

Abstract
Introduction
A gene–environment interaction between expression genotypes of the monoamine oxidase A (MAOA) and adverse childhood experience increases the risk of antisocial behavior. However, the neural underpinnings of this interaction remain uninvestigated. A cortico‐limbic circuit involving the prefrontal cortex (PFC) and the amygdala is central to the suppression of aggressive impulses and is modulated by serotonin (5‐HT). MAOA genotypes may modulate the vulnerability of this circuit and increase the risk for emotion regulation deficits after specific life events. Acute tryptophan depletion (ATD) challenges 5‐HT regulation and may identify vulnerable neuronal circuits, contributing to the gene–environment interaction.
Methods
Functional magnetic resonance imaging measured the resting‐state state activity in 64 healthy males in a double‐blind, placebo‐controlled study. Cortical maps of amygdala correlation identified the impact of ATD and its interaction with low‐ (MAOA‐L) and high‐expression variants (MAOA‐H) of MAOA on cortico‐limbic connectivity.
Results
Across all Regions of Interest (ROIs) exhibiting an ATD effect on cortico‐limbic connectivity, MAOA‐L carriers were more susceptible to ATD than MAOA‐H carriers. In particular, the MAOA‐L group exhibited a larger reduction of amygdala connectivity with the right prefrontal cortex and a larger increase of amygdala connectivity with the insula and dorsal PCC.
Conclusion
MAOA‐L carriers were more susceptable to a central 5‐HT challenge in cortico‐limbic networks. Such vulnerability of the cortical serotonergic system may contribute to the emergence of antisocial behavior after systemic challenges, observed as gene–environment interaction. Hum Brain Mapp 38:1622–1635, 2017. © 2016 Wiley Periodicals, Inc.
Keywords: amygdala, serotonin, MAOA, aggression, resting state fMRI
Abbreviations
- 5‐HT
serotonin;
- ACC
anterior cingulate cortex;
- ANOVA
analysis of variance;
- ATD
acute tryptophan depletion;
- BAL
balanced;
- CN
caudate nucleus;
- HCl
hydroxychloride;
- IFG
inferior frontal gyrus;
- KCl
potassiumchloride;
- LN
lentiform nucleus;
- MAOA
monoaminoxidase A;
- MAOA‐L
low expressing allele of the MAOA gene;
- MAOA‐H
high expressing allele of the MAOA gene;
- MgCl2
magnesium dichloride;
- PCC
posterior cingulate cortex;
- PCR
polymerase chain reaction;
- PFC
prefrontal cortex;
- PG
precentral gyrus;
- ROI
region of interest;
- RS
resting state;
- SFG
superior frontal gyrus;
- STG
superior temporal gyrus
INTRODUCTION
Behavioral and personality traits can emerge on the grounds of gene–environment interactions [South and Krueger, 2008]. In particular, antisocial behaviors may result from childhood maltreatment, specifically in carriers of the low expressing variant of the monoamine oxidase A gene [MAOA‐L; Caspi et al., 2002; meta‐analysis in Byrd and Manuck, 2014]. Antisocial behavior is related to dysfunctions in cortico‐limbic inhibition [Buckholtz et al., 2008]. In particular, amygdala responses to aversive stimuli are modulated by the prefrontal cortex [PFC, Frodl et al., 2009; Lemogne et al., 2011], which in turn receives dense serotonergic projections [Challis and Berton, 2015; Peyron et al., 1998]. MAOA‐L carriers may be more sensitive to disturbances of serotonin (5‐HT) metabolism, which may affect connectivity between PFC and limbic structures. One approach to investigate the neural processes of altered 5‐HT availability in MAOA‐L carriers is acute tryptophan depletion (ATD). This method decreases the availability of the essential 5‐HT precursor tryptophan [Delgado et al., 1990, 1991] and may test for serotonergic vulnerability of the cortico‐limbic network in different MAOA genotypes.
Regulation of the amygdala seems highly relevant for behavioral disorders. The amygdala is hyperactive in MAOA‐L carriers during the recall of negative events [Meyer‐Lindenberg et al., 2006] and anger control [Denson et al., 2014]. In particular, during aggressive behavior, amygdala regulation is pivotal for emotion processing [Klasen et al., 2013; Mathiak and Weber, 2006]. Conceivably, vulnerable amygdala regulation may contribute to dysfunctional emotion regulation, thereby increasing the risk for antisocial behavior in MAOA‐L carriers. Clinical effects of 5‐HT enhancing drugs on aggressive behavior suggest an involvement of the serotonergic system [Siever, 2008].
The PFC regulates amygdala responses to emotional stimuli [Lemogne et al., 2011]. This cortico‐limbic network is associated with impulse control and reduced activation in the ACC during a Go/NoGo task, emerging in MAOA‐L carriers only [Buckholtz and Meyer‐Lindenberg, 2008; Meyer‐Lindenberg et al., 2006]. Indeed, impaired cortico‐limbic regulation is supposed to contribute to antisocial behavior in MAOA‐L carriers [Chester et al., 2015; Davidson et al., 2000]. Therefore, a potential neural mechanism of a higher vulnerability in MAOA‐L carriers may originate from impaired regulation in the outlined network.
ATD is an established method to experimentally challenge the central nervous 5‐HT system [Delgado et al., 1990; Zimmermann et al., 2012]. This procedure reduces central 5‐HT availability [Nishizawa et al., 1997; Shoaf et al., 1998] and allows investigations on the effects of reduced central 5‐HT on the cortico‐limbic circuitry. Lowering brain 5‐HT synthesis yielded a bias to aversive emotional stimuli [Klaassen et al., 2002; Passamonti et al., 2012], and increased state aggression [Cleare and Bond, 1995; Pihl et al., 1995]. These factors probably contribute to developing aggressive behavior. Further, ATD affected the activity of the amygdala and the PFC [Bremner et al., 1997; Morris et al., 1999; Smith et al., 1999a; Neumeister et al., 2004; Lemogne et al., 2011]. In particular, emotionally salient stimuli yielded higher responses in the PFC as a result of impaired evaluation after ATD [Roiser et al., 2009b]. Thus, PFC regulation of emotion processing at the amygdala seems to depend on 5‐HT availability.
On the neural level, ATD reduced connectivity of the amygdala with the PFC and perturbed emotion regulation to aggressive social stimuli [Crockett et al., 2009; Hindi Attar et al., 2012; Passamonti et al., 2012; Roiser et al., 2009a; Robinson et al., 2012]. Passamonti et al. [2012] showed that ATD reduced PFC‐amygdala connectivity specifically for angry faces, supporting neurobiological models that postulate facilitation of the suppression of negative affect via the PFC‐amygdala circuit [Davidson et al., 2000; Miczek et al., 2007; Siever, 2008]. Such negative affect bias is a specific feature of Borderline and impulsive aggressive personality disorders in which serotonergic neurotransmission is disturbed [Lee et al., 2012; Leyton et al., 2001]. Moreover, activation of brain areas related to aggression and impulsivity (i.e., such as the orbitofrontal cortex) were correlated with ATD‐related depletion magnitude and trait‐impulsivity [Helmbold et al., 2015]. In summary, genetic and pharmacological alterations of 5‐HT levels affect neural correlates of emotion regulation and impulse control. Therefore, ATD may test the vulnerability to dysregulation of the cortico‐limbic network in MAOA‐L carriers.
The regulation networks for emotions have been widely investigated [see meta‐analysis in Kohn et al., 2014]. Task‐dependent paradigms focus on differential aspects of emotion regulation but may miss fundamental network changes. Therefore, one beneficial strategy to assess network activity is resting state [RS; Roy et al., 2009]. RS‐fMRI measurements identify neural networks independently from specific task demands or emotion‐eliciting stimuli [Beckmann et al., 2005]. During RS, functionally connected networks show synchronized activity even in the absence of a task and can be identified [Damoiseaux et al., 2006] and dysfunctional emotion regulation networks can be detected [e.g., in depression; Anand et al., 2009]. Therefore, RS‐fMRI may investigate the vulnerability of the cortico‐limbic network in MAOA genotypes to ATD.
We investigated whether the cortico‐limbic circuit is more vulnerable to changes of brain 5‐HT in MAOA‐L than in MAOA‐H carriers. Reducing central 5‐HT availability by ATD should reveal neural mechanisms of this vulnerability. Therefore, our study aimed to identify the influence of ATD on functional connectivity in MAOA‐L and ‐H carriers during resting state fMRI. First, we aimed to test whether the finding by Passamonti et al. [2012], that ATD reduces amygdala connectivity with the PFC to aggressive faces, extends to connectivity during stimulus‐independent resting state. Second, diminished central 5‐HT availability may affect MAOA‐L stronger than MAOA‐H carriers. In particular, we hypothesized an interaction between the MAOA genotype and ATD on cortico‐limbic connectivity, which may contribute to a higher vulnerability in MAOA‐L carriers.
METHODS
Participants
Sixty‐four male volunteers (25.0 ± 3.8 years, all German mother tongue, and of Caucasian ethnicity) participated in the present study. In a standardized assessment, all volunteers reported normal vision, normal hearing, no contraindications against MRI investigations, and no history of neurological or psychiatric disorders. The subjects were drug‐screened, right‐handed as assessed with the Edinburgh Handedness Inventory [Oldfield, 1971], and had normal intelligence according to the multiple‐choice‐word test [Lehrl, 2005]. The experiment was conducted in accordance with the Code of Ethics of the World Medical Association [Declaration of Helsinki, 2008], and the study protocol was approved by the local ethics committee. The subjects were financially compensated for their participation.
Genotyping
Prior to the fMRI scanning, all participants underwent a 9 mL venous blood sampling. Genomic DNA was isolated from peripheral lymphocytes with a routine salting‐out procedure. For the determination of the MAOA genotype, standard polymerase chain reaction (PCR) amplification was performed in a 25 μL volume containing 80 ng of genomic DNA, unit of recombinant Taq polymerase (Invitrogen, Darmstadt, Germany), PCR buffer (10 mM Tris‐HCl, 50 mM KCl, 2.5 mM MgCl2, pH 8.3), 200 mM deoxynucleotides, and 20 pmol of each primer. MAOA primer sequences were obtained from Sabol et al. [1998]. The PCR was run on an MJ PTC200 Temperature Cycler (Biozym, Hessisch‐Oldendorf, Germany), and each of the 35 cycles consisted of a 95°C denaturation step for 45 s, a 62°C annealing step for 30 s, and, finally, a 72°C elongation step for 90 s. PCR products were run on an automated sequencing system (AB3130, Applied Biosystems, Darmstadt, Germany), and the electropherograms were analyzed with gene mapping software. According to Sabol et al. [1998], 3 and 5 repeats were classified as representing a low expression (MAOA‐L) and 3.5 and 4 repeats as representing a high MAOA expression (MAOA‐H).
Serotonergic Challenge
We studied effects of ATD in a cross‐over, double‐blind design with blocked‐random group allocation. Two conditions were compared: (1) body weight‐adapted ATD according to the Moja‐De scheme [Demisch et al., 2002; Moja et al., 1988]; and (2) an L‐Tryptophan–balanced control drink (BAL). For ATD, a tryptophan‐free amino acid beverage was applied: for each 10 kg of body weight, 0.084 g L‐Isoleucine, 0.132 g L‐Leucine, 0.12 g L‐Lysine‐HCL, 0.05 g L‐Methionine, 0.132 g L‐Phenylalanine, 0.06 g L‐Threonine, and 0.096 g L‐Valine were introduced. In the BAL condition, an extra amino acid mixture was provided, which included an additional 0.7 g of tryptophan per 10 kg body weight (BAL). The latter composition acts as a control and does not impact 5‐HT synthesis in the brain [Biskup et al., 2012].
As a manipulation check, blood serum was collected immediately before beverage administration and 3 h later, that is, before the start of the fMRI resting state measurement. With an ANOVA, we tested effects of the within‐subject factors “Condition” (ATD and BAL) and “Time” (Before beverage administration and before resting state) as well as the between‐subject factors “Genotype” (MAOA‐H and MAOA‐L) and “Order” (first session ATD and BAL) on tryptophan levels.
Mood Assessment
ATD effects on mood across healthy men are inconsistent [Evers et al., 2006; Young and Leyton, 2002]. Therefore, we assessed mood using the Positive And Negative Affect Scale [PANAS; Watson et al., 1988] before beverage administration and before resting state in both conditions. In the ATD and BAL condition, differences between before beverage administration and before resting state fMRI were calculated separately for positive and negative affect, corresponding to baseline‐corrected effects on mood. Positive and negative affect changes were analyzed using repeated measures ANOVAs with the within‐subject factor “Condition” (ATD and BAL) and the between‐subject‐factor “Genotype” (MAOA‐H and MAOA‐L) in SPSS 21 (IBM Corp., Armonk, New York).
Task and Procedure
Participants were instructed to adhere to a tryptophan‐free diet from 8 pm the evening before each measurement. In the morning, the participants arrived at 8 am at the study site. At 8:30 am, an amino acid beverage (ATD or BAL) was administered blind to the experimenter and the participants. At 11:30 am, a known time point associated with a robust decline of TRP influx in the brain [Dingerkus et al., 2012], a resting state fMRI session was recorded for 8 min with eyes open (white fixation cross on a black background). The participants were instructed to relax, to think of nothing in particular, and to stay awake. The measurement was followed by a high resolution anatomical T1 scan. Individual measurements were separated 14.82 days on average (minimum 7 days, maximum 99 days). To assess the effect size of tryptophan depletion, we estimated Cohen's d for the effect of ATD on tryptophan depletion (compared with BAL).
Brain Imaging
Magnetic resonance imaging was conducted on a 3 Tesla Siemens Trio scanner (Siemens Medical, Erlangen, Germany). For fMRI, echo‐planar imaging (EPI) acquired 34 transversal slices for a total of 240 volumes (TE = 28 ms, TR = 2,000 ms, flip angle = 77°, voxel size = 3 × 3 mm with 64 × 64 matrix, 3 mm slice thickness, 0.75 mm gap). After the RS measurements, a high resolution, whole brain anatomical image was acquired (MPRAGE, T1‐weighted, TE = 2.52 ms; TI = 900 ms; TR = 1,900 ms; flip angle = 9°; FOV 256 × 256 mm2; 1‐mm isotropic voxels; 176 sagittal slices).
fMRI Preprocessing
Resting state measurements were analyzed with BrainVoyager QX 2.8 (Brain Innovation, Maastricht, The Netherlands). To avoid T1 saturation effects, the first five images of each session were discarded. Preprocessing of functional MR images included slice scan time correction, 3D motion correction, spatial smoothing (6 mm FWHM), and high‐pass filtering including linear trend removal. Additionally, rigid‐body motion parameters as well as cerebrospinal fluid and white matter time courses were regressed out to reduce physiological noise [Hutton et al., 2011]. Functional images were co‐registered individually to the anatomical images and transformed into Talairach space [Talairach and Szikla, 1980].
Seed‐Based Functional Connectivity Analysis
Amygdala maps were imported from the Anatomy Toolbox [Eickhoff et al., 2005] in SPM8 (http://www.fil.ion.ucl.ac.uk/spm/software/spm8) to BrainVoyager software (V 2.8) and transformed into Talairach space. Functional connectivity using the bilateral amygdala seed was calculated for each voxel of the whole brain. The t‐tests determined the effect of ATD on amygdala connectivity after Fisher's z‐transformation. Further t‐tests explored the effects of MAOA expression variants and the differences between left and right amygdala connectivity. The resulting maps for the amygdala connectivity were thresholded at a voxelwise P < 0.001 and corrected for multiple comparisons using Monte–Carlo simulation at P < 0.05, resulting in a cluster threshold of k > 12 voxels.
ROIs were functionally defined, based on the main effect of ATD on amygdala connectivity, and labeled according to the location of their peak voxels (Table 1). At these peak voxels, connectivity values were extracted for both conditions to investigate the influence of MAOA expression variants and to control for carry‐over effects. ROI analyses were conducted in SPSS 21 (IBM Corp., Armok, New York). Peak voxels of ROIs that exhibited increased amygdala connectivity (positive connectivity change) after the ATD challenge and those of ROIs with reduced amygdala connectivity (negative connectivity change) were averaged separately. We tested for effects of MAOA expression on these average scores in an ANOVA with the within‐subject factor “Direction‐ATD‐effect” (positive connectivity change and negative connectivity change) and the between‐subject factors “Genotype” (MAOA‐H and MAOA‐L) and “Order” (first session ATD and BAL). Post hoc t‐tests determined directionality of the effects. Further exploratory paired t‐tests compared MAOA genotype effects on connectivity change of the amygdala, after the ATD challenge to each ROI. We estimated Cohen's d to evaluate the effect sizes of MAOA genotype effects on amygdala connectivity.
Table 1.
Clusters of ATD effect on amygdala connectivity
| Anatomical regions | Brodmann areas | Peak voxel x y z | Cluster size (mm³) | Tpeak |
|---|---|---|---|---|
| Right PFC | ||||
| Middle frontal gyrus R | 6, 9, 32 | 20 31 39 | 6067 | −5.39 |
| Cingulate gyrus R | ||||
| Inferior frontal gyrus R | 9 | |||
| Medial frontal gyrus R | 6, 8, 9, 10 | |||
| Precentral gyrus R | 6, 9 | |||
| Superior frontal gyrus R | 6, 8, 9, 10 | |||
| Left PFC | ||||
| Middle frontal gyrus L | 6, 8, 9, 46 | −43 13 30 | 1705 | −4.22 |
| Inferior frontal gyrus L | 9, 46 | |||
| Precentral gyrus L | 9 | |||
| Superior frontal gyrus L | 8 | |||
| Right IFG | ||||
| Inferior frontal gyrus R | 11, 47 | 35 28 −16 | 1743 | −5.14 |
| Middle frontal gyrus | 11, 47 | |||
| Superior frontal gyrus R | 11 | |||
| Left SFG | ||||
| Superior frontal gyrus L | 8, 9, 10 | −22 13 39 | 1750 | −4.13 |
| Cingulate gyrus L | 32 | |||
| Medial frontal gyrus L | 6, 8, 9, 32 | |||
| Middle frontal gyrus L | 6, 8, 9 | |||
| Left PG | ||||
| Precentral gyrus L | 6, 9 | −46 4 36 | 592 | −3.94 |
| Inferior frontal gyrus L | 6, 9 | |||
| Middle frontal gyrus L | 6, 8, 9 | |||
| Right CN | ||||
| Caudate nucleus R | 11 −2 24 | 1756 | −5.16 | |
| Cingulate gyrus R | 24 | |||
| Right LN | ||||
| Cingulate gyrus R | 25 | 8 1 −9 | 757 | −4.45 |
| Caudate R | ||||
| Lentiform nucleus R | ||||
| Subcallosal gyrus R | 25 | |||
| Left CN | ||||
| Caudate nucleus L | −16 10 9 | 722 | −3.85 | |
| Claustrum L | ||||
| Lentiform nucleus L | ||||
| Right insula | ||||
| Claustrum R | 32, 17 15 | 1650 | 4.82 | |
| Insula R | ||||
| Lentiform nucleus R | ||||
| Superior temporal gyrus R | ||||
| Thalamus R | ||||
| Right ventral PCC | ||||
| Posterior cingulate cortex R | 29,30 | 14 −56 9 | 1132 | 4.43 |
| Lingual gyrus R | 18, 19 | |||
| Parahippocampal gyrus R | ||||
| Left ventral PCC | ||||
| Posterior cingulate L R | 23, 29, 30 | −4 −35 18 | 3468 | 4.71 |
| Caudate nucleus R | ||||
| Cingulate gyrus L R | 23, 31 | |||
| Parahippocampal gyrus R | 30 | |||
| Thalamus | ||||
| Left dorsal PCC | ||||
| Cingulate gyrus L | 31 | −13 −26 33 | 744 | 5.85 |
| Caudate nucleus L | ||||
| Posterior cingulate L | 23 | |||
| Left precuneus | ||||
| Precuneus L | 7, 31 | −19 −59 27 | 593 | 4.10 |
| Cingulate gyrus L | 31 | |||
| Left lingual gyrus | ||||
| Declive | 11 −80 −15 | 1376 | 4.84 | |
| Inferior occipital gyrus R | ||||
| Lingual gyrus R | ||||
| Right STG | ||||
| Caudate nucleus R | 35 −41 6 | 726 | 5.28 | |
| Middle temporal gyrus R | 22 | |||
| Parahippocampal gyrus R | 19 | |||
| Hippocampus R | ||||
| Superior temporal gyrus R | 22, 41 |
Abbreviations for cluster labels: PFC, prefrontal cortex; CN, caudate nucleus; IFG, inferior frontal gyrus; SFG, superior frontal gyrus; LN, lentiform nucleus; PG, precentral gyrus; PCC, posterior cingulate cortex; STG, superior temporal gyrus.
RESULTS
Genotype Results
In line with previously reported distributions of MAOA allele expression [Sabol et al., 1998], 21 out of 64 participants (32.8%) were classified as MAOA‐L (20 × 3 repeats; 1 × 5 repeats) and 43 (67.2%) as MAOA‐H carriers (2 × 3.5 repeats; 41 × 4 repeats).
Serotonin Challenge and Mood
Five participants were excluded from the analysis due to drop‐outs or technical problems. Of the remaining sample, 28 participants received the BAL condition and 31 participants the ATD condition first. We found significant main effects for the factors “Condition” (F(1,57) = 1014.3, P < 0.0001) and “Time” (F(1,57) = 700.8, P < 0.0001) as well as a significant “Condition*Time” interaction (F(1,57) = 1107.4, P < 0.0001). Post‐hoc t‐tests showed a significant increase of tryptophan levels, comparing before beverage administration and before resting state in the BAL condition (before beverage: 65.58 ± 1.39 mmol/L; before resting state: 446.63 ± 11.96 mmol/L; t(59) = −32.6, P < 0.0001; Cohen's d = 5.83) and significantly reduced tryptophan serum levels comparing before beverage administration and before resting state in the ATD condition (before beverage: 65.9 ± 1.65 mmol/L; before resting state: 22.51 ± 0.99 mmol/L; t(59) = −30.3, P < 0.0001; Cohen's d = −4.15). According to our hypotheses, we focused on the change of tryptophan levels for the “Condition*Time” interaction between before beverage administration and before resting state in the ATD and BAL conditions. The t‐test confirmed significant reduction of tryptophan levels in the ATD condition compared with the BAL condition before resting state (ATD: −40.08 ± 6.36 mmol/L, BAL: 356.14 ± 16.56 mmol/L; t(60) = −22.4, P < 0.0001; Cohen's d = 4.35). Neither the factors “Genotype” or “Order” nor any further interaction yielded significance (all F < 3.2).
Due to incomplete PANAS forms, seven participants who received ATD first and seven participants who received BAL first were excluded from the behavioral analysis. In line with previous studies [Booij et al., 2005; Roiser et al., 2009b], we found no main effect for “Condition” or “Genotype,” or any interaction on mood (all P > 0.5).
Seed‐Based Functional Connectivity Analysis
After the ATD challenge, amygdala connectivity was reduced to the bilateral prefrontal cortex (right/left PFC), the bilateral caudate nucleus (right/left CN), the right inferior frontal gyrus (right IFG), the left superior frontal gyrus (left SFG), the left precentral gyrus (left PG), and the right lentiform nucleus (right LN). Furthermore, t‐tests revealed increased amygdala connectivity to the right insula, the bilateral ventral and left dorsal posterior cingulate cortex (right/left ventral and left dorsal PCC), the right superior temporal gyrus (right STG), the right lingual gyrus, and the left precuneus after ATD challenge (Table 1, Fig. 1). No differences emerged between the MAOA genotypes or between left and right amygdala connectivity at the same statistical threshold.
Figure 1.
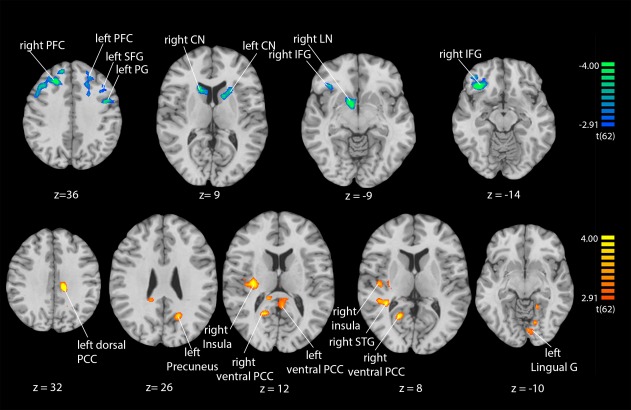
ATD effects on amygdala connectivity. Functional connectivity with the amygdala was reduced in prefrontal and striatal structures (cold colors; upper row) and increased to right insula, right STG, and the PCC (warm color; lower row). For abbreviations see Table 1. [Color figure can be viewed at http://wileyonlinelibrary.com]
Functional connectivity changes were extracted and averaged from the peaks of the seven positive (higher connectivity after ATD) and the eight negative ROIs (lower connectivity after ATD). In addition to the trivial main effect “Direction‐ATD‐effect” (F(1,60) = 88.0; P < 0.0001), the interaction “Direction‐ATD‐effect*Genotype” (F(1,60) = 12.7; P < 0.001) yielded significance. There was no significant effect of “Genotype” or “Order” or any further interaction on connectivity change (all F < 0.6, P > 0.4). Post hoc t‐tests revealed a higher negative (t(62) = −2.8, P < 0.014, after Bonferroni correction; d = −2.269; Fig. 2) as well as a higher positive connectivity change (t(62) = 2.8, P < 0.014, after Bonferroni correction; d = 1.928; Fig. 2) in the MAOA‐L group compared with the MAOA‐H group.
Figure 2.
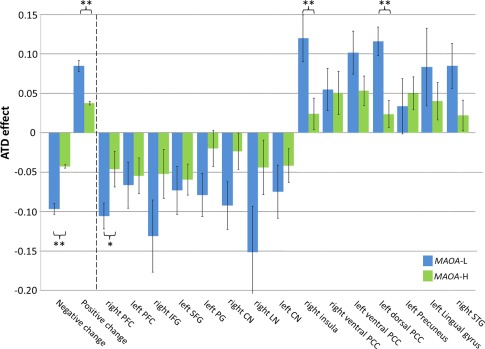
ATD effect on amygdala connectivity in MAOA‐L and ‐H carriers. Risk allele carriers (MAOA‐L) were more affected by ATD across ROIs (left‐most bars: “Negative change” in ROIs with reduced connectivity after ATD and “Positive change” in the other ROIs) and for the single ROIs yielding significance in right PFC, right insula, and left dorsal PCC. (Changes of correlation coefficients are shown after Fisher's z‐transformation with 95%‐confidence interval as error bars; MAOA‐L: blue; MAOA‐H: green; ** = P < 0.01; * = P < 0.05). [Color figure can be viewed at http://wileyonlinelibrary.com]
Further, an exploratory analysis considered the connectivity values in all ROIs separately. The paired t‐tests yielded significant differences in the right PFC, the right insula, and the left dorsal PCC (right PFC: t(61.85) = −2.1, P = 0.037 after Greenhouse–Geisser correction, d =−0.15; right insula: t(62) = 2.7; P = 0.009, d = 0.72; left dorsal PCC: t(62) = 3.3, P = 0.002, d = 0.93; Fig. 2 and Table 2).
Table 2.
ATD effects in MAOA‐H and ‐L carriers
| Average ATD effect on amygdala connectivity (±SD) | MAOA‐L (N = 21) | MAOA‐H (N = 43) |
|---|---|---|
| Right PFC | −0.11 ± 0.08* | −0.05 ± 0.15 |
| Left PFC | −0.07 ± 0.13 | −0.05 ± 0.15 |
| Right IFG | −0.13 ± 0.21 | −0.05 ± 0.20 |
| Left SFG | −0.07 ± 0.14 | −0.06 ± 0.13 |
| Left PG | −0.08 ± 0.12 | −0.02 ± 0.15 |
| Right CN | −0.09 ± 0.14 | −0.02 ± 0.15 |
| Right LN | −0.15 ± 0.27 | −0.04 ± 0.23 |
| Left CN | −0.07 ± 0.15 | −0.04 ± 0.14 |
| Right insula | 0.12 ± 0.14* | 0.02 ± 0.13 |
| Right ventral PCC | 0.05 ± 0.12 | 0.05 ± 0.18 |
| Left ventral PCC | 0.10 ± 0.13 | 0.05 ± 0.12 |
| Left dorsal PCC | 0.11 ± 0.08* | 0.02 ± 0.11 |
| Left precuneus | 0.03 ± 0.16 | 0.05 ± 0.14 |
| Left lingual gyrus | 0.08 ± 0.23 | 0.04 ± 0.16 |
| Right STG | 0.02 ± 0.13 | 0.08 ± 0.13 |
SD, Standard deviation; *: significant group difference, see text; abbreviations: see Table 1.
DISCUSSION
The present study investigated the effects of ATD and its interaction with MAOA expression alleles on amygdala connectivity. After the ATD challenge, amygdala connectivity was reduced to frontal areas, whereas connectivity to the insula and the posterior cingulate cortex was increased. On average, these ATD effects were more than twice as strong in the MAOA‐L as in the MAOA‐H carriers. Thus, the ATD challenge unmasked a vulnerability of cortico‐limbic connectivity in MAOA‐L carriers to reduced 5‐HT availability. In this genotype, the instable PFC‐amygdala connectivity may confer a risk for impaired emotion regulation, which is associated with antisocial behaviors.
Impact of ATD and BAL on Tryptophan Levels
Tryptophan levels in blood serum decreased to about a third after ATD. Such a reduction of tryptophan confers to diminished central 5‐HT availability [Sánchez et al., 2015]. In the BAL condition, serum tryptophan after BAL increased to about the 6‐fold level of before beverage intake, similarly to a previous study by Dingerkus et al. [2002]. However, such marked increase in the serum did not affect 5‐HT levels in the brain [Biskup et al., 2012].
Behavioral Effects of ATD and MAOA
The ATD challenge investigates the role of 5‐HT on mood in healthy subjects [Evers et al., 2010] and in patients with psychiatric disorders [Booij et al., 2003; Golightly et al., 2001; Zimmermann et al., 2012]. Apparently, mood decreased in healthy men after ATD [Young and Leyton, 2002]; although, recent research suggests only small, if any, effects of the ATD challenge on the mood in healthy men [Young, 2013]. Our study confirmed no ATD effect on mood; positive and negative affect neither differed between ATD and BAL conditions nor did an interaction with the genotype emerge. In previous research, mood effects emerged primarily in patient groups and their relatives [Benkelfat et al., 1994]. A different 5‐HT‐related gene—the 5‐HT transporter polymorphism (5‐HTTLPR)—yielded an effect in women only [Neumeister et al., 2002]. Thus, mood effects of ATD may depend on sex and genes regulating 5‐HT. We observed no such interaction for expression variants of the MAOA gene in healthy males.
Altered central nervous 5‐HT neurotransmission may contribute to the emergence of antisocial behavior. In a previous ATD challenge study, healthy men responded with more aggression to repeated provocation as 5‐HT levels decreased [Bjork et al., 1999; Young and Leyton, 2002]. Subsequent investigations replicated an increase of aggression after an ATD challenge [Bjork et al., 2000; Moeller et al., 1996]. Thus, the ATD challenge provides an interesting model to investigate the neural correlates involved in aggressive behavior. Conceivably, our participants have had an increased potential for aggressive behavior after the ATD challenge as well. Our study, however, chose not to trigger or test aggressive behavior, to obtain unbiased RS‐fMRI measurements.
Neural Effects of ATD
Consistent with our hypothesis, cortico‐limbic connectivity of the resting‐state networks was lower after the ATD challenge. In particular, the PFC exhibited reduced functional connectivity with the amygdala after the ATD challenge. Conceivably, 5‐HT mediates the communication between the PFC and the amygdala [Davidson et al., 2000; Siever, 2008]. Our study supports this idea and shows that functional connectivity between the PFC and the amygdala depends on central 5‐HT. In a study on impulsive behavior, the central 5‐HT availability influenced the cortico‐amygdala connectivity as well as the inhibitory control [Passamonti et al., 2006]. A few further studies in this field confirmed that reduced central 5‐HT was associated with decreased right prefrontal activity and increased impulsive behavior [Lamar et al., 2009; Rubia et al., 2005]. Although functional implications remain speculative, we hypothesize that lower PFC‐amygdala connectivity after the ATD challenge may reflect the neural basis of impaired impulse control and reduced emotion regulation as observed in behavioral studies.
Attempts were made to specify the roles of the regions of the PFC in emotion regulation. In our study, we found reduced IFG‐amygdala connectivity during resting state after the ATD challenge. The IFG achieved top–down‐regulation of negative emotional responses [Ochsner et al., 2004] and impulse control [Coccaro et al., 2007; Smith et al., 1999b]. This frontal region seems to be a core area for emotion regulation, in particular for negative emotions and impulse control [Kohn et al., 2014]. Studies in patients with borderline personality disorder reported an association between reduced IFG activation, and aggressive and impulsive behavior during a Go/NoGo task in contrast to controls without an emotion regulation disorder [Jacob et al., 2013]. Our study may provide a new perspective for the influence of 5‐HT on the IFG in cortico‐limbic regulation. The IFG's function during emotion regulation critically depends on 5‐HT levels [Rubia et al., 2005]. Indeed, after the ATD challenge, IFG activation was reduced and aggression levels were increased [Allen et al., 2006; Rubia et al., 2005]. Conceivably, IFG‐amygdala connectivity may reflect an additional mechanism for emotion regulation and impulse inhibition modulated by central 5‐HT.
Insula–Emotion Processing and Aggression
In contrast to frontal regions, amygdala connectivity with the insula was increased after the ATD challenge. The insula has abundant reciprocal connections to the amygdala [Nieuwenhuys, 2012] and is central to emotion processing [for a review, see Klasen et al., 2014]. Enhanced activation in the insula emerged particularly after provocation and is associated with aggressive retaliation [Krämer et al., 2007], and damage to the insula may reduce impulsive behavior [Naqvi et al., 2007]. Therefore, the insula may be considered as a modulating factor for aggressive and impulsive behavior. Indeed, insula activation was reduced after ATD challenge during the decision phase in Taylor's aggression paradigm [Krämer et al., 2011], which may be in line with a tighter coupling of limbic responses, as disclosed by our connectivity analysis.
The insula is related to the detection of, and the reaction to aggressive stimuli. On the one hand, during the Taylor aggression paradigm, insula activation increased after aggressive reactions to the opponent [Dambacher et al., 2015]. On the other hand, the insula is part of the salience network [Seeley et al., 2007] and contributes to the allocation of attention to salient environmental stimuli [Menon and Uddin, 2010] by integrating information for higher cognitive processing [Damasio et al., 2000]. Indeed, the insula is consistently involved in the processing and regulation of emotions, in particular of negative affect [Ochsner et al., 2004; Sambataro et al., 2006]. Hypothetically, an alteration of the insula–amygdala connectivity may reflect modulated integration and cognitive regulation of negative affective information derived from the environment.
Posterior Cingulate Cortex Connectivity after ATD
The PCC is the core hub of the default mode network [DMN; Raichle et al., 2001], and the DMN activity in the PCC and precuneus was influenced by the ATD challenge [Kunisato et al., 2011]. In our study, the amygdala connectivity with the PCC increased after the ATD challenge. This is in line with the lowered DMN activity under reduced 5‐HT metabolism [Scharinger et al., 2014] and tighter coupling to limbic activity. Indeed in patients with depression, the DMN activity during resting state was found to extend further into the limbic system [Sheline et al., 2009], and was restored to normal levels after increasing 5‐HT availability [Cullen et al., 2016].
The DMN and the PCC are involved in emotion processing, regulation, and introspection [Klasen et al., 2011; Maddock et al., 2003; Otto et al., 2014; Singer, 2006; Teasdale, 1999]. Similar to the insula, the PCC is involved in processing and control of salient negative emotional stimuli [Vogt, 2005], and increased activation is associated to situations with immediately harmful outcome for specific opponents [Helmbold et al., 2015; White et al., 2014]. On the molecular level, local and autoinhibitory 5‐HT1A binding inversely modulated the posterior cingulate cortex [Hahn et al., 2012], which may render this region particularly sensitive to changes in 5‐HT availability. Conceivably, the disturbed PCC‐amygdala network may reflect impaired processing of negative affect after the ATD challenge. Therefore, the increased amygdala connectivity with the PCC and the insula may show that the attention bias to negative and aggressive emotions processed in the amygdala depends on 5‐HT availability.
Vulnerability in MAOA‐L Carriers
In the current study, MAOA‐L carriers showed much stronger ATD effects as compared with MAOA‐H carriers, presumably reflecting a higher vulnerability of their serotonergic system. A similar interaction between a nutritional and a genetic factor was reported previously by for phenylketonuria—first described by Følling [1934]. In this disorder, the consumption of the amino acid phenylalanine led to severe impairments of brain development in carriers of a dysfunctional phenylketon‐hydroxylase gene [Pietz et al., 1999]. The gene–nutritional or –environmental interactions are not limited to somatic phenomena but extends to behavior and its neural correlates as well [for a review, see Burt, 2008].
In our study, the PFC, the insula, and the PCC were more susceptible to the ATD challenge in MAOA‐L than in MAOA‐H carriers. We discussed the functional relevance of these regions for emotion regulation above. Hypothetically, these regions may contribute to poor emotion regulation in MAOA‐L carriers after adverse events. MAOA‐L carriers are more likely to engage in delinquent behaviors [Beaver et al., 2014; Brunner et al., 1993; Cases et al., 1995; Guo et al., 2008] and constitute a higher proportion of violent offenders among incarcerated felons [Stetler et al., 2014]. Though several studies replicated the association between antisocial behavior and carriers of the MAOA‐L gene, a direct link has not been reported consistently [for a review, see Vassos et al., 2014]. These contradictory reports need to be seen in the light of neglecting environmental influences. Caspi et al. [2002] first reported that the interaction of MAOA‐L and childhood maltreatment contributed significantly to the emergence of antisocial behavior in males. This gene–environment interaction was repeatedly replicated in larger samples for the male sex only [see meta‐analysis in Byrd and Manuck, 2014]; although, some skepticism emerged as well [for a comprehensive review, see Ficks and Waldman, 2014]. Low expression of MAOA may lead to impaired inhibitory control of impulsive behaviors after developmental stress. However, the neural mechanisms underlying the vulnerability in MAOA‐L carriers are unknown. Our results may reflect alterations of the serotonergic system to the outlined cortico‐limbic network that become apparent after external stress. Therefore, the outlined cortico‐limbic network may represent the neural basis of 5‐HT contributing to the risk of developing impulsive‐aggressive behavior in MAOA‐L carriers when exposed to adverse environmental challenges [Caspi et al., 2002; Kim‐Cohen et al., 2006]. Further monoaminergic neurotransmitters such as adrenaline may contribute to this risk as well, as they are also affected by MAOA expression. These neurotransmitters are out of the scope of this study and need to be addressed in further research.
In previous studies, ATD and low expression of MAOA affected 5‐HT availability in opposite directions but both factors were associated with impulsive aggressive behavior [Caspi et al., 2002; Chester et al. 2015; Manuck et al., 2006]. Although a comprehensive explanation for this phenomenon is still lacking, adaptation during development may reverse the effect of high 5‐HT. Thus, the same cortico‐limbic network may contribute to higher impulsivity after the ATD challenge and after aversive child hood experiences in MAOA‐L carriers. Mouse models [see Uchida et al., 2005, 2007] may help to disentangle short‐ and long‐term effects of altered 5‐HT levels within specific neural circuits.
LIMITATIONS
Notably, the gene–environment interaction usually refers to an adverse environmental condition during development, and the current study employs the ATD challenge as an acute model. In a mouse model, chronic tryptophan depletion (1 month) induced lower anxiety and higher social dominance behavior [Uchida et al., 2005, 2007]. In a similar vein, increased impulsivity in humans may arise from reduced anxiety, and an increased stress response after reduced central 5‐HT during development. Furthermore, both—MAOA‐L and ATD—challenged the regulatory resources of the cortico‐limbic system in our study. Therefore, we suggest that the synergistic effects of both conditions may affect neural correlates, promoting impulsive behavior.
Contributions of distinct neurotransmitter systems to the development of antisocial behavior are rather unclear so far. MAOA degrades not only 5‐HT but other monoamines such as norepinephrine and dopamine as well. These confounds cannot be dismissed but we argue that the observed effects arise due to effects on 5‐HT, since MAOA influenced the 5‐HT levels most strongly compared with norepinephrine and dopamine [Nair et al., 1993]. Conceivably, the observed neural effects were most likely mediated by effects on 5‐HT availability rather than norepinephrine or dopamine.
The statistical power to detect medium‐sized effects (d = 0.5) in the fMRI experiment was limited by the size of the MAOA‐L group [MAOA‐L d = 0.58; MAOA‐H d = 0.89; calculated in GPower; Faul et al., 2007, 2009]. Therefore, the detection of other group differences may have failed in the current study. Nevertheless, statistics showed stable effects of MAOA genotype on amygdala connectivity after the ATD challenge.
In addition to 5‐HT, ATD affects the kynurenine system as well. Kynurenine is a metabolite of tryptophan and thus reduced after ATD. In a controversially discussed article, Van Donkelaar et al. [2011] speculated that memory impairments after ATD may also be due to reduced kynurenine. In our study, we investigated neural correlates of emotion regulation. Emotion regulation and impulse control has been consistently associated with 5‐HT. We argue that lowered 5‐HT synthesis was the main factor for altered cortico‐limbic connectivity.
CONCLUSION
The cortico‐limbic connectivity was affected by the ATD challenge during resting state. MAOA‐L carriers were more vulnerable to the depletion effects and are a known risk group for impulsive antisocial behavior. Altered cortico‐limbic connectivity is a promising candidate for the neural mechanism enhancing vulnerability to external stressors in MAOA‐L carriers and may contribute to the emergence of antisocial behaviors.
The authors report no further financial disclosures or potential conflicts of interest.
REFERENCES
- Allen PP, Cleare AJ, Lee F, Fusar‐Poli P, Tunstall N, Fu CH, Brammer MJ, McGuire PK (2006): Effect of acute tryptophan depletion on pre‐frontal engagement. Psychopharmacology 187:486–497. [DOI] [PubMed] [Google Scholar]
- Anand A, Li Y, Wang Y, Lowe MJ, Dzemidzic M (2009): Resting state corticolimbic connectivity abnormalities in unmedicated bipolar disorder and unipolar depression. Psychiatry Res: Neuroimaging 171:189–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaver KM, Barnes JC, Boutwell BB (2014): The 2‐repeat allele of the MAOA gene confers an increased risk for shooting and stabbing behaviors. Psychiatr Q 85:257–265. [DOI] [PubMed] [Google Scholar]
- Beckmann CF, DeLuca M, Devlin JT, Smith SM (2005): Investigations into resting‐state connectivity using independent component analysis. Philos Trans R Soc Lond B Biol Sci 360:1001–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkelfat C, Ellenbogen MA, Dean P, Palmour RM, Young SN (1994): Mood‐lowering effect of tryptophan depletion enhanced susceptibility in young men at genetic risk for major affective disorders. Arch Gen Psychiatry 51:687–697. [DOI] [PubMed] [Google Scholar]
- Biskup CS, Sánchez CL, Arrant A, Van Swearingen AE, Kuhn C, Zepf FD (2012): Effects of acute tryptophan depletion on brain serotonin function and concentrations of dopamine and norepinephrine in C57BL/6J and BALB/cJ mice. PLoS One 7:e35916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjork JM, Dougherty DM, Moeller FG, Cherek DR, Swann AC (1999): The effects of tryptophan depletion and loading on laboratory aggression in men: Time course and a food‐restricted control. Psychopharmacology (Berl.) 142:24–30. [DOI] [PubMed] [Google Scholar]
- Bjork JM, Dougherty DM, Moeller FG, Swann AC (2000): Differential behavioral effects of plasma tryptophan depletion and loading in aggressive and nonaggressive men. Neuropsychopharmacology 22:357–369. [DOI] [PubMed] [Google Scholar]
- Booij L, Van der Does AJ, Riedel WJ (2003): Monoamine depletion in psychiatric and healthy populations: Review. Mol Psychiatry 8:951–973. [DOI] [PubMed] [Google Scholar]
- Booij L, van der Does AJ, Haffmans PM, Spinhoven P, McNally RJ (2005): Acute tryptophan depletion as a model of depressive relapse: Behavioural specificity and ethical considerations. Br J Psychiatry 187:148–154. [DOI] [PubMed] [Google Scholar]
- Bremner JD, Innis RB, Salomon RM, Staib LH, Ng CK, Miller HL, Bronen RA, Krystal JH, Duncan J, Rich D, Price LH, Malison R, Dey H, Soufer R, Charney DS (1997): Positron emission tomography measurement of cerebral metabolic correlates of tryptophan depletion‐induced depressive relapse. Arch Gen Psychiatry 54:364–374. [DOI] [PubMed] [Google Scholar]
- Brunner HG, Nelen M, Breakefield XO, Ropers HH, van Oost BA (1993): Abnormal behavior associated with a point mutation in the structural gene for monoamine oxidase A. Science 22:578–580. [DOI] [PubMed] [Google Scholar]
- Buckholtz JW, Meyer‐Lindenberg A (2008): MAOA and the neurogenetic architecture of human aggression. Trends Neurosci 31:120–129. [DOI] [PubMed] [Google Scholar]
- Buckholtz JW, Callicott JH, Kolachana B, Hariri AR, Goldberg TE, Genderson M, Egan MF, Mattay VS, Weinberger DR, Meyer‐Lindenberg A (2008): Genetic variation in MAOA modulates ventromedial prefrontal circuitry mediating individual differences in human personality. Mol Psychiatry 13:313–324. [DOI] [PubMed] [Google Scholar]
- Burt SA (2008): Gene–environment interactions and their impact on the development of personality traits. Psychiatry 7:507–510. [Google Scholar]
- Byrd AL, Manuck SB (2014): MAOA, childhood maltreatment, and antisocial behavior: Meta‐analysis of a gene–environment interaction. Biol Psychiatry 75:9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cases O, Seif I, Grimsby J, Gaspar P, Chen K, Pournin S, Müller U, Aguet M, Babinet C, Shih JC, De Maeyer E (1995): Aggressive behavior and altered amounts of brain serotonin and norepinephrine in mice lacking MAOA. Science 268:1763–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi A, McClay J, Moffitt TE, Mill J, Martin J, Craig IW, Taylor A, Poulton R (2002): Role of genotype in the cycle of violence in maltreated children. Science 297:851–854. [DOI] [PubMed] [Google Scholar]
- Challis C, Berton O (2015): Top‐down control of serotonin systems by the prefrontal cortex: A path toward restored socioemotional function in depression. ACS Chem Neurosci 6:1040–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chester DS, DeWall CN, Derefinko KJ, Estus S, Peters JR, Lynam DR, Jiang Y (2015): Monoamine oxidase A (MAOA) genotype predicts greater aggression through impulsive reactivity to negative affect. Behav Brain Res 283:97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleare AJ, Bond AJ (1995): The effect of tryptophan depletion and enhancement on subjective and behavioural aggression in normal male subjects. Psychopharmacology (Berl) 118:72–81. [DOI] [PubMed] [Google Scholar]
- Coccaro EF, McCloskey MS, Fitzgerald DA, Phan KL (2007): Amygdala and orbitofrontal reactivity to social threat in individuals with impulsive aggression. Biol Psychiatry 62:168–178. [DOI] [PubMed] [Google Scholar]
- Crockett MJ, Clark L, Robbins TW (2009): Reconciling the role of serotonin in behavioral inhibition and aversion: Acute tryptophan depletion abolishes punishment‐induced inhibition in humans. J Neuroscience 29:11993–11999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen KR, Klimes‐Dougan B, Pham Vu D, Schreiner MW, Mueller BA, Eberly LE, Camchong J, Westervelt A, Lim KO (2016): Neural correlates of antidepressant treatment response in adolescents with major depressive disorder. J Child Adolesc Psychopharmacol 26:705–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damasio AR, Grabowski TJ, Bechara A, Damasio H, Ponto LLB, Parvizi J, Hichwa RD (2000): Subcortical and cortical brain activity during the feeling of self‐generated emotions. Nat Neurosci 3:1049–1056. [DOI] [PubMed] [Google Scholar]
- Dambacher F, Sack AT, Lobbestael J, Arntz A, Brugman S, Schuhmann T (2015): Out of control: evidence for anterior insula involvement in motor impulsivity and reactive aggression. Soc Cogn Affect Neurosci 10:508–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damoiseaux JS, Rombouts SA, Barkhof F, Scheltens P, Stam CJ, Smith SM, Beckmann CF (2006): Consistent resting‐state networks across healthy subjects. Proc Natl Acad Sci USA 103:13848–13853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson RJ, Putnam KM, Larson CL (2000): Dysfunction in the neural circuitry of emotion regulation ‐ a possible prelude to violence. Science 289:591–594. [DOI] [PubMed] [Google Scholar]
- Delgado PL, Charney DS, Price LH, Aghajanian GK, Landis H, Heninger GR (1990): Serotonin function and the mechanism of antidepressant action Reversal of antidepressant‐induced remission by rapid depletion of plasma tryptophan. Arch Gen Psychiatry 47:411–418. [DOI] [PubMed] [Google Scholar]
- Delgado PL, Price LH, Miller HL, Salomon RM, Licinio J, Krystal JH, Heninger GR, Charney DS (1991): Rapid serotonin depletion as a provocative challenge test for patients with major depression: Relevance to antidepressant action and the neurobiology of depression. Psychopharmacol Bull 27:321–330. [PubMed] [Google Scholar]
- Denson TF, Dobson‐Stone C, Ronay R, von Hippel W, Schira MM (2014): A functional polymorphism of the MAOA gene is associated with neural responses to induced anger control. J Cogn Neurosci 26:1418–1427. [DOI] [PubMed] [Google Scholar]
- Demisch L, Kewitz A, Schmeck K, Sadigorsky S, Barta S, Dierks T, Poustka F (2002): Methodology of rapid tryptophan depletion (RTD): Impact of gender and body weight. Eur Arch Psychiatry Clin Neurosci 252:1–25. 12056575 [Google Scholar]
- Dingerkus VL, Gaber TJ, Helmbold K, Bubenzer S, Eisert A, Sánchez CL, Zepf FD (2012): Acute tryptophan depletion in accordance with body weight: Influx of amino acids across the blood‐brain barrier. J Neural Transm (Vienna) 119:1037–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eickhoff SB, Stephan KE, Mohlberg H, Grefkes C, Fink GR, Amunts K, Zilles K (2005): A new SPM toolbox for combining probabilistic cytoarchitectonic maps and functional imaging data. Neuroimage 25:1325–1335. [DOI] [PubMed] [Google Scholar]
- Evers EA, van der Veen FM, van Deursen JA, Schmitt JA, Deutz NE, Jolles J (2006): The effect of acute tryptophan depletion on the BOLD response during performance monitoring and response inhibition in healthy male volunteers. Psychopharmacology (Berl) 187:200–208. [DOI] [PubMed] [Google Scholar]
- Evers EA, Sambeth A, Ramaekers JG, Riedel WJ, van der Veen FM (2010): The effects of acute tryptophan depletion on brain activation during cognition and emotional processing in healthy volunteers. Curr Pharm Des 16:1998–2011. [DOI] [PubMed] [Google Scholar]
- Faul F, Erdfelder E, Lang AG, Buchner A (2007): G*Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods 39:175–191. [DOI] [PubMed] [Google Scholar]
- Faul F, Erdfelder E, Buchner A, Lang AG (2009): Statistical power analyses using G*Power 3.1: Tests for correlation and regression analyses. Behav Res Methods 41:1149–1160. [DOI] [PubMed] [Google Scholar]
- Ficks CA, Waldman ID (2014): Candidate genes for aggression and antisocial behavior: A meta‐analysis of association studies of the 5HTTLPR and MAOA‐µVNTR. Behav Genet 44: 427–444. [DOI] [PubMed] [Google Scholar]
- Fölling A (1934): Über Ausscheidung von Phenylbrenztraubensäure in den Harn als Stoffwechselanomalie in Verbindung mit Imbezillität. Hoppe‐Seyler's Z Physiol Chem 227:169–181. [Google Scholar]
- Frodl T, Scheuerecker J, Albrecht J, Kleemann AM, Müller‐Schunk S, Koutsouleris N, Möller HJ, Brückmann H, Wiesmann M, Meisenzahl E (2009): Neuronal correlates of emotional processing in patients with major depression. World J Biol Psychiatry 10:202–208. [DOI] [PubMed] [Google Scholar]
- Golightly KL, Lloyd JA, Hobson JE, Gallagher P, Mercer G, Young AH (2001): Acute tryptophan depletion in schizophrenia. Psychol Med 31:75–84. [DOI] [PubMed] [Google Scholar]
- Guo G, Ou XM, Roettger M, Shih JC (2008): The VNTR 2 repeat in MAOA and delinquent behavior in adolescence and young adulthood: Associations and MAOA promoter activity. Eur J Hum Genet 16:626–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn A, Wadsak W, Windischberger C, Baldinger P, Höflich AS, Losak J, Nics L, Philippe C, Kranz GS, Kraus C, Mitterhauser M, Karanikas G, Kasper S, Lanzenberger R (2012): Differential modulation of the default mode network via serotonin‐1A receptors. Proc Natl Acad Sci U S A 109:2619–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmbold K, Zvyagintsev M, Dahmen B, Bubenzer‐Busch S, Gaber TJ, Crockett MJ, Klasen M, Sánchez CL, Eisert A, Konrad K, Habel U, Herpertz‐Dahlmann B, Zepf FD (2015): Effects of serotonin depletion on punishment processing in the orbitofrontal and anterior cingulate cortices of healthy women. Eur Neuropsychopharmacol 25:846–856. [DOI] [PubMed] [Google Scholar]
- Hindi Attar C, Finckh B, Büchel C (2012): The influence of serotonin on fear learning. PLoS One 7:e42397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutton C, Josephs O, Stadler J, Featherstone E, Reid A, Speck O, Bernarding J, Weiskopf N (2011): The impact of physiological noise correction on fMRI at 7 T. Neuroimage 57:101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob GA, Zvonik K, Kamphausen S, Sebastian A, Maier S, Philipsen A, Tebartz van Elst L, Lieb K, Tüscher O (2013): Emotional modulation of motor response inhibition in women with borderline personality disorder: An fMRI study. J Psychiatry Neurosci 38:164–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim‐Cohen J, Caspi A, Taylor A, Williams B, Newcombe R, Craig IW, Moffitt TE (2006): MAOA, maltreatment, and gene–environment interaction predicting children's mental health: New evidence and a meta‐analysis. Mol Psychiatry 11:903–913. [DOI] [PubMed] [Google Scholar]
- Klaassen T, Riedel WJ, Deutz NE, Van Praag HM (2002): Mood congruent memory bias induced by tryptophan depletion. Psychol Med 32:167–172. [DOI] [PubMed] [Google Scholar]
- Klasen M, Kenworthy CA, Mathiak KA, Kircher TT, Mathiak K (2011): Supramodal representation of emotions. J Neurosci 31:13635–13643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klasen M, Zvyagintsev M, Schwenzer M, Mathiak KA, Sarkheil P, Weber R, Mathiak K (2013): Quetiapine modulates functional connectivity in brain aggression networks. Neuroimage 75:20–26. [DOI] [PubMed] [Google Scholar]
- Klasen M, Chen YH, Mathiak K (2014): Multisensory emotions: Perception, combination and underlying neural processes. Rev Neurosci 23:381–392. [DOI] [PubMed] [Google Scholar]
- Kohn N, Eickhoff SB, Scheller M, Laird AR, Fox PT, Habel U (2014): Neural network of cognitive emotion regulation–an ALE meta‐analysis and MACM analysis. Neuroimage 87:345–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krämer UM, Jansma H, Tempelmann C, Münte TF (2007): Tit‐for‐tat: The neural basis of reactive aggression. Neuroimage 38:203–211. [DOI] [PubMed] [Google Scholar]
- Krämer UM, Riba J, Richter S, Münte TF (2011): An fMRI study on the role of serotonin in reactive aggression. PLoS ONE 6:e27668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunisato Y, Okamoto Y, Okada G, Aoyama S, Demoto Y, Munakata A, Nomura M, Onoda K, Yamawaki S (2011): Modulation of default‐mode network activity by acute tryptophan depletion is associated with mood change: A resting state functional magnetic resonance imaging study. Neurosci Res 69:129–134. [DOI] [PubMed] [Google Scholar]
- Lamar M, Cutter WJ, Rubia K, Brammer M, Daly EM, Craig MC, Cleare AJ, Murphy DG (2009): 5‐HT, prefrontal function and aging: fMRI of inhibition and acute tryptophan depletion. Neurobiol Aging 30:1135–1146. [DOI] [PubMed] [Google Scholar]
- Lee RJ, Gill A, Chen B, McCloskey M, Coccaro EF (2012): Modulation of central serotonin affects emotional information processing in impulsive aggressive personality disorder. J Clin Psychopharmacol 32:329–335. [DOI] [PubMed] [Google Scholar]
- Lehrl S (2005) Mehrfachwahl‐Wortschatz‐Intelligenztest MWT‐B. Balingen: Spitta Verlag. [Google Scholar]
- Lemogne C, Gorwood P, Bergouignan L, Pélissolo A, Lehéricy S, Fossati P (2011): Negative affectivity, self‐referential processing and the cortical midline structures. Soc Cogn Affect Neurosci 6:426–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyton M, Okazawa H, Diksic M, Paris J, Rosa P, Mzengeza S, Young SN, Blier P, Benkelfat C (2001): Brain Regional alpha‐[11C]methyl‐L‐tryptophan trapping in impulsive subjects with borderline personality disorder. Am J Psychiatry 158:775–782. [DOI] [PubMed] [Google Scholar]
- Maddock RJ, Garrett AS, Buonocore MH (2003): Posterior cingulate cortex activation by emotional words: fMRI evidence from a valence decision task. Hum Brain Mapp 18:30–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuck SB, Kaplan JR, Lotrich FE. (2006) Brain serotonin and aggressive disposition in humans and nonhuman primates In: Nelson RJ, editor. Biology of Aggression. New York: Oxford University Press. [Google Scholar]
- Mathiak K, Weber R (2006): Toward brain correlates of natural behavior: fMRI during violent video games. Hum Brain Mapp 27:948–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon V, Uddin LQ (2010): Saliency, switching, attention and control: A network model of insula function. Brain Struct Funct 214:655–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer‐Lindenberg A, Buckholtz JW, Kolachana B, Hariri AR, Pezawas L, Blasi G, Wabnitz A, Honea R, Verchinski B, Callicott JH, Egan M, Mattay V, Weinberger DR (2006): Neural mechanisms of genetic risk for impulsivity and violence in humans. Proc Natl Acad Sci USA 103:6269–6274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miczek KA, de Almeida RM, Kravitz EA, Rissman EF, de Boer SF, Raine A (2007): Neurobiology of escalated aggression and violence. J Neurosci 27:11803–11806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller FG, Dougherty DM, Swann AC, Collins D, Davis CM, Cherek DR (1996): Tryptophan depletion and aggressive responding in healthy males. Psychopharmacology (Berl) 126:97–103. [DOI] [PubMed] [Google Scholar]
- Morris JS, Smith KA, Cowen PJ, Friston KJ, Dolan RJ (1999): Covariation of activity in habenula and dorsal raphe nuclei following tryptophan depletion. Neuroimage 10:163–172. [DOI] [PubMed] [Google Scholar]
- Moja EA, Stoff DM, Gessa GL, Castoldi D, Assereto R, Tofanetti O (1988): Decrease in plasma tryptophan after tryptophan‐free amino acid mixtures in man. Life Sci 42:1551–1556. [DOI] [PubMed] [Google Scholar]
- Nair NP, Ahmed SK, Kin NM (1993): Biochemistry and pharmacology of reversible inhibitors of MAO‐A agents: focus on moclobemide. J Psychiatry Neurosci 18:214–225. [PMC free article] [PubMed] [Google Scholar]
- Naqvi NH, Rudrauf D, Damasio H, Bechara A (2007): Damage to the insula disrupts addiction to cigarette smoking. Science 315:531–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumeister A, Konstantinidis A, Stastny J, Schwarz MJ, Vitouch O, Willeit M, Praschak‐Rieder N, Zach J, de Zwaan M, Bondy B, Ackenheil M, Kasper S (2002): Association between serotonin transporter gene promoter polymorphism (5HTTLPR) and behavioral responses to tryptophan depletion in healthy women with and without family history of depression. Arch Gen Psychiatry 59:613–620. [DOI] [PubMed] [Google Scholar]
- Neumeister A, Nugent AC, Waldeck T, Geraci M, Schwarz M, Bonne O, Bain EE, Luckenbaugh DA, Herscovitch P, Charney DS, Drevets WC (2004): Neural and behavioral responses to tryptophan depletion in unmedicated patients with remitted major depressive disorder and controls. Arch Gen Psychiatry 61:765–773. [DOI] [PubMed] [Google Scholar]
- Nieuwenhuys R (2012): The insular cortex: A review. Prog Brain Res 195:123–163. [DOI] [PubMed] [Google Scholar]
- Nishizawa S, Benkelfat C, Young SN, Leyton M, Mzengeza S, de Montigny C, Blier P, Diksic M (1997): Differences between males and females in rates of serotonin synthesis in human brain. Proc Natl Acad Science U S A 94:5308–5313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochsner KN, Ray RD, Cooper JC, Robertson ER, Chopra S, Gabrieli JD, Gross JJ (2004): For better or for worse: Neural systems supporting the cognitive down‐ and up‐regulation of negative emotion. Neuroimage 23:483–499. [DOI] [PubMed] [Google Scholar]
- Oldfield RC (1971): The assessment and analysis of handedness: The Edinburgh inventory. Neuropsychologia 9:97–113. [DOI] [PubMed] [Google Scholar]
- Otto B, Misra S, Prasad A, McRa K (2014): Functional overlap of top‐down emotion regulation and generation: An fMRI study identifying common neural substrates between cognitive reappraisal and cognitively generated emotions. Cogn Affect Behav Neurosci 14:923–938. [DOI] [PubMed] [Google Scholar]
- Passamonti L, Fera F, Magariello A, Cerasa A, Gioia MC, Muglia M, Nicoletti G, Gallo O, Provinciali L, Quattrone A (2006): Monoamine oxidase‐a genetic variations influence brain activity associated with inhibitory control: New insight into the neural correlates of impulsivity. Biol Psychiatry 59:334–340. [DOI] [PubMed] [Google Scholar]
- Passamonti L, Crockett MJ, Apergis‐Schoute AM, Clark L, Rowe JB, Calder AJ, Robbins TW (2012): Effects of acute tryptophan depletion on prefrontal‐amygdala connectivity while viewing facial signals of aggression. Biol Psychiatry 71:36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyron C, Petit JM, Rampon C, Jouvet M, Luppi PH (1998): Forebrain afferents to the rat dorsal raphe nucleus demonstrated by retrograde and anterograde tracing methods. Neuroscience 82:443–468. [DOI] [PubMed] [Google Scholar]
- Pietz J, Kreis R, Rupp A, Mayatepek E, Rating D, Boesch C, Bremer HJ (1999): Large neutral amino acids block phenylalanine transport into brain tissue in patients with phenylketonuria. J Clin Investig 103:1169–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pihl RO, Young SN, Harden P, Plotnick S, Chamberlain B, Ervin FR (1995): Acute effect of altered tryptophan levels and alcohol on aggression in normal human males. Psychopharmacology (Berl) 119:353–360. [DOI] [PubMed] [Google Scholar]
- Raichle ME, MacLeod AM, Snyder AZ, Powers WJ, Gusnard DA, Shulman GL (2001): A default mode of brain function. Proc Natl Acad Sci U S A 98:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy AK, Shehzad Z, Margulies DS, Kelly AMC, Uddin LQ, Gotimer K, Biswal BB, Castellanos FX, Milham MP (2009): Functional connectivity of the human amygdala using resting state fMRI. Neuroimage 45:614–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson OJ, Cools R, Sahakian BJ (2012): Tryptophan depletion disinhibits punishment but not reward prediction: Implications for resilience. Psychopharmacology 219:599–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roiser JP, de Martino B, Tan GC, Kumaran D, Seymour B, Wood NW, Dolan RJ (2009a): A genetically mediated bias in decision making driven by failure of amygdala control. J Neurosci 29:5985–5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roiser JP, Levy J, Fromm SJ, Nugent AC, Talagala SL, Hasler G, Henn FA, Sahakian BJ, Drevets WC (2009b): The effects of tryptophan depletion on neural responses to emotional words in remitted depression. Biol Psychiatry 66:441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubia K, Lee F, Cleare AJ, Tunstall N, Fu CH, Brammer M, McGuire P (2005): Tryptophan depletion reduces right inferior prefrontal activation during response inhibition in fast, event‐related fMRI. Psychopharmacology 179:791–803. [DOI] [PubMed] [Google Scholar]
- Sabol SZ, Hu S, Hamer D (1998): A functional polymorphism in the monoamine oxidase A gene promoter. Hum Genet 103:273–279. [DOI] [PubMed] [Google Scholar]
- Sambataro F, Dimalta S, Di Giorgio A, Taurisano P, Blasi G, Scarabino T, Giannatempo G, Nardini M, Bertolino A (2006): Preferential responses in amygdala and insula during presentation of facial contempt and disgust. Eur J Neurosci 24:2355–2362. [DOI] [PubMed] [Google Scholar]
- Sánchez CL, Van Swearingen AE, Arrant AE, Biskup CS, Kuhn CM, Zepf FD (2015): Simplified dietary acute tryptophan depletion: Effects of a novel amino acid mixture on the neurochemistry of C57BL/6J mice. Food Nutr Res 59:27424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharinger C, Rabl U, Kasess CH, Meyer BM, Hofmaier T, Diers K, Bartova L, Pail G, Huf W, Uzelac Z, Hartinger B, Kalcher K, Perkmann T, Haslacher H, Meyer‐Lindenberg A, Kasper S, Freissmuth M, Windischberger C, Willeit M, Lanzenberger R, Esterbauer H, Brocke B, Moser E, Sitte HH, Pezawas L (2014): Platelet serotonin transporter function predicts default‐mode network activity. PLoS One 9:e92543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeley WW, Menon V, Schatzberg AF, Keller J, Glover GH, Kenna H, Reiss AL, Greicius MD (2007): Dissociable intrinsic connectivity networks for salience processing and executive control. J Neurosci 27:2349–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheline YI, Barch DM, Price JL, Rundle MM, Vaishnavi SN, Snyder AZ, Mintun MA, Wang S, Coalson RS, Raichle ME (2009): The default mode network and self‐referential processes in depression. Proc Natl Acad Sci U S A 106:1942–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoaf SE, Carson R, Hommer D, Williams W, Higley JD, Schmall B, Herscovitch P, Eckelman W, Linnoila M (1998): Brain serotonin synthesis rates in rhesus monkeys determined by [11C]alpha‐methyl‐L‐tryptophan and positron emission tomography compared to CSF 5‐hydroxyindole‐3‐acetic acid concentrations. Neuropsychopharmacology 19:345–353. [DOI] [PubMed] [Google Scholar]
- Siever LJ (2008): Neurobiology of aggression and violence. Am J Psychiatry 165:429–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer T (2006): The neuronal basis and ontogeny of empathy and mind reading: Review of literature and implications for future research. Neurosci Biobehav Rev 30:855–863. [DOI] [PubMed] [Google Scholar]
- Smith KA, Morris JS, Friston KJ, Cowen PJ, Dolan RJ (1999a): Brain mechanisms associated with depressive relapse and associated cognitive impairment following acute tryptophan depletion. Br J Psychiatry 174:525–529. [DOI] [PubMed] [Google Scholar]
- Smith KA, Fairburn CG, Cowen PJ (1999b): Symptomatic relapse in bulimia nervosa following acute tryptophan depletion. Arch Gen Psychiatry 56:171–176. [DOI] [PubMed] [Google Scholar]
- South SC, Krueger RF (2008): An interactionist perspective on genetic and environmental contributions to personality. Soc Pers Compass 2:929–948. [Google Scholar]
- Stetler DA, Davis C, Leavitt K, Schriger I, Benson K, Bhakta S, Wang LC, Oben C, Watters M, Haghnegahdar T, Bortolato M (2014): Association of low‐activity MAOA allelic variants with violent crime in incarcerated offenders. J Psychiatr Res 58:69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talairach J, Szikla G (1980): Application of stereotactic concepts to the surgery of epilepsy. Acta Neurochir Suppl (Wien) 30:35–54. [DOI] [PubMed] [Google Scholar]
- Teasdale JD, Howard RJ, Cox SG, Ha Y, Brammer MJ, Williams SC, Checkley SA (1999): Functional MRI study of the cognitive generation of affect. Am J Psychiatry 156:209–215. [DOI] [PubMed] [Google Scholar]
- Uchida S, Kitamoto A, Umeeda H, Nakagawa N, Masushige S, Kida S (2005): Chronic reduction in dietary tryptophan leads to changes in the emotional response to stress in mice. J Nutr Sci Vitaminol (Tokyo) 51:175–181. [DOI] [PubMed] [Google Scholar]
- Uchida S, Umeeda H, Kitamoto A, Masushige S, Kida S (2007): Chronic reduction in dietary tryptophan leads to a selective impairment of contextual fear memory in mice. Brain Res 1149:149–156. [DOI] [PubMed] [Google Scholar]
- Van Donkelaar EL, Blokland A, Ferrington L, Kelly PA, Steinbusch HW, Prickaerts J (2011): Mechanism of acute tryptophan depletion: Is it only serotonin? Mol Psychiatry 16:695–713. [DOI] [PubMed] [Google Scholar]
- Vassos E, Collier DA, Fazel S (2014): Systematic meta‐analyses and field synopsis of genetic association studies of violence and aggression. Mol Psychiatry 19:471–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt BA (2005): Pain and emotion interactions in subregions of the cingulate gyrus. Nat Rev Neurosci 6:533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson D, Clark LA, Tellegen A (1988): Development and validation of brief measures of positive and negative affect: The PANAS scales. J Pers Soc Psychol 54:1063–1070. [DOI] [PubMed] [Google Scholar]
- White SF, Brislin SJ, Sinclair S, Blair JR (2014): Punishing unfairness: Rewarding or the organization of a reactively aggressive response? Hum Brain Mapp 35:2137–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young SN (2013): The effect of raising and lowering tryptophan levels on human mood and social behaviour. Philos Trans R Soc Lond B Biol Sci 368:20110375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young SN, Leyton M (2002): The role of serotonin in human mood and social interaction. Insight from altered tryptophan levels. Pharmacol Biochem Behav 71:857–865. [DOI] [PubMed] [Google Scholar]
- Zimmermann M, Grabemann M, Mette C, Abdel‐Hamid M, Uekermann J, Kraemer M, Wiltfang J, Kis B, Zepf FD (2012): The effects of acute tryptophan depletion on reactive aggression in adults with attention‐deficit/hyperactivity disorder (ADHD) and healthy controls. PLoS One 7:e32023. [DOI] [PMC free article] [PubMed] [Google Scholar]