

Abstract
The aim of the study was to investigate the molecular mechanisms in childhood adrenocortical tumors (ACTs), which is still unclear.
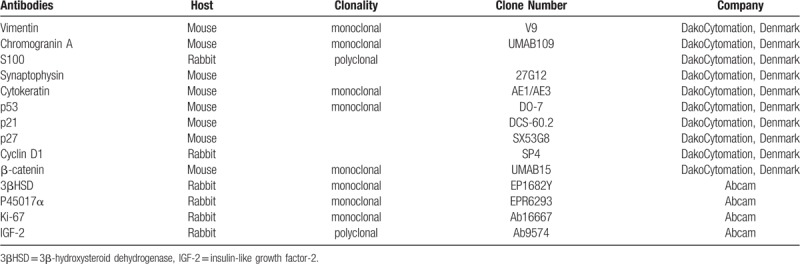
A total of 9 girls and 4 boys with ACTs were enrolled. Relevant clinical features were obtained from records. Immunohistochemistry of vimentin, chromogranin A, S100, synaptophysin, cytokeratin (CK), type 2 3β-hydroxysteroid dehydrogenase (3βHSD), cytochrome P45017α, p53, p21, p27, cyclin D1, Ki-67, insulin growth facter-2 (IGF-2), and β-catenin were undertaken for 13 tumors and 3 adjacent normal tissues. TP53 mutations in exon 2-11 were analyzed for 6 tumors and 3 blood samples.
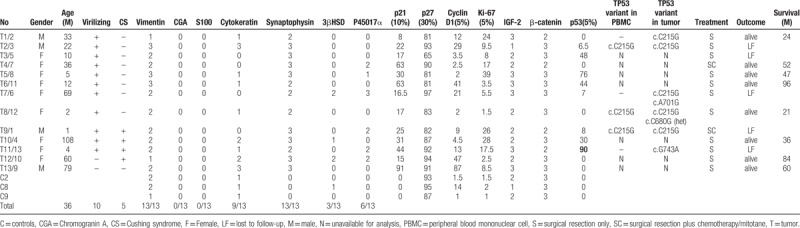
Virilization was the most common presentation (8/13, 61.5%). Immunohistochemically, p53 was positive in 8 of 13 ACTs and none in controls while p21 was positive in 12 of 13 ACTs and none in controls (P = .0036). Ki-67 was positive in 10 of 13 ACTs, but not in normal tissues (P = .0089). Although the expression of p27, cyclin D1, IGF-2 and β-catenin were similar between the ACTs and controls, β-catenin was noted in nuclear of 3 ACTs but not in controls. The difference of type 2 3βHSD and P450c17α was not significant (P > .05, respectively). Four variants of TP53 were identified in the 6 tumors. C215G variant was found in 5 of 6 while A701G and G743A variants were found in 1 case, respectively. A novel C680G variant was also noted in 1 case. It was notable that C215G variant was found in the blood mononuclear cell of 3 patients.
In conclusion, p53 variant and p21 overexpression, and abnormal β-catenin distribution may be involved in the etiology and mechanism of childhood ACTs.
Keywords: adrenocortical tumors, child, Ki-67, p21, TP53
1. Introduction
Childhood adrenocortical tumors (ACTs) are rare and comprise a heterogeneous group of hormone active or inactive disorders with the most common presentation being virilization with or without Cushing syndrome.[1] The exceptional incidence of childhood ACTs in Southern Brazil is probably attributed to the high frequency of germline mutation (p.R337H) of the TP53 tumor-suppressor gene in their population.[2] Childhood ACTs may occur as a component of several hereditary tumor syndromes resulting from abnormalities in genes that encode transcription factors implicated in cell proliferation, differentiation, senescence, apoptosis, and genomic instability.[3] Importantly, the genes involved in these syndromes, such as TP53 in Li-Fraumeni syndrome, insulin-like growth factor-2 (IGF-2) gene in Beckwith-Wiedemann syndrome and β-catenin, appear to contribute to sporadic tumor cases as well.[4] Genomic profiling of human tumors has found activation of cell-cycle programs,[5] supporting the notion that p53-sensitive checkpoints is a critical step in the process of adrenal tumorigenesis.[4,6–9] However, most molecular analysis of ACT of children and adolescents are from Brazilian origin, rare study was reported in China.
Herein, we highlight the molecular events of pediatric adrenocortical tumorigenesis, which may provide additional diagnostic, prognostic utility and guide the development of therapeutic targets.
2. Materials and methods
2.1. Patients
This study comprised 13 pediatric patients with clinically and pathologically confirmed diagnosis of ACT between 2009 and 2018. They were 9 females and 4 males with a female to male ratio of 2.25:1. Their age of diagnosis ranged from 1 to 108 months with a median age of 22 months. Clinical data, including patient age, gender, symptoms, laboratory results, and radiological findings, were collected. A patient was considered to have virilization if a girl had clitorimegaly, hirsutism, deepened voice or male musculature, and if a boy had precocious puberty. Cushing syndrome was considered if he or she had moon facies, weight gain, centripetal distribution of fat, plethora, hypertension, striae and easy bruising.
Informed consent was obtained from parents. This study was approved by the Ethical Committee of the Children Hospital of Zhejiang University School of Medicine.
2.2. Immunohistochemistry
The resected adrenal glands were examined in the pathology laboratory. In brief, after deparaffination and rehydration, sections were subjected to a pre-treatment by heat-induced epitope retrieval in 10 mM EDTA buffer in a pressure cooker. Endogenous peroxidases activity was quenched by 10 minutes incubation in 3% H2O2. Staining for vimentin, chromogranin A, S100, synaptophysin, and cytokeratin (CK), which may helpful for distinguishing the origin of the tumors, were undertaken.[10,11] Immunostaining for enzyme function markers, including cytochrome P45017α and type 2 3β-hydroxysteroid dehydrogenase (3βHSD), and markers for cell cycle and proliferation, including p53, p21, p27, cyclin D1, Ki-67, IGF-2, and β-catenin were performed as well. Slides were incubated for 1 hour at room temperature in appropriately diluted primary antibody (Table 1). Signal amplification was achieved with anti-rabbit or anti-mouse polymer-HRP for 30 minutes at room temperature and developed for 2 minutes with DAB substrate Kit according to the manufacturer's instructions. Nuclei were counterstained with Harris’ hematoxylin for one minute. Immunostaining results were analyzed using a light microscope at high magnification. A semiquantitative H score was calculated by multiplying the staining intensity score with the percentage of positive cells score as described previously.[12] Cytoplasmic or/and nuclear staining intensity was categorized as no staining (0), weak (1), moderate (2), and strong (3). The percentage of positive cells was assessed and scored 0 if 0% were positive, 0.5 if 10% to 49%, and 1 if 50% or more cells were positive. The result was a consensus between reviewing investigators. Three pediatric adrenal glands which had been removed together with renal tumors were used as controls.
Table 1.
Primary antibodies used in immunohistochemical analysis.
2.3. TP53 sequencing
TP53 variant was analyzed by PCR and Sanger sequencing for 6 tumor tissues, 3 corresponding peripheral blood mononuclear cells (PBMC). DNA was extracted according to standard protocols. Primers for exon 2 to 11 of TP53 gene were synthetized (Table 2). PCR amplification was performed in a volume of 20 μl according KAPA2G Robust PCR kit (KAPA Biosystems) protocol. Sequence analysis (ABI 3730XL sequencer) and pathogenic variant identification were performed with Mutation Surveyor DNA variant analysis software (Softgenetics, USA). Variants were checked against the International Agency for Research on Cancer (IARC) p53 database (http://p53.iarc.fr).
Table 2.
Primers of TP53.

2.4. Statistical analysis
Results are given as mean or median and range, or number of patients and proportion of patients. The comparison of IHC features between the tumor and control groups or between tumors with different endocrine phenotype were performed using the Mann–Whitney U test. A P value <.05 was regarded statistically significant. Two-tailed tests were used. Analyses were performed using the GraphPad Prism 4.0 (GraphPad, San Diego, CA).
3. Results
3.1. Clinical features
Among 13 patients, 12 (92.3%) presented hormone-secreting features, including 8 (61.5%) androgen-secreting tumors with virilizing features (Fig. 1), 3 (23.0%) mixed cortisol- and androgen-secreting tumors with both virilizing features and Cushing syndrome, and 1 (7.7%) cortisol-secreting tumor with Cushing syndrome. Only 1 patient (7.7%) did not present any endocrine clinical sign or symptom (Table 3).
Figure 1.
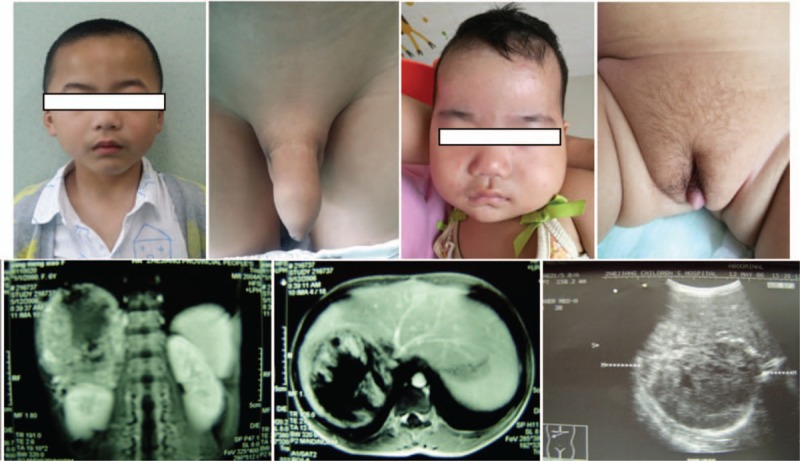
Clinical features and images of patients. (A). Beard presented in case 1. (B). Pubic hair and enlargement of penis in case 1. (C). Acne on the face of case 3. (D). Clitoromegaly and pubic hair in case 3. (E). Coronal magnetic resonance imaging with contrast agent showed a large right heterogeneous mass with a diameter of about 9 cm × 11 cm × 12 cm arising in the right hepatorenal recess. Compression of liver, kidney, inferior vena cava, and venae portal were noted in case 6. (F). Horizontal magnetic resonance imaging for case 6 showed a large right heterogeneous mass in the right hepatorenal recess. (G). Ultrasound for case 6 showed a mass in the right adrenal cortex.
Table 3.
Clinical data, immunohistochemical and TP53 variant analysis results of childhood ACTs.
3.2. Pathologic and immunohistochemical markers
Pathology was performed for all 13 cases (Fig. 2A). Vimentin, marker of adrenal gland, was positive in the cytoplasm with intermediate to strong staining intensity in all 13 tumorous samples and 3 adjacent normal tissues (Fig. 2B). Chromogranin A and S100, markers of the adrenal medullary cells, was negative in all tumors and adjacent normal tissue (Fig. 2C–D). Synaptophysin, regarded as a marker of neuroendocrine cells or tumor, was positive with intermediate or strong staining intensity in all tumorous samples (Fig. 2E), but negative in all 3 adjacent normal tissue. CK was detected in 9 of 13 tumorous samples and all 3 adjacent normal tissues (Fig. 2F) without significant difference (P > .9999), as shown in Tables 3 and 4. Only 3 of 13 tumors and one of 3 controls were positive of 3βHSD (Fig. 2G) without significant difference (P > .05). P450c17α was positive in 6 tumorous samples (Fig. 2H) and negative in all of 3 adjacent normal tissue without significant difference (P > .05), as shown in Tables 3 and 4.
Figure 2.
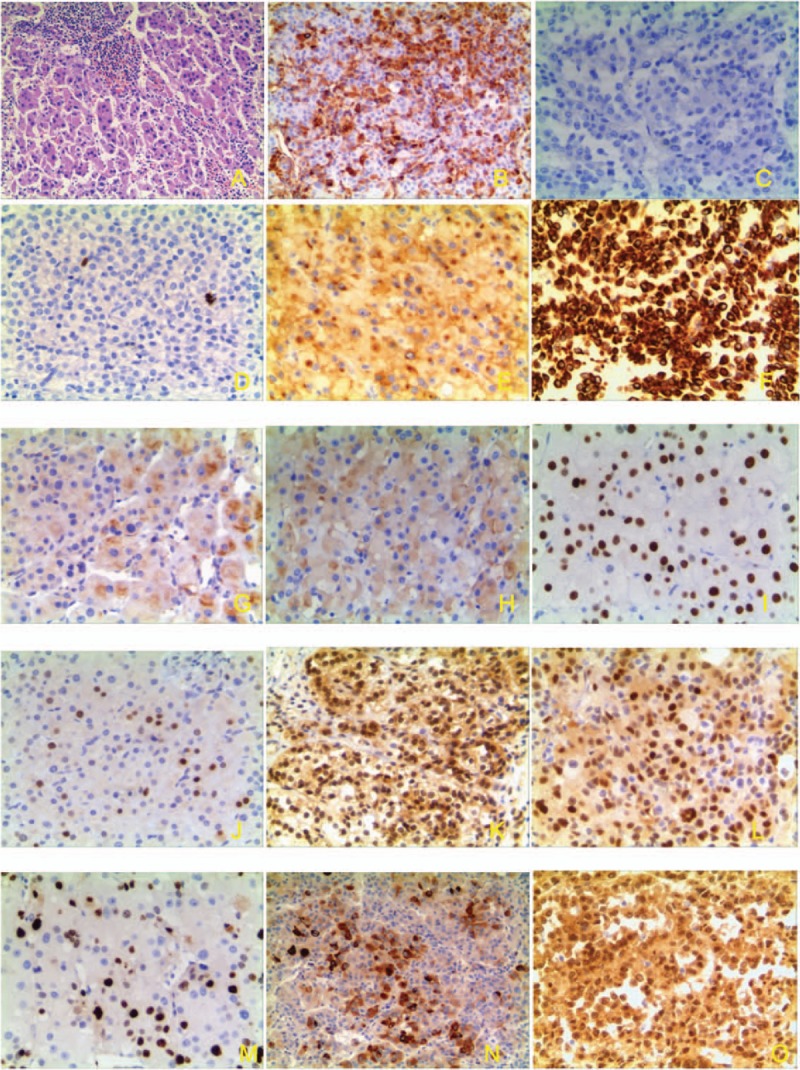
Pathology and immunohistochemistry of ACTs. (A) HE strain of adrenocortical tumors (×100). (B) Vimentin strong staining (cytoplasm) (×100). (C) Negative of chromogranin A (×100). (D) Negative of S100 (×100). (E) Positive of synaptophysin in ACT (cytoplasm) (×100). (F) CK strong staining (nucleus and cytoplasm) (×100). (G). Type 2 3βHSD moderate staining (×100). (H) P45017α weak staining in ACT (×100). (I) P53 stain in nucleus of ACT, (×100). (J) p21 stain in nucleus of ACT (×100). (K). p27 stain in nucleus and cytoplasm of ACT (×100). (L). Cyclin D1 in part nucleus and cytoplasm of ACT (×100). (M). Ki-67 stain (nucleus) in ACT (×100). (N). IGF-2 moderate staining in part cells (nucleus and cytoplasm) of ACT (×100). (O). β-catenin nuclear staining in ACTs (×100).
Table 4.
Immunohistochemical expression analysis in the ACT and control groups.
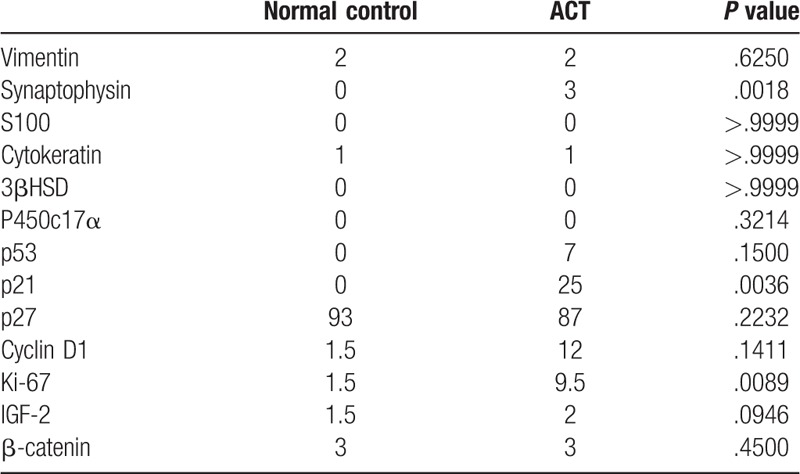
Cell cycle regulators and proliferation markers were evaluated. p53 was positive in 8 of 13 tumors (Fig. 2I) while p21 was positive in 12 of 13 tumors (Fig. 2J) and none of control tissue (P = .0036). Cyclin D1, the regulator of the G1 checkpoint of the cell cycle, was identified in 8 of 13 tumors (Fig. 2L) and 1 of 3 control tissue without significant difference (P = .1411). Ki-67 was positive in 10 of 13 ACTs (Fig. 2M), but not in normal tissues (P = .0089). Although the p27 (Fig. 2K), IGF-2 (Fig. 2N) and β-catenin (Fig. 2O) were similarly expressed in all the ACTs and controls (Tables 3 and 4), we noted that β-catenin accumulation in the nuclear of 3 ACTs samples but not in control.
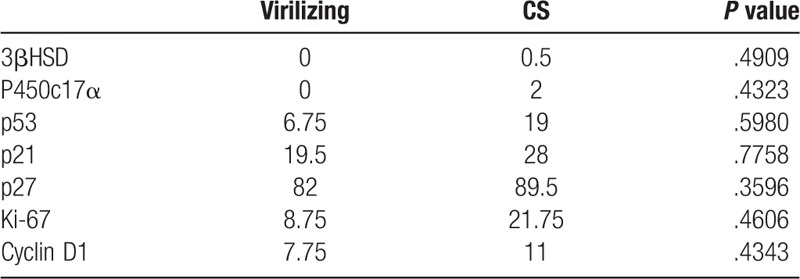
We did not find significant difference of immunohistochemical makers between patients with virilizing and without virilizing, as shown in Table 5.
Table 5.
Immunohistochemical expression analysis in the ACT and control groups with different ACTs.
3.3. TP53 variants
A total of 4 variants of TP53 gene were identified in the 6 timorous tissues (Table 3). A c.C215G (p.Pro72Arg) homozygous variant in exon 4 was found in 5 of 6 cases while c.A701G (p.Tyr234Cys) and c.G743A (p.Arg248Gln) homozygous variant and c.C680G (p.Ser227Cys) heterozygous variant in exon 7 was found in one case, respectively.
3.4. Treatment and outcome
All patients underwent primary surgical excision in our unit and 2 received chemotherapy after resection of the tumor. After discharge, 5 patients were lost to follow-up. The other 8 patients survived without evidence of recurrence with a median follow-up of 49.5 months (range, 21–96 months), shown in Table 1.
4. Discussion
Childhood ACT is relatively common tumor in children with endocrine abnormalities even though this is a rare disease. The etiology and molecular mechanisms of pediatric ACT are still unclear. Involvement of TP53 mutations,[13] Ki-67 and IGF-2 overexpression, and activity of β-catenin signaling[9] have been reported. TP53, a familiar negative regulatory factor in cell growth cycle and tumor suppressor gene, was analyzed by immunostaining and sequencing. In our series, we noted that p53 was immunostaining positive in 61.5% (8/13) tumors while 3 control tissues were all negative. Moreover, TP53 variants were detected in 4 of 6 tumors (66.67%) by sequencing in our series. It was interesting that TP53 variants were found in patients with p53 immunostaining negative (e.g., case 1 and 8). This was consistent with previous report showing a 52-year-old French Canadian man with TP53 germline heterozygous exon-4 polymorphism c.C215G but completely negative immunohistochemical staining for TP53,[14] suggesting that TP53 variants involved in the mechanism of ACTs, but cannot always prolong the half-life of p53. This difference may also be associated the immunohistochemical method, duration from resection to immunohistochemistry, or different variants.[15] Among the 4 variants found in our series, C215G in exon 4 was reported as a most common polymorphism and found in 5 of 6 ACTs samples. It was notable that C215G variant in exon 4 was also found in 3 peripheral mononuclear cells of patients and multiple variant was noted in 2 of 6 cases.
Cell-cycle regulators and proliferative markers have been involved in the mechanism of ACT. Although p27 and IGF-2 maybe involved in the mechanism of tumorigenesis,[16–19] we found they expressed in both the ACT and control tissues without significant difference. This is consistent with a study on transgenic mice that adrenal cortex specific overexpression of IGF-2 did not show tumor formation despite a mild increase in cortical cell proliferation.[18,20] Although no significant difference of cyclin D1 was noted, p21, the cyclin-dependent kinase inhibitor, was remarkably elevated in ACTs. p21 has been thought to contribute p53-mediated tumor suppression. This was consistent with previous report[19] and suggested that p21 was involved in the mechanism of ACT. Ki-67 was regarded as a cellular marker for proliferation and a marker of tumor proliferative and progression as well. In this study, we noted that high positive (10/13) of Ki-67 expression in ACT, but not in normal tissues. A study of 17 adrenocortical carcinomas implied that a Ki-67 index ≥7% was associated with shortened disease-free survival.[21] Although we did not analyze the relationship between Ki-67 index and survival due to small size, our data supported that Ki-67 may be involved in the mechanism of ACT as well. Moreover, we noted that β-catenin accumulation in the nuclear of 3 ACTs samples.
There are several limitations in this study. First, sample size is small. Second, sequencing of p53 was only performed for part of patients. Further investigation with a larger cohort may provide a more definitive picture of the true diagnostic and prognostic value of these potential markers.
In summary, p53 variant, p21 and Ki-67 over expression, and abnormal β-catenin distribution may be involved in the etiology and mechanism of childhood ACTs.
Acknowledgments
We thank the patient and their parents for allowing us to use the data.
Author contributions
Conceptualization: Chaochun Zou.
Data curation: Jun Xu.
Formal analysis: Xiaohui Wu, Chaochun Zou.
Funding acquisition: Chaochun Zou.
Investigation: Jun Xu, Weizhon Gu.
Methodology: Chaochun Zou.
Project administration: Chaochun Zou.
Resources: Jinhu Wang.
Supervision: Chaochun Zou.
Validation: Chaochun Zou.
Writing – original draft: Xiaohui Wu.
Writing – review & editing: Jinhu Wang, Chaochun Zou.
Footnotes
Abbreviations: 3βHSD = 3β-hydroxysteroid dehydrogenase, ACTs = adrenocortical tumors, CK = cytokeratin, IGF-2 = insulin growth facter-2, PBMC = peripheral blood mononuclear cell.
How to cite this article: Wu X, Xu J, Wang J, Gu W, Zou C. Childhood adrenocortical tumor: a clinical and immunohistochemical study of 13 cases. Medicine. 2019;98:46(e17921).
XW and JX contributed equally to this work.
All procedures performed in the studies involving human participants were in accordance with the ethical standards of the institutional committee.
This work is supported by National Natural Science Foundation (81371215 and 81670786) and Zhejiang Provincial Program for the Cultivation of High-Level Innovative Health Talents.
The authors have no conflicts of interests to disclose.
References
- [1].Ribeiro RC, Pinto EM, Zambetti GP, et al. The International Pediatric Adrenocortical Tumor Registry initiative: contributions to clinical, biological, and treatment advances in pediatric adrenocortical tumors. Mol Cell Endocrinol 2012;351:37–43. [DOI] [PubMed] [Google Scholar]
- [2].Faria AM, Almeida MQ. Differences in the molecular mechanisms of adrenocortical tumorigenesis between children and adults. Mol Cell Endocrinol 2012;351:52–7. [DOI] [PubMed] [Google Scholar]
- [3].Soon PS, McDonald KL, Robinson BG, et al. Molecular markers and the pathogenesis of adrenocortical cancer. Oncologist 2008;13:548–61. [DOI] [PubMed] [Google Scholar]
- [4].Pinto EM, Chen X, Easton J, et al. Genomic landscape of paediatric adrenocortical tumours. Nature communications 2015;6:6302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Zheng S, Cherniack AD, Dewal N, et al. Comprehensive pan-genomic characterization of adrenocortical carcinoma. Cancer Cell 2016;30:363. [DOI] [PubMed] [Google Scholar]
- [6].Hantel C, Beuschlein F. Mouse models of adrenal tumorigenesis. Best Pract Res Clin Endocrinol Metab 2010;24:865–75. [DOI] [PubMed] [Google Scholar]
- [7].Pinto EM, Rodriguez-Galindo C, Pounds SB, et al. Identification of clinical and biologic correlates associated with outcome in children with adrenocortical tumors without germline TP53 mutations: a St Jude Adrenocortical Tumor Registry and Children's Oncology Group Study. J Clin Oncol 2017;35:3956–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lefevre L, Bertherat J, Ragazzon B. Adrenocortical growth and cancer. Compr Physiol 2015;5:293–326. [DOI] [PubMed] [Google Scholar]
- [9].Leal LF, Mermejo LM, Ramalho LZ, et al. Wnt/beta-catenin pathway deregulation in childhood adrenocortical tumors. J Clin Endocrinol Metab 2011;96:3106–14. [DOI] [PubMed] [Google Scholar]
- [10].Saeger W. New aspects of tumor pathology of the adrenal glands. Pathologe 2015;36:301–9. [DOI] [PubMed] [Google Scholar]
- [11].Wick MR, Cherwitz DL, McGlennen RC, et al. Adrenocortical carcinoma. An immunohistochemical comparison with renal cell carcinoma. Am J Pathol 1986;122:343–52. [PMC free article] [PubMed] [Google Scholar]
- [12].Olaussen KA, Dunant A, Fouret P, et al. DNA repair by ERCC1 in non-small-cell lung cancer and cisplatin-based adjuvant chemotherapy. N Engl J Med 2006;355:983–91. [DOI] [PubMed] [Google Scholar]
- [13].Kelly M. Corticosteroids and carcinogenesis: a clinical survey. Acta Rheumatol Scand 1959;5:286–90. [DOI] [PubMed] [Google Scholar]
- [14].El Ghorayeb N, Rondeau G, Latour M, et al. Rapid and complete remission of metastatic adrenocortical carcinoma persisting 10 years after treatment with mitotane monotherapy: case report and review of the literature. Medicine 2016;95:e3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wasserman JD, Novokmet A, Eichler-Jonsson C, et al. Prevalence and functional consequence of TP53 mutations in pediatric adrenocortical carcinoma: a children's oncology group study. J Clin Oncol 2015;33:602–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Stojadinovic A, Brennan MF, Hoos A, et al. Adrenocortical adenoma and carcinoma: histopathological and molecular comparative analysis. Mod Pathol 2003;16:742–51. [DOI] [PubMed] [Google Scholar]
- [17].Sakai Y, Yanase T, Hara T, et al. Mechanism of abnormal production of adrenal androgens in patients with adrenocortical adenomas and carcinomas. J Clin Endocrinol Metab 1994;78:36–40. [DOI] [PubMed] [Google Scholar]
- [18].Altieri B, Colao A, Faggiano A. The role of insulin-like growth factor system in the adrenocortical tumors. Minerva Endocrinol 2019;44:43–57. [DOI] [PubMed] [Google Scholar]
- [19].Stojadinovic A, Ghossein RA, Hoos A, et al. Adrenocortical carcinoma: clinical, morphologic, and molecular characterization. J Clin Oncol 2002;20:941–50. [DOI] [PubMed] [Google Scholar]
- [20].Drelon C, Berthon A, Val P. Adrenocortical cancer and IGF2: is the game over or our experimental models limited? J Clin Endocrinol Metab 2013;98:505–7. [DOI] [PubMed] [Google Scholar]
- [21].Morimoto R, Satoh F, Murakami O, et al. Immunohistochemistry of a proliferation marker Ki67/MIB1 in adrenocortical carcinomas: Ki67/MIB1 labeling index is a predictor for recurrence of adrenocortical carcinomas. Endocr J 2008;55:49–55. [DOI] [PubMed] [Google Scholar]