

Abstract
Complex circuit interactions within the nucleus accumbens (NAc) facilitate goal-directed behavior. Medium spiny neurons (MSNs) mediate NAc output by projecting to functionally divergent brain regions, a property conferred, in part, by the differential projection patterns of D1- and D2 dopamine receptor-expressing MSNs. Glutamatergic afferents to the NAc direct MSN output by recruiting feedforward inhibitory microcircuits comprised of parvalbumin (PV)-expressing interneurons (INs). Furthermore, the GABAB heteroreceptor (GABABR), a Gi/o-coupled G-protein-coupled receptor, is expressed at glutamatergic synapses throughout the mesolimbic network, yet its physiological context and synaptic mechanism within the NAc remains unknown. Here, we explored GABABR function at glutamatergic synapses within PV-IN-embedded microcircuits in the NAc core of male mice. We found that GABABR is expressed presynaptically and recruits a noncanonical signaling mechanism to reduce glutamatergic synaptic efficacy at D1(+) and D1(−) (putative D2) MSN subtypes. Furthermore, PV-INs, a robust source of neuronal GABA in the NAc, heterosynaptically target GABABR to selectively modulate glutamatergic transmission onto D1(+) MSNs. These findings elucidate a new mechanism of feedforward inhibition and refine mechanisms by which GABAB heteroreceptors modulate mesolimbic circuit function.
SIGNIFICANCE STATEMENT Glutamatergic transmission in the nucleus accumbens (NAc) critically contributes to goal-directed behaviors. However, intrinsic microcircuit mechanisms governing the integration of these synapses remain largely unknown. Here, we show that parvalbumin-expressing interneurons within feedforward microcircuits heterosynaptically target GABAB heteroreceptors (GABABR) on glutamate terminals. Activation of presynaptically-expressed GABABR decreases glutamatergic synaptic strength by engaging a non-canonical signaling pathway that interferes with vesicular exocytotic release machinery. These findings offer mechanistic insight into the role of GABAB heteroreceptors within reward circuitry, elucidate a novel arm to feedforward inhibitory networks, and inform the growing use of GABABR-selective pharmacotherapy for various motivational disorders, including addiction, major depressive disorder, and autism (Cousins et al., 2002; Kahn et al., 2009; Jacobson et al., 2018; Stoppel et al., 2018; Pisansky et al., 2019).
Keywords: feedforward inhibition, GABAB, nucleus accumbens, parvalbumin interneurons, synaptic plasticity
Introduction
The nucleus accumbens (NAc) is a critical node within the mesolimbic reward network implicated in maladaptive motivational states, including addiction and major depressive disorder (Lüscher and Malenka, 2011; Koob and Volkow, 2016). The NAc orchestrates goal-directed motivational behavior by integrating glutamatergic input from cortical and limbic brain structures (Kalivas, 2009; Turner et al., 2018a). Whereas experience-driven adaptations at glutamatergic inputs drive reward-related behavioral outcomes (Pascoli et al., 2014; LeGates et al., 2018), microcircuit mechanisms governing excitatory gain in the NAc remain largely unidentified. A putative gain control mechanism in the NAc are feedforward inhibitory microcircuits mediated by fast-spiking parvalbumin (PV)-expressing interneurons (PV-INs). Glutamatergic afferents onto medium spiny projection neurons (MSNs), differentiated based on the expression of D1 [D1(+) MSNs] or D2 dopamine receptors [D1(−) MSNs], collateralize onto PV-INs, which exert robust GABAergic control over MSN output (Wright et al., 2017; Scudder et al., 2018). PV-IN-directed feedforward inhibition gates NAc-dependent behavioral output by coordinating time-contingent changes in MSN action potential activity (Yu et al., 2017).
A potential candidate bridging glutamatergic transmission in the NAc to PV-IN-embedded feedforward microcircuits is the GABAB heteroreceptor (GABABR), a Gi/o-coupled G-protein-coupled receptor (GPCR) expressed highly at synapses throughout mesolimbic and striatal networks (Lacey et al., 2005; Edwards et al., 2017). Clinical and preclinical studies of addiction indicate that baclofen (BAC), a selective GABABR agonist, attenuates drug-seeking behavior, drug craving, and relapse (Hotsenpiller and Wolf, 2003; Kahn et al., 2009). In vivo BAC treatment attenuates cocaine-induced dopamine (DA) efflux into the NAc and is accompanied by decreased psychostimulant-induced hyperlocomotion, self-administration, and conditioned place preference (CPP; Roberts and Andrews, 1997; Li et al., 2001; Di Ciano and Everitt, 2003; Voigt et al., 2011). Congruent with these findings, GABABR activity recruits postsynaptic inward-rectifying K+ channels (Kir) channels in the ventral tegmental area to hyperpolarize NAc-projecting DA neurons, reducing functional mesoaccumbens DA output (Cruz et al., 2004; Labouèbe et al., 2007; Edwards et al., 2017). In the NAc, GABABR is likely targeted by GABA from contiguous GABAergic circuits, such as PV-IN microcircuits, to elicit heterosynaptic changes in neurotransmission (Uchimura and North, 1991). In parallel with MSNs, PV-INs receive robust glutamatergic inputs that are required to drive activity-dependent feedforward inhibition (Yu et al., 2017; Scudder et al., 2018). Despite making up 0.5–1.0% of cells in the NAc, PV-INs extensively innervate MSN ensembles to regulate NAc-directed motivational output (Winters et al., 2012; Wright et al., 2017). For example, silencing PV-INs impairs amphetamine-induced locomotor sensitization and CPP, whereas strengthening of synapses onto PV-INs expedites cocaine self-administration (Yu et al., 2017; Wang et al., 2018). Although PV-INs critically regulate NAc-dependent motivational behavior, the synaptic repertoire used by these cells to entrain MSN output is unclear.
We hypothesized that PV-IN-embedded feedforward microcircuits regulate glutamatergic transmission in the NAc by heterosynaptically targeting GABABR. Utilizing transgenic mice, optogenetics, and whole-cell patch-clamp electrophysiology, in combination with rigorous pharmacology, we demonstrate that presynaptic GABABR activity in the NAc core reduces glutamate release probability non-canonically in a SNAP-25-dependent manner that is distinct from similar Gi/o-GPCRs in the NAc core. We find that PV-INs within feedforward inhibitory circuits are a heterosynaptic source of GABA regulating glutamatergic synapses by targeting presynaptically-expressed GABABR. Congruent with the absence of autonomous PV-IN action potential activity, our findings indicate a lack of tonic GABABR activity, suggesting that heterosynaptic targeting of GABABR is activity-dependent. Together, our results provide insight into mechanisms by which GABABR is recruited within a novel feedforward microcircuit to regulate glutamatergic transmission in the NAc.
Materials and Methods
Animals.
Animals were bred and housed at Vanderbilt University Medical Center in accordance to IACUC. Male mice 8–12 weeks of age were used for all electrophysiological experiments. Mice were housed according to sex in groups of 2–5/cage on a 12 h light/dark cycle with ad libitum access to food and water. Breeding cages were given 5LOD chow (PicoLab, LabDiet; 28.7% protein, 13.4% fat, 57.9% carbohydrate) to improve litter viability. For all electrophysiological experiments, C57BL/6J mice were bred to harbor a BAC carrying the tdTomato fluorophore under control of the Drd1a (D1 receptor) promoter. For a subset of experiments, PV-IRES-Cre mice (Pvalbtm1(cre)Arbr) were crossed with conditional channelrhodopsin-2 (ChR2) mice (Ai32(RCL-ChR2(H134R)/EYFP) and Drd1a-tdTomato mice, generating triple transgenic PVCre-cChR2-D1tdTomato (abbreviated PVCre) mice. SNAP25Δ3 transgenic mice lacking the Gβγ-binding motif at the C-terminus of SNAP-25 and wild-type (WT) littermate controls were generously donated to our laboratory by the Heidi Hamm laboratory and colleagues (Vanderbilt University).
Electrophysiology.
Whole-cell voltage-clamp recordings were obtained from D1tdTomato or PVCre mice, as described previously (Joffe and Grueter, 2016; Turner et al., 2018b). Mice were killed under isoflurane anesthesia. Briefly, parasagittal slices (250 μm) containing the NAc core were prepared from whole brain tissue using a Leica Vibratome in oxygenated (95% O2; 5%CO2) ice-cold N-methyl-d-glucamine (NMDG)-based solution (in mm: 2.5 KCl, 20 HEPES, 1.2 NaH2PO4, 25 glucose, 93 NMDG, 30 NaHCO3, 5.0 sodium ascorbate, 3.0 sodium pyruvate, 10 MgCl2, and 0.5 CaCl2-2H2O). Slices were then recovered in NMDG-based recovery solution for 10–15 min at 32°C before being transferred to a chamber containing artificial CSF (ACSF; in mm: 119 NaCl, 2.5 KCl, 1.3 MgCl2-6H2O, 2.5 CaCl2-2H2O, 1.0 NaH2PO4-H2O, 26.2 NaHCO3, and 11 glucose). All experiments were performed using a Scientifica SliceScope Pro System with continuously-perfused 32°C ACSF at 2 ml/min. MSNs in the NAc core were visualized using Scientifica PatchVision software and patched with 3–6 MΩ recording pipettes (P1000 Micropipette Puller) filled with a cesium (Cs+)-based internal solution [in mm: 120 CsMeSO3, 15 CsCl, 8 NaCl, 10 HEPES, 0.2 EGTA, 10 tetraethylammonium (TEA)-Cl, 4.0 Mg-ATP, 0.3 Na-GTP, 0.1 spermine, and 5.0 QX 314 bromide].
D1(+) and D1(−) MSNs were differentiated according to the expression of the tdTomato fluorophore via 530 nm LED light. D1(−) MSNs were distinguished from interneuron cell types based on morphological (size, shape) and biophysical properties (e.g., capacitance, membrane resistance, and AMPAR decay kinetics). In SNAP25Δ3 and WT littermate mice, MSNs were unlabeled and carefully differentiated from other NAc cell types according to the above criteria. Isolated electrically-evoked EPSCs (eEPSCs) were performed in the continuous presence of GABAAR antagonist, picrotoxin (PTX; 50 μm). In PVCre mice, optically-evoked IPSCs (oIPSCs) were isolated by continuously superfusing pan-AMPAR antagonist, NBQX (5 μm), and NMDAR antagonist, d-APV (50 μm), into the ACSF bath. Paired-pulse ratios (PPRs) were obtained within-experiment by delivering two 0.3 ms duration pulses with a 50 ms interstimulus interval and calculating the amplitude ratio of the second eEPSC to the first eEPSC (eEPSC2/eEPSC1). Coefficient of variance (CV) analysis was conducted within-experiment by calculating σ/μ of PSC amplitudes during specified time intervals. To assess cannabinoid receptor type-1 (CB1R) short-term plasticity, depolarization-induced suppression of excitation (DSE) was performed by depolarizing the postsynaptic cell from −70 to +40 mV for 10 s. eEPSCs obtained pre- and post-DSE were obtained with a 5 s interstimulus interval to capture synaptically-evoked short-term plasticity. To quantify the kinetics of the Cd2+-induced blockade of eEPSC amplitude, each experiment was fit with a nonlinear curve to capture the specific time point, T, at which eEPSC amplitude was 50% from baseline. T was then subtracted from the time point coinciding with the end of the baseline to obtain T1/2. mEPSC analysis was performed with Clampfit 10.4 using a stringent best-fit template obtained from preliminary 10 min recording bouts in D1(+) and D1(−) MSNs. Each recording bout yielded a rise/d time (≤3 ms) and amplitude (≥5 pA) selection criteria that was reflected in the overall template score. Series resistance (RS) was monitored continuously during all experiments, with >20% change in RS resulting in the omission of that experiment. Execution of experimental protocols, stimulus control, and data collection were accomplished using Molecular Devices pClamp 10 Analysis software. Monitoring electrical properties of cells was achieved using AxoPatch 500B MultiClamp amplifier and Axon Digidata 1550 low-noise data acquisition digitizer. Responses were filtered at 2 kHz and digitized at 10 kHz. Optical stimulation of ChR2-expressing cells was achieved using a CoolLED pE-100 LED excitation system. Four hundred and eighty nanometers of light at variable intensities (5–40%) was pulsed through the 40× high-power objective at 0.1 Hz with a duration of 0.3–0.5 ms.
Pharmacology.
(RS)-Baclofen, SCH 50911, CGP 7930, forskolin, CdCl2, BaCl2, LY 341495, LY 379268, ω-conotoxin GVIA(ω-CTx), ω-agatoxin IVA(ω-AgTx), WIN 55,212–2, H89, 4-aminopyridine, and tiagabine were purchased from Tocris Bioscience. PTX and N-ethylmaleimide were purchased from Sigma-Aldrich.
Statistics and data analysis.
Electrophysiological experiments were analyzed using Clampfit 10.4 and GraphPad Prism v7.0. Changes in baseline eEPSC/oIPSC amplitude, CV, and PPR were calculated by comparing mean values during 5 min intervals specified in each time course to baseline PPR and CV values. A depression was defined as a significant difference in eEPSC or oIPSC amplitude from baseline calculated during the time interval specified in the recording. For specific oIPSC experiments at PV-IN-to-MSN synapses, cells were rendered BAC-responsive (+) if BAC application resulted in a significant depression in oIPSC amplitude from baseline. To separate BAC(+) from BAC(−) negative synapses, a threshold criterion was set at >35% depression from baseline. Long-term depression (LTD) was defined as a significant difference in eEPSC or oIPSC amplitude from baseline that persisted in the presence of GABABR antagonist, SCH 50911. After obtaining each dataset, Shapiro–Wilk tests were performed to assess normality. Data depicted in Figures 1–8 were determined to be normally distributed. Thus, paired or unpaired t tests were used to analyze statistical differences between datasets. Sidak's post hoc analyses were used for analyses requiring multiple comparisons. Figure 9 depicts data that were determined to not be normally distributed, consistent with separable populations of PV-IN-to-MSN synapses. Power analyses were performed with preliminary data during the acquisition of each new dataset. The sample size obtained from each power analysis calculation was then compared with sample sizes reported in the literature for similar experiments. Errors bars depicted in figures represent SEM. For all analyses, α was set as 0.05, with p values < α indicating a statistically significant difference.
Figure 1.

GABABR activity reduces synaptic efficacy at glutamatergic synapses onto D1(+) and D1(−) MSNs in the NAc core. A, Schematic of parasagittal D1tdTomato mouse brain slice outlining the recording area within the dorsomedial NAc core. B, C, Representative experiments and traces of eEPSCs obtained from D1(+) MSNs (blue circles) and D1(−) MSNs (open circles). Scale bars: left, 300 pA/50 ms; right, 100 pA/50 ms. D, Time course of normalized eEPSCs obtained from D1(+) and D1(−) MSNs in the presence of GABABR agonist, BAC (3 μm), followed by GABABR antagonist, SCH 50911 (5 μm). BAC decreased eEPSC amplitude that returns to baseline in the presence of SCH 50911. E, Average eEPSC amplitude following BAC (t = 20–25) and SCH 50911 (t = 35–40). F, BAC dose–response curve (200 nm, 600 nm, 3 and 10 μm) obtained from D1(+) MSNs and D1(−) MSNs showing increased sensitivity to BAC at D1(+) MSNs. Note: 3 μm values obtained from averaged eEPSC values in D. G, Rough traces NMDAR-mediated eEPSCs obtained at +40 mV from D1(+) and D1(−) MSNs in the continuous presence of NBQX. Scale bars, 100 pA/100 ms. H, Time course summary and average NMDAR eEPSCs following BAC (t = 15–20 min). I, Prior application of SCH 50911 alone does not significantly alter eEPSC amplitude and blocks the actions of BAC. Error bars indicate SEM. *p < 0.05.
Figure 8.

PV-expressing interneurons preferentially decrease glutamatergic transmission onto D1(+) MSNs via heterosynaptic GABABR signaling. A, Left: model depicting breeding scheme of triple transgenic mouse line used to examine heterosynaptic plasticity in the NAc core. Right: synaptic model of heterosynaptic electrophysiological configuration of eEPSCs at D1(+) and D1(−) MSN synapses obtained via voltage-clamp at −70 mV. PV-INs were stimulated optically at 30 Hz for 5 min. B, Top, Representative traces of PTX (50 μm)-sensitive oIPSCs recorded in D1(+) and D1(−) MSNs. Scale bar, 300 pA/50 ms. Bottom, High-fidelity train of oIPSCs delivered at 30 Hz recorded in MSNs. Scale bar, 30 pA/0.5 s. C, D, Representative experiments and traces of eESPCs obtained from D1(+) MSNs (blue circles) and D1(−) MSNs (open circles) pre- and post-LFSPV (in PTX). LFS denotes optically-evoked stimulation at 30 Hz for 5 min. Scale bar, 200 pA/50 ms. E, Time course of normalized eEPSCs obtained from D1(+) and D1(−) MSNs pre- and post-LFSPV. F, Graph of average eEPSC amplitude post-LFS (blue; t = 15–20 min). G, H, Time course summary and graph of normalized eEPSCs obtained from D1(+) MSNs in normal ACSF (line), SCH 50911 (black dots), and CGP 7930 (blue). I, Graph depicting CV post-LFSPV (t = 15–20 min) in normal aCSF and CGP 7930 (n = 4). J, K, Time course summary and graph of normalized eEPSCs obtained from D1(+) MSNs in normal aCSF (line) and CGP 7930 (open circles). L, Graph depicting CV post-LFSPV (t = 15–20 min) in normal ACSF and CGP 7930. Error bars indicate SEM. *p < 0.05.
Figure 9.
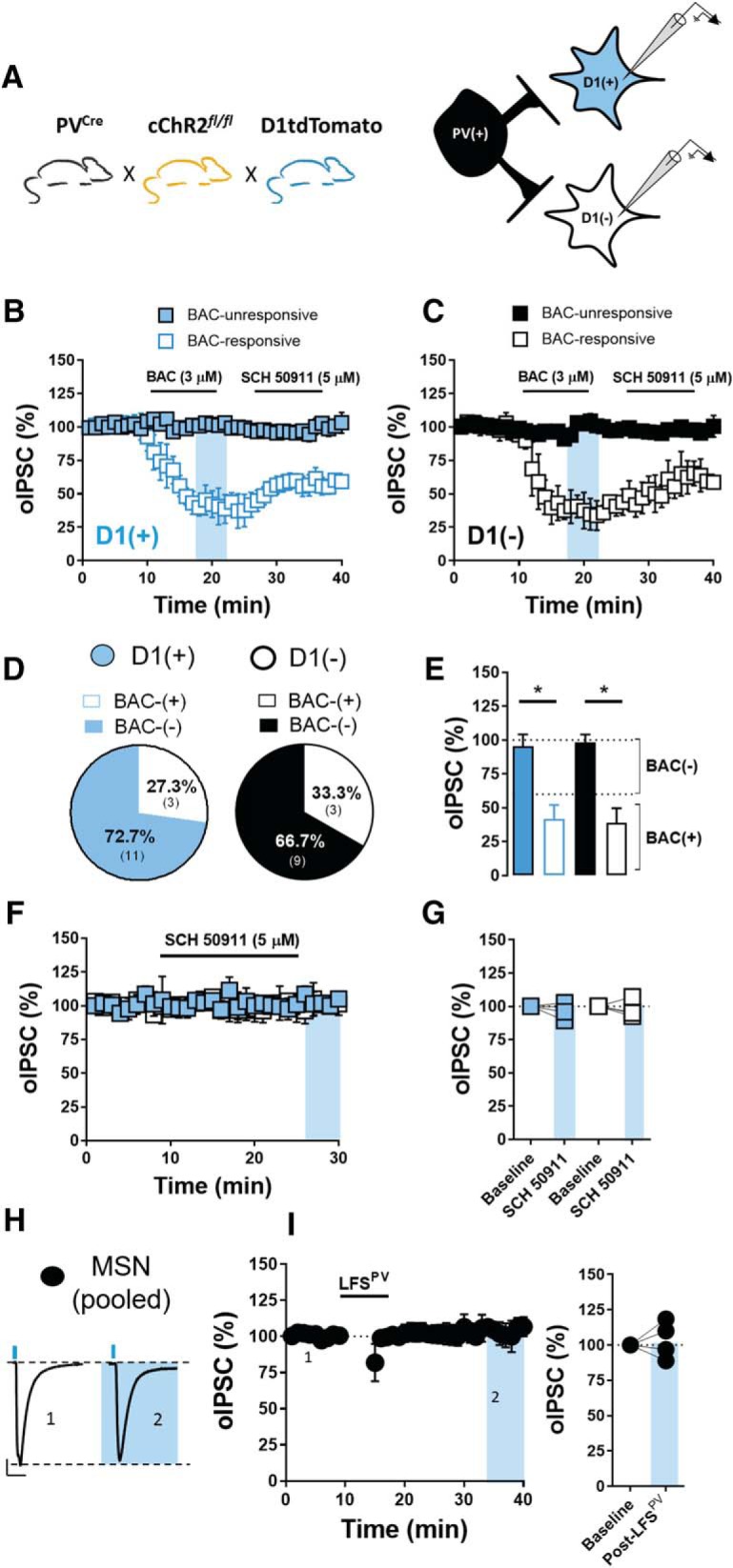
GABAergic transmission at PV-IN-to-D1(+) and D1(−) MSN synapses is largely BAC-insensitive and unchanged by LFS. A, Schematic of breeding scheme and electrophysiological configuration permitting optogenetic examination of GABAergic transmission at PV-IN-to-D1(+) MSN synapses (blue squares) and D1(−) MSN synapses (black squares) in the NAc core. B, Time course summary at PV-IN-to-D1(+) MSN synapses showing that BAC decreases oIPSC amplitude to elicit inhibitory long-term depression at a minority of synapses [BAC-response (BAC+), open squares; BAC-unresponsive (BAC−), blue filled squares]. C, Time course summary at PV-IN-to-D1(−) MSN synapses showing that BAC decreases oIPSC amplitude to elicit inhibitory long-term depression at a minority of synapses (BAC+, open squares; BAC−, black filled squares). D, Pie chart representation of BAC+/BAC− synapses onto D1(+) and D1(−) MSNs. Note that the pie-chart data were obtained following pharmacological characterization of BAC(+) and BAC(−) at PV-IN-to-MSN synapses. E, Quantification of average oIPSC amplitude depicting separation between BAC+ and BAC− PV-IN-to-MSN synapses. F, G, Time course summary and average oIPSC amplitude showing that bath application of GABABR antagonist, SCH 50911, alone does not alter oIPSC amplitude at PV-IN-to-D1(+) and D1(−) MSN synapses. H, Representative traces of oIPSCs pre- and post-LFSPV at pooled MSN synapses. I, Time course summary and quantification of average oIPSC amplitude pre- and post-LFSPV showing that LFSPV does not alter oIPSC amplitude at PV-IN-to-MSN (pooled) synapses. Error bars indicate SEM. *p < 0.05.
Results
Presynaptic GABABR activity reduces synaptic efficacy at glutamatergic synapses onto D1(+) and D1(−) MSNs in the NAc core
To determine whether GABABR activity modulates synaptic efficacy at glutamatergic synapses in the NAc core, we performed whole-cell voltage-clamp recordings in ex vivo brain slice preparations from D1tdTomato BAC transgenic reporter mice. Expression of the red-florescent protein, tdTomato (tdT), is driven by the D1 dopamine receptor promoter, with tdT-expressing cells indicating D1(+) MSNs and tdT-lacking cells indicating D1(−) MSNs (putative D2 receptor-expressing MSNs; Fig. 1A; Lim et al., 2012; Rothwell et al., 2014; Joffe and Grueter, 2016; Kashima and Grueter, 2017; Turner et al., 2018b). Electrically-evoked EPSCs (eEPSCs) were isolated by incorporating GABAA receptor (GABAAR) antagonist, PTX (50 μm), into the ACSF bath. Following a stable 10 min eEPSC baseline, GABABR agonist, BAC (3 μm), was superfused into the bath for 10 min, resulting in a robust depression in eEPSC amplitude at D1(+) and D1(−) MSNs that was indistinct between cell types (Fig. 1B–E; D1(+) BAC: 35.7 ± 4.1%, n = 7, p < 0.0001; D1(−) BAC: 32.9 ± 3.8%, n = 7, p < 0.0001). Subsequent application of GABABR antagonist, SCH 50911 (5 μm), reversed the BAC-induced depression to baseline at D1(+) and D1(−) MSNs, indicating a lack of GABABR-induced LTD (GABABR-LTD) at local glutamatergic synapses (Fig. 1B–E; D1(+): 103.5 ± 6.9%, n = 6, p = 0.61; D1(−): 99.6 ± 1.7%, n = 6, p = 0.80).
We next examined whether GABABR activity modulates pharmacologically-isolated NMDA receptor (NMDAR)-mediated eEPSCs obtained at +40 mV in D1(+) and D1(−) MSNs. Indeed, BAC application resulted in a robust decrease in NMDAR eEPSC amplitude at D1(+) and D1(−) MSN synapses (Fig. 1G,H; D1(+): 35.29 ± 10.2%, n = 4, p = 0.0052; D1(−): 36.27 ± 9.36%, n = 6, p = 0.0007). To determine whether synapses onto D1(+) and D1(−) MSNs are differentially sensitive to GABABR activation, we obtained a dose–response curve with BAC concentrations ranging from 200 nm-10 μm. Whereas higher concentrations (3–10 μm) resulted in an equivalent decrease in eEPSC amplitude at both MSN subtypes, lower concentrations (200–600 nm) resulted in a greater decrease in eEPSC amplitude at D1(+) than D1(−) MSN synapses (Fig. 1F; 200 nm, D1(+): 55.55 ± 4.80%, n = 5; D1(−): 72.46 ± 4.35%, n = 6; 600 nm, D1(+): 43.10 ± 5.72%, n = 5; D1(−): 59.82 ± 2.76%, n = 4; 3 μm, reported above; 10 μm, D1(+): 18.21 ± 4.16%, n = 3; D1(−): 21.38 ± 1.29%, n = 4; two-way ANOVA, effect of MSN subtype: F(1,28) = 8.688, p = 0.006). Prior application of SCH 50911 did not alter basal eEPSC amplitude and completely blocked the BAC-induced depression at both MSN subtypes (Fig. 1I, D1(+): 99.39 ± 3.95%, n = 5, p = 0.74; 100.66 ± 3.78%, n = 4, p = 0.21).
GABABR is expressed throughout the mesolimbic reward network with diverse presynaptic and postsynaptic sites of action (Cruz et al., 2004; Pitman et al., 2014; Edwards et al., 2017). Given that GABABR is presynaptically-expressed at MSN-to-MSN collateral synapses (Dobbs et al., 2016), we performed multiple electrophysiological measurements to determine the synaptic locus of GABABR at glutamatergic synapses in the NAc core. We first assessed changes in paired-pulse ratio (PPR) and coefficient of variance (CV), metrics which inversely correlate with presynaptic neurotransmitter release probability. BAC significantly increased PPR and CV at D1(+) and D1(−) MSN synapses that returned to baseline in the presence of SCH 50911 (Fig. 2A,B; PPR = D1(+) baseline: 1.19 ± 0.04, D1(+) BAC: 1.8 ± 0.2, D1(+) SCH: 1.16 ± 0.08, n = 8, one-way RM ANOVA, drug effect: F(2,21) = 13.85, p = 0.0001; Sidak's post hoc analysis, BAC: p = 0.004; D1(−) baseline: 0.97 ± 0.07, D1(−) BAC: 1.69 ± 0.20, D1(−) SCH: 1.02 ± 0.08, n = 8, one-way RM ANOVA, drug effect: F(2,21) = 11, p = 0.0005; Sidak's post hoc analysis, BAC: p = 0.008; CV = D1(+) baseline: 0.06 ± 0.01, D1(+) BAC: 0.51 ± 0.07, D1(+) SCH: 0.09 ± 0.02, n = 8, one-way RM ANOVA, drug effect: F(2,18) = 37.48, p < 0.0001; Sidak's post hoc analysis, BAC: p = 0.001; D1(−) baseline: 0.05 ± 0.01, D1(−) BAC: 0.55 ± 0.05, D1(+) SCH: 0.13 ± 0.03, n = 8, one-way RM ANOVA, drug effect: F(2,18) = 68.36, p < 0.0001; Sidak's post hoc analysis, BAC: p = 0.001). This experiment performed in a Cs+/TEA-free, K+-loaded internal solution resulted in a similar change in PPR and CV without altering holding current or membrane resistance, suggesting a presynaptic change in glutamate release probability that is unaccompanied by a postsynaptic K+ conductance (data not shown). We next examined the effects of BAC on tetrodotoxin (500 nm)-insensitive miniature EPSCs (mEPSCs). Consistent with a presynaptic site of action, BAC significantly decreased mEPSC frequency without altering mEPSC amplitude (Fig. 2C–F; mEPSC frequency = D1(+) baseline: 1.38 ± 0.022 Hz, D1(+) BAC: 0.82 ± 0.08 Hz, n = 9, p = 0.027; D1(−) baseline: 2.53 ± 0.0332 Hz, D1(−) BAC: 1.28 ± 0.15 Hz, n = 9, p = 0.0014; mEPSC amplitude = D1(+) baseline: −19.61 ± 0.88 pA, D1(+) BAC: −19.35 ± 0.82 pA, n = 9, p = 0.846; D1(−) baseline: −20.49 ± 0.61 pA, D1(−) BAC: −20.13 ± 0.82 pA, n = 9, p = 0.666). These data rigorously support a presynaptic localization of GABABR at glutamatergic synapses onto D1(+) and D1(−) MSNs in the NAc core.
Figure 2.
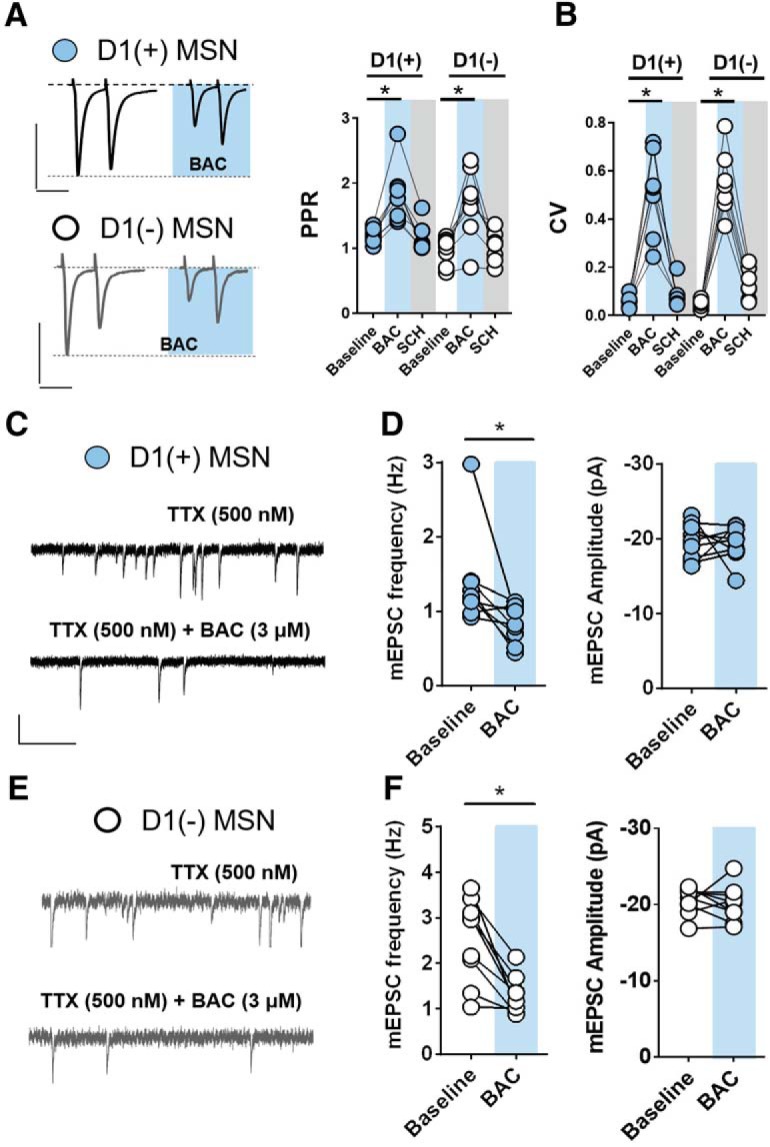
GABABR is functionally expressed at presynaptic loci at glutamatergic synapses onto both MSN subtypes in the NAc core. A, Representative traces of 50 ms ISI paired pulse eEPSCs obtained from D1(+)MSNs (blue circles) and D1(−) MSNs (open circles) at baseline and in the presence of BAC. Scale bars: top, 200 pA/50 ms; bottom, 100 pA/50 ms. BAC application increases PPR and (B) CV at D1(+) and D1(−) MSNs. C, E, Representative traces of TTX-insensitive mEPSCs pre- and post-BAC application at D1(+) MSNs (black) and D1(−) MSNs (gray). Scale bar, 20 pA/1 s. D, F, BAC decreases mEPSC frequency but not amplitude at D1(+) MSNs and D1(−) MSNs. Error bars indicate SEM. *p < 0.05.
GABABR activation recruits non-canonical intracellular signaling mechanisms to reduce glutamate release probability at both MSN subtypes
Presynaptic GABAB heteroreceptors canonically reduce neurotransmitter release probability by shifting the voltage dependence of voltage-gated Ca2+ channels (VGCCs; Kupferschmidt and Lovinger, 2015; Terunuma, 2018). To begin to interrogate this mechanism in the NAc core, we first tested whether prior GABABR activation diminishes broad-spectrum blockade of VGCCs by cadmium (Cd2+; 100 μm). Prior application of BAC had no effect on the amplitude or kinetics (Cd2+-blockade T1/2; see Materials and Methods) with which Cd2+ blocked evoked glutamatergic transmission (Fig. 3A–D; pooled MSNs, BAC: 9.08 ± 3.15%, n = 5; ACSF: 10.11 ± 1.35%, n = 5, p = 0.509; BAC Cd2+-T1/2: 0.67 ± 0.15 min, n = 6; ACSF Cd2+-T1/2: 0.65 ± 0.13 min, n = 6, p = 0.907). To specifically rule out subtype-selective actions at VGCCs, we tested whether GABABR reduces glutamate release probability by inhibiting N- or P/Q-type VGCCs. We examined the contribution of N-type VGCCs by superfusing selective N-type VGCC blocker, ω-CTx (800 nm), into the ACSF bath before BAC. ω-CTx significantly reduced eEPSC amplitude at D1(+) and D1(−) MSNs but failed to occlude the effects of BAC (Fig. 3E,J; D1(+): 35.11 ± 3.35%, n = 4, p = 0.9953; D1(−) 34.83 ± 3.99%, n = 4, p = 0.9999). To determine whether GABABR instead couples selectively to P/Q-type VGCCs, we repeated the above experiment with selective P/Q-type VGCC blocker, ω-AgTx (200 nm). ω-AgTx also resulted in a significant reduction in baseline eEPSC amplitude at D1(+) and D1(−) MSN synapses, consistent with previous reports showing that glutamatergic transmission in the NAc is mediated by N- and P/Q-type VGCCs. Prior ω-AgTx application also had no effect on the BAC-induced decrease in eEPSC amplitude at both MSN subtypes (Fig. 3E,J; pooled MSNs: 29.08 ± 3.15%, n = 3, p = 0.593). These findings suggest that GABABR reduces glutamate release probability independently of N- and P/Q-type VGCCs. Collectively, these findings suggest that the presynaptic mechanism of GABABR at glutamatergic synapses in the NAc core is largely VGCC-independent.
Figure 3.

GABABR activation recruits a non-canonical intracellular signaling mechanism to reduce glutamate release probability onto MSNs in the NAc core. A, Representative experiment and rough traces of the Cd2+-induced blockade of eEPSC from pooled MSNs the time interval from which T1/2 is obtained. B, Time course summary in ACSF and BAC (3 μm) -infused ACSF. C, D, Quantified kinetics (pink-shaded region) and magnitude of the Cd2+ block (blue-shaded region). Scale bar (for all traces), 100 pA/50 ms. E, Time course summary and representative traces showing that N-type VGCC blocker, ω-CTx (800 μm), failed to occlude the effects of BAC on eEPSC amplitude at D1(+)MSNs (blue circles) and D1(−) MSNs (open circles; t = 45–50 min, blue-shaded region, averaged). Black circles indicate time course summary for pooled MSNs of experiments performed with P/Q-type VGCC blocker, ω-AgTx (200 nm), instead of ω-CTx. ω-AgTx also failed to occlude the effects of BAC. F, Time course summary and representative traces showing effect of Kir channel blocker, Ba2+ (1 mm), on BAC-induced decrease in eEPSC amplitude at D1(+) and D1(−) MSNs. G, Time course summary and representative traces showing effect of AC activator, forskolin (10 μm), on BAC-induced decrease in eEPSC amplitude at D1(+) and D1(−) MSNs. H, Time course summary and representative traces showing effect of cell-permeant PKA inhibitor, H89 (10 μm), on BAC-induced decrease in eEPSC amplitude at D1(+) and D1(−) MSNs. I, Time course summary representative traces showing that NEM (50 μm) blunted but did not block the effects of BAC on eEPSC amplitude at D1(+) and D1(−) MSNs. J, Summary graph of BAC-induced decrease in eEPSC amplitude at D1(+) and D1(−) MSN synapses following various pharmacological manipulations. Error bars indicate SEM. *p < 0.05, n.s., not significant.
We next asked whether GABABR activity reduces synaptic efficacy by activating G-protein-coupled Kir channels, a downstream effector targeted by several classes of Gi/o-coupled GPCRs, including GABABR (Ladera et al., 2008). BAC application in the presence of Ba2+ (1 mm), a nonselective Kir channel blocker, resulted in a decrease in eEPSC amplitude at D1(+) and D1(−) MSNs that was similar to control conditions (Fig. 3F,J; D1(+): 27.12 ± 5.67%, n = 4, p = 0.4038; D1(−) 31.71 ± 6.67, n = 5, p = 0.9976). Having ruled out mechanisms mediated by classical Gβγ signaling, we next determined whether GABABR activation mobilizes Gαi to inhibit adenylyl cyclase (AC) function. Bath-application of AC activator, forskolin (1 μm), had no effect on the BAC-induced decrease in eEPSC amplitude at both MSN subtypes (Fig. 3G,J; D1(+): 35.99 ± 5.11%, n = 4, p = 0.9994; D1(−) 32.72 ± 3.91%, n = 4, p = 0.9999). Furthermore, prior application of cell-permeant protein kinase A (PKA) inhibitor, H89 (10 μm), did not occlude the effects of BAC at D1(+) and D1(−) MSN synapses (Fig. 3H,J; D1(+): 30.65 ± 3.36%, n = 5, p < 0.001; D1(−): 26.89 ± 6.04%, n = 5, p = 0.8871). These data collectively suggest a mechanism by which GABABR engages a non-canonical signaling pathway to influence presynaptic function. In line with this hypothesis, disabling Gi/o-GPCR function with N-ethylmaleimide (NEM; 50 μm) diminished but did not block the BAC-induced decrease in eEPSC amplitude (Fig. 4I,J; D1(+): 58.38 ± 10.27%, n = 6, one-way RM ANOVA, BAC effect: F(2,16) = 33.49, p < 0.001; D1(+) Sidak's post hoc analysis for BAC in ACSF vs BAC in NEM, p = 0.035; D1(−): 63.49 ± 6.54%, n = 6, one-way RM ANOVA, BAC effect: F(2,16) = 73.13, p < 0.001; D1(−) Sidak's post hoc analysis for BAC in ACSF vs BAC in NEM, p = 0.001).
Figure 4.

GABABR is functionally distinct from mGluRs and CB1R at glutamatergic synapses in the NAc core. A, Representative experiment and traces showing that group II mGluR agonist, LY379268 (200 nm), fails to occlude the effects of BAC on eEPSC amplitude at D1(+) and D1(−) MSNs. Scale bar (for all traces), 100 pA/50 ms. B, LY379268 application increases PPR and CV at D1(+) and D1(−) MSNs, consistent with a presynaptic locus of expression. C, Time course summary showing the effects of BAC on eEPSC amplitude renormalized to stabilized baseline in the presence of LY379268 (gray). D, Representative traces and time course summary showing that prior application of pan-mGluR antagonist, LY341495, does not prevent the BAC-induced decrease in eEPSC amplitude. E, Summary graph quantifying the effects of mGluR-specific pharmacological manipulations on GABABR function (averaged at t = 35–40 min; blue). F, Representative experiment and traces showing that CB1/2R agonist, WIN 55–212 (1 μm), depresses eEPSC amplitude at D1(+) and D1(−) MSNs but fails to occlude the effects of BAC. Scale bar (for all traces), 50 pA/50 ms. G, Bath application of WIN 55–212 increased PPR and CV at glutamatergic synapses onto D1(+) and D1(−) MSNs, confirming the canonical presynaptic expression of CB1R in the NAc. H, Time course summary and quantification showing the effects of BAC on eEPSC amplitude renormalized to stabilized baseline in the presence of WIN 55–212 (gray). I–L, Time course summaries and quantification (obtained at t = 60 s) of DSE followed by DSE in the presence of BAC for D1(+) and D1(−) MSNs. All DSE experiments performed in pairs such that DSE was repeated within-cell in the presence of BAC. Error bars indicate SEM. *p < 0.05, n.s., not significant.
We next examined whether group II mGluRs interact functionally with GABABR at presynaptic loci to reduce glutamate release probability. Presynaptically-expressed mGluRs in the NAc have previously been shown to decrease vesicular release by selectively targeting P/Q-type VGCCs (Robbe et al., 2002; Mato et al., 2008). Given that ω-AgTx failed to occlude the effects of BAC, we predicted that GABABR functions through a different pathway than group II mGluRs. Bath-application of selective group II mGluR agonist, LY379268 (200 nm), resulted in a significant reduction in eEPSC amplitude that was accompanied by an increase in PPR and CV, consistent with the presynaptic localization of group II mGluRs in the NAc core (Fig. 4A,B; PPR = D1(+) baseline: 1.19 ± 0.07, D1(+) LY, 1.70 ± 0.14, n = 4, p = 0.007; D1(−) baseline: 1.08 ± 0.064, D1(−) LY: 1.64 ± 0.14, n = 5, p = 0.0246; CV = D1(+) baseline: 0.03 ± 0.01, D1(+) LY: 0.41 ± 0.09, n = 5, p = 0.0198; D1(−) baseline: 0.12 ± 0.01, D1(−) LY: 0.35 ± 0.07, n = 4, p = 0.0141). Subsequent application of BAC decreased eEPSC amplitude at D1(+) and D1(−) MSNs that was also indistinct from control conditions, suggesting that presynaptic group II mGluRs and GABABR recruit distinct intracellular effectors to reduce glutamatergic transmission in the NAc core (Fig. 4C,E; D1(+): 34.45 ± 6.91%, n = 4, p = 0.6412; D1(−): 32.02 ± 4.53%, n = 4, p = 0.1032). In the cerebellum, GABABR couples to presynaptic metabotropic glutamate receptors (mGluR1) such that GABABR activation augments mGluR sensitivity to synaptic glutamate levels (Tabata and Kano, 2006, 2010). To determine whether a similar relationship exists in the NAc, LY3431495 was superfused at a concentration that antagonizes all mGluRs (100 μm). In the presence of LY3431495, BAC application resulted in a similar reduction in eEPSC amplitude at D1(+) and D1(−) MSNs relative to control conditions (Fig. 4D,E; D1(+): 39.74 ± 4.2%, n = 5, p = 0.999; D1(−) 43.07 ± 5.55%, n = 5, p = 0.4951). These data strongly suggest that GABABR functions independently of mGluRs in the NAc core.
We next examined whether GABABR shares a common intracellular mechanism with CB1R, the cognate receptor for endogenous cannabinoids (eCBs), such as 2-arachidonylglycerol (2-AG) and anandamide. We selected CB1R because it is the most ubiquitously expressed Gi/o-GPCR in the mammalian CNS, generally restricted to presynaptic domains, and critically regulates excitatory transmission in the striatum (Robbe et al., 2003; Grueter et al., 2010; Castillo et al., 2012). To ascertain the functional difference between CB1R and GABABR, we first looked at whether CB1/2R agonist, WIN 55–212 (1 μm), occludes the effects of BAC on eEPSC amplitude. WIN 55–212 resulted in a significant decrease in eEPSC amplitude that was accompanied by an increase in PPR and CV (Fig. 4F,G; PPR = D1(+) baseline: 1.31 ± 0.09, D1(+) WIN: 1.55 ± 0.15, n = 4, p = 0.0439); D1(−) baseline: 1.44 ± 0.80, D1(−) WIN: 1.75 ± 0.18, n = 5, p = 0.0336); CV = D1(+) baseline: 0.010 ± 0.003, D1(+) WIN 0.22 ± 0.05, n = 4, p = 0.02771; D1(−) baseline: 0.12 ± 0.018, D1(−) WIN: 0.29 ± 0.28, n = 5, p = 0.0079). However, WIN 55–212 failed to occlude BAC at synapses onto both MSN subtypes, suggesting that presynaptically-expressed CB1R and GABABR also modulate presynaptic release probability via distinct intracellular mechanisms (Fig. 4H; D1(+): 35.60 ± 6.78%, n = 4, p = 0.6412; D1(−) 38.07 ± 4.71%, n = 5, p = 0.1032). We next tested the effects of BAC on DSE, a CB1R-dependent form of short-term plasticity that transiently decreases glutamate release probability. N- and P/Q-type VGCCs have been implicated in the expression of CB1R-induced DSE in various regions, providing us with an additional means to assess the contribution of VGCCs in the mechanism of GABABR (Kreitzer and Regehr, 2001; Heifets et al., 2008; Castillo et al., 2012). Postsynaptic depolarization from −70 to +40 mV for 10 s resulted in a significant reduction in eEPSC amplitude at D1(+) and D1(−) MSN synapses, confirming the presence of DSE (Fig. 4I–L; D1(+) DSE-ACSF: 64.97 ± 2.69%, n = 8, p < 0.0001; D1(−) DSE-ACSF: 74.13 ± 1.61%, n = 9, p < 0.0001). Following a triplicate DSE baseline, BAC was superfused into the ACSF bath, resulting in a significant decrease in eEPSC amplitude that stabilized at the 20 min time point. Subsequent trials of DSE were then performed in the presence of BAC. DSE remained completely intact following BAC application at D1(+) and D1(−) MSN synapses, indicating that BAC did not occlude the expression of DSE (Fig. 4I–L; D1(+) DSE-BAC: 59.1 ± 4.38, n = 8, p = 0.2321; D1(−) DSE-BAC: 71.56 ± 1.69, n =, p = 0.1779).
Elevating presynaptic Ca2+ influx abrogates downstream GABABR effector function
The above data suggest that presynaptic GABABR function at glutamatergic synapses in the NAc core is distinct from the actions of GABABR elsewhere in the CNS. One possibility is that GABABR activation interferes with vesicular release machinery. To gain insight into how GABABR may be involved in vesicular release, we asked whether increasing presynaptic Ca2+ influx during the electrically-evoked fiber volley modulates the effects of BAC. We accomplished this by bath-applying K+ channel blocker, 4-aminopyridine (4-AP; 100 μm), at a concentration selective for voltage-gated K+ channels (KV; Fig. 5A). Selective blockade of KV channels elongates action potential half-width, increasing VGCC open probability and presynaptic Ca2+ conductance (Solís and Nicoll, 1992; Iremonger and Bains, 2009). Application of 4-AP alone significantly increased eEPSC amplitude at D1(+) and D1(−) MSNs (Fig. 5B,C; D1(+) 4-AP: 142.26 ± 12.38, n = 6, p < 0.001; D1(−) 4-AP: 143.55 ± 11.34, n = 6, p < 0.001) and was accompanied by a reduction in PPR, consistent with a Ca2+-dependent enhancement of presynaptic release probability (Fig. 5D; PPR = D1(+) baseline: 1.26 ± 0.13, D1(+) 4-AP: 0.52 ± 0.28, n = 5, p = 0.0468; D1(−) baseline: 1.16 ± 0.09, D1(−) 4-AP: 0.76 ± 0.13, n = 4, p = 0.0169). Subsequent application of BAC in the presence of 4-AP resulted in a depression in eEPSC amplitude at D1(+) and D1(−) MSNs that was significantly attenuated relative to control conditions (Fig. 5A–C; D1(+): 66.71 ± 3.09%, n = 5, p = 0.0001; D1(−): 69.35 ± 7.02%, n = 4, p = 0.0003). 4-AP also negated BAC-induced changes in PPR and fully reversed the depression in eEPSC amplitude elicited by BAC (Fig. 5D–F; PPR = D1(+) 4-AP + BAC: 0.79 ± 0.12, n = 5, p = 0.214; D1(−) 4-AP±BAC: 0.73 ± 0.11, n = 4, p = 0.4374; D1(+) 4-AP reversal: 116.40 ± 7.89%, n = 5, p = 0.0033; D1(−) 4-AP reversal: 107.08 ± 10.85%, n = 4, p = 0.0093). To verify that 4-AP increased Ca2+ influx secondary to its action at KV channels, we repeated this experiment in low-Ca2+ ACSF (1 mm Ca2+/3 mm Mg2+). Prior application of 4-AP in low-Ca2+ ACSF returned the BAC-induced decrease in eEPSC amplitude back to baseline conditions at MSN [pooled D1(+) and D1(−)] synapses, suggesting that increased Ca2+ influx via KV channel blockade can successfully overcome the inhibitory actions of BAC at glutamatergic synapses in the NAc core (Fig. 5C; pooled MSNs: 40.58 ± 9.28%, n = 11, p = 0.3533).
Figure 5.

Elevating presynaptic Ca2+ influx abrogates downstream GABABR effector function. A, 1: Representative traces showing that 4-AP application increases eEPSC amplitude and decay time at glutamatergic synapses onto D1(+) and D1(−) MSNs. 2: eEPSC amplitude at D1(+) and D1(−) MSN synapses in the presence of BAC. Scale bar: 200 pA/20 ms. B, Prior bath application of 4-AP blunts the BAC-induced decrease in eEPSC amplitude at D1(+) and D1(−) MSNs). Low Ca2+ (1 mm) ACSF returns BAC-induced decrease in eEPSC amplitude to control conditions (pooled D1(+) and D1(−) MSNs, line with shaded gray area within SEM). C, Graph of average eEPSC amplitude in the presence of BAC with 4-AP, 4-AP + low Ca2+ ACSF, and ACSF-only control conditions (t = 25–30 min). D, 4-AP application alone decreases PPR and negates the increase in PPR elicited by BAC. E, Time course summary showing that 4-AP application reverses eEPSC amplitude in the presence of BAC at D1(+) and D1(−) MSNs. F, Graph of average eEPSC amplitude following 4-AP reversal in the presence of BAC (blue, t = 20–25 min) and 4-AP + BAC (gray, t = 45–50) for D1(+) and D1(−) MSNs. Error bars indicate SEM. *p < 0.05.
Genetically disrupting the Gβγ-SNAP-25 interaction blunts the synaptic effect of GABABR on glutamatergic transmission in the NAc core
Gi/o-coupled GPCR-induced mobilization of Gβγ at the presynaptic active zone has been shown to interfere with exocytotic fusion mediated by ternary SNARE complexes. Gβγ-dependent targeting of membrane-associated target SNARE proteins (t-SNAREs), such as SNAP-25, has been shown to prevent the association of synaptotagmin with the SNARE complex to reduce vesicular release probability (Wells et al., 2012; Zurawski et al., 2019). The Gβγ-SNARE interaction is Ca2+-sensitive, as pharmacological enhancement of presynaptic [Ca2+] can offset Gβγ binding to restore baseline synaptic transmission (Gerachshenko et al., 2005). Given that GABABR heteroreceptor function in the NAc core is (1) putatively VGCC, Kir, AC, PKA, and mGluR-independent and (2) blunted by 4-AP-induced increases in presynaptic Ca2+ influx, we hypothesized that GABABR engages a presynaptic Gβγ-SNARE mechanism at glutamatergic synapses onto MSNs. To test this hypothesis, we used transgenic mice lacking the C-terminally-located Gβγ-binding motif of SNAP-25 (Fig. 6A). Inserting the SNAP25Δ3 allele into the WT SNAP-25 locus attenuates Gβγ-SNAP-25 complex formation by ∼47% without significantly disrupting evoked synaptic transmission (Zurawski et al., 2019).
Figure 6.

Genetically reducing the Gβγ-SNAP-25 interaction blunts the synaptic effect of GABABR at glutamatergic synapses in the NAc core. A, Schematic of presynaptic terminal with C-terminally truncated SNAP-25 in transgenic SNAP25Δ3 mouse. Syn indicates SNAP-25 binding partner, synaptotagmin-1. B, Representative traces of averaged 50 ms paired-pulse eEPSCs in SNAP25Δ3 (left, blue circle) and WT mice (right, open circle). Basal PPR at 50 ms is significantly increased in SNAP25Δ3 mice relative to WT littermate controls. Scale bars, 100 pA/50 ms. C, Time course summary of eEPSC amplitude obtained from unlabeled MSNs in SNAP25Δ3 mutant mice (blue circles) and WT littermate control mice (open circles) in the presence 3 μm BAC. D, PPR time course summary and averages pre-BAC (gray area) and post-BAC (blue area) at 3 μm. E, Time course summary of eEPSC amplitude obtained from unlabeled MSNs in SNAP25Δ3 mutant mice (blue circles) and WT littermate control mice (open circles) in the presence 600 nm BAC. F, PPR time course summary and averages pre-BAC (gray area) and post-BAC (blue area) at 600 nm. G, Time course summary of eEPSC amplitude obtained from unlabeled MSNs in SNAP25Δ3 mutant mice (blue circles) and WT littermate control mice (open circles) in the presence 200 nm BAC. H, PPR time course summary and averages pre-BAC (gray area) and post-BAC (blue area) at 200 nm. Error bars indicate SEM. *p < 0.05.
We prepared acute ex vivo brain slices from SNAP25Δ3 and WT mice and recorded eEPSCs in unlabeled MSNs. Interestingly, baseline PPR of glutamatergic transmission in SNAP25Δ3 mice was significantly elevated relative to WT controls, consistent with a SNAP-25-specific reduction in vesicular exocytosis [Fig. 6B; MSNs (unlabeled), 3 μm, WT baseline 50 ms ISI, PPR: 0.99 ± 0.14, n = 7; SNAP25Δ3: 1.56 ± 0.09, n = 5, p = 0.0021]. Bath-application of BAC (3 μm) in slices obtained from littermate (WT) control mice decreased eEPSC amplitude comparably to D1tdTomato mice. However, BAC application (3 μm) in SNAP25Δ3 mice resulted in a significantly blunted decrease in eEPSC amplitude [Fig. 6C; MSNs (unlabeled), WT: 27.54 ± 1.14%, n = 10; SNAP25Δ3: 55.45 ± 4.94%, n = 5, p = 0.0003]. To discern the functional relationship between GABABR and SNAP25 more clearly, we superfused BAC at lower concentrations to elicit submaximal GABABR activity. Although the BAC-induced decrease in eEPSC amplitude was modestly reduced at higher BAC concentrations (3 μm, 10 μm), the effect was significantly attenuated at lower concentrations (200 nm, 600 nm) in SNAP25Δ3 mice (Fig. 6E,G; 200 nm, WT: 79.35 ± 2.09, n = 6; SNAP25Δ3: 92.64 ± 7.92, n = 4, p = 0.0083; 600 nm, WT: 62.71 ± 4.10, n = 7; SNAP25Δ3: 83.03 ± 4.12%, n = 5, p = 0.0074). Furthermore, BAC application in WT mice was accompanied by a time-locked increase in PPR at all concentrations tested, whereas BAC application in SNAP25Δ3 mice did not significantly alter PPR (Fig. 6D,F,H; 200 nm, WT baseline: 1.12 ± 0.05, BAC: 1.34 ± 0.08, n = 4, p = 0.0208; SNAP25Δ3 baseline: 1.35 ± 0.11, BAC: 1.44 ± 0.19, n = 4,p = 0.0964; 600 nm, WT baseline: 1.16 ± 0.11, BAC: 1.36 ± 0.15, n = 6, p = 0.0306; SNAP25Δ3 baseline: 1.46 ± 0.17, BAC: 1.61 ± 0.16, n = 5, p = 0.1419; 3 μm, WT baseline: 1.04 ± 0.19, BAC: 1.73 ± 0.13, n = 4, p = 0.0054; SNAP25Δ3 baseline: 1.76 ± 0.16, BAC: 1.79 ± 0.14, n = 8, p = 0.6367). These findings indicate that presynaptic GABABR function at glutamatergic synapses in the NAc core is impaired in mice lacking the Gβγ-targeting motif of SNAP-25.
GAT-1-regulated ambient GABA acts on GABABR to decrease glutamatergic transmission in the NAc core
GABABR expressed on glutamate terminals may serve as a heteroreceptor for GABA released from contiguous GABAergic synapses in the NAc. To assess whether circuit-wide elevations in ambient GABA modulate glutamatergic synapses in the NAc core, we bath-applied tiagabine (20 μm), a blocker of GABA reuptake transporter type-1 (GAT-1), the CNS expression of which is high in the striatum relative to other subcortical regions (Augood et al., 1995; Kirmse et al., 2008). Tiagabine resulted in a significant decrease in eEPSC amplitude at D1(+) and D1(−) MSNs (Fig. 7A–C,F; D1(+): 62.00 ± 3.61%, n = 5, p = 0.0143; D1(−): 63.67 ± 5.13%, n = 6, p = 0.00244). To determine whether this effect was due to GABABR heteroreceptor function, we incorporated SCH 50911 into the superfusate before the application of tiagabine. SCH 50911 significantly decreased the tiagabine-induced decrease in eEPSC amplitude in both MSN subtypes (Fig. 7D,F; D1(+) SCH: 88.93 ± 10.16%, n = 9, one-way RM ANOVA, tiagabine effect: F(3,19) = 13.03, p = 0.0009; Sidak's post hoc analysis, ACSF vs SCH: p = 0.0256; D1(−) SCH: 88.57 ± 5.60%, n = 10, one-way RM ANOVA, tiagabine effect: F(3,20) = 18.66, p < 0.0001; Sidak's post hoc analysis, ACSF vs SCH: p = 0.0134). If neuronal GAT-1 blockade promotes heterosynaptic crosstalk between GABA- and glutamatergic synapses via GABABR, then CGP 7930, a potent, well characterized GABABR-positive allosteric modulator (PAM; Adams and Lawrence, 2007), should enhance the tiagabine-induced decrease in eEPSC amplitude. Indeed, prior application of CGP 7930 (30 μm) enhanced the tiagabine-induced decrease in eEPSC amplitude at D1(+) and D1(−) MSN synapses (Fig. 7E,F; D1(+) CGP: 42.81 ± 3.51%, n = 5, one-way RM ANOVA; Sidak's post hoc analysis, ACSF vs CGP: p = 0.0486; D1(−) CGP: 44.77 ± 4.26%, n = 7; Sidak's post hoc analysis, ACSF vs CGP: p = 0.0144). CGP 7930 alone had no effect on basal eEPSC amplitude at either MSN subtype (data not shown), consistent with a lack of effect of SCH 50911 alone on basal eEPSC amplitude. These findings suggest that elevations in GAT-1-regulated GABA levels in the NAc core modulate glutamatergic transmission onto D1(+) and D1(−) MSNs via GABABR.
Figure 7.

GAT-1-regulated ambient GABA acts on GABABR to decrease glutamatergic transmission in the NAc core. A, B, Representative experiment and traces of eEPSCs obtained from D1(+) MSNs (blue circles) and D1(−) MSNs (open circles) in the NAc core. Scale bar, 200 pA/50 ms. C, Time course of normalized eEPSC amplitude from D1(+) and D1(−) MSNs in the presence of GABA reuptake (GAT-1) inhibitor, tiagabine (30 μm). D, Time course of normalized eEPSC amplitude from D1(+)and D1(−) MSNs in the presence of tiagabine + SCH 50911 (5 μm). E, Time course of normalized eEPSC amplitude from D1(+) and D1(−) MSNs in the presence of tiagabine (30 μm) and GABABR-PAM, CGP 7930 (30 μm). F, Quantification of average eEPSC amplitude (t = 55–60 min) of tiagabine in normal ACSF, CGP 7930, and SCH 50911 at D1(+)and D1(−) MSNs. Error bars indicate SEM. *p < 0.05.
Parvalbumin-expressing interneurons preferentially decrease glutamatergic transmission onto D1(+) MSNs via heterosynaptic GABABR signaling
Although various sources of GABA exist in the NAc, we hypothesized that fast-spiking interneurons, the majority of which express the Ca2+-binding protein parvalbumin, contribute to elevations in extrasynaptic GABA concentration as a consequence of their fast-spiking activity. PV-INs in the NAc coordinate MSN spiking activity through feedforward inhibition, wherein glutamatergic synapses onto MSNs collateralize onto nearby PV-INs, which exert robust GABAergic control over MSN output (Wright et al., 2017; Yu et al., 2017; Scudder et al., 2018). Compared with MSNs and other GABAergic interneuron subtypes in the NAc, PV-INs receive stronger and more extensive excitatory input from afferents that drive MSN activity, providing them with greater temporal authority over information propagated through the NAc (Yu et al., 2017; Scudder et al., 2018). Furthermore, the lack of tonic GABABR activity at glutamatergic synapses suggests a neuronal source of GABA driven by acute shifts in circuit activity. To determine whether PV-INs exert heterosynaptic control over glutamatergic synapses onto D1(+) and D1(−) MSNs, we bred PV-Cre mice, in which Cre recombinase expression is driven by the PV promoter, with Ai32 conditional ChR2 (cChR2) and D1tdTomato BAC transgenic mouse lines, generating PVCre mice (Fig. 8A). This breeding strategy confers optogenetic control over PV-expressing cells in the NAc, which are GABAergic PV-INs. Indeed, oIPSCs recoded from D1(+) and D1(−) MSNs were abolished by PTX (50 μm), indicating that PV-IN-to-MSN transmission in the NAc core is mediated by GABAAR (Fig. 8B; D1(+) PTX: 4.5 ± 3.11%, n = 5, p = 0.8851; D1(−) 5.96 ± 2.07%, n = 6, p = 0.6500).
To determine whether PV-IN activity modulates glutamatergic transmission onto D1(+) and D1(−) MSNs, we prepared acute brain slices from PVCre mice and recorded eEPSCs from D1(+) and D1(−) MSNs in PTX-containing ACSF. PV-INs were stimulated optically at 30 Hz to resemble a PV-IN-directed, low-range gamma frequency stimulation (LFSPV) pattern. Before using this induction protocol, we first wanted to confirm that optical excitation of PV-INs at 30 Hz yielded high-fidelity oIPSCs in MSNs. Indeed, LFSPV resulted in oIPSCs in MSNs throughout the induction period, indicating that ChR2(H134R) is a suitable opsin for these experiments (Fig. 8B). Following a stable 10 min eEPSC baseline, LFSPV was delivered for 5 min, after which eEPSCs were recorded up to 30 min post-LFS. LFSPV resulted in a significant decrease in eEPSC amplitude at D1(+) but not D1(−) MSNs that was accompanied by an increase in CV, consistent with a presynaptic locus of action (Fig. 8C–F; D1(+): 79.71 ± 5.12%, n = 5, p = 0.0481; D1(−): 102.86 ± 5.26%, n = 5, p = 0.5762; CV = D1(+) baseline: 0.05 ± 0.01, D1(+) post-LFSPV: 0.072 ± 0.015, n = 5, p = 0.0352; D1(−) baseline: 0.05 ± 0.01, D1(−) post-LFSPV: 0.04 ± 0.004, n = 5, p = 0.3665). The effect of LFSPV on eEPSC amplitude was significantly different between D1(+) and D1(−) MSNs (two-way RM ANOVA, LFSPV-MSN subtype interaction: F(1,4) = 14.48, p = 0.0170). To determine whether this plasticity was mediated by GABABR, SCH 50911 was incorporated into the ACSF bath before the induction protocol. SCH 50911 completely abolished the LFSPV-induced depression of eEPSC amplitude at D1(+) MSN synapses (Fig. 8G–I; D1(+) SCH: 91.46 ± 7.63%, n = 6, p = 0.2751). Furthermore, prior application of CGP 7930 enhanced the LFSPV-induced depression at D1(+) MSNs and unmasked a significant depression in D1(−) MSNs that was accompanied by an increase in CV (Fig. 8G–L; D1(+) CGP: 48.14 ± 8.56%, n = 6, one-way RM ANOVA, Sidak's post hoc analysis ACSF vs CGP: p = 0.0043; D1(−) CGP: 73 ± 10.03%, n = 6, p = 0.0325; CV = D1(+) baseline: 0.048 ± 0.008, D1(+) post-LFSPV CGP: 0.226 ± 0.053, n = 5, p = 0.0069; D1(−) baseline: 0.048 ± 0.11, D1(−) post-LFSPV CGP: 0.18 ± 0.067, n = 7, p = 0.0475). These data indicate that PV-INs can heterosynaptically regulate glutamatergic transmission onto D1(+) MSNs and, less so, D1(−) MSNs by targeting presynaptically-expressed GABABR, identifying a novel arm within feedforward inhibitory microcircuits in the NAc core.
PV-IN-to-MSN synapses in the striatum maintain synaptic efficacy across a broad dynamic range of firing frequencies (Gittis et al., 2010). We hypothesized that this property may permit heterosynaptic crosstalk between PV-INs and glutamate terminals without altering GABAergic transmission at PV-IN-to-MSN synapses. To address this, we recorded oIPSCs elicited from PV-INs in D1(+) and D1(−) MSNs (Fig. 9A). We first tested whether GABABR autoregulates PV-IN-to-D1(+) and D1(−) MSN synapses. Interestingly, only a subset of PV-IN-to-MSN synapses were responsive to BAC [BAC(+)], with BAC(+) synapses exhibiting a significant decrease in oIPSC amplitude at both MSN subtypes and BAC(−) synapses exhibiting no change in oIPSC amplitude. Subsequent application of SCH 50911 incompletely reversed oIPSC amplitude to baseline at BAC(+) synapses, indicating the presence of inhibitory long-term depression at a subset of synapses (Fig. 9B–E; BAC(+) cells = D1(+) BAC: 41.73 ± 6.03%, D1(+) SCH: 58.76 ± 3.49%, n = 4, one-way RM ANOVA, drug effect: F(2,10) = 70.94, p < 0.0001, Sidak's post hoc analysis, baseline vs BAC: p < 0.001, vs SCH: p < 0.001; D1(−) BAC: 38.96 ± 6.17%, D1(−) SCH: 63.09 ± 5.37%, n = 4, one-way RM ANOVA, drug effect: F(2,10) = 93.97, p < 0.0001, Sidak's post hoc analysis, baseline vs BAC: p < 0.001, vs SCH: p < 0.001; BAC(−) cells = D1(+): 98.42 ± 2.76%, n = 3 of 11 cells, p = 0.1169; D1(−): 99.36 ± 2.86%, n = 3 of 9 cells, p = 0.2690). We next asked whether GABABR activity tonically inhibits PV-IN-to-D1(+) and D1(−) MSN synapses via presynaptic autoreceptor function. SCH 50911 added to the ACSF following a 10 min oIPSC baseline did not significantly alter oIPSC amplitude at D1(+) or D1(−) MSN synapses (Fig. 9F,G; D1(+): 100.05 ± 7.64%, n = 4, p = 0,9981; D1(−) 101.56 ± 4.23%, n = 5, p = 0.6900). Given these findings, we predicted that LFSPV would not elicit a GABABR-dependent change in synaptic strength at PV-IN-to-MSN synapses. Indeed, GABAergic transmission at PV-IN-to-MSN (pooled) synapses remained completely unchanged 40 min post-LFSPV (Fig. 9H,I; pooled MSNs: 103.56 ± 7.62%, n = 4, p = 0.3138). Together, these data suggest that PV-INs are well suited to functionally regulate glutamatergic transmission via GABAB heteroreceptors on glutamate terminals in the NAc core.
Discussion
We elucidate cell-type-specific and microcircuit-specific mechanisms by which GABABR dynamically regulates glutamatergic synapses in the NAc core. We report that presynaptically-expressed GABABR activation elicits a robust decrease in glutamatergic synaptic transmission onto D1(+) and D1(−) MSNs by engaging a non-canonical signaling mechanism that is distinct from other Gi/o-coupled GPCRs in the NAc, including CB1R and group II mGluRs. Instead, GABABR activity interferes with vesicular exocytosis in a SNAP-25-dependent manner. Furthermore, we show that GABAB heteroreceptors at D1(+) MSN synapses are preferentially recruited by PV-INs within feedforward inhibitory microcircuits. Alongside data showing a lack of GABABR tone at glutamatergic synapses, these data offer rigorous support for a new, activity-dependent GABABR-contingent arm within feedforward circuits in the NAc core.
Presynaptic GABABR recruits a non-canonical, SNAP-25-dependent signaling mechanism to regulate glutamatergic transmission in the NAc core
We find that GABABR activation elicits a robust decrease in synaptic efficacy at glutamatergic synapses onto D1(+) and D1(−) MSNs. Following pharmacological examination of GABABR function, our findings suggest that GABABR recruits a signaling mechanism that is N- and P/Q-type VGCC, Kir, mGluR, and AC/cAMP-independent, suggesting that GABABR recruits a non-canonical signaling pathway at these synapses. Anomalous GABABR function has been described in regions where the synaptic effects of BAC are insensitive to Gi/o-GPCR-disabling agents, such as pertussis toxin or NEM (Colmers and Pittman, 1989). We hypothesized that GABABR directly interferes with vesicular release machinery, such as membrane-associated t-SNARE, SNAP-25, to reduce glutamate release probability onto MSNs. SNAP-25 facilitates transmitter exocytosis by binding Ca2+-sensing SNARE protein, synaptotagmin, to establish ternary SNARE complexes that authorize vesicular release (Wells et al., 2012; Zurawski et al., 2019). The C-terminally-located botulinum toxin type-A cleavage site of SNAP-25 has previously been shown to mediate the inhibitory actions of Gβγ on SNAP-25 (Gerachshenko et al., 2005). Thus, we used SNAP25Δ3 transgenic mice partially deficient (∼47%) in the SNAP-25-Gβγ interaction. Consistent with reduced synaptic efficacy of GABABR, the actions of BAC were dose-dependently reduced in SNAP25Δ3 mutant mice. While targeted mutations in SNAP-25 will affect other neuromodulatory systems in the NAc, the BAC-induced reduction in mEPSC frequency supports a direct synaptic effect of GABABR on glutamatergic transmission. The pronounced increase in PPR induced by BAC was also abolished in SNAP25Δ3 mice, strongly supporting the hypothesis that GABABR-induced decrease in glutamate release probability is partially mediated by the SNAP-25-Gβγ interaction.
Several key findings also support a VGCC-independent interaction with vesicular release machinery. Low Ca2+ ACSF and pharmacological blockade of N- and P/Q-type VGCCs failed to occlude the effects of BAC on glutamatergic transmission. The nonlinear relationship between VGCCs and transmitter release at corticostriatal synapses complicates the interpretation of this finding, as a modest reduction in presynaptic Ca2+ influx can substantially impact neurotransmitter release (Kupferschmidt and Lovinger, 2015). Therefore, we examined the functional interaction between GABABR and VGCC-targeting Gi/o-coupled GPCRs in the NAc, such as CB1R and group II mGluRs. Presynaptically-expressed CB1R and mGluRs trigger short- and long-term plasticity can be attributed in part to their actions on presynaptic Ca2+ entry via VGCCs. For example, group II mGluRs in the NAc trigger presynaptic LTD by attenuating downstream P/Q-type VGCC function (Robbe et al., 2002). Similarly, activity-dependent release of eCBs can transiently activate presynaptic CB1Rs on glutamate terminals to elicit VGCC-dependent reductions in neurotransmitter release (Hoffman and Lupica, 2000; Kreitzer and Regehr, 2001). Our data collectively suggest that GABABR recruits different intracellular effectors than either group II mGluRs or CB1R, corroborating the hypothesis that GABABR mobilizes Gβγ to interact directly with exocytotic machinery at the presynaptic active zone.
Blocking KV channels significantly attenuated and reversed the actions of GABABR, consistent with previous studies showing that Gβγ-SNARE interactions can be overcome by increasing presynaptic Ca2+ levels (Wells et al., 2012; Li et al., 2016). Although we cannot definitively rule out GABABR-induced changes in K+ channel function, the observation that GABABR activity returns to normal in 4-AP-containing low-Ca2+ ACSF, is resistant to extracellular Ba2+ application, and produces no detectible change in PPR following 4-AP treatment points to a 4-AP-induced enhancement in presynaptic Ca2+ conductance. Although it is possible that GABABR initiates functionally redundant mechanisms to reduce glutamate release, this hypothesis is less likely given that the effects of BAC on eEPSC amplitude remained intact following each pharmacological manipulation. Together, we propose a novel mechanism by which GABABR in the NAc core reduces presynaptic glutamate release probability downstream of VGCCs by interfering with the assembly of core SNARE complexes in a SNAP-25-dependent manner.
PV-INs heterosynaptically regulate glutamatergic transmission via GABABR
Although heterosynaptic regulation of GABAergic synapses by glutamate has been characterized in the striatum, the reverse relationship in which GABA regulates glutamatergic transmission has not been demonstrated (Mathur et al., 2013; Patton et al., 2019). We report that optogenetic stimulation of PV(+) cells reduces glutamatergic transmission onto D1(+) and, less so, D1(−) MSNs in a GABABR-dependent manner, offering physiological context for GABAB heteroreceptor function within the NAc. This finding is striking for several reasons. First, striatal PV-INs form monosynaptic connections with MSNs at somatic and proximal dendritic domains, whereas glutamatergic afferents target distal dendrites (Hu et al., 2014; Yu et al., 2017). The privileged anatomical positioning of PV-IN-to-MSN synapses confers optimal GABAergic control over MSN spiking activity, supporting the role of PV-INs in feedforward inhibition. Second, PV-INs, unlike somatostatin-expressing (SST) INs, lack autonomous action potential (AP) firing that could contribute to tonic GABAB heteroreceptor function (Smith et al., 2017; Trouche et al., 2019). However, PV-INs receive significantly greater excitatory innervation than SOM-INs and sustain afferent-evoked firing rates of up to 250 Hz, indicating that acute shifts in circuit activity are likely to recruit PV-INs over other GABAergic cell types in the NAc (Scudder et al., 2018; Tepper et al., 2018; Trouche et al., 2019). SST-INs also exhibit biophysical limitations that prevent them from responding to time-locked changes in glutamatergic transmission, including a lower AP frequency, prolonged latency to spike, and accommodating AP firing pattern (Scudder et al., 2018; Tepper et al., 2018). Whereas tonic GABAAR and GABABR activity is observed at heterosynaptic loci on DA terminals in the dorsal striatum, NAc DA release is comparatively unaffected by GABABR blockade (Tritsch et al., 2014; Melchior et al., 2015; Lopes et al., 2019). Similarly, we do not detect GABABR tone at glutamatergic synapses onto D1(+) or D1(−) MSNs, supporting a distinct role for GABAergic neurons requiring afferent excitatory input to fire. Therefore, PV-INs may be better equipped than other GABAergic cell types in the NAc to modulate activity-dependent increases in glutamatergic activity.
PV-INs entrain principal neuron output via gamma frequency oscillations (25–100 Hz) that are sustained by the coordinated recruitment of electrically-connected PV-INs (Hu et al., 2014). We elected to activate PV-INs at 30 Hz (LFSPV) to resemble a physiologically relevant gamma frequency stimulation pattern that can be achieved with the transgenically-encoded opsin, ChR2(H134R). Our data suggest that LFSPV for 5 min is sufficient to heterosynaptically target GABABR at glutamatergic synapses onto D1(+) MSNs. Interestingly, we find that LFSPV targets GABABR at synapses onto D1(−) MSNs only in the presence of a GABABR PAM, suggesting that LFSPV may be subthreshold to elicit changes in glutamatergic transmission at synapses ono D1(−) MSNs. One possibility is that PV-IN-to-D1(+) and D1(−) MSN synapses are differentially regulated by GABABR, with synapses onto D1(−) MSNs exhibiting greater GABABR-dependent autoinhibitory feedback than synapses onto D1(+) MSNs. Differences in autoreceptor function at these synapses would limit the efficacy with which prolonged PV-IN stimulation elevates extracellular GABA levels. However, the majority of PV-IN-to-D1(+) and D1(−) MSN synapses were BAC-insensitive and LFSPV of oIPSCs failed to elicit homosynaptic plasticity at PV-IN-to-MSN synapses. Having ruled out intrinsic mechanisms at PV-IN-to-MSN synapses, the most probable explanation is a synapse-specific difference in GABA sensitivity. This hypothesis is strongly supported by the BAC dose–response relationship in which glutamatergic synapses onto D1(+) MSNs exhibited increased sensitivity to BAC at lower concentrations (200–600 nm) relative to D1(−) MSNs. Another intriguing possibility is whether differences in GABA reuptake kinetics between D1(+) and D1(−) synapses create synapse-specific microdomains that modulate glutamatergic inputs onto distinct cell types in the NAc. Future studies will be needed to elucidate fully the intricate mechanisms by which PV-INs sculpt cell-type-specific circuit output in the NAc core.
Concluding remarks
The NAc coordinates motivated behaviors by integrating PV-IN-directed inhibitory networks with glutamatergic inputs from various salience-encoding brain regions. While GABABR function is well characterized at upstream loci within the mesolimbic reward network, a comprehensive analysis of GABAB heteroreceptor function in the NAc is lacking. Here, we revise and expand knowledge on the role of GABABR function within PV-IN-embedded feedforward microcircuits in the NAc core. Our findings rigorously support a novel mechanism by which GABABR modulates glutamatergic transmission and define an activity-dependent source of GABA within PV-IN-embedded feedforward circuits. Understanding the role of GABABR within NAc microcircuits paints a more complete picture of how GABABR-specific pharmacological agents can be used to treat neuropsychiatric disorders characterized by aberrant motivational states, including addiction, major depressive disorder, and autism (Cousins et al., 2002; Kahn et al., 2009; Jacobson et al., 2018; Stoppel et al., 2018; Pisansky et al., 2019).
Footnotes
This work was supported by National Institute on Drug Abuse Grant R01DA040630 (B.A.G). We thank members of the Grueter laboratory for their helpful comments and Dr. Roger Nicoll for generously reviewing and offering feedback on this project.
The authors declare no competing financial interests.
References
- Adams CL, Lawrence AJ (2007) CGP7930: a positive allosteric modulator of the GABAB receptor. CNS Drug Rev 13:308–316. 10.1111/j.1527-3458.2007.00021.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augood SJ, Herbison AE, Emson PC (1995) Localization of GAT-1 GABA transporter mRNA in rat striatum: cellular coexpression with GAD67 mRNA, GAD67 immunoreactivity, and parvalbumin mRNA. J Neurosci 15:865–874. 10.1523/JNEUROSCI.15-01-00865.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo PE, Younts TJ, Chávez AE, Hashimotodani Y (2012) Endocannabinoid signaling and synaptic function. Neuron 76:70–81. 10.1016/j.neuron.2012.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colmers WF, Pittman QJ (1989) Presynaptic inhibition by neuropeptide Y and baclofen in hippocampus: insensitivity to pertussis toxin treatment. Brain Res 498:99–104. 10.1016/0006-8993(89)90403-4 [DOI] [PubMed] [Google Scholar]
- Cousins MS, Roberts DC, de Wit H (2002) GABAB receptor agonists for the treatment of drug addiction: a review of recent findings. Drug Alcohol Depend 65:209–220. 10.1016/S0376-8716(01)00163-6 [DOI] [PubMed] [Google Scholar]
- Cruz HG, Ivanova T, Lunn ML, Stoffel M, Slesinger PA, Lüscher C (2004) Bi-directional effects of GABAB receptor agonists on the mesolimbic dopamine system. Nat Neurosci 7:153–159. 10.1038/nn1181 [DOI] [PubMed] [Google Scholar]
- Di Ciano P, Everitt BJ (2003) The GABAB receptor agonist baclofen attenuates cocaine-and heroin-seeking behavior by rats. Neuropsychopharmacology 28:510–518. 10.1038/sj.npp.1300088 [DOI] [PubMed] [Google Scholar]
- Dobbs LK, Kaplan AR, Lemos JC, Matsui A, Rubinstein M, Alvarez VA (2016) Dopamine regulation of lateral inhibition between striatal neurons gates the stimulant actions of cocaine. Neuron 90:1100–1113. 10.1016/j.neuron.2016.04.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards NJ, Tejeda HA, Pignatelli M, Zhang S, McDevitt RA, Wu J, Bass CE, Bettler B, Morales M, Bonci A (2017) Circuit specificity in the inhibitory architecture of the VTA regulates cocaine-induced behavior. Nat Neurosci 20:438–448. 10.1038/nn.4482 [DOI] [PubMed] [Google Scholar]
- Gerachshenko T, Blackmer T, Yoon EJ, Bartleson C, Hamm HE, Alford S (2005) Gβγ acts at the C terminus of SNAP-25 to mediate presynaptic inhibition. Nat Neurosci 8:597–605. 10.1038/nn1439 [DOI] [PubMed] [Google Scholar]
- Gittis AH, Nelson AB, Thwin MT, Palop JJ, Kreitzer AC (2010) Distinct roles of GABAergic interneurons in the regulation of striatal output pathways. J Neurosci 30:2223–2234. 10.1523/JNEUROSCI.4870-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grueter BA, Brasnjo G, Malenka RC (2010) Postsynaptic TRPV1 triggers cell type-specific long-term depression in the nucleus accumbens. Nat Neurosci 13:1519–1525. 10.1038/nn.2685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heifets BD, Chevaleyre V, Castillo PE (2008) Interneuron activity controls endocannabinoid-mediated presynaptic plasticity through calcineurin. Proc Natl Acad Sci U S A 105:10250–10255. 10.1073/pnas.0711880105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman AF, Lupica CR (2000) Mechanisms of cannabinoid inhibition of GABAA synaptic transmission in the hippocampus. J Neurosci 20:2470–2479. 10.1523/JNEUROSCI.20-07-02470.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotsenpiller G, Wolf ME (2003) Baclofen attenuates conditioned locomotion to cues associated with cocaine administration and stabilizes extracellular glutamate levels in rat nucleus accumbens. Neuroscience 118:123–134. 10.1016/S0306-4522(02)00951-X [DOI] [PubMed] [Google Scholar]
- Hu H, Gan J, Jonas P (2014) Fast-spiking, parvalbumin+ GABAergic interneurons: from cellular design to microcircuit function. Science. 345:1255263. 10.1126/science.1255263 [DOI] [PubMed] [Google Scholar]
- Iremonger KJ, Bains JS (2009) Retrograde opioid signaling regulates glutamatergic transmission in the hypothalamus. J Neurosci 29:7349–7358. 10.1523/JNEUROSCI.0381-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson LH, Vlachou S, Slattery DA, Li X, Cryan JF (2018) The gamma-aminobutyric acid B receptor in depression and reward. Biol Psychiatry 83:963–976. 10.1016/j.biopsych.2018.02.006 [DOI] [PubMed] [Google Scholar]
- Joffe ME, Grueter BA (2016) Cocaine experience enhances thalamo-accumbens N-methyl-d-aspartate receptor function. Biol Psychiatry 80:671–681. 10.1016/j.biopsych.2016.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn R, Biswas K, Childress AR, Shoptaw S, Fudala PJ, Gorgon L, Montoya I, Collins J, McSherry F, Li SH, Chiang N, Alathari H, Watson D, Liberto J, Beresford T, Stock C, Wallace C, Gruber V, Elkashef A (2009) Multi-center trial of baclofen for abstinence initiation in severe cocaine-dependent individuals. Drug Alcohol Depend 103:59–64. 10.1016/j.drugalcdep.2009.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW. (2009) The glutamate homeostasis hypothesis of addiction. Nat Rev Neurosci 10:561–572. 10.1038/nrn2515 [DOI] [PubMed] [Google Scholar]
- Kashima DT, Grueter BA (2017) Toll-like receptor 4 deficiency alters nucleus accumbens synaptic physiology and drug reward behavior. Proc Natl Acad Sci U S A 114:8865–8870. 10.1073/pnas.1705974114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirmse K, Dvorzhak A, Kirischuk S, Grantyn R (2008) GABA transporter 1 tunes GABAergic synaptic transmission at output neurons of the mouse neostriatum. J Physiol 586:5665–5678. 10.1113/jphysiol.2008.161943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Volkow ND (2016) Neurobiology of addiction: a neurocircuitry analysis. The Lancet Psychiatry 3:760–773. 10.1016/S2215-0366(16)00104-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG (2001) Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron 29:717–727. 10.1016/S0896-6273(01)00246-X [DOI] [PubMed] [Google Scholar]
- Kupferschmidt DA, Lovinger DM (2015) Inhibition of presynaptic calcium transients in cortical inputs to the dorsolateral striatum by metabotropic GABAB and mGlu2/3 receptors. J Physiol 593:2295–2310. 10.1113/JP270045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labouèbe G, Lomazzi M, Cruz HG, Creton C, Luján R, Li M, Yanagawa Y, Obata K, Watanabe M, Wickman K, Boyer SB, Slesinger PA, Lüscher C (2007) RGS2 modulates coupling between GABAB receptors and GIRK channels in dopamine neurons of the ventral tegmental area. Nat Neurosci 10:1559–1568. 10.1038/nn2006 [DOI] [PubMed] [Google Scholar]
- Lacey CJ, Boyes J, Gerlach O, Chen L, Magill PJ, Bolam JP (2005) GABAB receptors at glutamatergic synapses in the rat striatum. Neuroscience 136:1083–1095. 10.1016/j.neuroscience.2005.07.013 [DOI] [PubMed] [Google Scholar]
- Ladera C, del Carmen Godino M, José Cabañero M, Torres M, Watanabe M, Luján R, Sánchez-Prieto J (2008) Pre-synaptic GABAB receptors inhibit glutamate release through GIRK channels in rat cerebral cortex. J Neurochem 107:1506–1517. 10.1111/j.1471-4159.2008.05712.x [DOI] [PubMed] [Google Scholar]
- LeGates TA, Kvarta MD, Tooley JR, Francis TC, Lobo MK, Creed MC, Thompson SM (2018) Reward behaviour is regulated by the strength of hippocampus–nucleus accumbens synapses. Nature 564:258–262. 10.1038/s41586-018-0740-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Liu ZL, Zhang WN, Yang K (2016) Blockade of presynaptic 4-aminopyridine-sensitive potassium channels increases initial neurotransmitter release probability, reinstates synaptic transmission altered by GABAB receptor activation in rat midbrain periaqueductal gray. Neuroreport 27:50–55. 10.1097/WNR.0000000000000499 [DOI] [PubMed] [Google Scholar]
- Li SM, Yin LL, Ren YH, Pan LS, Zheng JW (2001) GABAB receptor agonist baclofen attenuates the development and expression of d-methamphetamine-induced place preference in rats. Life Sci 70:349–356. 10.1016/S0024-3205(01)01397-2 [DOI] [PubMed] [Google Scholar]
- Lim BK, Huang KW, Grueter BA, Rothwell PE, Malenka RC (2012) Anhedonia requires MC4R-mediated synaptic adaptations in nucleus accumbens. Nature 487:183–189. 10.1038/nature11160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes EF, Roberts BM, Siddorn RE, Clements MA, Cragg SJ (2019) Inhibition of nigrostriatal dopamine release by striatal GABAA and GABAB receptors. J Neurosci 39:1058–1065. 10.1523/JNEUROSCI.2028-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, Malenka RC (2011) Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron 69:650–663. 10.1016/j.neuron.2011.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur BN, Tanahira C, Tamamaki N, Lovinger DM (2013) Voltage drives diverse endocannabinoid signals to mediate striatal microcircuit-specific plasticity. Nat Neurosci 16:1275–1283. 10.1038/nn.3478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mato S, Lafourcade M, Robbe D, Bakiri Y, Manzoni OJ (2008) Role of the cyclic-AMP/PKA cascade and of P/Q-type Ca++ channels in endocannabinoid-mediated long-term depression in the nucleus accumbens. Neuropharmacology 54:87–94. 10.1016/j.neuropharm.2007.04.014 [DOI] [PubMed] [Google Scholar]
- Melchior JR, Ferris MJ, Stuber GD, Riddle DR, Jones SR (2015) Optogenetic versus electrical stimulation of dopamine terminals in the nucleus accumbens reveals local modulation of presynaptic release. J Neurochem 134:833–844. 10.1111/jnc.13177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascoli V, Terrier J, Espallergues J, Valjent E, O'Connor EC, Lüscher C (2014) Contrasting forms of cocaine-evoked plasticity control components of relapse. Nature 509:459–464. 10.1038/nature13257 [DOI] [PubMed] [Google Scholar]
- Patton MH, Padgett KE, McKeon PN, Qadir H, Patton MS, Mu C, Roberts BM, Mathur BN (2019) TrkB-dependent disinhibition of the nucleus accumbens is enhanced by ethanol. Neuropsychopharmacology 44:1114–1122. 10.1038/s41386-019-0341-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisansky MT, Lefevre EM, Retzlaff CL, Trieu B, Rothwell PE (2019) Nucleus accumbens fast-spiking interneurons constrain impulsive action. Biol Psychiatry, in press 10.1016/j.biopsych.2019.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitman KA, Puil E, Borgland SL (2014) GABAB modulation of dopamine release in the nucleus accumbens core. Eur J Neurosci 40:3472–3480. 10.1111/ejn.12733 [DOI] [PubMed] [Google Scholar]
- Robbe D, Alonso G, Chaumont S, Bockaert J, Manzoni OJ (2002) Role of P/Q-Ca2+ channels in metabotropic glutamate receptor 2/3-dependent presynaptic long-term depression at nucleus accumbens synapses. J Neurosci 22:4346–4356. 10.1523/JNEUROSCI.22-11-04346.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbe D, Alonso G, Manzoni OJ (2003) Exogenous and endogenous cannabinoids control synaptic transmission in mice nucleus accumbens. Ann N Y Acad Sci 1003:212–225. 10.1196/annals.1300.013 [DOI] [PubMed] [Google Scholar]
- Roberts DC, Andrews MM (1997) Baclofen suppression of cocaine self-administration: demonstration using a discrete trials procedure. Psychopharmacology 131:271–277. 10.1007/s002130050293 [DOI] [PubMed] [Google Scholar]
- Rothwell PE, Fuccillo MV, Maxeiner S, Hayton SJ, Gokce O, Lim BK, Fowler SC, Malenka RC, Südhof TC (2014) Autism-associated neuroligin-3 mutations commonly impair striatal circuits to boost repetitive behaviors. Cell 158:198–212. 10.1016/j.cell.2014.04.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scudder SL, Baimel C, Macdonald EE, Carter AG (2018) Hippocampal-evoked feedforward inhibition in the nucleus accumbens. J Neurosci 38:9091–9104. 10.1523/JNEUROSCI.1971-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ACW, Scofield MD, Heinsbroek JA, Gipson CD, Neuhofer D, Roberts-Wolfe DJ, Spencer S, Garcia-Keller C, Stankeviciute NM, Smith RJ, Allen NP, Lorang MR, Griffin WC 3rd, Boger HA, Kalivas PW (2017) Accumbens nNOS interneurons regulate cocaine relapse. J Neurosci 37:742–756. 10.1523/JNEUROSCI.2673-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solís JM, Nicoll RA (1992) Pharmacological characterization of GABAB-mediated responses in the CA1 region of the rat hippocampal slice. J Neurosci 12:3466–3472. 10.1523/JNEUROSCI.12-09-03466.1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppel LJ, Kazdoba TM, Schaffler MD, Preza AR, Heynen A, Crawley JN, Bear MF (2018) R-baclofen reverses cognitive deficits and improves social interactions in two lines of 16p11.2 deletion mice. Neuropsychopharmacology 43:513–524. 10.1038/npp.2017.236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabata T, Kano M (2006) GABAB receptor-mediated modulation of glutamate signaling in cerebellar Purkinje cells. Cerebellum 5:127–133. 10.1080/14734220600788911 [DOI] [PubMed] [Google Scholar]
- Tabata T, Kano M (2010) GABAB receptor-mediated modulation of metabotropic glutamate signaling and synaptic plasticity in central neurons. Adv Pharmacol 58:149–173. 10.1016/S1054-3589(10)58007-4 [DOI] [PubMed] [Google Scholar]
- Tepper JM, Koós T, Ibanez-Sandoval O, Tecuapetla F, Faust TW, Assous M (2018) Heterogeneity and diversity of striatal GABAergic interneurons: update 2018. Front Neuroanat 12:91. 10.3389/fnana.2018.00091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terunuma M. (2018) Diversity of structure and function of GABAB receptors: a complexity of GABAB-mediated signaling. Proc Jpn Acad Ser B Phys Biol Sci 94:390–411. 10.2183/pjab.94.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritsch NX, Oh WJ, Gu C, Sabatini BL (2014) Midbrain dopamine neurons sustain inhibitory transmission using plasma membrane uptake of GABA, not synthesis. eLife 3:e01936. 10.7554/eLife.01936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trouche S, Koren V, Doig NM, Ellender TJ, El-Gaby M, Lopes-Dos-Santos V, Reeve HM, Perestenko PV, Garas FN, Magill PJ, Sharott A, Dupret D (2019) A hippocampus-accumbens tripartite neuronal motif guides appetitive memory in space. Cell 176:1393–1406.e16. 10.1016/j.cell.2018.12.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner BD, Kashima DT, Manz KM, Grueter CA, Grueter BA (2018a) Synaptic plasticity in the nucleus accumbens: lessons learned from experience. ACS Chem Neurosci 9:2114–2126. 10.1021/acschemneuro.7b00420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner BD, Rook JM, Lindsley CW, Conn PJ, Grueter BA (2018b) mGlu1 and mGlu5 modulate distinct excitatory inputs to the nucleus accumbens shell. Neuropsychopharmacology 43:2075–2082. 10.1038/s41386-018-0049-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchimura N, North RA (1991) Baclofen and adenosine inhibit synaptic potentials mediated by gamma-aminobutyric acid and glutamate release in rat nucleus accumbens. J Pharmacol Exp Ther 258:663–668. [PubMed] [Google Scholar]
- Voigt RM, Herrold AA, Riddle JL, Napier TC (2011) Administration of GABAB receptor positive allosteric modulators inhibit the expression of previously established methamphetamine-induced conditioned place preference. Behav Brain Res 216:419–423. 10.1016/j.bbr.2010.08.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Gallegos DA, Pogorelov VM, O'Hare JK, Calakos N, Wetsel WC, West AE (2018) Parvalbumin interneurons of the mouse nucleus accumbens are required for amphetamine-induced locomotor sensitization and conditioned place preference. Neuropsychopharmacology 43:953–963. 10.1038/npp.2017.178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells CA, Zurawski Z, Betke KM, Yim YY, Hyde K, Rodriguez S, Alford S, Hamm HE (2012) Gβγ inhibits exocytosis via interaction with critical residues on SNAP-25. Mol Pharmacol 82:1136–1149. 10.1124/mol.112.080507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winters BD, Krüger JM, Huang X, Gallaher ZR, Ishikawa M, Czaja K, Krueger JM, Huang YH, Schlüter OM, Dong Y (2012) Cannabinoid receptor 1-expressing neurons in the nucleus accumbens. Proc Natl Acad Sci U S A 109:E2717–E2725. 10.1073/pnas.1206303109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright WJ, Schlüter OM, Dong Y (2017) A feedforward inhibitory circuit mediated by CB1-expressing fast-spiking interneurons in the nucleus accumbens. Neuropsychopharmacology 42:1146–1156. 10.1038/npp.2016.275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Yan Y, Li KL, Wang Y, Huang YH, Urban NN, Nestler EJ, Schlüter OM, Dong Y (2017) Nucleus accumbens feedforward inhibition circuit promotes cocaine self-administration. Proc Natl Acad Sci U S A 114:E8750–E8759. 10.1073/pnas.1707822114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurawski Z, Thompson Gray AD, Brady LJ, Page B, Church E, Harris NA, Dohn MR, Yim YY, Hyde K, Mortlock DP, Jones CK, Winder DG, Alford S, Hamm HE (2019) Disabling the Gβγ-SNARE interaction disrupts GPCR-mediated presynaptic inhibition, leading to physiological and behavioral phenotypes. Sci Signal 12:eaat8595. 10.1126/scisignal.aat8595 [DOI] [PMC free article] [PubMed] [Google Scholar]