

Abstract
Photodynamic therapy (PDT) is a promising treatment modality for cancer management. So far, most PDT studies have focused on delivery of photosensitizers to tumors. O2, another essential component of PDT, is not artificially delivered but taken from the biological milieu. However, cancer cells demand a large amount of O2 to sustain their growth and that often leads to low O2 levels in tumors. The PDT process may further potentiate the oxygen deficiency, and in turn, adversely affect the PDT efficiency. In the present study, a new technology called red blood cell (RBC)-facilitated PDT, or RBC-PDT, is introduced that can potentially solve the issue. As the name tells, RBC-PDT harnesses erythrocytes, an O2 transporter, as a carrier for photosensitizers. Because photosensitizers are adjacent to a carry-on O2 source, RBC-PDT can efficiently produce 1O2 even under low oxygen conditions. The treatment also benefits from the long circulation of RBCs, which ensures a high intraluminal concentration of photosensitizers during PDT and hence maximizes damage to tumor blood vessels. When tested in U87MG subcutaneous tumor models, RBC-PDT shows impressive tumor suppression (76.7%) that is attributable to the codelivery of O2 and photosensitizers. Overall, RBC-PDT is expected to find wide applications in modern oncology.
1. Introduction
Photodynamic therapy (PDT) is a relatively new treatment modality that has gained widespread attention. PDT has been used in the clinic for treatment of Barrett’s esophagus, age-related macular degeneration, and esophageal cancer.[1] It is also under intensive investigation for cancer treatment, including cancers of head and neck, brain, skin, breast, lung, bladder, and prostate.[2] There are three essential components for PDT: light, photosensitizers, and oxygen. Individually, none of the components is toxic, but together they initiate a photochemical reaction, producing reactive oxygen species, most importantly singlet oxygen (1O2),[2a,3] which is cytotoxic. While PDT can kill cancer cells directly, it is a common approach to use PDT to damage tumor blood vessels, leading to vessel blockage, tissue ischemia, and eventually cancer cell death.[3b,4] In addition, PDT is immunostimulatory and can induce an immune response against tumors. [5]
Over the years, there have been continuous efforts on improving the delivery efficiency of photosensitizers.[6] O2, another essential component of PDT, is not artificially delivered but usually taken from the biological milieu. However, O2 is a precious resource in tumors. Uncontrolled proliferation of cancer cells often outgrows their blood supply, leading to low oxygen levels and even hypoxia.[7] Tumor hypoxia renders cancer cells more resistant to radiotherapy and anticancer chemotherapy, and increases the tendency of metastasis.[8] It also poses an obstacle for PDT, whose efficacy is heavily oxygen-dependent. Previous studies have shown that the efficacy of PDT decreases when partial oxygen pressure is below 40 mmHg,[9] and abolished in the event of tissue ischemia.[10] These facts underscore the importance of O2 in PDT and suggest the benefits for delivering both O2 and photosensitizers in such a treatment. So far, however, there have been few tentatives along this direction.
Herein we report a novel, red blood cell enhanced PDT (or RBC-PDT) approach that can potentially address the need. RBC-PDT uses RBCs as photosensitizer carriers; but unlike common drug carriers, RBCs bear with them a second cargo, O2. RBCs carry 270 million hemoglobin molecules per cell (each hemoglobin binds up to four O2 molecules), and are the primary source of oxygen to our body tissues. [11] Even in hypoxic tumor regions, there are O2 molecules released from RBCs to the surroundings,[12] but they are rapidly consumed by cancer cells adjacent to capillaries, leaving distant cells poorly oxygenated.[11] Our hypothesis is that while oxygen liberated from RBCs is not sufficient to pump up whole tumors, there exists an oxygen-rich zone close to the surface of RBCs. If photosensitizers are located within the zone or resided on the RBC surface, they may benefit from the oxygen flow from RBCs to continuously produce 1O2, even in hypoxic regions. As discussed above, PDT can not only directly kill cancer cells, but also damage tumor vasculatures, leading to tissue ischemia. From this perspective, the facts that RBCs afford a long circulation half-life and minimal extravasation are extra benefits to the RBC-PDT approach, ensuring maximized photodynamic impact onto the lumen and endothelium of tumor vasculatures. It is noted that unlike chemotherapy, PDT is a focal treatment modality that is largely regulated by photoirradiation. This means that efficient vascular PDT can be achieved when there is a high intraluminal concentration of photosensitizer at the time of irradiation, even when the photosensitizers are not internalized in cancer or endothelial cells. For instance, it was observed that for verteporfin-based PDT, a short drug-light interval (e.g., 15 min) led to efficient vasculature damage whereas a long drug-light interval (e.g., 3 h) induced direct cancer cell death due to extravasation of the photosensitizer. [13]
One challenge, however, is how to load a large amount of photosensitizers to the surface of RBCs. Most photosensitizers are porphyrin-like molecules, which are hydrophobic, prone to aggregation, and do not provide conjugation-friendly functional groups.[4] This problem can be solved by using nanocapsules to encapsulate photosensitizers and tether the conjugates onto RBC surface.[6c] In the present study, we used ferritin, a protein-based nanocage, for the purpose. Our recent studies showed that ferritins could load photosensitizers such as ZnF16Pc (maximum excitation at ≈671 nm, ΦΔ = 0.85 in THF[15]), by 40 wt% without inducing significant self-quenching or compromised colloidal stability. [6c] Using biotin–neutravidin-mediated coupling, we were able to conjugate ≈5 × 105 ZnF16Pc-loaded ferritins onto each RBC. Carrying a large amount of photosensitizers and oxygen, the resulting conjugates show efficient 1O2 production even under low oxygen conditions. The merit of having a carry-on oxygen reservoir translates to enhanced PDT capacity to kill cancer cells, which is confirmed both in vitro and in vivo with hypoxia tumor models. The results suggest a great potential of RBC-PDT in cancer therapy and shed light on a new avenue to enhance the efficacy of PDT.
2. Results
2.1. Preparation of P-FRT-RBCs
Ferritin preparation and ZnF16Pc loading were conducted by following our published protocols.[6c,16] We used a ZnF16Pc- loaded ferritin (P-FRT) formulation with a ferritin to ZnF16Pc molar ratio of 1:40 for the studies. According to our previous research, at this loading rate, there is little aggregation of the loaded ferritins and the self-quenching among the encapsulated ZnF16Pc molecules is minimal.[6c]
To conjugate P-FRT to RBCs, we biontinylated both ferritins (24 biotin molecules per ferritin cage; Figure S1, Supporting Information) and RBCs using biotin-X-NHS (Calbiochem), and then crosslinked them with neutravidin (Figure 1b, details of the procedures were described in the Experimental section). The resulting, P-FRT conjugated RBCs, or P-FRT-RBCs (Figure 1a), were characterized by scanning electron microscopy (SEM, Figure 1c). P-FRT-RBCs displayed a coarse surface, adorned with a large number of nanoparticles with diameters of 15–18 nm. The size is in good agreement with photosensitizer-loaded ferritins.[6c] By contrast, native RBCs presented a smooth surface (Figure 1c). To quantify the coupling efficiency of P-FRTs to RBCs, we labeled P-FRTs with a fluorescent dye molecule, IRDye800 (ex/em: 775/790 nm). By analyzing the 775 nm absorption and comparing it to a predetermined standard curve, it was determined that ≈5 × 105 P-FRTs (that is, ≈2 × 107 ZnF16Pc molecules) were tethered to the surface of each RBC. The high loading was validated by fluorescence microscopy, which found intense and evenly distributed fluorescence signals on the RBC membrane (Figure 1d).
Figure 1.
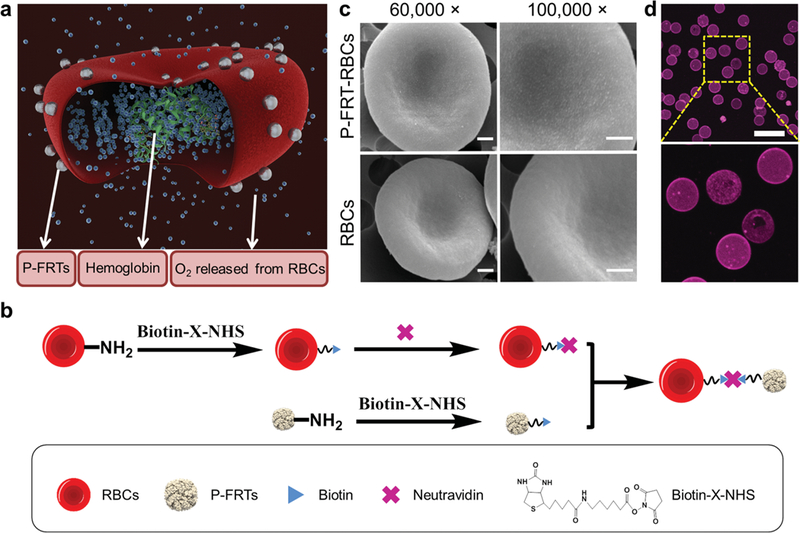
Construction of P-FRT-RBCs. a) Schematic illustration of the formation and working mechanism of P-FRT-RBCs. b) Schematic illustration of the preparation of P-FRT-RBCs (not to scale). c) SEM images of P-FRT-RBCs and RBCs. While the surface of RBCs is relatively smooth, the surface P-FRT-RBCs is adorned with small particles with sizes around 15−18 nm. Scale bars: 500 nm. d) Confocal microscopy images of Cy5.5-labeled P-FRT- RBCs. Ex/em: 673/707 nm. Scale bar: 20 μm.
There was little ZnF16Pc released from P-FRT-RBCs in the serum (Figure S2, Supporting Information), indicating minimal premature photosensitizer release. Due to the facts that the lifetime of 1O2 is very short in aqueous solutions and that the P-FRT-RBCs are not soluble in organic solvents, it is not possible to measure 1O2 quantum yield of them. Using singlet oxygen sensor green (SOSG) assays, however, we observed comparable 1O2 production between ZnF16Pc and P-FRTs in our previous studies, [6c] and between P-FRT-RBCs and P-RBCs in the present study (Figure S3, Supporting Information).
2.2. Cytotoxicity Studies with P-FRT-RBCs
The cytotoxicity of P-FRT-RBCs was evaluated with U87MG cells (human glioma) using 3-(4,5-dimethythiazon-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assays. In the absence of photoirradiation, P-FRT-RBCs showed little cytotoxicity (Figure S4, Supporting Information). When P-FRT-RBCs were applied in conjunction with a 671 nm laser radiation, however, the treatment led to extensive cell death (Figure S5, Supporting Information), which is a hallmark of PDT-induced toxicity. The PDT toxicity was validated by SOSG assays, which found widespread production of 1O2 (Figure S6, Supporting Information); meanwhile, ethidium homodimer-III assay (also known as dead assay, [17] Figure S6, Supporting Information) detected extensive positive staining, indicating severe breakage of cell membrane integrity upon the treatment. Interestingly, both MTT and ethidium homodimer-III staining results found more effective cell killing with P-FRT-RBCs than with P-FRTs. This again was attributed to a relatively high O2 level in P-FRT-RBC containing solutions [18] and corroborated with the 1O2 production result (Figure S3, Supporting Information).
RBCs lack nuclei and many cellular organelles and they do not undergo commonly recognized apoptosis.[19] Instead, aged RBCs manifest increased externalization of phosphatidylserine,[20] leading to their fast clearance from the circulation. Our flow cytometry analysis found no significant increase of RBC phosphatidylserine levels over their coupling with P-RBCs (Figure S7, Supporting Information). Moreover, we observed little phosphatidylserine level enhancement when subjecting P-FRT-RBCs under continuous photoirradiation (100 mW cm−2, up to 1 h; Figure S8, Supporting Information). In other words, conjugation and PDT processes have relatively minor impact on the circulation half-lives of RBCs, which is important to the in vivo applications. Notably, the relatively robustness of RBCs against PDT has been observed by others as well.[21]
2.3. P-FRT-RBC-Based PDT Can Efficiently Produce 1O2, Even under Low Oxygen Conditions
We next evaluated whether O2 released from RBCs can benefit P-FRT-RBCs for sustained 1O2 production under low oxygen conditions. This was first evaluated in solutions using SOSG as a 1O2 marker. Specifically, we injected P-FRT-RBCs into a sealed cuvette that had been loaded with PBS and bubbled with Ar. We then irradiated the cuvette by a 671 nm laser (100 mW cm−2); at selected time points, the irradiation was stopped and the amount of 1O2 in the solution was assessed by measuring fluorescence increase at 525 nm.[6c] For comparison, we also studied P-FRTs at the same ZnF16Pc concentration and irradiation doses. We observed little 1O2 production with P-FRTs, likely due to the oxygen-free condition. Meanwhile, there was continuous 1O2 generated in the cuvette loaded with P-FRT-RBCs (Figure 2a); this was attributed to the RBCs in the closed system, which functioned as an O2 reservoir. Notably, such RBC-facilitated 1O2 production worked best when photosensitizers were attached to the RBC surface. In a control group, we mixed (but not conjugated) the same amounts of P-FRTs and RBCs and injected them into a cuvette; in this case, we observed increased 1O2 production but the 1O2 level was significant less than that with P-FRT-RBCs (Figure 2a, P < 0.05). This may be due to the fact that an effective oxygen-rich zone only exists at the immediate surrounding of an RBC. In the case of a mixture of P-FRTs and RBCs, photosensitizers were distributed randomly in the solution and were less efficiently benefited from the O2 released.
Figure 2.

P-FRT-RBCs showed enhanced PDT effect under hypoxic environments. a) Comparison of 1O2 generation among P-FRT-RBCs, a mixture of RBCs and free P-FRTs, and free P-FRTs, conducted in an Ar-filled cuvette. The cuvette was irradiated by a 671 nm laser (0.1 W cm −2) for up to 60 min. SOSG was used as an indicator of 1O2 production. b) HIF-1± staining on tumor samples taken from the acute hypoxia tumor model and normal mice. Green, HIF-1±. Blue, DAPI. Scale bars: 100 μm. c) H&E staining on hypoxic tumors treated by PDT-mediated by different formulations. While P-FRT-RBCs and O2-treated P-FRT-RBCs induced signifi cant damage to tumors under irradiation, CO-treated P-FRT-RBCs and P-FRTs caused minimal cancer cell death. Scale bars: 100 μm. d) TUNEL assays on hypoxic tumors treated by PDT-mediated by different formulations. Similar to observations in (c), extensive cell death was only observed in animals treated with P-FRT-RBCs and O2-treated P-FRT-RBCs. Green, TUNEL. Blue, DAPI. Scale bars: 100 μm.
To further investigate the RBC-enhanced PDT under a low oxygen environment, we conducted an in vivo study with a murine acute hypoxia tumor model. [22] Briefly, we first implanted U87MG tumors to the hind legs of nude mice. When the tumors reached a size of 250 mm3, a tourniquet was placed onto the tumor-bearing leg for 15 min to temporally suppressed the blood flow.[22] The treatment led to widespread tumor hypoxia, which was confirmed by HIF-1α staining with tumor tissues (Figure 2b). For treatment, P-FRT-RBCs (0.075 mg ZnF16Pc kg−1) were intra-tumorally injected into the hypoxic tumors (n = 3), followed by photoirradiation to the tumor areas (100 mW cm−2 for 30 min).
Notably, the tourniquet remained bound to the animals during the PDT process and was removed only after the end of the irradiation. Twenty-four hours after the PDT, we euthanized the animals and analyzed the tumor tissues by hematoxylin and eosin (H&E) staining (Figure 2c) as well as terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay (Figure 2d). We observed widespread cell death and severely damaged tumor architectures in the P-FRT-RBC-treated group (Figure 2d). This is in stark contrast to the P-FRT-treated animals at the same ZnF16Pc dose, in which case little damage to tumors was observed.
To validate that the difference in therapy was due to oxygen from RBCs, we conducted two more studies with P-FRT-RBCs. In the first study, we exposed P-FRT-RBCs to 100% O2 (30 min) before the intratumoral injection. In the second group, P-FRT-RBCs were soaked in CO (N2/CO, 95:5 v:v, at a pressure of 1.5 kg cm−2, 30 min) before the injection. It is known that CO binds more strongly to hemoglobin than O2, and the binding can temporally suspend the O2-carrying ability of RBCs[23] The successful CO binding was verified by the blueshift of the characteristic absorption peaks of hemoglobin (Figure S9, Supporting Information). Compared to normal P-FRT-RBCs, the O2- soaked P-FRT-RBCs induced little enhancement of tumor treatment efficacy. This is not surprising because the oxygen carrying capacity of RBCs has reached the maximum under a normal oxygen partial pressure and further increasing O2 levels barely contributes to O2 loading (≈160 mmHg).,[24] CO-treated P-FRT- RBCs, on the other hand, almost lost their ability to induce PDT damage to tumors (Figure 2d). These results confirm that O2, released from RBCs is a key player in P-FRT-RBC-based PDT.
2.4. Circulation Half-Lives and Biodistribution of P-FRT-RBCs
We next intravenously (i.v.) injected P-FRT-RBCs to U87MG-bearing mice (0.75 mg ZnF16Pc kg−1) and investigated their circulation and biodistribution. In order to facilitate the tracking of P-FRT-RBCs, we labeled them with IRDye800 before the injection. As a comparison, IRDye800 labeled P-FRTs at the same ZnF16Pc dose were also injected. For P-FRT-RBCs injected mice, signals were distributed relatively evenly in the host at early time points and faded rather slowly (Figure 3a). The long circulation was also confirmed by florescence analysis on blood samples taken at different time points (Figure 3b).
Figure 3.

In vivo behaviors of P-FRT-RBCs. a) Representative fluorescence images taken at different time points after P-FRT-RBC injection (labeled with IRDye800). Tumors were circled with yellow dashed lines. P-FRT-RBCs were distributed relatively evenly throughout the animals and there was slow decay of signals over time. b) Circulation of P-FRT-RBCs, analyzed in healthy nude mice ( n = 3) by measuring remaining fluorescence activities in the circulation. P-FRT-RBCs had been labeled by IRDye800 before injection. c) Biodistribution study. P-FRT-RBCs were labeled with 64 Cu-PTMS and injected into U87MG tumor bearing mice. The distribution was analyzed by gamma counting with tissues taken from the animals at 24 h.
Fast clearance of photosensitizers is common with conventional, small molecule-based PDT,[6b] and leads to low intravascular photosensitizer concentrations or drop of photosensitizer concentrations during the course of PDT (each PDT session takes 15−30 min) [3b] These, however, are less of concerns for P-FRT-RBCs. According to region of interest (ROI) analysis (Figure S10, Supporting Information), the P-FRT-RBC contents in tumors were 6.9 ± 0.4%, 7.5 ± 0.2%, 7.2 ± 0.5%, 7.0 ± 0.4%, and 11.4 ± 0.8% of the injection dose (ID) at 10 min, 30 min, 1h, 4 h, and 24 h, respectively. As stated above, our RBC-PDT approach targets tumor vasculatures.[3b,4] The long circulation of P-FRT-RBCs and their minimal extravasation at tumor sites represent advantages in this regard.
After 24 h of imaging, the animals were euthanized. Tumors, along with the major organs, were harvested for ex vivo imaging (Figures S11 and S12, Supporting Information). In addition to the high IRDye800 signals in tumors, fluorescence activities were also observed in organs, such as the liver, spleen, and lung, but low in the skin. Given that ZnF16Pc is not toxic in the dark and PDT is largely regulated by focal photoradiation, such accumulation in internal organs poses few toxicity concerns. In addition to fluorescence imaging, we also investigated the circulation and tumor contents of P-FRT-RBCs by two other methods. One was to label P-FRT-RBCs with iron oxide nanoparticles (IONPs, details of the labeling are described in the Experimental section) and monitor signal change in tumors by T2-weighted magnetic resonance imaging (MRI, Figure S13a, Supporting Information). Based on ROI analysis, it was derived that the signal-to-background ratio (SBR) in tumors was dropped by 10.6 ± 2.1%, 15.6 ± 3.2%, 3.3 ± 1.6%, and 4.1 ± 1.9% at 4, 24, 48 and 72 h, respectively (Figure S13b, Supporting Information). The results confirmed a high content of P-FRT- RBCs in tumors at early time points. The second method was to label P-FRT-RBCs with 64Cu-PTSM (copper-64-pyruvaldehyde-bis(N4-methylthiosemicarbazone))[25] and assess the biodistribution of P-FRT-RBCs by ex vivo gamma counting (Figure 3c, labeling details are described in Experimental section). Consistent with the fluorescence results, there was a high radioactivities in the blood at early time points (7.6 ± 2.1%ID g−1 at 1 h) and the signals remained strong for 24 h (3.2 ± 0.4%ID g−1 at 24 h); meanwhile, high 64Cu activities were also found in hyper-vascular tissues, including the liver, spleen, lung, and tumors (Figure 3c). It was noted that although the distribution profiles were similar between the fluorescence and gamma counting analyses, the tumor ID% g−1 values from gamma counting were relatively low (e.g., 5.4 ± 1.0%ID g−1 for the 64Cu-PTSM analysis at 24 h, compared to that of 11.4 ± 0.8%ID g−1 for fluorescence imaging). The difference could be attributed to autofluorescence interference, and/or transmellation-induced 64Cu falloff in 64Cu-PTSM-based cell tracking.[25,261
2.5. Fluence-Dependent RBC-PDT Damage to Tumors
One important parameter in PDT is irradiation dose, or fluence. Too low a fluence may be insufficient to induce vascular damage, while too high a fluence may cause side effects to surrounding normal tissues. Hence, before a systematic therapy study, we investigated the impact of fluence on RBC-PDT. Specifically, we i.v. injected P-FRT-RBCs (0.75 mg ZnF16Pc kg−1) into U87MG subcutaneous tumor models and irradiated the tumors by a 671 nm laser 5 min after the injection. The irradiation time was maintained at 30 min but the fluence rate, or irradiance, was elevated from 20 to 300 mW cm−2. All of the animals were sacrificed 24 h after the treatment and the tumors were collected for histological analysis. H&E staining showed a clear fluence-dependent treatment efficacy (Figure 4a). While there was almost no therapy effect at 20 mW cm−2, there were large areas of necrosis and bleeding in tumors when the irradiance was raised to 100 mW cm−2 and above. Meanwhile, there was only marginal difference in efficacy among animals treated by 100, 200, and 300 mW cm−2 irradiations (Figure 4a,b). Based on these observations, we chose an irradiance of 100 mW cm−2 and irradiation time of 30 min for subsequent therapy studies. Notably, the chosen fluence and fluence rate are within the range commonly used for clinical PDT [27]
Figure 4.

Irradiance-dependent PDT effect. P-FRT-RBCs were i.v. injected first (0.75 mg ZnF16Pc kg−1). A 671 nm laser was applied to tumors for 30 min at different irradiances. a) H&E and b) TUNEL assay results, conducted on tumor samples taken 24 h after the PDT. There is an irradiance-dependent PDT effect that caused cancer cell death. While there was a large difference in efficacy between 100 mW cm−2 and 40 or 20 mW cm−2 treated animals, there was no significant improvement of efficacy when further increasing the irradiance. Green, TUNEL. Blue, DAPI. Scale bars: 100 μm.
2.6. Tumor Therapy Studies with RBC-PDT
The therapy study was conducted with U87MG tumor bearing mice (n = 5). P-FRT-RBCs were administered at a dose of 0.75 mg ZnF16Pc kg−1 into the animals; photoirradiation (671 nm, 100 mW cm−2 for 30 min, over a 1 cm diameter beam to cover tumors) was applied 5 min after the injection. For comparison, we also performed PDT with P-FRT-RBCs that had been soaked in O2 or CO for 30 min before injection. Moreover, PDT with P-FRTs alone at the same ZnF16Pc dose was also investigated (in which case the photoirradiation was applied at 4 h instead of 5 min after the injection, based on imaging study results, Figure S14, Supporting Information). For controls, animals were injected with P-FRT-RBCs or PBS but received no photoirradiation.
P-FRT-RBC-mediated PDT induced significant tumor growth suppression (Figure 5a); relative to the PBS control group, a tumor growth inhibition (TGI) rate of 63.6 ± 12.6% was observed on day 15. The treatment efficacy was slightly improved for O2-treated P-FRT-RBCs (TGI rate of 76.7 ± 22.6% on day 15). As a comparison, animals received PDT with CO-presoaked P-FRT-RBCs or P-FRTs alone showed significantly lower treatment efficiency (TGI values being 41.2 ± 4.4% and 38.7 ± 3.7%, respectively; P < 0.001).
Figure 5.
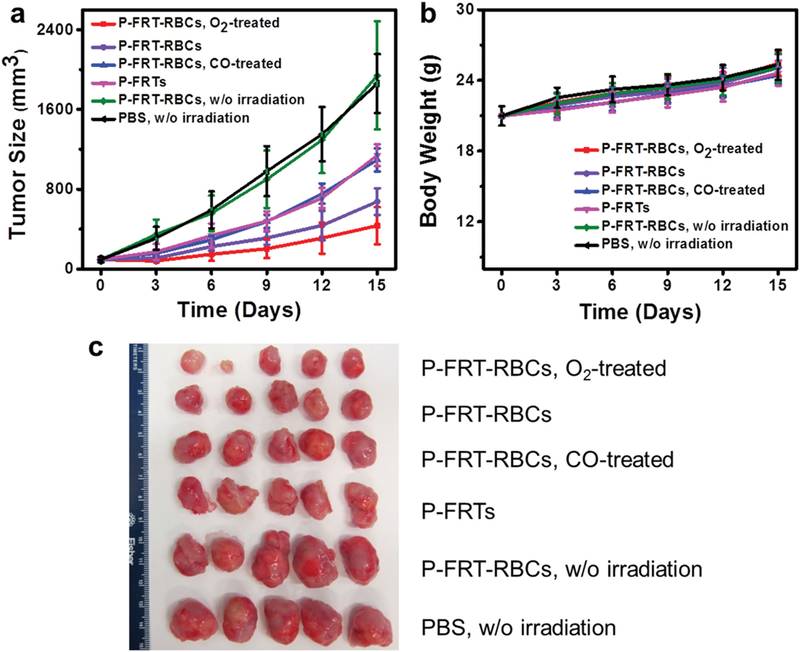
Therapy studies on U87MG tumor models (n = 5). a) Tumor growth curves. Significant tumor suppression was found with animals injected with O2-treated P-FRT-RBCs and P-FRT-RBCs, showing TGI rates of 76.7% and 63.6%, respectively, on day 15. These are much more significant than PDT-mediated by P-FRTs or CO-treated P-FRT-RBCs. b) Body weight growth curves. No significant weight drop was found with all the treatment groups. c) Photographs of all the dissected tumors.
Despite the efficient cancer cell killing, there was no sign of toxicities with P-FRT-RBC-mediated PDT (Figure S15, Supporting Information), including no weight loss (Figure 5b). All of the animals were euthanized after 15 d. The intergroup difference in tumor size (Figure 5c) and weight (Figure S16, Supporting Information) agreed well with those observed from caliper measurements. For RBC-PDT-treated animals, H&E staining found no significant off-target damage to normal tissues (Figure 6). To better assess the treatment efficacy, we repeated the study with P-FRT-RBCs (no CO or O2 pretreatment) and P-FRTs, but this time euthanized the animals 24 h after PDT; the tumor tissues were harvested for histology analysis. H&E and TUNEL assays found dramatic difference between the two treatment groups, with the P-FRT-RBC group showing more extensive cell killing, including to those lying deep under the skin (Figure S17a,b, Supporting Information).
Figure 6.

H&E staining on tumors and major organs after therapy. The tumors from the O2-treated P-FRT-RBCs (G1) and P-FRT-RBCs (G2) groups showed extensive cell destruction in tumors. No abnormalities were observed in major organs. Scale bars: 100 μm.
3. Discussions and Conclusion
Owing to good biocompatibility and long circulation, RBCs have been widely investigated as carriers for chemotherapeutics.[28] Using RBCs as carriers for delivery of photosensitizers, however, has seldom been explored. Very recently, the Liu group demonstrated that RBCs can be engineered to load doxorubincin, chlorine e6, and IONPs for magnetic-field-guided PDT and chemotherapy.[20] However, there has been no investigation into the oxygen-providing benefits of RBCs. To the best of our knowledge, our study is the first to show that the codelivery of oxygen and photosensitizers can lead to improved PDT efficacy.
As shown in our studies, tethering photosensitizers onto the surface of RBCs is critical for our approach. Conventionally, there are two strategies to load drugs onto or into RBCs. One is to encapsulate drug molecules into the interiors of RBCs, for instance, by hypotonic dialysis.[29] This is inappropriate for PDT considering the short lifetime of 1O2 (<0.04 μs).[30] The second is to conjugate drugs to RBC surface through covalent linking.[31] This approach is also not desirable because it is often associated with low loading efficiency, not to mention that most photo sensitizers do not possess a conjugation-friendly functional group. In the current study, we first load photosensitizers into a ferritin nanocapsule and then couple the photosensitizer-loaded ferritins onto RBC membranes. Affording high ZnF16Pc loading efficiency, good biocompatibility, and facile surface conjugation chemistry, ferritins provides a perfect solution to the otherwise challenging task.
We have observed both in vitro and in hypoxic tumor models that using RBCs as photosensitizer carriers can enhance the efficacy of PDT. It was shown that the sustained O2 supply by RBCs enabled efficient PDT even under hypoxic conditions (Figure 2c,d). It was also found that tethering photosensitizers to RBC surface was important for making the best out of the O2 released (Figure 2a). The merits of using RBCs as carriers were also confirmed in a PDT study, where P-FRT-RBCs were systematically injected. Compared to the P-RBC and CO-treated P-FRT-RBC groups, significantly improved therapy efficacy was observed with P-FRT-RBC or O2-treated P-FRT-RBC groups, validating the contribution of O2 released from RBCs in the enhanced treatment. It is noted that tumor hypoxia is not equal to zero oxygen or no RBC access (in which case cancer cells would die). In tumors, O2 molecules can diffuse shortly from blood vessels because they are quickly consumed by fast growing cancer cells adjacent to blood vessels.[32] Cancer cells that are ≈70−100 μm from blood vessels are hypoxic and those beyond the distance become necrotic.[33] Such location-dependent oxygen levels underscore the significance of having photosensitizers very close to an oxygen source (RBCs) for efficient PDT, which is satisfied in the RBC-PDT approach. By breaking down tumor blood vessels and cutting off blood supply, RBC-PDT can induce extensive cancer necrosis, which was observed in our therapy studies (Figure S17, Supporting Information).
According to the biodistribution study (Figure 3c), the concentration of P-FRT-RBCs in tumors was higher at 24 h than at 4 h, indicating a possible tumor targeting mechanism (Figure 3a). This is interesting because there was no targeting ligand artificially attached to the RBC surface. A possible explanation is that RBCs are gradually opsonized by a serum protein, lactadherin, during the circulation;[34] possessing an RGD motif, lactadherin-attached RBCs become sticky to tumor angiogenic endothelial cells, which overexpress integrin αvβ3.[34a,35] It was observed that artificially aged RBCs can home to the tumor endothelium and even get internalized by tumor endothelial cells. [34a] Future studies are needed to understand the mechanism behind the tumor accumulation and explore the possibility to further enhance the effect.
In conclusion, we have developed a novel, RBC-facilitated PDT methodology. Using ferritins as nanocapsules, we were able to tether a large amount of ZnF16Pc to the surface of RBCs. RBCs, as photosensitizer carriers, extend the circulation half-lives of photosensitizers. The resulting PDT focuses on the tumor vasculatures, ensuring maximum treatment efficacy. More importantly, we found that RBCs can provide O2 to enable sustained 1O2 production even when P-FRT-RBCs were under low oxygen conditions. All together, these characteristics lead to enhanced treatment efficacy against tumors. This novel technology addresses one critical limitation of conventional PDT and is expected to find wide applications in modern oncology.
4. Experimental Section
Preparation, Dye-Labeling, and Biotinylation of P-FRTs:
The protocols for producing FRTs and loading ZnF16Pc into FRTs were published previously. [6c,16] Briefly, 490 μl ferritin (0.5 mg mL−1 PBS) was gently mixed with 10 μL ZnF16Pc (5 mg mL−1 in DMSO) for ≈45 min at room temperature. The mixture was then purified on a NAP-5 column to remove the unloaded ZnF16Pc. For dye labeling, P-FRTs were incubated with IRDye800-NHS (LI-COR) or Cy5.5-NHS (Amersham) at a starting molar ratio of 1:2 for 30 min and then purified through a NAP-5 column to remove the uncoupled dye molecules. The coupling efficiency was assessed spectroscopically by comparing to a predetermined standard curve. It was determined that the final conjugates have on average one IRDye800 or Cy5.5 per P-FRT particle. To obtain biotinylated P-FRTs, the P-FRTs were mixed with 3 μL of 0.1 M biotin-X-NHS (Calbiochem) at room temperature for 1 h and then purified through a NAP-5 column to remove the uncoupled biotin molecules. The coupling efficiency was analyzed by matrix assisted laser desorption ionization.
Preparation of P-FRT-RBCs:
Whole blood was collected from heathy nude mice. RBCs were separated by centrifugation (1000 G, 5 min) and washed twice with cold phosphate-buffered saline (PBS, pH = 7.4). Then 100 μL of packed RBCs were resuspended in 900 μL PBS and mixed with 3 μL of 0.1 M biotin-X-NHS at 4 °C for 20 min. Subsequently, the cells were washed three times with PBS (1000 G, 5 min) to remove unbounded biotin molecules. The resulting biotinylated RBCs were mixed with 200 μL of 1 mg mL−1 neutravidin (Thermo Scientific) at 4 °C for 1 h. After another wash cycle with PBS (1000 G, 5 min, 3 times), the RBCs-biotin-neutravidin were mixed with the biotinylated P-FRTs for 1 h to yield P-FRT-RBCs. To quantify the molar ratio of P-FRTs to RBCs, IRDye800-labeled P-FRTs were used for the preparation of P-FRT-RBCs; the absorbance at 780 nm (maximum absorbance for IRDye800) was measured and the reading was subtracted from the absorbance of parent RBCs. The integrity of RBC cell membranes in P-FRT-RBCs was assessed by Annexin V binding assay based on a protocol provided by the vendor (FITC conjugate, Life Technologies) with a Beckman Coulter CyAn flow cytometer. The 1O2 quantum yield of P-FRT-RBCs was measured with a singlet oxygen sensor green reagent (S36002, Invitrogen) as a 1O2 indicator, by comparing to that of P-FRTs.
Preparation of IONP-P-FRT-RBCs:
IONPs and P-FRT dual-labeled RBCs (IONP-P-FRT-RBCs) were prepared for the in vivo magnetic resonance (MR) imaging. 1 mL of lONPs with surface amine groups (Ocean Nanotech, SHA-10, 1mg Fe mL−1) was mixed with 10 μL of 0.1 M biotin-X-NHS at 4 °C for 1 h and then purified on a NAP-5 column to remove the unconjugated biotin-X-NHS. The resulting biotinylated lONPs were mixed with the biotinylated P-FRTs at a 1:1 molar ratio. Subsequently, RBCs-biotin-neutravidin were added to the mixture solution and mixed for another 1 h to yield the IONP-P-FRT-RBCs. The final product was purified by centrifugation for three times (1000 G, 5 min).
Scanning Electron Microscopy:
The P-FRT-RBCs were dropped onto a filter paper with a pore size of 1 μm. The samples were primarily fixed by 2% glutaraldehyde in PBS at 4 °C for 1 h. After washing with PBS, the samples were incubated in 1% OsO4 for 1 h. Next, ethanol of gradient concentrations (25%, 50%, 75%, 90%, and 100%) was applied to the samples at room temperature (30 min for each concentration) to dehydrate the samples. Subsequently, the samples were critical-point dried in a SAM-DRl-790 CPD and diffuse-coated with a gold/palladium mix with a thickness of 5 nm in a low vacuum coater (Leica EM ACE 200). Finally, the samples were analyzed using a field emission gun SEM (FEI Inspect F FEG-SEM).
Confocal Microscopy:
The Cy5.5-labeled P-FRTs were conjugated to RBCs. The confocal florescence images of the resulting Cy5.5- labeled P-FRT-RBCs were imaged on a Zeiss LSM 510 META Confocal Microscope.
ZnF16Pc Release from the P-FRT-RBCs:
Photosensitizer release was investigated on a slide-A-lyzer dialysis device (10K MWCO, Pierce), which we used in our previous studies to analyze drug release.[16] Specifically, P-FRT-RBCs in 10% human serum albumins were loaded onto the dialysis device, and the device was immersed in a 15 mL tube filled with the same solvent. The whole system was incubated at 37 °C with gentle shaking. At different time points, 0.5 mL medium was taken from the tube for analysis, and the tube was replenished with 0.5 mL of the fresh medium. The amounts of ZnF16Pc in the releasing media were measured on a micro reader and then compared to a standard curve. Photosensitizer release in PBS was studied similarly.
Cell Line and Animal Models:
The U87MG human glioblastoma cell line was obtained from the American Type Culture Collection and cultured with Dulbecco’s Modified Eagle Medium in a cell culture flask. Athymic nude mice were purchased from Harlan laboratories. The animal model was established by subcutaneously injecting ≈5 × 106 U87MG cells into the right hind limb of each mouse. All of the experimental procedures had been conducted following a protocol approved by the University of Georgia lnstitutional Animal Care and Use Committee.
MTT Assay:
≈1 × 104 U87-MG cells were seeded in each well of a 96-well plate. After a 24 h incubation, P-FRT-RBCs with different ZnF16Pc concentrations (0, 1, 5, 10, 20, and 50 μg mL−1) were added to the plate. For comparison, RBCs and P-FRTs mixture, RBCs, and P-FRTs were used in control studies. A 100 mW cm−2 irradiation was immediately applied to each well for 200 s. Subsequently, the medium of each well was removed; the U87MG cells were washed twice with PBS and the well was then replenished with fresh medium. After incubating for another 24 h, MTT assay was performed to determine the cell viability. For controls, no irradiation was applied.
1O2 Production and Live and Dead Assay:
A singlet oxygen sensor green reagent (S36002, lnvitrogen) was used as a 1O2 indicator, and live and dead assays (lnvitrogen, only dead assay was used in the study) were used to assess cytotoxicity. Specifically, ≈1 × 105 U87MG cells were seeded in each well of a four-well cell culture chamber and were incubated overnight. Next, 2 μL of SOSG working solution were added to incubate with the U87MG cells 30 min prior to the addition of P-FRT-RBCs or P-FRTs (both at 10 μg ZnF16Pc mL−1). A 100 mW cm−2 irradiation was then applied to the chamber for 200 s. Subsequently, the incubation medium was removed. The U87MG cells were washed twice with PBS and the well was then replenished with fresh medium. After 2 h incubation, the cells were stained with EthD-III at 37 °C for 30 min and were observed under a fluorescence microscope.
PDT Effect to RBC Carriers:
A solution of P-FRT-RBCs (at a concentration of 10 μg ZnF16Pc mL−1) was irradiated with a 671 nm laser (100 mW cm−2) for different durations (0, 200 s, 10 min, 30 min, and 60 min) in separate experiments. Subsequently, the integrity of RBC cell membranes in P-FRT-RBCs was assessed by Annexin V binding assay based on the protocol provided by the vendor (FITC conjugate, Life Technologies) with a Beckman Coulter CyAn flow cytometry. RBC ghosts were also examined as positive controls.
1O2 Generation under Low Oxygen Environments:
To mimic a low oxygen environment, a cuvette preloaded with 1 mL PBS was pumped with ultrahigh pure Ar (Airgas, AR UHP300) gas for 30 min to deoxidize; the cuvette was then sealed. Next, a solution containing SOSG reagent and RBC-P-FRTs (at a final concentration of 50 μg ZnF16Pc mL−1) was injected into the cuvette. The cuvette was kept in the dark and irradiated with a 671 nm laser (100 mW cm−2). At selected time points (200 s, 10 min, 30 min, and 60 min), the irradiation was stopped and the amount of 1O2 generated was assessed by measuring fluorescence activities at 525 nm. For controls, free P-FRT and a RBCs and P-FRTs mixture at the same photosensitizer concentrations were also tested.
Acute Hypoxia Tumor Models:
We followed a published protocol with slight modifications to generate the acute hypoxia tumor model.[22] Briefly, U87MG-bearing subcutaneous models were first established. When the tumor reached about 250 mm3, a tourniquet was placed onto the tumor-bearing leg with a clamp for 15 min. To confirm the successful induction of hypoxia, the mice were euthanized and the tumors were subjected to HIF-1α staining. For therapy studies, O2-treated P-FRT-RBCs, P-FRT-RBCs, CO-treated P-FRT-RBCs, P-FRTs (all at 0.075 mg ZnF16Pc kg−1), or PBS were intratumorally injected and the tumors were irradiated by a 671 nm laser (100 mW cm−2, 30 min). After 24 h, the mice were euthanized, and the tumors were collected for H&E staining and TUNEL assays.
Pretreatment of P-FRT-RBCs by O2 or CO:
The protocol was modified from a published one.[36] Briefly, P-FRT-RBCs were soaked in an industrial grade O2 or a gaseous mixture of N2/CO (95:5 V:V) for 30 min at a pressure of 1.5 kg cm−2, The absorbance spectra of the resulting O2- or CO-treated P-FRT-RBCs were immediately measured by a microreader.
Circulation Half-Lives:
To determine the circulation half-lives, IRDye800-labeled P-FRT-RBCs or IRDye800-labeled P-FRTs (0.75 mg ZnF16Pc kg−1) were i.v. injected into healthy nude mice. At selected time points, 10–20 μL of blood were collected from the tail vein and dissolved in heparin solution (1000 U mL−1 in PBS). The IRDye800 fluorescence activities in the blood were measured and subtracted by auto-fluorescence of the blood from an untreated mouse.
In Vivo Imaging Studies:
The imaging studies started when tumors reached a size between 200 and 500 mm3. IRDye800 labeled P-FRT-RBCs or IRDye800 labeled P-FRTs (0.75 mg ZnF16Pc kg−1) were i.v. injected into U87MG-bearing mice (n = 3). Images were taken on a Maestro II imaging system (Perkin Elmer Inc) at 5, 10, 15, and 30 min, as well as 1, 4, and 24 h post injection (p.i.) time points. The images were unmixed by the vendor provided software. ROIs were circled around the tumors, and the optical intensities (in total scaled counts per second) were read by the Maestro software. After the 24 h imaging, all of the mice were euthanized. Tumors as well as major organs were harvested and subjected to ex vivo imaging. The images were analyzed with the Maestro software.
Magnetic Resonance Imaging:
The studies were conducted on U87MG tumor models. The animals (n = 3) were intravenously injected with IONP-P-FRT-RBCs (10 mg Fe kg−1). T2-weighted images were acquired on a 7T Varian small animal MRI system pre- and 4, 24, 48, and 72 h post the IONP-P-FRT-RBC injection, with the following scan parameters: TR = 2.5 s, TE = 48 ms, ETL = 8, FOV 402 mm2, matrix size = 2562, and 11 axial slices with 1 mm slice thickness. To quantify the signal drop, we calculated the SBR by finely analyzing regions of interest of the MR images and calculated the values of (SBRpre − SBRpost)/SBRpre to represent to signal changes.[37] Signal intensity (SI) of tumor and muscle were measured before and after injection of IONP-P-FRT-RBCs. The SBR values were calculated according to SBR = SItumor/SImuscle.
64Cu-PTSM Labeling:
10 μL PTSM (1 mg mL−1 in DMSO) was added to 10 μL 0.1 M NaOAc and the pH adjusted to 6. One mCi solution of 64Cu-CuCl2 was then added and vortexed for 10 min. The resulting mixture was added to Sep-Pak C18 column (Waters, MA) and eluted with ethanol. Solvent was removed by rotary evaporation and the residue was reconstituted in 500 μL 1× PBS to form 64Cu-PTSM. The freshly prepared RBCs (100 μL) were then incubated with 64Cu-PTSM for 30 min at 4 °C and washed three times with 1× PBS. 64Cu labeled RBCs were resuspended in 300 μL 1× PBS and mixed with 3 μL of 0.1 M biotin-X-NHS for 20 min. Then the mixture was washed three times with 1× PBS to remove the unbounded biotin. Subsequently, the biotinylated 64Cu-RBCs were mixed with 200 μL of 1 mg mL−1 neutravidin (Thermo Scientific) at 4 °C for 1 h. The cells were washed three times with 1× PBS to remove the unbounded neutravidin molecules. Finally, the 64Cu-RBCs-biotin-neutravidin were mixed with the 500 μL biotinylate P-FRTs for 1 h to yield 64Cu-PTSM labeled P-FRT-RBCs. The conjugates were washed with 1× PBS for three times to remove effluxed 64Cu before injection.
Biodistribution Studies:
64Cu-PTSM labeled P-FRT-RBCs (1.1−1.3 MBq) was injected intravenously into the animals. Blood samples were collated by remove up to 1 cm of the mice tail under isoflurane anesthesia at 1, 4, and 24 h after injection. At 24 h post-injection, the mice were sacrificed and the organs and blood were collected. All the blood samples and organs were weighed and radioactivities were counted by a gamma counter 2480 WIZARD2 Automatic Gamma Counter (PerkinElmer, Waltham, MA). Decay-corrected uptake was expressed as the percentage of injected dose per gram (%ID g−1) to represent the accumulation of 64Cu-PTMS labeled P-FRT-RBCs in major organs. Values are expressed as the means ± SD for a group of three animals.
Fluence-Dependent In Vivo PDT:
P-FRT-RBCs (0.75 mg ZnF16Pc kg−1) were i.v. injected into U87MG bearing mice. After 5 min, the tumors were irradiated by a 671 nm laser at different fluence rates (300, 200, 100, 40, 20, 0 mW cm−2) for 30 min. 24 h after the irradiation, the mice were euthanized and tumors were collected for H&E staining and TUNEL assay.
Therapy Studies:
The therapy studies were performed on U87MG tumor models. Thirty 4−6-week-old female nude mice were subcutaneously injected with 5 × 106 U87-MG cells on the right hind limbs. The 30 mice were randomly divided into six groups, five mice per group. The study started when the tumor size reached around 100 mm3 (average tumor size of 93.92 ± 27.22 mm3). For the treatment group, the mice were first i.v. injected with O2-treated P-FRT-RBCs (0.75 mg ZnF16Pc kg−1). After 5 min, the tumors were irradiated by a 671 nm laser (100 mW cm−2, over a 1 cm diameter beam) for 30 min. The five control groups received: (1) P-FRT-RBCs and irradiation; (2) CO-pretreated P-FRT-RBCs and irradiation; (3) P-FRTs and irradiation; (4) P-FRT-RBCs only; and (5) PBS only. All groups of animals were irradiated 5 min after the injections, except for the P-FRTs group, which were irradiated 4 h after the irradiation. Every three days, photographs of the mice were taken, and the tumor sizes and body weights were measured. Tumor sizes were measured by a caliper and computed following the formula: size (mm3) = length (mm) × width (mm)2/2. After therapy, the tumors as well as the major organs were collected and subjected to paraffin-embedded H&E staining. Photographs of the tumors were taken and the weights of tumors were measured. To better understand the treatment, we repeated the treatment with P-FRT-RBCs and free P-FRTs in a separate study but euthanized the animals 24 h after irradiation. The tumors were dissected for H&E staining and TUNEL assays.
Hematoxylin and Eosin Staining:
H&E staining for cryogenic and paraffin-embedded sections were both performed by following a protocol provided by the vendor (Oklahoma Medical Research Foundation). For cryogenic sections, 8 μm slides were prepared and fixed with 10% formalin for about 30 min at room temperature. After washing with running water for 5 min, the slides were treated with gradient concentrations of alcohol (100%, 95%, and 70%), each for 20 s. For paraffin-embedded tumor and organ sections in the therapy studies, 6 μm slides were prepared and treated with gradient xylene and/ or alcohol (100% xylene, 100% xylene, 100% xylene, 50:50 xylene/100% EtOH, 100% EtOH, 100% EtOH, 95% EtOH, and 95% EtOH), each for 3 min. After washing with running water for 5 min, the slides were ready for the staining. The hematoxylin staining was performed for about 5 min, and the slides were washed with water for 1 min. The eosin staining was performed for about 1 min. The slides were then washed, treated with xylene, and mounted with Canada balsam. The images were acquired on a Nikon Eclipse 90i microscope.
Immunofluorescence Staining:
The cryogenic slides were fixed with cold acetone for 10 min, washed by running water for 5 min, and blocked by 10% goat serum for 1 h. FITC-labeled anti-HIF-1-alpha antibody (ab187786, Abcam) was incubated with the slides at 4 °C overnight. After gently rinsing with PBS, the slides were mounted and ready for microscopic imaging. TUENL assays were performed by following a protocol provided by the vendor (FITC-labeled POD, GenScript).
Statistical Methods:
Quantitative data were expressed as mean ± SD. Means were compared using Student’s t-test. P < 0.05 were considered statistically significant.
Supplementary Material
Acknowledgements
The study was supported by an NCI/NIH R00 grant (R00CA153772), a DoD CDMRP grant (CA140666), a UGA startup grant, a UGA-GRU Seed Research grant, an Elsa U. Pardee foundation grant, and an American Cancer Society grant (MSRG-12–034-01-CCE). The authors thank Dr. Muthugapatti K. Kandasamy at UGA for helping us with the confocal microscopy imaging and Dr. John Shields and Stefan Eberhard at UGA for helping us with the critical point dry and the diffused coating of the SEM samples. The authors also thank Julie Nelson for helping us with the flow cytometry analysis.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Wei Tang, Department of Chemistry, University of Georgia, Athens, GA 30602, USA, jinxie@uga.edu.
Dr Zipeng Zhen, Department of Chemistry, University of Georgia, Athens, GA 30602, USA, jinxie@uga.edu.
Dr. Mengzhe Wang, Department of Radiology and Biomedical Research Imaging Center, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA, zibo_li@med.unc.edu
Dr. Hui Wang, Department of Radiology and Biomedical Research Imaging Center, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA, zibo_li@med.unc.edu
Dr. Yen-Jun Chuang, College of Engineering, University of Georgia, Athens, GA 30602, USA
Weizhong Zhang, Department of Chemistry, University of Georgia, Athens, GA 30602, USA, jinxie@uga.edu.
Geoffrey D Wang, Department of Chemistry, University of Georgia, Athens, GA 30602, USA, jinxie@uga.edu.
Trever Todd, Department of Chemistry, University of Georgia, Athens, GA 30602, USA, jinxie@uga.edu.
Taku Cowger, Department of Chemistry, University of Georgia, Athens, GA 30602, USA, jinxie@uga.edu.
Dr. Hongmin Chen, Department of Chemistry, University of Georgia, Athens, GA 30602, USA, jinxie@uga.edu
Lin Liu, Department of Radiology, China-Japan Union Hospital, Jilin University, Changchun, Jilin 130033, China
Zibo Li, Department of Radiology and Biomedical Research Imaging Center, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA, zibo_li@med.unc.edu
Jin Xie, Department of Chemistry, University of Georgia, Athens, GA 30602, USA, jinxie@uga.edu
References
- [1].a) Bessler NM, Vam C Study Writing, Retina 2004, 24, 512; [DOI] [PubMed] [Google Scholar]; b) Qumseya BJ, David W, Wolfsen HC, Clin. Endosc. 2013, 46, 30; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Weiss AA, Wiesinger HA, Owen D, Can. J. Gastroenterol 2006, 20, 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Dolmans DE, Fukumura D, Jain RK, Nat. Rev. Cancer 2003,3, 380; [DOI] [PubMed] [Google Scholar]; b) Eljamel MS, Goodman C, Moseley H, Lasers Med. Sci. 2008, 23, 361; [DOI] [PubMed] [Google Scholar]; c) Fisher AM, Murphree AL, Gomer CJ, Lasers Surg. Med. 1995, 17, 2; [DOI] [PubMed] [Google Scholar]; d) Huang Z, Technol. Cancer Res. Treat. 2005, 4, 283; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Moore CM, Pendse D, Emberton M, Nat. Clin. Pract. Urol. 2009, 6, 18; [DOI] [PubMed] [Google Scholar]; f) Nishiyama N, Morimoto Y, Jang WD, Kataoka K, Adv. Drug Deliv. Rev. 2009, 61, 327. [DOI] [PubMed] [Google Scholar]
- [3].a) Agostinis P, Berg K, Cengel KA, Foster TH, Girotti AW, Gollnick SO, Hahn SM, Hamblin MR, Juzeniene A, Kessel D, Korbelik M, Moan J, Mroz P, Nowis D, Piette J, Wilson BC, Golab J, CA-Cancer J. Clin. 2011, 61, 250; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dougherty TJ, Gomer CJ, Henderson BW, Jori G, Kessel D, Korbelik M, Moan J, Peng Q, J. Natl. Cancer Inst. 1998, 90, 889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Fingar VH, Wieman TJ, Doak KW, Cancer Res. 1990, 50, 2599. [PubMed] [Google Scholar]
- [5].Castano AP, Mroz P, Hamblin MR, Nat. Rev. Cancer 2006, 6, 535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Jin CS, Zheng G, Lasers Surg. Med. 2011, 43, 734; [DOI] [PubMed] [Google Scholar]; b) Konan YN, Gurny R, Allemann E, J. Photochem. Photobiol. B 2002, 66, 89; [DOI] [PubMed] [Google Scholar]; c) Zhen Z, Tang W, Guo C, Chen H, Lin X, Liu G, Fei B, Chen X, Xu B, Xie J, ACS Nano 2013, 7, 6988‥ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Brown JM, Wilson WR, Nat. Rev. Cancer 2004, 4, 437; [DOI] [PubMed] [Google Scholar]; b) Minchinton AI, Tannock IF, Nat. Rev. Cancer 2006, 6, 583‥ [DOI] [PubMed] [Google Scholar]
- [8].Harris AL, Nat. Rev. Cancer 2002, 2, 38. [DOI] [PubMed] [Google Scholar]
- [9].a) Henderson BW, Fingar VH, Cancer Res. 1987, 47, 3110; [PubMed] [Google Scholar]; b) Lee See K, Forbes IJ, Betts WH, Photochem. Photobiol. 1984, 39, 631. [DOI] [PubMed] [Google Scholar]
- [10].Gomer CJ, Razum NJ, Photochem. Photobiol. 1984, 40, 435. [DOI] [PubMed] [Google Scholar]
- [11].Jensen FB, J. Exp. Biol. 2009, 212, 3387. [DOI] [PubMed] [Google Scholar]
- [12].Di Gregorio E, Ferrauto G, Gianolio E, Lanzardo S, Carrera C, Fedeli F, Aime S, ACS Nano 2015, 9, 8239. [DOI] [PubMed] [Google Scholar]
- [13].a) Chen B, Pogue BW, Goodwin IA, O'Hara JA, Wilmot CM, Hutchins JE, Hoopes PJ, Hasan T, Radiat. Res. 2003, 160, 452; [DOI] [PubMed] [Google Scholar]; b) Chen B, Pogue BW, Hoopes PJ, Hasan T, Int. J. Radiat. Oncol. Biol. Phys. 2005, 61, 1216. [DOI] [PubMed] [Google Scholar]
- [14].Bonnett R, Chem. Soc. Rev. 1995, 24, 19. [Google Scholar]
- [15].Garcia AM, Alarcon E, Munoz M, Scaiano JC, Edwards AM, Lissi E, Photochem. Photobiol. Sci. 2011, 10, 507. [DOI] [PubMed] [Google Scholar]
- [16].Zhen Z, Tang W, Chen H, Lin X, Todd T, Wang G, Cowger T, Chen X, Xie J, ACS Nano 2013, 7, 4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Tang W, Zhen Z, Yang C, Wang L, Cowger T, Chen H, Todd T, Hekmatyar K, Zhao Q, Hou Y, Xie J, Small 2014, 10, 1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Baim DS, Grossman W, Grossman’s Cardiac Catheterization, Angiography, and Intervention, Lippincott Williams & Wilkins, Philadelphia, PA, USA: 2006. [Google Scholar]
- [19].Berg CP, Engels IH, Rothbart A, Lauber K, Renz A, Schlosser SF, Schulze-Osthoff K, Wesselborg S, Cell Death Differ. 2001. 8, 1197. [DOI] [PubMed] [Google Scholar]
- [20].Wang C, Sun X, Cheng L, Yin S, Yang G, Li Y, Liu Z, Adv. Mater. 2014, 26, 4794. [DOI] [PubMed] [Google Scholar]
- [21].Yin H, Zhang G, Chen H, Wang W, Kong D, Li Y, Artif Organs 2014, 38, 510. [DOI] [PubMed] [Google Scholar]
- [22].Jin CS, Lovell JF, Chen J, Zheng G, ACS Nano 2013, 7, 2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Blumenthal I,J. R. Soc. Med. 2001, 94, 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wagner MH, Berry RB, J. Clin. Sleep Med. 2007, 3, 313. [PMC free article] [PubMed] [Google Scholar]
- [25].Li Z-B, Chen K, Wu Z, Wang H, Niu G, Chen X, Mol. Imag. Biol. 2009, 11, 415. [DOI] [PubMed] [Google Scholar]
- [26].Boros E, Rybak-Akimova E, Holland JP, Rietz T, Rotile N, Blasi F, Day H, Latifi R, Caravan P, Mol. Pharm. 2014, 11, 617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Liu Y, Hou G, Zhang X, Liu JJ, Zhang S, Zhang J, J. Breast Cancer 2014, 17, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].a) Ihler GM, Glew RH, Schnure FW, Proc. Natl. Acad. Sci. USA 1973, 70, 2663; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Magnani M, Rossi L, Fraternale A, Bianchi M, Antonelli A, Crinelli R, Chiarantini L, Gene Ther. 2002, 9, 749; [DOI] [PubMed] [Google Scholar]; c) Murciano JC, Medinilla S, Eslin D, Atochina E, Cines DB, Muzykantov VR, Nat. Biotechnol. 2003, 21, 891; [DOI] [PubMed] [Google Scholar]; d) Muzykantov VR, Murciano JC, Taylor RP, Atochina EN, Herraez A, Anal. Biochem. 1996, 241, 109‥ [DOI] [PubMed] [Google Scholar]
- [29].a) Hamidi M, Zarrin A, Foroozesh M, Mohammadi-Samani S, J. Control. Release 2007, 118, 145; [DOI] [PubMed] [Google Scholar]; b) Rossi L, Serafini S, Pierige F, Antonelli A, Cerasi A, Fraternale A, Chiarantini L, Magnani M, Expert Opin. Drug Deliv. 2005, 2, 311‥ [DOI] [PubMed] [Google Scholar]
- [30].Moan J, Berg K, Photochem. Photobiol. 1991, 53, 549. [DOI] [PubMed] [Google Scholar]
- [31].a) Hall SS, Mitragotri S, Daugherty PS, Biotechnol. Prog. 2007, 23, 749; [DOI] [PubMed] [Google Scholar]; b) Krantz A, Blood Cells Mol. Dis. 1997, 23, 58; [DOI] [PubMed] [Google Scholar]; c) Muzykantov VR, Taylor RP, Anal. Biochem. 1994, 223, 142. [DOI] [PubMed] [Google Scholar]
- [32].a) Thomlinson RH, Gray LH, Br. J. Cancer 1955, 9, 539; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kizaka-Kondoh S, Inoue M, Harada H, Hiraoka M, Cancer Sci. 2003, 94, 1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Yeom CJ, Goto Y, Zhu Y, Hiraoka M, Harada H, Int.J. Mol. Sci. 2012, 13, 13949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].a) Fens MH, Mastrobattista E, de Graaff AM, Flesch FM, Ultee A, Rasmussen JT, Molema G, Storm G, Schiffelers RM,Blood 2008, 111, 4542; [DOI] [PubMed] [Google Scholar]; b) Grimsley C, Ravichandran KS, Trends Cell Biol. 2003, 13, 648; [DOI] [PubMed] [Google Scholar]; c) Stubbs JD, Lekutis C, Singer KL, Bui A, Yuzuki D, Srinivasan U, Parry G, Proc. Natl. Acad. Sci. USA 1990, 87, 8417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].a) Andersen MH, Graversen H, Fedosov SN, Petersen TE, Rasmussen JT, Biochemistry 2000, 39, 6200; [DOI] [PubMed] [Google Scholar]; b) Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S, Nature 2002, 417, 182. [DOI] [PubMed] [Google Scholar]
- [36].Zwart A, Buursma A, van Kampen EJ, Zijlstra WG, Clin. Chem. 1984, 30, 373. [PubMed] [Google Scholar]
- [37].Huang J, Bu L, Xie J, Chen K, Cheng Z, Li X, Chen X, ACS Nano 2010, 4, 7151. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.