

Abstract
Bilateral vestibular failure (BVF) is a severe chronic disorder of the labyrinth or the eighth cranial nerve characterized by unsteadiness of gait and disabling oscillopsia during head movements. According to animal data, vestibular input to the hippocampus is proposed to contribute to spatial memory and spatial navigation. Except for one seminal study showing the association of impaired spatial navigation and hippocampal atrophy, patient data in BVF are lacking. Therefore, we performed a voxel‐wise comparison of the hippocampal gray matter volume (GMV) in a clinically representative sample of 27 patients with incomplete BVF and 29 age‐ and gender‐matched healthy controls to test the hypothesis of hippocampal atrophy in BVF. Although the two groups did not generally differ in their hippocampal GMV, a reduction of GMV in the bilateral hippocampal CA3 region was significantly correlated with increased vestibulopathy‐related clinical impairment. We propose that GMV reduction in the hippocampus of BVF patients is related to the severity of vestibular‐induced disability which is in line with combined hippocampal atrophy and disorders of spatial navigation in complete vestibular deafferentation due to bilateral nerve section. Clinically, however, the most frequent etiologies of BVF cause incomplete lesions. Accordingly, hippocampus atrophy and deficits in spatial navigation occur possibly less frequently than previously suspected. Hum Brain Mapp 37:1998–2006, 2016. © 2016 Wiley Periodicals, Inc.
Keywords: voxel‐based morphometry, magnetic resonance imaging, bilateral vestibular failure, hippocampus
INTRODUCTION
Bilateral vestibular failure (BVF) is a severe chronic disorder of the labyrinth or the eighth cranial nerve characterized by unsteadiness of gait and disabling oscillopsia during head movements [Brandt, 1996]. BVF has a wide spectrum of etiologies [Zingler et al., 2007]. The most common causes of BVF are vestibulotoxicity of ototoxic drugs (specifically aminoglycosides), polyneuropathy, and unknown origin. New technical improvements allowing precise and easy assessment of vestibular function by video‐oculography have shown that BVF is much more common than previously believed [Machner et al., 2013].
Vestibular deafferentation induces several plastic functional [Bense et al., 2004a; Becker‐Bense et al., 2013; Helmchen et al., 2014] and structural [Helmchen et al., 2009, 2011; zu Eulenburg et al., 2010] changes in the brain. It is a matter of debate whether these changes are clinically beneficial, subserve vestibular compensation, or purely reflect the consequence of a lack of vestibular input. One prominent example of the latter is atrophy of the hippocampus secondary to peripheral BVF [Brandt et al., 2005]. This may be of potential clinical significance for the following reasons: First, BVF patients in the latter study showed impaired spatial memory and navigation during a virtual Morris water navigation task. Second, vestibular compensation in BVF is distinctly different from unilateral vestibular loss as peripheral recovery is usually poor and cerebral vestibular compensatory mechanisms are confined to adaptive mechanisms of sensory substitution, that is, by changing thresholds of other sensory processing and/or reciprocal intersensory interaction [Bense et al., 2004b; Dieterich et al., 2007; Deutschlander et al., 2008; Kalla et al., 2011]. Accordingly, clinical prognosis in BVF is often poor.
However, it may be promising that deafferentation from bilateral peripheral vestibular signals leads to even more fundamental changes: resting‐state activity in the brain, irrespective of any vestibular task, shows profound changes of functional connectivity in BVF patients [Göttlich et al., 2014]. There was lower bilateral connectivity in the posterior insula and parietal operculum but higher connectivity in the posterior cerebellum. Remarkably, functional connectivity in the supramarginal gyrus of the inferior parietal lobe and posterior cerebellum correlated with adaptive, head‐movement‐related changes of the vestibulo‐ocular reflex indicating adaptive cortical plasticity, which may be important for vestibular rehabilitation.
In contrast, structural brain volume changes in BVF patients have only sparsely been investigated. The seminal study by Brandt et al. [2005] indicated bilateral atrophy in the hippocampus by conventional tissue segmentation volumetry which was associated with spatial memory and navigation deficits. Spatial memory and navigation require multisensory integration, particularly of visual and vestibular signals [Brandt et al., 2014]. The hippocampus is a core structure for learning and memory, which receives vestibular information [Jacob et al., 2014] and reveals spatial separation of visual (posterior) and vestibular (anterior hippocampal formation) processing [Hufner et al., 2011b]. Moreover, the hippocampus and parahippocampus establish “cognitive” maps based on place cells, head direction cells, and grid cells activity, the latter encoding multiple specific locations in space [Hitier et al., 2014]. Grid‐like activity in entorhinal cortex, the main input to the hippocampus, has recently been recorded during virtual spatial navigation in epilepsy patients [Jacobs et al., 2013] but their role in human navigation in subjects lacking vestibular input to the hippocampus is not known yet.
There are several lines of evidence from animal data that the hippocampus is involved in spatial memory [Stackman et al., 2002; Wallace et al., 2002; Zheng et al., 2012]. In rodents, it has been shown that electrical [Cuthbert et al., 2000] or natural [Wiener et al., 1995] vestibular stimulation modulates the firing of hippocampal place cells which, in turn, is disrupted in bilateral peripheral vestibular lesions [Russell et al., 2006; Stackman and Herbert, 2002; Zheng et al., 2003]. BVF in rats leads to disruption of location‐specific firing in hippocampal place cells and severely impairs spatial memory [Besnard et al., 2012; Baek et al., 2010; Russell et al., 2003; Smith et al., 2010; Wallace et al., 2002; Zheng et al., 2006]. Some neurophysiological changes underlying BVF‐induced spatial learning and memory deficits are suspected to be due to up‐ and down‐regulation or changes in the affinity/efficacy of glutamate receptors at the membrane level [Zheng et al., 2013]. Moreover, galvanic vestibular stimulation can modulate cell proliferation and neurogenesis in the hippocampus, without affecting spatial memory, i.e., place recognition, object exploration, and Morris water maze tasks [Zheng et al., 2014].
In contrast to these animal data, evidence for behavioral (navigational deficits) and structural (standardized morphometry) abnormalities in BVF patients is sparse. We therefore applied voxel‐based morphometry (VBM) to investigate differences in the local composition of brain tissue, that is, voxel‐wise gray matter volume (GMV) [Ashburner and Friston, 2000]. In this first VBM study investigating gray matter density in BVF patients, we followed the guidelines for reporting VBM studies provided by Ridgway et al. [2008] to ease comparability across studies.
Structural volumetric brain imaging in patients with chronic complete unilateral vestibular failure (acoustic neuroma surgery) neither showed spatial memory and navigation deficits in a virtual Morris water task nor hippocampal atrophy [Hufner et al., 2007]. More recently, a longitudinal study in incomplete unilateral vestibular failure patients (vestibular neuritis) showed a trend toward an increase of GMV over time (3 months) in the left hippocampus and bilateral parahippocampal gyri, which correlated with clinical improvement [Hong et al., 2014] and thus has been interpreted as being due to vestibular rehabilitation therapy.
Excessive visual‐vestibular training in healthy professional dancers induced differential plastic morphometric changes of GMV in the anterior and posterior hippocampus, which receive vestibular and visual signals, respectively: smaller GMV in the anterior hippocampus but larger volumes in the posterior hippocampal formation [Hufner et al., 2011a]. Therefore, deficient afferent vestibular input may lead not only to a stronger dependence on visual cues but also to anterior hippocampal atrophy. Thus, we pursued the hypothesis of hippocampal atrophy in BVF [Brandt et al., 2005] by performing this voxel‐based morphometry study to investigate structural GMV changes in hippocampus of patients with the clinically frequent incomplete BVF [Ridgway et al., 2008].
MATERIALS AND METHODS
Participants
The study was approved by the institutional ethics committee of the University of Lübeck. All participants gave written informed consent before their inclusion into the study. The study was performed in agreement with the Declaration of Helsinki. Participants were recruited from an outpatient neurology clinic in a tertiary care academic medical center at the University of Lübeck (Center for Vertigo and Balance Disorder in Lübeck, Germany). Patients complained about dizziness, gait unsteadiness, and oscillopsia during locomotion and head movements. All participants were right‐handed and underwent extensive neurologic, neuro‐ophthalmologic, and neuro‐otologic examinations. BVF patients were on no regular medication known to affect central nervous system processing. None of the patients took any antivertiginous medication during the examination day. Patients were diagnosed to have BVF based on clinical examinations by experienced neurologists and neuro‐otologist of the University Dizziness Center in Lübeck and electrophysiological recordings [bithermal cold (27°) and warm (44°) caloric irrigation, quantitative head impulse test] analyzed by a co‐worker with longstanding experience in assessing vestibular function by caloric and quantitative head impulse videooculography who did not know about the history and clinical findings of the patients. Inclusion criteria for BVF were the following: (1) clinical assessment of a bilaterally pathologic HIT [Jorns‐Haderli et al., 2007], (2) bilaterally reduced gain of the horizontal VOR (<0.72) assessed by video‐HIT [Machner et al., 2013], 3) bilateral caloric hyporesponsiveness (mean peak slow phase velocity (SPV) of <5°/s on both sides), and (4) cranial magnetic resonance imaging without structural brain lesions. Patients with depression (as assessed by the Beck depression score), dementia (assessed by the MOCA scale), and those with additional evidence for cerebellar disease, polyneuropathy, or autoimmune and paraneoplastic diseases were excluded from the study. Participants subjectively rated their level of disease‐related impairment by the vertigo handicap questionnaire (VHQ) [Tschan et al., 2010], Vertigo Symptom Scale (VSS) [Tschan et al., 2008], the Clinical Vestibular Score (CVS; range: 0–28; with 0 for no objective impairment) [Helmchen et al., 2009], and the Subjective Dizziness Score (SDS; range: 0–44; with 0 for no subjective disability) [Helmchen et al., 2009]. A total number of 33 patients with chronic (>3 months, range: 3 months to 20 years) BVF were examined. Six patients had to be excluded due to comorbidities and/or coincidental brain lesions on MRI examinations. This resulted in 27 eligible BVF patients (16 male; age: 65.2 ± 9.7 years; disease duration: range 3 months to 20 years; mean 4.2 years). The most common etiology of BVF was antibiotic ototoxicity (n = 13; 48.1%), followed by BVF of unknown cause (n = 12; 44.4%), and sequential vestibular neuritis (n = 2; 7.4%). Thirty healthy control subjects were recruited. All participants had normal structural MR images showing no signs of cerebral atrophy. One control subject was excluded due to neuropsychiatric symptoms. Twenty‐nine healthy control subjects (16 male; age: 64.8 ± 9.8 years) were included in the study. The patient and the control group did not differ significantly in age (two‐sample t‐test p = 0.89), gender (chi‐square test p = 0.76), or MOCA score (two‐sample t‐test p = 0.52).
Electrophysiological Recordings
All participants were examined by a battery of vestibular investigations. Semicircular canal function was investigated by electronystagmography with caloric irrigation and quantitative head impulse testing (qHIT) and otolith function by static (background stationary) and dynamic (moving visual background) subjective visual vertical (SVV) [Dieterich and Brandt, 1993] and vestibular evoked myogenic potentials (VEMP). Psychophysical perception of the visual vertical was assessed by the subject's adjustment of a bar to the perceived visual vertical without any spatial orientation clues in a dotted hemispherical dome, which is stationary or moving around the line of sight [Dieterich and Brandt, 1993]. The normal range of SVV was defined as deviation of <2.5°. Several but not all patients and all healthy participants were investigated by ocular and cervical vestibular evoked myogenic potentials (oVEMP, cVEMP) [Curthoys et al., 2012; Rosengren and Kingma, 2013]. We used a vibration stimulus at the mid‐forehead delivered by a minishaker (Brüel & Kjaer, Denmark) to elicit oVEMPs recorded from surface EMG electrodes beneath the lower orbital rim above the obliquus inferior muscle. To activate this muscle, subjects fixated a target above their forehead. oVEMP was taken at a latency of about 10 ms from the stimulus‐locked ocular vestibular‐evoked myogenic potential and the first negative component. Cervical VEMP were delivered by short‐tone bursts of 500 Hz to either ear (headphone) and recorded over tensed sternocleidomastoid (SCM) muscles bilaterally. This results in a stimulus‐locked short‐latency myogenic potential recorded (cVEMP) with an initial positive (inhibitory) potential (p13) followed by a negative potential (n23) [Rosengren et al., 2010].
All participants were examined by quantitative head impulse test (qHIT) using video‐oculography [Machner et al., 2013]. Eye and head movements were recorded by the EyeSeeCam® HIT System (Autronics, Hamburg, Germany) at a sampling rate of 220 Hz. VOR gain was determined by robust linear regression of eye and head velocity starting at head velocity >10°/s to 95% of peak head velocity using Matlab®. Quantitative HIT was delivered by passive head impulses (HIT) with rapid, small‐amplitude (10–15°), horizontal head rotations (3000–4500°/s2) while the participant was sitting on a chair fixating a red LED at a distance of 100 cm.
Image Acquisition
Structural MRI images were recorded on a Philips Achieva 3 T scanner (Philips Healthcare, the Netherlands). A standard 8‐channel phase array head coil was used. High‐resolution structural images were obtained applying a T1‐weighted 3D turbo gradient‐echo sequence with SENSE (image matrix 240 × 240; 180 sagittal slices; isotropic 1 × 1 × 1 mm3 spatial resolution, TR = 8.1 ms; TE = 3.7 ms).
VBM Data Analysis
The VBM data analysis was performed using the SPM12 software package (University College London, Wellcome Trust Centre for Neuroimaging, http://www.fil.ion.ucl.ac.uk/spm/). VBM allows identifying differences in local gray and white matter volume. The following preprocessing steps were applied to the high‐resolution T1‐weighted structural images prior to the statistical analysis: (i) The T1‐weighted scans of all subjects were classified into different tissue types via the SPM segmentation routine. This resulted in tissue class images which were rigidly aligned to the SPM MNI template and in native space gray matter images. (ii) The Dartel suite [Ashburner, 2007] was used to estimate the nonlinear deformations that best align all images together. This resulted in a group template and tissue class maps registered to the template. During this preprocessing step, we adopted the default SPM settings for all options. (iii) Jacobian scaled (“modulated”) tissue class images normalized to MNI space were created. This step incorporates an affine transformation of the tissue class maps from the Dartel template space to MNI space, as well as a spatial smoothing step. The spatial smoothing was set to 6 mm FWHM, which is reasonable for subcortical regions with less variability compared to the cortex. The voxel size for the spatially normalized images was 1.5 × 1.5 × 1.5 mm3.
The data analysis was performed on “modulated” gray matter images. This means that differences in signal intensity represent volumetric differences.
Statistical Analysis
Differences in regional GMV between healthy controls and BVF patients were investigated applying voxel‐wise two‐sample t‐tests. The statistical analysis was limited to the hippocampus and the parahippocampus, that is, we applied a small‐volume correction using a mask including the hippocampus and the parahippocampus. The mask was created using the automated anatomical labeling (AAL) atlas [Tzourio‐Mazoyer et al., 2002]. In addition, all modulated gray matter images were thresholded by 0.15. The statistical analysis included N = 8475 voxels (voxel size 1.5 × 1.5 × 1.5 mm3; small‐volume correction). An FDR procedure was applied at the voxel level to correct for multiple comparisons (p < 0.05 corrected). Age, gender, MOCA score, and total intracranial volume were added as covariates to the GLM.
Regression analyses were used to relate changes in voxel‐wise GMV in BVF patients to behavioral data using the same covariates and the same procedure to correct for multiple testing were used. We investigated relations to five clinical parameters, that is, CVS, SDS, VHQ, VSS score and the VOR gain, and tested for positive and negative correlation. Taking into account the number of statistical test, we corrected our results at the level of α = 0.05/10 = 0.005.
The statistical analysis of behavioral data was performed using Matlab®. If not stated otherwise, we report the mean and standard deviation of the data. The results were visualized and FDR corrected using the xjView toolbox (http://www.alivelearn.net/xjview).
Anatomical Localization
We used cytoarchitectonic probability maps of the hippocampus [Amunts et al., 2005] and the SPM Anatomy toolbox [Eickhoff et al., 2005] to specify the anatomical location of our findings.
RESULTS
Behavioral and Electrophysiological Data
The mean gain of the patients' horizontal vestibulo‐ocular reflex (VOR) was severely reduced (0.25 ± 0.18; healthy controls: 0.94 ± 0.13). Ocular vestibular evoked myogenic potentials (oVEMP, n1/p1, peak‐to‐peak amplitude) were smaller in patients (3.4 ± 0.3 μV; z = −2.78; p = 0.003) as compared to healthy control participants (6.7 ± 0.69 μV); that is, patients showed pathologically reduced oVEMP amplitudes. In addition, cervical vestibular evoked myogenic potentials (cVEMP) were significantly smaller in patients (11.4 ± 2.5 μV) than in healthy control subjects (27.89 ± 4.7 μV; z = −2.00, p = 0.043).
The mean clinical vestibular score (CVS) was 10.5 ± 4.8 indicating a clinically visible functional impairment [Helmchen et al., 2009]. Subjective disease‐related impairment for the Vertigo Handicap Questionnaire (VHQ) revealed on average 40.0 ± 8.2, for the Vertigo Symptom Scale (VSS) on average 26.9 ± 18.7 and the Subjective Dizziness Score (SDS) on average 12.8 ± 7.7 indicating considerable subjective disability in daily life. There was no correlation of any of these scores with disease duration. The Montreal Cognitive Assessment test (MoCA) did not differ between both groups (scores ranged from 22 to 30 points for patients and 23 to 30 points for healthy controls; z = −0.89).
Gray Matter Volume
There was no significant difference between GMV in the hippocampus between BVF patients and healthy controls. We refrained from performing a post‐hoc power analysis as it is not informative because of the relationship between Type I error rate and power [Hoenig and Heisey, 2001; Mumford, 2012]. Instead, we provide unthresholded t‐maps (Fig. 1, NIfTI images as Supporting Information) and present uncorrected data (p < 0.05; uncorrected; Table 1) to allow for comparisons across studies and to assess the quality of the data from a continuous map. T‐maps (3D NIfTI images) are available as Supporting Information. We systematically altered the smoothing kernel (4 and 8 mm FWHM) but this did not change our results.
Figure 1.
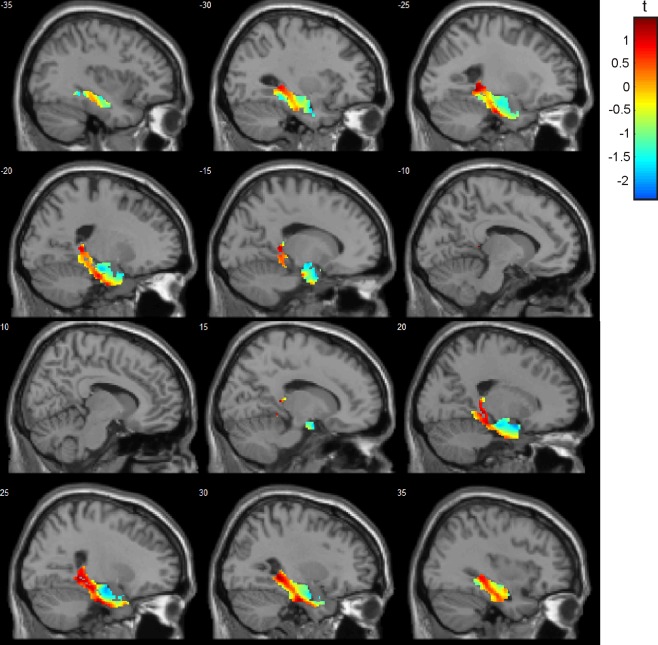
Gray matter volume in BVF patients compared to healthy controls. An unthresholded t‐map is shown as we did not find evidence for altered gray matter volume in BVF patients with incomplete BVF. There is a posterior–anterior gradient of GMV loss in the hippocampus, parahippocampus, and entorhinal cortex bilaterally (blue indicates stronger GMV reduction than red).
Table 1.
Local grey matter volume in healthy controls compared to BVF patients (HC > BVF)
| Cluster size k | Peak T(50) | Peak p(unc.) | MNI coordinates [mm] | ||
|---|---|---|---|---|---|
| x | y | z | |||
| 75 | 2.35 | 0.011 | 18 | 3 | −20 |
| 1.80 | 0.038 | 27 | 8 | −24 | |
| 36 | 2.15 | 0.018 | −17 | 2 | −20 |
| 138 | 2.09 | 0.020 | 24 | −11 | −17 |
| 11 | 1.98 | 0.026 | −14 | −8 | −15 |
| 29 | 1.89 | 0.032 | −18 | −17 | −18 |
The table shows a maximum of 3 local maxima more than 8.0 mm apart.
Relationship between vestibular function and clinical scores
There was a significant negative correlation of local GMV with the CVS score (α = 0.005 FDR corrected at the voxel level within the hippocampus mask) in the hippocampus region CA3 (bilateral; Fig. 2 and Table 2), i.e., GMV in patients decreased with increasing vestibular‐induced impairment. Figure 2B shows the three‐dimensional view of the hippocampus (visualization using MRIcroGL). We extracted the mean GMV in this region and tested for significant group differences between patients and healthy controls. Patients showed a significantly decreased GMV (left: p = 0.035; right: p = 0.023). The mean GMV in patients and controls and the standard error is shown in the histograms in Figure 2C. Note that Figure 2C shows the raw data, that is, the mean local gray matter volume within the clusters displayed in Figure 2A which was directly retrieved from the modulated structural images. The scatter plots show that the negative correlation was not driven by outliers and that healthy control subjects had a larger GMV in the hippocampal region, in which GMV was significantly correlated with the CVS score (histograms). We do not report correlation coefficients and corresponding p‐values for the ROI‐based analysis to avoid a circular analysis [Kriegeskorte et al., 2009]. The purpose of the scatter plot is to visualize the effect and to show that the result is not driven by outliers. There was neither a correlation of GMV in hippocampus with the other scores of subjective, vestibulopathy‐related impairment in daily life (VHQ, VSS, SDS), nor with the quantitative assessment of the vestibulo‐ocular reflex (VOR gain). It should be stressed that a correction for multiple testing at the voxel level was performed taking into account the number of correlation analyses and statistical test.
Figure 2.
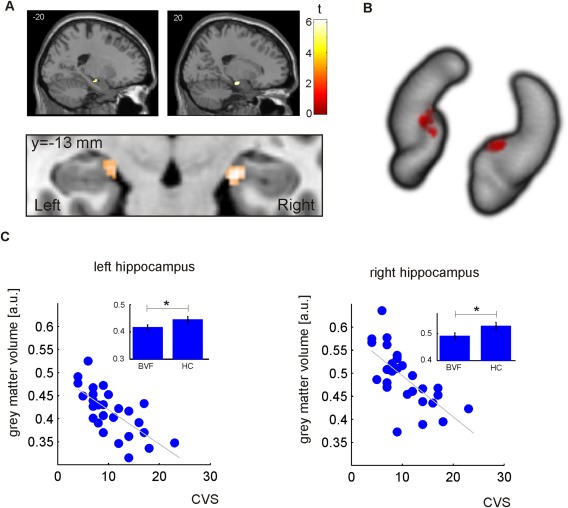
Relationship between CVS and hippocampal grey matter volume in BVF patients. (A) Anatomical location of clusters in the left and right hippocampus showing a significant negative correlation in the patient population (0.005 FDR corrected; taking into account ten statistical tests). (B) Three‐dimensional view of the hippocampus indicating the localization of the cluster showing GMV reduction with increasing clinical impairment. (C) With increasing disease severity (CVS), a decrease in grey matter volume is observed (scatter plot). Overall, BVF patients showed a decreased grey matter volume in these clusters (histograms, standard error is shown).
Table 2.
Relationship between CVS score and grey matter volume in BVF patients
| Localization | Cluster size k | Peak T(21) | MNI coordinates [mm] | |||
|---|---|---|---|---|---|---|
| x | y | Z | ||||
| Hippocampus CA3 | R | 63 | 6.16 | 21 | −14 | −17 |
| Hippocampus CA3 | L | 32 | 5.50 | −18 | −15 | −17 |
DISCUSSION
We performed a voxel‐wise comparison of hippocampal gray matter volume in a clinically representative sample of n = 27 patients with incomplete BVF and n = 29 age‐ and gender‐matched healthy controls. In contrast to our hypothesis, we did not find evidence for altered hippocampal GMV comparing the patient and the control group. Driven by our strong a priori hypothesis based on previous animal and human studies, we tested in this first VBM study of patients with BVF for correlations between relative GMV changes and subjective (SDS, VHQ, VSS) and clinical objective scores (CVS) as well as quantitative data of the vestibular function (VOR gain). There was a significant GMV reduction in the CA3 region of the hippocampus bilaterally in BVF patients with increasing vestibulopathy‐related disability (CVS). The clinical vestibular score (CVS) encompasses clinical signs of vestibular imbalance as it tests head‐motion‐related VOR‐deficits and ataxia of gait and stance, that is, it increases with vestibulopathy‐induced disability. The hippocampal region CA3 plays an important role in the formation of episodic memory [Rolls, 2013] and the acquisition of spatial information within short‐term memory in tasks that require rapid encoding, novelty detection, working memory, and recall primarily for spatial information [Kesner, 2013].
Our finding may be related to hippocampal atrophy and impaired spatial navigation in humans who had undergone bilateral nerve section and subsequently suffered from chronic complete bilateral vestibular loss [Brandt et al., 2005] and to animal studies of complete vestibular deafferentation [Russell et al., 2006; Stackman and Herbert, 2002; Zheng et al., 2003].
The seminal work by Brandt et al. [2005] on bilateral atrophy in the hippocampus was performed with manual segmentation of the head, body, and tail of the hippocampus based on a protocol by Bernasconi et al. [2003]. Their hypothesis‐driven study of hippocampus volume reduction was based on several preceding animal studies of complete vestibular deafferentation [Russell et al., 2006; Stackman and Herbert, 2002; Zheng et al., 2003]. In contrast to these studies, we did not find significant GMV changes in the hippocampus of patients and healthy controls correcting for multiple testing. This might be explained by the fact that our patients had severe but still incomplete BVF. In contrast to the rare nerve section etiology, our cohort represents the more frequently encountered bilateral vestibulopathy of toxic, idiopathic etiology and following recurrent episodic vertigo with chronic bilateral vestibular hypofunction. Our observation of GMV reduction in the CA3 region of hippocampus with increasing vestibulopathy‐related clinical impairment supports this view as patients who had undergone bilateral vestibular nerve section are likely to show strong vestibular‐induced disability and are therefore expected to show stronger hippocampal GMV reduction. The size of our cohort (n = 27 BVF patients) is considerably larger than in previous studies of patients with bilateral (n = 10) [Brandt et al., 2005] and unilateral vestibulopathies [Helmchen et al., 2009, 2011; Hong et al., 2014]. It is thus likely that differences in the clinical characteristics (i.e., incomplete lesions) of the patient cohorts rather than power in statistics are the reason why we did not observe significant differences between the patient and the control group.
Morphometric changes in vestibular deafferentation have been shown to be time‐dependent as they might reflect adaptive mechanisms [zu Eulenburg et al., 2010; Hong et al., 2014]. The disease duration in our BVF patients was shorter than in patients who had undergone surgical nerve section due to acoustic neuroma [Brandt et al., 2005]. However, disease duration is clearly long enough for changes in gray matter and even white matter fractional anisotropy to occur, which may develop even within months during exercise in healthy subjects [Draganski et al., 2004; zu Eulenburg et al., 2010; Hummel et al., 2014]. Conceivably, morphological GMV reduction could have been counterbalanced by compensatory mechanisms, that is, vestibular rehabilitation or altered visual sensitivity in hippocampus with GMV increase in posterior but GMV decrease in anterior hippocampus [Hufner et al., 2011b]. In our BVF patients, there was some trend (unthresholded t‐map) toward a gradient of GMV reduction with more pronounced volume reduction in the anterior hippocampus and entorhinal cortex, which receive vestibular input via vestibulo–thalamic–cortical pathways [Hitier et al., 2014; Kirsch et al., 2015].
One major inherent limitation of VBM analyses in general is the accuracy of the intersubject registration, that is, intersubject anatomical correspondence. The normalization accuracy varies over different brain regions and is generally better in subcortical structures as the hippocampus compared to the cortex. This justifies rather small Gaussian kernels for spatial smoothing to alleviate the problems of intersubject correspondence. Here, we applied spatial smoothing at different scales (4, 6, and 8 mm FWHM) and found consistent results at all scales.
We rigorously introduced covariates in our statistical analyses which are known to influence voxel‐wise gray matter volume, that is, age, gender, total intracranial volume, and the MOCA score. Repeating the analyses without using age, gender, and MOCA score as covariates had no impact on our results. This can be understood as the two experimental cohorts were closely matched regarding these demographic factors. The total intracranial volume (TICV) has a strong influence on voxel‐wise GMV and has to be considered in VBM analyses. Here, we decided to include the TICV as a covariate in our model as proportional scaling using this parameter led to strong artefacts in the statistical maps.
CONCLUSIONS
According to our data, GMV reductions in the hippocampus of BVF patients are related to the severity of vestibulopathy‐induced clinical impairment. Thus, we propose that hippocampal atrophy does not become evident unless BVF is severe. This includes cases of complete nerve section, that is, complete BVF. This might explain why up to now there is hardly any clinical evidence for spatial navigation disorders in incomplete BVF, which constitutes the most common form of BVF. In this clinically representative sample, hippocampus atrophy seems to be less frequent than previously suspected.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Conflicts of interest: The authors declare that there are no conflicts of interest.
REFERENCES
- Amunts K, Kedo O, Kindler M, Pieperhoff P, Mohlberg H, Shah NJ, Habel U, Schneider F, Zilles K (2005): Cytoarchitectonic mapping of the human amygdala, hippocampal region and entorhinal cortex: Intersubject variability and probability maps. Anat Embryol (Berl) 210:343–352. [DOI] [PubMed] [Google Scholar]
- Ashburner J (2007): A fast diffeomorphic image registration algorithm. NeuroImage 38:95–113. [DOI] [PubMed] [Google Scholar]
- Ashburner J, Friston KJ (2000): Voxel‐based morphometry–the methods. Neuroimage 11:805–821. [DOI] [PubMed] [Google Scholar]
- Baek JH, Zheng Y, Darlington CL, Smith PF (2010): Evidence that spatial memory deficits following bilateral vestibular deafferentation in rats are probably permanent. Neurobiol Learn Mem 94:402–413. [DOI] [PubMed] [Google Scholar]
- Becker‐Bense S, Dieterich M, Buchholz HG, Bartenstein P, Schreckenberger M, Brandt T (2013): The differential effects of acute right‐ vs. left‐sided vestibular failure on brain metabolism. Brain Struct Funct [DOI] [PubMed] [Google Scholar]
- Bense S, Bartenstein P, Lochmann M, Schlindwein P, Brandt T, Dieterich M (2004a): Metabolic changes in vestibular and visual cortices in acute vestibular neuritis. Ann Neurol 56:624–630. [DOI] [PubMed] [Google Scholar]
- Bense S, Deutschlander A, Stephan T, Bartenstein P, Schwaiger M, Brandt T, Dieterich M (2004b): Preserved visual‐vestibular interaction in patients with bilateral vestibular failure. Neurology 63:122–128. [DOI] [PubMed] [Google Scholar]
- Bernasconi N, Bernasconi A, Caramanos Z, Antel SB, Andermann F, Arnold DL (2003): Mesial temporal damage in temporal lobe epilepsy: A volumetric MRI study of the hippocampus, amygdala and parahippocampal region. Brain 126:462–469. [DOI] [PubMed] [Google Scholar]
- Besnard S, Machado ML, Vignaux G, Boulouard M, Coquerel A, Bouet V, Freret T, Denise P, Lelong‐Boulouard V (2012): Influence of vestibular input on spatial and nonspatial memory and on hippocampal NMDA receptors. Hippocampus 22:814–826. [DOI] [PubMed] [Google Scholar]
- Brandt T (1996): Bilateral vestibulopathy revisited. Eur J Med Res 1:361–368. [PubMed] [Google Scholar]
- Brandt T, Schautzer F, Hamilton DA, Bruning R, Markowitsch HJ, Kalla R, Darlington C, Smith P, Strupp M (2005): Vestibular loss causes hippocampal atrophy and impaired spatial memory in humans. Brain 128:2732–2741. [DOI] [PubMed] [Google Scholar]
- Brandt T, Strupp M, Dieterich M (2014): Towards a concept of disorders of “higher vestibular function”. Front Integr Neurosci 8:47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curthoys IS, Vulovic V, Manzari L (2012): Ocular vestibular‐evoked myogenic potential (oVEMP) to test utricular function: Neural and oculomotor evidence. Acta Otorhinolaryngol Ital 32:41–45. [PMC free article] [PubMed] [Google Scholar]
- Cuthbert PC, Gilchrist DP, Hicks SL, MacDougall HG, Curthoys IS (2000): Electrophysiological evidence for vestibular activation of the guinea pig hippocampus. Neuroreport 11:1443–1447. [DOI] [PubMed] [Google Scholar]
- Deutschlander A, Hufner K, Kalla R, Stephan T, Dera T, Glasauer S, Wiesmann M, Strupp M, Brandt T (2008): Unilateral vestibular failure suppresses cortical visual motion processing. Brain 131:1025–1034. [DOI] [PubMed] [Google Scholar]
- Dieterich M, Bauermann T, Best C, Stoeter P, Schlindwein P (2007): Evidence for cortical visual substitution of chronic bilateral vestibular failure (an fMRI study). Brain 130:2108–2116. [DOI] [PubMed] [Google Scholar]
- Dieterich M, Brandt T (1993): Ocular torsion and tilt of subjective visual vertical are sensitive brainstem signs. Ann Neurol 33:292–299. [DOI] [PubMed] [Google Scholar]
- Draganski B, Gaser C, Busch V, Schuierer G, Bogdahn U, May A (2004): Neuroplasticity: Changes in grey matter induced by training. Nature 427:311–312. [DOI] [PubMed] [Google Scholar]
- Eickhoff SB, Stephan KE, Mohlberg H, Grefkes C, Fink GR, Amunts K, Zilles K (2005): A new SPM toolbox for combining probabilistic cytoarchitectonic maps and functional imaging data. Neuroimage 25:1325–1335. [DOI] [PubMed] [Google Scholar]
- Göttlich M, Jandl NM, Wojak JF, Sprenger A, der Gablentz J, Munte TF, Krämer UM, Helmchen C (2014): Altered resting‐state functional connectivity in patients with chronic bilateral vestibular failure. Neuroimage Clin 4:488–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmchen C, Klinkenstein J, Machner B, Rambold H, Mohr C, Sander T (2009): Structural changes in the human brain following vestibular neuritis indicate central vestibular compensation. Ann N Y Acad Sci 1164:104–115. [DOI] [PubMed] [Google Scholar]
- Helmchen C, Klinkenstein JC, Kruger A, Gliemroth J, Mohr C, Sander T (2011): Structural brain changes following peripheral vestibulo‐cochlear lesion may indicate multisensory compensation. J Neurol Neurosurg Psychiatry 82:309–316. [DOI] [PubMed] [Google Scholar]
- Helmchen C, Ye Z, Sprenger A, Munte TF (2014): Changes in resting‐state fMRI in vestibular neuritis. Brain Struct Funct 219:1889–1900. [DOI] [PubMed] [Google Scholar]
- Hitier M, Besnard S, Smith PF (2014): Vestibular pathways involved in cognition. Front Integr Neurosci 8:59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoenig JM, Heisey DM (2001): The abuse of power: The pervasive fallacy of power calculations for data analysis. Am Stat 55:19–24. [Google Scholar]
- Hong SK, Kim JH, Kim HJ, Lee HJ (2014): Changes in the gray matter volume during compensation after vestibular neuritis: A longitudinal VBM study. Restor Neurol Neurosci 32:663–673. [DOI] [PubMed] [Google Scholar]
- Hufner K, Binetti C, Hamilton DA, Stephan T, Flanagin VL, Linn J, Labudda K, Markowitsch H, Glasauer S, Jahn K, Strupp M, Brandt T (2011a): Structural and functional plasticity of the hippocampal formation in professional dancers and slackliners. Hippocampus 21:855–865. [DOI] [PubMed] [Google Scholar]
- Hufner K, Hamilton DA, Kalla R, Stephan T, Glasauer S, Ma J, Bruning R, Markowitsch HJ, Labudda K, Schichor C, Strupp M, Brandt T (2007): Spatial memory and hippocampal volume in humans with unilateral vestibular deafferentation. Hippocampus 17:471–485. [DOI] [PubMed] [Google Scholar]
- Hufner K, Strupp M, Smith P, Brandt T, Jahn K (2011b): Spatial separation of visual and vestibular processing in the human hippocampal formation. Ann N Y Acad Sci 1233:177–186. [DOI] [PubMed] [Google Scholar]
- Hummel N, Hufner K, Stephan T, Linn J, Kremmyda O, Brandt T, Flanagin VL (2014): Vestibular loss and balance training cause similar changes in human cerebral white matter fractional anisotropy. PLoS ONE 9: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob PY, Poucet B, Liberge M, Save E, Sargolini F (2014): Vestibular control of entorhinal cortex activity in spatial navigation. Front Integr Neurosci 8:38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs J, Weidemann CT, Miller JF, Solway A, Burke JF, Wei XX, Suthana N, Sperling MR, Sharan AD, Fried I, Kahana MJ (2013): Direct recordings of grid‐like neuronal activity in human spatial navigation. Nat Neurosci 16:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorns‐Haderli M, Straumann D, Palla A (2007): Accuracy of the bedside head impulse test in detecting vestibular hypofunction. J Neurol Neurosurg Psychiatry 78:1113–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalla R, Muggleton N, Spiegel R, Bueti D, Claassen J, Walsh V, Bronstein A (2011): Adaptive motion processing in bilateral vestibular failure. J Neurol Neurosurg Psychiatry 82:1212–1216. [DOI] [PubMed] [Google Scholar]
- Kesner RP (2013): A process analysis of the CA3 subregion of the hippocampus. Front Cell Neurosci 7:78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch V, Keeser D, Hergenroeder T, Erat O, Ertl‐Wagner B, Brandt T, Dieterich M (2015): Structural and functional connectivity mapping of the vestibular circuitry from human brainstem to cortex. Brain Struct Funct [DOI] [PubMed] [Google Scholar]
- Kriegeskorte N, Simmons WK, Bellgowan PS, Baker CI (2009): Circular analysis in systems neuroscience: The dangers of double dipping. Nat Neurosci 12:535–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machner B, Sprenger A, Fullgraf H, Trillenberg P, Helmchen C (2013): [Video‐based head impulse test. Importance for routine diagnostics of patients with vertigo]. Nervenarzt 84:975–983. [DOI] [PubMed] [Google Scholar]
- Mumford JA (2012): A power calculation guide for fMRI studies. Soc Cogn Affect Neurosci 7:738–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridgway GR, Henley SM, Rohrer JD, Scahill RI, Warren JD, Fox NC (2008): Ten simple rules for reporting voxel‐based morphometry studies. Neuroimage 40:1429–1435. [DOI] [PubMed] [Google Scholar]
- Rolls ET (2013): A quantitative theory of the functions of the hippocampal CA3 network in memory. Front Cell Neurosci 7:98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosengren SM, Kingma H (2013): New perspectives on vestibular evoked myogenic potentials. Curr Opin Neurol 26:74–80. [DOI] [PubMed] [Google Scholar]
- Rosengren SM, Welgampola MS, Colebatch JG (2010): Vestibular evoked myogenic potentials: Past, present and future. Clin Neurophysiol 121:636–651. [DOI] [PubMed] [Google Scholar]
- Russell NA, Horii A, Smith PF, Darlington CL, Bilkey DK (2003): Long‐term effects of permanent vestibular lesions on hippocampal spatial firing. J Neurosci 23:6490–6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell NA, Horii A, Smith PF, Darlington CL, Bilkey DK (2006): Lesions of the vestibular system disrupt hippocampal theta rhythm in the rat. J Neurophysiol 96:4–14. [DOI] [PubMed] [Google Scholar]
- Smith PF, Geddes LH, Baek JH, Darlington CL, Zheng Y (2010): Modulation of memory by vestibular lesions and galvanic vestibular stimulation. Front Neurol 1:141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stackman RW, Clark AS, Taube JS (2002): Hippocampal spatial representations require vestibular input. Hippocampus 12:291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stackman RW, Herbert AM (2002): Rats with lesions of the vestibular system require a visual landmark for spatial navigation. Behav Brain Res 128:27–40. [DOI] [PubMed] [Google Scholar]
- Tschan R, Wiltink J, Best C, Bense S, Dieterich M, Beutel ME, Eckhardt‐Henn A (2008): Validation of the German version of the Vertigo Symptom Scale (VSS) in patients with organic or somatoform dizziness and healthy controls. J Neurol 255:1168–1175. [DOI] [PubMed] [Google Scholar]
- Tschan R, Wiltink J, Best C, Beutel M, Dieterich M, Eckhardt‐Henn A (2010): Validation of the German version of the Vertigo Handicap Questionnaire (VHQ) in patients with vestibular vertigo syndromes or somatoform vertigo and dizziness. Psychother Psychosom Med Psychol 60:e1–12. [DOI] [PubMed] [Google Scholar]
- Tzourio‐Mazoyer N, Landeau B, Papathanassiou D, Crivello F, Etard O, Delcroix N, Mazoyer B, Joliot M (2002): Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single‐subject brain. Neuroimage 15:273–289. [DOI] [PubMed] [Google Scholar]
- Wallace DG, Hines DJ, Pellis SM, Whishaw IQ (2002): Vestibular information is required for dead reckoning in the rat. J Neurosci 22:10009–10017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiener SI, Korshunov VA, Garcia R, Berthoz A (1995): Inertial, substratal and landmark cue control of hippocampal CA1 place cell activity. Eur J Neurosci 7:2206–2219. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Balabhadrapatruni S, Baek JH, Chung P, Gliddon C, Zhang M, Darlington CL, Napper R, Strupp M, Brandt T, Smith PF (2012): The effects of bilateral vestibular loss on hippocampal volume, neuronal number, and cell proliferation in rats. Front Neurol 3:20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Darlington CL, Smith PF (2006): Impairment and recovery on a food foraging task following unilateral vestibular deafferentation in rats. Hippocampus 16:368–378. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Geddes L, Sato G, Stiles L, Darlington CL, Smith PF (2014): Galvanic vestibular stimulation impairs cell proliferation and neurogenesis in the rat hippocampus but not spatial memory. Hippocampus 24:541–552. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Kerr DS, Darlington CL, Smith PF (2003): Unilateral inner ear damage results in lasting changes in hippocampal CA1 field potentials in vitro. Hippocampus 13:873–878. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Wilson G, Stiles L, Smith PF (2013): Glutamate receptor subunit and calmodulin kinase II expression, with and without T maze training, in the rat hippocampus following bilateral vestibular deafferentation. PLoS ONE 8:e54527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingler VC, Cnyrim C, Jahn K, Weintz E, Fernbacher J, Frenzel C, Brandt T, Strupp M (2007): Causative factors and epidemiology of bilateral vestibulopathy in 255 patients. Ann Neurol 61:524–532. [DOI] [PubMed] [Google Scholar]
- zu Eulenburg P, Stoeter P, Dieterich M (2010): Voxel‐based morphometry depicts central compensation after vestibular neuritis. Ann Neurol 68:241–249. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information