

Abstract
Increased amygdala reactivity might lead to negative bias during emotional processing that can be reversed by antidepressant drug treatment. However, little is known on how N‐methyl‐d‐aspartate (NMDA) receptor antagonism with ketamine as a novel antidepressant drug target might modulate amygdala reactivity to emotional stimulation. Using functional magnetic resonance imaging (fMRI) and resting‐state fMRI (rsfMRI), we assessed amygdalo‐hippocampal reactivity at baseline and during pharmacological stimulation with ketamine (intravenous bolus of 0.12 mg/kg, followed by a continuous infusion of 0.25 mg/kg/h) in 23 healthy subjects that were presented with stimuli from the International Affective Picture System (IAPS). We found that ketamine reduced neural reactivity in the bilateral amygdalo‐hippocampal complex during emotional stimulation. Reduced amygdala reactivity to negative pictures was correlated to resting‐state connectivity to the pregenual anterior cingulate cortex. Interestingly, subjects experienced intensity of psychedelic alterations of consciousness during ketamine infusion predicted the reduction in neural responsivity to negative but not to positive or neutral stimuli. Our findings suggest that the pharmacological modulation of glutamate‐responsive cerebral circuits, which is associated with a shift in emotional bias and a reduction of amygdalo‐hippocampal reactivity to emotional stimuli, represents an early biomechanism to restore parts of the disrupted neurobehavioral homeostasis in MDD patients. Hum Brain Mapp 37:1941–1952, 2016. © 2016 Wiley Periodicals, Inc.
Keywords: ketamine, antidepressants, glutamate, depression, emotional processing, fMRI
INTRODUCTION
The amygdala is generally implicated in coordinating the function of cortical networks during evaluation of the biological significance of affective stimuli [Herry et al., 2007; Pessoa and Adolphs, 2010]. During associative learning processes, interactions between sensory cortices and the amygdala are modulated by input from hippocampal and orbitofrontal cortical projections [Kringelbach and Rolls, 2004; Sarter et al., 2001]. Due to extensive connections, not only the amgdalae but also the hippocampus and parahippocampal gyri show extended significant activation during emotional stimulation that conveys information regarding environmental contexts that hold emotional salience [Aldhafeeri et al., 2012]. The amygdala and its projection to the anterior cingulate cortex (ACC) form another important affective neurocircuitry for mood regulation since it allows for downregulation of the amygdala [Drevets, 2003]. In general, an “affective subdivision” encompassing pregenual (pgACC) and subgenual areas (sgACC) and a dorsal “cognitive subdivision” of the ACC (dACC) have been distinguished [Devinsky et al., 1995; Pizzagalli, 2011]. The extended network formed by these structures integrates limbic and sensory input as part of assessing stimulus valence within a particular context. As a major hub of the default‐mode network (DMN), pgACC activity appears to be suppressed during demanding tasks for adaptive regulation of cognition [Pizzagalli, 2011]. Increased resting pgACC activity as well as functional coupling between the pgACC and amygdala are observed during resting states [Margulies et al., 2007] and self‐referential processing [Schmitz and Johnson, 2006]. The failure to deactivate the pgACC and amygdala during affective and cognitive challenges represents important components of frontocingulate dysfunction linked to maladaptive emotional and self‐focused processing [Pizzagalli, 2011].
Current pathophysiological models suggest that specifically overactivation of this affective processing network, in combination with a failing modulatory influence from prefrontal areas, may underlie the negative perceptual and mnemonic processing biases typically seen in major depressive disorder (MDD) [Beck, 2008; Drevets, 2000; Elliott et al., 2011; Mayberg, 1997; Phillips et al., 2003]. Relative to controls, depressed patients show stronger functional coupling between the pgACC and the amygdala during negative self‐referential processing [Yoshimura et al., 2010] as well as reduced structural connectivity between these two regions [Cullen et al., 2010]. Compared to healthy subjects, functional connectivity of the right amygdala with the hippocampus was increased in depressed patients during encoding of subsequently remembered negative but not neutral or positive stimuli [Hamilton and Gotlib, 2008]. A recent voxel‐wise meta‐analysis of studies using functional magnetic resonance imaging (fMRI) to estimate neural response to negative and/or positive stimuli confirmed that individuals diagnosed with MDD had reliably greater responses to negative stimuli in the amygdala [Hamilton et al., 2012]. This is further supported by a recent study linking cognitive vulnerability to depression with hyperactivation of the amygdala during matching of negative facial expressions [Zhong et al., 2011].
Several studies have reported reductions in pathologically exaggerated amygdala activation [Delaveau et al., 2011; Fu et al., 2004; Sheline et al., 2001; Victor et al., 2010] and changes in amygdalar–cortical connectivity [Chen et al., 2008; McCabe and Mishor, 2011] with antidepressant treatment. Decreased amygdala responses during emotional stimulation were also observed in healthy subjects following subchronic administration of citalopram [Harmer et al., 2006], (S)‐citalopram [Sladky et al., 2015], and duloxetine [van Marle et al., 2011]. These results suggest that serotonergic antidepressants attenuate the bottom–up processing of biologically salient information in an extended amygdala circuitry, while at the same time possibly potentiating the effective communication between its subparts [van Marle et al., 2011]. However, little is known about how nonserotonergic antidepressants affect the function of limbic brain structures and whether its therapeutic effects can be assessed on the neurocircuitry level by pharmacological imaging.
The N‐methyl‐d‐aspartate (NMDA) receptor antagonist ketamine has been shown to exhibit antidepressant properties and is currently used as a probe for the investigation of novel antidepressant drug targets [Aan Het Rot et al., 2012; Machado‐Vieira et al., 2009] since it shows a much faster onset of action and is particularly efficacious in otherwise treatment resistant patients [Aan Het Rot et al., 2010; Murrough et al., 2013; Zarate et al., 2006]. However, little is known about how ketamine as an NMDA‐antagonist modulates limbic reactivity during emotional processing as a model for antidepressant action. Glutamatergic NMDA receptors are most densely located throughout hippocampal and parahippocampal structures and have rich projections through the ventral perforant pathways, for example, to amygdala and entorhinal cortex [Walter et al., 2014]. Hence, modulating glutamatergic signaling via NMDA receptors might have an impact on neural processing of emotionally relevant information in the limbic system. A previous study examined the neural processing of facial emotion recognition in healthy subjects and found that the limbic response pattern to fearful faces was actually abolished during ketamine infusion compared to placebo [Abel et al., 2003b]. Hence, putative links among glutamatergic agents and task‐induced activity in the amygdalo–hippocampal complex during emotional stimulation have to be further elucidated.
Given the relevance of the cortico‐limbic neurocircuitry in emotional processing and its dysfunction in mood disorders, we aimed at directly probing the effect of a glutamatergic agent on functional reactivity of the amygdalo–hippocampal complex during emotional processing and cortico‐limbic resting‐state connectivity in healthy subjects. Based on the previously mentioned findings, we hypothesized that during ketamine infusion reactivity in the amygdalo–hippocampal complex will decrease during processing of negative relative to positive valenced stimuli. Based on cortico‐limbic neurocircuitry models of mood regulation, we further hypothesized that reductions in amygdalo–hippocampal reactivity are reflected by modulatory changes in functional connectivity to the pgACC.
MATERIALS AND METHODS
Ethics Statement
The study was approved by the University of Zurich institutional review board and all subjects gave written informed consent before screening.
Participants
Healthy subjects (n = 23, mean age, 25.5 ± 5 y [SD]; 12 males) without any psychiatric, neurological, or medical illness were self‐referred from online study advertisements. All subjects underwent a psychiatric interview and medical examination at the Internal Medicine Service of the Zurich University Hospital for Psychiatry. Only medication‐free subjects that were healthy according to physical examination, electrocardiogram, blood, and urine analyses were included in the study. Exclusion criteria were any present or previous history of psychiatric/neurological diseases, drug abuse, concurrent medication, cardiovascular disease, anemia, thyroid disease, any somatic disease affecting drug metabolism and excretion (e.g., renal or liver disease), MR exclusion criteria, pregnancy, and left‐handedness.
Experimental Design
Subjects completed two separate fMRI sessions on a Philips Achieva 3 T whole‐body MR unit equipped with an 8‐channel head coil (Philips Medical Systems, Best, The Netherlands). The first session served as physiological baseline and subjects were not infused with any pharmacological agent. In the second session, S‐ketamine (Ketanest® S, Pfizer, Zurich, Switzerland) was administered as an intravenous bolus of 0.12 mg/kg approximately 15 min prior to the fMRI task for rapid induction of an altered state of consciousness, followed by a continuous infusion of 0.25 mg/kg/h during the entire MR scanning period. Previous clinical trials mostly used an i.v. dose of 0.5 mg/kg of racemic ketamine (R/S enantiomer ratio of 1:1). In this study, the S(+)‐isomer of ketamine was used due to its 3–4 times higher affinity or potency at NMDA receptors that are critically involved in its antidepressant action, thus a dose reduction of 50% is recommended [Sinner and Graf, 2008]. The interval between the sessions was 3 days on average.
During the fMRI task, subjects were presented in a randomized manner with different versions of matched sets of photographs from the International Affective Picture System (IAPS) (Lang et al., 2008). To maintain attention during the entire task period, subjects were instructed to continuously rate the pictures by button press according to their valence. In total, 90 IAPS pictures (30 stimuli per positive, negative, and neutral valence) were presented in a blocked design (5 blocks per valence in pseudorandomized order, 5–7 pictures per block, block durations: 21–27 s). Each block was followed by resting periods (fixation cross, duration: 9 s). The stronger effect size associated with experiments using a block design compared to those using an event‐related design supports the notion that block designs are more efficient from a statistical point of view in terms of its sensitivity to detect small signal changes [Josephs and Henson, 1999]. To control for repetition or habituation effects, test–retest fMRI data were obtained in an independent sample of n = 16 subjects that completed two baseline sessions of the IAPS task without pharmacological intervention.
Each session contained a resting‐state fMRI (rsfMRI) scan to assess changes in functional connectivity approximately 25 min after the start of the ketamine infusion. During the acquisition of resting state, data subjects were told to lie still in the scanner with their eyes closed. The functional images were collected in 10 min runs (200 volumes).
MRI Data Acquisition
Functional time series were acquired with a sensitivity‐encoded single‐shot echo‐planar imaging sequence (SENSE‐sshEPI) (Schmidt et al., 2005). The following acquisition parameters were used in the fMRI protocol: TE = 35 ms, TR = 3000 ms (=82°), FOV = 22 cm, acquisition matrix = 80 × 80 interpolated to 128 × 128, voxel size = 2.75 × 2.75 × 4 mm, 32 contiguous axial slices (placed along the anterior–posterior commissure plane), and sensitivity‐encoded acceleration factor R = 2.0. A 3‐dimensional T1‐weighted anatomical scan was obtained for structural reference (T1 3D FFE sequence: TR/TE = 9.3/4.6 ms, flip angle = 8°, 160 sagittal slices, FOV 240 × 240 mm, voxel size 1 × 1 × 1 mm).
fMRI Data Analysis
The functional imaging data were analyzed using SPM8 (http://www.fil.ion.ucl.ac.uk/spm /). Functional data were corrected for motion artifacts by realignment to the mean image, mean‐adjusted by proportional scaling, normalized according to the unified segmentation normalization approach (2 × 2 × 2 mm), and spatially smoothed using a 6‐mm full‐width at half‐maximum Gaussian kernel. Statistical analysis was performed by modeling the different conditions (fixation cross; positive, negative, and neutral pictures), convolved with a hemodynamic response function as explanatory variables within the context of the general linear model on a voxel‐by‐voxel basis. Realignment parameters were included as additional regressors in the statistical model. A fixed‐effect model at a single‐subject level was performed to create images of parameter estimates, which were then entered into a second‐level random effects analysis. For the fMRI data group analysis, the contrast images from the analysis of the individual participants were analyzed using paired t tests. Clusters of activation were identified with a global height threshold of p < 0.05 (FDR‐corrected) and a minimal threshold of k > 100 voxels.
For the ROI analysis, mean %‐signal changes for the different conditions (positive, negative, neutral, fixcross) were extracted for each subject separately using Marsbar (Brett et al., 2002). Based on our hypothesis of ketamine‐associated reduction of limbic reactivity, we extracted %‐BOLD signal changes in a priori determined regions of interest (ROIs) including the pregenual anterior cingulate cortex (sphere at ± 0 38 0 with 6 mm radius), the bilateral amygdala, as well as the bilateral hippocampus taken from the AAL atlas (Tzourio‐Mazoyer et al., 2002). The effect of ketamine on task‐induced BOLD responses within these ROIs was analyzed using repeated measures ANOVAs with the factors valence (positive, negative, neutral) and treatment (baseline, ketamine). If ANOVAs revealed significant main or interaction effects, further statistical analyses were conducted using post hoc t‐tests. All tests were two‐tailed and the significant threshold was set at a probability of p ≤ 0.05. All statistical analyses were carried out using SPSS v. 19 (SPSS Inc. Chicago, IL, USA). Whole‐brain paired t significance images were thresholded in SPM8 and visualized with MRIcron (http://www.mccauslandcenter.sc.edu/mricro/mricron /). Graphs of the mean changes in %‐BOLD signal change were created using SigmaPlot v. 12 (Systat Software Inc. Chicago, IL, USA).
Resting‐State fMRI Data Analysis
Resting‐state data were analyzed using an SPM8‐based processing assistant for rsfMRI (DPARSF, Yan Chao‐Gan, State Key Laboratory of Cognitive Neuroscience and Learning, Beijing Normal University, China), which includes an rsfMRI data analysis toolkit (REST, Song Xiao‐Wei et al., 2011). The preprocessing steps followed the standard protocol described by Yan and Zang (2010). Functional resting‐state data was corrected for differences in slice acquisition time, motion‐corrected using a least squares approach and a six‐parameter (rigid body) linear transformation, spatially normalized (to 3 × 3 × 3 mm isovoxels in standard space) and smoothed using a 4‐mm fill‐width‐at‐half‐maximum Gaussian kernel. The data was linearly detrended and filtered by a band pass filter (0.01–0.08 Hz) to suppress cardiac and respiratory motion induced effects. An additional regression of nuisance covariates was applied during which the functional data was corrected for six head movement parameters and for global mean signal as well for white matter and cerebrospinal fluid signal (defined according to Yan and Zang, 2010). To specifically address changes in corticolimbic connectivity, we limited our analysis to the a priori defined seed regions from the fMRI analysis including the pregenual anterior cingulate cortex (sphere at ± 0 38 0 with 6 mm radius) as well as the bilateral hippocampus and amygdala, taken from the Automated Anatomical Labeling (AAL) atlas [55]. Statistical tests on ROI‐based functional connectivities were computed after application of Fisher's r‐to‐z transform, which yields variates that are approximately normally distributed. To test for effects of ketamine on resting‐state connectivity, the same ANOVA was used as for the fMRI data.
Psychometric Measures
Reaction times of all fMRI task responses were analyzed using two‐way repeated measures analyses of variance (ANOVAs) with the factors valence (positive, negative, and neutral) and treatment (baseline and ketamine). If ANOVAs revealed significant main or interaction effects, further statistical analyses were conducted using post hoc t‐tests. All tests were two‐tailed and the significance threshold was set at a probability of p ≤ 0.05. All statistical analyses were carried out using PASW (Predicitive Analysis SoftWare, Version 18.0, Chicago: SPSS Inc., Illinois, USA).
Subjective state and mood were assessed before and after the scanning sessions using the state‐trait anxiety inventory (STAI X1) (Spielberger et al., 1970). Subjective psychological effects during ketamine infusion were assessed post hoc using the five‐dimensional altered states of consciousness (5D‐ASC) self‐rating scale (Dittrich, 1998), that was shown to reliably measure ketamine‐induced altered states of consciousness (Vollenweider and Kometer, 2010). Subjects had to indicate the subjective alteration of consciousness compared to their general condition on a visual analogue scale. Individual 5D‐ASC scores were correlated with BOLD signal changes using Pearson correlation analysis. Subjects were kept under surveillance for at least 40 min after treatment until psychotomimetic side effects subsided.
RESULTS
Behavioral and Psychometric Measures
Behavioral data revealed significantly faster reaction times during the ketamine compared to the baseline session [F(1,21) = 8.33, p = .009, η 2 = 0.284].
Anxiety levels (STAI X1 ratings) did not differ significantly from pre‐ (35.47 ± 9.12) to postinfusion (33.96 ± 9.15) [paired t test, n = 23, p = 0.541]. Psychedelic alterations of consciousness during ketamine infusion were assessed post‐hoc using the Altered States of Consciousness self‐rating scale (5D‐ASC) containing 94 items (Dittrich, 1998). Subjects had to indicate the subjective alteration of consciousness compared to their general condition on a visual analog scale. Ketamine treatment increased scores in the scales of oceanic boundlesness (OBL: 39.65 ± 1.39% [SD]), reduction of vigilance (VR: 37.67 ± 1.19%), anxious ego‐dissolution (AED: 18.69 ± 1.17%), visionary restructuralization (VRE: 29.56 ± 1.36%), and acoustic alterations (AA: 14.22 ± 0.8%). Additional correlation analyses revealed a significant correlation between reaction times and increased scores in the scales of oceanic boundlessness in the ketamine condition (n = 20, r = −0.637, p = 0.003; Fig. 1).
Figure 1.
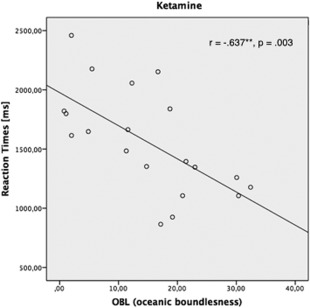
Significant negative correlation between reaction times and increased scores in the scales of oceanic boundlessness in the ketamine condition (n = 20, r = −0.637, p = 0.003).
fMRI Data
Pairwise comparison of the whole‐brain statistical parametric maps of the baseline and ketamine sessions (paired t test, n = 23, baseline > ketamine, p < 0.05, FDR‐corrected, k > 100) revealed drug‐induced BOLD signal reductions in bilateral amygdala, hippocampus, calcarine, fusiform, lingual, and parahippocampal gyrus, superior occipital gyrus, superior and inferior parietal lobule, and precentral gyrus (Table 1). At the whole‐brain level, BOLD signal reductions in the amygdalo–hippocampal complex were more pronounced for negative and neutral compared to positive stimuli after ketamine administration (Fig. 2). No brain region showed significant drug‐induced increases in BOLD signal reactivity. Test–retest baseline fMRI data from an independent sample of n = 16 healthy subjects did not reveal any significant habituation effects due to repeated exposure to the behavioral paradigm at the whole brain level.
Table 1.
Brain regions with significant BOLD signal reductions after ketamine administration compared to baseline
| Region | Side | Coordinates X Y Z | k | T | Z | Contrast |
|---|---|---|---|---|---|---|
| Calcarine, fusiform, lingual, hippocampal and parahippocampal gyrus | L/R | −10 −82 −8 | 9618 | 7.40 | 5.19 | Positive > fixation |
| Superior occipital gyrus | R | 28 −84 22 | 172 | 5.52 | 4.33 | |
| Precentral gyrus | L | −26 −22 66 | 193 | 4.89 | 3.98 | |
| Hippocampus | R | 38 −12 −16 | 121 | 4.41 | 3.69 | |
| Superior parietal gyrus | R | 16 −58 66 | 228 | 6.13 | 4.63 | Negative > fixation |
| Amygdala | L | −28 −10 −14 | 759 | 5.96 | 4.55 | |
| Hippocampus, calcarine, fusiform and lingual gyrus | L/R | 38 −20 −18 | 3393 | 5.50 | 4.32 | |
| Inferior parietal lobule | R | 46 −18 38 | 350 | 5.11 | 4.11 | |
| Calcarine and lingual gyrus | L/R | 36 −50 −12 | 536 | 4.75 | 3.90 | |
| Calcarine, fusiform, and lingual gyrus | L/R | −12 −78 10 | 6448 | 5.78 | 4.46 | Neutral > fixation |
| Hippocampus | R | 30 −6 −12 | 165 | 5.52 | 4.33 | |
| Hippocampus | L | −32 −10 −14 | 735 | 5.03 | 4.06 |
The cluster with the largest number of voxels within each region is reported for the different task conditions (positive, negative, and neutral stimuli). The MNI coordinates for the centroid of each cluster are presented. Significant voxel clusters were identified by paired t test (baseline > ketamine; n = 23). The whole‐brain probability threshold was set to p < 0.05, FDR‐corrected, k > 100.
Figure 2.
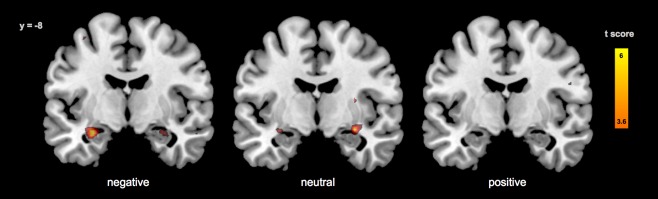
Reduced BOLD reactivity in the amygdalo‐hippocampal complex during processing of positive, negative, and neutral stimuli after ketamine administration compared to baseline (whole brain paired t test, n = 23, FDR‐corrected, k > 25).
ROI analyses confirmed ketamine‐associated BOLD signal changes with significant effects of treatment in right [F(1,22) = 9.77, p = 0.005, η 2 = 0.308] and left amygdala [F(1,22) = 16.88, p < 0.001, η 2 = 0.434] as well as right [F(1,22) = 6.84, p = 0.016, η 2 = 0.237] and left hippocampus [F(1,22) = 11.67, p = 0.002, η 2 = 0.347]. No significant effect of treatment could be found in the pregenual anterior cingulate cortex. Furthermore, there was a significant effect of emotional valence on BOLD signal changes in all investigated ROIs [right amygdala: F(2,21) = 7.03, p = 0.020, η 2 = 0.310; left amygdala: F(2,21) = 7.57, p = 0.003, η 2 = 0.419; right hippocampus: F(2,21) = 5.35, p = 0.013, η 2 = 0.338; left hippocampus: F(2,21) = 9.08, p = 0.001, η 2 = 0.464; pregenual anterior cingulate cortex: F(2,21) = 12.47, p < 0.001, η 2 = 0.543]. Post‐hoc comparisons revealed significant BOLD signal reductions after ketamine as well as higher BOLD signal changes after positive and negative compared to neutral stimuli in the respective ROIs (Fig. 3; for detailed statistics, see Table 2). There was no significant interaction between treatment and emotional valence in the investigated ROIs. Additional repeated measures ANOVA and pairwise comparisons of the test–retest fMRI datasets did not reveal any significant changes in the amygdalo–hippocampal ROIs between two repeated baseline measurements (Supporting Information), indicating that the BOLD signal differences that we observe here are likely due to pharmacological intervention and not a result of repeated task exposure.
Figure 3.
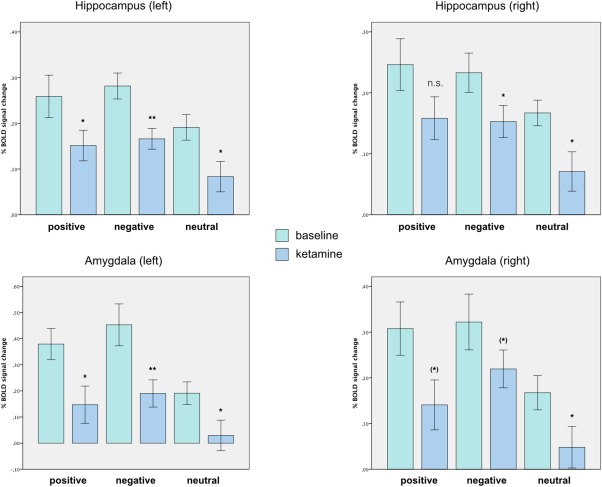
Reduced BOLD reactivity to positive, negative, and neutral stimuli in the bilateral amygdala and hippocampus (extracted using AAL MNI ROI library and Marsbar SPM8 ROI toolbox). Bars indicate task‐specific %‐BOLD signal changes (positive, negative, and neutral) for the baseline (green) and ketamine (blue) session (paired t test, n = 23, error bars: ±SEM). [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Table 2.
Effect of ketamine on %‐BOLD signal change during processing of positive, negative, and neutral pictures
| Valence | Region of interest (ROI) | Side | Mean difference | t value | p value |
|---|---|---|---|---|---|
| Positive | Amygdala | L | 0.23502 | 2.817 | 0.010* |
| R | 0.1678 | 2.006 | 0.057(*) | ||
| Hippocampus | L | 0.10744 | 2.268 | 0.033* | |
| R | 0.08781 | 1.542 | 0.137 | ||
| Negative | Amygdala | L | 0.26227 | 3.928 | 0.001** |
| R | 0.09999 | 1.804 | 0.085(*) | ||
| Hippocampus | L | 0.116 | 4.051 | 0.001** | |
| R | 0.08054 | 2.405 | 0.025* | ||
| Neutral | Amygdala | L | 0.16171 | 2.23 | 0.036* |
| R | 0.11753 | 2.159 | 0.042* | ||
| Hippocampus | L | 0.10779 | 2.186 | 0.040* | |
| R | 0.09632 | 2.145 | 0.043* |
Values indicate mean differences, t values, and p values for the pairwise comparison (n = 23) of task‐induced BOLD reductions from baseline to ketamine. %‐BOLD signal from the ROIs was obtained using Marsbar and the AAL library [Brett et al., 2002].
Correlation Analyses
A significant positive relationship between psychedelic alterations of consciousness and ketamine‐related %‐BOLD signal reductions to negative stimuli were found in the left and right amygdala. After exclusion of an outlier that showed very high self‐rating scores in all subscales and thus was biasing the results toward high correlation coefficients, we still found a significant positive relationship (Fig. 4) between psychedelic alterations of consciousness and ketamine‐related %‐BOLD signal reductions to negative stimuli in left (n = 22; OBL: r = 0.582, p = 0.004; VRE: r = 0.556, p = 0.007; AED: r = 0.446, p = 0.037; VR: r = 0.425, p = 0.049) and right amygdala (n = 22; OBL: r = 0.477, p = 0.025; AED: r = 0.428, p = 0.047) as well as in the left hippocampus (AA: n = 22; r = 0.588, p = 0.004). While there were no correlations between psychedelic alterations of consciousness and ketamine‐related %‐BOLD signal changes to positive and neutral stimuli, further statistical comparison of correlation coefficients revealed significant differences between valences exclusively for the left amygdala and hippocampus (n = 22; p < 0.05). Here, ketamine‐related %‐BOLD signal reductions to negative stimuli were associated with changes in OBL and AA.
Figure 4.
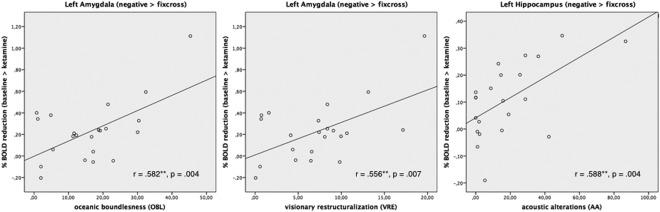
Significant positive correlations between %BOLD signal reductions to negative stimuli and psychedelic alterations of consciousness for the left amygdala and the left hippocampus.
Resting‐State Functional Connectivity
Additional resting‐state analysis including seed regions for the pgACC, the bilateral hippocampus, and bilateral amygdala did not reveal any significant effect of drug on cortico‐limbic functional connectivities. For further correlation analyses, the %BOLD signal changes to positive, negative, and neutral pictures under baseline and ketamine conditions were related to the cortico‐limbic connectivity measures. Only for %BOLD signal changes to negative pictures a significant positive correlation with functional connectivities to the pregenual cingulate cortex was found for both amygdalae during the ketamine condition (AMG left: n = 23, r = 0.530, p = 0.009; AMG right: n = 23, r = 0.501, p = 0.015; Fig. 5). No significant correlations were found for the positive or neutral stimuli in the ketamine condition. During the baseline condition, no correlations were found for all emotional valences.
Figure 5.
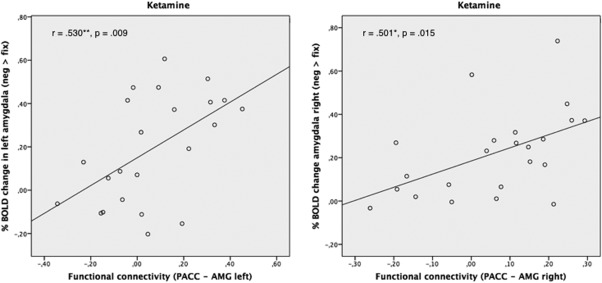
Significant positive correlation between %BOLD signal changes to negative pictures in both amygdalae with functional connectivities to the pgACC during the ketamine condition (AMG left: n = 23, r = 0.530, p = 0.009; AMG right: n = 23, r = 0.501, p = 0.015).
DISCUSSION
In this study, we found that ketamine attenuated task‐induced activation in the amygdalo–hippocampal complex during an emotional processing task. Specifically, reduction in BOLD reactivity was more marked in response to negative compared to neutral or positive pictures but did not reach statistical significance for a valence‐specific effect of ketamine on amygdal–hippocampal reactivity. However, the experienced intensity of psychedelic alterations of consciousness during ketamine infusion was related to the ketamine‐induced attenuation of amygdalo–hippocampal reactivity in response to negative but not to positive or neutral stimuli. These findings are complemented by the observation that during ketamine infusion BOLD reactivity of the amygdala was positively correlated with functional connectivity to the pgACC during processing of negative but not positive or neutral stimuli. This suggests that the processing of negative information is specifically altered in response to ketamine both on the experiential as well as at the neurocircuitry level.
Our results of attenuated amygdala reactivity after acute intravenous administration of ketamine are in line with previous findings of relative attenuation of amygdala reactivity after administration of serotonergic antidepressants in healthy subjects [Harmer et al., 2006; Henry et al., 2013; Outhred et al., 2014; Sladky et al., 2015; van Marle et al., 2011]. Two weeks of administration of the combined serotonin and norepinephrine reuptake inhibitor duloxetine reduced neural responses in affect processing regions including the amygdala and the ventral aspect of the anterior cingulate cortex, while functional coupling between the amygdala and the anterior insula was enhanced by the drug [van Marle et al., 2011]. In another recent study, modulatory effects of (S)‐citalopram on cortico‐limbic connections could be demonstrated in healthy subjects by connectivity methods revealing an increased inhibition of amygdala activation by the orbitofrontal cortex [Sladky et al., 2015]. This supports the notion that effects of antidepressants on cortico‐limbic circuitry can be assessed even in healthy subjects to investigate the neural mechanisms of antidepressant treatment response. Correspondingly, we found that acute NMDA‐receptor blockade with ketamine attenuates the reactivity of the human amygdalo–hippocampal complex during emotional processing, while functional coupling between the amgdala and the pregenual cingulate cortex was correlated to the BOLD reactivity of the amygdala to negative emotional valence specifically. These findings suggest an attenuation of bottom–up processing of biologically salient information in an extended cortico‐limbic neurocircuitry, while at the same time possibly potentiating the effective communication between its subparts. This is further supported by two recent magnetoencephalography (MEG) studies probing pretreatment activity and functional connectivity of the anterior cingulate cortex and the amygdala as potential biomarkers of treatment response to ketamine [Salvadore et al., 2009; Salvadore et al., 2010]. In MDD, reductions in depressive symptoms were associated with increased functional connectivity between frontocingulate regions and the amygdala [Anand et al., 2005; Chen et al., 2007; Kong et al., 2013] and decreased fMRI amygdalar activation to negative stimuli [Anand et al., 2007]. Recently, ketamine‐induced increases in functional connectivity were also observed in the cortico‐hippocampal networks that are vulnerable to mood and cognitive disorders [Khalili‐Mahani et al., 2015]. Overall, rebalancing subcortical–cortical network dynamics seems to emerge as a physiologic profile shared by various subtypes of antidepressant interventions. Based on these findings and the high density of NMDA‐receptors in the amygdalo–hippocampal complex, we hypothesize that the rapid onset of relief from depressive symptoms following NMDA‐receptor antagonism with ketamine might rely on the rapid decrease in amygdalo–hippocampal reactivity, while chronic blockade of serotonergic neurotransmission might be linked to more complex adaptive mechanisms involving the extended amygdala network as suggested by the prolonged onset of action of SSRIs.
However, besides the amygdala and hippocampus as main target regions, ketamine also reduced BOLD reactivity in other task‐relevant brain regions such as the bilateral calcarine, lingual, and fusiform gyrus, as well as the precentral, parietal, and occipital gyrus. Recently, a revised view of the amygdala was proposed by Pessoa et al. highlighting the crucial role of the amygdala in coordinating of cortical visual networks during the evaluation of the biological significance of affective visual stimuli. Feedback loops between subcortical and cortical visual networks synchronize the neuronal profile of activation throughout the visual cortex, and likewise the amygdala responses [Pessoa and Adolphs, 2010]. Hence, more widespread reductions in cortico–subcortical networks are likely to be expected in functionally coupled systems following pharmacological perturbations. On the other hand, a disrupted neurovascular coupling under NMDA‐receptor antagonism may render the interpretation of the effects of ketamine more difficult [Iannetti and Wise, 2007]. Interestingly, a recent pharmacological MRI study comes to the conclusion that subanesthetic doses of ketamine in humans do not appear to produce a disturbed coupling between cerebral blood flow and metabolism [De Simoni et al., 2013]. Another PET study concludes that ketamine‐induced increases in cerebral blood flow parallel global increases in glucose metabolism, hence ketamine‐induced disturbance of coupling between cerebral blood flow and metabolism is highly unlikely [Långsjö et al., 2004]. Together with the fact that ketamine induces primarily focal and task‐dependent BOLD changes [Abel et al., 2003a], this indicates that BOLD changes likely correspond to underlying neuronal activity.
Otherwise, a reduced limbic reactivity to emotional stimuli is also well compatible with the symptom cluster of emotional blunting that is frequently associated with the dissociative mental state during ketamine infusion. An fMRI study using a facial recognition task found that the normal pattern of neural response to fearful faces involving limbic and visual cortex activation was partially abolished by ketamine in healthy subjects [Abel et al., 2003b]. These authors conclude that emotional blunting during ketamine infusion may be associated with reduced limbic responses to emotional stimuli. Interestingly, in our study, reaction times as behavioral response to the pictorial stimuli were significantly faster during ketamine infusion and positively correlated with increased scores in the scales of oceanic boundlessness. Hence, dissolving the ego‐boundaries with ketamine might influence behavioral performance by reducing the complexity of information processing that is related to self‐referential processes. Another phenomenologically based assumption would be that a reduction of limbic reactivity might indeed be experienced as emotional blunting in healthy subjects, while in depressed patients, the attenuation of limbic hyperactivity might in contrast be experienced as alleviation of negative emotional bias. Although the reduction of limbic reactivity was more pronounced to negative compared to positive or neutral pictures at the whole‐brain level, our findings cannot support the hypothesis of a valence‐specific ketamine effect since additional ROI analyses did not reveal any significant drug‐by‐valence interaction. This suggests a more general shift in emotional processing on the level of neurodynamic reactivity also including attenuation to neutral stimuli. However, attenuated amygdalo–hippocampal reactions to negative (but not to positive or neutral stimuli) were predicted by the experienced intensity of psychedelic alterations of consciousness during ketamine infusion. This close relationship between reduced amygdala reactivity to negative stimuli and psychedelic experiences such as “oceanic boundlessness” (referring to positively experienced loss of ego boundaries that are associated with changes in the sense of time and emotions) and “visionary restructuralization” (referring to altered meaning of percepts) raises interesting new hypotheses regarding the therapeutic mechanisms of psychedelics and their clinical applications [Vollenweider and Kometer, 2010]. For instance, it was shown that the 5‐HT2A serotonin receptor agonist psilocybin biases emotional processing toward positive relative to negative information [Kometer et al., 2012]. Correspondingly, amygdala reactivity to negative and neutral stimuli was lowered by the administration of psilocybin compared to placebo [Kraehenmann et al., 2014]. Drug‐induced psychological experience might thus have an essential facilitatory effect that enables neurobehavioral change in patients suffering from severe mood disorders: a transitory attenuation of limbic hyper‐reactivity combined with altered perceptions may promote a change in mental perspective that alleviates depressed mood [Holtzheimer and Mayberg, 2011].
In conclusion, our results of reduced amygdalo–hippocampal reactivity suggest a putative neural mechanism through which ketamine could normalize typical negativity biases in depression. In line with pharmacological imaging studies that mainly report a reduction in amygdala and perihippocampal reactivity following different antidepressant drugs both in healthy and depressed subjects [Abel et al., 2003b; Chen et al., 2008; Delaveau et al., 2011; Fu et al., 2004; McCabe and Mishor, 2011; Sheline et al., 2001; van Marle et al., 2011; Victor et al., 2010], our findings support the notion that ketamine has a comparable potential to attenuate limbic hyperactivity. Clearly, further studies are needed to explore whether ketamine may also induce long‐lasting adaptive processes that mediate the sustained antidepressant effect. Interestingly, a recent translational imaging study demonstrates a cross‐species consistency of ketamine‐induced increases in prefrontal–hippocampal functional coupling in rats and healthy subjects [Grimm et al., 2015]. An increased prefrontal–hippocampal connectivity 20 min post ketamine infusion as reported by this study supports the notion of a dynamic reorganization of functional networks as a result of downstream effects of ketamine on synaptic plasticity [Duman and Aghajanian, 2012]. In contrast, we recently reported a subacute modulation in resting‐state functional connectivity in healthy subjects 24 h following intravenous ketamine administration [Scheidegger et al., 2012] that temporally aligns with the peak of efficacy in treatment resistant depression [Aan Het Rot et al., 2012; Zarate et al., 2006]. Hence, it is likely that functional connectivity patterns show multistage effects in the temporal domain and further studies are needed to disentangle the homeostatic adaptations in the different subsystems. Correspondingly, the relevance of an acute reduction in limbic reactivity as an early biomechanism of antidepressant drug action has to be evaluated in postchallenge fMRI studies. Recently, the neural effects of ketamine in depressed patients were examined using two different emotion perception tasks 24 h following administration of a single intravenous dose of ketamine [Murrough et al., 2015]. The authors did not report any changes in limbic reactivity but show that ketamine specifically enhances neural responses to positive emotion within the right caudate in depressed individuals [Murrough et al., 2015]. Hence, generalizability of our findings to MDD patients is limited. As an exploratory model based on observations in healthy subjects, it has to be further confirmed in randomized‐controlled clinical trials in MDD patients receiving ketamine.
Supporting information
Supporting Information
REFERENCES
- Aan Het Rot M, Collins KA, Murrough JW, Perez AM, Reich DL, Charney DS, Mathew SJ (2010): Safety and efficacy of repeated‐dose intravenous ketamine for treatment‐resistant depression. Biol Psychiatry 67:139–145. [DOI] [PubMed] [Google Scholar]
- Aan Het Rot M, Zarate CA, Charney DS, Mathew SJ (2012): Ketamine for depression: Where do we go from here? Biol Psychiatry 72:537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abel KM, Allin MPG, Kucharska‐Pietura K, Andrew C, Williams S, David AS, Phillips ML (2003a): Ketamine and fMRI BOLD signal: distinguishing between effects mediated by change in blood flow versus change in cognitive state. Hum Brain Mapp 18:135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abel KM, Allin MPG, Kucharska‐Pietura K, David A, Andrew C, Williams S, Brammer MJ, Phillips ML (2003b): Ketamine alters neural processing of facial emotion recognition in healthy men: an fMRI study. Neuroreport 14:387–391. [DOI] [PubMed] [Google Scholar]
- Aldhafeeri FM, Mackenzie I, Kay T, Alghamdi J, Sluming V (2012): Regional brain responses to pleasant and unpleasant IAPS pictures: different networks. Neurosci Lett 512:94–98. [DOI] [PubMed] [Google Scholar]
- Anand A, Li Y, Wang Y, Gardner K, Lowe MJ (2007): Reciprocal effects of antidepressant treatment on activity and connectivity of the mood regulating circuit: an FMRI study. J Neuropsychiatry Clin Neurosci 19:274–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand A, Li Y, Wang Y, Wu J, Gao S, Bukhari L, Mathews VP, Kalnin A, Lowe MJ (2005): Activity and connectivity of brain mood regulating circuit in depression: a functional magnetic resonance study. BPS 57:1079–1088. [DOI] [PubMed] [Google Scholar]
- Beck AT (2008): The evolution of the cognitive model of depression and its neurobiological correlates. Am J Psychiatry 165:969–977. [DOI] [PubMed] [Google Scholar]
- Brett M, Anton J, Valabregue R, Poline J (2002): Region of interest analysis using an SPM toolbox. Presented at the 8th International Conferance on Functional Mapping of the Human Brain, June 2–6, 2002, Sendai, Japan: Available on CD‐ROM in NeuroImage, Vol. 16, No. 2, abstract 497.
- Chao‐Gan Y, Yu‐Feng Z (2010): DPARSF: A MATLAB Toolbox for “Pipeline” Data Analysis of Resting‐State fMRI. Front Syst Neurosci 4:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C‐H, Ridler K, Suckling J, Williams S, Fu CHY, Merlo‐Pich E, Bullmore E (2007): Brain imaging correlates of depressive symptom severity and predictors of symptom improvement after antidepressant treatment. Biol Psychiatry 62:407–414. [DOI] [PubMed] [Google Scholar]
- Chen C‐H, Suckling J, Ooi C, Fu CHY, Williams SCR, Walsh ND, Mitterschiffthaler MT, Pich EM, Bullmore E (2008): Functional coupling of the amygdala in depressed patients treated with antidepressant medication. Neuropsychopharmacology 33:1909–1918. [DOI] [PubMed] [Google Scholar]
- Cullen KR, Klimes‐Dougan B, Muetzel R, Mueller BA, Camchong J, Houri A, Kurma S, Lim KO (2010): Altered white matter microstructure in adolescents with major depression: a preliminary study. Ed. Ben J Harrison. J Am Acad Child Adolesc Psychiatry 49:173–183.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Simoni S, Schwarz AJ, O'Daly OG, Marquand AF, Brittain C, Gonzales C, Stephenson S, Williams SCR, Mehta MA (2013): Test‐retest reliability of the BOLD pharmacological MRI response to ketamine in healthy volunteers. NeuroImage 64:75–90. [DOI] [PubMed] [Google Scholar]
- Delaveau P, Jabourian M, Lemogne C, Guionnet S, Bergouignan L, Fossati P (2011): Brain effects of antidepressants in major depression: a meta‐analysis of emotional processing studies. J Affect Disord 130:66–74. [DOI] [PubMed] [Google Scholar]
- Devinsky O, Morrell MJ, Vogt BA (1995): Contributions of anterior cingulate cortex to behaviour. Brain 118:279–306. [DOI] [PubMed] [Google Scholar]
- Dittrich A (1998): The standardized psychometric assessment of altered states of consciousness (ASCs) in humans. Pharmacopsychiatry 31 Suppl 2:80–84. [DOI] [PubMed] [Google Scholar]
- Drevets WC (2000): Functional anatomical abnormalities in limbic and prefrontal cortical structures in major depression. Prog Brain Res 126:413–431. [DOI] [PubMed] [Google Scholar]
- Drevets WC (2003): Neuroimaging abnormalities in the amygdala in mood disorders. Ann N Y Acad Sci 985:420–444. [DOI] [PubMed] [Google Scholar]
- Duman RS, Aghajanian GK (2012): Synaptic dysfunction in depression: potential therapeutic targets. Science 338:68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott R, Zahn R, Deakin JFW, Anderson IM (2011): Affective cognition and its disruption in mood disorders. Neuropsychopharmacology 36:153–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu CHY, Williams SCR, Cleare AJ, Brammer MJ, Walsh ND, Kim J, Andrew CM, Pich EM, Williams PM, Reed LJ, Mitterschiffthaler MT, Suckling J, Bullmore ET (2004): Attenuation of the neural response to sad faces in major depression by antidepressant treatment: a prospective, event‐related functional magnetic resonance imaging study. Arch Gen Psychiatry 61:877–889. [DOI] [PubMed] [Google Scholar]
- Grimm O, Gass N, Weber‐Fahr W, Sartorius A, Schenker E, Spedding M, Risterucci C, Schweiger JI, Böhringer A, Zang Z, Tost H, Schwarz AJ, Meyer‐Lindenberg A (2015): Acute ketamine challenge increases resting state prefrontal‐hippocampal connectivity in both humans and rats. Psychopharmacology 232:4231–4241. [DOI] [PubMed] [Google Scholar]
- Hamilton JP, Etkin A, Furman DJ, Lemus MG, Johnson RF, Gotlib IH (2012): Functional neuroimaging of major depressive disorder: a meta‐analysis and new integration of base line activation and neural response data. Am J Psychiatry 169:693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton JP, Gotlib IH (2008): Neural substrates of increased memory sensitivity for negative stimuli in major depression. Biol Psychiatry 63:1155–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmer CJ, Mackay CE, Reid CB, Cowen PJ, Goodwin GM (2006): Antidepressant drug treatment modifies the neural processing of nonconscious threat cues. BPS 59:816–820. [DOI] [PubMed] [Google Scholar]
- Henry ME, Lauriat TL, Lowen SB, Churchill JH, Hodgkinson CA, Goldman D, Renshaw PF (2013): Effects of citalopram and escitalopram on fMRI response to affective stimuli in healthy volunteers selected by serotonin transporter genotype. Psychiatry Res 213:217–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herry C, Bach DR, Esposito F, Di Salle F, Perrig WJ, Scheffler K, Lüthi A, Seifritz E (2007): Processing of temporal unpredictability in human and animal amygdala. J Neurosci 27:5958–5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzheimer PE, Mayberg HS (2011): Stuck in a rut: rethinking depression and its treatment. Trends Neurosci 34:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannetti GD, Wise RG (2007): BOLD functional MRI in disease and pharmacological studies: room for improvement? Magn Reson Imaging 25:978–988. [DOI] [PubMed] [Google Scholar]
- Josephs O, Henson RN (1999): Event‐related functional magnetic resonance imaging: modelling, inference and optimization. Philos Trans R Soc Lond B Biol Sci 354:1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalili‐Mahani N, Niesters M, van Osch MJ, Oitzl M, Veer I, de Rooij M, van Gerven J, van Buchem MA, Beckmann CF, Rombouts SARB Dahan A (2015): Ketamine interactions with biomarkers of stress: a randomized placebo‐controlled repeated measures resting‐state fMRI and PCASL pilot study in healthy men. NeuroImage 108:396–409. [DOI] [PubMed] [Google Scholar]
- Kometer M, Schmidt A, Bachmann R, Studerus E, Seifritz E, Vollenweider FX (2012): Psilocybin biases facial recognition, goal‐directed behavior, and mood state toward positive relative to negative emotions through different serotonergic subreceptors. Biol Psychiatry 72:898–906. [DOI] [PubMed] [Google Scholar]
- Kong L, Chen K, Tang Y, Wu F, Driesen N, Womer F, Fan G, Ren L, Jiang W, Cao Y, Blumberg H, Xu K, Wang F (2013): Functional connectivity between the amygdala and prefrontal cortex in medication‐naive individuals with major depressive disorder. J Psychiatry Neurosci 38:417–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraehenmann R, Preller KH, Scheidegger M, Pokorny T, Bosch OG, Seifritz E, Vollenweider FX (2014): Psilocybin‐induced decrease in amygdala reactivity correlates with enhanced positive mood in healthy volunteers. Biol Psychiatry 78:572–581. [DOI] [PubMed] [Google Scholar]
- Kringelbach ML, Rolls ET (2004): The functional neuroanatomy of the human orbitofrontal cortex: evidence from neuroimaging and neuropsychology. Prog Neurobiol 72:341–372. [DOI] [PubMed] [Google Scholar]
- Lang P, Bradley M, Cuthbert BN (2008): International affective picture System (IAPS): Affective ratings of pictures and instruction manual. Technical Report A‐8. University of Florida, Gainesville, FL.
- Långsjö JW, Salmi E, Kaisti KK, Aalto S, Hinkka S, Aantaa R, Oikonen V, Viljanen T, Kurki T, Silvanto M, Scheinin H (2004): Effects of subanesthetic ketamine on regional cerebral glucose metabolism in humans. Anesthesiology 100:1065–1071. [DOI] [PubMed] [Google Scholar]
- Machado‐Vieira R, Salvadore G, Diazgranados N, Zarate CA (2009): Ketamine and the next generation of antidepressants with a rapid onset of action. Pharmacol Ther 123:143–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies DS, Kelly AMC, Uddin LQ, Biswal BB, Castellanos FX, Milham MP (2007): Mapping the functional connectivity of anterior cingulate cortex. NeuroImage 37:579–588. [DOI] [PubMed] [Google Scholar]
- Mayberg HS (1997): Limbic‐cortical dysregulation: a proposed model of depression. J Neuropsychiatry Clin Neurosci 9:471–481. [DOI] [PubMed] [Google Scholar]
- McCabe C, Mishor Z (2011): Antidepressant medications reduce subcortical‐cortical resting‐state functional connectivity in healthy volunteers. NeuroImage 57:1317–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrough JW, Collins KA, Fields J, DeWilde KE, Phillips ML, Mathew SJ, Wong E, Tang CY, Charney DS, Iosifescu DV (2015): Regulation of neural responses to emotion perception by ketamine in individuals with treatment‐resistant major depressive disorder. Transl Psychiatry 5:e509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrough JW, Iosifescu DV, Chang LC, Jurdi Al RK, Green CE, Perez AM, Iqbal S, Pillemer S, Foulkes A, Shah A, Charney DS, Mathew SJ (2013): Antidepressant efficacy of ketamine in treatment‐resistant major depression: a two‐site randomized controlled trial. Am J Psychiatry 170:1134–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Outhred T, Das P, Felmingham KL, Bryant RA, Nathan PJ, Malhi GS, Kemp AH (2014): Impact of acute administration of escitalopram on the processing of emotional and neutral images: a randomized crossover fMRI study of healthy women. J Psychiatry Neurosci 39:267–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessoa L, Adolphs R (2010): Emotion processing and the amygdala: from a 'low road' to “many roads” of evaluating biological significance. Nat Rev Neurosci 11:773–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips ML, Drevets WC, Rauch SL, Lane R (2003): Neurobiology of emotion perception II: Implications for major psychiatric disorders. Biol Psychiatry 54:515–528. [DOI] [PubMed] [Google Scholar]
- Pizzagalli DA (2011): Frontocingulate dysfunction in depression: toward biomarkers of treatment response. Neuropsychopharmacology 36:183–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvadore G, Cornwell BR, Colon‐Rosario V, Coppola R, Grillon C, Zarate CA, Manji HK (2009): Increased anterior cingulate cortical activity in response to fearful faces: a neurophysiological biomarker that predicts rapid antidepressant response to ketamine. Biol Psychiatry 65:289–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvadore G, Cornwell BR, Sambataro F, Latov D, Colon‐Rosario V, Carver F, Holroyd T, Diazgranados N, Machado‐Vieira R, Grillon C, Drevets WC, Zarate CA (2010): Anterior cingulate desynchronization and functional connectivity with the amygdala during a working memory task predict rapid antidepressant response to ketamine. Neuropsychopharmacology 35:1415–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarter M, Givens B, Bruno JP (2001): The cognitive neuroscience of sustained attention: where top‐down meets bottom‐up. Brain Res Brain Res Rev 35:146–160. [DOI] [PubMed] [Google Scholar]
- Scheidegger M, Walter M, Lehmann M, Metzger C, Grimm S, Boeker H, Boesiger P, Henning A, Seifritz E (2012): Ketamine decreases resting state functional network connectivity in healthy subjects: implications for antidepressant drug action. PLoS ONE 7:e44799–e44799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt CF, Degonda N, Luechinger R, Henke K, Boesiger P (2005): Sensitivity‐encoded (SENSE) echo planar fMRI at 3T in the medial temporal lobe. NeuroImage 25:625–641. [DOI] [PubMed] [Google Scholar]
- Schmitz TW, Johnson SC (2006): Self‐appraisal decisions evoke dissociated dorsal‐ventral aMPFC networks. NeuroImage 30:1050–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheline YI, Barch DM, Donnelly JM, Ollinger JM, Snyder AZ, Mintun MA (2001): Increased amygdala response to masked emotional faces in depressed subjects resolves with antidepressant treatment: an fMRI study. BPS 50:651–658. [DOI] [PubMed] [Google Scholar]
- Sinner B, Graf BM (2008): Ketamine. Handb Exp Pharmacol 313–333. [DOI] [PubMed] [Google Scholar]
- Sladky R, Spies M, Hoffmann A, Kranz G, Hummer A, Gryglewski G, Lanzenberger R, Windischberger C, Kasper S (2015): (S)‐citalopram influences amygdala modulation in healthy subjects: a randomized placebo‐controlled double‐blind fMRI study using dynamic causal modeling. NeuroImage 108:243–250. [DOI] [PubMed] [Google Scholar]
- Song X‐W, Dong Z‐Y, Long X‐Y, Li S‐F, Zuo X‐N, Zhu C‐Z, He Y, Yan C‐G, Zang Y‐F (2011): REST: a toolkit for resting‐state functional magnetic resonance imaging data processing. PLoS ONE 6:e25031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielberger CD, Gorsuch RL, Edward LR (1970): STAI manual for the state‐trait anxiety inventory (“Self‐evaluation questionnaire”). Consulting Psychologists Press, Inc. [Google Scholar]
- Tzourio‐Mazoyer N, Landeau B, Papathanassiou D, Crivello F, Etard O, Delcroix N, Mazoyer B, Joliot M (2002): Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single‐subject brain. NeuroImage 15:273–289. [DOI] [PubMed] [Google Scholar]
- van Marle HJF, Tendolkar I, Urner M, Verkes RJ, Fernández G, van Wingen G (2011): Subchronic duloxetine administration alters the extended amygdala circuitry in healthy individuals. NeuroImage 55:825–831. [DOI] [PubMed] [Google Scholar]
- Victor TA, Furey ML, Fromm SJ, Ohman A, Drevets WC (2010): Relationship between amygdala responses to masked faces and mood state and treatment in major depressive disorder. Arch Gen Psychiatry 67:1128–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollenweider FX, Kometer M (2010): The neurobiology of psychedelic drugs: implications for the treatment of mood disorders. Nat Rev Neurosci 11:642–651. [DOI] [PubMed] [Google Scholar]
- Walter M, Li S, Demenescu LR (2014): Multistage drug effects of ketamine in the treatment of major depression. Eur Arch Psychiatry Clin Neurosci 264(Suppl 1):55–65. [DOI] [PubMed] [Google Scholar]
- Yoshimura S, Okamoto Y, Onoda K, Matsunaga M, Ueda K Suzuki S‐I, Shigetoyamawaki (2010): Rostral anterior cingulate cortex activity mediates the relationship between the depressive symptoms and the medial prefrontal cortex activity. J Affect Disord 122:76–85. [DOI] [PubMed] [Google Scholar]
- Zarate CA, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, Charney DS, Manji HK (2006): A randomized trial of an N‐methyl‐D‐aspartate antagonist in treatment‐resistant major depression. Arch Gen Psychiatry 63:856–864. [DOI] [PubMed] [Google Scholar]
- Zhong M, Wang X, Xiao J, Yi J, Zhu X, Liao J, Wang W, Yao S (2011): Amygdala hyperactivation and prefrontal hypoactivation in subjects with cognitive vulnerability to depression. Biol Psychol 88:233–242. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information