

Abstract
While the HTT CAG‐repeat expansion mutation causing Huntington's disease (HD) is highly correlated with the rate of pathogenesis leading to disease onset, considerable variance in age‐at‐onset remains unexplained. Therefore, other factors must influence the pathogenic process. We asked whether these factors were related to natural biological variation in the sensory‐motor system. In 243 participants (96 premanifest and 35 manifest HD; 112 controls), sensory‐motor structural MRI, tractography, resting‐state fMRI, electrophysiology (including SEP amplitudes), motor score ratings, and grip force as sensory‐motor performance were measured. Following individual modality analyses, we used principal component analysis (PCA) to identify patterns associated with sensory‐motor performance, and manifest versus premanifest HD discrimination. We did not detect longitudinal differences over 12 months. PCA showed a pattern of loss of caudate, grey and white matter volume, cortical thickness in premotor and sensory cortex, and disturbed diffusivity in sensory‐motor white matter tracts that was connected to CAG repeat length. Two further major principal components appeared in controls and HD individuals indicating that they represent natural biological variation unconnected to the HD mutation. One of these components did not influence HD while the other non‐CAG‐driven component of axial versus radial diffusivity contrast in white matter tracts were associated with sensory‐motor performance and manifest HD. The first component reflects the expected CAG expansion effects on HD pathogenesis. One non‐CAG‐driven component reveals an independent influence on pathogenesis of biological variation in white matter tracts and merits further investigation to delineate the underlying mechanism and the potential it offers for disease modification. Hum Brain Mapp 37:4615–4628, 2016. © 2016 Wiley Periodicals, Inc.
Keywords: effective connectivity, cortical thickness, somatosensory evoked potentials, grip force, principal component analysis, biological variation, biological trait
INTRODUCTION
In Huntington's disease (HD), the length of the expanded CAG tract in HTT explains about half of the variability of motor age‐at‐onset and is therefore the main determinant of biological events prior to clinical diagnosis (GeM‐HD Consortium, 2015). The remaining variability is independent of CAG repeat length and reflects the modifying influence of genetic and environmental factors on the pathogenic process (GeM‐HD Consortium, 2015). Structural differences in cerebral white and grey matter may be detectable as far as 15–20 years before onset of unequivocal signs of HD and have been reported using VBM (Tabrizi et al., 2013) and DTI (Matsui et al., 2015) (Aylward, 2007; Georgiou‐Karistianis et al., 2013; Rosas et al., 2008; Thieben et al., 2002). Similar to structural abnormalities, cross‐sectional task‐based, or resting‐state functional MRI (fMRI) studies have documented widespread cortical and subcortical changes of brain function in manifest HD (Wolf, et al., 2014). TRACK‐HD, a large longitudinal study combining clinical and structural imaging data from both premanifest and early manifest HD participants, showed that clinical measures—e.g., motor and cognitive tasks—and volumetric imaging tracked disease evolution were associated with manifest HD (Tabrizi et al., 2013). PREDICT‐HD, a longitudinal study in premanifest HD (preHD), demonstrated that motor and cognitive task performance, and structural imaging (in particular putamen volume), improved predictions of motor diagnosis compared with models using HTT CAG repeat length and age (Paulsen et al., 2014).
Some of the effects reported in TRACK‐HD and PREDICT‐HD were independent of HTT CAG repeat length and age. This indicates that biological variation exists that is not related to the cause of HD but may be common in the population and, when present in an individual with the HTT CAG‐repeat expansion, exerts an influence on HD pathogenesis. The identification of such traits could help reveal an interaction between the biology underlying the biological trait and the pathogenesis or manifestation of HD. The trait itself or the genetics underlying its variation could suggest a route to disease modification (genetic or pharmaceutical). Thus, HD‐independent work on mechanisms underlying the trait could provide specific pathways/processes to be tested as candidates for HD modification. It remains unresolved which additional biological factors are associated with task performance and clinical disease stage. We addressed this question in an a priori defined neuronal network. We focused on sensory‐motor circuits that involve brain areas for which macro‐ (volumetric) and microstructural (DTI) abnormalities have consistently been reported in HD. These include cortical somatosensory projections, abnormal in manifest disease (Jech et al., 2007), the primary sensory cortex, in which there is evidence of thinning prior to symptom onset (Tabrizi et al., 2009), white matter changes assessed using DTI (Tabrizi et al., 2009; Weaver et al., 2009), and SEPs where amplitudes of cortical components were reduced (Abbruzzese et al., 1990; Lefaucheur et al., 2006; Kuwert et al., 1993; Topper et al., 1993).
Most previous analyses—including analyses of the Track‐HD data—have examined these measures one at a time. The fact that differences to controls are expected for many of these in HD is a premise of this study in which we investigate the patterns by which they differ. Analyzing data from different modalities one at a time does not reveal these patterns. We aimed to first describe patterns of the HTT CAG‐repeat expansion associated pathology of the sensory‐motor circuit using multimodal measurements, i.e., sensory‐motor network brain structure (VBM, sensory‐motor cortex thickness and DTI tractography), function (fMRI and electrophysiology), and sensory‐motor task performance (grip force and motor score). We hypothesized that non‐CAG‐repeat‐driven factors influence the pathogenic process and thus predict task performance and clinical disease stage and examined whether these factors were related to natural biological variation in the sensory‐motor system.
METHODS
We investigated sensory‐motor clinical measures, and used neuroimaging and electrophysiological techniques to investigate macrostructure, microstructure, and network function. Following independent single modality evaluation, we employed principal component analysis (PCA) as a descriptive tool to identify data patterns using multimodal assessments. Finally, we asked whether those PCA patterns (1) were associated with pathology that is also known to be linked to HTT CAG‐repeat expansion, (2) were able to discriminate between controls, preHD and early HD participants, and (3) were associated with clinical measures of sensory‐motor network performance (grip force and UHDRS motor score).
PARTICIPANTS
All individuals who met baseline eligibility criteria for the TRACK‐HD premanifest cohort in 2008 (for inclusion/exclusion criteria, see Tabrizi et al., 2009) were eligible to participate in Track‐On regardless of current disease status. A premanifest gene carrier was either (a) an existing premanifest gene carrier previously enrolled in TRACK‐HD or (b) a newly recruited premanifest gene carrier with CAG repeat length ≥40 and burden of pathology score (CAG‐35.5) × age >250. All participants had to be able to tolerate MRI, and sample donation, and control participants were age and gender frequency matched to the premanifest gene carrier group.
Participants were assessed at baseline and 12 months at four study sites in London (61 participants; 25.1%), Paris (64; 26.3%), Leiden (60; 24.7%), and Vancouver (58; 23.9%). The study was approved by local ethical committees, and written informed consent was obtained from each participant.
The United Huntington's Disease Rating Scale (UHDRS) motor part was administered at both visits. Participants with a UHDRS diagnostic confidence score of 4 on the motor scale (criteria for clinical diagnosis of early HD) at 12 months were defined as the early (diagnosed) group. The remaining HD group was divided into preHD‐A (further from predicted diagnosis age; ≥10.8 years at baseline) and preHD‐B (nearer; <10.8 years) based on the survival analysis formula (Langbehn et al., 2004).
CLINICAL ASSESSMENT AND ELECTROPHYSIOLOGY
Participants were assessed clinically, including grip force analysis (Reilmann, et al., 2010) and HTT CAG repeat length determined as described for the TRACK‐HD study (Tabrizi et al., 2009; Tabrizi et al., 2011; Tabrizi et al., 2013). At all four sites, somatosensory‐evoked potentials (SEP) were recorded following median nerve stimulation with surface electrodes, and at three sites, transcranial magnetic stimulation (TMS) was done as previously described using established techniques (Fischer and Orth, 2011; Orth and Rothwell, 2004). The protocol included M. abductor pollicis brevis (APB) hot‐spot and motor threshold determination, motor‐evoked potential latencies and amplitudes, input/output curves at rest (110%, 130%, 150% resting motor threshold) and with preactivation (125%, 150%, 175% active motor threshold), and silent period determination.
NEUROIMAGING
Voxel‐Based Morphometry
Voxel‐based morphometry data were acquired using previously validated protocols for multisite use on two different 3 T MRI scanner systems (Philips Achieva at Leiden and Vancouver and Siemens TIM Trio at London and Paris). Cortical thickness measures were generated for each participant using Freesurfer version 5.3.0 applying default parameters and optimized for 3 T data (Fischl and Dale, 2000). Measures were extracted from Brodmann areas: BA4a/4b (motor cortex, M1); BA6 (premotor cortex, PMC); and BA3a/3b, BA1, BA2 (somatosensory cortex, S1) https://surfer.nmr.mgh.harvard.edu/fswiki/BrodmannAreaMaps. All segmentations were visually inspected for accuracy, blind to participant status.
Diffusion Tensor Imaging
Diffusion‐weighted images with 42 unique gradient directions (b = 1,000 s/mm2) were acquired from both Siemens and Phillips scanners. Eight images with no diffusion weighting (b = 0 s/mm2) and one image with no diffusion weighting (b = 0 s/mm2) were acquired from the Siemens and Philips scanners respectively. For the Siemens scanners, TE = 88 ms and TR = 13 s; for the Phillips scanners, TE = 56 ms and TR = 11 s. Voxel size for the Siemens scanners was 2 × 2 × 2 mm and for the Phillips scanners 1.96 × 1.96 × 2. 75 slices were collected for each diffusion‐weighted and non‐diffusion‐weighted volume. The diffusion data were preprocessed using standard FSL pipelines (Smith et al., 2004). Data were corrected for eddy current distortions, diffusion tensors fitted using DTFIT, and all metrics derived.
For DTI, the no‐gradient (B0) image was then skull‐stripped using the Brain Extraction Tool (BET) and was manually examined and corrected. (For Siemens data B0 images were merged to create a mean.) Diffusion tensors were then fit to the corrected data using dtifit. FA, AD, and RD values were derived from the tensors. For registration, we first created a high‐quality T1 brain mask by combining and dilating a thresholded segmented image (created using the VBM toolbox http://www.neuro.uni-jena.de/vbm /) with an eroded T1 mask from BET. The mask was then applied to the original, brain‐extracted, T1 image. The resultant T1 image was then registered to the B0 image using FLIRT (Jenkinson and Smith, 2001). Within‐voxel crossing fibers were modeled using a Bayesian probabilistic method implemented in Bedpostx (Behrens, et al., 2007).
For fiber‐tracking, pathways selected were between S1 and somatosensory thalamus; M1 and motor thalamus; PMC and the motor thalamus and the corticospinal tract (CST), connecting M1 and the cerebral peduncle. For all regions except the cerebral peduncle, we used a region of interest (ROI) approach. The main advantage in using a single voxel is that the signal measured fully represents the activity or structure within that voxel and is not impacted by the signal in neighboring voxels. However, it is widely accepted that neighboring voxels are likely to have a similar signal, be it resting‐state activity or white matter microstructure. In fact, averaging over an ROI can significantly increase the signal‐to‐noise ratio (SNR) compared to that of a single voxel. A single voxel, for example, may be heavily impacted by the presence of other tissues or non‐neuronal noise which would lower the SNR and the quality of the extracted signal. The location of a single voxel can also vary considerably between individuals. Using a cluster of voxels rather than an individual voxel within a region would provide greater confidence that the “nodes” used in the analyses were located in the anatomical or functional ROI. Furthermore, when defining a “node” based on the literature or a previous study, it is possible to use both standardized atlases or masks to create an ROI. This ensures across‐participant consistency in terms of location. It is important to note, however, that for instance for DTI, effects can be masked or diluted by reverse‐sense changes in different voxels within a single ROI. There is evidence that, for example, FA can increase in some voxels due to selective degradation of one of two fiber populations, but decreases due to the same effect in neighboring voxels that contain only one fiber population (Groeschel et al, 2014). In averaging over an ROI, it is, therefore, possible to mix voxels with differing effects, which may be partially cancelled out on averaging. Finally, for DCM (see below), the timeseries extraction was based on a principal components approach. Instead of signal averaging, we therefore extracted the signal that explains the majority of the variance in that ROI. For the tractography analyses, all metrics are weighted to ensure that those streamlines or fibers which contribute most toward the formation of a tract are most represented.
ROIs were created using the Anatomy Toolbox; thalamic regions for DTI and DCM analyses were therefore identical. The cortical regions were, however, different from those used in the DCM analyses as larger regions were more suitable for fiber‐tracking. The cerebral peduncle region was created using the FSL Montreal Neurological Institute template and the Johns Hopkins University White Matter Labels atlas. All regions were defined in standard space and warped into native space for each participant using the inverse deformation parameters that were outputted from the DARTEL registration for the resting‐state fMRI images (see resting‐state fMRI methods section). Masks were used to exclude any streamlines that tracked via the contralateral hemisphere or posteriorly to the thalamus and peduncle and to ensure tracts did not extend beyond the white matter into grey matter, CSF, or dura.
For each participant, and each set of tracts, probabilistic tractography was then performed using probtrackx (Behrens et al., 2003). Connectivity distributions were generated from our seed regions in native space. The resulting tract images were then warped into diffusion space using the FLIRT tool and overlaid onto the B0 image for quality checking. FA, AD, and RD values were extracted for each participant for each tract.
Resting‐State Functional MRI
Resting‐state fMRI data were collected using 48 continuously acquired ascending axial slices covering the whole cortex and cerebellum (slice thickness: 2.8 mm, gap: 1.5 mm, in plane resolution 3.3 × 3.3 mm, field of view (FOV) 212 mm) with a T2*‐weighted echo‐planar imaging (EPI) sequence (repetition time (TR) 3,000 ms, echo time (TE) 30 ms, flip angle (FA) 80°). One hundred and sixty‐five volumes were acquired in a single 8:20 min run. Preprocessing and subsequent statistical analyses were performed using SPM8 (Wellcome Trust Centre for Neuroimaging, http://www.fil.ion.ucl.ac.uk/spm). Dynamic Causal Modelling (DCM) using a stochastic framework, which models the randomness inherent within resting‐state brain activity, was then employed to investigate causal interactions between five regions within the sensorimotor network (Friston et al., 2003; Friston et al., 2011). Model specification and estimation were conducted using DCM10 (SPM12bWellcome Trust Centre for Neuroimaging) and all connectivity parameters extracted for further analysis.
For resting‐state fMRI, the first four EPI images were discarded to allow for steady‐state equilibrium. The T1 scan was segmented into grey and white matter using the VBM8 toolbox (http://dbm.neuro.uni-jena.de/vbm/) and deformation parameters extracted using DARTEL (Ashburner, 2007). The segmented images were used to create an improved anatomical scan for coregistration. Functional images were first realigned and field maps used for inhomogeneity correction. EPI images were then co‐registered to the new anatomical image and normalized using the DARTEL deformation parameters. Finally the data were smoothed using a 6mm full‐width at half‐maximum Gaussian kernel. Using a first‐level design consisting of smoothed images only, the principal eigenvariate of the white matter and CSF time series were extracted from a single voxel located within the pons (0, −24, −33) or lateral ventricle (−1, 45, 3) respectively. The white matter and CSF signals were then included with movement regressors as nuisance covariates in a second general linear model (GLM) at the individual level. The timeseries for each region within our model was then extracted using this GLM. The model consisted of five regions within the sensory and motor networks within the left hemisphere (dominant). Cortical regions were derived using coordinates from previous Track‐HD studies: S1 (−40, −34, 61); M1 (−40, −18, 60); and the PMC (−24, 0, 54). For the thalamic regions, see DTI methods. For cortical regions, timeseries were extracted by placing an 8 mm sphere around the specified coordinates, localized to the nearest local maximum for peak activity, within a regional anatomical mask generated by either the WFUPickatlas (Eickhoff, et al., 2005) or the Anatomy Toolbox (Maldjian, et al., 2003). For the thalamic ROIs, the principal eigenvariate of the timeseries was extracted from the predefined mask. Connections between and within all regions were modeled except the bidirectional connections between the two thalamic regions, the cortical motor regions and the sensory thalamic region and the S1 and the motor thalamic composite region; this totaled 17 connections. DCM specification and estimation was carried out with DCM10 in SPM software (SPM12b; Wellcome Trust Centre for Neuroimaging, http://www.fil.ion.ucl.ac.uk/spm) and connectivity parameters for each participant were extracted. The DCM estimation and model convergence were examined using SPM‐derived quality assurance.
STATISTICAL ANALYSIS
We first inspected all data for implausible outliers (between 0% and 2% of data, depending on the measure), which were either corrected or discarded after joint review with the subject‐matter investigators. We used log or square root transforms when appropriate to achieve approximate residual normality, as is assumed by inference procedures.
For modeling group differences in outcomes among early HD, preHD‐B, preHD‐A, and controls, we used general weighted least square (GWLS) regression with restricted maximum likelihood estimation (Diggle, et al., 1994). The method accounts for repeated measure correlations among participants with measures at two visits (Tabrizi et al., 2013). Participants with only one observation were also included however. This increases statistical power and ensures unbiased longitudinal analyses if visit 2 data is missing at random.
Individual measures from the various modalities were treated, one‐at‐a‐time, as outcome variables. The main effects of interest were the above HD groups and their potential interaction with time between visits (“time”). Longitudinal group effects were tested via the time interactions with group.
The “cross‐sectional” effects of interest were mean group values, averaged over both visits. These were estimated by a linear combination of main effects of group and group‐by‐time interaction, such that estimated baseline and 12 month values were given equal weight. These averaged cross‐sectional comparisons reduce statistical noise due to factors such as measurement error and accurate but irrelevant measurement of short term (e.g., day‐to‐day) fluctuations, leading to smaller standard errors for group comparisons.
Because we also attempted to measure longitudinal change over 1 year, one might question the validity of using both the baseline and follow‐up measures for a mean cross‐sectional analysis. The justification is empirical and due to the slow evolution of most HD phenomena. Even for measures that exhibit statistically significant longitudinal changes over 1 year, the magnitude of change is quite small in comparison to the cross‐sectional changes exhibited between HD groups and controls. This cross‐sectional difference is the net result of many years of longitudinal change (Tabrizi et al., 2009; Tabrizi et al., 2013). We also note that this is an observational study. Within the broad ranges defined by our groupings, the time of the baseline measurement is arbitrary. For measures that are not subject to practice effects, there is no reason to prefer time 1 or time 2 as representative of premanifest or control states.
We controlled for age at study entry, gender, study site (scanner), and education level, as well as their interaction with time. For TMS thresholds, we additionally controlled for main effects of skull to cortex (M1) distance and for Freesurfer measures of mean cortical thickness in the left Brodmann areas B4a and B4b (the cortical areas stimulated). Some models also controlled for underlying HD progression risk determined by CAG repeat length and age via the cumulative probability of onset (CPO) statistic from a survival analysis of onset ages (Tabrizi et al., 2013; Langbehn et al., 2004). Because variance of outcome measures sometimes changes notably in early HD versus preHD versus controls, separate residual covariance was estimated for each group. We used the Kenward–Rogers correction to estimate denominator degrees of freedom (Kenward and Roger, 1997). All models were fit using Proc Mixed from SAS 9.4 (Littell et al., 2006). Within modalities (electrophysiology, DTI, DCM, etc.), we also calculated false discovery rates (FDR), abbreviated by q, using the Benjamini and Hochberg method (Benjamini and Hochberg, 1995).
We created a composite grip force motor score from the sum of sample‐standardized orientation and position indices, collected while grip force heavy‐load task was performed with the dominant hand.
For the final steps of the statistical regression analyses, which involved composite scores created from multiple variables, we addressed the issue of missing data by use of multiple imputation (Schafer, 1997) done separately for cases and controls. MI was based on outcome measures at baseline and follow‐up (treated as separate variables) along with all demographic covariates, including HD group and study site interactions with time. Five imputations were generated using Proc MI and subsequent model inferences were adjusted using Proc MIAnalyze (SAS 9.4).
PCA was performed on outcome variables of interest as identified by their between‐group differences in the GWLS regressions. The PCA input was the maximum likelihood estimate of the correlation matrix, derived during the multiple imputation procedure. Nonrotated component scores were then used in to model the relationships to grip force, UHDRS motor score (Pearson correlations and linear models), and manifest versus premanifest HD discrimination (logistic discrimination).
For the PCA, we selected measures from each of the modalities. We were guided by the results of individual modality analyses, i.e., we chose measures with the greatest difference between controls and HD. Some of the measures within a given modality were highly correlated, for instance, the DTI measures from all tracts or cortical thickness from all regions. Hence we only included for instance diffusivity measures from 2 of the 4 tracts in the multimodel PCA. Overall, we included only 15 variables within the multimodal PCA to ensure representation of the different modalities was roughly equal. We included VBM data (volumes of white matter, grey matter, and caudate), cortical thickness data (BA6 (premotor cortex), BA3a + 2 (somatosensory cortex)), diffusion metrics from tractography (AD and RD of S1–thalamus and PMC–thalamus tracts), effective connectivity parameters from DCM (PMC–thalamus and PMC–PMC connections), and SEPs (N20/P25 amplitude and N20 latency) into the PCA (multimodal‐PCA). In a second PCA that focused solely on DTI, we included RD and AD measures from all 4 available DTI tracts. We were interested in the question of whether patterns in HD participants differed from those in controls or were similar. For this reason, we did the PCAs separately in HD and healthy controls.
RESULTS
The total Track‐On cohort comprised 131 HD (101 former TRACK‐HD and 30 new premanifest) and 112 controls (79 former TRACK‐HD and 33 new) recruited between April 16, 2012 and December 10, 2012 (Table 1). At the 12 month follow‐up visit, 11 participants from the HD group and 7 from the control group did not take part. The most common reasons for withdrawal were intolerance to MRI, personal time constraints, and the burden of the study day.
Table 1.
Demographic and clinical characteristics of study participants by subgroup
| Variable | Control N = 112 | preHD A N = 41 | preHD B N = 55 | preHD all N = 96 | Early HD N = 35 |
|---|---|---|---|---|---|
| Gender N (%F) | 67 (59.8) | 24 (58.5) | 26 (47.3) | 50 (52.1) | 19 (54.3) |
| Age | 48.1 (10.7) | 40.4 (8.8) | 44.0 (9.0) | 42.4 (9.0) | 45.3 (8.4) |
| Education | 3.9 (1.0) | 4.0 (1.0) | 4.0 (1.0) | 4.0 (1.0) | 3.9 (0.9) |
| CAG repeat length | – | 42.3 (2.2) | 43.4 (2.3) | 42.9 (2.3) | 43.5 (2.5) |
| Disease burden | – | 257.2 (30.7) | 330.0 (39.8) | 298.9 (51.1) | 348.2 (61.6) |
| Motorscore | 1.3 (1.6) | 5.4 (3.3) | 5.0 (3.8) | 5.2 (3.6) | 12.6 (7.1) |
We defined a priori a total of 53 measures for analysis (17 DCM, 3 brain volume, 7 cortical thickness, 12 DTI, and 14 electrophysiology; Supporting Information, Table 1). First we asked if there was detectable change in any of the measures from baseline to the 12 month visit. Longitudinal change was small and not statistically significant (data not shown). In contrast, cross‐sectional differences among the groups, averaged over time, were evident and are presented in Table 2.
Table 2.
Principal component analysis with data from all modalities
| HD | Controls | |||||
|---|---|---|---|---|---|---|
| PC | Eigenvalue | % Variance explained | Cumulative | Eigenvalue | % Variance explained | Cumulative |
| 1 | 3.102 | 20.7 | 20.7 | 3.432 | 22.9 | 22.9 |
| 2 | 2.267 | 15.1 | 35.8 | 2.353 | 15.7 | 38.6 |
| 3 | 1.703 | 11.3 | 47.1 | 1.850 | 12.3 | 50.9 |
| 4 | 1.442 | 9.7 | 56.8 | 1.103 | 7.4 | 58.2 |
| 5 | 1.084 | 7.2 | 64.0 | 1.081 | 7.2 | 65.5 |
| 6 | 0.995 | 6.6 | 70.6 | 0.968 | 6.5 | 71.9 |
| 7 | 0.891 | 6.0 | 76.6 | 0.901 | 6.0 | 77.9 |
| 8 | 0.795 | 5.3 | 81.9 | 0.829 | 5.5 | 83.4 |
| 9 | 0.704 | 4.7 | 86.6 | 0.706 | 4.7 | 88.2 |
| 10 | 0.531 | 3.5 | 90.1 | 0.581 | 3.9 | 92.0 |
| 11 | 0.522 | 3.5 | 93.6 | 0.446 | 3.0 | 95.0 |
| 12 | 0.468 | 3.1 | 96.7 | 0.335 | 2.2 | 97.2 |
| 13 | 0.211 | 1.4 | 98.1 | 0.229 | 1.5 | 98.8 |
| 14 | 0.161 | 1.1 | 99.2 | 0.128 | 0.9 | 99.6 |
| 15 | 0.125 | 0.8 | 100 | 0.059 | 0.4 | 100 |
PCA was performed on all gene‐carrier participants (combined HD and preHD), and, independently, in controls because we were interested in comparing independently derived descriptive patterns between the HD and the control groups.
In the combined HD group, multimodal‐PCA reduced the dimensionality of the data to three important dimensions or principal components (PCs) based on their eigenvalues (Table 2; Supporting Information, Table 2). In HD, the first multimodal‐PC explained 21% of the variance in the data. In the multimodal‐PC1 dimension, lower caudate, grey, and white matter volumes, and lower BA6 and BA1 and 2 cortical thickness were associated with higher AD and RD of S1‐thalamus and PMC‐thalamus tracts (Fig. 1A). The first PC was the only multimodal‐PC that correlated significantly with the age‐CAG cumulative probability of onset (r = −0.58, P < 0.0001, Fig. 1B). Furthermore, this first multimodal‐PC showed association with sensory‐motor network specific performance (grip force orientation and position index; Fig. 1C and Table 3). Multimodal‐PC1 substantially mediated, and improved upon, the predictive effect of age‐CAG on grip force (P = 0.006) such that there was no residual association of age‐CAG (P = 0.468) after this PC was included in the model. Multimodal‐PC1 was also associated with clinical motor score (P = 0.041), and probability of manifest versus preHD status (P = 0.003, Fig. 1D and Table 3). However, unlike for grip force associations, the influence of age‐CAG on clinical motor score or HD status remained significant, though weaker, when the first PC was included in the model. This indicates that the first multimodal‐PC does not completely mediate the age–CAG relationship to motor score.
Figure 1.
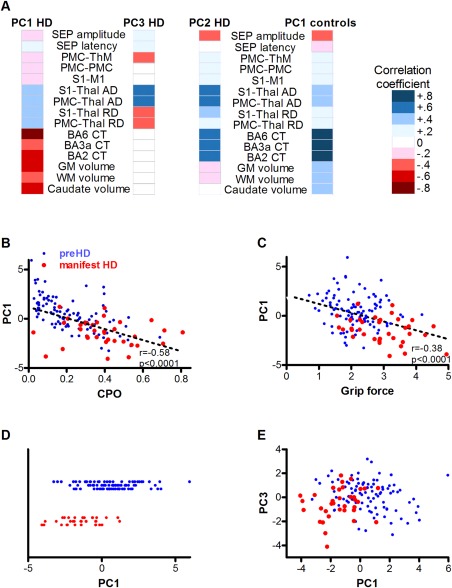
Multimodal principal component analysis. (A) Heat map of correlation coefficients of each modality with dimensions derived from principal component analysis done independently in healthy controls and HD participants. The first multimodal principal component (PC) in HD contains higher axial (AD) and radial diffusivity (RD) in S1‐Thalamus (Thal) and PMC‐Thalamus tracts; less cortical thickness in the PMC (BA6) and S1 (BA3a; BA2); and less total brain grey matter (GM), white matter (WM), and caudate volume. Multimodal‐PC3 in HD reflects the difference between axial and radial diffusivity. Multimodal‐PC2 in HD and PC1 in controls show a similar pattern of thicker cortex, higher axial diffusivity, and lower SEP amplitudes. (B) HD multimodal‐PC1 scores negatively correlate with cumulative probability of onset (CPO) and grip force orientation and position index (C; pre HD blue dots, manifest HD red dots). (D) Multimodal‐PC1 scores distinguish manifest HD (red dots) from preHD participants (blue dots; the Y‐axis just separates HD and controls). (E) Multimodal‐PC3 is a substantial additional predictor of manifest HD (P = 0.021) improving the separation of manifest HD (red dots) and preHD participants (blue dots). Abbreviations: PMC‐ThM, effective connectivity PMC to motor thalamus parcellation; PMC‐PMC, effective connectivity PMC to PMC; S1‐M1, effective connectivity S1 to motor cortex.
Table 3.
PCA (on data from all modalities) multivariate model of grip force position and orientation composite score, UHDRS motor score, and diagnosis status, i.e., premanifest versus manifest Huntington's disease
| Regression coefficient | Standard error | T value | CPO mediated (%) | P | |
|---|---|---|---|---|---|
| Grip force | |||||
| CPO only (a) | |||||
| Intercept | −0.3831 | 0.1560 | −2.46 | 0.015 | |
| CPO (a) | −0.1462 | 0.0498 | 2.93 | 0.004 | |
| PC1 added | |||||
| Intercept | −0.1156 | 0.1812 | −0.64 | 0.525 | |
| CPO (a) | −0.0442 | 0.0606 | 0.73 | 75.1 | 0.468 |
| PC1 (b) | −0.1742 | 0.0614 | −2.84 | 0.006 | |
| PC3 added | |||||
| Intercept | −0.0939 | 0.1767 | −0.53 | 0.596 | |
| CPO (a) | −0.0357 | 0.0602 | 0.59 | 79.9 | 0.555 |
| PC1 | −0.1805 | 0.0617 | −2.93 | 0.005 | |
| PC3 | −0.1519 | 0.0700 | −2.17 | 0.036 | |
| UHDRS motor score | |||||
| CPO only (a) | |||||
| Intercept | 1.853 | 0.154 | 12.02 | <0.0001 | |
| CPO (a) | −0.273 | 0.049 | 5.55 | <0.0001 | |
| PC1 added | |||||
| Intercept | 2.043 | 0.178 | 11.46 | <0.0001 | |
| CPO (a) | −0.201 | 0.060 | 3.35 | 39.6 | 0.001 |
| PC1 (b) | −0.124 | 0.060 | −2.06 | 0.041 | |
| PC3 added | |||||
| Intercept | 2.068 | 0.173 | 11.95 | <0.0001 | |
| CPO (a) | −0.191 | 0.059 | 3.25 | 41.4 | 0.002 |
| PC1 | −0.131 | 0.058 | −2.24 | 0.027 | |
| PC3 | −0.180 | 0.065 | −2.77 | 0.007 | |
| Premanifest versus manifest Huntington's disease | |||||
| CPO only (a) | ln(OR) | ||||
| 0.679 | 0.147 | 4.61 | <0.0001 | ||
| PC1 added | |||||
| CPO (a) | 0.422 | 0.172 | 2.54 | 44.9 | 0.011 |
| PC1 (b) | 0.627 | 0.211 | 2.98 | 0.003 | |
| PC3 added | |||||
| CPO (a) | 0.444 | 0.172 | 2.58 | 44.0 | 0.010 |
| PC1 | 0.653 | 0.206 | 3.17 | 0.002 | |
| PC3 | 0.496 | 0.214 | 2.32 | 0.021 | |
The second multimodal‐PC in HD explained 15% of data variance (Fig. 1A). Higher AD was correlated with smaller SEP amplitudes and greater cortical thickness. Multimodal‐PC2 was not predicted by age and CAG length (r = 0.12, P = 0.178), nor was it associated with HD outcomes (data not shown). This second PC may represent an element of biological variation unrelated to HD, an interpretation supported by partial similarity to the first multimodal‐PC in an independent PCA in healthy controls. The patterns were similar except that volumes of grey and white matter and caudate also correlated with the component in controls but not HD (Fig. 1A; Supporting Information, Table 3). Consistent with their known central role in the illness, these volumes instead contributed substantially to the first principal component of variation among the HD groups.
The third multimodal‐PC in HD, explaining 11% of data variance, revealed a dimension of contrast between AD and RD (Fig. 1A). The third PC was a substantial additional predictor of grip force score (P = 0.036), of UHDRS motor score (P = 0.007) and of diagnosis (P = 0.021, Fig. 1E and Table 3). Caudate, grey, and white matter volumes contributed very little to this third multimodal‐PC, and its values were not predicted by age and CAG length (r = 0.04; P = 0.643). By definition, this AD versus RD component is uncorrelated with the more general brain structural effects of the first multimodal‐PC, which are driven by genetic HD load. Furthermore, multimodal‐PCA of the normal controls revealed a similar AD versus RD contrasting component that accounted for 12% of the variance within the control group (Supporting Information, Table 3). Therefore, higher AD compared to RD is associated with HD pathogenesis in the presence of a CAG expansion mutation, but is also observed as natural variation in healthy controls. It is independent of the unique HD brain structural effects that are related to HTT CAG repeat length.
We then asked if the relationship of AD and RD extended to the other white matter tracts that we had measured but excluded from the initial PCA. There was a very high correlation of AD, and RD, values among the four tracts. The finding was consistent in separate analyses of the controls (Fig. 2A,B) and the HD group (Fig. 2D,E). This was further reflected in a PCA restricted to AD and RD values from all 4 tracts (ADRD‐PCA) and performed independently in healthy controls and HD participants. Two dimensions (illustrated for controls in Fig. 2C and for HD in Fig. 2F) explained 94% of the variability in controls and 90% of variability in HD participants (Fig. 2G). ADRD‐PC1 contains a dimension of common correlation among all diffusivity measures while ADRD‐PC2, consistent with the multimodal PCA, reflects the contrast between AD and RD values (Fig. 2G and Table 4). AD measures were significantly correlated with grip force, but the RD measures were not (Fig. 2H and Table 5). There was no correlation between age and grip force performance in controls, and correlation of AD and RD with grip force was not significantly affected if controlled for age. In HD participants, the patterns were similar to controls. The correlation of both ADRD‐PC1 and AD with grip force was stronger, and the relationship of ADRD‐PC2 and grip force was slightly stronger than in controls (Fig. 2I and Table 5).
Figure 2.
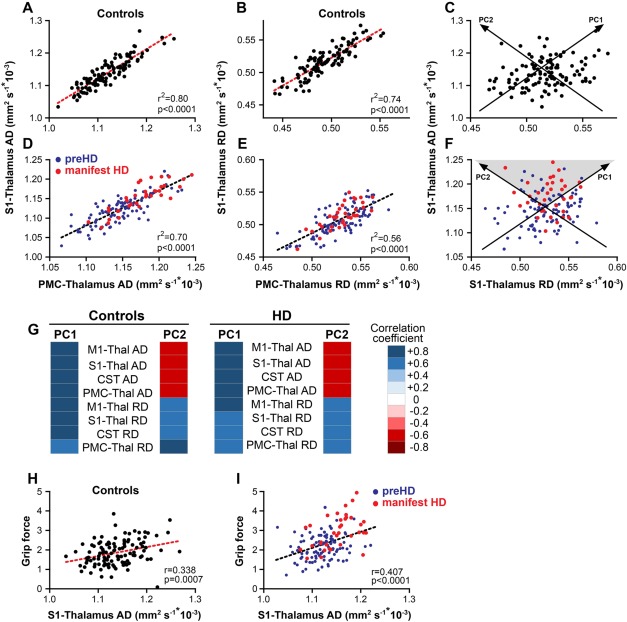
Principal component analysis with axial and radial diffusivity (ADRD‐PCA). In controls, (A) axial diffusivity values or (B) radial diffusivity values from different tracts are highly correlated. (C). ADRD principal component (PC) 1 reflects that greater, or smaller, axial diffusivity is associated with greater, or smaller, radial diffusivity. There is additional variability in the relationship of axial and radial diffusivity, which is reflected in ADRD‐PC2. In HD participants, the relationship between (D) axial diffusivity (AD) or (E) radial diffusivity (RD) in the two tracts is similar to controls. (F) Manifest HD participants (red dots) have higher than average axial and radial diffusivity values and higher axial relative to radial diffusivity values (grey triangle) than preHD (blue dots). (G) A principal component analysis with axial and radial diffusivity values from all 4 tracts done independently in healthy controls and HD participants reveals that PC1 and PC2 explain 94% of data variability in controls and 90% in HD participants. Heat maps of correlation coefficients show that in controls and HD participants in PC1 axial and radial diffusivity are positively correlated while PC2 reflects the difference between axial and radial diffusivity. In (H) controls and (I) HD participants, higher axial diffusivity is associated with higher grip force orientation and position index scores. NB. C and F contain approximate representations of the PCs relative to paired, observed AD and RD measures. Abbreviations: M1: motor cortex; S1: somatosensory cortex; CST: cortico‐spinal tract; PMC: premotor cortex.
Table 4.
Correlations of observed axial and radial diffusivity in independent PCAs in controls and Huntington's disease participants
| Controls | Huntington's Disease | |||
|---|---|---|---|---|
| Variable | PC1 | PC2 | PC1 | PC2 |
| M1 – Thal AD | 0.785 | −0.598 | 0.793 | −0.577 |
| S1 – Thal AD | 0.812 | −0.531 | 0.784 | −0.548 |
| CST AD | 0.797 | −0.544 | 0.782 | −0.542 |
| PMC – Thal AD | 0.747 | −0.625 | 0.770 | −0.581 |
| M1 – Thal RD | 0.749 | 0.631 | 0.706 | 0.666 |
| S1 – Thal RD | 0.770 | 0.562 | 0.626 | 0.662 |
| CST RD | 0.741 | 0.595 | 0.644 | 0.702 |
| PMC – Thal RD | 0.634 | 0.718 | 0.619 | 0.684 |
Table 5.
Correlations of grip force with axial (AD) and radial diffusivity (RD) in independent principal component analyses (PCAs) in controls and Huntington's disease participants
| Grip force correlation | Controls | Huntington | ||
|---|---|---|---|---|
| Correlation coefficient | P | Correlation coefficient | P | |
| PCA1 mean | 0.273 | 0.0065 | 0.381 | <0.0001 |
| PCA2 mean | −0.168 | 0.0978 | −0.201 | 0.0352 |
| M1 – Thalamus AD | 0.342 | 0.0006 | 0.420 | <0.0001 |
| S1 – Thalamus AD | 0.338 | 0.0007 | 0.407 | <0.0001 |
| CST AD | 0.277 | 0.0058 | 0.408 | <0.0001 |
| PMC – Thalamus AD | 0.291 | 0.0036 | 0.410 | <0.0001 |
| M1 – Thalamus RD | 0.087 | 0.3946 | 0.136 | 0.1577 |
| S1 – Thalamus RD | 0.136 | 0.1825 | 0.077 | 0.422 |
| CST RD | 0.070 | 0.4921 | 0.127 | 0.1857 |
| PMC – Thalamus RD | 0.075 | 0.4629 | 0.103 | 0.2864 |
DISCUSSION
In this study, we went beyond the analysis of data from single modalities one at a time to investigate data patterns reflecting the structural and functional state of an a priori defined sensory‐motor circuit that is relevant for motor control. We aimed to delineate CAG‐repeat‐length‐dependent and ‐independent patterns that influence the pathogenic process before disease onset. We did not detect longitudinal differences over 12 months. Cross‐sectionally, in HD, dimension reduction using PCAs revealed one dimension specific to its cause, i.e., HTT CAG repeat‐length expansion, that included a correlated loss of caudate, grey, and white matter volume, cortical thickness, and disturbed diffusivity in white matter tracts. Within our data, this component predicted sensory‐motor performance and a diagnosis of manifest HD. Two further dimensions were not unique to HD but were also present in controls. The first of these dimensions represented natural variation in axonal diffusivity, cortical thickness, and SEP amplitude but did not influence HD. The second non‐CAG dimension consisting of an AD versus RD contrast in white matter tracts did influence HD in that it was an additional predictor of sensory‐motor performance in HD and controls, and a clinical diagnosis of HD. This pattern of diffusivity is consistent with naturally occurring variability in a biological trait that influences HD manifestations.
The HTT mutation is the main determinant of pathogenesis that leads to the structural and functional neurodegeneration that underlies HD and the emergence of unequivocal clinical signs. We examined this pathogenic process in that we investigated the structural and functional state in an a priori defined sensory‐motor circuit relevant for motor control. Presumably owing to the slow progression of HD and perhaps also the sensitivity limits of the methods employed, there was no detectable change in any of the sensory‐motor modalities within the 12 month observation period. This may appear inconsistent with the findings in the original TRACK‐HD study at 12 months (Tabrizi et al., 2011). It is important to note, though, that many of the measures that we used in this study were not part of TRACK‐HD. It is therefore possible that, e.g., electrophysiological measures or DTI are less suitable for the detection of change over 12 months because either network brain function or white matter microstructure do not change much or because measurement error for these measures is large relative to the change that happens. In addition, in TRACK‐HD, most detectable changes were observed only in the diagnosed, early HD group, which was both substantially larger and more advanced in disease than in our newly diagnosed HD subgroup. It is therefore conceivable that the ability to detect change over time depends on the measurements themselves, on the time that elapsed between measurements, and also on the stage of HD. Cross‐sectionally, however, our data confirmed that, compared with controls, in HTT gene expansion carriers white and grey matter brain structure is diminished, and sensory afferent signal transmission is concomitantly slower and less efficient. Furthermore, these differences were measurable before unequivocal motor signs of HD emerged. Brain structure and electrophysiology remained abnormal following the emergence of diagnostic motor signs. Further, in early HD, in addition to RD, AD was substantially worse and effective connectivity from PMC to thalamus reduced. The analyses used to examine differences at the individual modality level the patterns by which the data differ across modalities. We therefore next employed multivariate analysis using PCA to reveal such data patterns. PCA reduced data to two dimensions that were relevant for HD and one that represents natural variation that does not influence HD. The first dimension relevant to HD contained a correlated loss of caudate, grey and white matter volume, cortical thickness, and disturbed diffusivity in white matter tracts. These changes increased with an increase of the age‐CAG‐length HD risk, indicating that structural abnormalities relate to the primary cause of pathogenesis. For performance that is related to the sensory‐motor network, we show that the current structure of white and grey matter within the network is a more relevant (and presumably more proximal) predictor than the key HD biological variables, age, and CAG repeat length. Our data indicate that structural changes in white and grey matter are relevant for sensory‐motor task performance and are an integral part of HD biology.
We then examined whether we could identify biological variation that was independent of genetic risk but still associated with sensory‐motor performance and clinical category, i.e., preHD versus manifest HD. PCA identified two dimensions not connected to CAG repeat length in which data variance was similar in controls and HD participants. One of these dimensions was not associated with clinical status. It contrasted more AD with smaller SEP amplitudes and greater cortical thickness, and, in controls, increased grey matter volume as well. In HD participants, this dimension was not related to age‐CAG repeat length. The dimension seems to capture an element of natural biological variation unrelated to HD.
Axonal damage is known to be associated with a loss of amplitude of evoked potentials so that it is conceivable that variation of axonal microstructure relates to SEP amplitude. In addition, the distance between the soma of the cell receiving sensory afferent inputs within S1 and the recording electrode on the scalp can also influence SEP amplitude. With a thicker cortex that distance may be slightly greater, and subsequently the SEP amplitude smaller.
A second dimension unrelated to age‐CAG repeat length in both controls and HD participants revealed differences between AD and RD in white matter tracts. This dimension was also, by definition, uncorrelated with either the previous non‐CAG‐repeat dimension or with the primary dimension related to CAG‐repeat length found only in HD. This axial–radial DTI dimension was independently associated with sensory‐motor performance and a clinical diagnosis of HD. Somewhat surprisingly, it was also associated with task performance in controls. The diffusivity contrast was evident in all the tracts we analyzed, and, thus, is not specific to a single white matter tract. Therefore, the diffusivity patterns constitute a quantitative phenotype that reflects naturally occurring variability in white matter tract biology.
AD reflects water movement in tracts parallel to the main fiber organization while RD measures water movement perpendicular to this organization.
In animal models, there is evidence suggesting that AD reflects axonal integrity and RD myelin integrity (Song et al., 2002; Song et al., 2003; Tobin et al., 2011). Myelin‐producing glial cells and neurons form a unit in which axon integrity depends on intact myelin and axons help maintain that myelin (Bremer et al., 2010; Nave, 2010). In diseases primarily affecting white matter myelin, such as multiple sclerosis, clinical signs seem to manifest when axonal damage arises from the loss of myelin trophic support (Nave, 2010). Naturally occurring variability in the axon–myelin unit in itself does not cause disease. However, with a genetically sensitized background such as in HD or in other diseases naturally occurring variability in the make‐up of the axon–myelin unit may influence onset and the course of the disease. DTI diffusivity patterns need to be interpreted with caution, as we cannot infer with confidence the anatomical significance based on the diffusivity changes we observed in our participants. Nonetheless, given the prominent involvement of white matter in HD (Tabrizi et al., 2009), and since mutant huntingtin has been implicated in oligodendrocyte function and axonal transport (Huang et al., 2015; Li and Conforti, 2013), our observational data suggest that it is worth exploring the role of the differential loss of myelin and axons in HD pathogenesis further.
In contrast to brain structure, sensory‐motor functional measures such as effective connectivity or SEPs did not contribute significantly to models of clinical status. While some functional measures differed significantly between preHD or early HD versus controls, task performance, and clinical status. One possible explanation could be the use of resting‐state measures. Brain activity differs substantially when performing a task compared to when at rest even within our carefully chosen sensory‐motor network model. It is also possible that the precision of functional measurements is inadequate to reliably detect relevant underlying phenomena.
In summary, we went beyond the analysis of single modality data one at a time to assess patterns of structural and functional properties of the sensory‐motor system in HD and healthy controls using multivariate analysis. While there was very little, if any, change in these modalities over 12 months, the cross‐sectional analysis revealed a dimension of white and grey matter loss attributable to HD biology that is associated with network‐specific task performance and clinical diagnosis. Our data suggest that the microstructural anatomy influencing water diffusivity perpendicular to white matter tracts, e.g., myelin, is already changed in preHD. In contrast, the anatomical compartment that determines water diffusivity parallel to the tracts, e.g., the axon, is not substantially affected until early HD. Although these patterns are amplified with HD, we also show that these measures of white matter organization consistently vary among healthy controls across different white matter tracts. Given the absence of an age–CAG correlation, this indicates that naturally occurring variability in white matter tract microanatomy may represent a biological trait with a disease modifying influence on the pathogenesis of HD. These findings need to be confirmed, e.g., examining more tracts with DTI in conjunction with more task performance outcomes.
No disease‐modifying treatment is known that can delay the onset or slow the progression of HD, although there are several promising approaches about to reach clinical testing (Wild and Tabrizi, 2014). These approaches directly target the production or regulation of the huntingtin protein. However, there is still a need to identify other important disease‐modifying biological factors in HD as these may eventually provide additional drug development targets for this devastating disease (GeM‐HD Consortium, 2015).
AUTHOR CONTRIBUTIONS
Conception and design of the study: MO, AD, RACR, BRL, GBL, RR, BB, LB, RIS, SK, GR, JCR, DL, and SJT.
Acquisition and analysis of data: MO, ISMM, SG, KKS, LM, RIS, GR, and DL.
Drafting of the manuscript, tables, and figures: MO and SG.
All co‐authors reviewed and critiqued the manuscript.
TRACK‐ON investigators: Canada: Kate Brown, Joji Decolongon, Mannie Fan, Tamara Koren, and Terri Patkau (University of British Columbia, Vancouver, Canada). France: Celine Jauffret, Sabine Meunier, and Traian Popa (ICM Institute, Paris). Germany: Christian Sass and Nathalia Weber (GHI, Münster). Netherlands: Omar Odish and Anne Schoonderbeek (Leiden University Medical Center, Leiden). UK: Claire Berna, Helen Crawford, Mahaleskshmi Desikan, Davina J Hensman Moss, Nicola Z Hobbs, Eileanoir Johnson, Gail Owen, Adeel Razi, Joy Read, and Gionata Strigaro (University College London, London). USA: Hans Johnson and Jim Mills (University of Iowa, Iowa).
CONFLICTS OF INTEREST AND FINANCIAL DISCLOSURES
The authors have no conflicts of interest in relation to the material presented in this article.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
The authors thank the Track‐On study participants and their families; the CHDI/High Q Foundation, a not‐for‐profit organization dedicated to finding treatments for HD; and Richard Frackowiak, Christian Wolf, Christoph Kaller, Karl Friston, Nikolaus Weiskopf, Daniel S. O'Leary, and Stephane Lehericy for helpful advice. We thank Ray Young for his assistance with artwork.
REFERENCES
- Abbruzzese G, Dall'Agata D, Morena M, Reni L, Favale E (1990): Abnormalities of parietal and prerolandic somatosensory evoked potentials in Huntington's disease. Electroencephalogr Clin Neurophysiol 77:340–346. [DOI] [PubMed] [Google Scholar]
- Ashburner J (2007): A fast diffeomorphic image registration algorithm. NeuroImage 38:95–113. [DOI] [PubMed] [Google Scholar]
- Aylward EH (2007): Change in MRI striatal volumes as a biomarker in preclinical Huntington's disease. Brain Res Bull, 72(2‐3):152–158. [DOI] [PubMed] [Google Scholar]
- Behrens TE, Berg HJ, Jbabdi S, Rushworth MF, Woolrich MW (2007): Probabilistic diffusion tractography with multiple fibre orientations: What can we gain? NeuroImage 34:144–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens TE, Woolrich MW, Jenkinson M, Johansen‐Berg H, Nunes RG, Clare S, Matthews PM, Brady JM, Smith SM (2003): Characterization and propagation of uncertainty in diffusion‐weighted MR imaging. Magn Reson Med 50:1077–1088. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y (1995): Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc B 57:289–300. [Google Scholar]
- Bremer J, Baumann F, Tiberi C, Wessig C, Fischer H, Schwarz P, Steele AD, Toyka KV, Nave KA, Weis J, Aguzzi A (2010): Axonal prion protein is required for peripheral myelin maintenance. Nat Neurosci 13:310–318. [DOI] [PubMed] [Google Scholar]
- Diggle PJ, Liang K‐Y, Zeger SL (1994): Analysis of Longitudinal Data. Oxford: Clarendon Press. [Google Scholar]
- Eickhoff SB, Stephan KE, Mohlberg H, Grefkes C, Fink GR, Amunts K, Zilles K (2005): A new SPM toolbox for combining probabilistic cytoarchitectonic maps and functional imaging data. NeuroImage 25:1325–1335. [DOI] [PubMed] [Google Scholar]
- Fischer M, Orth M (2011): Short‐latency sensory afferent inhibition: Conditioning stimulus intensity, recording site, and effects of 1 Hz repetitive TMS. Brain Stimulation 4:202–209. [DOI] [PubMed] [Google Scholar]
- Fischl B, Dale AM (2000): Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proc Natl Acad Sci USA 97:11050–11055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friston KJ, Harrison L, Penny W (2003): Dynamic causal modelling. NeuroImage 19:1273–1302. [DOI] [PubMed] [Google Scholar]
- Friston KJ, Li B, Daunizeau J, Stephan KE (2011): Network discovery with DCM. NeuroImage 56:1202–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genetic Modifiers of Huntington's Disease (GeM‐HD) Consortium (2015): Identification of genetic factors that modify clinical onset of Huntington's disease. Cell 162:516–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiou‐Karistianis N, Scahill R, Tabrizi SJ, Squitieri F, Aylward E (2013): Structural MRI in Huntington's disease and recommendations for its potential use in clinical trials. Neurosci Biobehav Rev 37:480–490. [DOI] [PubMed] [Google Scholar]
- Groeschel S, Tournier JD, Northam GB, Baldeweg T, Wyatt J, Vollmer B, Connelly A (2014): Identification and interpretation of microstructural abnormalities in motor pathways in adolescents born preterm. Neuroimage 87:209–219. [DOI] [PubMed] [Google Scholar]
- Huang B, Wei W, Wang G, Gaertig MA, Feng Y, Wang W, Li XJ, Li S (2015): Mutant huntingtin downregulates myelin regulatory factor‐mediated myelin gene expression and affects mature oligodendrocytes. Neuron 85:1212–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jech R, Klempir J, Vymazal J, Zidovska J, Klempirova O, Ruzicka E, Roth J (2007): Variation of selective gray and white matter atrophy in Huntington's disease. Mov Disord 22:1783–1789. [DOI] [PubMed] [Google Scholar]
- Jenkinson M, Smith S (2001): A global optimisation method for robust affine registration of brain images. Med Image Anal 5:143–156. [DOI] [PubMed] [Google Scholar]
- Kenward MG, Roger JH (1997): Small sample inference for fixed effects from restricted maximum likelihood. Biometrics 53:983–997. [PubMed] [Google Scholar]
- Kuwert T, Noth J, Scholz D, Schwarz M, Lange HW, Topper R, Herzog H, Aulich A, Feinendegen LE (1993): Comparison of somatosensory evoked potentials with striatal glucose consumption measured by positron emission tomography in the early diagnosis of Huntington's disease. Mov Disord 8:98–106. [DOI] [PubMed] [Google Scholar]
- Langbehn DR, Brinkman RR, Falush D, Paulsen JS, Hayden MR (2004): A new model for prediction of the age of onset and penetrance for Huntington's disease based on CAG length. Clin Genet 65:267–277. [DOI] [PubMed] [Google Scholar]
- Lefaucheur JP, Menard‐Lefaucheur I, Maison P, Baudic S, Cesaro P, Peschanski M, Bachoud‐Levi AC (2006): Electrophysiological deterioration over time in patients with Huntington's disease. Mov Disord 21:1350–1354. [DOI] [PubMed] [Google Scholar]
- Li JY, Conforti L (2013): Axonopathy in Huntington's disease. Exp Neurol 246:62–71. [DOI] [PubMed] [Google Scholar]
- Littell RC, Milliken GA, Stroup WW, Wolfinger RD, Schabenberger O (2006): SAS for Mixed Models. Cary, NC: SAS Institute Inc. [Google Scholar]
- Maldjian JA, Laurienti PJ, Kraft RA, Burdette JH (2003): An automated method for neuroanatomic and cytoarchitectonic atlas‐based interrogation of fMRI data sets. NeuroImage 19:1233–1239. [DOI] [PubMed] [Google Scholar]
- Matsui JT, Vaidya JG, Wassermann D, Kim RE, Magnotta VA, Johnson HJ, Paulsen JS, Investigators, P.‐H., Coordinators of the Huntington Study, G (2015): Prefrontal cortex white matter tracts in prodromal Huntington disease. Hum Brain Mapp 36:3717–3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nave KA (2010): Myelination and the trophic support of long axons. Nat Rev Neurosci 11:275–283. [DOI] [PubMed] [Google Scholar]
- Orth M, Rothwell JC (2004): The cortical silent period: Intrinsic variability and relation to the waveform of the transcranial magnetic stimulation pulse. Clin Neurophysiol 115:1076–1082. [DOI] [PubMed] [Google Scholar]
- Paulsen JS, Long JD, Ross CA, Harrington DL, Erwin CJ, Williams JK, Westervelt HJ, Johnson HJ, Aylward EH, Zhang Y, Bockholt HJ, Barker RA (2014): Prediction of manifest Huntington's disease with clinical and imaging measures: A prospective observational study. Lancet Neurol 13:1193–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilmann R, Bohlen S, Klopstock T, Bender A, Weindl A, Saemann P, Auer DP, Ringelstein EB, Lange HW (2010): Grasping premanifest Huntington's disease ‐ shaping new endpoints for new trials. Mov Disord 25:2858–2862. [DOI] [PubMed] [Google Scholar]
- Rosas HD, Salat DH, Lee SY, Zaleta AK, Pappu V, Fischl B, Greve D, Hevelone N, Hersch SM (2008): Cerebral cortex and the clinical expression of Huntington's disease: Complexity and heterogeneity. Brain 131:1057–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer JL (1997): Analysis of Incomplete Multivariate Data. New York: Chapman & Hall. [Google Scholar]
- Smith SM, Jenkinson M, Woolrich MW, Beckmann CF, Behrens TE, Johansen‐Berg H, Bannister PR, De Luca M, Drobnjak I, Flitney DE, Niazy RK, Saunders J, Vickers J, Zhang Y, De Stefano N, Brady JM, Matthews PM (2004): Advances in functional and structural MR image analysis and implementation as FSL. NeuroImage 23:S208–S219. [DOI] [PubMed] [Google Scholar]
- Song SK, Sun SW, Ju WK, Lin SJ, Cross AH, Neufeld AH (2003): Diffusion tensor imaging detects and differentiates axon and myelin degeneration in mouse optic nerve after retinal ischemia. NeuroImage 20:1714–1722. [DOI] [PubMed] [Google Scholar]
- Song SK, Sun SW, Ramsbottom MJ, Chang C, Russell J, Cross AH (2002): Dysmyelination revealed through MRI as increased radial (but unchanged axial) diffusion of water. NeuroImage 17:1429–1436. [DOI] [PubMed] [Google Scholar]
- Tabrizi SJ, Langbehn DR, Leavitt BR, Roos RA, Durr A, Craufurd D, Kennard C, Hicks SL, Fox NC, Scahill RI, Borowsky B, Tobin AJ, Rosas HD, Johnson H, Reilmann R, Landwehrmeyer B, Stout JC (2009): Biological and clinical manifestations of Huntington's disease in the longitudinal TRACK‐HD study: Cross‐sectional analysis of baseline data. Lancet Neurol 8:791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabrizi SJ, Scahill RI, Durr A, Roos RA, Leavitt BR, Jones R, Landwehrmeyer GB, Fox NC, Johnson H, Hicks SL, Kennard C, Craufurd D, Frost C, Langbehn DR, Reilmann R, Stout JC, TRACK‐HD Investigators (2011): Biological and clinical changes in premanifest and early stage Huntington's disease in the TRACK‐HD study: The 12‐month longitudinal analysis. Lancet Neurol 10:31–42. [DOI] [PubMed] [Google Scholar]
- Tabrizi SJ, Scahill RI, Owen G, Durr A, Leavitt BR, Roos RA, Borowsky B, Landwehrmeyer B, Frost C, Johnson H, Craufurd D, Reilmann R, Stout JC, Langbehn DR (2013): Predictors of phenotypic progression and disease onset in premanifest and early‐stage Huntington's disease in the TRACK‐HD study: Analysis of 36‐month observational data. Lancet Neurol 12:637–649. [DOI] [PubMed] [Google Scholar]
- Thieben MJ, Duggins AJ, Good CD, Gomes L, Mahant N, Richards F, McCusker E, Frackowiak RS (2002): The distribution of structural neuropathology in pre‐clinical Huntington's disease. Brain 125:1815–1828. [DOI] [PubMed] [Google Scholar]
- Tobin JE, Xie M, Le TQ, Song SK, Armstrong RC (2011): Reduced axonopathy and enhanced remyelination after chronic demyelination in fibroblast growth factor 2 (Fgf2)‐null mice: Differential detection with diffusion tensor imaging. J Neuropathol Exp Neurol 70:157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topper R, Schwarz M, Podoll K, Domges F, Noth J (1993): Absence of frontal somatosensory evoked potentials in Huntington's disease. Brain 116:87–101. [DOI] [PubMed] [Google Scholar]
- Weaver KE, Richards TL, Liang O, Laurino MY, Samii A, Aylward EH (2009): Longitudinal diffusion tensor imaging in Huntington's Disease. Exp Neurol 216:525–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild EJ, Tabrizi SJ (2014): Targets for future clinical trials in Huntington's disease: What's in the pipeline? Mov Disord 29:1434–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf RC, Sambataro F, Vasic N, Depping MS, Thomann PA, Landwehrmeyer GB, Sussmuth SD, Orth M (2014): Abnormal resting‐state connectivity of motor and cognitive networks in early manifest Huntington's disease. Psychol Med 44:3341–3356. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information