaux et al., 2006] has made this technique a common choice for identifying the functional topography of networks in an individual or in a group of subjects. It is common to perform a group ICA (gICA) by concatenating the data across subjects, in order to obtain a single representation of the spatiotemporally independent functional topographies. The gICA maps are then used as a template in a two‐step multivariate regression analysis to identify subject‐specific spatial maps and timecourses that capture any group or condition differences (e.g., network changes between placebo and drug). This process is called dual regression (DR): the first regression defines the timecourse of fluctuations in each network; and the second regression examines the relation between these network‐specific fluctuations and the fluctuations within each voxel of the brain. In gICA/DR studies, the choice of networks of interest (NOIs) is usually limited to 8–20 networks that resemble the most common network anatomies. In theory, by increasing the number of independent components, one might explore finer‐grained network topographies. However, component ordering and selection in ICA is very challenging, as this methods is also sensitive to detecting structured noise, and physiological and motion artifacts that diminish the reliability of component detection [Zuo et al., 2010a]. Techniques such as ICASSO [Himberg and Hyvarinen, 2003] and RELICA [Artoni et al., 2014] have been proposed to help identify the most reliable independent components, or to reject the most likely spurious ones [Salimi‐Khorshidi et al., 2014]. Yet, to fully automate the selection of functionally relevant components is not easy. A proposed workaround is to use predefined NOIs (instead of seeds) as template networks, and then proceed with DR. Group/condition differences in network connectivity are then assessed by higher‐level statistical analysis on these subject‐specific spatial maps.
GICA/DR and NOI/DR approaches are very common, and they are vastly used in pharma‐RSfMRI studies [Boveroux et al., 2010; Cole et al., 2013; Esposito et al., 2013b; Greicius et al., 2008; Guldenmund et al., 2013; Jordan et al., 2013; Khalili‐Mahani et al., 2012; Klaassens et al., 2015; Klumpers et al., 2012; Licata et al., 2013; Mueller et al., 2014; Niesters et al., 2012, 2014; Roseman et al., 2014; Tanabe et al., 2011]. Similar to seed‐based analysis, NOI/DR‐based analyses are easy to standardize and accommodates replication studies, however issues related to nonstationarity of functional networks needs to be further explored (see “Challenges and Limitations”).
Graph theoretical approaches
Almost 15% of studies surveyed here have explored drug‐induced effects on network properties. Graph‐based metrics are derived from mathematical equations that examine the global relations between regions whose connectivity is determined from seed/NOI or ICA based analysis. Whereas seed‐ and ICA‐based approaches reveal network topographies by finding areas that fluctuate in a similar and correlated manner, graph theoretical methods reflect the net value of global interaction in the brain. Metrics such as local and global network efficiency, network modularity and integration, path length, clustering coefficients, and small‐worldness serve as surrogate markers of the efficiency with which neural information propagates across brain regions. Compared with other metrics (connectivity, coherence or spectral power), which reflect the functional anatomy of the drug effect, graph metrics are more suitable for studying whole‐brain dynamics. This interpretation is supported by the observation of network‐wide variations linked to cerebral blood flow [Liang et al., 2013] and glucose metabolism [Tomasi et al., 2013].
As will be discussed in “Pharmacological Probing of Neural Networks,” pharmacological studies involving anesthetics have been critical in validation of these graph metrics, by finding association between their features and states of conscious mental activity. For example, reduced local and global efficiency of cortical regions are detected during light chloral hydrate sedation in children [Wei et al., 2013] and during loss of consciousness in propofol sedation in adults [Monti et al., 2013], indicating that global network efficiency can serve as a biomarker of the state of consciousness. Although these global effects may reflect the global state of consciousness and metabolism, the regional network properties do not change in a single direction. For example, it has been shown that nicotine causes significant variations in local efficiency of the brain networks, with significant increases in the regional efficiency of thalamus and putamen and large decreases in the regional efficiency of basal ganglia, while leaving the primary sensory and motor regions unchanged [Wylie et al., 2012]. Another graph‐metric is eigenvector centrality (EC) that indicates how each brain region connects to the most important brain hubs [Lohmann et al., 2010]. A significant increase in hypothalamic EC has been detected in response to viewing food pictures under Exenatide (vs. placebo condition) a drug that is expected to alter energy uptake by blocking the glucagon‐like peptide‐1 receptors in the hypothalamus [Schlogl et al., 2013]. Significant decreases in EC in the thalamus and the brainstem have been observed in response to mild propofol sedation [Gili et al., 2013]. Using the method developed by [Lohmann et al., 2010], anatomically distinct and drug‐specific effects of morphine (decreases in EC in cerebellum, caudate and putamen, and increases in anterior cingulate and retrosplenium) and alcohol (decreases in EC in cerebellum, hippocampus and subthalamic nuclei and an increase in anterior cingulate cortex) have been discovered in regions other than those detected from dual regression and parametric CBF mapping (see Fig. 6).
Figure 6.
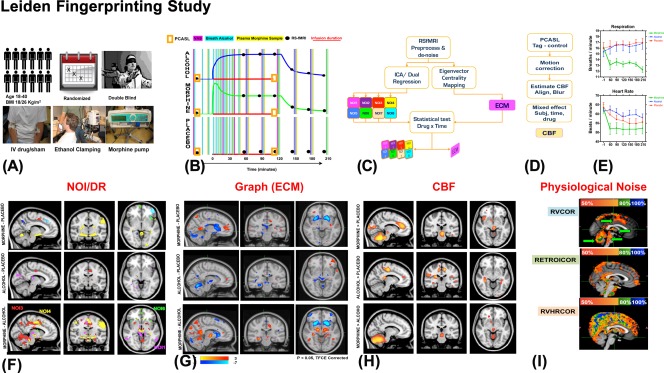
Schematic summary of the Leiden Fingerprinting Study, which aimed to investigate sensitivity and drug‐specificity of biomarkers derived from RSfMRI and ASL data. (A) 12 healthy young men participated in a double blind, crossover, placebo controlled study and were scanned several times under morphine, alcohol or sham placebo conditions; (B) Drugs were administered according to pharmacokinetic models and repeated scans were made; (C) After common preprocessing, drug with time interactions with RSfMRI were studied using ICA/DR and EC methods [Khalili‐Mahani et al., 2012]; (D) Impact of drugs on CBF was also studied [Khalili‐Mahani et al., 2012]; (E) Physiological rates were measured and the data was integrated in statistical analysis [Khalili‐Mahani et al., 2013]; (F) Drug specific changes in the cortical areas in relation to some of the canonical NOIs were detected. (G) ECM revealed drug‐distinct changes in subcortical hub regions, which differed from effects detected by ICA/DR; (H) Drug effects on CBF maps were also distinguishable in morphine and in alcohol (vs. placebo); (I) The topography of different physiological noise regressors was examined to rule out confounding neurovascular artifacts.
Different graph metrics can be explored to describe different properties of brain networks. For instance, Schroter et al. [2012] have demonstrated that, compared with wakeful state, propofol‐induced loss of consciousness is associated with a decrease in general connectivity strength particularly in the hub regions (thalamus, putamen and associative areas) and decrease in whole brain integration. In contrast, propofol treatment has been associated with increases in clustering ratio and small worldness. These effects have been interpreted as a decoupling of cortical processing from information integration in the brain [Schroter et al., 2012]. While corroborating these findings in another similar study, Monti et al. [2013] have noted that clustering ratio and small worldness are not specific to states of consciousness, and remain elevated through post‐sedation recovery, suggesting that the change in these metrics are not solely related to the drug action in the brain, but perhaps also related to the state of brain in response to loss of consciousness [Monti et al., 2013].
These variables are explored in conscious states as well. It has been shown that catecholaminergic depletion of dopamine is associated with reduced global and local efficiency of brain networks, within a range that is compatible with a small‐world topology, and reduced regional efficiency of amygdala and orbitofrontal networks [Carbonell et al., 2014b]. These network‐wide changes associated with dopamine down‐regulation have been interpreted as underlying cognitive impairments that accompany dopaminergic dysfunction in Parkinson's disease or schizophrenia. In the same vein, Wylie et al. [2012] have studies the effect of cholinergic modulation with nicotine, and have reported a significant increase in local efficiency and significant increases in regional efficiency of the limbic and paralimbic areas, suggesting that nicotine influences cognition by increasing the efficiency of communication within the brain [Wylie et al., 2012]. These examples, underline the potential of pharmacological studies in illuminating the large‐scale network properties of the brain and how they relate to higher behavioral or cognitive states. However, as it will be discussed in “Challenges and Limitations” reproducibility and reliability of graph metrics is contingent on strict preprocessing and analytical assumptions.
Hierarchical clustering
Techniques such as s/tICA parcellate the brain into spatio/temporally independent networks, and graph theoretical metrics reflect the states of integration of neuronal activity in brain networks. Hierarchical clustering techniques combine the two by generating information about the scale space of functional specialization and functional integration [Marrelec et al., 2008], yielding a more fine‐grained representation of how different brain sub‐networks interact and organize under normal or abnormal (e.g., drug‐influenced) conditions. (For a comprehensive review, see [Craddock et al., 2015].) Hierarchical clustering is a model‐free approach that parcellates the brain into biologically and anatomically meaningful spatial scales [Bellec et al., 2010; Kelly et al., 2012; Power et al., 2011; Smith et al., 2013]. Hierarchical clustering can be used to study functional modularity, that is, the extent to which the brain is divided into communities with connections of different strength. Clinical viability of studying network hierarchy has been established in demonstrating reduced hierarchical organization in functional brain connectivity of schizophrenic patients versus healthy controls [Bassett et al., 2008], in Alzheimer's patients versus healthy controls [Xia et al., 2014], in epileptic patients [Dansereau et al., 2014] and even in relation to impulsivity [Davis et al., 2013]. These techniques have not been extensively reported in pharmacological neuroimaging, however promising examples exist. Schrouff et al. showed that sedation with propofol reduced integration of information within and between six well‐known ICA‐identified networks, excluding the ventral attention network, thus suggesting functional specificity of the method [Schrouff et al., 2011]. Hierarchical clustering has been used to illustrate a reduction in modularity of resting‐state brain networks under acute dopamine depletion, consistent with the expected role of dopamine in functional integration [Carbonell et al., 2014b].
Network hierarchies can also be explored to study the dynamics of brain function and non‐stationarity. Methods such as BASC [Bellec et al., 2010] perform multi‐level bootstrap analysis to identify stable cluster hierarchies, and aberrations in epileptic patients versus healthy controls [Dansereau et al., 2014]. Network hierarchies can be plotted as multi‐resolution curves over a range of temporal, spatial, geometric and structural scales [Lohse et al., 2014]. To study the dynamics of reconfigurations of network hierarchies in relation to mental states [Bassett et al., 2011; Cole et al., 2014; Jones et al., 2012] will be a potentially important method in PK/PD modeling (discussed in “Experimental Objectives and Clinical Relevance” and “Challenges and Limitations”).
Arterial Spin Labeling and Cerebral Perfusion
Cerebral perfusion is an important pharmacodynamic endpoint. It depends on a number of factors including the viscosity of blood, vascular anatomy and pathology, systemic factors like cardiac output, end‐tidal CO2, neuronal activity and the encompassing cerebrovascular autoregulation [Ainslie and Duffin, 2009]. Autoregulation refers to the ability to maintain constant cerebral perfusion over a wide range of perfusion pressures. Cerebrovascular resistance is, however, influenced by many more (physiological) parameters, including arterial carbon dioxide levels, cerebral metabolic rate of oxygen or glucose consumption, neural activation, activity of the sympathetic nervous system, posture and other physiological variables. Under normal resting conditions, it is expected that global cerebral blood flow would be relatively stable but under pharmacological stimulation, variations in CBF become an important index of how the drug interacts with cerebrovascular autoregulation.
Cerebral perfusion can be measured with arterial spin labeling (ASL) techniques, which use radiofrequency pulses to magnetically label the water molecules in the arterial blood at the base of the brain for several seconds (1.5–2.0 s), followed by a readout of the signal within the brain after a few seconds of delay to allow blood to flow to the upstream tissue. Subtracting this labeled image from a second image acquired without labeling the inflowing blood, results in a perfusion‐weighted image that can be used to calculate cerebral blood flow in units of mL/100 mL tissue/min, using a biophysical model based on parameters of acquisition, physiological constants and validated models of “normal” cerebral hemodynamics [Alsop et al., 2014; Buxton et al., 1998; Buxton, 2005].
Quantitative methods such as ASL are reliable if acquisition protocols are well standardized [Alslop et al., 2014]. Consistency of CBF estimation with background‐suppressed pCASL has been established in multi‐center and multi‐acquisition reproducibility studies [Gevers et al., 2011; Vidorreta et al., 2013]. Intersession, intrasession and inter‐scanner reproducibility across different vendors suggests that normalizing acquisition parameters on different machines yields stable measurements of the gray matter perfusion. However, in presence of differences in acquisitions sequences, spatial differences caused by different smoothing behavior of the readout module become problematic [Mutsaerts et al., 2014; Mutsaerts, 2015; Vidorreta et al., 2013].
Various ASL acquisition schemes exist which differ in implementation and compatibility with scanner hardware. The most commonly available sequences are pulsed, (pseudo)‐continuous and velocity‐selective ASL (PASL, pCASL and VS‐ASL, respectively) [Wong, 2014]. See Figure 4 for a schematics representation of labeling differences in each method. Currently, pCASL in combination with background suppression and a (segmented) 3D readout has been proposed as the work‐horse technique for clinical experiments [Alsop et al., 2014]. Many of the arguments that led to the advice to use pCASL in radiological applications, such as signal‐to‐noise ratio (SNR), robustness, and availability [Deibler et al., 2008a, 2008b], also hold true for applications in pharmacological imaging experiments. More complex methods such as time‐encoded ASL [Teeuwisse et al., 2014], and velocity‐ and acceleration selective ASL [Schmid et al., 2014; Wong et al., 2006], are likely to become valuable assets in PK/PD modeling, but no feasibility studies have been presented so far. Here, we focus on three of the most common applications of resting state‐ASL measures used in pharmacological studies.
Figure 4.
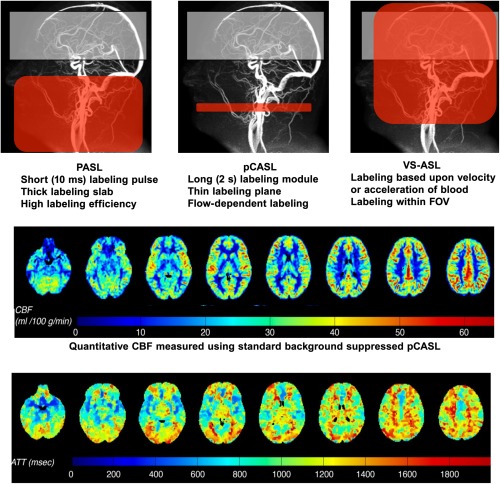
Schematic representation of ASL tagging and acquisition frames. Top row: Pulsed ASL (PASL) which provides high labeling efficiency, but low SNR; pseudo‐Continuous ASL (pCASL) has higher SNR, but is susceptible to magnetic field disturbances, arterial velocities, arterial anatomy and tag position; velocity‐selective ASL (VS‐ASL) does not require spatial selectivity as it tags all inflowing spins as long as they are above a certain velocity threshold. By including a second velocity selective module just before signal readout, signal from venous blood is suppressed, since that normally accelerates upon return. Middle row, an example of quantified CBF measured using background‐suppressed pCASL. Lower Row, Regional differences in arterial transit time.
Global cerebral blood flow
Global CBF is computed by averaging the quantitative CBF values over the entire brain, or more commonly, over the gray matter (GM) regions, which have much higher baseline CBF than white matter (WM) (∼60 mL/100g/min versus ∼20 mL/100g/min). The global CBF is a single value estimated over a specific period of time, that constitutes a pharmacodynamic endpoint in dose‐response experiments [Fernandez‐Seara et al., 2011; Marxen et al., 2014; Strang et al., 2015].
The reliability and reproducibility of global CBF over a 1‐year period (4 time points) has been demonstrated in cognitively normal subjects [Jiang et al., 2010]. Recently, in an O15‐H2O PET/pCASL validation study involving hypercapnia in the same population, precision, accuracy and high correspondence of pCASL‐based quantification of global CBF with O15‐H2O PET (the current gold standard) has been established [Heijtel et al., 2014]. It has been shown that within‐subject, between‐time or between‐session values measured on the same scanner are stable in the absence of drugs, but change significantly with drug administration [Khalili‐Mahani et al., 2011], and return to baseline levels, for example, after ketamine washout [Khalili‐Mahani et al., 2015]. Although global CBF is a reliable marker for within‐subject comparisons, it should not be treated as a quantitative measure for between‐subject comparisons. Between‐subject baseline values are expected to vary within a normal range predicted from the physical modeling of the ASL signal, however differences in acquisition parameters, modeling, and stereotaxic positioning of tag pulses with respect to arteries can influence the absolute quantitative values. Therefore, it is important to be vigilant about validation protocols that establish limits of the model in presence of acquisition noise or between‐subject variations in physiology or anatomy. For this reason, it is common to examine relative CBF, normalized to a within‐subject global CBF.
CBF mapping
Voxel‐based perfusion maps yield a snapshot of changes of the spatial distribution of CBF, and are well suited for drug fingerprinting. A handful of pharma‐fMRI studies to date have employed ASL for examining regional changes relative to placebo, in response to metoclopramide [Fernandez‐Seara et al., 2011], alcohol [Khalili‐Mahani et al., 2011; Marxen et al., 2014; Strang et al., 2015; Tolentino et al., 2011] morphine [Khalili‐Mahani et al., 2011], ketamine [Khalili‐Mahani et al., 2015], psilocybin [Carhart‐Harris et al., 2012], MDMA [Carhart‐Harris et al., 2015], THC [Van Hell et al., 2011], remifentanil [MacIntosh et al., 2008] and caffeine [Mutsaerts, 2015]. Because most CNS drugs show both a global and a local CBF response, it is interesting to study inter‐regional differences in CBF maps by isolating the global CBF effects. There are several approaches to achieve this. The simplest method is to normalize CBF voxel‐wise by taking the ratio of blood flow at each voxel to global CBF or to CBF in a control region of interest [Fernandez‐Seara et al., 2011]. More complex approaches to computing relative CBF involve modeling the effects of a drug on cerebral perfusion with respect to variations in label‐control CBF pairs over time and in relation to expected pharmacodynamics [Marxen et al., 2014], for example, using a general linear models (GLM) approach.
Although many of pharmacological ASL studies report regional CBF variations in brain areas with higher affinity for drug's target receptors, this does not necessarily mean an increased activation in the co‐localized excitatory systems [Wagner et al., 2007]. Numerous cofactors such as interneurons, inhibitory receptors, as well as adaptive functional changes due to higher‐level processing of the interoceptive or proprioceptive states (e.g., placebo) [Wager et al., 2007] may impact regional effects. For example, factors such as fatigue [Lim et al., 2010] and respiration rates [Khalili‐Mahani et al., 2011] can influence both regional and global CBF values. Thus, relation between CBF and drug effects must be interpreted carefully.
Arterial transit time
ASL can also be used to map other physiological parameters, such as the arterial transit time (ATT, or arterial time of arrival), which is the time it takes labeled blood to travel from the labeling plane to the imaging regions. Changes in ATT can be thought of both as a hemodynamic parameter that can help localize cerebrovascular effects of a drug, and as a nuisance parameter that introduces errors in the CBF quantification model. MacIntosh et al. [2008] found a spatially variable pre‐drug arterial transit time, ranging from 340 ms in the superior frontal region, to 600 ms in the putamen and insula. Brain regions with the largest ATT had the largest change in response to drug (slowing down by 10–11 ms after drug infusion). ATT measurements are easily obtained in PASL, but not with the standard pCASL acquisition protocols [MacIntosh et al., 2008]. Combined perfusion and ATT measurements are possible with multi‐timepoint ASL as well as time‐encoded pCASL [Teeuwisse et al., 2014]. These advanced techniques come, however, at a price, as they can suffer from lower SNR, lower temporal resolution, increased technological complexity, poorer robustness or a combination of these.
Combined ASL/BOLD fMRI and Neurovascular Coupling
The BOLD signal is a combination of variations in blood oxygenation, metabolic rate of oxygen consumption, cerebral blood volume and cerebral blood flow. The complexity of neurovascular coupling in RSfMRI studies has been previously reviewed [Liu, 2013], and using a combined ASL/BOLD fMRI acquisition, together with metabolic calibration methods have been proposed to disambiguate the neuronal and vascular components of drug effects. Only a handful of pharma‐fMRI experiments have acquired both ASL and BOLD‐fMRI in the same experiment, and in most cases not simultaneously [Carhart‐Harris et al., 2015; Denier et al., 2012; Griffeth et al., 2011; Khalili‐Mahani et al., 2015, 2014; Nasrallah et al., 2012; Perthen et al., 2008; Qiu et al., 2008; Rack‐Gomer et al., 2009; Van Hell et al., 2011].
In the simplest form, ASL and BOLD‐RSfMRI acquisitions are made consecutively and analyzed jointly. For example, Carhart‐Harris et al. [2015] showed in humans that changes in CBF after administration of 3,4‐methylenedioxymethamphetamine (MDMA) were also accompanied by changes in BOLD resting‐state functional connectivity in brain regions such as the amygdala and hippocampus and that the functional changes in CBF and BOLD functional connectivity were related to the global subjective effects of MDMA. Conversely, using a conjunction analysis approach, Khalili‐Mahani et al. [2014] showed that BOLD‐related connectivity and CBF correlations under a morphine condition were most consistent in the prefrontal networks and most uncorrelated in the sensorimotor area.
More elaborate acquisition techniques such as dual echo PCASL [Dai et al., 2008] allow to acquire BOLD/CBF measurements simultaneously in order to investigate the neurovascular coupling with higher temporal precision [Fukunaga et al., 2008; Gauthier and Hoge, 2012; Tak et al., 2015; Wu et al., 2009]. In one such pharmacological experiment, Qiu et al. [2008] estimated both BOLD and relative CBF after sevoflurane anesthesia induction or after withdrawal of sevoflurane in healthy subjects, and showed that CBF and BOLD remained coupled under anesthesia although the coupling rates varied spatially.
To disentangle the neurovascular component of brain activity from the measured BOLD signals, the simultaneous BOLD/CBF data collected during rest are “calibrated” (via a biophysical model) using a separate scan with the same acquisition parameters and under controlled administration of gases (CO2 or O2) or breath‐holding. This, a form of phfMRI itself, is the gold standard for evaluating the rate of change in neuronal activity, perfusion and cerebral metabolic rate of oxygen consumption (CMRO2). For review, see [Hoge, 2012]. Pharmacological interventions further complicate the “calibration” of the hemodynamic response. For example, in studies of caffeine, it has been shown that factors such as dosage and history of exposure determine activation of different receptor subtypes [Chen and Parrish, 2009b, 2009a], which cause opposite vascular responses depending on dose or exposure history [Diukova et al., 2012; Griffeth et al., 2011]. The need for gas challenges (CO2 for hypercapnia, or O2 for hyperoxia) or breath holding [Bright and Murphy, 2013a; Lipp et al., 2015; Madjar et al., 2012] for calibration makes this technique impractical for most pharma‐fMRI studies. However a promising new technique by Blockley et al. [2012, 2015] utilizes MR pulse sequences as a means of estimating the parameters in the BOLD biophysical model, obviating the need for a gas calibration scan. Such quantitative approaches in drug studies will be important for better modeling of neuronal, vascular and metabolic factors that modulate brain activity under different drugs.
EXPERIMENTAL OBJECTIVES AND CLINICAL RELEVANCE
Neuropharmacological research encompasses a broad range of exploratory, proof‐of‐concept and clinical objectives. Drug classes and expected pharmacodynamics often influence experimental design—also the cost. Generally, pharma‐fMRI experiments investigate relationships between drug actions and brain function in terms of (1) neurophysiological changes resulting from drug interactions with specific target receptors; (2) alteration of activity of other neuropeptides in downstream pathways and (3) peripheral and nonspecific physiological responses in organs other than the brain. Figure 5 illustrates the proportion of different drug classes studied to date in relation to common study objectives in the literature surveyed here.
Figure 5.
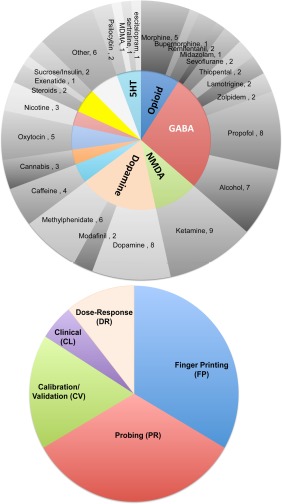
A visual summary of the distribution of drugs tested and research objectives. [Color figure can be viewed at http://wileyonlinelibrary.com]
Drug Fingerprinting
Drug fingerprinting (FP) refers to characterizing the drug‐specific topography of brain activation, assuming that drugs modulate the neuronal activity in brain regions that have a preferred neurotransmitter affinity (i.e., higher receptor density, higher affinity or higher binding potential) for that drug. However, drugs rarely act on just one neurotransmitter, and often produce overlapping side effects (e.g., most antipsychotic drugs are also sedative, or drugs used to treat Parkinson's Disease may lead to psychosis or anxiety side effects). For this reason, it is important to define fingerprinting realistically: FP does not refer to the ability to identify a unique signature of a particular drug effect in the brain, rather it refers to methods that help tease apart the drug‐specific actions from common effects. To identify whether a drug acts on a specific neural pathway is important for target identification and clinical design. As such, FP requires a model‐free and generalizable analytical technique that helps characterize the full profile of brain's drug‐distinct and physiologically‐common responses to different drugs across populations. To localize the effect of drugs in the brain is the primary objective of more than 30% of studies surveyed here (see Table 1 and Fig. 5).
In an early FP study (Leiden fingerprinting study, Fig. 6), it was shown that the NOI‐based dual regression (DR) method provides a sensitive framework for detecting compound‐specific effects in resting‐state brain networks [Khalili‐Mahani et al., 2012]. A similar analytical approach has been used to explore the effects of psilocybin versus MDMA [Roseman et al., 2014]. The NOI/DR method has been used to study the effects of a single compound on the topography of resting‐state networks under PK‐controlled administration of THC [Klumpers et al., 2012; Van Hell et al., 2011], ketamine [Niesters et al., 2012] and bupivacaine [Niesters et al., 2014]. ASL has proven to be a sensitive method for FP studies of alcohol [Khalili‐Mahani et al., 2011; Marxen et al., 2014; Strang et al., 2015], opioids [Khalili‐Mahani et al., 2011; Kofke et al., 2007; MacIntosh et al., 2008] and THC [Van Hell et al., 2011].
Other examples of FP experiments without pharmacokinetic modeling include mapping the effects of nicotine [Tanabe et al., 2011], methylphenidate [Mueller et al., 2014] or amphetamine [Esposito et al., 2013b] and serotonin [Klaassens et al., 2015]. The objective of FP has also been explored in studying the effect of receptor agonist and antagonists in cross sectional designs, for example, comparing functional connectivity in response to levodopa versus haloperidol [Cole et al., 2013], or the NMDA antagonist ketamine versus the glutamate suppressant lamotrigine [Deakin et al., 2008; Doyle et al., 2013].
Dose/Response in Anesthesiology
PhfMRI was first conceived to address the questions related to dose responses in the brain during administration of different doses of anesthetics and analgesics. In these studies (depicted in Fig. 1B), the magnitude of change in the BOLD signals measured over the timecourse of drug infusion and washout is fit to the pharmacokinetic profile of anesthetics such as morphine [Becerra et al., 2006; Leppa et al., 2006], buprenorphine [Upadhyay et al., 2011] and ketamine [De Simoni et al., 2013]. Seed‐ or network‐based connectivity analyses of Pharma‐RSfMRI data have also proven sensitive to detecting dose response effects in the brain. Dose dependent variations in the motor network (identified by a seed in M1 cortex) under different stages of anesthesia with different doses of sevoflurane were first demonstrated by [Peltier et al., 2005]. Using the putamen as a seed, Upadhyay et al. showed a dose‐dependent decreases in functional connectivity of the somatosensory and sensorimotor networks [Upadhyay et al., 2012] under buprenorphine, with a topography that closely resembled an NOI‐based analysis of the morphine effect on these networks in an independent study [Khalili‐Mahani et al., 2012] prominent effects in the hippocampus and sensorimotor networks. A dose‐dependent increase in functional connectivity of the frontoparietal and thalamocortical regions has been shown to correlate with pain perception in spinal anesthesia using bupivacaine [Niesters et al., 2014]. A dose‐dependent ketamine‐induced increase in the functional connectivity of the corticohippocampal subnetworks has also been reported concurrent with reduced alertness states [Khalili‐Mahani et al., 2015].
Anesthetics have often been used to gain a better understanding of the clinical relevance of resting‐state fMRI biomarkers (see “Methods Validation and Calibration” for details). Derivative biomarkers such as network properties, coherence or spectral power, have been used to study the global effects of drugs on the brain, in terms of the reconfiguration of resting‐state networks under sedation with propofol [Gili et al., 2013; Jordan et al., 2013; Monti et al., 2013; Schroter et al., 2012; Schrouff et al., 2011], chloral hydrate [Wei et al., 2013] and sevoflurane [Peltier et al., 2005; Qiu et al., 2008].
As will be discussed in “Challenges and Limitations,” dose/response evaluation, also known as pharmacokinetic/pharmacodynamic (PK/PD) modeling is one of the most challenging, but also most important goals of pharmacological neuroimaging.
Pharmacological Probing of Neural Networks
In pharmacological probing, the effects of a given dose of a neuroreceptor agonist/antagonist or blocker are investigated in order to test an a priori hypothesis about the relationship between the drugs mechanism of action on a region or a system, and the neuropsychological function targeted by that drug. Pharmacological probing experiments can serve several research objectives.
Systems neuroscience
In the simplest designs, pharmacological probing can be used to investigate the neuro‐circuitry of a given neurotransmitter‐signaling pathway in animals or humans. (See Fig. 5 for a summary of the most commonly tested neurotransmitter systems.)
Pharmacological probing of the dopaminergic system with direct or indirect targeting of the dopamine receptors has been well‐studied [Carbonell et al., 2014b; Cole et al., 2013; Fernandez‐Seara et al., 2011; Flodin et al., 2012; Kelly et al., 2009; Mueller et al., 2014; Ramaekers et al., 2013; Vytlacil et al., 2014]. For instance, Cole et al. compared the effects of the dopamine agonist levodopa and the dopamine antagonist haloperidol against placebo on large‐scale network connectivity using group ICA and dual regression, and found a dichotomous pattern of activity in the connectivity of the anteromedial cortical regions to the DMN: compared with levodopa, haloperidol administration was associated with higher positive correlations between the DMN and left precentral and middle frontal gyri, and higher negative correlations between the DMN and the supramarginal gyrus and intraparietal sulcus. Halopridol induced modulation of the connectivity in these regions was also associated with behavioral scores, with higher negative connectivity predicting higher impulsivity [Cole et al., 2013].
With a growing understanding of the role of glutamatergic dysregulation in various neurological and psychiatric disorders [Krystal et al., 2003], pharmacological probing of the NMDA receptor system is becoming a more active area of research as well [Becerra et al., 2009; De Simoni et al., 2013; Deakin et al., 2008; Doyle et al., 2013; Driesen et al., 2013; Gass et al., 2014; Grimm et al., 2015; Khalili‐Mahani et al., 2015; Li et al., 2014; Niesters et al., 2012; Scheidegger et al., 2012]. In the first study of its kind, Deakin et al. [2008] probed the glutamatergic system using an NMDA‐receptor antagonist to test an experimental model of schizophrenia and demonstrated a significant ketamine‐induced reduction of the BOLD signal in the antero‐ and posteromedial regions, that was significantly associated with psychosis scales [Deakin et al., 2008]. Taking a different analytical approach, Driesen et al. [2013] showed a significant correlation between increase in global connectivity and increased psychotic‐like and social withdrawal and cognitive distraction scores [Driesen et al., 2013]. In an elaborate partial‐crossover design, Doyle et al. [2013] used two different pre‐treatments with lamotrigine (300 mg, oral single dose, an anticonvulsant drug that directly inhibits glutamate release) and risperidone (2 mg, oral single dose, an antipsychotic drug that acts on the serotonergic system and indirectly inhibits glutamate release) to provide evidence that ketamine modulation of the resting‐state BOLD amplitude is linked to glutamate release [Doyle et al., 2013]. Importantly, by showing distinct regional interactions of risperidone and lamotrigine with ketamine (compared with placebo), this study offered further evidence for the plausibility of such experimental designs in drug fingerprinting.
From a different perspective, Khalili‐Mahani et al. [2015] used ketamine to test the “hippocampal negative feedback inhibition of the HPA axis” model [Khalili‐Mahani et al., 2010; Pruessner et al., 2008], and asked whether a pharmacological blockade of NMDA‐receptors would cause a de‐activation of the hippocampus and trigger a neuroendocrine stress response due to disinhibition of the hippocampal controlled HPA axis. In fact a reduction of hippocampal CBF relative to the rest of the brain, concomitant with increased cortisol secretion, and increased cortico‐hippocampal connectivity that correlated with changes in states of alertness was observed [Khalili‐Mahani et al., 2015]. An independent research group has reported a similar increase in corticohippocampal connectivity in both men and mice [Grimm et al., 2015].
The GABAergic system is often associated with inhibitory neuronal activity, and pharmacological stimulation with drugs such as zolpidem and alcohol that act on GABA receptors. For example, it has been shown that alcohol and zolpidem decrease activation of visual cortex during visual stimulation [Calhoun et al., 2004; Levin et al., 1998; Licata et al., 2011]. However GABAergic drugs have been shown to increase functional connectivity within many cortical regions [Licata et al., 2013, Esposito et al., 2010; Greicius et al., 2008; Khalili‐Mahani et al., 2012; Kiviniemi et al., 2005]. Specifically, zolpidem increased resting‐state functional connectivity within auditory, visual, motor, and limbic networks [Licata et al., 2013]; midazolam enhanced BOLD signal synchrony within visual, auditory, and motor cortices during conscious sedation [Greicius et al., 2008; Kiviniemi et al., 2005], and alcohol also increased the connectivity strength in the auditory, somatosensory, sensorimotor, cerebellar, and visual RSNs [Esposito et al., 2010; Khalili‐Mahani et al., 2011]. Although interpretation of these studies is still tentative, they illustrate that RSfMRI connectivity analysis provides a complementary view of brain's activation states, which challenges the assumptions of deactivation at the receptor site, and suggests wide spread effects of GABAergic signaling on the entire brain.
Disease mechanisms
To differentiate disease‐related aberrations in neural function, by using drugs that target brain regions or neurotransmitters affected by the disease, is another objective of probing studies. Pharmacological stimulation of the dopaminergic system has been tested in search of biomarkers that differentiate the states of brain activity in ADHD [An et al., 2013], in Parkinson's disease versus normal [Esposito et al., 2013a], and in addiction to heroine [Denier et al., 2013; Schmidt et al., 2015] and cocaine [Konova et al., 2015, 2013; Kufahl et al., 2005]. With growing interest in the glutamatergic underpinnings of mood and anxiety disorders, several RSfMRI studies have attempted to probe variations in resting‐state functional connectivity during pharmacological perturbation of the NMDA receptor system [De Simoni et al., 2013; Doyle et al., 2013; Khalili‐Mahani et al., 2015; Scheidegger et al., 2012]. Yet in other studies, differences in cognitive control between healthy adult and alcohol‐dependent individuals have been probed using stimulants such as modafinil [Schmaal et al., 2013], and differences in emotional systems in patients with anxiety disorder and healthy controls have been probed with oxytocin [Dodhia et al., 2014]. Even sucrose has been used to investigate differences in the neural correlates of metabolic regulation between healthy obese and lean individuals [Kilpatrick et al., 2014].
Methods Validation and Calibration
Resting‐State fMRI has exponentially grown in popularity; however, the majority of studies to date are correlational or methodological investigations. Pharmacological experiments offer an important opportunity to study causal relationships, and increase the biological impact of RSfMRI findings. Greicius et al. [2003] validated their theory about the role of the DMN in sustaining the states of consciousness [Greicius et al., 2003] by comparing functional connectivity during administration of midazolam in a sedated but awake state versus a resting unsedated state [Greicius et al., 2008]. The link between the DMN and states of consciousness has also been studied under propofol [Boveroux et al., 2010; Stamatakis et al., 2010]. The general resting‐state brain activity related to consciousness states has also been investigated in other relay hubs such as the thalamus and brainstem [Guldenmund et al., 2013].
Similarly, drugs can be used to “calibrate” neuroimaging variables, such as the BOLD signal and blood flow. For example, based upon a priori hypotheses about the vasoconstrictive properties of caffeine [Rack‐Gomer et al., 2009; Tal et al., 2013; Wong et al., 2012; Wu et al., 2014], and the vasodilatory effects of CO2 [Gauthier et al., 2012; Wise et al., 2004b; Xu et al., 2011], these substances have served as “non‐invasive” neurochemical modulators of the neurovascular components of the BOLD signal to study neurovascular coupling.
Finally, pharmacological calibration may lead to more accurate models of the BOLD/CBF relationship. Although the correlation between BOLD fluctuations and cerebral blood flow in brain networks is significant [Chuang et al., 2008], this relationship is not uniform across the brain [Khalili‐Mahani et al., 2014; Li et al., 2012]. Using drugs to manipulate brain physiology may help clarify to what extent this heterogeneity arises from spatial variations in cerebrovascular physiology [Liu, 2013] or from regional chemoarchitecture affecting neuromodulation [Zilles and Amunts, 2009]. This insight will be critical for developing more accurate models to describe the relationships between brain metabolism, neuronal activity and vascular reactivity.
Animal Research
Drug development and basic neuroscience discovery hinge on animal‐to‐human translation. Methodological issues related to animal phfMRI studies have been long investigated [Gozzi et al., 2007, 2005; Schwarz et al., 2006, 2003]. In vivo mapping of the functional connectivity of neurotransmitter networks of the animal brain under drugs was first introduced by Schwarz et al., who have since explored and validated various approaches to animal phfMRI analysis [Schwarz et al., 2007a, 2007b, 2007c, d]. With methods being established and availability of ultra‐high field (>9 Tesla) scanners, rodent pharma‐RSfMRI is likely to become a very active research field. A full review of challenges of animal fMRI is beyond the scope of the current review. For a recent review of the promising implications of this method, see Jonckers et al. [2013]. However, some animal studies have been specifically conducted to validate several of the RSfMRI biomarkers used in humans. To date, animal pharma‐RSfMRI studies have:
established a link between spontaneous electrophysiological and hemodynamic fluctuations measured in rat brain by measuring EEG and BOLD signal [Liu et al., 2011; Nasrallah et al., 2014a; Otte et al., 2014], and by pharmacological manipulation of the mitochondrial Ca++ influx [Sanganahalli et al., 2013];
detected reproducible set of anatomically distinct resting‐state network from ICA of BOLD signal fluctuations in rats [Becerra et al., 2011] and mice [Mechling et al., 2014] and characterized similarities and differences between rats and mice [Jonckers et al., 2011], thus providing a methodological framework for standardized model‐free exploration of drug effects on the brain of different species;
illustrated different patterns of resting‐state connectivity with different anesthetics (isoflurane, alpha‐chloralose and medetomidine) [Williams et al., 2010], or adrenergic agonists versus antagonists [Nasrallah et al., 2014b], thus providing evidence for the sensitivity of the methods to detecting receptor‐specific effects. These findings confirm potential biases that are expected from interactions of the anesthetic drugs with the test compounds [Gozzi et al., 2008].
illustrated that the functional network topography [Liu et al., 2013b], and spectral power [Magnuson et al., 2014] of the spontaneous fluctuation in the BOLD signal were dynamically altered by different doses of the anesthetics isoflurane and medetomidine [Nasrallah et al., 2014a], even at different stages of coming out of anesthesia during ketamine washout [Bettinardi et al., 2015];
And last but not least, illustrated the efficacy of the technique in fingerprinting and pharmacological probing by providing evidence of homologous effects of an opiodergic drug, buprenorphine, in humans and in rats [Becerra et al., 2013], by detecting changes in the dopaminergic system after haloperidol [Gass et al., 2013] administration, and by detecting changes in the glutamatergic system (including hippocampus and medial prefrontal cortex) with the NMDA antagonist memantine [Sekar et al., 2013].
These findings strengthen the case for applicability of RSfMRI in translational neuroscience.
CHALLENGES AND LIMITATIONS
For pharma‐RSfMRI to be clinically viable, three aspects of reliability, reproducibility and sensitivity are critical. First, it must be established that the biomarkers are reliable and robustly reproducible. Next, those biomarkers must be able to measure drug effects with a certain degree of statistical reliability within a standardized framework, and third, the models used to assess effects must be clinically meaningful.
So far, we have shown applications of different fMRI biomarkers and provided evidence from existing studies to illustrate the clinical plausibility of these metrics to characterize regional and global effects of drugs in the brain. However, several limitations in study design and analysis that remain to study further.
Biological Confounds
As discussed in “Basics and Biomarkers,” both BOLD and ASL measurements relate to cerebral blood flow, which is tied to autoregulation and is affected not only by the direct modulation of neuronal activity at the site of drug receptors, but also by indirect modulation of the endocrine system, overall metabolic rate and the cardiopulmonary function that may demand an autoregulatory response from the brain (Fig. 1). These interdependencies challenge a simple interpretation of neuroimaging findings as pure neuronal activity, and necessitate a multivariate approach to assess the impact of different confounds. Most critical biological confounds include:
Baseline (psycho)physiological states
One of the most challenging aspects of designing functional neuroimaging studies is to establish and define a “baseline” state. In task‐based fMRI, the problem is to some extent circumvented by the explicit design of a “resting” block, against which the activation in the task‐blocks is compared. In RSfMRI, however, inferences are made based on the spatiotemporal features of the spontaneous hemodynamic fluctuations over time. Slight psychophysiological variations such as keeping eyes open or closed [Patriat et al., 2013], or diurnal phases [Hodkinson et al., 2014] may impact the reliability of functional network detection. It has been shown that circadian rhythm (with respect to the time of awakening) influences fluctuations in DMN and sensorimotor areas over the course of ten hours of wakefulness [Blautzik et al., 2013]. Interestingly, the functional connectivity of the posterior component of the DMN, and the insula have the highest interclass correlation coefficients if assessed from BOLD signals (but not from similar analysis of perfusion‐weighted timeseries) [Jann et al., 2015]. It is not yet clear whether this BOLD‐specific interclass correlation is explained by the consistency of physiological pulsations [Liu et al., 2013a], by the neuronal or cerebrovascular physiology [Liu, 2013] or by anatomy [Vigneau‐Roy et al., 2014]. However by adding a control baseline condition (i.e., a pre‐drug scan) and including an additional placebo session to the design, might make it more likely to tease apart such confounds.
The placebo effect on neuroimaging biomarkers in pharma‐fMRI studies is not negligible [Meissner et al., 2011]. To add a placebo session can help identify drug‐unrelated effects related to the experience of being in the scanner (e.g., fatigue, phobia, anticipation). However, variations in psychological states over time (during drug administration or placebo session) can contribute to the nonstationarity of the resting‐state brain fluctuations (further discussed in “Reliability, Reproducibility”). It is therefore important to include repeated measurements of in both placebo and drug conditions in order to evaluate the influence of interinidividual variations in brain's psychopharmacodynamics.
Cardiopulmonary function
As will be discussed in “Structured Noise and Artifact Removal,” most fMRI studies treat respiration‐ or heart‐related signals as nuisance factors and try to eliminate them, although some argue that by removing these variations information about neural correlates of proprioceptive functions or autonomic and metabolic control may be erroneously discarded [Bright and Murphy, 2015; Iacovella and Hasson, 2011; Vazquez et al., 2014].
Physiological correction in pharma‐fMRI is a more complicated issue because of the direct and indirect effects of the CNS drugs on the autonomic nervous system. Respiration and heart rate are regulated by the interplay between brain areas that integrate autonomic, somatomotor and proprioception, and by cerebrovascular autoregulation that adaptively maintains cerebral oxygen level via arterial inflow. Changes in the heart rate or respiration can be correlated with activation of the brainstem nuclei that control the autonomic system, or with neurovascular and neurochemical processes that fluctuate with the cardiac or respiratory rhythms, respectively. For instance, respiratory depression after opioidergic drugs causes hypercapnic conditions that trigger a strong vasodilatory response and create strong vascular reactivity, which in turn confounds the use of BOLD signal to detect neuronal responses [Pattinson et al., 2009]. Drugs that increase heart rate or blood pressure also increase pressure wave pulsatility in cerebral arteries, and may result in motion‐related spurious effects in the proximity of major arteries [Dagli et al., 1999]. Global physiological responses of the cardiorespiratory system during different drug conditions likely give rise to signal changes that resemble a global change in CBF—which will be indistinguishable between different drug classes. Efforts to tease apart these mechanisms on phMRI signals continue [Faull et al., 2015; Mitsis et al., 2009; Pattinson et al., 2009], but combined BOLD/ASL acquisitions discussed in “Combined ASL/BOLD fMRI and Neurovascular Coupling” will further improve our understanding of these mechanisms.
Either way, neuronal fluctuations related to physiological factors are essential pharmacodynamic endpoints and must not be discarded as “noise.” Physiological noise correction schemes may drastically alter the statistical outcome of the drug effects on the brain [Khalili‐Mahani et al., 2013]. Interestingly, the regions of precuneus and posterior cingulate cortex in the DMN seem to be most vulnerable to physiological effects especially in higher‐level statistical comparisons [Khalili‐Mahani et al., 2011]. However, regional susceptibility to physiological noise relates to the scale of drug‐induced change in respiration and heart rate. For example, these effects are very significant in the morphine condition that causes strong respiratory effects, and non‐significant in alcohol condition in the same subjects [Khalili‐Mahani et al., 2013]. The method chosen for physiological noise correction can also impact the extent of the statistical outcome. For example, the impact of time‐series correction using derived respiration and cardiac variation regressors, or RETROICOR on final statistical outcome is lower than using averaged respiration rates in the highest level group analysis [Khalili‐Mahani et al., 2013] (see Fig. 6). Unfortunately, many of existing pharmacological studies surveyed here do not report any physiological records (see Table 1). However, a rigorous characterization of the physiological interactions with vascular effects of the drugs is essential and informative in understanding specific and nonspecific effects of the drug on neuroimaging biomarkers.
Age and sex effects
As Figure 7 shows, the age range (20–45, with the exception of three studies) and sample sex (male > female) are biased in the studies we reviewed, reflecting the challenging nature of experimental designs. In older adults, the study design must provision for how a particular compound interacts with other medications or concomitant health conditions. To include females would require controlling for endocrinological factors related to menstrual cycle, birth control or hormonal replacement therapy (in older women) [Comasco et al., 2014]. Yet, without expanding demographic representation of females and age ranges in pharma‐fMRI studies, it remains a challenge to draw general conclusions about the CNS mechanisms of any drug. While challenging in humans, animal pharma‐RSfMRI studies might illuminate the significance of these variables.
Figure 7.
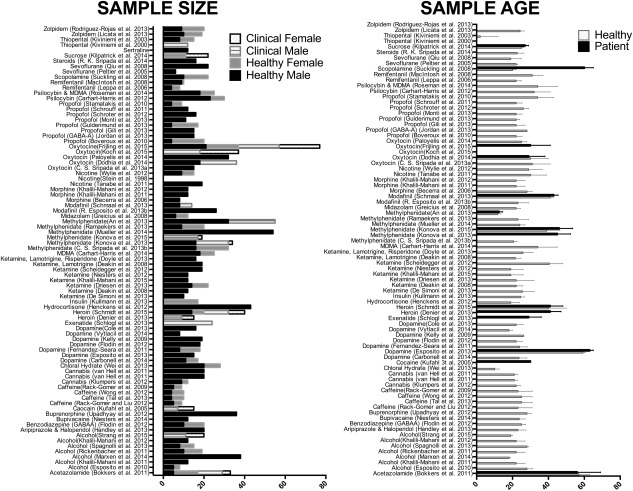
Sample sizes and age ranges distribution in the surveyed studies.
Anatomical variability
This survey illustrates a general neglect of the potential impact of anatomical variations on localizing drug effects. In particular, given that drugs reach the brain via the circulatory system, and given the high likelihood of cardiopulmonary physiological responses to drugs, the impact of the morphology of the cerebrovascular tree and flow territories on functional topography of drug effects may not be a negligible confound.
Gross anatomical features include variables such as the shape and the size of certain brain structures, cortical surface anatomy and gyrification. Cao and colleagues have shown that anatomical variations related to the choice of anatomical parcellation schemes used for network representation of the brain) do impact the reliability of graph‐metrics, although some local and global network properties are more robust to parcellation effects than other metrics [Cao et al., 2014].
Gross anatomical features also include configurations of the cerebrovascular tree and the white matter fiber tracts that underpin connectivity by “wiring” the brain. CNS drugs are transported into brain tissue from blood across the walls of capillaries or through CSF transportation into venous blood [Pardridge, 2012]. Variations in the cerebrovascular architecture are thus likely to influence variations in measured RSfMRI and ASL signals [Tak et al., 2015, 2014; Vigneau‐Roy et al., 2014]. In fact, one of the major challenges in reproducing ASL results stems from between‐subject variations in cervical anatomy that can alter model parameters used to quantify CBF [Aslan et al., 2010]. To what extent these model parameters vary with pharmacological stimulation, cerebrovascular anatomy or the layout of the arterial flow territories [Bokkers et al., 2011; Hartkamp et al., 2011] is not yet studied.
Structured Noise and Artifact Removal
Head motion
Rigid‐body head motion artifacts, that is, those caused by moving the head during acquisition, deteriorate the quality of the MRI data. If the movement happens during acquisition of a frame, the data becomes corrupted and must be discarded, but if motion happens between acquisition echoes, then it causes misalignment of frames and it can be corrected. The most common motion correction technique uses simple transformation (3 translation and 3 rotations, and maybe three stretches, using a standard cost minimizing function such as least square error) to align all frames of an EPI dataset with respect to an arbitrary frame of reference from the time series (usually the middle frame). This procedure often returns a matrix with an estimate of linear movement, stretch and rotation in a 3D space per each frame, or an estimate of excessive and artifactual signal spikes in adjacent frames. These motion parameters (and their derivatives) are often included as nuisance regressors, and included in models that estimate connectivity. Another approach is to “scrub” or “censor” frames that are characterized with excessive movement and to discard them out of the RSfMRI timeseries [Power et al., 2011], albeit with added temporal and spatial filtering and regression options [Carp, 2013; Power et al., 2014; Satterthwaite et al., 2013].
In an elegant study evaluating the impact of interactions between the temporal, spatial and spectral characteristics of motion and different preprocessing techniques, Satterthwaite et al. have demonstrated that different correction methods are not uniformly effective [Satterthwaite et al., 2013]. In fact, in a survey of various motion‐correction methods, Power and colleagues have concluded that regression‐based motion‐corrections “represent cosmetic improvements rather than true correction of the data” [Power et al., 2014] and that there is no definitive answer to the best way motion effects can be mitigated [Power et al., 2015]. Methods using ICA to isolate motion components from the data [Griffanti et al., 2014; Kundu et al., 2012; Salimi‐Khorshidi et al., 2014; Tohka et al., 2008] avoid breaking the temporal continuity of the data or creating inconsistent temporal degrees of freedom, which would be caused by scrubbing methods. But ICA‐denoising techniques depend either on subjective classification of the artifact components, or on automatic feature selection based on a priori assumptions about the pattern of motion artifacts [Pruim et al., 2015]. In a recent comparative analysis Pruim et al. have compared eight common motion correction strategies, and have concluded that ICA‐AROMA was more effective in reducing motion artifacts across datasets in retaining signal of interest and improving reproducibility.
Although these corrective measures seem to improve the consistency of detecting the functional architecture of the brain, the correction residuals of these regressors are not “white,” and remain anatomically specific [Power et al., 2012; Van Dijk et al., 2012; Yan et al., 2013b]. In fact the problem of motion may be different in patients versus healthy controls [Kong et al., 2014; Wylie et al., 2014]. This raises concern for pharmacological studies, if the drug causes variations in movement patterns, for example, with sedatives and anti‐tremor compounds that are likely to reduce movement as compared with placebo, or stimulants and hallucinogens that may increase it.
Physiological artifacts
Physiological signals are important biological confounds but they can give rise to artifacts. The simplest form of physiological artifact arises from arterovascular pulsation correlated with the heart beating, bulk movement due to breathing, or even a movement of the imaging plane due to magnetic susceptibility artifacts resulting from breathing in the magnet. These artifacts are often dealt with in addition to motion, either by modeling a movement‐related phase‐shift in each EPI acquisition, combined with a canonical hemodynamic response function to respiration and heart rate [Birn et al., 2008b; Chang et al., 2009]; by using a proxy correction by assuming that the BOLD signal fluctuations in the deep WM and CSF encode non‐neuronal variations in respiration and heart rate signals [Birn, 2012]; or by using dual‐echo acquisition methods that can help separate true noise from biologically relevant signals [Bright and Murphy, 2013b]. To date, the majority of work examining the impact of physiological correction on resting‐state fMRI has focused on improved consistency of the DMN [Marchitelli et al., 2016], although in some cases, the DMN and the respiration component identified from ICA may be overlapping [Birn et al., 2008a]. In fact, it has been shown that respiration‐related changes in end‐tidal CO2 create anatomically specific patterns in many temporal (e.g., Insula) and medial brain areas (e.g., cingulate cortex) that are commonly studied in pharmacological studies [Peng et al., 2013; Wise et al., 2004b].
In more than 60% of studies surveyed here, heart and respiration rates are not measured or reported. In only half of the rest, these variations are used as nuisance factors to “clean” artifacts from the data. In only one of these studies, the impact of various noise removal techniques on the statistical outcome of drug effect on the brain has been reported [Khalili‐Mahani et al., 2013]. This study shows that morphine doses that cause significant physiological response (in terms of respiratory depression) also cause significant changes in the topography of the group results—namely a significant reduction of the statistical significance of drug effect on the DMN. In this study, corrective methods that are applied at the level of individual RSfMRI preprocessing or DR (e.g., regressors corresponding to respiration or heart rate variations or retrospective image correction with RETROICOR and RVHRCOR), were not as impactful–even though their impact on within‐subject BOLD variations were significant and consistent [Khalili‐Mahani et al., 2013] (also see Fig. 5). These observations raise concern about the implications of unscrutinized physiological “noise” correction and their impact on interpretations of results.
Global signal regression
Global Signal Regression (GSR) aims to correct for combination noise arising from subject movement, physiological artifacts, as well as high frequency electronic noise, or amplitude drift [Power et al., 2014; Satterthwaite et al., 2013]. Similar to motion and physiological correction, there is also no consensus on whether this preprocessing step improves the results or gives rise to false positives and misinterpretation. The strongest argument against GSR is that it artificially shifts and zero‐centers the distribution of correlations, thus giving rise to anti‐correlated patterns “erroneously” interpreted as deactivation [Murphy et al., 2009; Weissenbacher et al., 2009]. It has also been shown that GSR can hinder analyses of inter‐individual differences [Saad et al., 2012], bias the topography of networks in relation to motion parameters [Satterthwaite et al., 2013], and confound activity that is closely correlated with signal in the gray matter [Yan et al., 2013a], including spontaneous fluctuations in local field potentials induced by changes of behavioral states of monkeys [Scholvinck et al., 2010]. Although the mathematical argument against zero‐centering holds, others argue that this process increases the detectability of physiologically meaningful phenomena from the cortex [Fox et al., 2009]. The impact of GSR on test‐retest reliability of metrics other than functional connectivity is also contentious. It has been shown that GSR improves the reliability of spectral analysis (using Alff) [Yan et al., 2013a], but worsens REHO [Zuo et al., 2013], although the influences of GSR on REHO reliability seem to be complex, anatomically varying, and related with neuronal mechanisms that underlie eyes‐closed or eyes‐open states [Qing et al., 2015]. It has also been shown that GSR alters the topography of graph representation of the brain by altering the network thresholds [Schwarz and McGonigle, 2011]. The effect of GSR on graph properties and test–retest reliability is made further complex by the mathematical assumptions underlying graph computations, for example, partial correlation or Pearson correlation, although differences in terms of improved reliability with and without GSR may be negligible [Liang et al., 2012]. A recent review indicates that GSR does not cause significant changes in degree, local and global efficiency, but lowers the reliability of clustering coefficient and assortivity [Andellini et al., 2015]. In presence of these uncertainties, newer methods advise performing post‐hoc estimation of the extent to which GSR changes the analysis outcomes [Carbonell et al., 2014a]. In general, although there is no consensus about including GSR in preprocessing, existing evidence suggest the necessity of considering the full scope of anatomical and spectral characteristics correlated with GSR [Liang et al., 2012; Qing et al., 2015]. Given the repeated‐measures design in most pharmacological studies here, it might be revealing to investigate the differences in the test outcomes with and without applying different preprocessing steps.
Reliability, Reproducibility
In pharmacological neuroimaging, biomarker test–retest reliability is critical and a determining factor in establishing a meaningful effect size. It is also the most challenging, because spontaneous brain oscillations are non‐stationary. Reliability and reproducibility in pharma‐RSfMRI is not well addressed yet. In one study involving pharmacological stimulation by ketamine, it was shown that reliable estimates of the pharmacodynamics in the ROIS were possible but only if nuisance variables such as motion and drift were well modeled [De Simoni et al., 2013]. Although an important first step, the conclusions of this study are not generalizable. In this section, we highlight the impact of non‐stationarity, data acquisition and analytical parameters on test–retest reliability in RSfMRI studies.
Non‐stationary neural networks
A major challenge in establishing a ground truth for RSfMRI metrics is the fact that resting‐state functional networks can be non‐stationary, i.e. exhibit time‐varying functional connectivity patterns [Allen et al., 2014; Hutchison et al., 2013; Jones et al., 2012; Liu and Duyn, 2013; Messe et al., 2014]. Non‐stationarity in the brain is not ubiquitous, and may vary depending on the mental state, network of interest, or the time window over which connectivity is assessed. Although mentation is an obvious source of non‐stationarity in brain physiology [Barttfeld et al., 2015], it can also exist in the absence of conscious or cognitive states. For example, Hutchison et al. [2013] have observed non‐stationary behavior in the resting‐state oculomotor network of anesthetized macaques that are comparable to those observed in awake human subjects [Hutchison et al., 2013]. It has been suggested that this dynamic change in network topographies relates to an adaptive change of states, in order to balance information processing and metabolic expenditure of the brain [Zalesky et al., 2014]. Using mathematical modeling in both simulated and empirical data, Messe et al. [2014] have shown that anatomical connections (derived from DWI) drive the stationary dynamics of functional connectivity. These findings create a compelling case for exploring non‐stationarity itself as a biomarker of disease [Jones et al., 2012], or as a pharmacological endpoint.
As will be discussed in “Opportunities and Future Directions,” re‐analyzing existing pharma‐RSfMRI data with attention to the non‐stationary nature of the spontaneous brain fluctuations and dynamics of network modularity might provide interesting information about cerebral pharmacodynamics.
BOLD RSfMRI test–retest reliability
The problem of non‐stationarity underpins challenges in test‐retest reliability of the BOLD response.
Moderate to high reliability is observed in seed‐based or NOI‐based analysis; however, the degree of reliability varies between networks suggesting that some networks are more susceptible to state changes in acquisition or (psycho)physiological [Birn et al., 2013; Jann et al., 2015; Shehzad et al., 2009]. REHO seems to offer high test–retest reliability but is also sensitive to motion, global signal, scan duration and anatomical variations, thus can drastically change with preprocessing options [Zuo et al., 2013]. Moderate to high intra‐ and inter‐session test–retest reliability of (f)ALFF has been shown although reliability varies in different brain regions, leaving the question of the impact of variations in mental states on reliability assessments [Zuo et al., 2010b]. Dual regression analysis using NOIs, seems to produce more reliable and reproducible results than seed‐based analysis [Schultz et al., 2014; Zuo et al., 2010a], however the reproducibility of results depend on which set of networks are included in the analysis [Schultz et al., 2014], and preprocessing options or physiological covariates can change the configuration of certain networks [Khalili‐Mahani et al., 2013]. The reliability of graph metrics is still under investigation. A systematic review of graph metrics failed to make definitive conclusions regarding the best technique, due to the heterogeneity of study designs and analytical choices [Welton et al., 2015]. Two studies [Braun et al., 2012; Cao et al., 2014] indicated that graph metrics such as path length, global efficiency, assortativity and modularity have higher test–retest reliability (0.57 < ICC's < 0.75) than clustering coefficient, local efficiency and degree. The consistency of several brain networks that emerge from ICA has been well established [Damoiseaux et al., 2006; Wisner et al., 2013; Zuo et al., 2010a], but structured noise, and physiological and motion artifacts diminish the reliability of component detection [Zuo et al., 2010a]. Also, inconsistencies in dimensionality of ICA (e.g., number of components, and component ordering) impact the reliability of individual [Esposito and Goebel, 2011] and group [Abou‐Elseoud et al., 2010] ICA. A relatively reliable group‐ICA does not guarantee the reliability of the same individual ICA [Wisner et al., 2013]. Lower individual reliability is likely to impact the reliability of the group inference testing as well, and therefore interpretation of effects. Several methods such as ICASSO [Himberg and Hyvarinen, 2003], RELICA [Artoni et al., 2014] and FOCIS [Wang and Li, 2014] have been proposed to help identify the most reliable independent components, however to automate the selection of functionally relevant components from spurious networks remains challenging. Interestingly, the impact of post‐processing noise regression on group level statistics varies between different biomarkers [Yan et al., 2013a].
Acquisition time is an important factor in test‐retest reliability. Gonzalez‐Castillo et al. [2014] have conducted a series of tests to investigate the impact of scan duration and window size on reliability of functional networks, both at the individual and group levels. They found that the primary sensory‐motor network was the most stable network across different scan time and analytical windows, compared with areas involved in higher cognitive processing. Tomasi et al. have recently shown that non‐stationarity affects various RSfMRI metrics differently [Tomasi et al., 2016b]. For example, they showed that functional connectivity density (FCD, a data‐driven graph theory metric that quantifies the local degree, the size of the local network cluster functionally connected to each brain network node) was more accurate than sICA or seed‐based connectivity, but that accuracy for each metric varied with acquisition time, that is, sICA relied on longer scans (>12 min) to achieve the same accuracy as FCD (7 min).
Reliability of different RSfMRI metrics can vary depending on the mathematical description of the biomarker, which hinge on various factors including data acquisition parameters, preprocessing and statistical approaches, as well as different approaches to establishing reliability (e.g., interclass correlations versus coefficient of variation). These complexities make it difficult to be conclusive yet but encourage adopting standardized protocols that will enable us to compare sensitivity, accuracy, and reliability of various metrics toward establishing a practical guideline.
ASL test‐retest reliability
Because ASL methods can provide a quantitative measure of cerebral perfusion, it is easier to evaluate their reliability. Consistency of CBF estimation with background‐suppressed pCASL has been established in multi‐center and multi‐acquisition reproducibility studies [Gevers et al., 2011; Vidorreta et al., 2013]. Intersession, intrasession and inter‐scanner reproducibility across different vendors suggests that normalizing acquisition parameters on different machines yields stable measurements of the gray matter perfusion. However, in presence of differences in acquisitions sequences, spatial differences caused by different smoothing behavior of the readout module become problematic [Mutsaerts et al., 2014; Mutsaerts, 2015; Vidorreta et al., 2013].
A major challenge in reproducing ASL results stems from between‐subject variations in cervical anatomy that can alter model parameters used to quantify CBF. The main limiting factor in between‐examination and ‐subject variability is probably differences in labeling efficiency [Aslan et al., 2010]. Labeling efficiency in pCASL is dependent on local off‐resonance effects, blood velocity as well as the angle of the artery with respect to the labeling plane [Dai et al., 2008]. Variations in labeling efficiency differences can be minimized by employing a pre‐scan [Jung et al., 2010] or by performing additional shimming of the labeling region. Finally, during post‐processing normalization by average whole brain CBF can partially correct for variations in labeling efficiency.
Another factor affecting the accuracy of CBF quantification by ASL is ATT (see “Arterial Transit Time”). The common model employed to quantify CBF assumes that all labeled blood has reached the imaging volume. Incomplete delivery of label would lead to underestimation of CBF. Furthermore, the loss of label due to longitudinal relaxation (T 1) depends on whether the label is located in blood where it will decay with the T 1 of arterial blood (∼1,650 ms), or whether the label is in tissue where it will decay with the T 1 of tissue (±1,200 ms). Shorter arterial transit times will often be accompanied with earlier arrival in tissue, and therefore faster decay of label. To what extent these model parameters can vary with pharmacological experiments, cerebrovascular anatomy or the layout of the arterial flow territories [Bokkers et al., 2011; Hartkamp et al., 2011] is not yet well understood. As discussed in “Arterial Spin Labeling and Cerebral Perfusion,” ASL methods are best suited for crossover designs where state‐dependent variations in baseline cerebral perfusion, or regional heterogeneities in perfusion caused by drug can serve as surrogate markers of neuronal activity.
Statistical Analysis
Besides data acquisition, preprocessing protocols and the biomarker type, reliability depends on the criteria of statistical analyses. Pharmacological studies, whether exploratory (i.e., finger printing and probing experiment) or intended for clinical trials, demand establishing reliable statistical criteria to assess interpretable effectiveness of a given intervention. Existing statistical modeling approaches and toolboxes available for the statistical analysis of RSfMRI and ASL data are by and large inherited from task‐based fMRI. As such, there are limitations in terms of estimating the effect size and statistical modeling, which we briefly discuss here.
Effect size estimation
Three main components are involved in the estimation of the statistical power: (1) the expected effect size, (2) the variability of the effect size and (3) the sample size of the study. To establish an a priori effect size is one of the most critical aspects of clinical pharmacology for making go‐no go decisions. In task‐fMRI, the percentage of the change in BOLD signal is estimated by fitting a canonical hemodynamic response function to epochs of rest and activation, at the subject level. As such it is possible to compute the effect size as a voxel‐wise averaged percentage of change. Usually, a 2% signal change from the no‐task condition reflects a task‐induced neuronal effect. Effect size can also be easily estimated in ASL studies, as it is possible to quantify the percentage of change in cerebral blood flow. In RSfMRI, effect size is estimated in terms of connectivity (z‐scores or correlation coefficients) or some graph metric, which are dependent on meaningful thresholding of connectivity or coherence metrics. Given the dynamic nature of drug effects on the brain and thus on RSfMRI fluctuations, interpretation of an effect depends not only on the magnitude of change (e.g., pre‐ vs. post‐drug, placebo versus drug), but also on the dynamics of change (increases and decreases) between different timepoints. Furthermore, as discussed above, the direction of observed effects do not represent absolute neurobiological phenomena, but relative changes which can vary with variations in global or regional corrections and noise thresholds. For this reason, it is essential to interpret results with attention to the mathematical underpinnings of each biomarker, and in the least plotting the temporal profile of data in a given region during the course of drug administration in relation to placebo—instead of just relying on test statistics and average activation maps.
In neuroimaging studies, power analysis may become possible by collecting information from one ROI, but the choice of the ROI runs the risk of biasing the estimated effect size [Mumford, 2012]. It is also possible to select multiple ROIs, although there are no generally established guidelines to account for multiple comparisons in this case. A major complication in power analysis for any fMRI experiments is that variability comes (in a hierarchical way) from several sources: intra‐run variability that reflects noise within auto‐correlated time series of each fMRI run; intra‐subject variability that comes from repeated runs within the same subject; and between‐subjects variability within a population. To this complicated picture, add the complexity of central and peripheral pharmacodynamic effects over time, and over different regions, which need to be accounted in power analysis as well.
Statistical modeling
There are three existing limitations in statistical modeling for pharmacological imaging experiments. First, drugs exert non‐linear and spatially heterogeneous effects (related to a combination of neurovascular factors or chemoarchitecture [Shulman and Rothman, 1998]). What covariates to include in an analysis can impact the topography of the results significantly [Khalili‐Mahani et al., 2013, 2012; Niesters et al., 2012]. We have shown that the relations between CBF and connectivity measures can vary from region to region, depending on what explanatory variables are included in the model [Khalili‐Mahani et al., 2013, 2014]. Secondly, pharmacological experiments ideally depend on integration of repeated measurements from different modalities, for example BOLD‐related connectivity maps and relative CBF maps, which are correlated but have different smoothing characteristics. Finally, and importantly, repeated measures analysis is essential to most pharmacological experiments concerned with finger‐printing, where the minimum number of scans is four (one pre‐, and one post drug scan per placebo and drug session), thus controlling for within‐subject correlations is important.
The majority of neuroimaging reports surveyed here rely on voxel‐wise T‐tests (between group or between time), (m)AN(c)OVA or multilevel GLM with mixed effects (time, group, drug, nuisance, behavior) analyses, often using standard software packages [Beckmann et al., 2003; Chen et al., 2012; Friston et al., 2005; Worsley et al., 2002]. Methods to address the first two problems are explored in the context of multivariate analysis and machine learning, which will be discussed later in this section. The third problem is addressed in newer methods such as multi‐scale adaptive generalized estimating equation (MA‐GEE) [Li et al., 2013] or Sandwich Estimator [Guillaume et al., 2014], which are more suitable to inference testing with repeated, longitudinal measurements. However, to our knowledge, these methods have not been used by any of the studies in this survey. A re‐analysis of the data using these methods will be informative.
Significance criteria
Neuroimaging studies create massive amounts of data, and rely on special adjustments for addressing the problem of multiple comparisons. The issue of multiple comparisons is well addressed in neuroimaging statistics. Most current fMRI studies surveyed here rely on cluster‐wise correction of family‐wise errors. In this approach voxels satisfying a nominal threshold (e.g., t‐value >1.7, to yield uncorrected threshold of 0.05, on assumption of a normally distributed t‐test) are selected, then a cluster‐level extent threshold, measured in units of contiguous voxels over resolution elements (resels, which depend on the search volume and the FWHM of the smoothing kernel) is determined based on the estimated distribution of cluster sizes under the null hypothesis of no activation to correct for family‐wise errors. The sampling distribution of the largest null cluster size can be estimated using random field theory [Worsley et al., 1992], Monte Carlo simulation [Forman et al., 1995], or nonparametric methods [Nichols and Holmes, 2002]. To avoid this arbitrary selection of the initial threshold, Smith and Nichols [2009] have proposed a threshold‐free‐cluster‐estimation (TFCE) method, which considers the entire statistical data in order to identify voxels that, judged by the magnitude of the signal and its spatial distribution, cluster together [Smith and Nichols, 2009]. Whereas these sets of techniques try to minimize the probability of false positives), another method, false discovery rate (FDR) provides more statistical power by setting thresholds based on tolerance for false positives, by ranking the probability of all observations and arbitrarily rejecting a percentage of those [Genovese et al., 2002]. Whether cluster‐ or FDR‐based correction methods are stringent or reliable enough has recently come under scrutiny [Eklund et al., 2016; Woo et al., 2014]. A recent analysis of 499 RSfMRI datasets has estimated that standard cluster‐based thresholding methods available in popular analysis packages (FSL, AFNI and SPM) are prone to inflating false positives [Eklund et al., 2016] and have proposed permutation testing to avoid errors arising from unmet assumptions about smoothness and normal distribution of the data.
Permutation testing provides a more stringent approach to determining statistical significance based on consistent estimates of means and variances for central and non‐central t‐tests [Nichols and Holmes, 2002; Zhou et al., 2014]. This is particularly important in pharmacological studies, which have small samples and thus may not satisfy the assumptions of normal distribution at the level of group analysis. Permutation testing is also more robust to cluster‐size changes due to non‐stationarity in fMRI signals, whether using RFT methods [Hayasaka et al., 2004] or TFCE [Salimi‐Khorshidi et al., 2011] to establish statistical significance. Multi‐level permutation testing will also give more stringent control of false positives [Winkler et al., 2015] in repeated measures longitudinal analysis, which would be necessary for most pharmacological experiments.
Multivariate analysis and machine learning
Univariate analyses (discussed above) aim to detect if and how different brain regions are affected by a given stimulus (regression), or by other independent variables (factors or covariate), while accounting for the problem of multiple comparisons. Multimodal data collection lends to more sophisticated multivariate analysis approaches. In the simplest implementation, multivariate models extend the GLM to allow for a single analysis with multiple dependent variables (as opposed to running several regressions on a single dependent variable in univariate analysis). As such a single analysis provides multiple levels of inference and a more suitable design for complex research such as pharmacoimaging.
Supervised Machine Learning (ML) techniques are increasingly important for multivariate analysis, and predictive pattern recognition. These methods aim to identify optimal mappings between the multivariate brain signals and external variables, such as symptom levels or drug conditions. ML algorithms might utilize any number of data features, such as network connectivity matrices, seed or dual regression maps, variance or REHO measures, or graph theoretical characterizations. These mappings can produce prediction accuracies in new data that greatly exceed what is possible from any individual feature of the data, and can reveal the modulation of subtle, spatially extended features of neural activity by pharmacological agents that would otherwise be missed by univariate analyses. As such ML techniques are likely to be crucial for potential translational applications of pharmacological imaging such as the identification of disease states in patients with limited ability to communicate [Wager, 2013], the assessment of pharmaceutical candidates for signatures of pharmacological efficacy [Doyle et al., 2013; Duff et al., 2015], or the characterization of the timecourse of drug action in individual subjects [Paloyelis et al., 2016].
Common ML pattern recognition techniques include support vector machine (SVM) and Gaussian process classification (GPC). SVM aims to classify individual observations (e.g., connectivity maps) into distinct groups or classes based on maximizing the margin between classes in a high‐dimensional space (i.e., multivariate data). This classification depends on a pre‐defined training data, which is obtained by exhaustive comparison of data from two groups (e.g., patient and control, or pre and post drug). This classifier is then used to identify to which group data from a new individual maps [Orru et al., 2012]. In contrast to SVM, where the certainty of the prediction is tested against the support vector, GPCs are based on a probabilistic distribution of functions which represent the training data. As such GPCs are suitable for providing a probabilistic prediction (as opposed to binary prediction in SVM) of states such as pain [Marquand et al., 2010]. Differences in SVM and GPC have implications for when to apply which. For example, linear SVMs discriminate according to a subset of samples (training set) that form the separating support vector. As such SVMs are robust to outliers and perform reasonably well with the typically low sample sizes of pharma‐fMRI studies. However, SVMs are most suitable to binary discrimination between two groups, and heterogeneous data may require more complex discrimination functions which can be difficult to define with SVMs. Because GPC utilizes the full sample distribution for predictions it allows using more complex decision functions and probabilistic outcomes, but these functions are more challenging to estimate than support vectors and, due to its probabilistic nature, GPC is more affected by outliers.
To date, Monti et al. [2013] have used network connectivity matrices as inputs to SVMs in an exploration of states associated with propofol anesthesia. In their study, connectivity matrices could reliably discriminate wakefulness from periods of sedation and unconsciousness. Similarly, Sripada et al. [2013a] trained SVMs to discriminate effects of methylphenidate from placebo scans using network connectivity matrices generated from 1,080 ROIs. The whole‐brain data was able to reliably identify the effects of methylphenidate. The results of an initial feature selection stage were used to identify predictive changes as being associated with reductions in coupling within a number of resting‐state networks (RSNs), and changes in the coupling of the default mode network (DMN) with other RSNs.
Classifications algorithms can also be used to characterize similarity. Using GPC, Doyle et al. [2013] first established the predictive probability of BOLD signal change in response to ketamine based on discriminating placebo–saline and ketamine–placebo) and then showed that pretreatment with lamotrigine and risperidone attenuated the effects of ketamine, making resting‐state ketamine–lamotrigine and ketamine–risperidone conditions more difficult to distinguish from placebo.
GPC has also been used to explore the dynamics of oxytocin action on the spatiotemporal profiles of brain activity, by examining changes in the sensitivity of connectivity‐based classifiers across an administration period [Paloyelis et al., 2016].
However, despite their strengths, all ML techniques have limitations and tradeoffs, and are not always the appropriate tool. Due to the multivariate nature of ML algorithms, it can be difficult to determine the nature of the signals driving predictive accuracy. ML algorithms may take advantage of subtle modulations of brain activity for prediction, but may also exacerbate the influence of subtle confounds. Choosing and tuning a classification algorithm can be complex, particularly with the low sample numbers of imaging, which make selection of parameters via approaches such as cross‐validation unreliable. The diversity of options for ML, and lack of standard software for neuroimaging applications, produce additional challenges for reproducibility. Also, multivariate analysis depends on complete sample and is not amenable to missing data. Therefore, in many cases, univariate analysis would be more appropriate.
Pharmacokinetic/Pharmacodynamic (PK/PD) Modeling
The ultimate goal of pharmacological neuroimaging is to establish a relation between drug dose, and a meaningful effect in neuronal activity that helps estimate the efficacy of the treatment. PK/PD modeling (which encompasses limitations discussed in “Statistical Analysis”) involves estimating piecewise‐linear transfer functions that describe the nonlinear profile of the drug uptake and washout. A mathematical description of a predictive transfer function is obtained from solving a partial differential equation that estimates pharmacokinetics model coefficients, that is, the time course of drug action at different doses in different compartments (e.g., gut, liver, blood, target tissue) in relation to different pharmacodynamic phenomena (i.e., the behavioral or symptomatic endophenotypes for which the drug is designed). Such predictive models are important for reproducing results, or for identifying deviations from the model in clinical populations. Within this mathematical framework, differences between subjects (e.g., in terms of age, gender, illness severity and comorbidities, medications, etc.) should also be accounted for.
PK/PD modeling has contributed significantly to a better understanding of drug effects (and their variability) in clinical studies, to the translation of drug effects between species or populations, and to the optimization of clinical trial designs. A clear PK/PD‐relationship also offers reliable information on which part of an observed effect is due to pharmacological activity—in addition to other sources of variation. For these reasons, the identification of PK/PD models that predict the relation between the rates of drug distribution, absorption, metabolism and excretion (pharmacokinetics) and changes of MRI parameters, will provide strong support for the significance of neuropharmacological imaging.
PK/PD modeling is a challenging method in general and even more so in fMRI experiments. The CNS effects of drugs are highly dependent on the concentrations of the pharmacologically active moiety at the action site. The time to reach an effect, the concentration at the site of action, and hence the intensity of the effect, are determined by the pharmacokinetic properties of a drug. The onset and the size of the effect can vary considerably among individuals and over time, depending on differences in absorption (related to formulation, mode of administration and influence of meals), metabolism to active or inactive moieties, and excretion and distribution of the compound. Penetration of the brain is regulated by the blood–brain barrier (BBB), which can limit the extent and rate of distribution of compounds into the CNS. In humans, it is almost always impossible to measure the concentrations of the active compounds (which can also be metabolites) at the action sites in the brain. In most cases, drug concentrations are measured in blood serum or plasma. The anatomy and density of blood vessels that deliver the drug to the BBB are likely to introduce variations in that measure as well.
The consequence of this complex cascade of pharmacokinetic and pharmacodynamic events is that there is nearly always a time lag between administration of the drug, its binding to its target, and the measurement of CNS effects. This can vary from seconds or minutes for intravenously injected drugs that penetrate rapidly into the brain and affect cell membrane receptors, to hours for compounds that are slowly absorbed or induce protein formation, and even longer for medicines that influence cell growth. Moreover, the relationships between concentrations and effects can change over time, by development of tolerance or by accumulation of lipophilic active metabolites in the effect compartment. Finally, most effects require a minimal level of drug exposure, and are limited to a maximum tolerable dose. As a consequence, the relationships between blood concentrations and effects are usually not linear, but can often be adequately described by a more complex mathematical function, incorporating measures of baseline activity, minimal and maximal effect and (cumulative) time delay.
The earlier phfMRI experiments (to remind, these are experiments where the drug serves as a stimulus and the hemodynamic response is estimated from variations in the drug dose) used pharmacokinetic modeling to estimate regional activations, for example with nicotine [Bloom et al., 1999; Stein et al., 1998], cocaine [Kufahl et al., 2005], remifentanil [Leppa et al., 2006], morphine [Becerra et al., 2006], citalopram [McKie et al., 2005], m‐Chlorophenylpiperazine (mCPP) and Mirtazapine [McKie et al., 2011] and ketamine [Deakin et al., 2008]. In these studies, duration of acquisitions was short and consistent with peak effects, for example, 7 minutes for remifentanil [Leppa et al., 2006], 8 [Deakin et al., 2008] and 10 minutes with ketamine [De Simoni et al., 2013], and 25–30 minutes for morphine [Becerra et al., 2006] and between 30 and 60 minutes for the serotonergic drugs citalopram and mPCC [McKie et al., 2005, 2011]. Pharmacokinetic modeling enables dose normalization in pharma‐RSfMRI studies and preliminary attempts have been made to illustrate a link between plasma drug concentrations of drugs such as ketamine [De Simoni et al., 2013; Doyle et al., 2013; Khalili‐Mahani et al., 2015], THC [Klumpers et al., 2012], opioidergic drugs [Becerra et al., 2006, 2013; Khalili‐Mahani et al., 2012; Wise et al., 2004a] and alcohol [Esposito et al., 2010; Khalili‐Mahani et al., 2012], and the amplitude of change in selective brain regions significantly affected by these drugs. However, no formal PK/PD models for predicting a cerebral dose‐response have been reported yet. There are two main reasons for this: one is the paucity of studies with detailed time profiles of drug concentrations and effects, and the second is the lack of simple MRI readouts.
Current attempts at PK modeling have been further limited by either fitting a canonical PK model to the brain data [Bloom et al., 1999; Doyle et al., 2013; Stein et al., 1998], or by using a summarized readout (e.g., global signal change or ROI signals) to identify and validate a model [De Simoni et al., 2013]. As statistical parametric maps of the chemoarchitecture of the brain are being developed [Zilles et al., 2002; Zilles et al., 2004; Zilles and Amunts, 2009], to impose a single model across the entire brain anatomy (e.g., in calibrated BOLD/ASL modeling of neurovascular coupling) becomes questionable. However, with evidence from fingerprinting dose‐response, and probing experiments it becomes feasible to integrate data from different modalities (e.g., cortical morphology, white matter tractography, and cerebrovascular anatomy) and apply advanced biomathematical modeling in a similar fashion as proposed for PET [Gunn et al., 2011]. (See section “Contributions From PET” for more details.)
OPPORTUNITIES AND FUTURE DIRECTIONS
The field of RSfMRI is relatively new, yet rapidly evolving. Many of the limitations mentioned above, such as optimal pre‐processing and data cleaning, biomarker accuracy and reliability, and the best statistical modeling and effect size estimation methods, are being actively investigated. So far, we have discussed existing methods that can be used for studying brain chemistry and have provided examples to illustrate how pharmacological experiments can help interpretability of RSfMRI studies in general. There are still unexplored opportunities provided by the current state‐of‐the‐art data acquisition and analysis strategies, which will guide future pharmacological studies.
Data‐Sharing and Meta‐Analysis of the Existing Data
In many areas of clinical neuroimaging research, collaborative research takes place within a common framework for data harmonization, sharing and standardized analysis [Das et al., 2016; Petersen et al., 2010; Schmaal et al., 2016; Thyreau et al., 2012; Zijdenbos et al., 2002]. In 2007, the Radiologic Society of North America (RSNA) organized the Quantitative Imaging Biomarker Alliance (QIBA) to develop standards for reducing variability across instruments and analysis, and they have now provided guidelines for uniformed terminology, statistical approach and performance measure [Kessler et al., 2015]. Pharmacoimaging researchers may provision for more impactful research through data‐sharing and data‐mining. In fact, FDA's Critical Path Initiative [2004] emphasizes the importance of using technology in drug discovery process. Since then, initiatives such as the Open Pharmacological Concepts Triple Store (Open PHACTS) have aimed to create a pharmacological data‐mining platform to address drug discovery problems including target identification, dose optimization, toxicology and drug‐drug interaction [Goldmann et al., 2014; Williams et al., 2012]. Existing neuroinformatics platforms for collaborative neuroimaging data‐sharing and data‐mining [Das et al., 2011; Glatard et al., 2013, 2015; Sherif et al., 2014] can be adapted to advance reliability and reproducibility of pharmacological trials.
With few exceptions of studies conducted in the same center, the studies surveyed here are very heterogeneous in experimental and analytical designs and because there are no standards for modeling and defining a meaningful effect size yet, it is difficult to perform a meta‐analyses and arrive at a conclusive interpretation of the sensitivity and reliability of a given biomarker to a given neurobiological process. Nevertheless, there are many commonalities in the existing datasets from multiple centers that it is plausible to aim for a future data‐mining exploration. The majority of pharmacological experiments reviewed here have collected data from healthy young men (Fig. 7) and T2*W images are collected in more than 75% of these studies. Two thirds of research objectives of studies reviewed are drug fingerprinting (33%) and probing specific neurochemical signaling systems (33%), with a majority also using functional connectivity metrics obtained from seed, ICA, clustering or graph analyses (>53%).
To pool, harmonize and (pre‐)process common data through a standardized pipeline, serve the objectives of an exploratory model‐free mega‐analysis which may shed light on current uncertainties about meaningful effect sizes and analytical and biological factors that differentiate the sensitivity of various metrics and models. This approach could increase the clinical impact of pharmacological studies. For example, different groups have studied pharmacological probing of the dopaminergic, opioidergic, GABAergic and glutamatergic systems using the same scanning parameters. Data processing parameters (e.g., choice of filters, blurring kernels and inclusion of common nuisance regressors) and statistical analysis criteria (e.g., standards for thresholding to assess statistical significance or inclusion of common covariates such as physiological rates or voxel‐based anatomical regressors in the group‐level models) can be established independent of drug class. Re‐analyzing these data with similar preprocessing options and statistical modeling choices will make direct comparison of results possible. For instance, Kleinloog et al. [2015] combined 323 identically‐acquired RSfMRI samples from four different pharmacological challenges (ethanol, morphine, Δ(9)‐tetrahydrocannabinol and ketamine) to examine the relation between the severity of drug induced psychomimetic effects and functional connectivity and showed that the subjective effect of perception was the only psychomimetic factor that was commonly correlated with the connectivity of the posterior cingulate cortex and precentral gyrus with the sensorimotor network [Kleinloog et al., 2015].
The issue of non‐stationarity and dynamics of network hierarchy and modularity is poorly studied, and a joint analysis of data under different drugs may help better characterize which neural networks are stationary and which vary according to drug‐dependent dynamics.
Standardizing Multivariate Data Acquisition
Pharmacological fMRI experiments are expensive and necessitate highly controlled monitoring setups, which generally make the datasets richer than non‐pharma RSfMRI studies. With the current drive for generating Open Science and data‐sharing platforms, it is important to provision for a longer life cycle for the data, by adhering to operational guidelines that help minimize data acquisition errors [Schwarz et al., 2011a, 2007b] and by developing community standards for data acquisition in such a way as to maximize the amount of data that can be gathered from a given study without compromising the main study objectives. Given the cost and practical difficulties associated with a pharmacological experiment, the benefits of collecting comprehensive datasets outweigh the cost of adding scan time. Furthermore, with growing availability of pharma‐RSfMRI data, and with unified and standardized analytical approaches to biomarker development and validation, multivariate data analysis, and machine learning pattern recognition are expected to become the paradigm of choice for response prediction and drug‐effect classification, which are critical objectives for CNS drug research.
We have summarized the minimum recommended requirements of a standard pharmacological imaging experiment in Table 2, and have listed all basic and advances design, acquisition and analysis options in Table 3, which can serve as a guideline.
Table 2.
Table of challenges and study objectives and related data acquisition solutions
| Placebo‐design | Repeated measures | Multimodal imaging | Physiological | Blood Sample | Saliva Cortisol | Psychometric | Mega‐analytical framework | Standardized MR acquisition | ||
|---|---|---|---|---|---|---|---|---|---|---|
| CHALLENGES | Psychophysiological States | X | X | X | X | X | ||||
| Cardiopulmunary confounds | X | X | X | X | X | |||||
| Age and Gender | X | X | X | X | X | X | ||||
| Anatomical variability | X | X | X | |||||||
| Artifact removal | X | X | X | |||||||
| Statistical modeling | X | X | X | X | X | X | X | X | X | |
| Reliability and reproducibility | X | X | X | X | ||||||
| Pharmacokinetic modeling | X | X | X | X | X | X | X | X | X | |
| OBJECTIVES | Fingerprinting | X | X | X | X | X | ||||
| Probing | X | X | X | X | X | X | X | X | ||
| Calibration Validation | X | X | X | X | X | |||||
| Clinical research | X | X | X | X | X | X | X | |||
| Animal research | X | X | X | X | X |
Table 3.
Guidelines for design and analysis of pharmacological RSfMRI studies
| Minimum standards | Advanced state‐of‐the‐art | Section in text | |
|---|---|---|---|
| Design Considerations | |||
| Study Objectives | Experimental Objectives and Clinical Relevance | ||
| Pharmacokinetics | 10‐min RSfMRIs, repeated as needed to cover the entire duration of drug administration and washout | Fast RSfMRI | Pharmacokinetic/Pharmacodynamic (PK/PD) Modeling |
| Baselines and Control | Placebo Control | Pre‐drug, post‐washout scans in both drug session and placebo session | Biological Confounds |
| Health condition | Full screening of prescriptions as well as medical screening of physiological and metabolic state | Biological Confounds | |
| Mode of administrations | Single or multi‐dose oral | PK‐controlled infusion | Table 1 |
| MRI Acquisition | |||
| Anatomical | T1W anatomical MRI | TOF‐MRA | Biological Confounds, Standardizing Multivariate Data Acquisition |
| Functional fMRI | T2*w EPI (TR/TE = 2,000/30), 5–10 min | Ultrafast T2* , dual‐echo BOLD/ASL | Pharmacological fMRI (Pharma‐fMRI), Spontaneous BOLD Signal Fluctuations and Functional Networks, Standardizing Multivariate Data Acquisition |
| Blood flow | pCASL | VS‐ASL, TE‐ASL | Arterial Spin Labeling and Cerebral Perfusion |
| Myelination | DWI | Standardizing Multivariate Data Acquisition | |
| Biomarkers | |||
| Functional connectivity | Seed‐based or NOI‐based networks | Dynamic FC, hierarchical clusters | Spontaneous BOLD Signal Fluctuations and Functional Networks and Reliability, Reproducibility |
| ICA | sICA, tICA, gICA | ICASSO, RELICA | Spontaneous BOLD Signal Fluctuations and Functional Networks and Reliability, Reproducibility |
| Graph | Centrality, efficiency, small worldness, path length | Dynamic modularity, assortativity | Spontaneous BOLD Signal Fluctuations and Functional Networks, Dose/Response in Anesthesiology, and Reliability, Reproducibility |
| spatial coherence | REHO | Spontaneous BOLD Signal Fluctuations and Functional Networks, and Reliability, Reproducibility | |
| Spectral features | f(ALFF) | Spontaneous BOLD Signal Fluctuations and Functional Networks, and Reliability, Reproducibility | |
| CBF | Average biophysical model of perfusion weighted images | ATT | Arterial Spin Labeling and Cerebral Perfusion and Combined ASL/BOLD fMRI and Neurovascular Coupling |
| BOLD signal | Fitting a hemodynamic response to drug's pharmacokinetic curve. | Pharmacological fMRI (Pharma‐fMRI), Dose/Response in Anesthesiology, and Methods Validation and Calibration | |
| Preprocessing | |||
| Motion | Realignment to a frame of reference | Scrubbing, including motion regressors, ICA‐based correction | Structured Noise and Artifact Removal and Reliability, Reproducibility |
| Physiological noise | Monitoring and regressing out (e.g., RETROICOR, RVHRCOR) | Post hoc analysis of neurobiological correlates | Biological Confounds and Structured Noise and Artifact Removal |
| Global signal regression | Controversial | Compare results with and without regressors | Structured Noise and Artifact Removal and Reliability, Reproducibility |
| Registration | Functional to anatomical | Biological Confounds | |
| Validity | |||
| Stability | Standardized acquisition and preprocessing | Characterization of non‐stationarity, dynamic connectivity and hierarchical clustering | Reliability, Reproducibility |
| Test‐retest reliability | Different subjects, similar biomarkers | Same subject, same biomarker from repeated measurements | Reliability, Reproducibility |
| Interpretation | Hybrid PET/MRI, hybrid BOLD/CBF, meta‐analysis | Experimental Objectives and Clinical Relevance, Biological Confounds, Data‐Sharing and Meta‐Analysis of the Existing Data, and Contributions From PET | |
| Statistics | |||
| Univariate analysis | Mixed effect multilevel GLM | Multi‐scale adaptive GEE, Sandwich estimator | Statistical Analysis |
| Multivariate analysis | MGLM | Machine learning (SVM, response prediction, GCP dose/response‐prediction) | Statistical Analysis , Data‐Sharing and Meta‐Analysis of the Existing Data |
| Statistical thresholding | FDR, cluster‐correction, TFCE | Permutation testing | Statistical Analysis |
| Covariates of interest | |||
| Structured noise | Anatomical correlates of Physiology and motion | Structured Noise and Artifact Removal | |
| Psychophysical states | Eyes open/close | Anxiety, arousal, physiological rates | Biological Confounds , Structured Noise and Artifact Removal |
| Circadian phases | Control time of day | Collect blood/saliva samples | Biological Confounds |
| Age and sex | Control for variations in age and sex group analysis | Include anatomical MRIs as a covariate in group analysis | Biological Confounds |
As discussed in “Challenges and Limitations,” RSfMRI signals are highly sensitive to psycophysiological states. Randomized and placebo controlled experiments that include repeated RSfMRI and ASL measurements help increase the power of the study and provide a picture of the dynamics of drug action in the brain during the pre‐drug baseline, the peak of drug dose, and the washout, if the drug is fast acting. Monitoring cardiac and respiration rates is critical for data interpretation and noise removal. Repeated psychosomatic readouts (e.g., nausea, weakness, sleepiness) and psychometric assessments (e.g., alertness, hallucination, calmness, anxiety), as well as serum samples from which to measure plasma concentrations of the drug, certain hormones, proteins and metabolites, will provide a richer parameter set for machine learning or PK/PD modeling. Furthermore, Including MR angiography or venography scans can help characterize the contribution of vascular factors to resting‐state functional variations and to learn how neurovascular features such as capillary density, tissue vascularization and arterial flow territories factor into the efficacy of drug transportation to its site of action. Also, since functional and structural connectivity are closely linked [Greicius et al., 2009], and disease‐related changes in white matter fiber connections might influence resting‐state functional biomarkers [Wang et al., 2015], including diffusion weighted tractography further enriches any phMRI dataset. More recent methods such as ultra‐fast fMRI [Feinberg et al., 2010], multi‐echo fMRI [Kundu et al., 2013], quantitative fMRI [Christen et al., 2013], and vessel‐encoded [Zhang et al., 2016] or time‐encoded [Teeuwisse et al., 2014] pCASL have not been tested in pharmacological studies yet, but are likely to improve the temporal resolution of data and circumvent some errors related to physiological or motion artifacts. Importantly, to acquire fMRI data at higher temporal resolution would be important for exploring non‐stationarity in spontaneous BOLD fluctuations—which can be studied in windows of 40–100 s [Leonardi and Van De Ville, 2015; Zalesky and Breakspear, 2015]. Such datasets provide a better opportunity to study the dynamics of functional connectivity toward a better understanding of the neural mechanisms of brain adaptation to intervention or disease [e.g., Bassett et al., 2011 ; Cole et al., 2014; Jones et al., 2012].
Of course, collecting multimodal data causes subject fatigue and increases the risks of excessive movement and discontinuation. A well‐designed pharmacological experiment is usually conducted in two independent sessions (one for drug and one for sham placebo). In each session, one set of anatomical scans (T1W, MRA and DTI) are acquired—usually at the beginning of each scanning session to help localize the field of view for RSfMRIs. Optimal acquisition time for detecting reliable functional networks is between 7 and 14 min, albeit dependent on the metric of interest [Anderson et al., 2011; Gonzalez‐Castillo et al., 2014; Tomasi et al., 2016b], and a reliable background suppressed whole‐brain pCASL (30–40 pairs) can be acquired in less than 5 min; thus each repetition of a post‐drug RSfMRI can be done in less than 20 min which is tolerable. In drug fingerprinting studies conducted at Leiden University Medical Centre that run up to 8 hours, the scanner areas are designed in such a way to allow rolling study participants in and out of the scanner (preferably in supine position) at given intervals. Psychometric and bio‐specimen data are thus collected outside the scanner in a dedicated private area. Such procedures are made possible by scanners that are equipped with a smart scout protocol (e.g., SMART in Philips or AutoAlign in Siemens scanners), including the scanner used in the Leiden studies, that help automatically reposition the field of view (FOV) in subsequent scans to facilitate breaking up scanning sessions when conducting repeated measurements. To provision for a comfortable scanning environment will increase participant compliance with longer scanning protocols accommodating multi‐modal data acquisition.
Contributions from PET
The arguments in favor of MRI versus PET, for mapping neuronal response to pharmacological interventions have already been presented at the very beginning of this article. Although BOLD‐fMRI effects are by and large indirect and nonspecific (confounding neuronal, metabolic, and hemodynamic responses), the convergence between RSfMRI biomarkers and metabolic activity detected by PET [Aiello et al., 2015; Passow et al., 2015; Soddu et al., 2016; Tomasi et al., 2016a, 2013; Toussaint et al., 2012] justifies substitution of some PET experiments with fMRI for localizing brain activity. However, in pharmacological applications, the value of PET rests with its exquisite sensitivity to radioactivity concentration variations, which makes it capable of measuring interactions of PET radioligands with different molecular targets in the physiologic range. To this day, no other non‐invasive imaging approach has been able to obtain the type of molecular information provide by PET, and none is expected to do so in the short to medium term. Recent advanced in PET/MRI imaging are thus critical to advancing pharma‐fMRI research.
After a relatively long period of development, PET/MR scanners have now reached the stage of commercial availability [Heiss, 2016], and are sold as whole‐body, dedicated brain and pre‐clinical (small animal) systems. The first immediate benefit of such systems is that they improve anatomical localization of PET anomalies in oncology [Bagade et al., 2015] and clinical studies in dementia and other neurodegenerative diseases[Barthel et al., 2015; Schutz et al., 2016] and epilepsy [Shin et al., 2015]. In the field of translational pharmacological research, the combination of simultaneous MRI and PET acquisitions allows for better interpretation of different resting‐state fMRI metrics [Aiello et al., 2016], which is of course critical to the interpretation of pharma‐fMRI. In one such study, Aiello et al. [2015] have provided evidence for a regionally specific correlation between regional glucose uptake (FDG‐PET) and simultaneously measured REHO and fALFF. Their findings confirm the importance of considering spatial heterogeneity of the relations between metabolism and blood‐flow‐driven measures obtained from fMRI [Aiello et al., 2015]—a concern that requires more careful modeling and interpretation of pharma‐RSfMRI studies [Khalili‐Mahani et al., 2014]. Particularly, hybrid PET/fMRI will help better understand the coupling [Riedl et al., 2014; Soddu et al., 2016] and the decoupling [Di et al., 2012] of metabolic and connectivity metrics against the pathological background over which pharmacological interventions may be performed.
The second, and by far the most important impact of combined PET/MRI technologies on pharmacological interventions will come from combining different but highly complementary types of information gleaned during the exact same time frame from the two techniques. Such studies have only recently become feasible. For example, in a recently reported study hybrid PET/MRI has been used to assess receptor occupancy variations and BOLD variations using different 5‐HT1a agonists, in order to evaluate whether it is possible to target different cellular pathways using molecules with similar receptor targeting‐properties, but slightly different binding which will affect molecular/pharmacological responses [Vidal et al., 2016].
Hybrid PET/MRI will also be critical for advancing both kinetic modeling for PET, and by extension PK/PD modeling for fMRI. Both PET and PK/PD modeling depend on accurate assessment of the arterial input functions (AIF) for full quantification of tracer kinetics or pharmacokinetics. In PET studies, this is typically obtained from arterial blood sampling, an invasive, cumbersome and often inaccurate approach. To address this problem, Su et al. [2016] have developed and validated an image‐derived AIF (IDAIF) estimation technique to simultaneously quantity brain hemodynamic parameters, by acquiring a TOF MRA in the same anatomical space as the PET data in order to assess image‐derived time‐activity curves in arterial regions of interest [Su et al., 2016].
Such interventions are, again, only beginning to be reported. However, by drawing on the strengths of fMRI (high spatial/temporal resolutions) and of PET (high sensitivity to even limited signal changes, access to a large variety of highly specific ligands), combined PET/MRI studies have the potential to bring pharmacological intervention studies to a new level of sophistication.
CONCLUSIONS
This survey provides compelling evidence for applicability of the pharma‐RSfMRI method in studying neurochemistry. Animal studies corroborate the viability of these methods in pharmacological research, but the ultimate goal of PK/PD modeling hinges on further reliability and reproducibility tests, characterizing confounding factors that determine the interpretability of RSfMRI biomarkers and establishing meaningful effect size. This review illustrates the great degree of heterogeneity in data acquisition, experimental and analytical designs, statistical analysis and modeling of existing reports, and highlights the need for further research and development to create a standardized framework for data acquisition and analysis of pharmacological studies. We share recent concerns about validity and reliability of fMRI studies, and emphasize the fact that most existing statistical analyses methods are oblivious to nonstationarity of functional networks, and complex dynamics of regionally heterogeneous neurochemical modulation of the neuroimaging signals. We have provided a summary table of the basic and advanced options for design and execution of pharma‐RSfMRI studies. To have these many options is itself a limitation in terms of what the optimum approach could be. However, the minimum requirements for such studies should include: (1) Placebo‐controlled, repeated measures, and cross‐over design; (2) multimodal data acquisition including data to characterize cerebral blood flow, and brain anatomy (myelin, and cerebrovascular architecture); (3) diligent monitoring of physiological pulses during the MRI acquisition as well as recording of somatosensory and awakeness states; (4) studying the impact of preprocessing and noise‐regression residuals on statistical inference tests; (5) permutation testing of a multivariate parameter space.
In conclusion, we emphasize the necessity of collaborative efforts in at least two areas:
First, the community (including pharmaceutical industry) must invest into developing practical and methodological frameworks that promote and facilitate data‐sharing and allow for meta‐ and mega‐analyses of existing pharma‐RSfMRI data. Such a framework will also enable the community to evaluate and optimize different preprocessing and modeling pipelines. Only within a standardized data‐processing framework would it be possible to investigate the commonality and differences sensitivity of different biomarkers to different drug classes.
Second, the community must adopt a set of standardized basic practices for data acquisition, which takes advantages of the state‐of‐the‐art imaging technologies for acquiring multispectral and quantitative MRI data, as well as various metabolic, physiological and psychometric samples. A rich multimodal data (for different drug types) can be explored using multivariate and univariate analysis methods, to answer a range of questions about the effect size, specificity, reliability and, most importantly, interpretation of the biomarkers. It can also help us investigate between‐subject variations in response to the same drug.
Collaborations between neuroimaging methodologists and clinical neuropharmacologists over such experiments will be mutually beneficial. To develop and validate non‐invasive methods and biomarkers for studying drug effects on the brain will also open a window of opportunity to study the chemical underpinnings of healthy brain development and aging, with implications for clinical diagnostics and interventions.
Conflict of interest: The authors do not declare any conflict of interest in conducting this survey.
J v G is the director of CNS drug research in Centre for Human Drug Research (CHDR).
A P Z, and A C E are both shareholders in Biospective Inc.
Footnotes
In this report, pharma‐fMRI refers to any fMRI study that includes a pharmacological manipulation (with or without a task). Pharma‐RSfMRI refers to a pharma‐fMRI study that does not include any active task. PhfMRI refers to a specific case of pharma‐RSfMRI where a drug is the stimulus of interest and dynamics of drug dosage and uptake are used for estimation of a hemodynamic response.
REFERENCES
- Abou‐Elseoud A, Starck T, Remes J, Nikkinen J, Tervonen O, Kiviniemi V (2010): The effect of model order selection in group PICA. Hum Brain Mapp 31:1207–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiello M, Cavaliere C, Salvatore M (2016): Hybrid PET/MR imaging and brain connectivity. Front Neurosci 10:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiello M, Salvatore E, Cachia A, Pappata S, Cavaliere C, Prinster A, Nicolai E, Salvatore M, Baron JC, Quarantelli M (2015): Relationship between simultaneously acquired resting‐state regional cerebral glucose metabolism and functional MRI: A PET/MR hybrid scanner study. Neuroimage 113:111–121. [DOI] [PubMed] [Google Scholar]
- Ainslie PN, Duffin J (2009): Integration of cerebrovascular CO2 reactivity and chemoreflex control of breathing: Mechanisms of regulation, measurement, and interpretation. Am J Physiol Regul Integr Comp Physiol 296:R1473–R1495. [DOI] [PubMed] [Google Scholar]
- Alkire MT, Haier RJ, Barker SJ, Shah NK, Wu JC, Kao YJ (1995): Cerebral metabolism during propofol anesthesia in humans studied with positron emission tomography. Anesthesiology 82:393–403. [DOI] [PubMed] [Google Scholar]
- Allen EA, Damaraju E, Plis SM, Erhardt EB, Eichele T, Calhoun VD (2014): Tracking whole‐brain connectivity dynamics in the resting state. Cereb Cortex 24:663–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsop DC, Detre JA, Golay X, Gunther M, Hendrikse J, Hernandez‐Garcia L, Lu H, MacIntosh BJ, Parkes LM, Smits M, van Osch MJ, Wang DJ, Wong EC, Zaharchuk G (2014): Recommended implementation of arterial spin‐labeled perfusion MRI for clinical applications: A consensus of the ISMRM perfusion study group and the European consortium for ASL in dementia. Magn Reson Med 73:102–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An L, Cao XH, Cao QJ, Sun L, Yang L, Zou QH, Katya R, Zang YF, Wang YF (2013): Methylphenidate normalizes resting‐state brain dysfunction in boys with attention deficit hyperactivity disorder. Neuropsychopharmacology 38:1287–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andellini M, Cannata V, Gazzellini S, Bernardi B, Napolitano A (2015): Test‐retest reliability of graph metrics of resting state MRI functional brain networks: A review. J Neurosci Methods 253:183–192. [DOI] [PubMed] [Google Scholar]
- Anderson JS, Ferguson MA, Lopez‐Larson M, Yurgelun‐Todd D (2011): Reproducibility of single‐subject functional connectivity measurements. AJNR Am J Neuroradiol 32:548–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artoni F, Menicucci D, Delorme A, Makeig S, Micera S (2014): RELICA: A method for estimating the reliability of independent components. Neuroimage 103:391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslan S, Xu F, Wang PL, Uh J, Yezhuvath US, van Osch M, Lu H (2010): Estimation of labeling efficiency in pseudocontinuous arterial spin labeling. Magn Reson Med 63:765–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagade S, Fowler KJ, Schwarz JK, Grigsby PW, Dehdashti F (2015): PET/MRI evaluation of gynecologic malignancies and prostate cancer. Semin Nucl Med 45:293–303. [DOI] [PubMed] [Google Scholar]
- Barthel H, Schroeter ML, Hoffmann KT, Sabri O (2015): PET/MR in dementia and other neurodegenerative diseases. Semin Nucl Med 45:224–233. [DOI] [PubMed] [Google Scholar]
- Barttfeld P, Uhrig L, Sitt JD, Sigman M, Jarraya B, Dehaene S (2015): Signature of consciousness in the dynamics of resting‐state brain activity. Proc Natl Acad Sci U S A 112:887–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett DS, Bullmore E, Verchinski BA, Mattay VS, Weinberger DR, Meyer‐Lindenberg A (2008): Hierarchical organization of human cortical networks in health and schizophrenia. J Neurosci 28:9239–9248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett DS, Wymbs NF, Porter MA, Mucha PJ, Carlson JM, Grafton ST (2011): Dynamic reconfiguration of human brain networks during learning. Proc Natl Acad Sci U S A 108:7641–7646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becerra L, Harter K, Gonzalez RG, Borsook D (2006): Functional magnetic resonance imaging measures of the effects of morphine on central nervous system circuitry in opioid‐naive healthy volunteers. Anesth Analg 103:208–216. [DOI] [PubMed] [Google Scholar]
- Becerra L, Pendse G, Chang PC, Bishop J, Borsook D (2011): Robust reproducible resting state networks in the awake rodent brain. PLoS One 6:e25701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becerra L, Schwartzman RJ, Kiefer RT, Rohr P, Moulton EA, Wallin D, Pendse G, Morris S, Borsook D (2009): CNS measures of pain responses pre‐ and post‐anesthetic ketamine in a patient with complex regional pain syndrome. Pain Med. [DOI] [PubMed] [Google Scholar]
- Becerra L, Upadhyay J, Chang PC, Bishop J, Anderson J, Baumgartner R, Schwarz AJ, Coimbra A, Wallin D, Nutile L, George E, Maier G, Sunkaraneni S, Iyengar S, Evelhoch JL, Bleakman D, Hargreaves R, Borsook D (2013): Parallel buprenorphine phMRI responses in conscious rodents and healthy human subjects. J Pharmacol Exp Ther 345:41–51. [DOI] [PubMed] [Google Scholar]
- Beckmann CF, DeLuca M, Devlin JT, Smith SM (2005): Investigations into resting‐state connectivity using independent component analysis. Philos Trans R Soc Lond B Biol Sci 360:1001–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellec P, Rosa‐Neto P, Lyttelton OC, Benali H, Evans AC (2010): Multi‐level bootstrap analysis of stable clusters in resting‐state fMRI. Neuroimage 51:1126–1139. [DOI] [PubMed] [Google Scholar]
- Bettinardi RG, Tort‐Colet N, Ruiz‐Mejias M, Sanchez‐Vives MV, Deco G (2015): Gradual emergence of spontaneous correlated brain activity during fading of general anesthesia in rats: Evidences from fMRI and local field potentials. Neuroimage 114:185–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birn RM (2012): The role of physiological noise in resting‐state functional connectivity. Neuroimage 62:864–870. [DOI] [PubMed] [Google Scholar]
- Birn RM, Molloy EK, Patriat R, Parker T, Meier TB, Kirk GR, Nair VA, Meyerand ME, Prabhakaran V (2013): The effect of scan length on the reliability of resting‐state fMRI connectivity estimates. Neuroimage 83:550–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birn RM, Murphy K, Bandettini PA (2008a): The effect of respiration variations on independent component analysis results of resting state functional connectivity. Hum Brain Mapp 29:740–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birn RM, Smith MA, Jones TB, Bandettini PA (2008b): The respiration response function: The temporal dynamics of fMRI signal fluctuations related to changes in respiration. Neuroimage 40:644–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswal B, Yetkin FZ, Haughton VM, Hyde JS (1995): Functional connectivity in the motor cortex of resting human brain using echo‐planar MRI. Magnetic resonance in medicine Magn Reson Med 34:537–541. [DOI] [PubMed] [Google Scholar]
- Biswal BB, Ulmer JL (1999): Blind source separation of multiple signal sources of fMRI data sets using independent component analysis. J Comput Assist Tomogr 23:265–271. [DOI] [PubMed] [Google Scholar]
- Blautzik J, Vetter C, Peres I, Gutyrchik E, Keeser D, Berman A, Kirsch V, Mueller S, Poppel E, Reiser M, Roenneberg T, Meindl T (2013): Classifying fMRI‐derived resting‐state connectivity patterns according to their daily rhythmicity. Neuroimage 71:298–306. [DOI] [PubMed] [Google Scholar]
- Blockley NP, Griffeth VE, Buxton RB (2012): A general analysis of calibrated BOLD methodology for measuring CMRO2 responses: Comparison of a new approach with existing methods. Neuroimage 60:279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blockley NP, Griffeth VE, Simon AB, Dubowitz DJ, Buxton RB (2015): Calibrating the BOLD response without administering gases: Comparison of hypercapnia calibration with calibration using an asymmetric spin echo. Neuroimage 104:423–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom AS, Hoffmann RG, Fuller SA, Pankiewicz J, Harsch HH, Stein EA (1999): Determination of drug‐induced changes in functional MRI signal using a pharmacokinetic model. Hum Brain Mapp 8:235–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokkers RP, van Osch MJ, Klijn CJ, Kappelle LJ, Hendrikse J (2011): Cerebrovascular reactivity within perfusion territories in patients with an internal carotid artery occlusion. J Neurol Neurosurg Psychiatry 82:1011–1016. [DOI] [PubMed] [Google Scholar]
- Borsook D, Bleakman D, Hargreaves R, Upadhyay J, Schmidt KF, Becerra L (2008): A BOLD experiment in defining the utility of fMRI in drug development. Neuroimage 42:461–466. [DOI] [PubMed] [Google Scholar]
- Borsook D, Hargreaves R, Becerra L (2011): Can functional magnetic resonance imaging improve success rates in cns drug discovery?. Expert Opin Drug Discov 6:597–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boveroux P, Vanhaudenhuyse A, Bruno MA, Noirhomme Q, Lauwick S, Luxen A, Degueldre C, Plenevaux A, Schnakers C, Phillips C, Brichant JF, Bonhomme V, Maquet P, Greicius MD, Laureys S, Boly M (2010): Breakdown of within‐ and between‐network resting state functional magnetic resonance imaging connectivity during propofol‐induced loss of consciousness. Anesthesiology 113:1038–1053. [DOI] [PubMed] [Google Scholar]
- Braun U, Plichta MM, Esslinger C, Sauer C, Haddad L, Grimm O, Mier D, Mohnke S, Heinz A, Erk S, Walter H, Seiferth N, Kirsch P, Meyer‐Lindenberg A (2012): Test‐retest reliability of resting‐state connectivity network characteristics using fMRI and graph theoretical measures. Neuroimage 59:1404–1412. [DOI] [PubMed] [Google Scholar]
- Bright MG, Murphy K (2013a): Reliable quantification of BOLD fMRI cerebrovascular reactivity despite poor breath‐hold performance. Neuroimage 83:559–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright MG, Murphy K (2013b): Removing motion and physiological artifacts from intrinsic BOLD fluctuations using short echo data. Neuroimage 64:526–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright MG, Murphy K (2015): Is fMRI “noise” really noise? Resting state nuisance regressors remove variance with network structure. Neuroimage 114:158–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes MJ, Woolrich M, Luckhoo H, Price D, Hale JR, Stephenson MC, Barnes GR, Smith SM, Morris PG (2011): Investigating the electrophysiological basis of resting state networks using magnetoencephalography. Proc Natl Acad Sci U S A 108:16783–16788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullmore E, Sporns O (2009): Complex brain networks: Graph theoretical analysis of structural and functional systems. Nat Rev Neurosci 10:186–198. [DOI] [PubMed] [Google Scholar]
- Buxton RB (2005): Quantifying CBF with arterial spin labeling. J Magn Reson Imaging 22:723–726. [DOI] [PubMed] [Google Scholar]
- Buxton RB, Frank LR, Wong EC, Siewert B, Warach S, Edelman RR (1998): A general kinetic model for quantitative perfusion imaging with arterial spin labeling. Magn Reson Med 40:383–396. [DOI] [PubMed] [Google Scholar]
- Calhoun VD, Adali T, Pearlson GD, Pekar JJ (2001): Spatial and temporal independent component analysis of functional MRI data containing a pair of task‐related waveforms. Hum Brain Mapp 13:43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun VD, Altschul D, McGinty V, Shih R, Scott D, Sears E, Pearlson GD (2004): Alcohol intoxication effects on visual perception: an fMRI study. Hum Brain Mapp 21:15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun VD, Eichele T, Adali T, Allen EA (2012): Decomposing the brain: components and modes, networks and nodes. Trends Cogn Sci 16:255–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun VD, Kiehl KA, Pearlson GD (2008): Modulation of temporally coherent brain networks estimated using ICA at rest and during cognitive tasks. Hum Brain Mapp 29:828–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H, Plichta MM, Schafer A, Haddad L, Grimm O, Schneider M, Esslinger C, Kirsch P, Meyer‐Lindenberg A, Tost H (2014): Test‐retest reliability of fMRI‐based graph theoretical properties during working memory, emotion processing, and resting state. Neuroimage 84:888–900. [DOI] [PubMed] [Google Scholar]
- Carbonell F, Bellec P, Shmuel A (2014a): Quantification of the impact of a confounding variable on functional connectivity confirms anti‐correlated networks in the resting‐state. Neuroimage 86:343–353. [DOI] [PubMed] [Google Scholar]
- Carbonell F, Nagano‐Saito A, Leyton M, Cisek P, Benkelfat C, He Y, Dagher A (2014b): Dopamine precursor depletion impairs structure and efficiency of resting state brain functional networks. Neuropharmacology 84:90–100. [DOI] [PubMed] [Google Scholar]
- Carhart‐Harris RL, Erritzoe D, Williams T, Stone JM, Reed LJ, Colasanti A, Tyacke RJ, Leech R, Malizia AL, Murphy K, Hobden P, Evans J, Feilding A, Wise RG, Nutt DJ (2012): Neural correlates of the psychedelic state as determined by fMRI studies with psilocybin. Proc Natl Acad Sci U S A 109:2138–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carhart‐Harris RL, Murphy K, Leech R, Erritzoe D, Wall MB, Ferguson B, Williams LT, Roseman L, Brugger S, De Meer I, Tanner M, Tyacke R, Wolff K, Sethi A, Bloomfield MA, Williams TM, Bolstridge M, Stewart L, Morgan C, Newbould RD, Feilding A, Curran HV, Nutt DJ (2015): The effects of acutely administered 3,4‐methylenedioxymethamphetamine on spontaneous brain function in healthy volunteers measured with arterial spin labeling and blood oxygen level‐dependent resting state functional connectivity. Biol Psychiatry 78:554–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carp J (2013): Optimizing the order of operations for movement scrubbing: Comment on Power et al. Neuroimage 76:436–438. [DOI] [PubMed] [Google Scholar]
- Chang C, Cunningham JP, Glover GH (2009): Influence of heart rate on the BOLD signal: The cardiac response function. Neuroimage 44:857–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Saad ZS, Nath AR, Beauchamp MS, Cox RW (2012): FMRI group analysis combining effect estimates and their variances. Neuroimage 60:747–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Parrish TB (2009b): Caffeine dose effect on activation‐induced BOLD and CBF responses. Neuroimage 46:577–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Parrish TB (2009a): Caffeines effects on cerebrovascular reactivity and coupling between cerebral blood flow and oxygen metabolism. Neuroimage 44:647–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christen T, Bolar DS, Zaharchuk G (2013): Imaging brain oxygenation with MRI using blood oxygenation approaches: Methods, validation, and clinical applications. AJNR Am J Neuroradiol 34:1113–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang KH, van Gelderen P, Merkle H, Bodurka J, Ikonomidou VN, Koretsky AP, Duyn JH, Talagala SL (2008): Mapping resting‐state functional connectivity using perfusion MRI. Neuroimage 40:1595–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole DM, Beckmann CF, Oei NY, Both S, van Gerven JM, Rombouts SA (2013): Differential and distributed effects of dopamine neuromodulations on resting‐state network connectivity. Neuroimage 78:59–67. [DOI] [PubMed] [Google Scholar]
- Cole DM, Smith SM, Beckmann CF (2010): Advances and pitfalls in the analysis and interpretation of resting‐state FMRI data. Front Syst Neurosci 4:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole MW, Bassett DS, Power JD, Braver TS, Petersen SE (2014): Intrinsic and task‐evoked network architectures of the human brain. Neuron 83:238–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comasco E, Frokjaer VG, Sundstrom‐Poromaa I (2014): Functional and molecular neuroimaging of menopause and hormone replacement therapy. Front Neurosci 8:388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craddock RC, Tungaraza RL, Milham MP (2015): Connectomics and new approaches for analyzing human brain functional connectivity. Gigascience 4:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagli MS, Ingeholm JE, Haxby JV (1999): Localization of cardiac‐induced signal change in fMRI. Neuroimage 9:407–415. [DOI] [PubMed] [Google Scholar]
- Dai W, Garcia D, de Bazelaire C, Alsop DC (2008): Continuous flow‐driven inversion for arterial spin labeling using pulsed radio frequency and gradient fields. Magn Reson Med 60:1488–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damoiseaux JS, Rombouts SA, Barkhof F, Scheltens P, Stam CJ, Smith SM, Beckmann CF (2006): Consistent resting‐state networks across healthy subjects. Proc Natl Acad Sci U S A 103:13848–13853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dansereau CL, Bellec P, Lee K, Pittau F, Gotman J, Grova C (2014): Detection of abnormal resting‐state networks in individual patients suffering from focal epilepsy: An initial step toward individual connectivity assessment. Front Neurosci 8:419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Glatard T, MacIntyre LC, Madjar C, Rogers C, Rousseau ME, Rioux P, MacFarlane D, Mohades Z, Gnanasekaran R, Makowski C, Kostopoulos P, Adalat R, Khalili‐Mahani N, Niso G, Moreau JT, Evans AC (2016): The MNI data‐sharing and processing ecosystem. Neuroimage 124:1188–1195. [DOI] [PubMed] [Google Scholar]
- Das S, Zijdenbos AP, Harlap J, Vins D, Evans AC (2011): LORIS: A web‐based data management system for multi‐center studies. Front Neuroinform 5:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis FC, Knodt AR, Sporns O, Lahey BB, Zald DH, Brigidi BD, Hariri AR (2013): Impulsivity and the modular organization of resting‐state neural networks. Cereb Cortex 23:1444–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Simoni S, Schwarz AJ, O'Daly OG, Marquand AF, Brittain C, Gonzales C, Stephenson S, Williams SC, Mehta MA (2013): Test‐retest reliability of the BOLD pharmacological MRI response to ketamine in healthy volunteers. Neuroimage 64:75–90. [DOI] [PubMed] [Google Scholar]
- Deakin JF, Lees J, McKie S, Hallak JE, Williams SR, Dursun SM (2008): Glutamate and the neural basis of the subjective effects of ketamine: A pharmaco‐magnetic resonance imaging study. Arch Gen Psychiatry 65:154–164. [DOI] [PubMed] [Google Scholar]
- Deibler AR, Pollock JM, Kraft RA, Tan H, Burdette JH, Maldjian JA (2008a): Arterial spin‐labeling in routine clinical practice, part 1: technique and artifacts. AJNR Am J Neuroradiol 29:1228–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deibler AR, Pollock JM, Kraft RA, Tan H, Burdette JH, Maldjian JA (2008b): Arterial spin‐labeling in routine clinical practice, part 2: hypoperfusion patterns. AJNR Am J Neuroradiol 29:1235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denier N, Gerber H, Vogel M, Klarhofer M, Riecher‐Rossler A, Wiesbeck GA, Lang UE, Borgwardt S, Walter M (2013): Reduction in cerebral perfusion after heroin administration: A resting state arterial spin labeling study. PLoS One 8:e71461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denier N, Walter M, Bendfeldt K, Lang U, Borgwardt S (2012): Resting state abnormalities in psychosis compared to acute cannabinoids and opioids challenges: A systematic review of functional imaging studies. Curr Pharm Des 18:5081–5092. [DOI] [PubMed] [Google Scholar]
- Di X, Biswal BB, Alzheimer's Disease Neuroimaging Initiative (2012): Metabolic brain covariant networks as revealed by FDG‐PET with reference to resting‐state fMRI networks. Brain Connect 2:275–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diukova A, Ware J, Smith JE, Evans CJ, Murphy K, Rogers PJ, Wise RG (2012): Separating neural and vascular effects of caffeine using simultaneous EEG‐FMRI: Differential effects of caffeine on cognitive and sensorimotor brain responses. Neuroimage 62:239–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodhia S, Hosanagar A, Fitzgerald DA, Labuschagne I, Wood AG, Nathan PJ, Phan KL (2014): Modulation of resting‐state amygdala‐frontal functional connectivity by oxytocin in generalized social anxiety disorder. Neuropsychopharmacology 39:2061–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle OM, De Simoni S, Schwarz AJ, Brittain C, O'Daly OG, Williams SC, Mehta MA (2013): Quantifying the attenuation of the ketamine pharmacological magnetic resonance imaging response in humans: A validation using antipsychotic and glutamatergic agents. J Pharmacol Exp Ther 345:151–160. [DOI] [PubMed] [Google Scholar]
- Driesen NR, McCarthy G, Bhagwagar Z, Bloch M, Calhoun V, D'Souza DC, Gueorguieva R, He G, Ramachandran R, Suckow RF, Anticevic A, Morgan PT, Krystal JH (2013): Relationship of resting brain hyperconnectivity and schizophrenia‐like symptoms produced by the NMDA receptor antagonist ketamine in humans. Mol Psychiatry 18:1199–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duff EP, Vennart W, Wise RG, Howard MA, Harris RE, Lee M, Wartolowska K, Wanigasekera V, Wilson FJ, Whitlock M, Tracey I, Woolrich MW, Smith SM (2015): Learning to identify CNS drug action and efficacy using multistudy fMRI data. Sci Transl Med 7:274ra16. [DOI] [PubMed] [Google Scholar]
- Eklund A, Nichols TE, Knutsson H (2016): Cluster failure: Why fMRI inferences for spatial extent have inflated false‐positive rates. Proc Natl Acad Sci U S A 113:7900–7905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito F, Goebel R (2011): Extracting functional networks with spatial independent component analysis: The role of dimensionality, reliability and aggregation scheme. Curr Opin Neurol 24:378–385. [DOI] [PubMed] [Google Scholar]
- Esposito F, Pignataro G, Di Renzo G, Spinali A, Paccone A, Tedeschi G, Annunziato L (2010): Alcohol increases spontaneous BOLD signal fluctuations in the visual network. Neuroimage 53:534–543. [DOI] [PubMed] [Google Scholar]
- Esposito F, Tessitore A, Giordano A, De Micco R, Paccone A, Conforti R, Pignataro G, Annunziato L, Tedeschi G (2013a): Rhythm‐specific modulation of the sensorimotor network in drug‐naive patients with Parkinson's disease by levodopa. Brain 136 (Pt 3):710–725. [DOI] [PubMed] [Google Scholar]
- Esposito R, Cilli F, Pieramico V, Ferretti A, Macchia A, Tommasi M, Saggino A, Ciavardelli D, Manna A, Navarra R, Cieri F, Stuppia L, Tartaro A, Sensi SL (2013b): Acute effects of modafinil on brain resting state networks in young healthy subjects. PLoS One 8:e69224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faull OK, Jenkinson M, Clare S, Pattinson KT (2015): Functional subdivision of the human periaqueductal grey in respiratory control using 7 tesla fMRI. Neuroimage 113:356–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg DA, Moeller S, Smith SM, Auerbach E, Ramanna S, Gunther M, Glasser MF, Miller KL, Ugurbil K, Yacoub E (2010): Multiplexed echo planar imaging for sub‐second whole brain FMRI and fast diffusion imaging. PLoS One 5:e15710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez‐Seara MA, Aznarez‐Sanado M, Mengual E, Irigoyen J, Heukamp F, Pastor MA (2011): Effects on resting cerebral blood flow and functional connectivity induced by metoclopramide: A perfusion MRI study in healthy volunteers. Br J Pharmacol 163:1639–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flodin P, Gospic K, Petrovic P, Fransson P (2012): Effects of L‐dopa and oxazepam on resting‐state functional magnetic resonance imaging connectivity: A randomized, cross‐sectional placebo study. Brain Connect 2:246–253. [DOI] [PubMed] [Google Scholar]
- Forman SD, Cohen JD, Fitzgerald M, Eddy WF, Mintun MA, Noll DC (1995): Improved assessment of significant activation in functional magnetic resonance imaging (fMRI): use of a cluster‐size threshold. Magn Reson Med 33:636–647. [DOI] [PubMed] [Google Scholar]
- Fowler JS, MacGregor RR, Wolf AP, Arnett CD, Dewey SL, Schlyer D, Christman D, Logan J, Smith M, Sachs H, et al. (1987): Mapping human brain monoamine oxidase A and B with 11C‐labeled suicide inactivators and PET. Science 235:481–485. [DOI] [PubMed] [Google Scholar]
- Fox MD, Zhang D, Snyder AZ, Raichle ME (2009): The global signal and observed anticorrelated resting state brain networks. J Neurophysiol 101:3270–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frijling JL, van Zuiden M, Koch SB, Nawijn L, Veltman DJ, Olff M (2016): Intranasal oxytocin affects amygdala functional connectivity after trauma script‐driven imagery in distressed recently trauma‐exposed individuals. Neuropsychopharmacology 41:1286–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friston KJ, Stephan KE, Lund TE, Morcom A, Kiebel S (2005): Mixed‐effects and fMRI studies. Neuroimage 24:244–252. [DOI] [PubMed] [Google Scholar]
- Fukunaga M, Horovitz SG, de Zwart JA, van Gelderen P, Balkin TJ, Braun AR, Duyn JH (2008): Metabolic origin of BOLD signal fluctuations in the absence of stimuli. J Cereb Blood Flow Metab 28:1377–1387. [DOI] [PubMed] [Google Scholar]
- Gass N, Schwarz AJ, Sartorius A, Cleppien D, Zheng L, Schenker E, Risterucci C, Meyer‐Lindenberg A, Weber‐Fahr W (2013): Haloperidol modulates midbrain‐prefrontal functional connectivity in the rat brain. Eur Neuropsychopharmacol 23:1310–1319. [DOI] [PubMed] [Google Scholar]
- Gass N, Schwarz AJ, Sartorius A, Schenker E, Risterucci C, Spedding M, Zheng L, Meyer‐Lindenberg A, Weber‐Fahr W (2014): Sub‐anesthetic ketamine modulates intrinsic BOLD connectivity within the hippocampal‐prefrontal circuit in the rat. Neuropsychopharmacology 39:895–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier CJ, Desjardins‐Crepeau L, Madjar C, Bherer L, Hoge RD (2012): Absolute quantification of resting oxygen metabolism and metabolic reactivity during functional activation using QUO2 MRI. Neuroimage 63:1353–1363. [DOI] [PubMed] [Google Scholar]
- Gauthier CJ, Hoge RD (2012): Magnetic resonance imaging of resting OEF and CMRO(2) using a generalized calibration model for hypercapnia and hyperoxia. Neuroimage 60:1212–1225. [DOI] [PubMed] [Google Scholar]
- Genovese CR, Lazar NA, Nichols T (2002): Thresholding of statistical maps in functional neuroimaging using the false discovery rate. Neuroimage 15:870–878. [DOI] [PubMed] [Google Scholar]
- Gevers S, van Osch MJ, Bokkers RP, Kies DA, Teeuwisse WM, Majoie CB, Hendrikse J, Nederveen AJ (2011): Intra‐ and multicenter reproducibility of pulsed, continuous and pseudo‐continuous arterial spin labeling methods for measuring cerebral perfusion. J Cereb Blood Flow Metab 31:1706–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gili T, Saxena N, Diukova A, Murphy K, Hall JE, Wise RG (2013): The thalamus and brainstem act as key hubs in alterations of human brain network connectivity induced by mild propofol sedation. J Neurosci 33:4024–4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatard T, Lartizien C, Gibaud B, da Silva RF, Forestier G, Cervenansky F, Alessandrini M, Benoit‐Cattin H, Bernard O, Camarasu‐Pop S, Cerezo N, Clarysse P, Gaignard A, Hugonnard P, Liebgott H, Marache S, Marion A, Montagnat J, Tabary J, Friboulet D (2013): A virtual imaging platform for multi‐modality medical image simulation. IEEE Trans Med Imaging 32:110–118. [DOI] [PubMed] [Google Scholar]
- Glatard T, Lewis LB, Ferreira da Silva R, Adalat R, Beck N, Lepage C, Rioux P, Rousseau ME, Sherif T, Deelman E, Khalili‐Mahani N, Evans AC (2015): Reproducibility of neuroimaging analyses across operating systems. Front Neuroinform 9:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldmann D, Montanari F, Richter L, Zdrazil B, Ecker GF (2014): Exploiting open data: A new era in pharmacoinformatics. Future Med Chem 6:503–514. [DOI] [PubMed] [Google Scholar]
- Gonzalez‐Castillo J, Handwerker DA, Robinson ME, Hoy CW, Buchanan LC, Saad ZS, Bandettini PA (2014): The spatial structure of resting state connectivity stability on the scale of minutes. Front Neurosci 8:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozzi A, Ceolin L, Schwarz A, Reese T, Bertani S, Crestan V, Bifone A (2007): A multimodality investigation of cerebral hemodynamics and autoregulation in pharmacological MRI. Magn Reson Imaging 25:826–833. [DOI] [PubMed] [Google Scholar]
- Gozzi A, Schwarz A, Crestan V, Bifone A (2008): Drug‐anaesthetic interaction in phMRI: The case of the psychotomimetic agent phencyclidine. Magn Reson Imaging 26:999–1006. [DOI] [PubMed] [Google Scholar]
- Gozzi A, Schwarz AJ, Reese T, Crestan V, Bertani S, Turrini G, Corsi M, Bifone A (2005): Functional magnetic resonance mapping of intracerebroventricular infusion of a neuroactive peptide in the anaesthetised rat. J Neurosci Methods 142:115–124. [DOI] [PubMed] [Google Scholar]
- Greicius MD, Kiviniemi V, Tervonen O, Vainionpaa V, Alahuhta S, Reiss AL, Menon V (2008): Persistent default‐mode network connectivity during light sedation. Hum Brain Mapp 29:839–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greicius MD, Krasnow B, Reiss AL, Menon V (2003): Functional connectivity in the resting brain: A network analysis of the default mode hypothesis. Proc Natl Acad Sci U S A 100:253–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greicius MD, Supekar K, Menon V, Dougherty RF (2009): Resting‐state functional connectivity reflects structural connectivity in the default mode network. Cereb Cortex 19:72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffanti L, Salimi‐Khorshidi G, Beckmann CF, Auerbach EJ, Douaud G, Sexton CE, Zsoldos E, Ebmeier KP, Filippini N, Mackay CE, Moeller S, Xu J, Yacoub E, Baselli G, Ugurbil K, Miller KL, Smith SM (2014): ICA‐based artefact removal and accelerated fMRI acquisition for improved resting state network imaging. Neuroimage 95:232–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffeth VE, Perthen JE, Buxton RB (2011): Prospects for quantitative fMRI: Investigating the effects of caffeine on baseline oxygen metabolism and the response to a visual stimulus in humans. Neuroimage 57:809–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm O, Gass N, Weber‐Fahr W, Sartorius A, Schenker E, Spedding M, Risterucci C, Schweiger JI, Bohringer A, Zang Z, Tost H, Schwarz AJ, Meyer‐Lindenberg A (2015): Acute ketamine challenge increases resting state prefrontal‐hippocampal connectivity in both humans and rats. Psychopharmacology (Berl) 232:4231–4241. [DOI] [PubMed] [Google Scholar]
- Guillaume B, Hua X, Thompson PM, Waldorp L, Nichols TE, Alzheimer's Disease Neuroimaging Initiative (2014): Fast and accurate modelling of longitudinal and repeated measures neuroimaging data. Neuroimage 94:287–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guldenmund P, Demertzi A, Boveroux P, Boly M, Vanhaudenhuyse A, Bruno MA, Gosseries O, Noirhomme Q, Brichant JF, Bonhomme V, Laureys S, Soddu A (2013): Thalamus, brainstem and salience network connectivity changes during propofol‐induced sedation and unconsciousness. Brain Connect 3:273–285. [DOI] [PubMed] [Google Scholar]
- Gunn RN, Guo Q, Salinas CA, Tziortzi AC, Searle GE (2011): Advances in biomathematical modeling for PET neuroreceptor imaging. Drug Discov Today Technol 8:e45–e51. [DOI] [PubMed] [Google Scholar]
- Handley R, Zelaya FO, Reinders AA, Marques TR, Mehta MA, O'Gorman R, Alsop DC, Taylor H, Johnston A, Williams S, McGuire P, Pariante CM, Kapur S, Dazzan P (2013): Acute effects of single‐dose aripiprazole and haloperidol on resting cerebral blood flow (rCBF) in the human brain. Hum Brain Mapp 34:272–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartkamp NS, Bokkers RP, van der Worp HB, van Osch MJ, Kappelle LJ, Hendrikse J (2011): Distribution of cerebral blood flow in the caudate nucleus, lentiform nucleus and thalamus in patients with carotid artery stenosis. Eur Radiol 21:875–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartvig P, Valtysson J, Lindner KJ, Kristensen J, Karlsten R, Gustafsson LL, Svensson JO, Øye I, Antoni G, Westerberg G, Långström B (1995): Central nervous system effects of subdissociative doses of (S)‐ketamine are related to plasma and brain concentrations measured with positron emission tomography in healthy volunteers. Clin Pharmacol Ther 58:165–173. [DOI] [PubMed] [Google Scholar]
- Hayasaka S, Phan KL, Liberzon I, Worsley KJ, Nichols TE (2004): Nonstationary cluster‐size inference with random field and permutation methods. Neuroimage 22:676–687. [DOI] [PubMed] [Google Scholar]
- Heijtel DF, Mutsaerts HJ, Bakker E, Schober P, Stevens MF, Petersen ET, van Berckel BN, Majoie CB, Booij J, van Osch MJ, Vanbavel E, Boellaard R, Lammertsma AA, Nederveen AJ (2014): Accuracy and precision of pseudo‐continuous arterial spin labeling perfusion during baseline and hypercapnia: A head‐to‐head comparison with (1)(5)O H(2)O positron emission tomography. Neuroimage 92:182–192. [DOI] [PubMed] [Google Scholar]
- Heiss WD (2016): Hybrid PET/MR imaging in neurology: Present applications and prospects for the future. J Nucl Med 57:993–995. [DOI] [PubMed] [Google Scholar]
- Henckens MJ, van Wingen GA, Joels M, Fernandez G (2012): Corticosteroid induced decoupling of the amygdala in men. Cereb Cortex 22:2336–2345. [DOI] [PubMed] [Google Scholar]
- Himberg J, Hyvarinen A (2003): ICASSO: Software for investigating the reliability of ICA estimates by clustering and visualization. In: 2003 IEEE XIII Workshop on Neural Networks for Signal Processing ‐ NNSP'03. pp 259−268.
- Hodkinson DJ, O'Daly O, Zunszain PA, Pariante CM, Lazurenko V, Zelaya FO, Howard MA, Williams SC (2014): Circadian and homeostatic modulation of functional connectivity and regional cerebral blood flow in humans under normal entrained conditions. J Cereb Blood Flow Metab 34:1493–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoge RD (2012): Calibrated FMRI. Neuroimage 62:930–937. [DOI] [PubMed] [Google Scholar]
- Hutchison RM, Womelsdorf T, Gati JS, Everling S, Menon RS (2013): Resting‐state networks show dynamic functional connectivity in awake humans and anesthetized macaques. Hum Brain Mapp 34:2154–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacovella V, Hasson U (2011): The relationship between BOLD signal and autonomic nervous system functions: Implications for processing of “physiological noise”. Magn Reson Imaging 29:1338–1345. [DOI] [PubMed] [Google Scholar]
- Iannetti GD, Wise RG (2007): BOLD functional MRI in disease and pharmacological studies: Room for improvement?. Magn Reson Imaging 25:978–988. [DOI] [PubMed] [Google Scholar]
- Jann K, Gee DG, Kilroy E, Schwab S, Smith RX, Cannon TD, Wang DJ (2015): Functional connectivity in BOLD and CBF data: Similarity and reliability of resting brain networks. Neuroimage 106:111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Kim M, Chodkowski B, Donahue MJ, Pekar JJ, Van Zijl PC, Albert M (2010): Reliability and reproducibility of perfusion MRI in cognitively normal subjects. Magn Reson Imaging 28:1283–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonckers E, Van Audekerke J, De Visscher G, Van der Linden A, Verhoye M (2011): Functional connectivity fMRI of the rodent brain: Comparison of functional connectivity networks in rat and mouse. PLoS One 6:e18876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonckers E, Van der Linden A, Verhoye M (2013): Functional magnetic resonance imaging in rodents: An unique tool to study in vivo pharmacologic neuromodulation. Curr Opin Pharmacol 13:813–820. [DOI] [PubMed] [Google Scholar]
- Jones DT, Vemuri P, Murphy MC, Gunter JL, Senjem ML, Machulda MM, Przybelski SA, Gregg BE, Kantarci K, Knopman DS, Boeve BF, Petersen RC, Jack CR, Jr. (2012): Non‐stationarity in the “resting brains” modular architecture. PLoS One 7:e39731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan D, Ilg R, Riedl V, Schorer A, Grimberg S, Neufang S, Omerovic A, Berger S, Untergehrer G, Preibisch C, Schulz E, Schuster T, Schroter M, Spoormaker V, Zimmer C, Hemmer B, Wohlschlager A, Kochs EF, Schneider G (2013): Simultaneous electroencephalographic and functional magnetic resonance imaging indicate impaired cortical top‐down processing in association with anesthetic‐induced unconsciousness. Anesthesiology 119:1031–1042. [DOI] [PubMed] [Google Scholar]
- Jung Y, Wong EC, Liu TT (2010): Multiphase pseudocontinuous arterial spin labeling (MP‐PCASL) for robust quantification of cerebral blood flow. Magn Reson Med 64:799–810. [DOI] [PubMed] [Google Scholar]
- Kelly C, de Zubicaray G, Di Martino A, Copland DA, Reiss PT, Klein DF, Castellanos FX, Milham MP, McMahon K (2009): L‐dopa modulates functional connectivity in striatal cognitive and motor networks: A double‐blind placebo‐controlled study. J Neurosci 29:7364–7378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly C, Toro R, Di Martino A, Cox CL, Bellec P, Castellanos FX, Milham MP (2012): A convergent functional architecture of the insula emerges across imaging modalities. Neuroimage 61:1129–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler LG, Barnhart HX, Buckler AJ, Choudhury KR, Kondratovich MV, Toledano A, Guimaraes AR, Filice R, Zhang Z, Sullivan DC, Group QTW (2015): The emerging science of quantitative imaging biomarkers terminology and definitions for scientific studies and regulatory submissions. Stat Methods Med Res 24:9–26. [DOI] [PubMed] [Google Scholar]
- Khalili‐Mahani N, Chang C, van Osch MJ, Veer IM, van Buchem MA, Dahan A, Beckmann CF, van Gerven JM, Rombouts SA (2013): The impact of “physiological correction” on functional connectivity analysis of pharmacological resting state fMRI. Neuroimage 65:499–510. [DOI] [PubMed] [Google Scholar]
- Khalili‐Mahani N, Dedovic K, Engert V, Pruessner M, Pruessner JC (2010): Hippocampal activation during a cognitive task is associated with subsequent neuroendocrine and cognitive responses to psychological stress. Hippocampus 20:323–334. [DOI] [PubMed] [Google Scholar]
- Khalili‐Mahani N, Niesters M, van Osch MJ, Oitzl M, Veer I, de Rooij M, van Gerven J, van Buchem MA, Beckmann CF, Rombouts SA, Dahan A (2015): Ketamine interactions with biomarkers of stress: A randomized placebo‐controlled repeated measures resting‐state fMRI and PCASL pilot study in healthy men. Neuroimage 108:396–409. [DOI] [PubMed] [Google Scholar]
- Khalili‐Mahani N, van Osch MJ, Baerends E, Soeter RP, de Kam M, Zoethout RW, Dahan A, van Buchem MA, van Gerven JM, Rombouts SA (2011): Pseudocontinuous arterial spin labeling reveals dissociable effects of morphine and alcohol on regional cerebral blood flow. J Cereb Blood Flow Metab 31:1321–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalili‐Mahani N, van Osch MJ, de Rooij M, Beckmann CF, van Buchem MA, Dahan A, van Gerven JM, Rombouts SA (2014): Spatial heterogeneity of the relation between resting‐state connectivity and blood flow: An important consideration for pharmacological studies. Hum Brain Mapp 35:929–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalili‐Mahani N, Zoethout RM, Beckmann CF, Baerends E, de Kam ML, Soeter RP, Dahan A, van Buchem MA, van Gerven JM, Rombouts SA (2012): Effects of morphine and alcohol on functional brain connectivity during “resting state”: a placebo‐controlled crossover study in healthy young men. Hum Brain Mapp 33:1003–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpatrick LA, Coveleskie K, Connolly L, Labus JS, Ebrat B, Stains J, Jiang Z, Suyenobu BY, Raybould HE, Tillisch K, Mayer EA (2014): Influence of sucrose ingestion on brainstem and hypothalamic intrinsic oscillations in lean and obese women. Gastroenterology 146:1212–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiviniemi V, Jauhiainen J, Tervonen O, Paakko E, Oikarinen J, Vainionpaa V, Rantala H, Biswal B (2000): Slow vasomotor fluctuation in fMRI of anesthetized child brain. Magn Reson Med 44:373–378. [DOI] [PubMed] [Google Scholar]
- Kiviniemi V, Kantola JH, Jauhiainen J, Hyvarinen A, Tervonen O (2003): Independent component analysis of nondeterministic fMRI signal sources. Neuroimage 19:253–260. [DOI] [PubMed] [Google Scholar]
- Kiviniemi V, Ruohonen J, Tervonen O (2005): Separation of physiological very low frequency fluctuation from aliasing by switched sampling interval fMRI scans. J Magn Reson Imaging 23:41–46. [DOI] [PubMed] [Google Scholar]
- Klaassens BL, van Gorsel HC, Khalili‐Mahani N, van der Grond J, Wyman BT, Whitcher B, Rombouts SA, van Gerven JM (2015): Single‐dose serotonergic stimulation shows widespread effects on functional brain connectivity. Neuroimage 122:440–450. [DOI] [PubMed] [Google Scholar]
- Kleinloog D, Rombouts S, Zoethout R, Klumpers L, Niesters M, Khalili‐Mahani N, Dahan A, van Gerven J (2015): Subjective effects of ethanol, morphine, delta(9)‐tetrahydrocannabinol, and ketamine following a pharmacological challenge are related to functional. Brain Connect 5:641–648. [DOI] [PubMed] [Google Scholar]
- Klumpers LE, Cole DM, Khalili‐Mahani N, Soeter RP, Te Beek ET, Rombouts SA, van Gerven JM (2012): Manipulating brain connectivity with delta(9)‐tetrahydrocannabinol: A pharmacological resting state FMRI study. Neuroimage 63:1701–1711. [DOI] [PubMed] [Google Scholar]
- Koch SB, van Zuiden M, Nawijn L, Frijling JL, Veltman DJ, Olff M (2016): Intranasal Oxytocin Administration Dampens Amygdala Reactivity towards Emotional Faces in Male and Female PTSD Patients. Neuropsychopharmacology 41:1495–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofke WA, Blissitt PA, Rao H, Wang J, Addya K, Detre J (2007): Remifentanil‐induced cerebral blood flow effects in normal humans: Dose and ApoE genotype. Anesth Analg 105:167–175. [DOI] [PubMed] [Google Scholar]
- Kong XZ, Zhen Z, Li X, Lu HH, Wang R, Liu L, He Y, Zang Y, Liu J (2014): Individual differences in impulsivity predict head motion during magnetic resonance imaging. PLoS One 9:e104989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konova AB, Moeller SJ, Tomasi D, Goldstein RZ (2015): Effects of chronic and acute stimulants on brain functional connectivity hubs. Brain Res 1628:147–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konova AB, Moeller SJ, Tomasi D, Volkow ND, Goldstein RZ (2013): Effects of methylphenidate on resting‐state functional connectivity of the mesocorticolimbic dopamine pathways in cocaine addiction. JAMA Psychiatry 70:857–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystal JH, D'Souza DC, Mathalon D, Perry E, Belger A, Hoffman R (2003): NMDA receptor antagonist effects, cortical glutamatergic function, and schizophrenia: Toward a paradigm shift in medication development. Psychopharmacology (Berl) 169:215–233. [DOI] [PubMed] [Google Scholar]
- Kufahl PR, Li Z, Risinger RC, Rainey CJ, Wu G, Bloom AS, Li SJ (2005): Neural responses to acute cocaine administration in the human brain detected by fMRI. Neuroimage 28:904–914. [DOI] [PubMed] [Google Scholar]
- Kullmann S, Frank S, Heni M, Ketterer C, Veit R, Haring HU, Fritsche A, Preissl H (2013): Intranasal insulin modulates intrinsic reward and prefrontal circuitry of the human brain in lean women. Neuroendocrinology 97:176–182. [DOI] [PubMed] [Google Scholar]
- Kundu P, Brenowitz ND, Voon V, Worbe Y, Vertes PE, Inati SJ, Saad ZS, Bandettini PA, Bullmore ET (2013): Integrated strategy for improving functional connectivity mapping using multiecho fMRI. Proc Natl Acad Sci U S A 110:16187–16192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu P, Inati SJ, Evans JW, Luh WM, Bandettini PA (2012): Differentiating BOLD and non‐BOLD signals in fMRI time series using multi‐echo EPI. Neuroimage 60:1759–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahti AC, Holcomb HH, Medoff DR, Tamminga CA (1995): Ketamine activates psychosis and alters limbic blood flow in schizophrenia. Neuroreport 6:869–872. [DOI] [PubMed] [Google Scholar]
- Leonardi N, Van De Ville D (2015): On spurious and real fluctuations of dynamic functional connectivity during rest. Neuroimage 104:430–436. [DOI] [PubMed] [Google Scholar]
- Leppa M, Korvenoja A, Carlson S, Timonen P, Martinkauppi S, Ahonen J, Rosenberg PH, Aronen HJ, Kalso E (2006): Acute opioid effects on human brain as revealed by functional magnetic resonance imaging. Neuroimage 31:661–669. [DOI] [PubMed] [Google Scholar]
- Levin JM, Ross MH, Mendelson JH, Kaufman MJ, Lange N, Maas LC, Mello NK, Cohen BM, Renshaw PF (1998): Reduction in BOLD fMRI response to primary visual stimulation following alcohol ingestion. Psychiatry Res 82:135–146. [DOI] [PubMed] [Google Scholar]
- Li J, Ishiwari K, Conway MW, Francois J, Huxter J, Lowry JP, Schwarz AJ, Tricklebank M, Gilmour G (2014): Dissociable effects of antipsychotics on ketamine‐induced changes in regional oxygenation and inter‐regional coherence of low frequency oxygen fluctuations in the rat. Neuropsychopharmacology 39:1635–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Gilmore JH, Shen D, Styner M, Lin W, Zhu H (2013): Multiscale adaptive generalized estimating equations for longitudinal neuroimaging data. Neuroimage 72:91–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Zhu Y, Childress AR, Detre JA, Wang Z (2012): Relations between BOLD fMRI‐derived resting brain activity and cerebral blood flow. PLoS One 7:e44556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, Wang J, Yan C, Shu N, Xu K, Gong G, He Y (2012): Effects of different correlation metrics and preprocessing factors on small‐world brain functional networks: A resting‐state functional MRI study. PLoS One 7:e32766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, Zou Q, He Y, Yang Y (2013): Coupling of functional connectivity and regional cerebral blood flow reveals a physiological basis for network hubs of the human brain. Proc Natl Acad Sci U S A 110:1929–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licata SC, Lowen SB, Trksak GH, Maclean RR, Lukas SE (2011): Zolpidem reduces the blood oxygen level‐dependent signal during visual system stimulation. Prog Neuropsychopharmacol Biol Psychiatry 35:1645–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licata SC, Nickerson LD, Lowen SB, Trksak GH, Maclean RR, Lukas SE (2013): The hypnotic zolpidem increases the synchrony of BOLD signal fluctuations in widespread brain networks during a resting paradigm. Neuroimage 70:211–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, Wu WC, Wang J, Detre JA, Dinges DF, Rao H (2010): Imaging brain fatigue from sustained mental workload: An ASL perfusion study of the time‐on‐task effect. Neuroimage 49:3426–3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipp I, Murphy K, Caseras X, Wise RG (2015): Agreement and repeatability of vascular reactivity estimates based on a breath‐hold task and a resting state scan. Neuroimage 113:387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Yan C, Ren J, Yao L, Kiviniemi VJ, Zang Y (2010): Using coherence to measure regional homogeneity of resting‐state FMRI signal. Front Syst Neurosci 4:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Hebrank AC, Rodrigue KM, Kennedy KM, Park DC, Lu H (2013a): A comparison of physiologic modulators of fMRI signals. Hum Brain Mapp 34:2078–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu TT (2013): Neurovascular factors in resting‐state functional MRI. Neuroimage 80:339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Duyn JH (2013): Time‐varying functional network information extracted from brief instances of spontaneous brain activity. Proc Natl Acad Sci U S A 110:4392–4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Zhu XH, Zhang Y, Chen W (2011): Neural origin of spontaneous hemodynamic fluctuations in rats under burst‐suppression anesthesia condition. Cereb Cortex 21:374–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Zhu XH, Zhang Y, Chen W (2013b): The change of functional connectivity specificity in rats under various anesthesia levels and its neural origin. Brain Topogr 26:363–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logothetis NK (2008): What we can do and what we cannot do with fMRI. Nature 453:869–878. [DOI] [PubMed] [Google Scholar]
- Lohmann G, Margulies DS, Horstmann A, Pleger B, Lepsien J, Goldhahn D, Schloegl H, Stumvoll M, Villringer A, Turner R (2010): Eigenvector centrality mapping for analyzing connectivity patterns in fMRI data of the human brain. PLoS One 5:e10232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse C, Bassett DS, Lim KO, Carlson JM (2014): Resolving anatomical and functional structure in human brain organization: Identifying mesoscale organization in weighted network representations. PLoS Comput Biol 10:e1003712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Stein EA (2014): Resting state functional connectivity: Its physiological basis and application in neuropharmacology. Neuropharmacology 84:79–89. [DOI] [PubMed] [Google Scholar]
- MacIntosh BJ, Pattinson KT, Gallichan D, Ahmad I, Miller KL, Feinberg DA, Wise RG, Jezzard P (2008): Measuring the effects of remifentanil on cerebral blood flow and arterial arrival time using 3D GRASE MRI with pulsed arterial spin labelling. J Cereb Blood Flow Metab 28:1514–1522. [DOI] [PubMed] [Google Scholar]
- Madjar C, Gauthier CJ, Bellec P, Birn RM, Brooks JC, Hoge RD (2012): Task‐related BOLD responses and resting‐state functional connectivity during physiological clamping of end‐tidal CO(2). Neuroimage 61:41–49. [DOI] [PubMed] [Google Scholar]
- Magnuson ME, Thompson GJ, Pan WJ, Keilholz SD (2014): Time‐dependent effects of isoflurane and dexmedetomidine on functional connectivity, spectral characteristics, and spatial distribution of spontaneous BOLD fluctuations. NMR Biomed 27:291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchitelli R, Minati L, Marizzoni M, Bosch B, Bartres‐Faz D, Muller BW, Wiltfang J, Fiedler U, Roccatagliata L, Picco A, Nobili F, Blin O, Bombois S, Lopes R, Bordet R, Sein J, Ranjeva JP, Didic M, Gros‐Dagnac H, Payoux P, Zoccatelli G, Alessandrini F, Beltramello A, Bargallo N, Ferretti A, Caulo M, Aiello M, Cavaliere C, Soricelli A, Parnetti L, Tarducci R, Floridi P, Tsolaki M, Constantinidis M, Drevelegas A, Rossini PM, Marra C, Schonknecht P, Hensch T, Hoffmann KT, Kuijer JP, Visser PJ, Barkhof F, Frisoni GB, Jovicich J (2016): Test‐retest reliability of the default mode network in a multi‐centric fMRI study of healthy elderly: Effects of data‐driven physiological noise correction techniques. Hum Brain Mapp 37:2114–2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquand A, Howard M, Brammer M, Chu C, Coen S, Mourao‐Miranda J (2010): Quantitative prediction of subjective pain intensity from whole‐brain fMRI data using Gaussian processes. Neuroimage 49:2178–2189. [DOI] [PubMed] [Google Scholar]
- Marrelec G, Bellec P, Krainik A, Duffau H, Pelegrini‐Issac M, Lehericy S, Benali H, Doyon J (2008): Regions, systems, and the brain: Hierarchical measures of functional integration in fMRI. Med Image Anal 12:484–496. [DOI] [PubMed] [Google Scholar]
- Marxen M, Gan G, Schwarz D, Mennigen E, Pilhatsch M, Zimmermann US, Guenther M, Smolka MN (2014): Acute effects of alcohol on brain perfusion monitored with arterial spin labeling magnetic resonance imaging in young adults. J Cereb Blood Flow Metab 34:472–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeown MJ, Makeig S, Brown GG, Jung TP, Kindermann SS, Bell AJ, Sejnowski TJ (1998): Analysis of fMRI data by blind separation into independent spatial components. Hum Brain Mapp 6:160–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKie S, Del‐Ben C, Elliott R, Williams S, del Vai N, Anderson I, Deakin JF (2005): Neuronal effects of acute citalopram detected by pharmacoMRI. Psychopharmacology (Berl) 180:680–686. [DOI] [PubMed] [Google Scholar]
- McKie S, Richardson P, Elliott R, Vollm BA, Dolan MC, Williams SR, Anderson IM, Deakin JF (2011): Mirtazapine antagonises the subjective, hormonal and neuronal effects of m‐chlorophenylpiperazine (mCPP) infusion: A pharmacological‐challenge fMRI (phMRI) study. Neuroimage 58:497–507. [DOI] [PubMed] [Google Scholar]
- Mechling AE, Hubner NS, Lee HL, Hennig J, von Elverfeldt D, Harsan LA (2014): Fine‐grained mapping of mouse brain functional connectivity with resting‐state fMRI. Neuroimage 96:203–215. [DOI] [PubMed] [Google Scholar]
- Meissner K, Bingel U, Colloca L, Wager TD, Watson A, Flaten MA (2011): The placebo effect: Advances from different methodological approaches. J Neurosci 31:16117–16124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messe A, Rudrauf D, Benali H, Marrelec G (2014): Relating structure and function in the human brain: Relative contributions of anatomy, stationary dynamics, and non‐stationarities. PLoS Comput Biol 10:e1003530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RL, Yaesoubi M, Calhoun VD (2014): Higher dimensional analysis shows reduced dynamism of time‐varying network connectivity in schizophrenia patients. Conf Proc IEEE Eng Med Biol Soc 2014:3837–3840. [DOI] [PubMed] [Google Scholar]
- Mitsis GD, Governo RJ, Rogers R, Pattinson KT (2009): The effect of remifentanil on respiratory variability, evaluated with dynamic modeling. J Appl Physiol (1985) 106:1038–1049. [DOI] [PubMed] [Google Scholar]
- Monti MM, Lutkenhoff ES, Rubinov M, Boveroux P, Vanhaudenhuyse A, Gosseries O, Bruno MA, Noirhomme Q, Boly M, Laureys S (2013): Dynamic change of global and local information processing in propofol‐induced loss and recovery of consciousness. PLoS Comput Biol 9:e1003271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller S, Costa A, Keeser D, Pogarell O, Berman A, Coates U, Reiser MF, Riedel M, Moller HJ, Ettinger U, Meindl T (2014): The effects of methylphenidate on whole brain intrinsic functional connectivity. Hum Brain Mapp 35:5379–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumford JA (2012): A power calculation guide for fMRI studies. Soc Cogn Affect Neurosci 7:738–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy K, Birn RM, Handwerker DA, Jones TB, Bandettini PA (2009): The impact of global signal regression on resting state correlations: Are anti‐correlated networks introduced?. Neuroimage 44:893–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutsaerts HJ, Steketee RM, Heijtel DF, Kuijer JP, van Osch MJ, Majoie CB, Smits M, Nederveen AJ (2014): Inter‐vendor reproducibility of pseudo‐continuous arterial spin labeling at 3 Tesla. PLoS One 9:e104108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutsaerts HJ (2015): Reproducibility of pharmacological ASL using sequences from different vendors: Implications for multicenter drug studies. MAGMA 28:427–436. [DOI] [PubMed] [Google Scholar]
- Nasrallah FA, Lew SK, Low AS, Chuang KH (2014a): Neural correlate of resting‐state functional connectivity under alpha2 adrenergic receptor agonist, medetomidine. Neuroimage 84:27–34. [DOI] [PubMed] [Google Scholar]
- Nasrallah FA, Low SM, Lew SK, Chen K, Chuang KH (2014b): Pharmacological insight into neurotransmission origins of resting‐state functional connectivity: Alpha2‐adrenergic agonist vs antagonist. Neuroimage 103:364–373. [DOI] [PubMed] [Google Scholar]
- Nasrallah FA, Tan J, Chuang KH (2012): Pharmacological modulation of functional connectivity: Alpha2‐adrenergic receptor agonist alters synchrony but not neural activation. Neuroimage 60:436–446. [DOI] [PubMed] [Google Scholar]
- Nichols TE, Holmes AP (2002): Nonparametric permutation tests for functional neuroimaging: A primer with examples. Hum Brain Mapp 15:1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niesters M, Khalili‐Mahani N, Martini C, Aarts L, van Gerven J, van Buchem MA, Dahan A, Rombouts S (2012): Effect of subanesthetic ketamine on intrinsic functional brain connectivity: A placebo‐controlled functional magnetic resonance imaging study in healthy male volunteers. Anesthesiology 117:868–877. [DOI] [PubMed] [Google Scholar]
- Niesters M, Sitsen E, Oudejans L, Vuyk J, Aarts LP, Rombouts SA, de Rover M, Khalili‐Mahani N, Dahan A (2014): Effect of deafferentation from spinal anesthesia on pain sensitivity and resting‐state functional brain connectivity in healthy male volunteers. Brain Connect 4:404–416. [DOI] [PubMed] [Google Scholar]
- Onoe H, Inoue O, Suzuki K, Tsukada H, Itoh T, Mataga N, Watanabe Y (1994): Ketamine increases the striatal N‐[11C]methylspiperone binding in vivo: Positron emission tomography study using conscious rhesus monkey. Brain Res 663:191–198. [DOI] [PubMed] [Google Scholar]
- Orru G, Pettersson‐Yeo W, Marquand AF, Sartori G, Mechelli A (2012): Using Support Vector Machine to identify imaging biomarkers of neurological and psychiatric disease: A critical review. Neurosci Biobehav Rev 36:1140–1152. [DOI] [PubMed] [Google Scholar]
- Otte WM, van der Marel K, Braun KP, Dijkhuizen RM (2014): Effects of transient unilateral functional brain disruption on global neural network status in rats: A methods paper. Front Syst Neurosci 8:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paloyelis Y, Doyle OM, Zelaya FO, Maltezos S, Williams SC, Fotopoulou A, Howard MA (2016): A spatiotemporal profile of in vivo cerebral blood flow changes following intranasal oxytocin in humans. Biol Psychiatry 79:693–705. [DOI] [PubMed] [Google Scholar]
- Pardridge WM (2012): Drug transport across the blood‐brain barrier. J Cereb Blood Flow Metab 32:1959–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passow S, Specht K, Adamsen TC, Biermann M, Brekke N, Craven AR, Ersland L, Gruner R, Kleven‐Madsen N, Kvernenes OH, Schwarzlmuller T, Olesen RA, Hugdahl K (2015): Default‐mode network functional connectivity is closely related to metabolic activity. Hum Brain Mapp 36:2027–2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patriat R, Molloy EK, Meier TB, Kirk GR, Nair VA, Meyerand ME, Prabhakaran V, Birn RM (2013): The effect of resting condition on resting‐state fMRI reliability and consistency: A comparison between resting with eyes open, closed, and fixated. Neuroimage 78:463–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattinson KT, Governo RJ, MacIntosh BJ, Russell EC, Corfield DR, Tracey I, Wise RG (2009): Opioids depress cortical centers responsible for the volitional control of respiration. J Neurosci 29:8177–8186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltier SJ, Kerssens C, Hamann SB, Sebel PS, Byas‐Smith M, Hu X (2005): Functional connectivity changes with concentration of sevoflurane anesthesia. Neuroreport 16:285–288. [DOI] [PubMed] [Google Scholar]
- Peng T, Niazy R, Payne SJ, Wise RG (2013): The effects of respiratory CO2 fluctuations in the resting‐state BOLD signal differ between eyes open and eyes closed. Magn Reson Imaging 31:336–345. [DOI] [PubMed] [Google Scholar]
- Perthen JE, Lansing AE, Liau J, Liu TT, Buxton RB (2008): Caffeine‐induced uncoupling of cerebral blood flow and oxygen metabolism: A calibrated BOLD fMRI study. Neuroimage 40:237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen RC, Aisen PS, Beckett LA, Donohue MC, Gamst AC, Harvey DJ, Jack CR, Jr. , Jagust WJ, Shaw LM, Toga AW, Trojanowski JQ, Weiner MW (2010): Alzheimers Disease Neuroimaging Initiative (ADNI): clinical characterization. Neurology 74:201–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posse S, Ackley E, Mutihac R, Zhang T, Hummatov R, Akhtari M, Chohan M, Fisch B, Yonas H (2013): High‐speed real‐time resting‐state FMRI using multi‐slab echo‐volumar imaging. Front Hum Neurosci 7:479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power JD, Barnes KA, Snyder AZ, Schlaggar BL, Petersen SE (2012): Spurious but systematic correlations in functional connectivity MRI networks arise from subject motion. Neuroimage 59:2142–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power JD, Cohen AL, Nelson SM, Wig GS, Barnes KA, Church JA, Vogel AC, Laumann TO, Miezin FM, Schlaggar BL, Petersen SE (2011): Functional network organization of the human brain. Neuron 72:665–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power JD, Mitra A, Laumann TO, Snyder AZ, Schlaggar BL, Petersen SE (2014): Methods to detect, characterize, and remove motion artifact in resting state fMRI. Neuroimage 84:320–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power JD, Schlaggar BL, Petersen SE (2015): Recent progress and outstanding issues in motion correction in resting state fMRI. Neuroimage 105:536–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruessner JC, Dedovic K, Khalili‐Mahani N, Engert V, Pruessner M, Buss C, Renwick R, Dagher A, Meaney MJ, Lupien S (2008): Deactivation of the limbic system during acute psychosocial stress: Evidence from positron emission tomography and functional magnetic resonance imaging studies. Biol Psychiatry 63:234–240. [DOI] [PubMed] [Google Scholar]
- Pruim RH, Mennes M, van Rooij D, Llera A, Buitelaar JK, Beckmann CF (2015): ICA‐AROMA: A robust ICA‐based strategy for removing motion artifacts from fMRI data. Neuroimage 112:267–277. [DOI] [PubMed] [Google Scholar]
- Qing Z, Dong Z, Li S, Zang Y, Liu D (2015): Global signal regression has complex effects on regional homogeneity of resting state fMRI signal. Magn Reson Imaging 33:1306–1313. [DOI] [PubMed] [Google Scholar]
- Qiu M, Ramani R, Swetye M, Constable RT (2008): Spatial nonuniformity of the resting CBF and BOLD responses to sevoflurane: In vivo study of normal human subjects with magnetic resonance imaging. Hum Brain Mapp 29:1390–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rack‐Gomer AL, Liau J, Liu TT (2009): Caffeine reduces resting‐state BOLD functional connectivity in the motor cortex. Neuroimage 46:56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rack‐Gomer AL, Liu TT (2012): Caffeine increases the temporal variability of resting‐state BOLD connectivity in the motor cortex. Neuroimage 59:2994–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raichle ME, MacLeod AM, Snyder AZ, Powers WJ, Gusnard DA, Shulman GL (2001): A default mode of brain function. Proc Natl Acad Sci U S A 98:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaekers JG, Evers EA, Theunissen EL, Kuypers KP, Goulas A, Stiers P (2013): Methylphenidate reduces functional connectivity of nucleus accumbens in brain reward circuit. Psychopharmacology (Berl) 229:219–226. [DOI] [PubMed] [Google Scholar]
- Rickenbacher E, Greve DN, Azma S, Pfeuffer J, Marinkovic K (2011): Effects of alcohol intoxication and gender on cerebral perfusion: an arterial spin labeling study. Alcohol 45:725–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl V, Bienkowska K, Strobel C, Tahmasian M, Grimmer T, Forster S, Friston KJ, Sorg C, Drzezga A (2014): Local activity determines functional connectivity in the resting human brain: A simultaneous FDG‐PET/fMRI study. J Neurosci 34:6260–6266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez‐Rojas R, Machado C, Alvarez L, Carballo M, Estevez M, Perez‐Nellar J, Pavon N, Chinchilla M, Carrick FR, DeFina P (2013): Zolpidem induces paradoxical metabolic and vascular changes in a patient with PVS. Brain Inj 27:1320–1329. [DOI] [PubMed] [Google Scholar]
- Roseman L, Leech R, Feilding A, Nutt DJ, Carhart‐Harris RL (2014): The effects of psilocybin and MDMA on between‐network resting state functional connectivity in healthy volunteers. Front Hum Neurosci 8:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saad ZS, Gotts SJ, Murphy K, Chen G, Jo HJ, Martin A, Cox RW (2012): Trouble at rest: How correlation patterns and group differences become distorted after global signal regression. Brain Connect 2:25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salimi‐Khorshidi G, Douaud G, Beckmann CF, Glasser MF, Griffanti L, Smith SM (2014): Automatic denoising of functional MRI data: Combining independent component analysis and hierarchical fusion of classifiers. Neuroimage 90:449–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salimi‐Khorshidi G, Smith SM, Nichols TE (2011): Adjusting the effect of nonstationarity in cluster‐based and TFCE inference. Neuroimage 54:2006–2019. [DOI] [PubMed] [Google Scholar]
- Sanganahalli BG, Herman P, Hyder F, Kannurpatti SS (2013): Mitochondrial functional state impacts spontaneous neocortical activity and resting state FMRI. PLoS One 8:e63317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satterthwaite TD, Elliott MA, Gerraty RT, Ruparel K, Loughead J, Calkins ME, Eickhoff SB, Hakonarson H, Gur RC, Gur RE, Wolf DH (2013): An improved framework for confound regression and filtering for control of motion artifact in the preprocessing of resting‐state functional connectivity data. Neuroimage 64:240–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheidegger M, Walter M, Lehmann M, Metzger C, Grimm S, Boeker H, Boesiger P, Henning A, Seifritz E (2012): Ketamine decreases resting state functional network connectivity in healthy subjects: Implications for antidepressant drug action. PLoS One 7:e44799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlogl H, Kabisch S, Horstmann A, Lohmann G, Muller K, Lepsien J, Busse‐Voigt F, Kratzsch J, Pleger B, Villringer A, Stumvoll M (2013): Exenatide‐induced reduction in energy intake is associated with increase in hypothalamic connectivity. Diabetes Care 36:1933–1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmaal L, Goudriaan AE, Joos L, Kruse AM, Dom G, van den Brink W, Veltman DJ (2013): Modafinil modulates resting‐state functional network connectivity and cognitive control in alcohol‐dependent patients. Biol Psychiatry 73:789–795. [DOI] [PubMed] [Google Scholar]
- Schmaal L, Veltman DJ, van Erp TG, Samann PG, Frodl T, Jahanshad N, Loehrer E, Tiemeier H, Hofman A, Niessen WJ, Vernooij MW, Ikram MA, Wittfeld K, Grabe HJ, Block A, Hegenscheid K, Volzke H, Hoehn D, Czisch M, Lagopoulos J, Hatton SN, Hickie IB, Goya‐Maldonado R, Kramer B, Gruber O, Couvy‐Duchesne B, Renteria ME, Strike LT, Mills NT, de Zubicaray GI, McMahon KL, Medland SE, Martin NG, Gillespie NA, Wright MJ, Hall GB, MacQueen GM, Frey EM, Carballedo A, van Velzen LS, van Tol MJ, van der Wee NJ, Veer IM, Walter H, Schnell K, Schramm E, Normann C, Schoepf D, Konrad C, Zurowski B, Nickson T, McIntosh AM, Papmeyer M, Whalley HC, Sussmann JE, Godlewska BR, Cowen PJ, Fischer FH, Rose M, Penninx BW, Thompson PM, Hibar DP (2016): Subcortical brain alterations in major depressive disorder: Findings from the ENIGMA Major Depressive Disorder working group. Mol Psychiatry. 21:806–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid S, Ghariq E, Teeuwisse WM, Webb A, van Osch MJ (2014): Acceleration‐selective arterial spin labeling. Magn Reson Med 71:191–199. [DOI] [PubMed] [Google Scholar]
- Schmidt A, Denier N, Magon S, Radue EW, Huber CG, Riecher‐Rossler A, Wiesbeck GA, Lang UE, Borgwardt S, Walter M (2015): Increased functional connectivity in the resting‐state basal ganglia network after acute heroin substitution. Transl Psychiatry 5:e533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholvinck ML, Maier A, Ye FQ, Duyn JH, Leopold DA (2010): Neural basis of global resting‐state fMRI activity. Proc Natl Acad Sci U S A 107:10238–10243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroter MS, Spoormaker VI, Schorer A, Wohlschlager A, Czisch M, Kochs EF, Zimmer C, Hemmer B, Schneider G, Jordan D, Ilg R (2012): Spatiotemporal reconfiguration of large‐scale brain functional networks during propofol‐induced loss of consciousness. J Neurosci 32:12832–12840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrouff J, Perlbarg V, Boly M, Marrelec G, Boveroux P, Vanhaudenhuyse A, Bruno MA, Laureys S, Phillips C, Pelegrini‐Issac M, Maquet P, Benali H (2011): Brain functional integration decreases during propofol‐induced loss of consciousness. Neuroimage 57:198–205. [DOI] [PubMed] [Google Scholar]
- Schultz AP, Chhatwal JP, Huijbers W, Hedden T, van Dijk KR, McLaren DG, Ward AM, Wigman S, Sperling RA (2014): Template based rotation: A method for functional connectivity analysis with a priori templates. Neuroimage 102 Pt 2:620–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutz L, Lobsien D, Fritzsch D, Tiepolt S, Werner P, Schroeter ML, Berrouschot J, Saur D, Hesse S, Jochimsen T, Rullmann M, Sattler B, Patt M, Gertz HJ, Villringer A, Classen J, Hoffmann KT, Sabri O, Barthel H (2016): Feasibility and acceptance of simultaneous amyloid PET/MRI. Eur J Nucl Med Mol Imaging 43:2236–2243. [DOI] [PubMed] [Google Scholar]
- Schwarz AJ, Becerra L, Upadhyay J, Anderson J, Baumgartner R, Coimbra A, Evelhoch J, Hargreaves R, Robertson B, Iyengar S, Tauscher J, Bleakman D, Borsook D (2011b): A procedural framework for good imaging practice in pharmacological fMRI studies applied to drug development #1: processes and requirements. Drug Discov Today 16:583–593. [DOI] [PubMed] [Google Scholar]
- Schwarz AJ, Becerra L, Upadhyay J, Anderson J, Baumgartner R, Coimbra A, Evelhoch J, Hargreaves R, Robertson B, Iyengar S, Tauscher J, Bleakman D, Borsook D (2011a): A procedural framework for good imaging practice in pharmacological fMRI studies applied to drug development #2: protocol optimization and best practices. Drug Discov Today 16:671–682. [DOI] [PubMed] [Google Scholar]
- Schwarz AJ, Danckaert A, Reese T, Gozzi A, Paxinos G, Watson C, Merlo‐Pich EV, Bifone A (2006): A stereotaxic MRI template set for the rat brain with tissue class distribution maps and co‐registered anatomical atlas: Application to pharmacological MRI. Neuroimage 32:538–550. [DOI] [PubMed] [Google Scholar]
- Schwarz AJ, Gozzi A, Reese T, Bifone A (2007a): Functional connectivity in the pharmacologically activated brain: Resolving networks of correlated responses to d‐amphetamine. Magn Reson Med 57:704–713. [DOI] [PubMed] [Google Scholar]
- Schwarz AJ, Gozzi A, Reese T, Bifone A (2007b): In vivo mapping of functional connectivity in neurotransmitter systems using pharmacological MRI. Neuroimage 34:1627–1636. [DOI] [PubMed] [Google Scholar]
- Schwarz AJ, Gozzi A, Reese T, Heidbreder CA, Bifone A (2007c): Pharmacological modulation of functional connectivity: The correlation structure underlying the phMRI response to d‐amphetamine modified by selective dopamine D3 receptor antagonist SB277011A. Magn Reson Imaging 25:811–820. [DOI] [PubMed] [Google Scholar]
- Schwarz AJ, McGonigle J (2011): Negative edges and soft thresholding in complex network analysis of resting state functional connectivity data. Neuroimage 55:1132–1146. [DOI] [PubMed] [Google Scholar]
- Schwarz AJ, Reese T, Gozzi A, Bifone A (2003): Functional MRI using intravascular contrast agents: Detrending of the relative cerebrovascular (rCBV) time course. Magn Reson Imaging 21:1191–1200. [DOI] [PubMed] [Google Scholar]
- Schwarz AJ, Whitcher B, Gozzi A, Reese T, Bifone A (2007d): Study‐level wavelet cluster analysis and data‐driven signal models in pharmacological MRI. J Neurosci Methods 159:346–360. [DOI] [PubMed] [Google Scholar]
- Sekar S, Jonckers E, Verhoye M, Willems R, Veraart J, Van Audekerke J, Couto J, Giugliano M, Wuyts K, Dedeurwaerdere S, Sijbers J, Mackie C, Ver Donck L, Steckler T, Van der Linden A (2013): Subchronic memantine induced concurrent functional disconnectivity and altered ultra‐structural tissue integrity in the rodent brain: revealed by multimodal MRI. Psychopharmacology 227:479–491. [DOI] [PubMed] [Google Scholar]
- Shehzad Z, Kelly AM, Reiss PT, Gee DG, Gotimer K, Uddin LQ, Lee SH, Margulies DS, Roy AK, Biswal BB, Petkova E, Castellanos FX, Milham MP (2009): The resting brain: Unconstrained yet reliable. Cereb Cortex 19:2209–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherif T, Rioux P, Rousseau ME, Kassis N, Beck N, Adalat R, Das S, Glatard T, Evans AC (2014): CBRAIN: A web‐based, distributed computing platform for collaborative neuroimaging research. Front Neuroinform 8:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin HW, Jewells V, Sheikh A, Zhang J, Zhu H, An H, Gao W, Shen D, Hadar E, Lin W (2015): Initial experience in hybrid PET‐MRI for evaluation of refractory focal onset epilepsy. Seizure 31:1–4. [DOI] [PubMed] [Google Scholar]
- Shulman RG, Rothman DL (1998): Interpreting functional imaging studies in terms of neurotransmitter cycling. Proc Natl Acad Sci U S A 95:11993–11998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SM, Fox PT, Miller KL, Glahn DC, Fox PM, Mackay CE, Filippini N, Watkins KE, Toro R, Laird AR, Beckmann CF (2009): Correspondence of the brains functional architecture during activation and rest. Proc Natl Acad Sci U S A 106:13040–13045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SM, Miller KL, Moeller S, Xu J, Auerbach EJ, Woolrich MW, Beckmann CF, Jenkinson M, Andersson J, Glasser MF, Van Essen DC, Feinberg DA, Yacoub ES, Ugurbil K (2012): Temporally‐independent functional modes of spontaneous brain activity. Proc Natl Acad Sci U S A 109:3131–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SM, Nichols TE (2009): Threshold‐free cluster enhancement: Addressing problems of smoothing, threshold dependence and localisation in cluster inference. Neuroimage 44:83–98. [DOI] [PubMed] [Google Scholar]
- Smith SM, Vidaurre D, Beckmann CF, Glasser MF, Jenkinson M, Miller KL, Nichols TE, Robinson EC, Salimi‐Khorshidi G, Woolrich MW, Barch DM, Ugurbil K, Van Essen DC (2013): Functional connectomics from resting‐state fMRI. Trends Cogn Sci 17:666–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soddu A, Gomez F, Heine L, Di Perri C, Bahri MA, Voss HU, Bruno MA, Vanhaudenhuyse A, Phillips C, Demertzi A, Chatelle C, Schrouff J, Thibaut A, Charland‐Verville V, Noirhomme Q, Salmon E, Tshibanda JF, Schiff ND, Laureys S (2016): Correlation between resting state fMRI total neuronal activity and PET metabolism in healthy controls and patients with disorders of consciousness. Brain Behav 6:e00424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spagnolli F, Cerini R, Cardobi N, Barillari M, Manganotti P, Storti S, Mucelli RP (2013): Brain modifications after acute alcohol consumption analyzed by resting state fMRI. J Magn Reson Imaging 31:1325–1330. [DOI] [PubMed] [Google Scholar]
- Sripada CS, Kessler D, Welsh R, Angstadt M, Liberzon I, Phan KL, Scott C (2013a): Distributed effects of methylphenidate on the network structure of the resting brain: A connectomic pattern classification analysis. Neuroimage 81:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sripada CS, Phan KL, Labuschagne I, Welsh R, Nathan PJ, Wood AG (2013b): Oxytocin enhances resting‐state connectivity between amygdala and medial frontal cortex. Int J Neuropsychopharmacol 16:255–260. [DOI] [PubMed] [Google Scholar]
- Sripada RK, Welsh RC, Marx CE, Liberzon I (2014): The neurosteroids allopregnanolone and dehydroepiandrosterone modulate resting‐state amygdala connectivity. Hum Brain Mapp 35:3249–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis EA, Adapa RM, Absalom AR, Menon DK (2010): Changes in resting neural connectivity during propofol sedation. PLoS One 5:e14224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein EA, Pankiewicz J, Harsch HH, Cho JK, Fuller SA, Hoffmann RG, Hawkins M, Rao SM, Bandettini PA, Bloom AS (1998): Nicotine‐induced limbic cortical activation in the human brain: A functional MRI study. Am J Psychiatry 155:1009–1015. [DOI] [PubMed] [Google Scholar]
- Strang NM, Claus ED, Ramchandani VA, Graff‐Guerrero A, Boileau I, Hendershot CS (2015): Dose‐dependent effects of intravenous alcohol administration on cerebral blood flow in young adults. Psychopharmacology (Berl) 232:733–744. [DOI] [PubMed] [Google Scholar]
- Su Y, Vlassenko AG, Couture LE, Benzinger TL, Snyder AZ, Derdeyn CP, Raichle ME (2016): Quantitative hemodynamic PET imaging using image‐derived arterial input function and a PET/MR hybrid scanner. J Cereb Blood Flow Metab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suckling J, Wink AM, Bernard FA, Barnes A, Bullmore E (2008): Endogenous multifractal brain dynamics are modulated by age, cholinergic blockade and cognitive performance. J Neurosci Methods 174:292–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui J, Huster R, Yu Q, Segall JM, Calhoun VD (2014): Function‐structure associations of the brain: Evidence from multimodal connectivity and covariance studies. Neuroimage 102 Pt 1:11–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tak S, Polimeni JR, Wang DJ, Yan L, Chen JJ (2015): Associations of resting‐state fmri functional connectivity with flow‐bold coupling and regional vasculature. Brain Connect 5:137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tak S, Wang DJ, Polimeni JR, Yan L, Chen JJ (2014): Dynamic and static contributions of the cerebrovasculature to the resting‐state BOLD signal. Neuroimage 84:672–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tal O, Diwakar M, Wong CW, Olafsson V, Lee R, Huang MX, Liu TT (2013): Caffeine‐induced global reductions in resting‐state bold connectivity reflect widespread decreases in meg connectivity. Front Hum Neurosci 7:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe J, Nyberg E, Martin LF, Martin J, Cordes D, Kronberg E, Tregellas JR (2011): Nicotine effects on default mode network during resting state. Psychopharmacology (Berl) 216:287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teeuwisse WM, Schmid S, Ghariq E, Veer IM, van Osch MJ (2014): Time‐encoded pseudocontinuous arterial spin labeling: Basic properties and timing strategies for human applications. Magn Reson Med 72:1712–1722. [DOI] [PubMed] [Google Scholar]
- Thyreau B, Schwartz Y, Thirion B, Frouin V, Loth E, Vollstadt‐Klein S, Paus T, Artiges E, Conrod PJ, Schumann G, Whelan R, Poline JB, Consortium I (2012): Very large fMRI study using the IMAGEN database: Sensitivity‐specificity and population effect modeling in relation to the underlying anatomy. Neuroimage 61:295–303. [DOI] [PubMed] [Google Scholar]
- Tian L, Kong Y, Ren J, Varoquaux G, Zang Y, Smith SM (2013): Spatial vs. temporal features in ica of resting‐state fmri ‐ a quantitative and qualitative investigation in the context of response inhibition. PLoS One 8:e66572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohka J, Foerde K, Aron AR, Tom SM, Toga AW, Poldrack RA (2008): Automatic independent component labeling for artifact removal in fMRI. Neuroimage 39:1227–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolentino NJ, Wierenga CE, Hall S, Tapert SF, Paulus MP, Liu TT, Smith TL, Schuckit MA (2011): Alcohol effects on cerebral blood flow in subjects with low and high responses to alcohol. Alcohol Clin Exp Res 35:1034–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasi D, Shokri‐Kojori E, Volkow ND (2016a): Temporal changes in local functional connectivity density reflect the temporal variability of the amplitude of low frequency fluctuations in gray matter. PLoS One 11:e0154407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasi D, Wang GJ, Volkow ND (2013): Energetic cost of brain functional connectivity. Proc Natl Acad Sci U S A 110:13642–13647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasi DG, Shokri‐Kojori E, Volkow ND (2016b): Temporal evolution of brain functional connectivity metrics: Could 7 min of rest be enough?. Cereb Cortex. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toussaint PJ, Perlbarg V, Bellec P, Desarnaud S, Lacomblez L, Doyon J, Habert MO, Benali H, Benali, Alzheimer's Disease Neuroimaging Initiative (2012): Resting state FDG‐PET functional connectivity as an early biomarker of Alzheimers disease using conjoint univariate and independent component analyses. Neuroimage 63:936–946. [DOI] [PubMed] [Google Scholar]
- Upadhyay J, Anderson J, Baumgartner R, Coimbra A, Schwarz AJ, Pendse G, Wallin D, Nutile L, Bishop J, George E, Elman I, Sunkaraneni S, Maier G, Iyengar S, Evelhoch JL, Bleakman D, Hargreaves R, Becerra L, Borsook D (2012): Modulation of CNS pain circuitry by intravenous and sublingual doses of buprenorphine. Neuroimage 59:3762–3773. [DOI] [PubMed] [Google Scholar]
- Upadhyay J, Anderson J, Schwarz AJ, Coimbra A, Baumgartner R, Pendse G, George E, Nutile L, Wallin D, Bishop J, Neni S, Maier G, Iyengar S, Evelhoch JL, Bleakman D, Hargreaves R, Becerra L, Borsook D (2011): Imaging drugs with and without clinical analgesic efficacy. Neuropsychopharmacology 36:2659–2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dijk KR, Sabuncu MR, Buckner RL (2012): The influence of head motion on intrinsic functional connectivity MRI. Neuroimage 59:431–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hell HH, Bossong MG, Jager G, Kristo G, van Osch MJ, Zelaya F, Kahn RS, Ramsey NF (2011): Evidence for involvement of the insula in the psychotropic effects of THC in humans: A double‐blind, randomized pharmacological MRI study. Int J Neuropsychopharmacol 14:1377–1388. [DOI] [PubMed] [Google Scholar]
- Vazquez AL, Murphy MC, Kim SG (2014): Neuronal and physiological correlation to hemodynamic resting‐state fluctuations in health and disease. Brain Connect 4:727–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal B, Villien M, Le Bars D, Fieux S, Newman‐Tancredi A, Costes N, Zimmer L (2016): Exploration of the concept of biased agonism: A serotonin 5‐HT1A PET‐MR receptor stud. In: 5th Conference on PET/MR and SPECT/MR. Cologne, Germany. p. 110.
- Vidorreta M, Wang Z, Rodriguez I, Pastor MA, Detre JA, Fernandez‐Seara MA (2013): Comparison of 2D and 3D single‐shot ASL perfusion fMRI sequences. Neuroimage 66:662–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigneau‐Roy N, Bernier M, Descoteaux M, Whittingstall K (2014): Regional variations in vascular density correlate with resting‐state and task‐evoked blood oxygen level‐dependent signal amplitude. Hum Brain Mapp 35:1906–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Fowler JS, Logan J, Gatley SJ, Dewey SL, MacGregor RR, Schlyer DJ, Pappas N, King P, wang GJ (1995): Carbon‐11‐cocaine binding compared at subpharmacological and pharmacological doses: A PET study. J Nucl Med 36:1289–1297. [PubMed] [Google Scholar]
- Volkow ND, Gillespie H, Mullani N, Tancredi L, Grant C, Ivanovic M, Hollister L (1991): Cerebellar metabolic activation by delta‐9‐tetrahydro‐cannabinol in human brain: A study with positron emission tomography and 18F‐2‐fluoro‐2‐deoxyglucose. Psychiatry Res 40:69–78. [DOI] [PubMed] [Google Scholar]
- Vollenweider FX, Leenders KL, Scharfetter C, Antonini A, Maguire P, Missimer J, Angst J (1997): Metabolic hyperfrontality and psychopathology in the ketamine model of psychosis using positron emission tomography (PET) and [18F]fluorodeoxyglucose (FDG). Eur Neuropsychopharmacol 7:9–24. [DOI] [PubMed] [Google Scholar]
- Vytlacil J, Kayser A, Miyakawa A, D'Esposito M (2014): An approach for identifying brainstem dopaminergic pathways using resting state functional MRI. PLoS One 9:e87109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wager TD, Atlas LY, Lindquist MA, Roy M, Woo CW, Kross E (2013): An fMRI‐based neurologic signature of physical pain. N Engl J Med 368:1388–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wager TD, Scott DJ, Zubieta JK (2007): Placebo effects on human mu‐opioid activity during pain. Proc Natl Acad Sci U S A 104:11056–11061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner KJ, Sprenger T, Kochs EF, Tolle TR, Valet M, Willoch F (2007): Imaging human cerebral pain modulation by dose‐dependent opioid analgesia: A positron emission tomography activation study using remifentanil. Anesthesiology 106:548–556. [DOI] [PubMed] [Google Scholar]
- Wagner KJ, Willoch F, Kochs EF, Siessmeier T, Tolle TR, Schwaiger M, Bartenstein P (2001): Dose‐dependent regional cerebral blood flow changes during remifentanil infusion in humans: A positron emission tomography study. Anesthesiology 94:732–739. [DOI] [PubMed] [Google Scholar]
- Wang Y, Li TQ (2015): Dimensionality of ICA in resting‐state fMRI investigated by feature optimized classification of independent components with SVM. Front Hum Neurosci 9:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Wang J, Zhang H, McHugh R, Sun X, Li K, Yang QX (2015): Interhemispheric functional and structural disconnection in alzheimers disease: A combined resting‐state fmri and dti study. PLoS One 10:e0126310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Z, Alcauter S, Jin K, Peng ZW, Gao W (2013): Graph theoretical analysis of sedations effect on whole brain functional system in school‐aged children. Brain Connect 3:177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissenbacher A, Kasess C, Gerstl F, Lanzenberger R, Moser E, Windischberger C (2009): Correlations and anticorrelations in resting‐state functional connectivity MRI: A quantitative comparison of preprocessing strategies. Neuroimage 47:1408–1416. [DOI] [PubMed] [Google Scholar]
- Welton T, Kent DA, Auer DP, Dineen RA (2015): Reproducibility of graph‐theoretic brain network metrics: A systematic review. Brain Connect 5:193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AJ, Harland L, Groth P, Pettifer S, Chichester C, Willighagen EL, Evelo CT, Blomberg N, Ecker G, Goble C, Mons B (2012): Open PHACTS: Semantic interoperability for drug discovery. Drug Discov Today 17:1188–1198. [DOI] [PubMed] [Google Scholar]
- Williams KA, Magnuson M, Majeed W, LaConte SM, Peltier SJ, Hu X, Keilholz SD (2010): Comparison of alpha‐chloralose, medetomidine and isoflurane anesthesia for functional connectivity mapping in the rat. Magn Reson Imaging 28:995–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler AM, Webster MA, Vidaurre D, Nichols TE, Smith SM (2015): Multi‐level block permutation. Neuroimage 123:253–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise RG, Ide K, Poulin MJ, Tracey I (2004b): Resting fluctuations in arterial carbon dioxide induce significant low frequency variations in BOLD signal. Neuroimage 21:1652–1664. [DOI] [PubMed] [Google Scholar]
- Wise RG, Tracey I (2006): The role of fMRI in drug discovery. J Magn Reson Imaging 23:862–876. [DOI] [PubMed] [Google Scholar]
- Wise RG, Williams P, Tracey I (2004a): Using fMRI to quantify the time dependence of remifentanil analgesia in the human brain. Neuropsychopharmacology 29:626–635. [DOI] [PubMed] [Google Scholar]
- Wisner KM, Atluri G, Lim KO, Macdonald AW, 3rd. (2013): Neurometrics of intrinsic connectivity networks at rest using fMRI: Retest reliability and cross‐validation using a meta‐level method. Neuroimage 76:236–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong CW, Olafsson V, Tal O, Liu TT (2012): Anti‐correlated networks, global signal regression, and the effects of caffeine in resting‐state functional MRI. Neuroimage 63:356–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong EC (2014): An introduction to ASL labeling techniques. J Magn Reson Imaging 40:1–10. [DOI] [PubMed] [Google Scholar]
- Wong EC, Cronin M, Wu WC, Inglis B, Frank LR, Liu TT (2006): Velocity‐selective arterial spin labeling. Magn Reson Med 55:1334–1341. [DOI] [PubMed] [Google Scholar]
- Woo CW, Krishnan A, Wager TD (2014): Cluster‐extent based thresholding in fMRI analyses: Pitfalls and recommendations. Neuroimage 91:412–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worsley KJ, Evans AC, Marrett S, Neelin P (1992): A three‐dimensional statistical analysis for CBF activation studies in human brain. J Cereb Blood Flow Metab 12:900–918. [DOI] [PubMed] [Google Scholar]
- Worsley KJ, Liao CH, Aston J, Petre V, Duncan GH, Morales F, Evans AC (2002): A general statistical analysis for fMRI data. Neuroimage 15:1–15. [DOI] [PubMed] [Google Scholar]
- Wu CW, Gu H, Lu H, Stein EA, Chen JH, Yang Y (2009): Mapping functional connectivity based on synchronized CMRO2 fluctuations during the resting state. Neuroimage 45:694–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu WC, Lien SH, Chang JH, Yang SC (2014): Caffeine alters resting‐state functional connectivity measured by blood oxygenation level‐dependent MRI. NMR Biomed 27:444–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wylie GR, Genova H, DeLuca J, Chiaravalloti N, Sumowski JF (2014): Functional magnetic resonance imaging movers and shakers: Does subject‐movement cause sampling bias?. Hum Brain Mapp 35:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wylie KP, Rojas DC, Tanabe J, Martin LF, Tregellas JR (2012): Nicotine increases brain functional network efficiency. Neuroimage 63:73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia M, Wang Z, Dai Z, Liang X, Song H, Shu N, Li K, He Y (2014): Differentially disrupted functional connectivity in posteromedial cortical subregions in Alzheimers disease. J Alzheimers Dis 39:527–543. [DOI] [PubMed] [Google Scholar]
- Xu F, Uh J, Brier MR, Hart J, Jr. , Yezhuvath US, Gu H, Yang Y, Lu H (2011): The influence of carbon dioxide on brain activity and metabolism in conscious humans. J Cereb Blood Flow Metab 31:58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan CG, Cheung B, Kelly C, Colcombe S, Craddock RC, Di Martino A, Li Q, Zuo XN, Castellanos FX, Milham MP (2013b): A comprehensive assessment of regional variation in the impact of head micromovements on functional connectomics. Neuroimage 76:183–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan CG, Craddock RC, Zuo XN, Zang YF, Milham MP (2013a): Standardizing the intrinsic brain: Towards robust measurement of inter‐individual variation in 1000 functional connectomes. Neuroimage 80:246–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan R, Di X, Kim EH, Barik S, Rypma B, Biswal BB (2013): Regional homogeneity of resting‐state fMRI contributes to both neurovascular and task activation variations. Magn Reson Imaging 31:1492–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalesky A, Breakspear M (2015): Towards a statistical test for functional connectivity dynamics. Neuroimage 114:466–470. [DOI] [PubMed] [Google Scholar]
- Zalesky A, Fornito A, Cocchi L, Gollo LL, Breakspear M (2014): Time‐resolved resting‐state brain networks. Proc Natl Acad Sci U S A 111:10341–10346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang Y, Jiang T, Lu Y, He Y, Tian L (2004): Regional homogeneity approach to fMRI data analysis. Neuroimage 22:394–400. [DOI] [PubMed] [Google Scholar]
- Zhang X, Ghariq E, Hartkamp NS, Webb AG, van Osch MJ (2016): Fast cerebral flow territory mapping using vessel encoded dynamic arterial spin labeling (VE‐DASL). Magn Reson Med. 75:2041–2049. [DOI] [PubMed] [Google Scholar]
- Zhou C, Zwilling CE, Calhoun VD, Wang MY (2014): Efficient Blockwise Permutation Tests Preserving Exchangeability. Int J Stat Med Res 3:145–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zijdenbos AP, Forghani R, Evans AC (2002): Automatic “pipeline” analysis of 3‐D MRI data for clinical trials: Application to multiple sclerosis. IEEE Trans Med Imaging 21:1280–1291. [DOI] [PubMed] [Google Scholar]
- Zilles K, Amunts K (2009): Receptor mapping: Architecture of the human cerebral cortex. Curr Opin Neurol 22:331–339. [DOI] [PubMed] [Google Scholar]
- Zilles K, Palomero‐Gallagher N, Grefkes C, Scheperjans F, Boy C, Amunts K, Schleicher A (2002): Architectonics of the human cerebral cortex and transmitter receptor fingerprints: Reconciling functional neuroanatomy and neurochemistry. Eur Neuropsychopharmacol 12:587–599. [DOI] [PubMed] [Google Scholar]
- Zilles K, Palomero‐Gallagher N, Schleicher A (2004): Transmitter receptors and functional anatomy of the cerebral cortex. J Anat 205:417–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou QH, Zhu CZ, Yang Y, Zuo XN, Long XY, Cao QJ, Wang YF, Zang YF (2008): An improved approach to detection of amplitude of low‐frequency fluctuation (ALFF) for resting‐state fMRI: Fractional ALFF. J Neurosci Methods 172:137–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo XN, Di Martino A, Kelly C, Shehzad ZE, Gee DG, Klein DF, Castellanos FX, Biswal BB, Milham MP (2010b): The oscillating brain: Complex and reliable. Neuroimage 49:1432–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo XN, Kelly C, Adelstein JS, Klein DF, Castellanos FX, Milham MP (2010a): Reliable intrinsic connectivity networks: Test‐retest evaluation using ICA and dual regression approach. Neuroimage 49:2163–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo XN, Xu T, Jiang L, Yang Z, Cao XY, He Y, Zang YF, Castellanos FX, Milham MP (2013): Toward reliable characterization of functional homogeneity in the human brain: Preprocessing, scan duration, imaging resolution and computational space. Neuroimage 65:374–386. [DOI] [PMC free article] [PubMed] [Google Scholar]