

Abstract
In this longitudinal study, we investigated the regional patterns of focal lesions accumulation, and gray (GM) and white matter (WM) atrophy progression over a five‐year follow‐up (FU) in multiple sclerosis (MS) patients and their association with clinical and cognitive deterioration. Neurological, neuropsychological and brain MRI (dual‐echo and 3D T1‐weighted sequences) assessments were prospectively performed at baseline (T0) and after a median FU of 4.9 years from 66 MS patients (including relapse‐onset and primary progressive MS) and 16 matched controls. Lesion probability maps were obtained. Longitudinal changes of GM and WM volumes and their association with clinical and cognitive deterioration were assessed using tensor‐based morphometry and SPM12. At FU, 36/66 (54.5%) MS patients showed a significant disability worsening, 14/66 (21.2%) evolved to a worse clinical phenotype, and 18/63 (28.6%) developed cognitive deterioration. At T0, compared to controls, MS patients showed a widespread pattern of GM atrophy, involving cortex, deep GM and cerebellum, and atrophy of the majority of WM tracts, which further progressed at FU (P < 0.001, uncorrected). Compared to stable patients, those with clinical and cognitive worsening showed a left‐lateralized pattern of GM and WM atrophy, involving deep GM, fronto‐temporo‐parieto‐occipital regions, cerebellum, and several WM tracts (P < 0.001, uncorrected).GM and WM atrophy of relevant brain regions occur in MS after 5 years. A different vulnerability of the two brain hemispheres to irreversible structural damage may be among the factors contributing to clinical and cognitive worsening in these patients. Hum Brain Mapp 38:5648–5665, 2017. © 2017 Wiley Periodicals, Inc.
Keywords: MRI, gray matter, white matter, atrophy progression, multiple sclerosis
INTRODUCTION
The identification of reliable markers charting the progression of clinical disability and cognitive deterioration is an urgent unmet need in multiple sclerosis (MS). In patients with clinically isolated syndromes (CIS), T2 hyperintense lesion measures obtained at disease onset predict the development of MS and the accumulation of irreversible clinical disability and cognitive deficits [Fisniku et al., 2008a; Summers et al., 2008; Tintore et al., 2015]. In patients with established MS, cross‐sectional and longitudinal studies have shown that measurements of brain atrophy correlate better with clinical disability [Filippi et al., 2013; Fisher et al., 2008; Fisniku et al., 2008b; Khaleeli et al., 2008; Lansley et al., 2013] and neuropsychological impairment [Amato et al., 2007; Filippi et al., 2013] than T2‐ and T1‐lesion volumes, with a prominent role played by gray matter (GM) atrophy [Filippi et al., 2013; Lansley et al., 2013].
Only a few longitudinal studies, with relatively short follow‐up (FU) duration, have investigated the dynamics of the regional evolution of focal lesions and atrophy in MS patients and their correlations with clinical worsening. Globally, these works have shown that early in the course of the disease, tissue loss prominently affects deep GM structures, with involvement of other cortical and subcortical GM regions within 2 years of FU [Audoin et al., 2006; Rocca et al., 2016; Sepulcre et al., 2006]. Regional GM atrophy progression is associated with the formation of new T2‐visible lesions in the white matter (WM) [Bendfeldt et al., 2012, 2009], with modest [Battaglini et al., 2009] or no [Bendfeldt et al., 2012] anatomical overlap. Evolution of atrophy in specific GM regions has been associated with worsening of disability over 2 years of FU in relapsing‐remitting (RR) MS [Audoin et al., 2006] and after 5 years in primary progressive (PP) MS patients [Eshaghi et al., 2014].
Here, we applied voxel‐based approaches to investigate the regional patterns of focal lesion accumulation, and GM and WM atrophy progression over a 5 year FU in MS patients and their association with clinical and cognitive deterioration.
MATERIALS AND METHODS
Ethics Committee Approval
Institutional Review Board approval and written informed consent were obtained.
Subjects
In this prospective study, from January 2007 to March 2009, 66 consecutive MS patients and 16 healthy controls (HC) underwent clinical and MRI evaluation at baseline (T0) and after a median FU of 4.9 years (till May 2014) (Table 1). Patients also underwent a neuropsychological assessment at the two time points.
Table 1.
Main demographic, clinical, and MRI characteristics of healthy controls (HC) and multiple sclerosis (MS) patients at baseline (T0) and follow‐up (FU)
| HC | MS | P value (MS vs HC) | |
|---|---|---|---|
| Male/female | 5/11 | 18/48 | 0.75b |
| Mean age at T0 (range) [years] | 40.1 (25–54) | 42.2 (19–60) | 0.49a |
| Median education at T0 (range) [years] | 13 (5–18) | 12 (4–18) | 0.68a |
| Median DD at T0 (range) [years] | ‐ | 10.8 (0.1–40) | ‐ |
| Median FU duration (range) [years] | 5.3 (4.5–5.7) | 4.7 (4.3–5.6) | 0.23a |
| Median EDSS score (range) | ‐ | ||
| T0 | ‐ | 3.0 (0.0–7.5) | |
| FU | ‐ | 4.0 (1.0–8.5) | |
| P | ‐ | 0.0005c | ‐ |
| Clinical phenotype | |||
| T0 | ‐ | 13 CIS/12 RR/9 SP/18 B/14 PP | ‐ |
| FU | ‐ | 6 CIS/17 RR/17 SP/12 B/14 PP | ‐ |
| Treatment | ‐ | ‐ | |
| (1) none | 49 (74.2%) | ||
| (2) First line DMD | 13 (19.7%) | ||
| (3) Second line and immunosuppressants | 4 (6.1%) | ||
| Patients with treatment change (%) | ‐ |
4 (6%) (1 [1.5%] 1st line DMD; 3 [4.5%]) 2nd line treatments) |
‐ |
| Mean brain T2 LV (SD) [ml] | ‐ | ||
| T0 | ‐ | 20.5 (23.4) | |
| FU | ‐ | 22.9 (24.6) | |
| P | ‐ | 0.01c | ‐ |
| Median # of new brain T2 lesions (range) | ‐ | 4 (0–103) | ‐ |
| Mean brain T1 LV (SD) [ml] | ‐ | ||
| T0 | ‐ | 8.9 (12.4) | |
| FU | ‐ | 11.7(14.9) | |
| P | ‐ | <0.0001c | ‐ |
| Median # of new brain T1 lesions (range) | ‐ | 2 (0–81) | ‐ |
| Mean NBV at T0 (SD) [ml] | 1635 (59) | 1518 (109) | 0.01a |
| Mean PBVC (SD) [%] | −1.9% (1.5) | −4.5% (4.2) | 0.02a |
| Mean GMV at T0 (SD) [ml] | 754 (88) | 668 (103) | 0.02a |
| Mean GMV change at FU (SD) [%] | −2.0% (0.8) | −6.3% (6.2) | 0.04a |
| Mean WMV at T0 (SD) [ml] | 846 (95) | 850 (98) | 0.80a |
| Mean WMV change at FU (SD) [%] | +0.5% (2.7) | +1.0% (9.1) | 0.87a |
Mann–Whitney test.
Pearson chi‐square test.
Wilcoxon signed rank test.
HC = healthy controls; MS = multiple sclerosis; T0 = baseline; FU = follow‐up; DD = disease duration; EDSS = Expanded Disability Status Scale; DMD = disease modifying drug; LV = lesion volume; SD = standard deviation; ml = milliliter; NBV = normalized brain volume; PBVC = percent brain volume change; GMV = gray matter volume; WMV = white matter volume; CIS = clinically isolated syndrome; RR = relapsing‐remitting; SP = secondary progressive; B = benign; PP = primary progressive. See text for further details.
To be included, subjects had to (1) be relapse‐ and steroid‐free for at least three months before study inclusion (patients only, 3 patients excluded); (2) have no significant medical illnesses or substance abuse that could interfere with cognitive functioning (1 patient excluded); (3) have no other major systemic, psychiatric or neurological disorders (2 patients and 1 HC excluded); (4) be right‐handed according to Edinburgh Handedness Inventory scale [Oldfield, 1971] (3 patients and 2 HC excluded); and (5) have a normal neurological examination (HC only).
Thirteen patients had CIS suggestive of MS [Polman et al., 2011], 12 RRMS [Lublin and Reingold, 1996], 9 secondary progressive (SP) MS [Lublin and Reingold, 1996], 18 benign (B) MS (Expanded Disability Status Scale [EDSS] score ≤ 3.0 and disease duration ≥ 15 years), and 14 PPMS [Polman et al., 2011].
Neurological and Neuropsychological Evaluation
Within 3 days from MRI acquisition, an experienced neurologist blinded to MRI findings performed a neurological examination, with EDSS rating (SM, 20 years of experience).
At FU, patients were considered clinically worsened if they had an EDSS score increase ≥ 1.0, when EDSS score at T0 was < 6.0, or an EDSS score increase ≥ 0.5, when EDSS score at T0 was ≥ 6.0 [Filippi et al., 2013]. Any EDSS change was always confirmed by a second visit after 3 months.
Phenotype evolution occurred when during the FU, a patient evolved to a more severe clinical phenotype according to the natural history of the disease (CIS to RRMS; CIS, RRMS, or BMS to SPMS) [Lublin and Reingold, 1996].
Cognitive performance was assessed at T0 and FU by an expert neuropsychologist, blinded to the clinical and MRI data, using two alternative versions of the Brief Repeatable Battery of Neuropsychological Tests (BRB‐N) [Rao and and the Cognitive Function Study Group of the National Multiple Sclerosis Society, 1990] in 63 of the 66 MS patients (JD, 8 years of experience).
The BRB‐N [Rao and and the Cognitive Function Study Group of the National Multiple Sclerosis Society, 1990] includes the Selective Reminding Test (SRT), to assess verbal memory; the 10/36 Spatial Recall Test (SPART), to assess visual memory; the Symbol Digit Modalities Test (SDMT) and the Paced Auditory Serial Addition Test (PASAT) 2″ and 3″, to assess attention and information processing speed; and the Word List Generation (WLG) test, to assess verbal fluency.
A global z‐score of cognitive functions (obtained by averaging z‐scores of all neuropsychological tests) was calculated [Sepulcre et al., 2006]. Patients with at least three abnormal tests (defined as a score more than 1.5 standard deviations [SD] below the normative value provided by Obradovic et al. [2012] were considered cognitively impaired (CI) at T0 [Benedict et al., 2007].
Variations of cognitive performances were assessed for each test and for global z‐score using the reliable change index (RCI), to correct for measurement error [Portaccio et al., 2013]. The RCI was calculated using the formula (X FU − X T0)/SDdiff, where X T0 = score of each test or global z‐score at T0, X FU = score of each test and global z‐score at FU, and SDdiff was defined as the SD of the mean difference score. Cognitive performances were considered significantly worsened if global z‐score RCI score was <−1.25 [Portaccio et al., 2013].
MRI Acquisition and Analysis
Using a 1.5 T scanner (Avanto, Siemens, Erlangen, Germany) under regular maintenance program (no major scanner hardware or software upgrades occurred during the study), the following brain sequences were acquired from all subjects at the two timepoints: (1) axial dual‐echo (DE) turbo spin‐echo (TSE) (repetition time [TR]=2650 ms, echo time [TE]=28–113 ms, echo train length [ETL]=5, number of slices = 50, slice thickness = 2.5 mm with no gap, matrix size = 256 × 256, field of view [FOV]= 250 × 250 mm2) for T2 lesion quantification; (2) sagittal 3D T1‐weighted magnetization prepared rapid acquisition gradient‐echo (MPRAGE) (TR = 2000 ms, TE = 3.93 ms, inversion time = 1100 ms, number of sections = 208, section thickness = 0.9 mm, matrix size = 256 × 224, FOV = 236 × 270 mm2) for T1‐hypointense lesion and atrophy quantification. For all scans, the slices were positioned to run parallel to a line that joins the most inferoanterior and inferoposterior parts of the corpus callosum (CC), with careful repositioning during the FU scan.
At T0 and FU, T2‐hyperintense and T1‐hypointense lesions were identified by consensus of two experienced observers blinded to patients' identity (PP and MAR, 8 and 20 years of experience, respectively) and lesion volumes (LV) were measured, using a local thresholding segmentation technique (Jim 6, Xinapse Systems, Colchester, UK). At FU, the number of new T2 and T1 lesions was also quantified.
Sagittal 3D T1‐weighted images were coregistered to DE sequence. The lesion refilling tool in FSL was applied to 3D T1‐weighted images using edited T2 lesion masks, to improve segmentation and registration processes in patients [Battaglini et al., 2012].
After T1‐hypointense lesion refilling, normalized brain volumes (NBV), GM volumes (GMV) and WM volumes (WMV), and longitudinal percent brain volume changes (PBVC) were assessed on 3D T1‐weighted images using the SIENAx and SIENA softwares. Longitudinal changes of GMV and WMV were calculated as the percent change vs T0.
T2 and T1 Lesion Probability Maps
From T2 lesions, binarized masks were obtained, coregistered to the 3D T1‐weighted scans (using the rigid transformation calculated between the T2‐weighted and the 3D T1‐weighted image), transformed to the mid‐point average template of tensor‐based morphometry (TBM) (see below for details), normalized to the standard space, and averaged to obtain T2 lesion probability maps (LPMs). The same procedure was applied on binarized masks from T1 lesions, to obtain T1‐hypointense LPMs. After the application of smoothing, FU lesional masks were subtracted from baseline ones to obtain maps of T2 and T1 lesional changes.
Regional GM and WM Volume Abnormalities
TBM, as implemented in SPM12 (http://www.fil.ion.ucl.ac.uk/spm), was used to map changes of regional GM and WM volumes over time within and between patients' subgroups. Baseline scan of each subject was coregistered to the FU one with a rigid transformation and vice versa. The absolute values of the two transformations were averaged, halved, and written in the image nifti header with opposite signs one to the other, to obtain a halfway coregistration without image reslicing. Pairwise longitudinal registration was used to align the first and second scan of each subject [Ashburner and Ridgway, 2012]. The rate of volume change, corrected for the FU duration, was quantified by saving the map of divergence of the velocity field, where positive values indicate expansion and negative values contraction. The mid‐point average template image was also saved. This was used for groupwise alignment: first, the mid‐point average template images were segmented into different tissue types via the Segmentation routine in SPM12 [Ashburner and Friston, 2005]. Then, GM and WM segmented images of all subjects, in the closest possible rigid‐body alignment with each other, were used to produce GM and WM templates and to drive the deformation to the templates. At each iteration, the deformations, calculated using the Diffeomorphic Anatomical Registration using Exponentiated Lie algebra (DARTEL) registration method [Ashburner, 2007], were applied to GM and WM, with an increasingly good alignment of subject morphology, to produce templates. Finally, an affine transformation that maps from the population average (DARTEL Template space) to Montreal Neurological Institute (MNI) space was calculated, images were spatially normalized and smoothed with a 3 mm Gaussian kernel. This amount of smoothing was decided to limit the partial volume effect in the GM from the surrounding CSF, as this was a relevant confounding factor when an 8 mm smoothing was used in a preliminary analysis. These last 3 steps are incorporated in a unique tool, called “Normalise to MNI Space.”
The steps described for groupwise alignment were repeated for baseline 3D T1‐weighted images to run a voxel‐based morphometry (VBM) analysis. The only difference in the procedure described above is that the normalization to MNI space was applied to GM and WM maps, and that, after transformation these were modulated to ensure that the overall amount of each region was not altered by the spatial normalization procedure.
Lateralization Index
To determine hemispheric lateralization of brain damage accumulation, a “lateralization index” (LI) of between‐group differences was calculated, according to the formula (L Vox − R Vox)/(L Vox + R Vox) × 100, where L Vox and R Vox were the number of voxels above the threshold (P < 0.001 uncorrected, cluster extent = 10 voxels) in the left and right hemisphere [Seghier, 2008]. A positive index corresponds to a left‐predominant lateralization (strong from +50% to +100%, weak from +25% to +50%), while a negative index corresponds to a right‐predominant lateralization (strong from −100% to −50%, weak from −50% to −25%). Symmetric distribution was considered between −25% and +25% [Lehericy et al., 2000].
Statistical Analysis
Between‐group comparisons of baseline variables between MS patients and HC were assessed using the Mann–Whitney U tests for continuous variables, and the Pearson chi‐square test for categorical variables. Within‐ and between‐group longitudinal changes of structural MRI variables in MS patients and HC were assessed with the Wilcoxon Signed Rank Test and the Mann–Whitney test (SPSS software, version 22.0).
The general linear model and theory of Gaussian fields were used to assess longitudinal regional modifications of T2 and T1 LPMs and of GM and WM volumes (one‐sample t tests and ANCOVA models) [Friston et al., 1995]. To limit the analysis to the GM (WM), an inclusive mask obtained from the GM (WM) DARTEL Template, transformed to the MNI space, smoothed and thresholded at 0.25, was used. All analyses were corrected for age and sex.
T2 and T1 lesion distribution, VBM and TBM results were assessed at a threshold of P < 0.05, family‐wise error corrected (FWE) for multiple comparisons and also tested at a P < 0.001, uncorrected (cluster extent = 10 voxels). The localization of areas of T2 and T1 lesion occurrence as well as of GM and WM atrophy was defined using available atlases.
RESULTS
Demographic, Clinical, Neuropsychological, and Conventional MRI Findings
Table 1 summarizes the main demographic, clinical, and structural MRI findings in HC and MS patients at T0 and their longitudinal changes. Age, gender, and years of education did not differ between MS patients and HC (P values ranging from 0.49 to 0.75). At T0, 13/63 (20.6%) MS patients were CI. Table 2 shows the number and percentage of MS patients having impairment at T0 and worsening of performance at FU at each neuropsychological test of the BRB‐N. During the FU, the median number of relapses was 1 (mean = 2.3, range = 0–16).
Table 2.
Prevalence of multiple sclerosis (MS) patients showing impairment at baseline (T0) and worsening of performance at follow‐up (FU) at each neuropsychological test of the Brief Repeatable Battery of Neuropsychological Tests
| Neuropsychological tests | Patients with impaired performance at T0a (%) | Patients with worsening of performance at FUb (%) | Patients with worsening of performance at FUb (%) | ||
|---|---|---|---|---|---|
| Cognitively stable (n = 45) | Cognitively worsened (n = 18) | ||||
| Verbal memory | SRT lts | 6 (9.5) | 13 (20.6) | 2 (4.4) | 11 (61.1) |
| SRT cltr | 15 (23.8) | 11 (17.5) | 3 (6.7) | 8 (44.4) | |
| SRT d | 7 (11.1) | 29 (46.0) | 13 (28.9) | 16 (88.9) | |
| Visual memory | SPART | 22 (34.9) | 7 (11.1) | 2 (4.4) | 5 (27.8) |
| SPART d | 27 (42.8) | 14 (22.2) | 2 (4.4) | 12 (66.7) | |
| Attention and processing speed | SDMT | 22 (34.9) | 11 (17.5) | 0 (0.0) | 11 (61.1) |
| PASAT 3 sec | 11 (17.5) | 15 (23.8) | 2 (4.4) | 13 (72.2) | |
| PASAT 2 sec | 18 (28.6) | 22 (34.9) | 5 (11.1) | 17 (94.4) | |
| Verbal fluency | WLG | 24 (38.1) | 14 (22.2) | 3 (6.7) | 11 (61.1) |
T0 = baseline; FU = follow‐up. SRT lts = Selective Reminding Test long‐term storage; SRT cltr = Selective Reminding Test consistent long‐term retrieval; SRT d = Selective Reminding Test delayed retrieval; SPART = Spatial Recall Test; SPART d = Spatial Recall Test delayed retrieval; SDMT = Symbol Digit Modalities Test; PASAT 3 sec = Paced Auditory Serial Addition Test 3 seconds; PASAT 2 sec = Paced Auditory Serial Addition Test 2 seconds; WLG = Word List Generation.
Defined as a score more than 1.5 standard deviation below the normative values.
Defined as a reliable change index (RCI) < −1.25.
At T0, compared to HC, MS patients had lower NBV (P = 0.01) and GMV (P = 0.02) (Table 1). At FU compared to T0, MS patients showed increased T2 LV (P = 0.01) and T1 LV (P < 0.0001) (Table 1). Compared to HC, they developed higher PBVC (P = 0.02) and GMV (P = 0.04) changes (Table 1).
Regional Damage Distribution
Figure 1 shows the T2 and T1 LPMs in MS patients at T0. Lesions were mostly located in the bilateral corona radiata (CR) and periventricular WM (P < 0.001, uncorrected). At T0, compared to HC, MS patients showed a diffuse pattern of regional GM and WM atrophy (Fig. 1) (P < 0.001, uncorrected). At FU, MS patients showed a higher frequency of T2 and T1 lesions in a few clusters mainly located in the posterior corona radiata, body, and splenium of the CC (Fig. 1) (P < 0.001, uncorrected). They also showed more significant GM atrophy of several fronto‐temporo‐parieto‐occipital regions, deep GM nuclei (thalamus, caudate nuclei, and putamen), and the cerebellum, and of the majority of WM tracts (Fig. 1) (P < 0.001, uncorrected). No volumetric changes were detected in HC.
Figure 1.
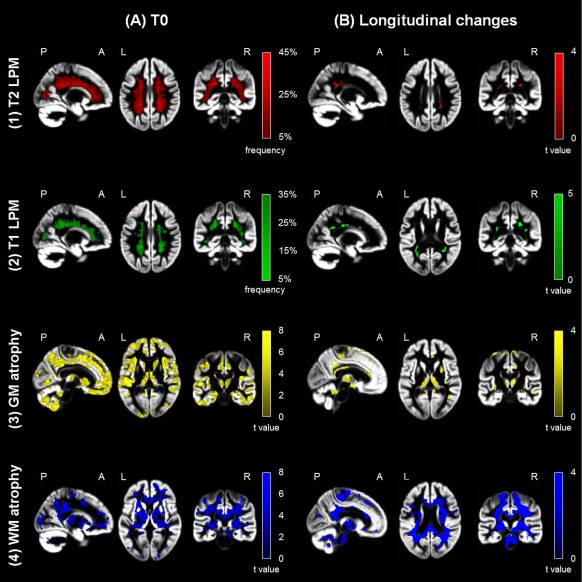
Regional damage in multiple sclerosis (MS) patients versus healthy controls (HC) at baseline (T0) and follow‐up. (a) Representative images showing (1) T2 (red color‐coded) and (2) T1 (green color‐coded) lesion probability maps (LPMs) and statistical parametric mapping (SPM) analysis showing regions of (3) gray matter (GM) (yellow color‐coded) and (4) white matter (WM) (blue color‐coded) volume loss superimposed on the customized GM template in MS patients versus HC at T0 (P < 0.001 uncorrected; cluster extent = 10 voxels). The LPMs are thresholded to show voxels in which lesion frequency of 5% is present, up to a maximum lesion frequency of 45% for T2 LPM and of 35% for T1 LPM. (b) Representative images showing areas of increased frequency of (1) T2 (red color‐coded) and (2) T1 (green color‐coded) lesions in MS patients and regions of (3) GM (yellow color‐coded) and (4) WM (blue color‐coded) atrophy after 5 years superimposed on the customized GM template in MS patients versus HC (P < 0.001 uncorrected; cluster extent = 10 voxels). Images are in neurological convention. See text for further details. [Color figure can be viewed at http://wileyonlinelibrary.com]
Regional modifications of lesions and atrophy in the different study groups according to clinical and cognitive deterioration at FU are shown in Figures 2 and 3 and summarized in Table 3 (P < 0.001, uncorrected). At visual inspection, no anatomical overlap was found between regions of volume reduction and areas of T2 lesion modifications (Fig. 3).
Figure 2.
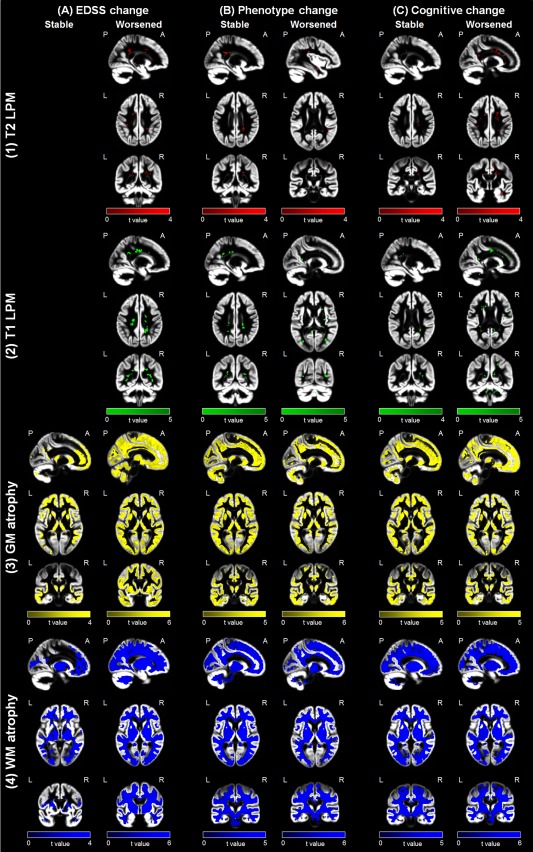
Evolution of regional damage in the main study subgroups. Representative images showing areas of increased frequency of (1) T2 (red color‐coded) and (2) T1 (green color‐coded) lesions and regions of (3) gray matter (GM) (yellow color‐coded) and (4) white matter (WM) (blue color‐coded) atrophy after 5 years superimposed on the customized GM template according to clinical (EDSS score and phenotype change) and cognitive evolution at follow‐up (P < 0.001 uncorrected; cluster extent = 10 voxels). Images are in neurological convention. See text for further details. [Color figure can be viewed at http://wileyonlinelibrary.com]
Figure 3.
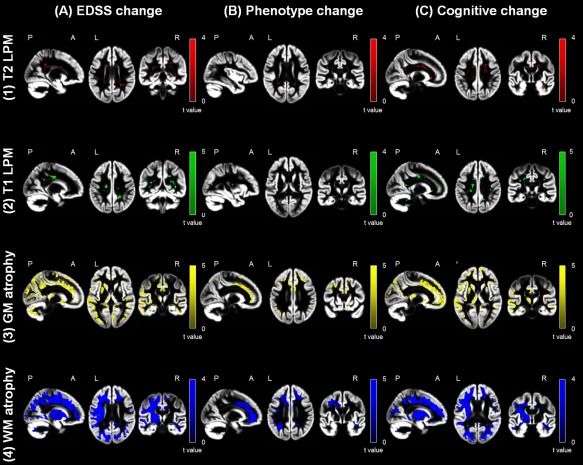
Difference of regional damage progression in worsened versus stable multiple sclerosis (MS) patients. Statistical parametric mapping (SPM) analysis showing areas of increased frequency of (1) T2‐hyperintense lesions (red color‐coded) and (2) T1‐hypointense lesions (green color‐coded), as well as regions of (3) gray matter (GM) (yellow color‐coded) and (4) white matter (WM) (blue color‐coded) atrophy after 5 years superimposed on the customized GM template in worsened versus stable MS patients according to the different outcomes. Left column (a): EDSS worsened versus stable; middle column (b): MS patients with vs without phenotype change; right column (c): cognitively worsened versus cognitively stable (P < 0.001 uncorrected; cluster extent = 10 voxels). Images are in neurological convention. See text for further details. [Color figure can be viewed at http://wileyonlinelibrary.com]
Table 3.
Brain regions with modifications of T2‐hyperintense and T1‐hypointense lesions and progression of gray matter (GM) and white matter (WM) atrophy, according to clinical (EDSS and phenotype change) and cognitive evolution at follow‐up (P < 0.001 uncorrected, ke = 10 voxels)
| Comparison | Variable | Cluster extent | Anatomical region | Side | MNI coordinates | t value | ||
|---|---|---|---|---|---|---|---|---|
| x | y | z | ||||||
| EDSS change: worsened vs stable MS patients | Increased T2 lesion frequency | 30 | Posterior CR | L | −26 | −39 | 21 | 4.03 |
| 18 | Posterior CR | R | 27 | −42 | 26 | 4.03 | ||
| 23 | IFOF | R | 40 | −34 | −6 | 3.71 | ||
| 18 | Body of the CC | L | −12 | 9 | 26 | 3.71 | ||
| 19 | Anterior CR | R | 22 | 4 | 27 | 3.71 | ||
| 42 | Splenium of the CC | R | 20 | −44 | 27 | 3.71 | ||
| Increased T1 lesion frequency | 31 | Forceps major | L | −27 | −64 | 3 | 4.89 | |
| 173 | Body of the CC | L | −16 | −15 | 34 | 4.83 | ||
| 105 | Posterior CR | R | 22 | −44 | 33 | 4.77 | ||
| 35 | Forceps major | R | 30 | −63 | 6 | 4.76 | ||
| 71 | IFOF | R | −32 | −48 | 15 | 4.53 | ||
| 11 | SLF | R | 36 | −40 | 20 | 4.38 | ||
| 35 | IFOF | L | −33 | −50 | 6 | 4.29 | ||
| 17 | Splenium of the CC | L | −21 | −51 | 21 | 4.23 | ||
| 17 | Posterior CR | L | −20 | −44 | 28 | 3.94 | ||
| 41 | SLF | L | −32 | −34 | 28 | 4.07 | ||
| 27 | CST | R | 20 | −21 | 36 | 3.92 | ||
| GM atrophy | 24194 | SPL | L | −14 | −72 | 52 | 5.95a | |
| PCC | R | 3 | −38 | 30 | 5.74a | |||
| Supramarginal gyrus | L | −46 | −45 | 12 | 5.37 | |||
| Precuneus | L | −14 | −46 | 36 | 5.37 | |||
| ITG | L | −42 | −48 | −14 | 5.21 | |||
| Cuneus | L | −6 | −81 | 26 | 5.21 | |||
| Fusiform gyrus | L | −33 | −44 | −14 | 5.15 | |||
| STG | L | −56 | −33 | 12 | 5.11 | |||
| IOG | L | −44 | −82 | 3 | 5.11 | |||
| Supramarginal gyrus | L | −57 | −44 | 16 | 5.09 | |||
| SFG | L | −16 | −2 | 68 | 5.04 | |||
| 2794 | Cerebellum (lobule 7) | L | −10 | −69 | −40 | 5.89a | ||
| Cerebellum Crus 2 | L | −27 | −82 | −44 | 5.38 | |||
| Cerebellum Crus 1 | L | −40 | −57 | −39 | 5.15 | |||
| 1369 | Cerebellum Crus 2 | R | 16 | −82 | −36 | 5.26 | ||
| 82 | SMA | L | −16 | −2 | 68 | 5.04 | ||
| 97 | Postcentral gyrus | R | 21 | −38 | 64 | 5.03 | ||
| 398 | Precuneus | R | 12 | −54 | 58 | 5.00 | ||
| 309 | Calcarine cortex | L | −15 | −57 | 10 | 4.99 | ||
| SPL | R | 12 | −51 | 69 | 3.71 | |||
| 485 | Cerebellum (lobule 3) | R | 6 | −46 | −20 | 4.92 | ||
| Cerebellar vermis | L | −2 | −50 | −10 | 4.20 | |||
| Cerebellum (lobule 6) | R | 8 | −63 | −21 | 3.93 | |||
| 4196 | Angular gyrus | R | 50 | −50 | 18 | 4.83 | ||
| MTG | R | 46 | −45 | 15 | 4.77 | |||
| ITG | R | 58 | −32 | −21 | 4.57 | |||
| Fusiform gyrus | R | 30 | −72 | 18 | 4.48 | |||
| MOG | R | 40 | −78 | 24 | 4.16 | |||
| 453 | Lingual gyrus | R | 26 | −45 | −6 | 4.67 | ||
| 1249 | Putamen | L | −21 | 9 | 3 | 4.61 | ||
| Caudate nucleus | L | −27 | 10 | 0 | 4.54 | |||
| 485 | ACC | R | 12 | 20 | 28 | 4.51 | ||
| 187 | Lingual gyrus | L | 6 | −69 | 6 | 4.50 | ||
| 1002 | Cuneus | R | 14 | −82 | 27 | 4.48 | ||
| Calcarine cortex | R | 8 | −84 | 16 | 4.29 | |||
| 226 | SFG | L | −14 | 20 | 54 | 4.46 | ||
| 67 | Calcarine cortex | R | 9 | −56 | 16 | 4.43 | ||
| 386 | STG | R | 46 | −20 | 6 | 4.42 | ||
| 29 | SMA | R | 4 | −15 | 57 | 4.39 | ||
| 112 | Parahippocampal gyrus | L | −26 | −8 | −26 | 4.37 | ||
| 150 | Cerebellum (lobule 9) | L | −16 | −50 | −52 | 4.28 | ||
| 92 | IFG | R | 48 | 33 | 0 | 4.18 | ||
| 226 | SFG | R | 18 | 33 | 39 | 4.12 | ||
| 110 | Lingual gyrus | R | 14 | −48 | 6 | 4.15 | ||
| 15 | Hippocampus | L | −24 | −34 | 2 | 4.12 | ||
| 26 | SPL | R | 34 | −40 | 56 | 4.07 | ||
| 158 | Insula | L | −36 | 2 | 16 | 4.06 | ||
| 41 | Precentral gyrus | L | −50 | −8 | 48 | 4.04 | ||
| 103 | Cerebellum (lobule 6) | L | −28 | −52 | −24 | 4.01 | ||
| 49 | Parahippocampal gyrus | R | 28 | −20 | −20 | 3.99 | ||
| 134 | IFG | L | −9 | 28 | −12 | 3.97 | ||
| 46 | Cerebellum (lobule 8) | R | 9 | −64 | −38 | 3.95 | ||
| 51 | Supramarginal gyrus | R | 36 | −39 | 38 | 3.93 | ||
| 11 | MFG | R | 34 | 6 | 57 | 3.86 | ||
| 158 | Thalamus | L | −10 | −21 | 8 | 3.85 | ||
| 14 | Cerebellum (lobule 7) | R | 40 | −51 | −44 | 3.85 | ||
| 127 | Insula | R | 38 | −3 | −15 | 3.76 | ||
| 70 | Cerebellum (lobule 9) | R | 14 | −52 | −50 | 3.67 | ||
| 10 | Precentral gyrus | R | 22 | −10 | 57 | 3.63 | ||
| 22 | MFG | R | 30 | 15 | 45 | 3.59 | ||
| 17 | Cerebellum (lobule 9) | L | −21 | −57 | −52 | 3.54 | ||
| 12 | Putamen | R | 27 | 14 | 4 | 3.49 | ||
| 10 | Hippocampus | R | 38 | −80 | −2 | 3.49 | ||
| 10 | Thalamus | R | 16 | −27 | 8 | 3.30 | ||
| WM atrophy | 444 | MCP | L | −10 | −69 | −40 | 5.87a | |
| 2140 | Posterior cingulum | R | 4 | −38 | 28 | 5.56a | ||
| IFOF | R | 24 | −82 | 28 | 4.06 | |||
| 43667 | SLF | L | −46 | −45 | 12 | 5.35a | ||
| Posterior cingulum | L | −14 | −48 | 36 | 5.27a | |||
| ILF | L | −44 | −82 | 2 | 5.09 | |||
| Anterior CR | L | −20 | 12 | 38 | 4.67 | |||
| Anterior cingulum | L | −8 | 21 | 24 | 4.50 | |||
| CST | L | −27 | −23 | 23 | 4.30 | |||
| Body of the CC | L | −12 | 17 | 27 | 4.00 | |||
| IFOF | L | −32 | −77 | −2 | 3.84 | |||
| OR | L | −32 | −66 | 0 | 3.75 | |||
| Splenium of the CC | L | −15 | −45 | 27 | 3.75 | |||
| Forceps minor | L | −15 | 30 | 20 | 3.89 | |||
| Uncinate fasciculus | L | −24 | 21 | −6 | 3.43 | |||
| 83 | Posterior CR | R | 21 | −38 | 64 | 4.97 | ||
| 4318 | SLF | R | 42 | −54 | 18 | 4.82 | ||
| 344 | ILF | R | 26 | −56 | −4 | 4.45 | ||
| 2679 | Anterior cingulum | R | 12 | 21 | 28 | 4.53 | ||
| Forceps minor | R | 14 | 46 | 4 | 4.18 | |||
| 66 | MCP | R | 15 | −80 | −38 | 4.34 | ||
| 73 | Forceps major | R | 14 | −46 | 6 | 4.07 | ||
| 241 | Uncinate fasciculus | R | 30 | −3 | −15 | 3.76 | ||
| 12 | CST | R | 20 | −20 | −6 | 3.53 | ||
| Phenotype change: worsened vs stable MS patients | Increased T2 lesion frequency | 24 | SLF | R | 40 | −39 | 18 | 4.10 |
| 10 | SLF | L | −28 | −32 | 39 | 4.10 | ||
| 19 | IFOF | R | 39 | −4 | −24 | 3.21 | ||
| Increased T1 lesion frequency | 61 | ILF | R | 34 | −69 | 9 | 4.64 | |
| 40 | SLF | L | −36 | −22 | 26 | 4.44 | ||
| 56 | Forceps major | L | −28 | −75 | 6 | 4.41 | ||
| 16 | Splenium of the CC | L | −12 | −48 | 12 | 3.87 | ||
| GM atrophy | 314 | Precentral gyrus | L | −34 | −2 | 44 | 5.61a | |
| MFG | L | −26 | 0 | 48 | 4.13 | |||
| 783 | SFG | R | 36 | 28 | 20 | 5.10 | ||
| MFG | R | 24 | 21 | 38 | 4.47 | |||
| 1012 | ACC | L | −3 | 28 | 12 | 5.00 | ||
| Orbitofrontal cortex | L | −12 | 44 | −10 | 4.06 | |||
| 94 | MFG | L | −38 | 18 | 32 | 4.82 | ||
| 900 | MOG | L | −36 | −66 | 30 | 4.71 | ||
| Supramarginal gyrus | L | −46 | −39 | 28 | 4.38 | |||
| 173 | SFG | L | −26 | 32 | 28 | 4.65 | ||
| 66 | ITG | L | −36 | −40 | −14 | 4.59 | ||
| 139 | Orbitofrontal gyrus | L | −27 | 38 | −8 | 4.49 | ||
| 514 | STG | R | 45 | −24 | −2 | 4.48 | ||
| MTG | R | 44 | −34 | 2 | 4.41 | |||
| 743 | ACC | R | 6 | 34 | 3 | 4.48 | ||
| 140 | Insula | R | 34 | −18 | −3 | 4.30 | ||
| 53 | SPL | R | 20 | −48 | 56 | 4.28 | ||
| 70 | Precuneus | L | −10 | −48 | 48 | 4.16 | ||
| 119 | SFG | L | −27 | 48 | −2 | 4.16 | ||
| 76 | ITG | R | 52 | −44 | −8 | 4.13 | ||
| 137 | PCC | L | −6 | −28 | 40 | 4.04 | ||
| 38 | Fusiform gyrus | R | 28 | −34 | −22 | 4.03 | ||
| 49 | SMC | L | −10 | 8 | 48 | 4.02 | ||
| 69 | IFG | L | −45 | 3 | 18 | 4.01 | ||
| 40 | Orbitofrontal cortex | R | 20 | 32 | −12 | 4.01 | ||
| 23 | Insula | L | −28 | 28 | 8 | 4.00 | ||
| 79 | Supramarginal gyrus | R | 62 | −42 | 28 | 3.99 | ||
| 23 | Angular gyrus | L | −62 | −51 | 21 | 3.98 | ||
| 50 | Caudate nucleus | R | 15 | 18 | −8 | 3.84 | ||
| 94 | SPL | L | −24 | −54 | 57 | 3.85 | ||
| 27 | MTG | L | −63 | −34 | −15 | 3.80 | ||
| 30 | Hippocampus | R | 21 | −2 | −22 | 3.79 | ||
| 30 | Precuneus | R | 12 | −54 | 39 | 3.77 | ||
| 14 | PCC | R | 15 | −42 | 38 | 3.77 | ||
| 30 | Lingual gyrus | R | 24 | −40 | −9 | 3.75 | ||
| 26 | Precuneus | L | −6 | −51 | 14 | 3.65 | ||
| 11 | MOG | L | −38 | −69 | −2 | 3.37 | ||
| WM atrophy | 12377 | SLF | L | −34 | −2 | 44 | 5.31a | |
| Forceps minor | L | −14 | 39 | 3 | 5.10 | |||
| SLF | R | 30 | 22 | 10 | 4.91 | |||
| IFOF | R | 36 | 28 | 20 | 4.83 | |||
| Genu of the CC | L | −3 | 28 | 12 | 4.79 | |||
| Forceps minor | R | 21 | 36 | 16 | 4.68 | |||
| Body of the CC | L | −15 | 6 | 32 | 4.64 | |||
| Uncinate fasciculus | R | 16 | 32 | −10 | 4.61 | |||
| Anterior CR | L | −12 | 3 | 38 | 4.49 | |||
| IFOF | L | −22 | 40 | −2 | 4.42 | |||
| Anterior cingulum | L | −10 | 33 | 10 | 4.39 | |||
| 116 | ILF | L | −38 | −40 | −14 | 4.59 | ||
| 2460 | ILF | R | 40 | −3 | −21 | 4.49 | ||
| 848 | Posterior cingulum | R | 18 | −50 | 32 | 3.83 | ||
| 22 | Body of the CC | R | 15 | −20 | 34 | 3.57 | ||
| Cognitive change: worsened vs stable MS patients | Increased T2 lesion frequency | 65 | Body of the CC | L | −12 | 9 | 26 | 5.43a |
| 17 | Anterior CR | R | 20 | −4 | 33 | 5.43a | ||
| 42 | Genu of the CC | L | −18 | 32 | 3 | 4.10 | ||
| 19 | Splenium of the CC | L | −15 | −50 | 21 | 4.10 | ||
| 35 | SLF | R | 33 | −27 | 27 | 4.10 | ||
| 25 | SLF | L | −26 | −8 | 33 | 4.10 | ||
| 11 | Anterior CR | L | −15 | −9 | 39 | 4.10 | ||
| 20 | MCP | L | −9 | −42 | −32 | 3.43 | ||
| 32 | ILF | R | 45 | −20 | −16 | 3.43 | ||
| 24 | IFOF | R | 26 | 16 | −3 | 3.43 | ||
| 13 | Uncinate fasciculus | L | −26 | 12 | 18 | 3.43 | ||
| 12 | CST | R | 28 | −27 | 22 | 3.43 | ||
| 26 | Posterior cingulum | R | 21 | −40 | 40 | 3.43 | ||
| 19 | CST | L | −18 | −26 | 42 | 3.43 | ||
| Increased T1 lesion frequency | 12 | Anterior cingulum | L | −12 | 32 | 27 | 5.30 | |
| 41 | Anterior cingulum | R | 14 | 16 | 34 | 4.95 | ||
| 122 | Body of the CC | L | −15 | −12 | 34 | 4.90 | ||
| 38 | SLF | R | 38 | −24 | 33 | 4.71 | ||
| 33 | Uncinate fasciculus | R | 34 | 4 | −30 | 4.63 | ||
| 41 | Posterior CR | L | −21 | −26 | 36 | 4.61 | ||
| 39 | SLF | L | −33 | −26 | 30 | 4.54 | ||
| 31 | IFOF | L | −30 | −74 | 3 | 4.40 | ||
| 26 | Anterior CR | R | 15 | 9 | 45 | 4.29 | ||
| 27 | MCP | L | −10 | −48 | −34 | 4.03 | ||
| 28 | Splenium of the CC | R | 14 | −48 | 16 | 4.02 | ||
| 47 | MCP | R | 28 | −46 | −30 | 3.98 | ||
| 33 | Posterior cingulum | R | 16 | −52 | 34 | 3.98 | ||
| 18 | Splenium of the CC | L | −14 | −48 | 14 | 3.92 | ||
| 11 | Anterior CR | L | −24 | −21 | 16 | 3.91 | ||
| 43 | Body of the CC | L | −3 | 4 | 26 | 3.90 | ||
| 13 | CST | L | −20 | −30 | 50 | 3.83 | ||
| 16 | Forceps minor | R | 16 | 27 | 10 | 3.81 | ||
| 26 | IFOF | R | 26 | 16 | −6 | 3.74 | ||
| GM atrophy | 9806 | ACC | L | −4 | 15 | 27 | 5.25 | |
| IFG | L | −39 | 20 | 12 | 5.05 | |||
| Putamen | L | −33 | 8 | −2 | 4.87 | |||
| Rectus gyrus | L | −33 | 8 | −2 | 4.87 | |||
| Insula | L | −32 | 15 | 0 | 4.85 | |||
| Amygdala | L | −21 | −9 | −10 | 4.73 | |||
| 5300 | Supramarginal gyrus | L | −48 | −44 | 14 | 5.41 | ||
| MTG | L | −62 | −50 | −3 | 5.22 | |||
| STG | L | −64 | −24 | 6 | 5.09 | |||
| Postcentral gyrus | L | −56 | −21 | 27 | 4.72 | |||
| 2542 | Orbitofrontal gyrus | R | 44 | 38 | −4 | 5.29 | ||
| Insula | R | 36 | −18 | −4 | 4.59 | |||
| IFG | R | 48 | 34 | 3 | 4.45 | |||
| MTG | R | 52 | −14 | −16 | 4.36 | |||
| ITG | R | 48 | −6 | −28 | 4.20 | |||
| 2710 | MOG | R | 40 | −80 | 22 | 5.24 | ||
| Angular gyrus | R | 40 | −56 | 18 | 4.86 | |||
| PCC | R | 4 | −39 | 27 | 4.84 | |||
| SOG | R | 20 | −86 | 38 | 4.67 | |||
| Cuneus | R | 9 | −86 | 30 | 4.45 | |||
| Precuneus | R | 10 | −54 | 16 | 4.36 | |||
| 148 | Lingual gyrus | R | 27 | −56 | −4 | 5.11 | ||
| 1352 | MOG | L | −27 | −74 | 16 | 5.01 | ||
| Cuneus | L | −8 | −81 | 24 | 4.35 | |||
| 1828 | Orbitofrontal cortex | R | 10 | 64 | −8 | 4.84 | ||
| MFG | R | 26 | 32 | 27 | 4.67 | |||
| ACC | R | 15 | 27 | 26 | 4.59 | |||
| SFG | R | 18 | 28 | 42 | 4.56 | |||
| 52 | Calcarine cortex | L | −14 | −100 | 0 | 4.77 | ||
| 124 | Precuneus | L | −9 | −64 | 27 | 4.64 | ||
| 695 | Parahippocampal gyrus | L | −18 | −38 | −10 | 4.64 | ||
| Fusiform gyrus | L | −34 | −42 | −10 | 4.52 | |||
| Hippocampus | L | −15 | −39 | 4 | 4.30 | |||
| Lingual gyrus | L | −24 | −50 | −3 | 4.24 | |||
| 76 | Cerebellum Crus 2 | R | 10 | −90 | −33 | 4.61 | ||
| 178 | IFG | R | 48 | −15 | 20 | 4.58 | ||
| 251 | Thalamus | L | −9 | −16 | 6 | 4.57 | ||
| 559 | Cerebellum Crus 2 | L | −27 | −82 | −44 | 4.40 | ||
| Cerebellum (lobule 7) | L | −21 | −75 | −52 | 3.91 | |||
| 46 | Postcentral gyrus | L | −24 | −36 | 56 | 4.32 | ||
| 120 | Precentral gyrus | R | 50 | 4 | 24 | 4.31 | ||
| 181 | ITG | L | −57 | −16 | −33 | 4.31 | ||
| 163 | Cerebellum Crus 1 | L | −46 | −62 | −36 | 4.30 | ||
| 90 | Calcarine cortex | R | 12 | −84 | 9 | 4.27 | ||
| 29 | Cerebellum Crus 1 | R | 22 | −74 | −21 | 4.22 | ||
| 21 | PAG | L | −0 | −28 | −12 | 4.09 | ||
| 73 | Parahippocampal gyrus | R | 32 | −18 | −24 | 4.00 | ||
| Hippocampus | R | 24 | −12 | −24 | 3.56 | |||
| 25 | Caudate nucleus | L | −6 | 14 | −8 | 4.00 | ||
| 15 | ITG | L | −42 | −4 | −45 | 3.92 | ||
| 45 | MFG | L | −38 | 12 | 48 | 3.89 | ||
| 14 | Angular gyrus | R | 51 | −48 | 34 | 3.86 | ||
| 29 | Putamen | R | 22 | 15 | 2 | 3.86 | ||
| 20 | Cerebellum (lobule 8) | L | −18 | −50 | −52 | 3.85 | ||
| 35 | STG | R | 54 | −27 | 3 | 3.78 | ||
| 23 | Precentral gyrus | L | −24 | −28 | 51 | 3.77 | ||
| 18 | Cerebellum (lobule 9) | R | 9 | −57 | −38 | 3.74 | ||
| 18 | Cerebellum (lobule 6) | R | 22 | −57 | −30 | 3.68 | ||
| 87 | Supramarginal gyrus | R | 38 | −38 | 38 | 3.66 | ||
| 42 | Cerebellum (lobule 5) | L | −26 | −39 | −28 | 3.66 | ||
| 15 | Cerebellum (lobule 6) | L | −32 | −44 | −30 | 3.63 | ||
| 10 | Postcentral gyrus | R | 45 | −21 | 38 | 3.63 | ||
| 14 | Cerebellum (lobule 8) | R | 10 | −66 | −38 | 3.62 | ||
| 14 | Cerebellar vermis | R | 2 | −72 | −27 | 3.59 | ||
| 12 | PCC | R | 12 | −38 | 39 | 3.58 | ||
| 12 | Thalamus | R | 20 | −28 | 8 | 3.51 | ||
| 13 | Caudate | R | 12 | 14 | −9 | 3.50 | ||
| 16 | PCC | L | −8 | −46 | 28 | 3.50 | ||
| 10 | Fusiform gyrus | R | 28 | −50 | −12 | 3.43 | ||
| WM atrophy | 30503 | Anterior CR | L | −15 | 9 | 39 | 5.38a | |
| Anterior cingulum | L | −12 | 46 | 18 | 5.30a | |||
| SLF | L | −48 | −44 | 14 | 5.26a | |||
| IFOF | L | −39 | 21 | 12 | 5.18a | |||
| Uncinate fasciculus | L | −32 | 45 | 16 | 5.08 | |||
| Forceps minor | L | −15 | 57 | 20 | 5.00 | |||
| CST | L | −26 | −27 | 24 | 4.98 | |||
| 1442 | IFOF | R | 36 | −15 | −8 | 5.01 | ||
| Uncinate fasciculus | R | 38 | −3 | −18 | 4.92 | |||
| 5230 | Forceps minor | R | 10 | 64 | −8 | 4.69 | ||
| Anterior cingulum | R | 15 | 24 | 27 | 4.65 | |||
| 3089 | Forceps major | R | 26 | −62 | 20 | 4.67 | ||
| 3116 | ILF | L | −40 | −62 | −2 | 4.57 | ||
| 298 | SLF | R | 48 | −15 | 20 | 4.43 | ||
| 293 | Posterior cingulum | R | 6 | −39 | 26 | 4.28 | ||
| Splenium of the CC | R | 18 | −42 | 26 | 4.01 | |||
| 443 | Anterior CR | R | 27 | 20 | 10 | 4.17 | ||
| 185 | MCP | R | 22 | −50 | −33 | 3.85 | ||
| 62 | Splenium of the CC | L | −8 | −40 | 24 | 3.70 | ||
| 10 | Uncinate fasciculus | L | −38 | 14 | −33 | 3.68 | ||
| 645 | Body of the CC | R | 14 | −8 | 40 | 3.60 | ||
| 16 | ILF | R | 36 | 16 | −34 | 3.57 | ||
| 20 | MCP | L | −16 | −74 | −38 | 3.47 | ||
| 20 | CST | R | 26 | −18 | 32 | 3.38 | ||
P < 0.05 FWE corrected for multiple comparisons.
MNI = Montreal Neurological Institute; MS = multiple sclerosis; LPM = lesion probability map; GM = gray matter; WM = white matter; L = left; R = right; CR = corona radiata; IFOF = inferior fronto‐occipital fasciculus; CC = corpus callosum; SLF = superior longitudinal fasciculus; CST = corticospinal tract; ILF = inferior longitudinal fasciculus; SPL = superior parietal lobule; PCC = posterior cingulate cortex; ITG = inferior temporal gyrus; STG = superior temporal gyrus; IOG = inferior occipital gyrus; SFG = superior frontal gyrus; SMA = supplementary motor area; MTG = middle temporal gyrus; IFG = inferior frontal gyrus; MOG = middle occipital gyrus; ACC = anterior cingulate cortex; MFG = middle frontal gyrus; OR = optic radiation; MCP = middle cerebellar peduncle; SMC = supplementary motor cortex; SOG = superior occipital gyrus; PAG = periacqueductal gray matter.
EDSS Deterioration at FU
At FU, 36/66 (54.5%) MS patients had EDSS worsening. Compared to stable patients, they showed an higher frequency of T2 and T1 lesions in a few clusters in the right corticospinal tract (CST), body and splenium of the CC, and, bilaterally, in the inferior fronto‐occipital fasciculus (IFOF), superior longitudinal fasciculus (SLF), forceps major, and CR (Fig. 3 and Table 3) (P < 0.001, uncorrected). They also showed more significant GM atrophy of several fronto‐temporo‐parieto‐occipital regions, deep GM nuclei and the cerebellum, and of the majority of WM tracts (P < 0.001, uncorrected). Although the progression of GM and WM atrophy in worsened MS patients occurred bilaterally, a left‐lateralized distribution of such findings was evident (Fig. 3 and Table 3) (P < 0.001, uncorrected). Due to this finding, we quantified an LI, which confirmed a left‐predominant lateralization (+41% for the GM and +56% for the WM) (Fig. 4).
Figure 4.
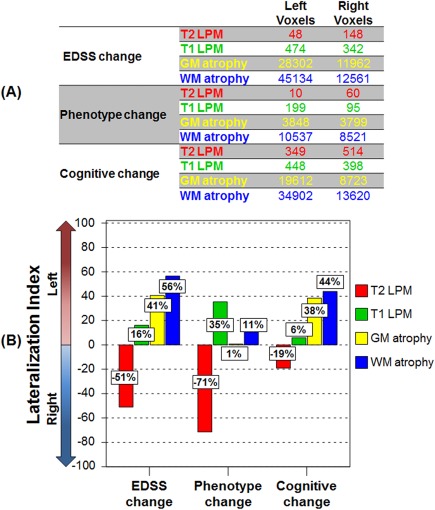
Lateralization index in the main study subgroups. Graphical representation of (a) total number of voxels and (b) lateralization index of number of voxels in the left and right hemispheres showing increased frequency of T2‐hyperintense lesions (red color‐coded) and T1‐hypointense lesions (green color‐coded), and gray matter (GM) (yellow color‐coded) and white matter (WM) (blue color‐coded) atrophy after 5 years in worsened versus stable multiple sclerosis (MS) patients according to the different outcomes. The lateralization index of between‐group differences was calculated as (total number of significant left hemisphere voxels − total number of significant right hemisphere voxels)/(total number of significant brain voxels) × 100. See text for further details. [Color figure can be viewed at http://wileyonlinelibrary.com]
Phenotype Change at FU
At FU, 14/66 (21.2%) MS patients evolved to a more severe clinical phenotype. Compared to stable patients, those with phenotype change showed a higher frequency of T2 lesions in the right IFOF and bilateral SLF, and higher frequency of T1 lesions in the right inferior longitudinal fasciculus (ILF), left SLF, forceps major, and splenium of the CC (Fig. 3 and Table 3) (P < 0.001, uncorrected). They also showed more significant GM atrophy in several fronto‐temporo‐parieto‐occipital regions, as well as of the majority of WM tracts, with a relative symmetric distribution of GM and WM atrophy in the two hemispheres (LI= +1% for the GM and +11% for the WM) (Fig. 4) (P < 0.001, uncorrected).
Cognitive Deterioration at FU
At FU, 18/63 (28.6%) MS patients had a worsening of cognitive functions. Neuropsychological tests showing the higher frequency of worsening were SRT delayed retrieval (46.0%), PASAT 2″ (34.9%), SPART delayed retrieval (22.2%), and WLG (22.2%). Compared to stable patients, cognitively worsened patients had a higher occurrence of T2 and T1 lesions in several regions involved in cognitive functions, including the CC, cingulum, SLF, ILF, IFOF, uncinate fasciculus, MCP, CST, and the forceps minor (Fig. 3 and Table 3) (P < 0.001, uncorrected). They also showed more significant GM atrophy in several fronto‐temporo‐parieto‐occipital regions, deep GM nuclei and the cerebellum, and of the majority of WM tracts (P < 0.001, uncorrected). The progression of GM and WM atrophy occurred bilaterally in worsened MS patients, but with a significant left‐lateralized pattern (Fig. 3 and Table 3) (P < 0.001, uncorrected). The LI quantification confirmed such a finding (+38% for the GM and +44% for the WM) (Fig. 4).
DISCUSSION
By combining lesional and volumetric MRI measurements and voxel‐based approaches to map the regional evolution of damage over a five‐year period in MS patients with the main MS clinical phenotypes, this multiparametric MRI study provides interesting pieces of information regarding the association between focal and diffuse brain damage accumulation and the progression of clinical disability and cognitive impairment. As the majority of MS patients received no treatment during the study, our results can be considered as representative of the natural progression of the disease.
Consistently with the literature, accumulation of WM focal lesions and progression of brain atrophy was found in MS patients after 5 years. The voxel‐wise analysis of T2 and T1 lesion changes showed that compared to stable patients, those with clinical and cognitive worsening formed significantly more new lesions in several clusters mainly located in supratentorial regions. Anatomically, such lesional clusters were located in clinically relevant WM tracts involved in motor (e.g., the CST and MCP) and cognitive functions (e.g., the IFOF, ILF, SLF, CR, and CC), thus confirming previous studies [Bodini et al., 2011; Wybrecht et al., 2012] which have suggested that not only the volume but also the topography of focal lesion accumulation, with the involvement of eloquent brain regions, might play an important role in determining disease clinical manifestations, possibly through a mechanism of disconnection.
The analysis of the longitudinal pattern of GM atrophy progression confirmed previous studies with shorter FU duration, which demonstrated the early involvement of deep GM structures by atrophic processes [Audoin et al., 2006; Rocca et al., 2016; Sepulcre et al., 2006], followed by atrophy of several cortical and subcortical structures [Audoin et al., 2006; Eshaghi et al., 2014; Rocca et al., 2016; Sepulcre et al., 2006]. We did not limit our analysis to GM volumetric changes, but included also the WM and found that widespread atrophy of several WM regions developed in MS patients after 5 years, with the involvement of several tracts located in the brain hemispheres, brainstem, and cerebellum. In line with previous studies with 1–3 years of FU [Battaglini et al., 2009; Bendfeldt et al., 2012], there was no anatomical overlap between areas affected by lesions and those affected by atrophy over time, suggesting that the accumulation of focal WM lesions plays only a partial role in explaining progressive tissue loss. Clearly, we cannot exclude that the evaluation of cortical lesions or the estimation of microstructural damage along the tracts affected by WM lesions would have allowed to characterize other possible mechanisms underlying atrophy development, as suggested by multiparametric, regional mapping studies [Bodini et al., 2016; Steenwijk et al., 2015].
The most interesting finding of our study derives from the analysis of the association between the regional patterns of lesions and atrophy progression and clinical deterioration in these patients, which showed that clinical relevant progression of irreversible GM/WM atrophy in MS patients occurred mainly in the left hemisphere, irrespective of focal lesion accumulation, which had a more symmetric pattern of distribution. Such a finding was confirmed by the quantification of LI, a measure already used in the literature [Lehericy et al., 2000; Seghier, 2008] to evaluate hemispheric lateralization. This suggests that a different vulnerability of the two brain hemispheres to irreversible structural damage may be among the factors contributing to clinical and cognitive deterioration in these patients.
While the left‐lateralized involvement for cognitive functions might be due to an asymmetry of representation of functions of the human brain and, therefore, depend from the battery of neuropsychological test administered, this is not the case for clinical deterioration, which was quantified with the EDSS (which is strongly influenced by locomotor ability). In addition, as we recruited only right‐handed [Oldfield, 1971] subjects, they cannot be attributed to different handedness among study subjects.
A growing number of studies has suggested that the two brain hemispheres have a different susceptibility to damage accumulation with aging and in course of diseases [Filippi et al., 1995; Lambrecq et al., 2013; Prinster et al., 2006; Thompson et al., 2003], even if these findings have not been confirmed by others [Araque Caballero et al., 2015; Audoin et al., 2006; Battaglini et al., 2009; Eshaghi et al., 2014; Rocca et al., 2016; Tosun et al., 2011]. In HC and patients with Alzheimer's disease, a two‐year longitudinal study detected faster GM loss in the left rather the right hemisphere [Thompson et al., 2003]. A meta‐analysis of volumetric studies in Huntington's disease found that neurodegenerative processes in this condition start in the left hemisphere and extend to the contralateral one over the course of the disease [Lambrecq et al., 2013].
Hemispheric asymmetry of damage accumulation in MS has been only partially investigated, with conflicting results [Audoin et al., 2006; Battaglini et al., 2009; Eshaghi et al., 2014; Filippi et al., 1995; Prinster et al., 2006; Rocca et al., 2016]. One study [Filippi et al., 1995] has demonstrated that hand dominance might be directly associated with interhemispheric lesion distribution, with a significant higher lesion burden in the dominant hemisphere. A cross‐sectional VBM study [Prinster et al., 2006] found a preferential cortical left‐sided GM loss in RRMS patients, which was not correlated with brain T2 LV.
Several factors can contribute to explain discrepancies among studies, including heterogeneous patients characteristics (disease duration, clinical phenotypes, treatments, etc.), methods used for the analyses (e.g., regional versus global), type of MRI measures investigated (lesion, microstructural tissue abnormalities, and/or irreversible tissue loss), and study settings (cross‐sectional vs longitudinal). Moreover, the majority of studies did not directly evaluate the presence of asymmetry in hemispheric involvement.
All together, these results suggest that the left hemisphere, which is dominant for both handedness and language in the majority of right‐handed subjects, might be more vulnerable to the accumulation of damage, independently from the underlying pathology. Several factors may contribute to explain this asymmetric distribution of damage, including higher susceptibility to neuronal and metabolic dysfunction of the left hemisphere, which might be the consequence of its overuse. The differential role of the two hemispheres in modulating immune function may also play a role, as suggested by studies in epileptic patients following surgery [Meador et al., 2004]. Structural involvement of the left hemisphere with a possible preservation of the right hemisphere might also explain recent results from functional MRI studies in aging, showing a decreased lateralization of sensorimotor, attentional, and frontal networks, which might reflect preserved compensatory mechanisms in the nondominant hemisphere [Agcaoglu et al., 2015]. Clearly further longitudinal studies, with larger samples of subjects and with the application of other MRI sequences sensitive and specific to different pathological substrates of the disease (e.g., diffusion tensor MRI) are necessary to confirm our results.
Our study is not without limitations. First, we limited our regional analysis to atrophy, which is an end‐stage phenomenon. The assessment of microstructural tissue abnormalities (for instance using diffusion tensor imaging) would have allowed us to tackle other processes responsible for disease worsening and measuring secondary degenerative phenomena along the main WM tracts. Second, our protocol did not include imaging of the spinal cord, which is a relevant structure whose damage might have a significant role in determining disability progression. Third, our cohort includes patients with heterogeneous clinical phenotypes. Owing to the relative small number of patients per group, we could not analyze progression of damage in the main clinical phenotypes, separately, which might be characterized by different pathological substrates (e.g., demyelination versus neurodegeneration).
CONFLICTS OF INTEREST
P. Preziosa received speakers honoraria from Biogen Idec, Novartis, and ExceMED. E. Pagani, S. Mesaros, G.C. Riccitelli, J. Dackovic, and J. Drulovic report no disclosures. M. Filippi serves on a scientific advisory board for Teva Pharmaceutical Industries; has received compensation for consulting services and/or speaking activities from Biogen Idec, ExceMED, Novartis, and Teva Pharmaceutical Industries; and receives research support from Biogen Idec, Teva Pharmaceutical Industries, Novartis, Italian Ministry of Health, Fondazione Italiana Sclerosi Multipla, Cure PSP, Alzheimer's Drug Discovery Foundation (ADDF), the Jacques and Gloria Gossweiler Foundation (Switzerland), and ARiSLA (Fondazione Italiana di Ricerca per la SLA). M.A. Rocca received speakers honoraria from Biogen Idec, Novartis, Genzyme, Sanofi‐Aventis, Teva, and Merk Serono and received research support from the Italian Ministry of Health and Fondazione Italiana Sclerosi Multipla.
REFERENCES
- Agcaoglu O, Miller R, Mayer AR, Hugdahl K, Calhoun VD (2015): Lateralization of resting state networks and relationship to age and gender. NeuroImage 104:310–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amato MP, Portaccio E, Goretti B, Zipoli V, Battaglini M, Bartolozzi ML, Stromillo ML, Guidi L, Siracusa G, Sorbi S, Federico A, De Stefano N (2007): Association of neocortical volume changes with cognitive deterioration in relapsing‐remitting multiple sclerosis. Arch Neurol 64:1157–1161. [DOI] [PubMed] [Google Scholar]
- Araque Caballero MA, Brendel M, Delker A, Ren J, Rominger A, Bartenstein P, Dichgans M, Weiner MW, Ewers M Alzheimer's Disease Neuroimaging, I (2015): Mapping 3‐year changes in gray matter and metabolism in Abeta‐positive nondemented subjects. Neurobiol Aging 36:2913–2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner J (2007): A fast diffeomorphic image registration algorithm. NeuroImage 38:95–113. [DOI] [PubMed] [Google Scholar]
- Ashburner J, Friston KJ (2005): Unified segmentation. NeuroImage 26:839–851. [DOI] [PubMed] [Google Scholar]
- Ashburner J, Ridgway GR (2012): Symmetric diffeomorphic modeling of longitudinal structural MRI. Front Neurosci 6:197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audoin B, Davies GR, Finisku L, Chard DT, Thompson AJ, Miller DH (2006): Localization of grey matter atrophy in early RRMS: A longitudinal study. J Neurol 253:1495–1501. [DOI] [PubMed] [Google Scholar]
- Battaglini M, Giorgio A, Stromillo ML, Bartolozzi ML, Guidi L, Federico A, De Stefano N (2009): Voxel‐wise assessment of progression of regional brain atrophy in relapsing‐remitting multiple sclerosis. J Neurol Sci 282:55–60. [DOI] [PubMed] [Google Scholar]
- Battaglini M, Jenkinson M, De Stefano N (2012): Evaluating and reducing the impact of white matter lesions on brain volume measurements. Hum Brain Mapp 33:2062–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendfeldt K, Hofstetter L, Kuster P, Traud S, Mueller‐Lenke N, Naegelin Y, Kappos L, Gass A, Nichols TE, Barkhof F, Vrenken H, Roosendaal SD, Geurts JJ, Radue EW, Borgwardt SJ (2012): Longitudinal gray matter changes in multiple sclerosis–differential scanner and overall disease‐related effects. Hum Brain Mapp 33:1225–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendfeldt K, Kuster P, Traud S, Egger H, Winklhofer S, Mueller‐Lenke N, Naegelin Y, Gass A, Kappos L, Matthews PM, Nichols TE, Radue EW, Borgwardt SJ (2009): Association of regional gray matter volume loss and progression of white matter lesions in multiple sclerosis ‐ A longitudinal voxel‐based morphometry study. NeuroImage 45:60–67. [DOI] [PubMed] [Google Scholar]
- Benedict RH, Bruce J, Dwyer MG, Weinstock‐Guttman B, Tjoa C, Tavazzi E, Munschauer FE, Zivadinov R (2007): Diffusion‐weighted imaging predicts cognitive impairment in multiple sclerosis. Multiple Sclerosis 13:722–730. [DOI] [PubMed] [Google Scholar]
- Bodini B, Battaglini M, De Stefano N, Khaleeli Z, Barkhof F, Chard D, Filippi M, Montalban X, Polman C, Rovaris M, Rovira A, Samson R, Miller D, Thompson A, Ciccarelli O (2011): T2 lesion location really matters: A 10 year follow‐up study in primary progressive multiple sclerosis. J Neurol Neurosurg Psychiatry 82:72–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodini B, Chard D, Altmann DR, Tozer D, Miller DH, Thompson AJ, Wheeler‐Kingshott C, Ciccarelli O (2016): White and gray matter damage in primary progressive MS: The chicken or the egg? Neurology 86:170–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshaghi A, Bodini B, Ridgway GR, Garcia‐Lorenzo D, Tozer DJ, Sahraian MA, Thompson AJ, Ciccarelli O (2014): Temporal and spatial evolution of grey matter atrophy in primary progressive multiple sclerosis. NeuroImage 86:257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippi M, Martino G, Mammi S, Campi A, Comi G, Grimaldi LM (1995): Does hemispheric dominance influence brain lesion distribution in multiple sclerosis? J Neurol Neurosurg Psychiatry 58:748–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippi M, Preziosa P, Copetti M, Riccitelli G, Horsfield MA, Martinelli V, Comi G, Rocca MA (2013): Gray matter damage predicts the accumulation of disability 13 years later in MS. Neurology 81:1759–1767. [DOI] [PubMed] [Google Scholar]
- Fisher E, Lee JC, Nakamura K, Rudick RA (2008): Gray matter atrophy in multiple sclerosis: A longitudinal study. Ann Neurol 64:255–265. [DOI] [PubMed] [Google Scholar]
- Fisniku LK, Brex PA, Altmann DR, Miszkiel KA, Benton CE, Lanyon R, Thompson AJ, Miller DH (2008a): Disability and T2 MRI lesions: A 20‐year follow‐up of patients with relapse onset of multiple sclerosis. Brain 131:808–817. [DOI] [PubMed] [Google Scholar]
- Fisniku LK, Chard DT, Jackson JS, Anderson VM, Altmann DR, Miszkiel KA, Thompson AJ, Miller DH (2008b): Gray matter atrophy is related to long‐term disability in multiple sclerosis. Ann Neurol 64:247–254. [DOI] [PubMed] [Google Scholar]
- Friston KJ, Holmes AP, Poline JB, Grasby PJ, Williams SC, Frackowiak RS, Turner R (1995): Analysis of fMRI time‐series revisited. NeuroImage 2:45–53. [DOI] [PubMed] [Google Scholar]
- Khaleeli Z, Ciccarelli O, Manfredonia F, Barkhof F, Brochet B, Cercignani M, Dousset V, Filippi M, Montalban X, Polman C, Rovaris M, Rovira A, Sastre‐Garriga J, Vellinga M, Miller D, Thompson A (2008): Predicting progression in primary progressive multiple sclerosis: A 10‐year multicenter study. Ann Neurol 63:790–793. [DOI] [PubMed] [Google Scholar]
- Lambrecq V, Langbour N, Guehl D, Bioulac B, Burbaud P, Rotge JY (2013): Evolution of brain gray matter loss in Huntington's disease: A meta‐analysis. Eur J Neurol 20:315–321. [DOI] [PubMed] [Google Scholar]
- Lansley J, Mataix‐Cols D, Grau M, Radua J, Sastre‐Garriga J (2013): Localized grey matter atrophy in multiple sclerosis: A meta‐analysis of voxel‐based morphometry studies and associations with functional disability. Neurosci Biobehav Rev 37:819–830. [DOI] [PubMed] [Google Scholar]
- Lehericy S, Cohen L, Bazin B, Samson S, Giacomini E, Rougetet R, Hertz‐Pannier L, Le Bihan D, Marsault C, Baulac M (2000): Functional MR evaluation of temporal and frontal language dominance compared with the Wada test. Neurology 54:1625–1633. [DOI] [PubMed] [Google Scholar]
- Lublin FD, Reingold SC (1996): Defining the clinical course of multiple sclerosis: Results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology 46:907–911. [DOI] [PubMed] [Google Scholar]
- Meador KJ, Loring DW, Ray PG, Helman SW, Vazquez BR, Neveu PJ (2004): Role of cerebral lateralization in control of immune processes in humans. Ann Neurol 55:840–844. [DOI] [PubMed] [Google Scholar]
- Obradovic D, Petrovic M, Antanasijevic I, Marinkovic J, Stojanovic T, Obradovic S (2012): The Brief Repeatable Battery: Psychometrics and normative values with age, education and gender corrections in a Serbian population. Neurol Sci 33:1369–1374. [DOI] [PubMed] [Google Scholar]
- Oldfield RC (1971): The assessment and analysis of handedness: The Edinburgh inventory. Neuropsychologia 9:97–113. [DOI] [PubMed] [Google Scholar]
- Polman CH, Reingold SC, Banwell B, Clanet M, Cohen JA, Filippi M, Fujihara K, Havrdova E, Hutchinson M, Kappos L, Lublin FD, Montalban X, O'Connor P, Sandberg‐Wollheim M, Thompson AJ, Waubant E, Weinshenker B, Wolinsky JS (2011): Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol 69:292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portaccio E, Stromillo ML, Goretti B, Hakiki B, Giorgio A, Rossi F, De Leucio A, De Stefano N, Amato MP (2013): Natalizumab may reduce cognitive changes and brain atrophy rate in relapsing‐remitting multiple sclerosis–a prospective, non‐randomized pilot study. Eur J Neurol 20:986–990. [DOI] [PubMed] [Google Scholar]
- Prinster A, Quarantelli M, Orefice G, Lanzillo R, Brunetti A, Mollica C, Salvatore E, Morra VB, Coppola G, Vacca G, Alfano B, Salvatore M (2006): Grey matter loss in relapsing‐remitting multiple sclerosis: A voxel‐based morphometry study. NeuroImage 29:859–867. [DOI] [PubMed] [Google Scholar]
- Rao SM, the Cognitive Function Study Group of the National Multiple Sclerosis Society (1990): A Manual for the Brief Repeteable Battery of Neuropsychological Tests in Multiple Sclerosis. Milwakee, WI: Medical College of Wisconsin. [Google Scholar]
- Rocca MA, Preziosa P, Mesaros S, Pagani E, Dackovic J, Stosic‐Opincal T, Drulovic J, Filippi M (2016): Clinically isolated syndrome suggestive of multiple sclerosis: Dynamic patterns of gray and white matter changes—A 2‐year MR imaging study. Radiology 278:841–853. [DOI] [PubMed] [Google Scholar]
- Seghier ML (2008): Laterality index in functional MRI: Methodological issues. Magn Reson Imag 26:594–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepulcre J, Sastre‐Garriga J, Cercignani M, Ingle GT, Miller DH, Thompson AJ (2006): Regional gray matter atrophy in early primary progressive multiple sclerosis: A voxel‐based morphometry study. Arch Neurol 63:1175–1180. [DOI] [PubMed] [Google Scholar]
- Steenwijk MD, Daams M, Pouwels PJ, L JB, Tewarie PK, Geurts JJ, Barkhof F, Vrenken H (2015): Unraveling the relationship between regional gray matter atrophy and pathology in connected white matter tracts in long‐standing multiple sclerosis. Hum Brain Mapp 36:1796–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers M, Swanton J, Fernando K, Dalton C, Miller DH, Cipolotti L, Ron MA (2008): Cognitive impairment in multiple sclerosis can be predicted by imaging early in the disease. J Neurol Neurosurg Psychiatry 79:955–958. [DOI] [PubMed] [Google Scholar]
- Thompson PM, Hayashi KM, de Zubicaray G, Janke AL, Rose SE, Semple J, Herman D, Hong MS, Dittmer SS, Doddrell DM, Toga AW (2003): Dynamics of gray matter loss in Alzheimer's disease. J Neurosci 23:994–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tintore M, Rovira A, Rio J, Otero‐Romero S, Arrambide G, Tur C, Comabella M, Nos C, Arevalo MJ, Negrotto L, Galan I, Vidal‐Jordana A, Castillo J, Palavra F, Simon E, Mitjana R, Auger C, Sastre‐Garriga J, Montalban X (2015): Defining high, medium and low impact prognostic factors for developing multiple sclerosis. Brain 138:1863–1874. [DOI] [PubMed] [Google Scholar]
- Tosun D, Schuff N, Mathis CA, Jagust W, Weiner MW, Alzheimer's Disease NeuroImaging I (2011): Spatial patterns of brain amyloid‐beta burden and atrophy rate associations in mild cognitive impairment. Brain 134:1077–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wybrecht D, Reuter F, Zaaraoui W, Faivre A, Crespy L, Rico A, Malikova I, Confort‐Gouny S, Soulier E, Cozzone PJ, Pelletier J, Ranjeva JP, Audoin B (2012): Voxelwise analysis of conventional magnetic resonance imaging to predict future disability in early relapsing‐remitting multiple sclerosis. Multiple Sclerosis 18:1585–1591. [DOI] [PubMed] [Google Scholar]