

Abstract
The tryptophan hydroxylase‐2 (TPH2) gene is considered a promising genetic candidate regarding its association with a predisposition to major depressive disorder (MDD). Local gyrification reflects the early neural development of cortical connectivity, and is regarded as a potential neural endophenotype in psychiatric disorders. They aimed to investigate the alterations in the cortical gyrification of the prefrontal cortex and anterior cingulate cortex and their association with the TPH2 rs4570625 polymorphism in patients with MDD. One hundred and thirteen patients with MDD and eighty‐six healthy controls underwent T1‐weighted structural magnetic resonance imaging and genotyping for TPH2 rs4570625. The local gyrification index of 22 cortical regions in the prefrontal cortex and anterior cingulate cortex was analyzed using the FreeSurfer. The patients with MDD showed significant hypergyria in the right rostral anterior cingulate cortex (P = 0.001), medial orbitofrontal cortex (P = 0.003), and frontal pole (P = 0.001). There was a significant genotype‐by‐diagnosis interaction for the local gyrification index in the right rostral anterior cingulate cortex (P = 0.003). Their study revealed significant hypergyria of the anterior cingulate cortex and prefrontal cortex and an interactive effect between the diagnosis of MDD and the genotype in the anterior cingulate cortex. This might be associated with the dysfunction of neural circuits mediating emotion processing, which could contribute to pathophysiology of MDD. Hum Brain Mapp 38:1299–1310, 2017. © 2016 Wiley Periodicals, Inc.
Keywords: TPH2 rs4570625, local gyrification index, major depressive disorder, prefrontal cortex, anterior cingulate cortex, orbitofrontal cortex
INTRODUCTION
The etiology of major depressive disorder (MDD) is multifaceted and is characterized by a complex interplay between genetic variants and environmental exposure that produces functional and structural alterations in the neural networks of emotion processing [Kupfer et al., 2012]. The neural‐circuit model of emotion dysregulation in MDD can be conceptualized as a hyper‐activation of subcortical limbic structures, and a failure of emotion control by the lateral and medial prefrontal cortex (PFC) and anterior cingulate cortex (ACC) [Phillips et al., 2008; Rive et al., 2013]. Cumulative evidence has supported this model by consistently reporting morphological and functional abnormalities of these brain regions in patients with MDD [Bora et al., 2012; Koolschijn et al., 2009; Rive et al., 2013]. This prefrontal–limbic‐network system is modulated by serotonin neurotransmission [Kupfer et al., 2012], and the influence of serotonin‐related genetic variants on structural and morphological changes in the PFC, ACC, and limbic regions has been reported in numerous imaging genetic studies [Won and Ham, 2016].
Because gyrification reflects the pattern and degree of cortical folding, several studies have proposed that abnormal gyrification might be a more stable indicator of cortical pathology than previous neuroanatomical parameters due to its putative state independence. In addition, gyrification can now be evaluated with an automatically reconstructed cortical surface model [Nenadic et al., 2015]. Local gyrification is known to reflect the early neural development of cortical connectivity in which the fiber tension of densely connected cortical regions forms gyri and sparsely connected regions move apart to become separated by sulci during the second trimester of pregnancy [Nanda et al., 2014]. Abnormal gyrification is thought to reflect disrupted functional connectivity of the brain cortex [Dauvermann et al., 2012; Nixon et al., 2014]. Recent research has suggested that the gyrification index could describe a useful endophenotype of several psychiatric disorders including schizophrenia, bipolar disorder, and schizoaffective disorder [Janssen et al., 2014; Nanda et al., 2014].
Several studies have investigated the regional folding pattern of the cortex and reported aberrant gyrification of the ACC, orbitofrontal cortex (OFC), posterior mid‐cingulate cortex, and cortical regions belonging to the default mode network (DMN) in patients with MDD [Nixon et al., 2014; Peng et al., 2015; Zhang et al., 2009]. Considering the important role of these cortical regions in emotion processing and mood regulation [Phillips et al., 2008; Rive et al., 2013], and their abnormal functional connectivity in patients with MDD [Kaiser et al., 2015b; Workman et al., 2016], an aberrant cortical folding pattern might be deeply implicated in the pathophysiology of MDD. The above‐mentioned analyses on gyrification and MDD were based on relatively small samples (16–20 patients with MDD), and generated controversy regarding the direction of the alteration in cortical gyrification (i.e., hyper‐ or hypogyria). Thus, replication of these results is required with a large sample size.
Even though cumulative evidence has suggested that local gyrification could be a potential neural endophenotype in psychiatric disorders, there have been no studies to date that have investigated the association between the cortical folding pattern and candidate variants of genes involved in serotonin neurotransmission in MDD. There is an increasing need for a comprehensive imaging genetic study of MDD that investigates both the level of local gyrification and its potential association with a serotonin‐related genetic candidate using a sufficiently large study sample. Among the various serotonergic genes, the tryptophan hydroxylase‐2 (TPH2) gene that encodes the rate‐limiting enzyme responsible for the biosynthesis of serotonin is considered a promising genetic candidate for an association with MDD [Gao et al., 2012; Serretti et al., 2011; Won and Ham, 2016]. Strong epidemiological evidence indicates that G‐703 T (rs4570625), which is a single‐nucleotide polymorphism (SNP) that is located in the putative promoter region of this gene, is correlated with MDD [Gao et al., 2012]. Furthermore, serotonergic neurotransmission is capable of modulating neuronal migration, cell proliferation, synaptogenesis and dendritic maturation during cortical development [Gaspar et al., 2003; Vitalis et al., 2007]. Considering that cell proliferation and migration greatly influence the driving force behind cortical folding during cortical development [Zilles et al., 2013], the TPH2 gene which affects serotonergic neurotransmission in the brain may be associated with changes in local gyrification observed in patients with MDD.
Therefore, we aimed to investigate altered cortical gyrification in patients with MDD by measuring the local gyrification index (LGI) in a relatively large sample. We also aimed to elucidate the association between TPH2 rs4570625 and LGI in patients with MDD. Based on the results of our previous imaging genetic study of this SNP [Yoon et al., 2012], we assumed that the G allele might be a risk allele for the MDD‐related LGI changes. We used the PFC and the ACC as our regions of interest (ROIs), because these regions are deeply involved in the pathophysiology of MDD and play a pivotal role in the emotion processing network [Phillips et al., 2015]. In addition, most imaging genetic studies on serotonin‐related genes have reported structural changes in these cortical regions [Won and Ham, 2016]. Our a priori hypotheses were as follows: (1) the patients with MDD will demonstrate significantly altered gyrification in the ACC and/or PFC compared with healthy controls; (2) the diagnosis of MDD and the TPH2 rs4570625 genotype will have significant interactive effects on the LGI of the ACC and/or PFC.
METHODS AND MATERIALS
Participants
A total of 113 patients with MDD were recruited from the outpatient psychiatric clinic of Korea University Anam Hospital, located in Seoul, Republic of Korea. The inclusion criteria for the MDD group were as follows: (1) adults aged 20–65 years; (2) meeting the diagnosis criteria of MDD in DSM‐IV. The diagnosis of MDD based on DSM‐IV criteria was made by a board‐certified psychiatrist, and an independent psychiatrist confirmed the diagnosis using Structured Clinical Interview for DSM‐IV Axis I disorders (SCID‐1) to improve its validity. The concordance rate for the diagnosis of MDD by the two board‐certified psychiatrists was above 0.95. Two psychiatrists assessed the duration of illness for the patients with MDD by conducting interviews using the life‐chart methodology. The exclusion criteria for patients with MDD were as follows: (1) presumptive primary comorbid diagnosis of any other major psychiatric illness (based on DSM‐IV criteria) on Axis I or Axis II, within the last 6 months; (2) MDD with psychotic features; (3) acute suicidal or homicidal patients requiring inpatient treatment; (4) history of serious or unstable medical illness; (5) abnormal findings in a physical examination and routine laboratory tests; (6) primary neurological illness; and (7) any contraindication for MRI including pacemakers, metal implants, and claustrophobia. A total of 86 healthy participants aged 20–65 years without any history of psychiatric illness were recruited from the community via advertisements as the healthy control group. Board‐certified psychiatrists evaluated the healthy control participants using SCID‐1 and confirmed that they had no current or previous psychiatric disorders. We also used the same exclusion criteria as those used for patients with MDD. We found that all participants in this study were confirmed as right‐handed using the Edinburgh Handedness Test [Oldfield, 1971]. All participants were self‐identified Koreans and their ethnicity was confirmed through the identification of the ethnicity of three generations of the participants' families. The patients with MDD and healthy control participants were evaluated for the severity of their depressive symptoms on the same day as the MRI scans using the 17‐item Hamilton Depression Rating Scale (HDRS) [Hamilton, 1960]. The details of the antidepressant treatment in the MDD group are described in Table I. Our study protocol was approved by the Institutional Review Board of Korea University Anam Hospital. In accordance with the Declaration of Helsinki, all participants gave written informed consent to participate in the study.
Table I.
Demographic and clinical characteristics of patients with major depressive disorder and healthy controls
| MDD (n = 113) | HC (n = 86) | P value | |
|---|---|---|---|
| Age | 42.78 ± 1.07 | 39.23 ± 1.46 | 0.051 |
| Gender (female/male) | 90/23 | 59/27 | 0.099 |
| Education level | |||
| Elementary and middle school | 25 | 11 | 0.128 |
| High school or college/university | 80 | 64 | |
| Above graduate school | 8 | 11 | |
| HDRS‐17 score | 15.05 ± 0.74 | 2.23 ± 0.23 | <0.001 |
| Duration of illness (months) | 45.81 ± 4.60 | ||
| TPH2 gene rs4570625 | |||
| TT | 26 | 30 | 0.174 |
| TG | 60 | 40 | |
| GG | 27 | 16 | |
| HWE | 0.510 | 0.680 | |
| TT + TG | 86 | 70 | 0.391 |
| GG | 27 | 16 | |
| T (allele frequency) | 112 | 100 | 0.105 |
| G (allele frequency) | 114 | 72 | |
| Drug‐naïve/Antidepressant | 52/61 | ||
| TT + TG | 38/48 | 0.514 | |
| GG | 14/13 | ||
| Antidepressant type | |||
| SSRI | 31 | ||
| SNRI | 9 | ||
| NDRI | 6 | ||
| NaSSA | 4 | ||
| Combination | 11 |
Data are means ± standard error for age, HDRS‐17 scores, and duration of illness.
The P values for distributions of gender, education level, TPH2 genotype, allele frequency, and drug‐naïve patients according to the genotype were obtained by chi‐square test.
The P values for comparisons of age and HDRS‐17 scores were obtained using independent t‐tests.
Allele frequencies (T/G): MDD patients 0.50/0.50, HC subjects 0.58/0.42.
MDD, major depressive disorder; HC, healthy controls; HDRS‐17, Hamilton Depression Rating Scale; HWE, Hardy‐Weinberg equilibrium; SSRI, selective serotonin reuptake inhibitor; SNRI, serotonin and norepinephrine reuptake inhibitor; NDRI, norepinephrine‐dopamine reuptake inhibitor; NaSSA, noradrenergic and specific serotonergic antidepressant; Combination, combinations of two or more types of antidepressants.
Genotyping Analysis
Genomic DNA samples extracted from the peripheral venous blood of each participant were used in the genotyping of TPH2 rs4570625 according to a standard protocol, as previously described [Canli et al., 2005]. Polymerase chain reaction was performed using the following primers: forward, 5′‐TGCATAGAGGCATCACAGGA‐3′; reverse, 5′‐CATTCCAATTCCACTCTTCCA‐3′; extension, 5′‐CTCACACATTTGCATGCACAAAATTA‐3′. The genotyping success rate in our study was above 95%. The details of the allele frequencies and Hardy–Weinberg equilibrium were described in Table I.
MRI Data Acquisition
MRI scans were acquired parallel to the anterior‐commissure–posterior‐commissure line using a 3.0 T Siemens Trio whole‐body imaging system (Siemens Medical Systems, Iselin, NJ), using 3D T1‐weighted magnetization‐prepared rapid gradient‐echo (MP‐RAGE) with the following parameters: 1,900 ms repetition time, 2.6 ms echo time, 220 mm field of view, 256 × 256 matrix size, 1 mm slice thickness, 176 coronal slices without gap, 0.86 × 0.86 × 1 mm3 voxels, 16° flip angle, number of excitations = 1. After individual's MRI scanning, the artifacts of MRI system and motion were visually checked, and when the artifacts were observed, the subject's MRI was rescanned.
Image Processing
The average LGI values of each cortical parcellation in the bilateral PFC and ACC were calculated in the 3D T1 images with the automated reconstruction process for whole brain structures that is implemented in the FreeSurfer 5.3 Development Version (Massachusetts General Hospital, Boston, http://surfer.nmr.mgh.harvard.edu). The average LGI was defined as the ratio of the buried cortex surface area to outer convex (hull) surface area in each parcellated cortical region. We calculated the LGI values according to a previously reported standard protocol [Nanda et al., 2014]. To calculate the LGI values, a three‐dimensional model of the cortical surface reconstructions computed from T1 images was pre‐processed in FreeSurfer. Detailed technical aspects of procedures used in the cortical reconstruction have been described in previous literature [Dale et al., 1999; Fischl et al., 2001, 1999, 2002, 2004; Segonne et al., 2007]. After the sequential processes of cortical reconstruction, the LGI was calculated by measuring the ratio of the sulcal and buried surface to the outer hull surface, which indicates the extent of cortical folding in spherical three‐dimensional regions of interest, using an automated FreeSurfer procedure, as described previously [Nanda et al., 2014]. Each hemisphere was automatically parcellated into 33 distinct cortical regions according to the Desikan–Killiany atlas [Desikan et al., 2006]. The mean LGI values of these regions were automatically calculated. Among the LGI values of the 33 cortical parcels, those of the 11 cortical regions (per hemisphere) comprising the PFC or the ACC were used in the final analysis. We selected the 11 cortical regions within Desikan–Killiany atlas according to a previous imaging study, which used the PFC and the ACC as their ROIs [Kremen et al., 2010]. The list of 11 cortical regions included in the PFC or ACC is described in Table II. The LGI maps of all subjects which were separately created in the analysis and the automated cortical parcellation according to the Desikan–Killiany atlas are shown in Figure 1.
Table II.
The differences in local gyrification indexes among groups determined by genotype and diagnosis
| Cortical regions | MDD vs. HC | GG vs. TT + TG | Diagnosis × Genotype interaction | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| F | P | Cohen's f | F | P | Cohen's f | F | P | Cohen's f | ||
| L superior frontal cortex | 0.273 | 0.602 | 0.038 | 1.214 | 0.272 | 0.080 | 0.147 | 0.702 | 0.028 | |
| L rostral anterior cingulate cortex | 2.817 | 0.095 | 0.121 | 0.196 | 0.659 | 0.032 | 0.684 | 0.409 | 0.060 | |
| L caudal anterior cingulate cortex | 0.184 | 0.668 | 0.031 | 0.185 | 0.668 | 0.031 | 0.475 | 0.492 | 0.050 | |
| L rostral middle frontal cortex | 0.245 | 0.621 | 0.036 | 0.893 | 0.346 | 0.068 | 2.349 | 0.127 | 0.111 | |
| L caudal middle frontal cortex | 0.005 | 0.945 | 0.005 | 2.063 | 0.153 | 0.104 | 0.517 | 0.473 | 0.052 | |
| L medial orbitofrontal cortex | 4.234 | 0.041 | 0.149 | MDD > HC | 0.657 | 0.419 | 0.059 | 1.451 | 0.230 | 0.087 |
| L lateral orbitofrontal cortex | 2.272 | 0.133 | 0.109 | 2.451 | 0.119 | 0.113 | 2.414 | 0.122 | 0.112 | |
| L pars opercularis | 1.534 | 0.217 | 0.090 | 1.788 | 0.183 | 0.097 | 1.275 | 0.260 | 0.082 | |
| L pars orbitalis | 0.009 | 0.923 | 0.007 | 3.797 | 0.053 | 0.141 | 0.028 | 0.866 | 0.012 | |
| L pars triangularis | 0.005 | 0.946 | 0.005 | 0.876 | 0.350 | 0.068 | 0.243 | 0.622 | 0.036 | |
| L frontal pole | 2.097 | 0.149 | 0.105 | 0.526 | 0.469 | 0.053 | 0.306 | 0.581 | 0.040 | |
| R superior frontal cortex | 2.617 | 0.107 | 0.117 | 0.110 | 0.740 | 0.024 | 2.059 | 0.153 | 0.104 | |
| R rostral anterior cingulate cortex | 11.576 | 0.001a | 0.246 | MDD > HC | <0.001 | 0.994 | 0.001 | 9.265 | 0.003a | 0.220 |
| R caudal anterior cingulate cortex | 1.969 | 0.162 | 0.102 | <0.001 | 0.982 | 0.002 | 0.328 | 0.567 | 0.041 | |
| R rostral middle frontal cortex | 2.470 | 0.118 | 0.114 | 0.129 | 0.720 | 0.026 | 0.057 | 0.811 | 0.017 | |
| R caudal middle frontal cortex | 0.778 | 0.379 | 0.064 | 0.632 | 0.428 | 0.058 | 3.325 | 0.070 | 0.132 | |
| R medial orbitofrontal cortex | 8.897 | 0.003a | 0.216 | MDD > HC | 0.137 | 0.712 | 0.027 | 5.609 | 0.019 | 0.171 |
| R lateral orbitofrontal cortex | 2.738 | 0.100 | 0.120 | 0.714 | 0.399 | 0.061 | 3.822 | 0.052 | 0.141 | |
| R pars opercularis | 0.457 | 0.500 | 0.049 | 0.026 | 0.873 | 0.012 | 1.345 | 0.248 | 0.084 | |
| R pars orbitalis | 2.489 | 0.116 | 0.114 | 1.435 | 0.232 | 0.087 | 2.347 | 0.127 | 0.111 | |
| R pars triangularis | 1.877 | 0.172 | 0.099 | 0.004 | 0.950 | 0.005 | 0.993 | 0.320 | 0.072 | |
| R frontal pole | 12.248 | 0.001a | 0.253 | MDD > HC | 0.615 | 0.434 | 0.057 | 3.677 | 0.057 | 0.139 |
The F and P values were obtained using two‐way analysis of covariance adjusted for age, gender, education level, and total surface area as covariates.
The False Discovery Rate (FDR) was applied in the analyses of diagnosis effect (MDD vs. HC) and diagnosis‐by‐genotype interaction.
Cortical regions that remained significant after FDR correction are marked with an asterisk.
MDD, major depressive disorder; HC, healthy controls; GG, GG genotype of rs4570625; TT + TG, TT or TG genotype of rs4570625; L, left hemisphere; R, right hemisphere.
Figure 1.
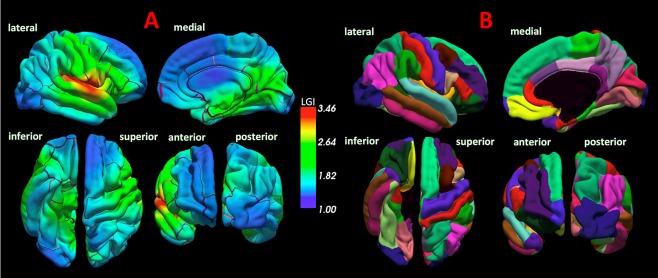
The local gyrification index (LGI) map and the automated cortical parcellation. The panel A represents the LGI maps of all subjects which were separately created in the analysis, and the color bar represents LGI scale. The panel B represents the cortical parcellation criteria of the Desikan–Killiany atlas. [Color figure can be viewed at http://wileyonlinelibrary.com]
Statistical Analyses
In the main analysis, the LGI values extracted from the 22 cortical regions comprising the PFC or the ACC in both hemispheres were compared between the MDD and healthy control groups and the TPH2 rs4570625 genotype groups (GG vs. TT + TG), and the genotype‐by‐diagnosis interaction was further investigated. The genotype groups were divided based on the dominant model (comparing risk‐allele homozygotes with carriers of the non‐risk allele) in accordance with previous imaging studies on the TPH2 gene [Brown et al., 2005; Canli et al., 2005; Inoue et al., 2010; Yoon et al., 2012]. The LGI values that were automatically calculated by the FreeSurfer were analyzed with a two‐way analysis of covariance (ANCOVA) that included the individual LGI values as the dependent variables; diagnosis and genotype as the independent variables; and age, gender, education level (scores of 1–3 according to level in the Korean educational system), and demeaned value of the total cortical surface area of the whole brain of each participant (calculated with an automated FreeSurfer procedure) as covariates according to the statistical methods used in previous imaging genetic studies [Carballedo et al., 2012; Fani et al., 2013; Fani et al., 2014; Kohli et al., 2011; Murphy et al., 2012]. In order to correct for multiple comparisons, we applied False Discovery Rate (FDR) correction [Benjamini and Hochberg, 1995] to both, the main effect of diagnosis and the diagnosis‐by‐genotype interaction using q < 0.05, a total of 44 comparisons (22 comparisons of LGI values according to the diagnosis group and 22 analyses of the diagnosis‐by‐genotype interaction). Age and HDRS scores were analyzed using independent t‐tests, and the distributions of gender, education level, genotype, and drug‐naïve patients according to genotype were analyzed using the chi‐square test. Statistical analyses were performed using SPSS version 18.0 (SPSS Inc., Chicago, IL).
RESULTS
Demographic and Genotypic Characteristics
We could not find any significant differences between the MDD and healthy control groups in terms of age, gender, education level, HDRS score, and genotype distribution except for the HDRS score (t (197) = 16.649, P < 0.001), as shown in the Table I. The proportion of drug‐naïve MDD patients did not differ significantly according to the genotype subgroup (χ 2 = 0.486, P = 0.514).
Differences in LGI Values According to Diagnosis and TPH2 rs4570625 Genotype
The patients with MDD showed significantly greater LGIs in the medial OFC (mOFC, F (1,198) = 8.897, P = 0.003, Cohen's f = 0.216; MDD = 2.215 ± 0.010, HC = 2.203 ± 0.013), rostral ACC (F (1,198) = 11.576, P = 0.001, Cohen's f = 0.246; MDD = 2.163 ± 0.009, HC = 2.154 ± 0.014), and frontal pole (F (1,198) = 12.248, P = 0.001, Cohen's f = 0.253; MDD = 2.233 ± 0.012, HC = 2.205 ± 0.013) in the right hemisphere compared with healthy controls (Table II and Fig. 2). We also observed significant diagnosis‐by‐genotype interactive effects on the LGI value of the right rostral ACC (F (1, 198) = 9.265, P = 0.003, Cohen's f = 0.220), as shown in Table II. The above‐mentioned findings remained significant after FDR correction. The genotype alone did not influence the LGI values in the main analysis (Table II).
Figure 2.
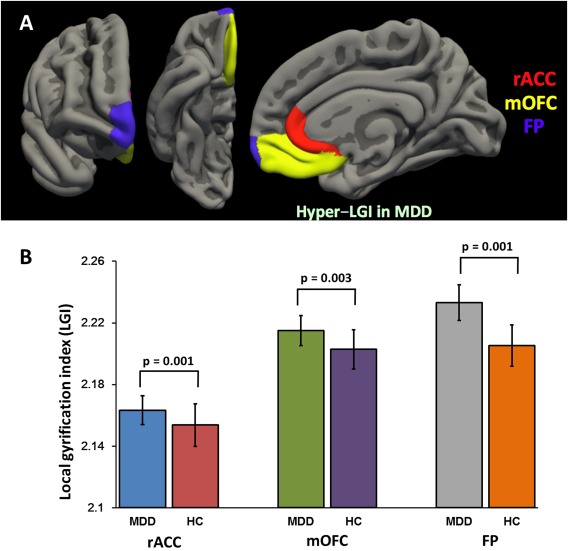
Significant difference of the mean local gyrification index (LGI) values between the patients with major depressive disorder (MDD) and healthy controls (HCs). The panel A represents the statistical comparison map of LGI values between the MDD and HC group, and the panel B shows significant difference of LGI values between the groups. Hypergyria in the right rostral anterior cingulate cortex (rACC, P = 0.001), medial orbitofrontal cortex (mOFC, P = 0.003), and frontal pole (FP, P = 0.001) was observed in the patients with MDD compared with HCs. [Color figure can be viewed at http://wileyonlinelibrary.com]
We performed post‐hoc analyses of the LGI value in the right rostral ACC which showed significant diagnosis‐by‐genotype interaction. We compared the LGI extracted from the right rostral ACC between MDD patients and healthy controls within each genotype group using a one‐way ANCOVA, controlling for the same covariates as those used in the main analysis. In the G‐homozygous group, MDD patients with G‐homozygote showed significantly increased LGI values in the right rostral ACC (F (1,42) = 13.551, P = 0.001, Cohen's f = 0.605; MDD = 2.195 ± 0.012, HC = 2.105 ± 0.026) compared with healthy controls with G‐homozygote, as shown in Table III and Figure 3. In the T allele carrier group, the LGI values of the right rostral ACC did not differ between MDD patients and healthy controls (P > 0.1). The detailed data are described in Table III and Figure 3.
Table III.
Post‐hoc analysis of the local gyrification index of right rostral anterior cingulate cortex according to diagnosis and the genotype
| Cortical regions | GG | TG + TT | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MDD | HC | F | P | Cohen's f | MDD | HC | F | P | Cohen's f | |
| R rostral anterior cingulate cortex | 2.195 ± 0.063 | 2.105 ± 0.102 | 13.551 | 0.001 | 0.605 | 2.154 ± 0.106 | 2.165 ± 0.132 | 0.365 | 0.547 | 0.049 |
Data are means ± standard error (values of local gyrification index).
The F and P values were obtained using one‐way analysis of covariance adjusted for age, gender, education level, and total surface area as covariates.
MDD, major depressive disorder; HC, healthy controls; GG, GG genotype of rs4570625; TT + TG, TT or TG genotype of rs4570625; L, left hemisphere; R, right hemisphere.
Figure 3.
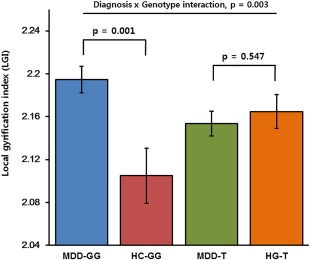
Comparison of the local gyrification index (LGI) values of the right rostral anterior cingulate cortex (rACC) among groups determined by genotype and diagnosis. Significant diagnosis‐by‐genotype interaction was observed in the right rACC (P = 0.003). In the G allele‐homozygous group, MDD patients showed significantly increased LGI value in the right rACC compared with healthy controls (P = 0.001), while in the T allele carrier group, LGI value did not differ between MDD patients and healthy controls (P = 0.547). (MDD‐GG, patients with MDD and the GG genotype; MDD‐T, patients with MDD and the T allele; HC‐GG, healthy controls with the GG genotype; HC‐T, healthy controls with the T allele; Error bars represent standard error of LGI value.). [Color figure can be viewed at http://wileyonlinelibrary.com]
As a secondary analysis, we performed the same statistical tests on LGI values extracted from cortical regions (44 cortical regions in both hemispheres) which are not included in the PFC or the ACC in the Desikan–Killiany atlas. No findings remained significant after correcting for multiple comparisons (Supporting Information Table SI). The details of the results are described in Supporting Information Table SI.
Association of LGIs with Medication, Depression Severity, and Duration of Illness
To assess the effects of psychotropic medication on gyrification, we compared LGI values of the ROIs between medicated and medication‐naïve patients using a one‐way ANCOVA with the same covariates as those used in the main analysis. We did not find any significant difference in LGI values between medication‐naïve and medicated patients (right superior frontal cortex, P > 0.05; all other ROIs, P > 0.1). The data regarding the influence of psychotropic medication use are described in detail in Supporting Information Table SII. We also investigated correlations between the HDRS score and the LGI values of 22 cortical regions in MDD patients using partial correlation analysis and adjusting for age, gender, education level, and total cortical surface area, but could not find any significant correlations (Supporting Information Table SIII; all P > 0.1, except for the right pas opercularis: P > 0.05). In the correlation analysis between duration of illness and LGI values of ROIs in patients with MDD using the same statistical methods as those used for the HDRS score, we observed a correlation in the right superior frontal cortex (r = −0.235, P = 0.033; Supporting Information Table SIV). However, this finding did not remain significant after FDR correction for 22 comparisons in both hemispheres. No other cortical regions showed significant findings (all P > 0.1; except for the left frontal pole, P > 0.05). The data for the severity of depression, duration of illness, and LGI values in MDD patients are described in detail in Supporting Information Tables SIII and SIV.
DISCUSSION
This study firstly demonstrated evidence for an association between aberrant cortical folding in the PFC and ACC and a candidate gene related to the pathophysiology of MDD. We observed significant hypergyrification of the right mOFC, rostral ACC, and frontal pole in patients with MDD. There were significant interactions between TPH2 rs4570625 and a diagnosis of MDD on the LGI of the right rostral ACC in our subjects. The G allele‐homozygous patients showed higher LGI values in the right rostral ACC compared with the patients with the G allele‐homozygous healthy controls, while no significant difference of LGI between the MDD and healthy control group within T allele carrier group.
Only three studies have explored alterations of cortical gyrification in patients with MDD, and there have been controversies regarding the direction of the changes in cortical gyrification related to MDD among the findings of these three studies. Zhang et al. firstly investigated LGI in medication‐naïve patients, and reported hypogyria in the OFC, ACC, and posterior mid‐cingulate cortex [Zhang et al., 2009]. One later study investigated LGI measurements of cortical regions included in the DMN using a sample of recovered‐state MDD patients, and found hypogyria of the bilateral precuneus and hypergyria of the left ACC [Nixon et al., 2014], which is consistent with our observation of an increased LGI in the rostral ACC. The most recent study investigating the LGI in patients experiencing their first episode of MDD suggested that the patients had higher LGIs in the precentral and supramarginal region compared with healthy controls [Peng et al., 2015]. The three studies mentioned above have several different sample characteristics including psychotropic‐medication status, and duration of illness. Peng et al. suggested that different illness durations might lead to different gyrification levels and this could account for differences between the results of these three studies. However, in our study, the factors that could reflect the clinical state of patients with MDD including severity of depression and duration of illness did not influence the LGI values in the PFC and ACC. Psychotropic medication also had no effect on cortical gyrification. Our findings are based on a relatively large sample size compared with previous studies (16–20 patients per group), along with conservative multiple comparison correction parameters and exclusion of confounding factors.
The tension‐based morphogenetic hypothesis, which is widely accepted, states that the process of gyrification is deeply implicated in cortical connectivity and that regional variations in tension during brain development induce specific cortical folding patterns and spatial organization of the connectome [Zilles et al., 2013]. The intensity of cortical folding is associated with the segregation of cortex into functional and cytoarchitectonic areas, and abnormalities in the LGI could lead to cerebral dysfunction by changing corticocortical and corticosubcortical connections [Zilles et al., 2013]. Multiple neuroimaging studies investigating functional connectivity in patients with MDD have suggested that MDD is characterized by abnormal corticocortical and corticosubcortical connectivity in the neural network for emotion processing [Kaiser et al., 2015a; Mulders et al., 2015; Rive et al., 2013]. According to the tension‐based morphogenetic hypothesis, the degree and pattern of axonal connectivity could influence cortical surface morphology through gyrification during brain development [Palaniyappan and Liddle, 2012], and the mOFC and ACC are the most important neural hubs and pivotal in the neural networks of emotion and mood regulation [Phillips et al., 2015; Rive et al., 2013]. Thus, aberrant gyrification in the ACC, mOFC, and frontal pole resulting from abnormal connectivity in the emotion‐processing neural circuits might lead to disturbances in mood regulation and, eventually, a predisposition to MDD. However, we could not verify this hypothesis because our study did not include functional connectivity as an imaging modality. The results of a study that simultaneously investigated cortical gyrification and functional connectivity suggested that regional hypergyrification is associated with reduced long‐range functional connectivity in patients with MDD [Nixon et al., 2014].
We observed hypergyria in the rostral ACC, mOFC, and frontal pole. Structural changes in these regions are the most replicable findings in neuroimaging or post‐mortem histopathological studies on MDD. The reduction in the volume of the ACC in patients with MDD has been reported consistently by meta‐analysis studies using voxel‐based morphometry [Arnone et al., 2016; Bora et al., 2012; Lai, 2013; Wise et al., 2016]. Investigations using FreeSurfer have revealed cortical thinning of the ACC in patients with MDD [Wagner et al., 2012], and a post‐mortem study of depressed patients has also reported a decrease in neuronal cell size and glial cell density in the ACC [Cotter et al., 2001]. The volume reduction of the mOFC in patients with MDD has been reported by several neuroimaging [Arnone et al., 2016; Koolschijn et al., 2009; Van Eijndhoven et al., 2013; Wise et al., 2016] and post‐mortem studies [Rajkowska, 2000; Rajkowska et al., 1999]. A recent study using microstructurally‐informed subdivision‐ and voxel‐wise morphometric analysis has found that patients with MDD showed a reduced volume in the medial frontal pole, and the decrease in the volume of the region was correlated with disease severity and duration [Bludau et al., 2015]. The ACC and mOFC are the core structures in neural circuits responsible for emotion regulation [Phillips et al., 2008]. According to the dysregulated cortico‐limbic network model of depression, the rostral and subgenual part of the ACC and mOFC are involved in the automatic and involuntary control of emotions that are generated by subcortical structures of emotion and reward processing including the amygdala, insula, and ventral striatum [Rive et al., 2013]. The top‐down cognitive and voluntary control of emotions by the lateral prefrontal cortical system including the dorsolateral and ventrolateral PFC is mediated by the ventral part of the ACC and mOFC [Phillips et al., 2008; Rive et al., 2013]. Based on our findings, we speculate that alteration of cortical gyrification in the rostral ACC and mOFC might be associated with dysfunctional emotion regulation which could contribute to pathophysiology of MDD. Previous studies indicating abnormal gyrification of the ACC and mOFC in patients with MDD also support our results [Nixon et al., 2014; Zhang et al., 2009].
In this study, along with the observation of hypergyria in the rostral ACC in depressed patients, we found a significant diagnosis‐by‐genotype interactive effect in the rostral ACC with regard to TPH2 rs4570625. The patients with MDD showed increased LGI values for the rostral ACC only in the G allele‐homozygote group. Our explanation for these results is that abnormalities in the cortical folding pattern together with the genetic risk conferred by TPH2 rs4570625 might be associated with the dysregulation of neural circuitry involved in emotion regulation which could lead to a depressive mood. Growing evidence has indicated that serotonin plays a critical role in refining the organization of the cerebral cortex during development [Vitalis et al., 2007]. We presumed that alterations in neuronal migration and cell proliferation modulated by serotonergic neurotransmission and TPH2 rs4570625 might affect the driving force behind fiber tension and eventual cortical folding during cortical development [Gaspar et al., 2003; Vitalis et al., 2007; Zilles et al., 2013]. Recently, an animal study indicated that serotonin depletion in TPH2 gene knockout mice was associated with delayed maturation of the upper cortical layers during postnatal cortical development [Narboux‐Neme et al., 2013]. However, further evidence establishing a direct correlation between the TPH2 gene and cortical folding is required.
Considering there is much evidence linking TPH2 rs4570625 with structural and functional alterations in emotion‐processing neural circuits [Won and Ham, 2016], we speculate that hypergyria in the rostral ACC could be a neural endophenotype of this gene regarding the development of MDD. Nanda et al. [2014] suggested that aberrant local gyrification could be an endophenotype of psychotic bipolar and schizoaffective disorders by demonstrating that LGI abnormalities had a significant illness association, heritability, and state independence. Although we observed that patients with MDD showed hyper‐gyrification in the rostral ACC, which was associated with TPH2 rs4570625, further studies on LGI and MDD are required to confirm that hypergyria in the rostral ACC has sufficient heritability and confers a greater predisposition to MDD in unaffected family members in order to meet the criteria for a useful endophenotype proposed by Gottesman and Gould and others [Bearden and Freimer, 2006; Gottesman and Gould, 2003; Kendler and Neale, 2010].
The association between TPH2 rs4570625 and a predisposition to MDD has been suggested by numerous genetic studies [Ma et al., 2015; Waider et al., 2011; Won and Ham, 2016]. A meta‐analysis study has indicated that, among the 28 SNPs in 12 independent loci of the TPH2 gene, rs4570625 has the strongest epidemiological evidence of a relationship with MDD [Gao et al., 2012]. In imaging genetic studies using healthy control participants, rs4570625 was associated with morphological changes in brain structures involved in emotion‐processing neural circuits including the hippocampus, amygdala, and OFC. Inoue et al. [2010] found that T allele carriers showed significantly smaller volumes in the bilateral amygdala and hippocampus and higher reward dependence than did homozygotes with the G allele. In our previous study investigating the whole brain of healthy controls using the VBM method, G homozygous subjects had reduced gray‐matter concentrations in the inferior OFC compared with T allele carriers, and this reduction was correlated with the degree of anger‐related personality traits [Yoon et al., 2012]. However, to our best knowledge, there has not yet been a study investigating the relationship between genetic variants of TPH2 rs4570625 and structural changes to neural circuitry including the PFC and ACC in patients with MDD.
There are certain limitations to consider in this study. First, sixty‐one patients (54.0%) were treated with antidepressants with or without benzodiazepine in this study, and we could not exclude the possibility that psychotropic‐medication status influenced our results. We did not find any effect of medication on the LGIs of the ROIs in a secondary analysis; similarly, a previous study on LGIs in MDD also found no significant correlation between medication status and local gyrification [Nixon et al., 2014], However, the correlation between the use of psychotropic medication and the changes in cortical folding pattern could be confirmed by future studies with larger sample sizes including medicated as well as un‐medicated depressed participants. Second, the age range of the participants in our study was 20–65 years, and we cannot exclude the possibility that the inclusion of participants who were aged 60 or older might have affected our results. However, the age range in our study was similar to those in recent imaging genetic studies on patients with MDD (18–65 range) [Carballedo et al., 2012; Frodl et al., 2010; Murphy et al., 2012; Tozzi et al., 2016] and two of the three previous studies on LGI and MDD [24–64 range, Nixon et al., 2014; 18–60 range, Zhang et al., 2009]. We suggest that the influence of age range on the results was minimal because the percentages of patients with MDD and healthy controls who were 60 or older were only 7.1% (8 patients with MDD) and 3.5% (3 healthy controls), respectively. In addition, the age of the participants was controlled in the study, and this factor was included in all the analyses of our study. Third, we did not include environmental factors such as childhood adversity or maltreatment, which could influence the cortical folding pattern [Kelly et al., 2013], and thus, we could not explore genetic–environmental interactive effects on local gyrification in patients with MDD. Finally, the cross‐sectional design of this study might have weakened our ability to find the presumptive causal relationship between the hypergyria of the PFC and ACC and a predisposition to MDD and limited our postulation that hypergyria is a stable indicator that is independent of the clinical state. Therefore, these findings need to be replicated in future studies with longitudinal designs.
CONCLUSION
In conclusion, our study revealed that patients with MDD had significant hypergyria of the rostral ACC, the mOFC, and the frontal pole, compared with healthy controls. We speculate that these changes might be associated with dysfunction in the emotion processing circuitry, which could contribute to pathophysiology of MDD. Our study first explored the effects of the interaction of a MDD diagnosis and TPH2 rs4570625 on the gyrification of the PFC and ACC, and we found that homozygosity for the G allele was associated with a higher LGI in the rostral ACC in the patients with MDD. We hope that this study has extended the neurobiological basis for alterations in cortical folding and provided additional evidence for the influence of TPH2 rs4570625 on structural changes to the neural networks involved in emotion regulation.
Supporting information
Supporting Information
The authors declare no conflict of interest.
Contributor Information
Woo‐Suk Tae, Email: wstae@korea.ac.kr.
Byung‐Joo Ham, Email: hambj@korea.ac.kr.
REFERENCES
- Arnone D, Job D, Selvaraj S, Abe O, Amico F, Cheng Y, Colloby SJ, O'Brien JT, Frodl T, Gotlib IH, Ham BJ, Kim MJ, Koolschijn PC, Perico CA, Salvadore G, Thomas AJ, Van Tol MJ, van der Wee NJ, Veltman DJ, Wagner G, McIntosh AM (2016): Computational meta‐analysis of statistical parametric maps in major depression. Hum Brain Mapp 37:1393–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bearden CE, Freimer NB (2006): Endophenotypes for psychiatric disorders: Ready for primetime?. Trends Genet: TIG 22:306–313. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y (1995): Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc Ser B (Method) 289–300. [Google Scholar]
- Bludau S, Bzdok D, Gruber O, Kohn N, Riedl V, Sorg C, Palomero‐Gallagher N, Muller VI, Hoffstaedter F, Amunts K, Eickhoff SB (2015): Medial prefrontal aberrations in major depressive disorder revealed by cytoarchitectonically informed voxel‐based morphometry. Am J Psychiatry 173:291–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bora E, Fornito A, Pantelis C, Yucel M (2012): Gray matter abnormalities in Major Depressive Disorder: A meta‐analysis of voxel based morphometry studies. J Affect Disord 138:9–18. [DOI] [PubMed] [Google Scholar]
- Brown SM, Peet E, Manuck SB, Williamson DE, Dahl RE, Ferrell RE, Hariri AR (2005): A regulatory variant of the human tryptophan hydroxylase‐2 gene biases amygdala reactivity. Mol Psychiatry 10:805–884‐8. [DOI] [PubMed] [Google Scholar]
- Canli T, Congdon E, Gutknecht L, Constable RT, Lesch KP (2005): Amygdala responsiveness is modulated by tryptophan hydroxylase‐2 gene variation. J Neural Transm (Vienna, Austria: 1996) 112:1479–1485. [DOI] [PubMed] [Google Scholar]
- Carballedo A, Amico F, Ugwu I, Fagan AJ, Fahey C, Morris D, Meaney JF, Leemans A, Frodl T (2012): Reduced fractional anisotropy in the uncinate fasciculus in patients with major depression carrying the met‐allele of the Val66Met brain‐derived neurotrophic factor genotype. Am J Med Genet B Neuropsychiatr Genet 159B:537–548. [DOI] [PubMed] [Google Scholar]
- Cotter D, Mackay D, Landau S, Kerwin R, Everall I (2001): Reduced glial cell density and neuronal size in the anterior cingulate cortex in major depressive disorder. Arch Gen Psychiatry 58:545–553. [DOI] [PubMed] [Google Scholar]
- Dale AM, Fischl B, Sereno MI (1999): Cortical surface‐based analysis. I. Segmentation and surface reconstruction. NeuroImage 9:179–194. [DOI] [PubMed] [Google Scholar]
- Dauvermann MR, Mukherjee P, Moorhead WT, Stanfield AC, Fusar‐Poli P, Lawrie SM, Whalley HC (2012): Relationship between gyrification and functional connectivity of the prefrontal cortex in subjects at high genetic risk of schizophrenia. Curr Pharm Des 18:434–442. [DOI] [PubMed] [Google Scholar]
- Desikan RS, Segonne F, Fischl B, Quinn BT, Dickerson BC, Blacker D, Buckner RL, Dale AM, Maguire RP, Hyman BT, Albert MS, Killiany RJ (2006): An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. NeuroImage 31:968–980. [DOI] [PubMed] [Google Scholar]
- Fani N, Gutman D, Tone EB, Almli L, Mercer KB, Davis J, Glover E, Jovanovic T, Bradley B, Dinov ID, Zamanyan A, Toga AW, Binder EB, Ressler KJ (2013): FKBP5 and attention bias for threat: Associations with hippocampal function and shape. JAMA Psychiatry 70:392–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fani N, King TZ, Reiser E, Binder EB, Jovanovic T, Bradley B, Ressler KJ (2014): FKBP5 genotype and structural integrity of the posterior cingulum. Neuropsychopharmacology 39:1206–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischl B, Liu A, Dale AM (2001): Automated manifold surgery: Constructing geometrically accurate and topologically correct models of the human cerebral cortex. IEEE Trans Med Imaging 20:70–80. [DOI] [PubMed] [Google Scholar]
- Fischl B, Sereno MI, Dale AM (1999): Cortical surface‐based analysis. II: Inflation, flattening, and a surface‐based coordinate system. NeuroImage 9:195–207. [DOI] [PubMed] [Google Scholar]
- Fischl B, Salat DH, Busa E, Albert M, Dieterich M, Haselgrove C, van der Kouwe A, Killiany R, Kennedy D, Klaveness S, Montillo A, Makris N, Rosen B, Dale AM (2002): Whole brain segmentation: Automated labeling of neuroanatomical structures in the human brain. Neuron 33:341–355. [DOI] [PubMed] [Google Scholar]
- Fischl B, van der Kouwe A, Destrieux C, Halgren E, Segonne F, Salat DH, Busa E, Seidman LJ, Goldstein J, Kennedy D, Caviness V, Makris N, Rosen B, Dale AM (2004): Automatically parcellating the human cerebral cortex. Cereb Cortex 14:11–22. [DOI] [PubMed] [Google Scholar]
- Frodl T, Reinhold E, Koutsouleris N, Donohoe G, Bondy B, Reiser M, Möller HJ, Meisenzahl EM (2010): Childhood stress, serotonin transporter gene and brain structures in major depression. Neuropsychopharmacology 35:1383–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Pan Z, Jiao Z, Li F, Zhao G, Wei Q, Pan F, Evangelou E (2012): TPH2 gene polymorphisms and major depression–a meta‐analysis. PloS One 7:e36721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar P, Cases O, Maroteaux L (2003): The developmental role of serotonin: News from mouse molecular genetics. Nat Rev Neurosci 4:1002–1012. [DOI] [PubMed] [Google Scholar]
- Gottesman II, Gould TD (2003): The endophenotype concept in psychiatry: Etymology and strategic intentions. Am J Psychiatry 160:636–645. [DOI] [PubMed] [Google Scholar]
- Hamilton M (1960): A rating scale for depression. J Neurol Neurosurg Psychiatry 23:56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue H, Yamasue H, Tochigi M, Takei K, Suga M, Abe O, Yamada H, Rogers MA, Aoki S, Sasaki T, Kasai K (2010): Effect of tryptophan hydroxylase‐2 gene variants on amygdalar and hippocampal volumes. Brain Res 1331:51–57. [DOI] [PubMed] [Google Scholar]
- Janssen J, Aleman‐Gomez Y, Schnack H, Balaban E, Pina‐Camacho L, Alfaro‐Almagro F, Castro‐Fornieles J, Otero S, Baeza I, Moreno D, Bargallo N, Parellada M, Arango C, Desco M (2014): Cortical morphology of adolescents with bipolar disorder and with schizophrenia. Schizophr Res 158:91–99. [DOI] [PubMed] [Google Scholar]
- Kaiser RH, Andrews‐Hanna JR, Wager TD, Pizzagalli DA (2015a): Large‐scale network dysfunction in major depressive disorder: A meta‐analysis of resting‐state functional connectivity. JAMA Psychiatry 72:603–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser RH, Whitfield‐Gabrieli S, Dillon DG, Goer F, Beltzer M, Minkel J, Smoski M, Dichter G, Pizzagalli DA (2015b): Dynamic resting‐state functional connectivity in major depression. Neuropsychopharmacology 41:1822–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly PA, Viding E, Wallace GL, Schaer M, De Brito SA, Robustelli B, McCrory EJ (2013): Cortical thickness, surface area, and gyrification abnormalities in children exposed to maltreatment: Neural markers of vulnerability?. Biol Psychiatry 74:845–852. [DOI] [PubMed] [Google Scholar]
- Kendler KS, Neale MC (2010): Endophenotype: A conceptual analysis. Mol Psychiatry 15:789–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohli MA, Lucae S, Saemann PG, Schmidt MV, Demirkan A, Hek K, Czamara D, Alexander M, Salyakina D, Ripke S, Hoehn D, Specht M, Menke A, Hennings J, Heck A, Wolf C, Ising M, Schreiber S, Czisch M, Muller MB, Uhr M, Bettecken T, Becker A, Schramm J, Rietschel M, Maier W, Bradley B, Ressler KJ, Nothen MM, Cichon S, Craig IW, Breen G, Lewis CM, Hofman A, Tiemeier H, van Duijn CM, Holsboer F, Muller‐Myhsok B, Binder EB (2011): The neuronal transporter gene SLC6A15 confers risk to major depression. Neuron 70:252–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koolschijn PC, van Haren NE, Lensvelt‐Mulders GJ, Hulshoff Pol HE, Kahn RS (2009): Brain volume abnormalities in major depressive disorder: A meta‐analysis of magnetic resonance imaging studies. Hum Brain Mapp 30:3719–3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremen WS, O'Brien RC, Panizzon MS, Prom‐Wormley E, Eaves LJ, Eisen SA, Eyler LT, Hauger RL, Fennema‐Notestine C, Fischl B, Grant MD, Hellhammer DH, Jak AJ, Jacobson KC, Jernigan TL, Lupien SJ, Lyons MJ, Mendoza SP, Neale MC, Seidman LJ, Thermenos HW, Tsuang MT, Dale AM, Franz CE (2010): Salivary cortisol and prefrontal cortical thickness in middle‐aged men: A twin study. NeuroImage 53:1093–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupfer DJ, Frank E, Phillips ML (2012): Major depressive disorder: New clinical, neurobiological, and treatment perspectives. Lancet (London, England) 379:1045–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai CH (2013): Gray matter volume in major depressive disorder: A meta‐analysis of voxel‐based morphometry studies. Psychiatry Res 211:37–46. [DOI] [PubMed] [Google Scholar]
- Ma J, Xiao H, Yang Y, Cao D, Wang L, Yang X, Qiu X, Qiao Z, Song J, Liu Y, Wang P, Zhou J, Zhu X (2015): Interaction of tryptophan hydroxylase 2 gene and life events in susceptibility to major depression in a Chinese Han population. J Affect Disord 188:304–309. [DOI] [PubMed] [Google Scholar]
- Mulders PC, van Eijndhoven PF, Schene AH, Beckmann CF, Tendolkar I (2015): Resting‐state functional connectivity in major depressive disorder: A review. Neurosci Biobehav Rev 56:330–344. [DOI] [PubMed] [Google Scholar]
- Murphy ML, Carballedo A, Fagan AJ, Morris D, Fahey C, Meaney J, Frodl T (2012): Neurotrophic tyrosine kinase polymorphism impacts white matter connections in patients with major depressive disorder. Biol Psychiatry 72:663–670. [DOI] [PubMed] [Google Scholar]
- Nanda P, Tandon N, Mathew IT, Giakoumatos CI, Abhishekh HA, Clementz BA, Pearlson GD, Sweeney J, Tamminga CA, Keshavan MS (2014): Local gyrification index in probands with psychotic disorders and their first‐degree relatives. Biol Psychiatry 76:447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narboux‐Neme N, Angenard G, Mosienko V, Klempin F, Pitychoutis PM, Deneris E, Bader M, Giros B, Alenina N, Gaspar P (2013): Postnatal growth defects in mice with constitutive depletion of central serotonin. ACS Chem Neurosci 4:171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nenadic I, Maitra R, Dietzek M, Langbein K, Smesny S, Sauer H, Gaser C (2015): Prefrontal gyrification in psychotic bipolar I disorder vs. schizophrenia. J Affect Disord 185:104–107. [DOI] [PubMed] [Google Scholar]
- Nixon NL, Liddle PF, Nixon E, Worwood G, Liotti M, Palaniyappan L (2014): Biological vulnerability to depression: Linked structural and functional brain network findings. Br J Psychiatry 204:283–289. [DOI] [PubMed] [Google Scholar]
- Oldfield RC (1971): The assessment and analysis of handedness: The Edinburgh inventory. Neuropsychologia 9:97–113. [DOI] [PubMed] [Google Scholar]
- Palaniyappan L, Liddle PF (2012): Aberrant cortical gyrification in schizophrenia: A surface‐based morphometry study. J Psychiatry Neurosci: JPN 37:399–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng D, Shi F, Li G, Fralick D, Shen T, Qiu M, Liu J, Jiang K, Shen D, Fang Y (2015): Surface vulnerability of cerebral cortex to major depressive disorder. PloS One 10:e0120704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips ML, Ladouceur CD, Drevets WC (2008): A neural model of voluntary and automatic emotion regulation: Implications for understanding the pathophysiology and neurodevelopment of bipolar disorder. Mol Psychiatry 13:833–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips ML, Chase HW, Sheline YI, Etkin A, Almeida JR, Deckersbach T, Trivedi MH (2015): Identifying predictors, moderators, and mediators of antidepressant response in major depressive disorder: Neuroimaging approaches. Am J Psychiatry 172:124–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajkowska G (2000): Postmortem studies in mood disorders indicate altered numbers of neurons and glial cells. Biol Psychiatry 48:766–777. [DOI] [PubMed] [Google Scholar]
- Rajkowska G, Miguel‐Hidalgo JJ, Wei J, Dilley G, Pittman SD, Meltzer HY, Overholser JC, Roth BL, Stockmeier CA (1999): Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol Psychiatry 45:1085–1098. [DOI] [PubMed] [Google Scholar]
- Rive MM, van Rooijen G, Veltman DJ, Phillips ML, Schene AH, Ruhe HG (2013): Neural correlates of dysfunctional emotion regulation in major depressive disorder. A systematic review of neuroimaging studies. Neurosci Biobehav Rev 37:2529–2553. [DOI] [PubMed] [Google Scholar]
- Segonne F, Pacheco J, Fischl B (2007): Geometrically accurate topology‐correction of cortical surfaces using nonseparating loops. IEEE Trans Med Imaging 26:518–529. [DOI] [PubMed] [Google Scholar]
- Serretti A, Chiesa A, Porcelli S, Han C, Patkar AA, Lee SJ, Park MH, Pae CU (2011): Influence of TPH2 variants on diagnosis and response to treatment in patients with major depression, bipolar disorder and schizophrenia. Psychiatry Res 189:26–32. [DOI] [PubMed] [Google Scholar]
- Tozzi L, Carballedo A, Wetterling F, McCarthy H, O'Keane V, Gill M, Morris D, Fahey C, Meaney J, Frodl T (2016): Single‐nucleotide polymorphism of the fkbp5 gene and childhood maltreatment as predictors of structural changes in brain areas involved in emotional processing in depression. Neuropsychopharmacology 41:487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Eijndhoven P, van Wingen G, Katzenbauer M, Groen W, Tepest R, Fernandez G, Buitelaar J, Tendolkar I (2013): Paralimbic cortical thickness in first‐episode depression: Evidence for trait‐related differences in mood regulation. Am J Psychiatry 170:1477–1486. [DOI] [PubMed] [Google Scholar]
- Vitalis T, Cases O, Passemard S, Callebert J, Parnavelas JG (2007): Embryonic depletion of serotonin affects cortical development. Eur J Neurosci 26:331–344. [DOI] [PubMed] [Google Scholar]
- Wagner G, Schultz CC, Koch K, Schachtzabel C, Sauer H, Schlosser RG (2012): Prefrontal cortical thickness in depressed patients with high‐risk for suicidal behavior. J Psychiatr Res 46:1449–1455. [DOI] [PubMed] [Google Scholar]
- Waider J, Araragi N, Gutknecht L, Lesch KP (2011): Tryptophan hydroxylase‐2 (TPH2) in disorders of cognitive control and emotion regulation: A perspective. Psychoneuroendocrinology 36:393–405. [DOI] [PubMed] [Google Scholar]
- Wise T, Radua J, Via E, Cardoner N, Abe O, Adams TM, Amico F, Cheng Y, Cole JH, de Azevedo Marques Perico C, Dickstein DP, Farrow TF, Frodl T, Wagner G, Gotlib IH, Gruber O, Ham BJ, Job DE, Kempton MJ, Kim MJ, Koolschijn PC, Malhi GS, Mataix‐Cols D, McIntosh AM, Nugent AC, O'Brien JT, Pezzoli S, Phillips ML, Sachdev PS, Salvadore G, Selvaraj S, Stanfield AC, Thomas AJ, van Tol MJ, van der Wee NJ, Veltman DJ, Young AH, Fu CH, Cleare AJ, Arnone D (2016): Common and distinct patterns of grey‐matter volume alteration in major depression and bipolar disorder: Evidence from voxel‐based meta‐analysis. Mol Psychiatry 254:41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won E, Ham BJ (2016): Imaging genetics studies on monoaminergic genes in major depressive disorder. Prog Neuropsychopharmacol Biol Psychiatry 64:311–319. [DOI] [PubMed] [Google Scholar]
- Workman CI, Lythe KE, McKie S, Moll J, Gethin JA, Deakin JF, Elliott R, Zahn R (2016): Subgenual cingulate‐amygdala functional disconnection and vulnerability to melancholic depression. Neuropsychopharmacology 41:2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon HK, Lee HJ, Kim L, Lee MS, Ham BJ (2012): Impact of tryptophan hydroxylase 2 G‐703T polymorphism on anger‐related personality traits and orbitofrontal cortex. Behav Brain Res 231:105–110. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Yu C, Zhou Y, Li K, Li C, Jiang T (2009): Decreased gyrification in major depressive disorder. Neuroreport 20:378–380. [DOI] [PubMed] [Google Scholar]
- Zilles K, Palomero‐Gallagher N, Amunts K (2013): Development of cortical folding during evolution and ontogeny. Trends Neurosci 36:275–284. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information