

Abstract
Regenerating axons often have to grow considerable distances to reestablish circuits, making functional recovery a lengthy process. One solution to this problem would be to co-opt the “temporal” guidance mechanisms that control the rate of axon growth during development to accelerate the rate at which nerves regenerate in adults. We have previously found that the loss of Limk1, a negative regulator of cofilin, accelerates the rate of spinal commissural axon growth. Here, we use mouse models to show that spinal motor axon outgrowth is similarly promoted by the loss of Limk1, suggesting that temporal guidance mechanisms are widely used during development. Furthermore, we find that the regulation of cofilin activity is an acute response to nerve injury in the peripheral nervous system. Within hours of a sciatic nerve injury, the level of phosphorylated cofilin dramatically increases at the lesion site, in a Limk1-dependent manner. This response may be a major constraint on the rate of peripheral nerve regeneration. Proof-of-principle experiments show that elevating cofilin activity, through the loss of Limk1, results in faster sciatic nerve growth, and improved recovery of some sensory and motor function.
SIGNIFICANCE STATEMENT The studies shed light on an endogenous, shared mechanism that controls the rate at which developing and regenerating axons grow. An understanding of these mechanisms is key for developing therapies to reduce painful recovery times for nerve-injury patients, by accelerating the rate at which damaged nerves reconnect with their synaptic targets.
Keywords: axon, development, limk1/cofilin, regeneration, sciatic nerve, spinal cord
Introduction
Damage to neural circuitry can be catastrophic for patients, leading to devastating deficits in critical motor, sensory, and cognitive functions. Studies of neural regeneration have focused on overcoming the inhibitory environment of the CNS (Fournier and Strittmatter, 2001); an additional problem is that axons in both the CNS and PNS must regenerate over considerable distances to reestablish circuits in human patients. In the PNS, regenerative growth is sufficiently slow (∼1 mm/d) (Hadlock et al., 2005), which denervated targets can atrophy during the recovery period, resulting in the loss of motor control and sensory dysfunction (Campbell, 2008; Gordon et al., 2011; Kang et al., 2011).
Peripheral nerve injuries are surprisingly common, accounting for 2%–5% of patients admitted to U.S. trauma centers (Evans, 2001; Kang et al., 2011), and can result in the degeneration of extensive segments of peripheral nerves (Lundborg, 2000). Ongoing studies to reverse peripheral nerve injuries have concentrated on the immediate reconnection of severed peripheral nerves (Britt et al., 2010; Spaeth et al., 2010), as well as supplying substrates that improve the regrowth of peripheral nerves (Kuffler, 2014). Our studies seek to address this often-unrecognized problem by considering an alternative strategy: manipulating the mechanisms controlling the rate at which axons grow.
Toward this goal, we are assessing how the speed of axon extension is controlled during development. Canonically, guidance mechanisms mediate the presentation of directional information to axons (Butler and Tear, 2007). Our studies have shown there are also “temporal” signals, which regulate the rate at which axons grow and encounter directional information (Phan et al., 2010; Yamauchi et al., 2013). Such signals could permit neural circuits to develop in synchrony with time-dependent events in the developing embryo. One possibility is that temporal guidance signals control the rate at which actin polymerizes, or “treadmills” (Bamburg and Bernstein, 2008) by acting through the cofilin/Limk1 pathway (Meberg and Bamburg, 2000; Endo et al., 2003; Gehler et al., 2004; Nishita et al., 2005; Wen et al., 2007; Marsick et al., 2010; Phan et al., 2010; Bravo-Cordero et al., 2013). Cofilin depolymerizes or severs actin (schematized in Fig. 1A), liberating the monomers needed for further rounds of actin polymerization (Bamburg and Bernstein, 2008). Limk1 phosphorylates, and thereby inactivates, cofilin (Arber et al., 1998) (see Fig. 1A) such that dynamic actin treadmilling occurs when there is a balance between the activities of Limk1 and cofilin (Meberg and Bamburg, 2000; Endo et al., 2003; Sarmiere and Bamburg, 2004; Tursun et al., 2005; Huang et al., 2006; Phan et al., 2010) (idealized in Fig. 1B).
Figure 1.

Cofilin activity in spinal MNs during axon extension. A, Cofilin severs actin to produce the actin monomers needed to generate dynamically “treadmilling” filamentous (F) actin. Limk1 phosphorylates, and thereby inactivates, cofilin leading to the stabilization of F-actin. This model has been simplified for clarity (for a review of cofilin regulation, see Bravo-Cordero et al., 2013). B, Idealized model suggesting how the activities of cofilin and Limk control the rate of actin polymerization. Dynamic actin treadmilling occurs when there is a balance between the activities of Limk and cofilin. C, ISH experiments for Limk1 on transverse sections of embryonic day (E) 11.5 brachial mouse spinal cord. Limk1 is expressed broadly in after mitotic spinal neurons, including MNs. D–N, Transverse forelimb (brachial, D–G, H, I, K, L) or hindlimb (lumbar, M, N) sections of E11.5 mouse spinal cord taken from Hb9:gfp (D–G), Limk1+/+ (H, K, M), or Limk1−/− (I, L, N) embryos and labeled with antibodies against total cofilin (D, E), (p)-cofilin (inactive) (red, F, G, H, I), FoxP1 (red, K–N), GFP (green, D, F), Isl1 (green, K–N), and Isl1/2 (blue, D, F). D–G, Both cofilin (D, E) and (p)-cofilin (F, G) are present broadly in after mitotic neurons in the spinal cord. (p)-Cofilin is found at the highest levels in the dorsal neurons and ventral intermediate zone and lower levels in the Hb9+, Isl1/2+ MNs (E). F, Boxed region represents higher confocal gain/offset levels in F′ and G′. H–J, Cofilin is more active in Limk1 mutant motor columns (I) compared with control littermates (H). Quantification of the intensity of cofilin and (p)-cofilin staining (J) demonstrated that Limk1−/− brachial Isl1+ MNs express slightly higher levels of cofilin (n = 51 sections from 2 embryos) and very significantly lower (∼50%) levels of lower (p)-cofilin (n = 26 sections from 4 embryos) compared with than control littermates (cofilin control, n = 33 sections from 2 embryos; probability of similarity between control and mutant, p < 0.0002, Mann–Whitney test; (p)-cofilin control; n = 24 sections from 6 embryos; probability of similarity between control and mutant, p < 0.0001, Mann–Whitney test). For both measurements, the intensity levels were normalized with respect to background. K–N, There were no observable differences between the timing and segregation of the LMC and MMC/HMC motor columns (summarized in K, inset) in control and Limk1 mutant spinal cords. Scale bars: C–E, K–N, 150 μm; F–I, 100 μm. Probability of similarity, ****p < 0.00005; ***p < 0.0005, Mann-Whitney test.
Our previous studies have shown that manipulating cofilin activity alters the rate of commissural axon extension in the chicken and mouse developing spinal cord (Phan et al., 2010; Yamauchi et al., 2013). Here, we demonstrate that regulation of cofilin activity also controls the rate of axon outgrowth of spinal motor neurons (MNs). Thus, the activation status of cofilin may more generally regulate temporal guidance decisions during development. We also show that the regulation of cofilin activity by Limk1 is relevant to the endogenous process of axon regeneration. There are substantially lower levels of phosphorylated cofilin [(p)-cofilin], in the mouse adult spinal cord compared postnatal stages. However, after a peripheral nerve injury, (p)-cofilin levels rapidly increase at the lesion site, in a Limk1-dependent manner. These observations suggest that an immediate consequence of being injured is to reassemble the regulators that previously controlled the rate of developing axon outgrowth. Moreover, the high levels of phosphorylated (i.e., inactive) cofilin activity may account for the slow speed of regenerative axon growth in the PNS. We find that increasing cofilin activity, through the loss of Limk1, promotes axon regrowth after injury, and improves the recovery of some motor and sensory functions. Thus, accelerating the rate of nerve regeneration may be a viable method of encouraging damaged nerves to reconnect with their synaptic targets before substantial atrophy occurs.
Materials and Methods
Generation and analysis of mutant mice
Mice expressing the Hb9::gfp transgene (purchased from The Jackson Laboratory) (Wichterle et al., 2002) were crossed with Limk1 mutant mice (Meng et al., 2002) to obtain Hb9-gfp+ Limk1+/− parents. Limk1 embryos were genotyped by PCR as described previously (Meng et al., 2002). The presence of the Gfp transgene was assessed either visually or by PCR (forward primer: 5′-GCA AGC TGA CCC TGA AGT TCA TC-3′; reverse primer: 5′-AAC TCC AGC AGG ACC ATG TGA TCG-3′).
Embryonic spinal cord.
E10.5-E12.5 control and Limk1−/− embryos from Hb9::gfp+ Limk1+/− mating pairs were processed for 30 μm transverse cryosections, whole-mount preparations, and dissociated cell culture. Length and pixel density measurements were taken using ImageJ (National Institutes of Health). Axon length was normalized using the average control length for each litter.
The cryosections were processed for immunohistochemistry (Hazen et al., 2011) and analyzed for MN length (E10.5) or the extent of MN development and protein expression (E11.5). The motor column was defined by the extent of ventral Isl1 staining. Protein expression within the motor pool was normalized against the background noise density.
For the whole-mount preparations, section E11.5–E12.5 embryos spanning the brachial to lumbar region, with limbs intact, were mounted dorsal side up in a depression slide in Vectashield mounting medium (Vector Laboratories) and analyzed for developmental stage, motor axon length (E11.5 only), or the pattern of motor nerve branching. Embryos were blindly binned into developmental stages using a predetermined timeline (see Fig. 3A–J). The length of motor axons extending into the hindlimb was normalized by dividing the length of each projection by the average control length for the relevant stage.
Figure 3.

Motor axon branching is advanced in E11.5 Limk1 mutant embryos compared with controls. A–T, Using whole-mount limb preparations, we established the timeline (Stages 1–7) over which motor nerves project into either the forelimb (A–R) or hindlimb (S, T; and data not shown) of E11.5 (A–N) to E12.5 (O–T) Hb9::gfp transgenic embryos, either alone (control, A–E, K, L, O, P, S) or in combination with the Limk1 mutation (mutant, F–J, M, N, Q, R, T). The pattern by which the GFP+ branches segregated was highly reproducible between embryos; however, the ventral nerve branches, especially the median (m) nerve, often extended out of the plane most easily visualized by confocal microscopy. A, At Stage 1, the earliest stage observed in E11.5 embryos, the MNs in the brachial lateral motor column have projected into and past the brachial plexus (arrowhead) and have just divided to innervate the dorsal and ventral forelimb. B, Stage 2 is marked by the ventral branch segregating into the cutaneous maximus (cm), ulnar (u), and median nerves (see also H). C, Stage 3 is marked by the emergence of the radial (r) nerve (open arrowhead). D, E, Stages 4 and 5 are distinguished by the extent of growth of the radial nerve (open arrowheads) and the increasing branching of the cutaneous maximus nerve. In the later stages, the axillary (a) nerve starts (Stage 4) and then completes (Stage 5) a highly reproducible turn (E, J, arrows). F–J, Limk1 mutant motor nerves branch with a similar pattern to those in control littermates. However, the GFP+ Limk1−/− axons in the axillary nerve tip appear to be less exploratory as they reach the turning point in stages 4 and 5 (J, box). K–N, High-magnification images of the control (K, L) or mutant (M, N) axillary nerve tip shown in the boxed region of E and J, respectively. L, N, Outline of the area of the tip used for quantification in V. O, P, Stages 6 and 7, the stages in our timeline occurring in E12.5 embryos, are marked by the increasing extent of the cutaneous maximus nerve and length of the axillary nerve. Both the radial and axillary nerves start to bifurcate in Stage 6; this bifurcation is pronounced at Stage 7. Q, R, By E12.5, the branching pattern of Stage 6 and Stage 7 GFP+ Limk1 mutant motor nerves is indistinguishable from controls. S, T, By E12.5 in the hindlimb, GFP+ motor nerves have projected from the lumbar plexus into three branches: the femoral (f), dorsal peroneal (p), and ventral tibial (t) nerves. There was no distinguishable difference between the branching patterns in the control (S; n = 10) and Limk1 mutant (T; n = 3) hindlimbs. U, E11.5 embryos are not evenly distributed between the five stages. Whereas control forelimbs (n = 43 limbs) are more likely to be either Stage 1 or Stage 2, mutant forelimbs (n = 27 limbs) are more likely (probability that the distribution of control stages is similar to the mutants: p < 0.0025, Fisher's exact test) to be either Stage 4 or Stage 5, strongly suggesting that the extension of Limk1−/− motor nerves is more advanced in development than controls. V, The total area of the axillary nerve tip is ∼35% smaller in Stage 3–5 Limk1 mutant forelimbs (n = 8 embryos) compared with Stage 3–5 control littermates (n = 9 embryos, probability of similar, p < 0.0022 Student's t test). Interestingly, the area of control nerve tips expands rapidly between Stage 3 and Stage 4; this expansion is observed in Limk1 mutant nerve tips, but not until Stage 5. This result suggests the response of the accelerated Limk1−/− axons to the surrounding environment is delayed until the appropriate time in development. Scale bars: A–J, 250 μm; K–N, 150 μm; O–T, 500 μm. Probability of similarity, **p < 0.005, Student's t-test.
For dissociated cell culture, the ventral region of E11.5 control and Limk1 mutant spinal cords were dissected and dissociated as previously described (Augsburger et al., 1999). After 20 h, the cultures were fixed and the GFP+ MNs analyzed.
Adult spinal cord.
Following perfusion (Gage et al., 2012), the ossified spinal cord was removed and postfixed overnight. After postfixation, the laminectomy method was used to remove the spinal cord from the spine using #2 forceps and iridectomy scissors (Fine Science Tools), cryosectioned in 30 μm transverse sections, and processed for immunohistochemistry. The level of cofilin was assessed in MNs in sections taken from the lumbar (L) 3-L6 levels of spinal cord at different times after a crush injury. Pixel density measurements were taken blindly from NeuN+ ChatN+ MNs; within each transverse section, the three MNs expressing the highest levels of cofilin on the injured side were normalized against their equivalents on the uninjured side.
Immunohistochemistry and ISH
Antibodies against the following proteins were used: mouse α-GFP at 1:2000 (3E6; Invitrogen), mouse α-neuronal Class III β-tubulin at 1:1000 (Tuj1; Covance), mouse α-neurofilament (α-NF) at 1:50 (3A10; Developmental Studies Hybridoma Bank), mouse α-neuronal nuclei at 1:100 (NeuN; Millipore), rat α-F4/80 at 1:500 (Ab6640; Abcam), chicken α-NF at 1:1000 (Ab5539; from Millipore), rabbit α-cofilin at 1:100 (Cytoskeleton), rabbit α-phosphorylated (p)-cofilin at 1:100 (Santa Cruz Biotechnology), rabbit α-GAP43 at 1:200 (D9C8; Cell Signaling Technology), rabbit α-Foxp1 (Abcam, Ab16645), sheep α-GFP at 1:2000 (Biogenesis), and goat α-islet1 (Isl1; R&D Systems). Cy3-, Cy5-, and FITC-coupled secondary antibodies were obtained from Jackson ImmunoResearch Laboratories. Fluorescence images were collected on a Carl Zeiss LSM510 confocal and LSM700 confocal.
An in situ probe against the 3′ untranslated region of the mouse Limk1 mRNA was prepared and hybridized to cryosections of E11.5 mouse tissue as described previously (Phan et al., 2010). Differential interference contrast images were collected on a Carl Zeiss Axiovert 200M.
Western analysis
Spinal cord tissue from P19 pups and 7-week-old mice was extracted as previously described (Richner et al., 2017). Briefly, the spinal cord was flushed from decapitated animals by the insertion of a 5 ml (adult) or 2.5 ml (for pups) syringe needle filled with PBS into the spinal cavity and the application of hydraulic pressure resulting in the extrusion of spinal tissue. Using the ventral crease as a guide, the spinal cord was dissected longitudinally to specifically isolate ventral spinal tissue. Flash-frozen spinal tissue was crushed using a pestle, and the proteins extracted into RIPA buffer for 2 h at 4°C. A standard Western analysis was performed using the following antibodies: primaries, rabbit α-cofilin at 1:500 (Cytoskeleton), rabbit α-cofilin at 1:500 (Santa Cruz Biotechnology), goat α-GAPDH at 1:200; secondaries, donkey α-rabbit HRP at 1:50,000 and donkey α-goat HRP at 1:50,000. The blots were developed with SuperSignal West Femto Chemiluminescent Substrate (Themo Fisher Scientific), imaged on a Sapphire Biomedical Imager (Azure Biosystems), and analyzed using AzureSpot software (Azure Biosystems). GAPDH was used a loading control to normalize the signal in the experimental lanes.
Sciatic nerve crush injury
Standard sciatic nerve crushes (Ma et al., 2011a) were performed on 6- to 8-week-old control and Limk1 mutant age-matched male and female mice. While the mice were under anesthesia, the sciatic nerve was located and crushed either for 30 s using #2 forceps (Fine Science Tools) or for two periods of 30 s using an ultra-fine hemistat tool (Fine Science Tools). In both methods, the area around the crush site was observed to be transparent, indicating that it was void of nerve fibers After injury, the surgical site was sutured, and the mice were allowed to recover for 0, 2, 4, 8, 16, 24, and 48 h. After recovery, the mice were perfused and the operated and unoperated (control) sciatic nerves were collected, marked with charcoal at the proximal end, postfixed for 1 h, and left in sucrose overnight. To keep conditions consistent between the injured and control nerves, the nerves from each mouse were cryo-sectioned into 20–30 μm longitudinal sections onto the same microscope slide and stained for image analysis using immunohistochemistry. The lesion site was defined by the upregulation of both GAP43 protein and NF at the site of injury. All quantifications (axon length, protein levels, and intensity) were performed blind using ImageJ. (p)-cofilin levels were normalized against the level of NF staining in the control sciatic nerve; NF levels are unaltered in Limk1 mutant mice compared with control littermates (data not shown). Intensity values for NF, GAP43, and F4/80 antibody staining were measured within the area of rectangular boxes (x = 50 μm, y = the height of the nerve) drawn sequentially along the length of the nerve using the FIJI module These measurements were then normalized to basal intensity levels, by measuring the corresponding average of the NF, GAP43, or F4/80 intensity per area in a control nerve (i.e., the uninjured nerve from the same animal).
Behavioral tests
In all cases, mice were scored blind to their genotype, and a 50:50 ratio of male and female mice were analyzed.
Gait analysis.
To assess the recovery of motor function after injury, stride length was measured using a simple gait analysis system (Carter et al., 2001). The right and left hindlimbs were painted a different color, and the mice were permitted to run on a paper-lined corridor into a dark enclosure, resulting in footprints from both the injured and uninjured limbs for analysis. WT and mutant mice were tested before injury to determine a baseline stride length and every day for 5 d after injury.
Plantar test (Hargreaves' method).
To assess the recovery of motor function after injury, paw withdrawal response time to radiant heat was measured as described previously (Hargreaves et al., 1988). Control and Limk1 mutant mice were tested before injury and then every 2–3 d after injury for the next 30 d. Both the injured and uninjured hindlimb paws were tested for a total of five trials per paw for each time point.
Statistical analyses were performed using a two-tailed Student's t test (when the data fit a normal distribution), the Mann–Whitney test, or Fisher's exact test.
Results
Our previous studies have shown that manipulating the balance between cofilin and Limk1 activity alters the rate of axon outgrowth during development (Phan et al., 2010; Yamauchi et al., 2013). Most notably, increasing the activity of cofilin indirectly, through the loss of Limk1, accelerated the rate of spinal commissural axon outgrowth by ∼20%. However, it has remained unresolved whether this mechanism of controlling the temporal development of neural circuits was specific to sensory axons or more generally applied to other classes of axons in the developing spinal cord.
Cofilin is inactivated by Limk1 in many classes of embryonic spinal neurons
During development, cofilin and Limk1 are present in a broad subset of after mitotic neurons in the marginal layer of the mouse embryonic spinal cord (Sarmiere and Bamburg, 2004; Phan et al., 2010) (Fig. 1C–E), suggesting that the cofilin/Limk1 pathway serves as a more general regulator of spinal axon extension. Limk1 appears to be active in many classes of spinal axons, since phosphorylated, inactive cofilin ((p)-cofilin) is present throughout the embryonic (E) stage 11.5 spinal cord in a pattern similar to that of Limk1. (p)-cofilin is found at highest levels in dorsal neurons and their processes (Fig. 1F,G), as observed previously (Phan et al., 2010), as well as at rather lower levels in spinal MNs (Fig. 1F′,G′). It is present throughout the motor columns, which were demarcated by the presence of an Hb9::gfp transgene (Fig. 1F,F′) (Wichterle et al., 2002). More specifically, labeling with antibodies against Islet (Isl) 1/2 (Fig. 1D,F,F′) and FoxP1 (data not shown) (Rousso et al., 2008) showed that (p)-cofilin is present in the medial, hypaxial, and lateral motor columns (MMC, HMC, and LMC) at both limb levels. The inactivation of cofilin requires Limk1: the level of inactive cofilin was reduced by ∼40% in the motor columns of Limk1 mutant embryos (Fig. 1H–J).
Limk1 and cofilin have not previously been found to have significant roles directing neuronal cell fate (Mizuno, 2013). Nonetheless, we examined whether spinal MNs differentiate on schedule, with their normal complement of identities, in Limk1 mutants. MNs start to differentiate at ∼E9.0; as they are generated, they segregate into different functional classes defined by the positions of their cell bodies within the spinal cord and the patterns of their axonal trajectories (Alaynick et al., 2011). We observed no significant differences between Limk1 mutant and control littermates in the timing or spatial segregation of the Isl1+ Foxp1− MMC and HMC, Isl1+ FoxP1+ medial LMC (LMCm), or Isl1− FoxP1+ lateral LMC (LMCl) (Fig. 1K–N). That the identities of the mutant MNs are indistinguishable from those of controls suggests that Limk1 does not function in the specification of MN fate.
Loss of Limk1 activity results in longer embryonic motor axons both in vitro and in vivo
To assess the consequences of decreasing Limk1 activity on motor axon growth, we analyzed mice mutant for Limk1 (Meng et al., 2002). Previous studies have shown that these mice have elevated levels of cofilin activity and accelerated growth rates of spinal commissural axons (Meng et al., 2002; Phan et al., 2010). To facilitate our analysis, we crossed the Limk1 mutant line to an Hb9::gfp genetically encoded reporter to enable the visualization of motor axons (Wichterle et al., 2002).
Starting from E9.5, motor axons in the LMC extend from the ventral horn of the spinal cord toward the developing limbs (Fig. 2A). LMC neurons initially project their axons along a common path, but at the base of the limb the motor nerve pauses at the plexus and then bifurcates to form distinct dorsal and ventral branches (Tosney and Landmesser, 1985) (Fig. 2C, bracket; Fig. 3A, arrow). Our studies show that motor axons deficient for Limk1 have an ∼15% increase in their extent of outgrowth in vivo compared with control littermates, a similar value to that observed for commissural axons (Phan et al., 2010) (Fig. 2H). This increase in axon outgrowth was observed in two embryonic stages as well as in culture in vitro. At lumbar levels in E10.5 spinal cords, Limk1−/− motor axons have extended ∼18% further from the ventral root compared with motor axons in the control littermates (Fig. 2A,B,H). This increase persists; at E11.5, Limk1−/− motor axons have extended ∼18% further from the lumbar plexus than controls (Fig. 2C,D,H). Supporting the model that signaling has been intrinsically altered within Limk1−/− MNs to permit them to extend faster-growing axons, Limk1−/− MNs grown in dissociated culture in vitro also extend ∼12% longer axons (Fig. 2F–H) than those dissociated from control littermates (Fig. 2E,H).
Figure 2.

Limk1 mutant motor axons extend consistently further than control axons. A, B, In transverse sections of E10.5 spinal cord, motor axons have started to exit the lumbar spinal cord through the ventral root. Motor axon growth (brackets) is more advanced in Limk1 mutants compared with littermate controls. Motor axons were labeled by the presence of an Hb9::gfp transgene (green), and axons were labeled with antibodies against NF (red). C, D, By E11.5, motor axons have started to project beyond the lumbar plexus. Whole-mount preparations of the lumbar spinal cord show that axon growth continues to be more advanced in Limk1 mutants compared with controls. E–G, MNs were dissociated from limb-level E11.5 Limk1+/+;Hb9::gfp and Limk1−/−;Hb9::gfp spinal cords. After 20 h in culture, the Limk1 mutant GFP+ neurons had extended longer processes than the control GFP+ neurons. H, Quantification of axon lengths demonstrated that the loss of Limk1 activity results in axons that are consistently ∼15% longer than control axons (i.e., similar to the figure previously seen for dorsal sensory interneurons) (Phan et al., 2010). Dissociated MNs: The Limk1−/− GFP+ neurons (n = 446 neurons from 5 embryos; probability different from control, p < 0.028, Student's t test) extended axons that were ∼12% longer than those from the GFP+ control neurons (n = 171 neurons from 4 embryos). E10.5 spinal cord sections: The Limk1−/− motor axons (n = 155 sections from 11 embryos; probability different from control, p < 0.03) had extended ∼18% further from the ventral root of the spinal cord compared with control motor axons (n = 90 sections from 10 embryos). E11.5 spinal cord whole mounts: The Limk1−/− motor axons (n = 13 limbs taken from 8 embryos; probability different from control, p < 0.046) had extended ∼18% further from the lumbar plexus compared with control motor axons (n = 12 limbs taken from 7 embryos). Scale bars: A, B: 100 μm; C, D, 400 μm; E–G: 25 μm. Probability of similarity, *p < 0.05, Student's t-test.
Motor axon branching patterns are relatively normal in Limk1 mutant embryos
After motor axons project from the plexus, they immediately start to branch into the motor nerves that will innervate the distinct musculature of each limb. Branching starts around E11.5 in the forelimb, and E12.5 in the hindlimb, with each limb containing a distinctive and highly stereotyped pattern of branches (Fig. 3). To assess whether the timing of motor nerve branching was altered in Limk1 mutants, we first characterized the normal branching pattern of GFP+ motor nerves in the E11.5–E12.5 forelimb (Fig. 3A–E,O,P). Forelimbs were assigned into one of seven stages, described in detail in Figure 3. Stages 1–5 span the observed stages in E11.5, from the first bifurcation into the dorsal and ventral halves of the limb, to the emergence of five characteristic branches, the radial (r), axillary (a), median (m), ulnar (u), and cutaneous maximus (cm) nerves (Fig. 3A–E). Stages 6 and 7 in E12.5 embryos were distinguished by further nerve growth and bifurcations (Fig. 3O,P).
The sequence in which the motor nerves branch is highly stereotyped between embryos. This stereotypy permitted us to assess the distribution of Stage 1–5 forelimbs in E11.5 embryos taken from litters of Limk1+/− parents. We assessed 15 litters and found that there was a significant bias (p < 0.0025, Fisher's exact test) toward the more advanced stages in the Limk1−/− embryos, compared with control littermates (Fig. 3U), suggesting that these axons were growing more rapidly than control axons. While Limk1−/− motor nerves appeared to be more advanced in their development, their pattern of branching was largely indistinguishable from that in control embryos (Fig. 3F–J,Q,R). However, there were some subtle morphological differences, most notably in the axillary nerve, which enlarges at Stage 3 and then starts a distinctive turn at Stage 4 (arrow, Fig. 3D,K,L,). In controls, the area of the growing end of the axillary nerve abruptly increases by ∼2-fold during Stage 4 (Fig. 3V), as if the axons defasciculate within the nerve to make the turn. In contrast, the area of the Limk1−/− axillary nerve is ∼50% smaller, only starting to increase in size during Stage 5 (Fig. 3V,M,N). This delay in defasciculation suggests that there may be a synchronization defect between the axillary nerve and the surrounding tissue, such that the nerve only expands once the forelimb is at the appropriate stage of development (i.e., the axons reach their intermediate target too early in development to correctly interpret it).
Together, these data suggest that increasing the activity of cofilin accelerates the rate of motor axon growth in a very similar manner to that observed for sensory commissural axons (Phan et al., 2010). Perturbing this temporal guidance mechanism has consequences for motor nerve morphology in the forelimb; however, these defects are transient. By Stage E12.5, the morphology and branching pattern of Limk1−/− motor nerves in both the fore and hindlimbs are indistinguishable from controls (Fig. 3O–T). Supporting this conclusion, Limk1 mutant adult mice walk with a normal alternating gait (data not shown).
Levels of inactivated cofilin are reduced in adult mice
Our studies have suggested that the cofilin pathway regulates the rate of axon outgrowth during development. Does cofilin also regulate the rate at which axons regenerate? Cofilin is present at high levels in the embryonic spinal cord (Fig. 1E) (Phan et al., 2010) and throughout the P19 postnatal spinal cord, including at very high levels in the ChAT+ NeuN+ MNs (Fig. 4A,B, box, C). Cofilin is also highly phosphorylated in P19 spinal cords (Fig. 4E,G,H,I). (p)-Cofilin is present in ChAT+ NeuN+ MNs in the ventral gray matter (Mullen et al., 1992; Friese et al., 2009) (Fig. 4F, boxed region, inset), and in Tuj1+ neurites in the white matter (Fig. 4H, inset), suggesting that it is mostly present in an inactive form in neurons and their processes. However, while cofilin continues to be expressed in the adult spinal cord (Fig. 4D,I), the levels of (p)-cofilin diminish by ∼50% in the adult spinal cord compared with the P19 stage (Fig. 4H,I), suggesting that less of the total cofilin is present in its inactive form. Since a ratio of high (p)-cofilin to total cofilin appears to be a signature of developing neuronal circuits, we further investigated whether this ratio was also a feature of regenerating circuits.
Figure 4.

Cofilin is largely inactive in the adult spinal cord. A–H, Limb level sections of P19 (A–C, E–G) or adult (D, H) spinal cords were labeled with antibodies against total cofilin (red, A, C, D), phosphorylated (p)-cofilin (red, E, G, H), NeuN (green, neuronal nuclei; A, E), ChAT (blue, mature MNs; A, B, E, F), and Tuj1 (green, neuronal processes; H, inset). A–C, Cofilin is present at high levels throughout the P19 postnatal spinal cord. In this lumbar section, cofilin is present at high levels in a population of ventral NeuN+ ChAT+ neurons, thought to be α MNs (magnified B, inset). D, Cofilin continues to be expressed in the adult spinal cord, albeit at significantly lower levels, particularly in the white matter. E–H, Cofilin is highly phosphorylated at both neonatal (E–G) and adult stages (H), including in the ventral NeuN+ ChaAT+ MNs (F, inset) and the Tuj1+ neural processes in the white matter (H, inset; and data not shown), suggesting that cofilin is mostly inactive in axons. I, The levels of cofilin and (p)-cofilin were quantified in a Western analysis of P19 and adult ventral spinal cord tissue, using GAPDH levels as a loading control. While cofilin levels are not significantly different (p > 0.17, Student's t test) between the P19 and adult spinal cord (three independent samples shown in I; n = 6 animals analyzed in 5 experiments), there was a ∼50% decrease in (p)-cofilin levels in adult spinal cord (n = 6 animals analyzed in 4 experiments; p < 0.0004, Student's t test) compared with P19 animals. Scale bars, 300 μm. Probability of similarity, ***p < 0.0005, Student's t-test.
Cofilin is inactivated after peripheral nerve injury
Motor circuitry is established relatively normally in Limk1−/− mice, permitting us to investigate whether the cofilin/Limk1 pathway has a role in regulating the regeneration of adult peripheral nerves. We first assessed the timeline over which axon regeneration occurs in a standard sciatic crush (Bauder and Ferguson, 2012). During this procedure, the sciatic nerve was clamped for 30 s, and then the mice were permitted to recover for up to 3.5 d after injury. The injured and uninjured sciatic nerves were then longitudinally thin-sectioned and labeled with antibodies against NF to reveal the total axon trajectories (Dodd et al., 1988), GAP43 to assess the regenerating axons (Li et al., 1996), and F4/80, which labels mouse macrophages (Austyn and Gordon, 1981).
NF+ axons are completely severed in the region of the crush at day 0 of our protocol (Fig. 5B). They start to degenerate distal to the crush site by day 2, such that the intensity of NF is reduced distal to the lesion (Fig. 5C,E, bracketed region), consistent with the timeline observed previously (Beirowski et al., 2005). The process of Wallerian degeneration, followed by regeneration of nerves across the lesion site, appears to be largely complete by day 3.5, when the distal NF levels become indistinguishable from day 0 (Fig. 5E). The regeneration process is remarkably fast in mice, and appears to be facilitated by first, the rapid mobilization of macrophages to the lesion site at day 0 (Fig. 5B,D), and the distal portion of the nerve by day 2 (Fig. 5C,D), where they phagocytose the degenerating axonal debris (Gaudet et al., 2011). This process is so fast, that it is observed in nerves immediately after being crushed (Fig. 5B) (i.e., the macrophages invade the lesion site in the time taken to process the mice for perfusion). Second, the regeneration of the proximal stump starts within hours of the crush injury. Consistent with previous observations (Shin et al., 2014), our time course experiments showed that GAP43 can be observed accumulating in severed axons on the proximal side of the lesion as early as 2 h after the crush injury (Fig. 5G,H). GAP43 levels are ∼2-fold higher 4 h after the crush (Fig. 5I, arrows) and increase to ∼5-fold higher after 8 h (Fig. 5J, arrows), suggesting that axon regeneration commences very rapidly after the crush.
Figure 5.

Timeline of regeneration in sciatic nerve crush model. A–E, Sciatic nerves of 6- to 8-week-old mice were crushed for 30 s and the mice permitted to recover for 0 d (i.e., immediate perfusion; A, B, D), 2 d (C, D), and 3.5 d (D). The extent of both NF+ (red, A–C) axon-severing, and F4/80+ (green, A–C) macrophage invasion was assessed in control (unoperated side) and crushed nerves. Control nerves (A, n = 11 sections from 4 mice) show the baseline numbers of F4/80+ macrophages, and an orderly array of NF+ axons. In contrast, immediately after the crush (n = 14 sections, from 4 mice), F4/80+ macrophages invade the lesion site (D), which is devoid of NF+ axons (E), suggesting that the crush is complete. By 2 d after the crush (n = 19 sections, from 5 mice), F4/80+ macrophages are elevated through the region distal to the crush (D; p < 0.0001 similar to day 0, Student's t test); these elevated levels continue to be observed at 3.5 d after crush (n = 6 sections, from 3 mice, p < 0.0001 similar to day 0, Student's t test). At 2 d, NF+ axons have started to degenerate distal to the crush (C); this process appears to be complete by 3.5 d, with the intensity of NF+ axons returning to day 0 levels (E). F–K, Mice with sciatic crush injuries were permitted to recover for 0 h (i.e., immediate perfusion; n = 11 sections from 4 mice; F, G), 2 h (n = 12 sections, from 4 mice; H), 4 h (n = 11 sections, from 4 mice; I), and 8 h (n = 8 sections, from 3 mice; J). We quantified the level of GAP43 intensity (K) to assess the timeline over which GAP43+ axon regeneration begins. Consistent with previous findings (Shin et al., 2014), we found that elevated levels of GAP43 are observed in proximal axons (I, K, arrowheads) by 4 h after the crush (p < 0.005 similar to 0 h, Student's t test), with GAP43+ axons apparently starting to regenerate trough the lesioned area (J, K, arrowheads) as soon as 8 h after the crush (p < 0.0001 similar to 0 h, Student's t test). Scale bars: A–F, 300 μm; G–J, 250 μm.
We next assessed the level of cofilin activity after a sciatic nerve crush injury. Remarkably, we found that (p)-cofilin levels increase in proximal axons at the lesion site, over a timeline that follows that of GAP43. (p)-Cofilin levels start to increase at the proximal-most edge of the lesion (L-P) at 8 h (Fig. 6G,T, arrowheads). This increase becomes significantly different (p < 0.005, Student's t test) by 16 h after the crush injury (Fig. 6H,T). Levels of (p)-cofilin progressively increase as the regenerating axons traverse the lesion site (Fig. 6I, arrowheads), with axons robustly growing past the distal-most edge of the lesion (L-D) 48 h after injury (Fig. 6B,T). By the 2 d time point, (p)-cofilin levels are ∼2-fold higher at the lesion site, compared with the levels either in the uninjured nerve or in the region distal to the lesion site (Fig. 6A,B,F,T). (p)-Cofilin levels also appear to be elevated specifically in the GAP43+ regenerating axons immediately distal to the lesion (Fig. 6B,C, compare arrows on contiguous sections from the same nerve).
Figure 6.

Cofilin is endogenously inactivated in regenerating peripheral axons in a Limk1-dependent manner. A–F, The sciatic nerves of Limk1+/+ (control, A–C; n = at least 39 sections from 10 mice) or Limk1−/− (mutant, D, E; n = at least 30 sections from 5 mice) 6- to 8-week-old mice were crushed and the mice permitted to recover for 2 d. Contiguous longitudinal sections of both injured and uninjured sciatic nerves were labeled with antibodies against NF (red, A, D), (p)-cofilin (green, A, B, D, E), and GAP43 (C), and the intensity of (p)-cofilin staining quantified on the uninjured side, the site of the crush lesion and distal to the lesion (F). By 2 d of recovery, (p)-cofilin levels were higher in the lesion site (A, B, F) compared with uninjured controls (probability of similarity p < 2.3 × 10−11, Student's t test) and in regenerating GAP43+ axons (B, C, arrows; B and C are serial sections). Significantly reduced (p)-cofilin levels were observed in the uninjured or distal region in Limk1 mutant peripheral nerves compared with controls (F, similarity with control lesion: proximal, p < 3.9 × 10−7; distal, p < 6.3 × 10−4). While cofilin was inactivated by ∼25% at the lesion site in Limk1 mutants (D, E), this level was significantly less than observed in control lesions (F, similarity with control lesion, p < 1.7 × 10−6). G–I, T, We examined the level of cofilin activity in the lesion site 8 h (G, Y, n = 16 sections, from 3 mice), 16 h (H, T, n = 14 sections, from 3 mice), 24 h (I, T, n = 10 sections, from 3 mice), and 48 h (T, n = 7 sections, from 5 mice) after injury. Phosphorylated cofilin levels were quantified in the regions immediately proximal and distal to the lesion, as well as at three levels within the lesion (for position, see I): proximal (L-P), middle (L-M), and distal (L-D). By 8 h, the first regenerating axons have reached the proximal edge of the crush site (L-P) where they start to upregulate (p)-cofilin levels (G, arrowheads; p < 0.081 significantly different from the 8 h proximal region, Student's t test). This process is robust by 16 h (H, arrowheads; p < 0.005 different from the 8 h proximal region), with regenerating axons reaching L-M by 24 h (I, arrowheads; p < 5 × 10−6) and L-D by 48 h (T, p < 6 × 10−9). J–R, We assessed total cofilin levels in large diameter Chat+ NeuN+ MNs in the L3–L6 spinal cord in control (J–M) and injured (N–Q) animals. While there was little deviation in cofilin levels in control MNs on the left and right side of the spinal cord (R, n = 30 MNs, from 3 mice), there was an average 15% increase in cofilin levels in MNs on the injured side, 4 h after injury (R, n = 36 MNs, from 3 mice, p < 5.0 × 10−4 significantly different from control) with up to twofold increases observed in some MNs. There is a corresponding ∼14% decrease in cofilin levels in MNs 8 h after injury (R, n = 24 MNs, from 2 mice, p > 0.068 significantly different from control), with cofilin levels normalizing at 16 h from injury (R, n = 33 MNs, from 3 mice, p > 0.38 that injured side is significantly different from the uninjured side). S, There was also an increase in total cofilin in the injured nerve (S). By 4 h after the injury, cofilin levels are elevated distal to the lesion (p < 0.0001 significantly different from 0 h, Mann–Whitney test); this elevation is robust by 8 h (p < 0.0001 significantly different from 0 h, Mann–Whitney test). For n, see Figure 5G, H. U, Model for the timeline over which the rate of peripheral axon regeneration is regulated. After a sciatic nerve crush injury (1; t = 0), a putative retrograde signal (2) increases cofilin levels first in injured spinal MNs (3; t = 4 h) and then in regenerating nerves (3; t = 4–8 h). Cofilin is inactivated in axons, which controls the rate at which they regenerate across and past the lesion site (4; t = 8–16 h). Scale bars: A–E, 300 μm; F–H, 225 μm; J–Q, 100 μm. Probability of similarity, ***p < 0.0005, Student's t-test.
The increase in inactivated cofilin in the regenerating axons is prefigured by an increase in total cofilin in the MNs at the L3-L6 levels of the spinal cord, which send axons into the sciatic nerve (Watson et al., 2009). In control spinal cords, there is no difference between the levels of cofilin in MNs in the left and right sides of the L3-L6 spinal cord (Fig. 6J–M,R). In contrast, 4 h after injury, there is a significant (p < 5.0 × 10−4, Student's t test) increase in the level of cofilin in MNs on the injured side of the spinal cord (Fig. 6N–Q,R). This increase is transient and is followed by a small but significant decrease in cofilin levels by 8 h after injury (Fig. 6R). Cofilin may subsequently be trafficked into the injured nerve: total cofilin levels increase by ∼40% in the axons flanking the lesion site by 4–8 h after injury (Fig. 6S). Together, these results suggest that an early response to a nerve injury is to reassemble the cytoskeletal regulators that were critical for controlling the rate of axon growth while neural circuits are being assembled during development.
The sciatic nerve regenerates more rapidly in Limk1 mutant mice
Previous studies have shown that cofilin activity is negatively regulated by Limk1 (Bernard, 2007); thus, we examined whether the increase in (p)-cofilin levels observed after a sciatic lesion is dependent on Limk1. In experiments performed in parallel with those described above, we also crushed the sciatic nerves of Limk1 mutant littermates. As predicted, the loss of Limk1 resulted in cofilin being less phosphorylated and presumably more active. This effect was particularly dramatic at the lesion site (Fig. 6D–F), where the level of (p)-cofilin was very similar to that observed either in uninjured nerves or in the regions distal to control lesions (Fig. 6A,B,F).
Our previous work suggested that activating cofilin in regenerating axons would result in increased axon outgrowth (Phan et al., 2010; Yamauchi et al., 2013). To test this hypothesis, we assessed whether the regeneration of the sciatic nerve is accelerated in Limk1 mutants after a crush injury. In a series of six independent experiments, 18 Limk1+/+ (control) and 17 Limk1−/− 6- to 8-week-old mice underwent the sciatic nerve crush procedure. The animals recovered for 2 d and then were perfused. Longitudinal sections of the injured sciatic nerve from both control and mutant mice were labeled with antibodies against GAP43 (Fig. 7A,B). After 2 d of recovery, the axons in control animals had grown ∼1.5 mm, the expected length (Hadlock et al., 2005) (Fig. 7A). In contrast, the Limk1−/− axons had grown ∼15% further (Fig. 7B,C), suggesting that their average growth rate was increased by the same extent observed for Limk1−/− axons in the developing spinal cord (Phan et al., 2010). Closer inspection of the individual lengths of regenerating GAP43+ axons suggested that there was a more dramatic 2- to 3-fold increase in the lengths of the longest axons: those ≥60 μm longer that the average length (Fig. 7D). This phenotype was highly reproducible; in all six experiments, we observed that the Limk1−/− axons had grown further than the control littermates. Together, these studies, along with those from the developing spinal cord, strongly suggest that increasing cofilin activity results in faster rates of regeneration of peripheral nerves.
Figure 7.
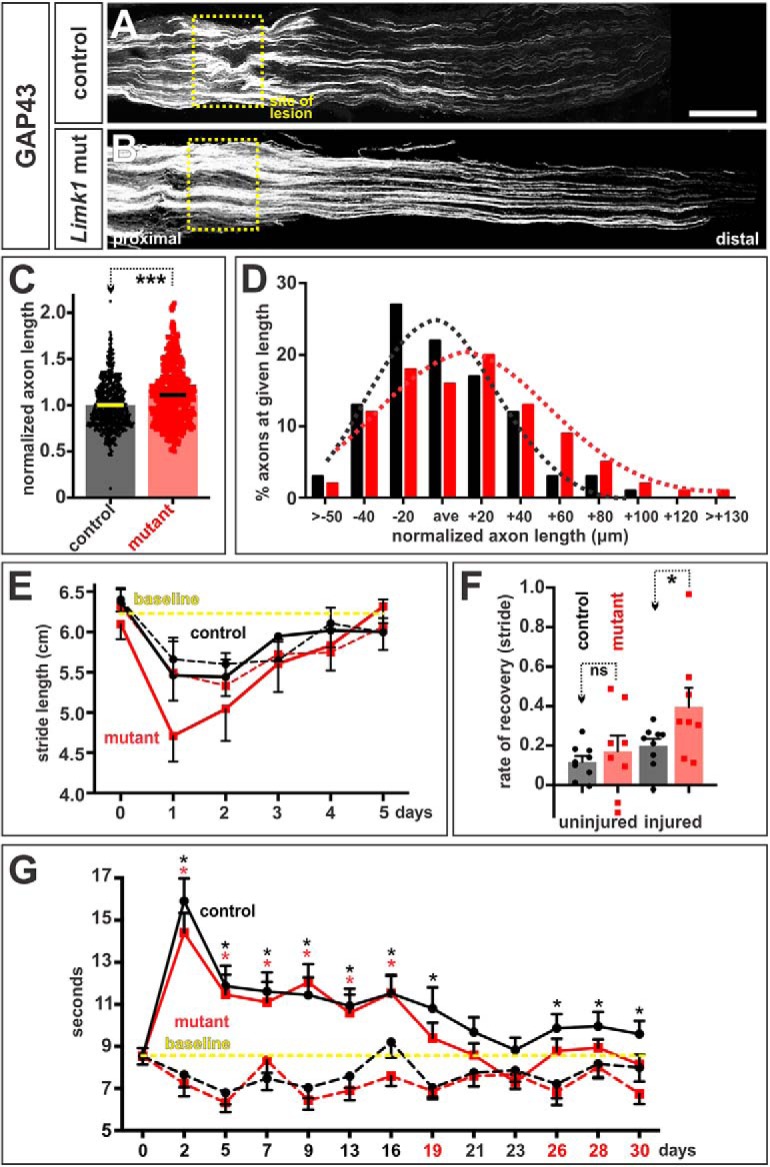
Promoting peripheral axon growth enhances recovery of motor and sensory function. A, B, The sciatic nerves of Limk1+/+ (control, A) or Limk1−/− (mutant, B) 6- to 8-week-old mice were crushed and permitted to regenerate for 2 d. Transverse sections of the sciatic nerves were labeled with antibodies against GAP43, which labels regenerating axons. C, After 2 d of recovery, axons had grown ∼15% further in the mutants (36 sections from 18 mice, 6 experiments) than the controls (33 sections from 17 mice, p < 4 × 10−13, Student's t test). D, The length of individual regenerating axons was compared with the average (ave) axon length for each experiment. Each bin represents a given axon length in ±20 μm increments with respect to the average length. Dotted lines indicate the normal distributions for the control and Limk1 mutant axon lengths and show that there is ∼3-fold increase in the number of longest axons in the Limk1 mutant compared with controls. E, Gait analysis (recovery of motor function). Stride length was measured from the footprints of operated (continuous line) versus unoperated (dotted lines) hindpaws painted different colors. Control (n = 9 mice from three experiments) and mutant mice (n = 8) were tested from day 0 (baseline) to day 5, immediately before and after the sciatic nerve crush injury. The crush injury more severely compromises the movement of the mutant mice; however, their rate of recovery (the gradient of the slope from day 1 to day 5) is apparently faster. F, The rate of recovery is ∼2-fold faster for the Limk1 mutant mice compared with control littermates (p < 0.032, Mann–Whitney test). G, Plantar test (recovery of sensory function). Sensitivity to heat was tested over a 30 d period using the Hargreaves methods by measuring the time taken for the mice to withdraw their operated (continuous line) versus unoperated (dotted line) hindpaws from a focused beam of radiant heat. Neither the control nor mutant unoperated paws showed a statistical difference compared with the day 0 baseline (data not shown). In contrast, the control (n = 6 mice from two experiments) and mutant (n = 6) operated paws lost sensitivity to heat. However, the Limk1 mutant mice more rapidly recovered sensitivity compared with control littermates returning and staying at the baseline level for paw withdrawal ∼2 d earlier. Although the controls returned to baseline levels of heat sensitivity at ∼3 weeks, unlike the mutants, they did not consistently stay there, suggesting that they may require >30 d to fully recover their preoperation withdrawal response to heat. Probability of similarity, ***p < 0.0005, *p < 0.05, Student's t-test or Mann Whitney test. Scale bar, 300 μm.
More robust sciatic nerve regeneration leads to improved sensory and motor recovery
The increase in regenerative axon growth also resulted in a modest improvement in the recovery of motor and sensory function. To assess motor function, we performed a gait analysis to compare the time course over which the stride length of control or Limk1 mutant-injured mice returned to baseline (i.e., preoperation) levels. Stride length was assessed by painting the hindpaws of the operated mice different colors, and then measuring the distance between the footprints left as they ran along a corridor toward an enclosed box (Fei et al., 2010). The stride of both control and Limk1 mice was most severely affected the day after the crush injury. Both the operated (Fig. 7E, continuous lines) and unoperated (Fig. 7E, dotted lines) hindlimbs have shortened stride lengths, which then recover to the baseline length by day 5. The dynamics of this recovery were very similar in the control (operated and unoperated) and Limk1 mutant (unoperated only) limbs. In contrast, the stride length on the operated Limk1 mutant hindlimb was slightly worse than either control or unoperated Limk1−/− hindlimbs, suggesting that the mutants were more compromised in their ability to move after the crush injury. However, the rate of recovery was twofold faster for the Limk1−/− mice compared with controls (Fig. 7F), such that they returned to baseline stride length between day 4 and 5, slightly before the control mice (Fig. 7E).
We also assessed recovery of sensory function by determining the time course over which the animals recovered heat sensitivity in the Plantar test (Hargreaves' method) (Hargreaves et al., 1988). Using a Plantar analgesia meter, we measured the time taken for a mouse to withdraw its operated versus unoperated hindpaw from a focused beam of radiant light. Both control and Limk1 mutant mice had lost significant sensitivity the day after injury, which gradually returned toward the baseline times over the course of a month. Control mice first recovered levels of heat sensitivity that were not significantly different from the baseline time at day 21 (Fig. 7G). However, this recovery was not robust: heat sensitivity levels clearly improved but were consistently significantly different from baseline levels between days 23–30. In contrast, Limk1−/− mice recovered faster, returning to the baseline times for paw withdrawal at day 19, ∼2 d earlier than the control littermates. Moreover, their recovery was more robust, with the heat sensitivity times remaining close to or at the baseline for the next ∼10 d (Fig. 7G). These results suggest that Limk1−/− mice can recover sensory function both faster and putatively more profoundly than the controls.
Discussion
Neural circuits are established in a constantly changing embryonic environment. If circuits are to form in synchrony with the rest of the developing embryo, axonal growth must be regulated temporally, as well as spatially, for axons to reach and innervate the correct targets at the right time in development. In contrast, regenerating axons in the adult nervous system grow back to their targets in a relatively static environment, such that it is unclear whether the rate of regrowth needs to be actively regulated. We have assessed these questions by examining the role of the cofilin/Limk1 pathway in spinal motor circuitry development and sciatic nerve regeneration.
We have found that the cofilin/Limk1 pathway regulates the rate of developing and regenerative axon growth in strikingly similar ways. High levels of cofilin, much of it in its inactive, phosphorylated form, is a feature of developing circuits that is rapidly recapitulated in injured nerves, presumably to facilitate the regrowth of nerves and/or regulate the endogenous rate of axon regeneration (∼1 mm/d). Together, our data suggested a model in which severing the nerve (Fig. 6U, 1) initiates a retrograde signal (Fig. 6U, 2) that directs spinal MNs to make more cofilin protein (Fig. 6U, 3). Cofilin is then transported into the injured nerve and inactivated in regenerating axons, in a Limk1-dependent mechanism, as they extend across the lesion site (Fig. 6U, 4).
The role of temporal guidance cues in development
While much is known about the mechanisms by which axons interpret spatial information (Butler and Tear, 2007), the means by which temporal information is decoded by growth cones have remained unclear. We have identified a key role for cofilin controlling the extension of axons in the developing spinal cord, presumably by regulating the rate at which actin polymerizes (Bamburg and Bernstein, 2008) (Fig. 1A). Many canonical axon guidance cues, such as netrin1 (Marsick et al., 2010), slit2 (Piper et al., 2006), semaphorins (Aizawa et al., 2001), and the bone morphogenetic proteins (Wen et al., 2007; Phan et al., 2010), can regulate the cofilin pathway and the pathways that control the local orientation of the cytoskeleton. Thus, a growth cone could use sequentially encountered guidance cues to establish the relevant rate of growth and orientation needed to extend along a particular segment of its trajectory. While the identity of the extrinsic cues that establish growth rate of motor axons are not known, our studies have shown that the bone morphogenetic proteins both orient (Augsburger et al., 1999; Butler and Dodd, 2003; Perron and Dodd, 2011) and slow (Phan et al., 2010; Yamauchi et al., 2013) the rate of commissural axon growth away from the dorsal midline.
Disrupting the process of temporal axon guidance results in axons being at the wrong place at a particular developmental time. Upregulating Limk1 in MNs results in defects in motor axon growth and pathfinding, which mimic the human clubfoot phenotype (Collinson et al., 2018). In contrast, downregulating Limk1 activity results in commissural axons reaching the floor plate too early (Phan et al., 2010) and precocious motor axon growth (this study). These latter guidance phenotypes are relatively modest; however, guidance defects are often transient (Serafini et al., 1996) and/or subtle (Lin et al., 1994) because the process of axon guidance tends to be regulative, or self-correcting. Nonetheless, Limk1 may be required to more finely sculpt motor circuits. Although the early branching pattern of motor nerves is grossly normal, and the adult mice walk relatively normally by eye, Limk1−/− mice have been reported to be hyperactive, showing increased rearing behaviors (Meng et al., 2002). Moreover, Limk1 is one of a number of key genes uncovered by the William's syndrome deletion, which leads to many cognitive and motor deficits (Scott and Olson, 2007). Thus, defects in the process of temporal guidance may lead to subtle changes in executive motor circuits and/or spinal motor circuits, including the regulatory interneurons that regulate MNs.
The role of the cofilin pathway in peripheral nerve regeneration
Both cofilin and (p)-cofilin are present throughout the spinal cord in late postnatal mice, with particularly high levels of cofilin observed both in MNs and dorsal sensory afferents. The function of cofilin/(p)-cofilin in postnatal mice is unclear, although it is notable that the mouse learns to walk during the final weeks of postnatal development. A regulated level of cofilin activity may be required to refine motor circuits as the mouse adjusts its gait, such that it may be informative to determine whether modulating cofilin affects the ability of mice to learn motor tasks.
Unexpectedly, we found that the levels of cofilin are rapidly upregulated after a sciatic crush injury, consistent with returning the nerve to a “younger,” more dynamic state. The timeline, proposed in Figure 6U, suggests that crushing the nerve initiates a retrograde signal, which directs MNs to make more cofilin protein, by regulating transcription and/or translation. The nature of this signal is unknown: possibilities include the loss of electrical connectivity, or the immediate influx of calcium known to occur after injury (for review, see van Niekerk et al., 2016). The surge in somal cofilin appears to result in increased levels of cofilin being transported into regenerating axons. The ratio of active to inactive cofilin is then calibrated by Limk1, putatively controlling the rate at which axons start to regenerate. The mechanism by which cofilin is specifically inactivated in axons regenerating across the lesion remains to be identified.
Our previous studies (Phan et al., 2010; Yamauchi et al., 2013) and those of others (Endo et al., 2003) suggest that the consequence of locally inactivating cofilin would be to slow the growth of axons through the lesion site and into the distal nerve. What is the biological role for regulating axon growth rate during axon regeneration? One possibility is that cofilin also regulates sprouting: the overexpression of cofilin leads to more complex filamentous growth cones (Phan et al., 2010), and the loss of Limk1 induces abnormalities in spine morphology and enhanced LTP (Meng et al., 2002). Many species tightly regulate axonal sprouting in the adult nervous system; it is observed during the induction of plasticity (Trachtenberg et al., 2002) but is limited under normal situations, given the potential for deleterious side effects. The benefit/risk of increased sprouting during the process of regeneration has not been fully investigated. However, it may be informative to compare how the levels or activity of cofilin are regulated in animals that show limited regenerative capacity compared those that regenerate neural circuitry more effectively.
Extent of behavioral recovery after injury in accelerated mice
Previous studies have demonstrated that the overexpression of the regeneration-associated protein Heat shock protein 27 (Hsp27) after a sciatic nerve transection increases the length of regenerating GAP43+ axons and improves motor, but not sensory, recovery times (Ma et al., 2011b). We find that the loss of Limk1 also accelerates both developing and regenerating axons by ∼15%, suggesting that there is conservation between the mechanisms that control axon growth rate at different ages. This relatively modest increase in axon growth rates also can have a modest, but significant, effect on the ability of the mouse to recover both motor and sensory function after injury. In the short term, we observed a more rapid improvement in the gait of Limk1−/− mouse compared with controls. This analysis was complicated by the observation that the Limk1−/− mice were more negatively impacted by the crush injury than age-matched control littermates. The basis for this effect is not clear; however, given that the effect of the injury on the Limk1−/− unoperated side is indistinguishable from the controls, it is unlikely to result from generally compromised motor circuitry in the Limk1−/− mice.
These studies are the also first to show that accelerating peripheral axon regeneration can result in the faster recovery of sensory function. These longer-term experiments were not complicated by any initial side effects in the Limk1−/− mice; rather, we observed that they more stably recover the ability to detect radiant heat ∼30% faster than control littermates. This result is significant because sensory function can take longer to recover than motor function after a traumatic injury, yet it is required for patients to recover their quality of life and protect themselves from noxious stimulus.
In conclusion, these studies have provided further insight into the role of the cofilin pathway during the establishment of motor circuitry and reveal that regulation of cofilin is an early response in the regeneration of peripheral nerves. They suggest that monitoring the effects of manipulating cofilin during development may be predictive of the effect of that manipulation during regeneration. It is now critical to determine whether the other modulations of cofilin activity/levels, which more effectively accelerate growth during development, are similarly more effective in a regenerative context.
Footnotes
This work was supported by National Institutes of Health National Research Service Award Fellowship NS 077619 to M.E.F., and Craig H. Neilsen Foundation Grant 284402, Merkin Foundation, and National Institutes of Health Grant NS 063999 to S.J.B. We thank Zhengping Zia for the Limk1 mutant mice line; Bennett Novitch for FoxP1 antibodies; Aaron McGee for teaching us the sciatic crush protocol; David McKemy and his laboratory for help with the sensory behavioral tests; Tanmoy Bhattacharya, Olivia Handley, Harley Kornblum, Francesca Mariani, Aaron McGee, Bennett Novitch, Caroline Pearson, and Larry Zipursky for invaluable discussions; and Reggie Edgerton, Bennett Novitch, and Larry Zipursky for comments on the manuscript.
The authors declare no competing financial interests.
References
- Aizawa H, Wakatsuki S, Ishii A, Moriyama K, Sasaki Y, Ohashi K, Sekine-Aizawa Y, Sehara-Fujisawa A, Mizuno K, Goshima Y, Yahara I (2001) Phosphorylation of cofilin by LIM-kinase is necessary for semaphorin 3A-induced growth cone collapse. Nat Neurosci 4:367–373. 10.1038/86011 [DOI] [PubMed] [Google Scholar]
- Alaynick WA, Jessell TM, Pfaff SL (2011) SnapShot: spinal cord development. Cell 146:178.e1. 10.1016/j.cell.2011.06.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arber S, Barbayannis FA, Hanser H, Schneider C, Stanyon CA, Bernard O, Caroni P (1998) Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature 393:805–809. 10.1038/31729 [DOI] [PubMed] [Google Scholar]
- Augsburger A, Schuchardt A, Hoskins S, Dodd J, Butler S (1999) BMPs as mediators of roof plate repulsion of commissural neurons. Neuron 24:127–141. 10.1016/S0896-6273(00)80827-2 [DOI] [PubMed] [Google Scholar]
- Austyn JM, Gordon S (1981) F4/80, a monoclonal antibody directed specifically against the mouse macrophage. Eur J Immunol 11:805–815. 10.1002/eji.1830111013 [DOI] [PubMed] [Google Scholar]
- Bamburg JR, Bernstein BW (2008) ADF/cofilin. Curr Biol 18:R273–R275. 10.1016/j.cub.2008.02.002 [DOI] [PubMed] [Google Scholar]
- Bauder AR, Ferguson TA (2012) Reproducible mouse sciatic nerve crush and subsequent assessment of regeneration by whole mount muscle analysis. J Vis Exp 60:3606. 10.3791/3606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beirowski B, Adalbert R, Wagner D, Grumme DS, Addicks K, Ribchester RR, Coleman MP (2005) The progressive nature of Wallerian degeneration in wild-type and slow Wallerian degeneration (WldS) nerves. BMC Neurosci 6:6. 10.1186/1471-2202-6-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard O. (2007) Lim kinases, regulators of actin dynamics. Int J Biochem Cell Biol 39:1071–1076. 10.1016/j.biocel.2006.11.011 [DOI] [PubMed] [Google Scholar]
- Bravo-Cordero JJ, Magalhaes MA, Eddy RJ, Hodgson L, Condeelis J (2013) Functions of cofilin in cell locomotion and invasion. Nat Rev Mol Cell Biol 14:405–415. 10.1038/nrm3609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britt JM, Kane JR, Spaeth CS, Zuzek A, Robinson GL, Gbanaglo MY, Estler CJ, Boydston EA, Schallert T, Bittner GD (2010) Polyethylene glycol rapidly restores axonal integrity and improves the rate of motor behavior recovery after sciatic nerve crush injury. J Neurophysiol 104:695–703. 10.1152/jn.01051.2009 [DOI] [PubMed] [Google Scholar]
- Butler SJ, Dodd J (2003) A role for BMP heterodimers in roof plate-mediated repulsion of commissural axons. Neuron 38:389–401. 10.1242/dev.02762 [DOI] [PubMed] [Google Scholar]
- Butler SJ, Tear G (2007) Getting axons onto the right path: the role of transcription factors in axon guidance. Development 134:439–448. 10.1016/S0896-6273(03)00254-X [DOI] [PubMed] [Google Scholar]
- Campbell WW. (2008) Evaluation and management of peripheral nerve injury. Clin Neurophysiol 119:1951–1965. 10.1016/j.clinph.2008.03.018 [DOI] [PubMed] [Google Scholar]
- Carter RJ, Morton J, Dunnett SB (2001) Motor coordination and balance in rodents. Curr Protoc Neurosci Chapter 8:Unit 8.12. 10.1002/0471142301.ns0812s15 [DOI] [PubMed] [Google Scholar]
- Collinson JM, Lindström NO, Neves C, Wallace K, Meharg C, Charles RH, Ross ZK, Fraser AM, Mbogo I, Oras K, Nakamoto M, Barker S, Duce S, Miedzybrodzka Z, Vargesson N (2018) The developmental and genetic basis of 'clubfoot' in the peroneal muscular atrophy mutant mouse. Development 145:dev160093. 10.1242/dev.160093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd J, Morton SB, Karagogeos D, Yamamoto M, Jessell TM (1988) Spatial regulation of axonal glycoprotein expression on subsets of embryonic spinal neurons. Neuron 1:105–116. 10.1016/0896-6273(88)90194-8 [DOI] [PubMed] [Google Scholar]
- Endo M, Ohashi K, Sasaki Y, Goshima Y, Niwa R, Uemura T, Mizuno K (2003) Control of growth cone motility and morphology by LIM kinase and slingshot via phosphorylation and dephosphorylation of cofilin. J Neurosci 23:2527–2537. 10.1523/JNEUROSCI.23-07-02527.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans GR. (2001) Peripheral nerve injury: a review and approach to tissue engineered constructs. Anat Rec 263:396–404. 10.1002/ar.1120 [DOI] [PubMed] [Google Scholar]
- Fei T, Xia K, Li Z, Zhou B, Zhu S, Chen H, Zhang J, Chen Z, Xiao H, Han JD, Chen YG (2010) Genome-wide mapping of SMAD target genes reveals the role of BMP signaling in embryonic stem cell fate determination. Genome Res 20:36–44. 10.1101/gr.092114.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier AE, Strittmatter SM (2001) Repulsive factors and axon regeneration in the CNS. Curr Opin Neurobiol 11:89–94. 10.1016/S0959-4388(00)00178-1 [DOI] [PubMed] [Google Scholar]
- Friese A, Kaltschmidt JA, Ladle DR, Sigrist M, Jessell TM, Arber S (2009) Gamma and alpha motor neurons distinguished by expression of transcription factor Err3. Proc Natl Acad Sci U S A 106:13588–13593. 10.1073/pnas.0906809106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage GJ, Kipke DR, Shain W (2012) Whole animal perfusion fixation for rodents. J Vis Exp 65:3564. 10.3791/3564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudet AD, Popovich PG, Ramer MS (2011) Wallerian degeneration: gaining perspective on inflammatory events after peripheral nerve injury. J Neuroinflammation 8:110. 10.1186/1742-2094-8-110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehler S, Shaw AE, Sarmiere PD, Bamburg JR, Letourneau PC (2004) Brain-derived neurotrophic factor regulation of retinal growth cone filopodial dynamics is mediated through actin depolymerizing factor/cofilin. J Neurosci 24:10741–10749. 10.1523/JNEUROSCI.2836-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon T, Tyreman N, Raji MA (2011) The basis for diminished functional recovery after delayed peripheral nerve repair. J Neurosci 31:5325–5334. 10.1523/JNEUROSCI.6156-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadlock TA, Heaton J, Cheney M, Mackinnon SE (2005) Functional recovery after facial and sciatic nerve crush injury in the rat. Arch Facial Plast Surg 7:17–20. 10.1001/archfaci.7.1.17 [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J (1988) A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 32:77–88. 10.1016/0304-3959(88)90026-7 [DOI] [PubMed] [Google Scholar]
- Hazen VM, Phan KD, Hudiburgh S, Butler SJ (2011) Inhibitory Smads differentially regulate cell fate specification and axon dynamics in the dorsal spinal cord. Dev Biol 356:566–575. 10.1016/j.ydbio.2011.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang TY, DerMardirossian C, Bokoch GM (2006) Cofilin phosphatases and regulation of actin dynamics. Curr Opin Cell Biol 18:26–31. 10.1016/j.ceb.2005.11.005 [DOI] [PubMed] [Google Scholar]
- Kang JR, Zamorano DP, Gupta R (2011) Limb salvage with major nerve injury: current management and future directions. J Am Acad Orthop Surg 19 [Suppl 1]:S28–S34. [DOI] [PubMed] [Google Scholar]
- Kuffler DP. (2014) An assessment of current techniques for inducing axon regeneration and neurological recovery following peripheral nerve trauma. Prog Neurobiol 116:1–12. 10.1016/j.pneurobio.2013.12.004 [DOI] [PubMed] [Google Scholar]
- Li GL, Farooque M, Holtz A, Olsson Y (1996) Increased expression of growth-associated protein 43 immunoreactivity in axons following compression trauma to rat spinal cord. Acta Neuropathol 92:19–26. 10.1007/s004010050484 [DOI] [PubMed] [Google Scholar]
- Lin DM, Fetter RD, Kopczynski C, Grenningloh G, Goodman CS (1994) Genetic analysis of fasciclin II in Drosophila: defasciculation, refasciculation, and altered fasciculation. Neuron 13:1055–1069. 10.1016/0896-6273(94)90045-0 [DOI] [PubMed] [Google Scholar]
- Lundborg G. (2000) A 25-year perspective of peripheral nerve surgery: evolving neuroscientific concepts and clinical significance. J Hand Surg 25:391–414. 10.1053/jhsu.2000.4165 [DOI] [PubMed] [Google Scholar]
- Ma CH, Brenner GJ, Omura T, Samad OA, Costigan M, Inquimbert P, Niederkofler V, Salie R, Sun CC, Lin HY, Arber S, Coppola G, Woolf CJ, Samad TA (2011a) The BMP coreceptor RGMb promotes while the endogenous BMP antagonist noggin reduces neurite outgrowth and peripheral nerve regeneration by modulating BMP signaling. J Neurosci 31:18391–18400. 10.1523/JNEUROSCI.4550-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma CH, Omura T, Cobos EJ, Latrémolière A, Ghasemlou N, Brenner GJ, van Veen E, Barrett L, Sawada T, Gao F, Coppola G, Gertler F, Costigan M, Geschwind D, Woolf CJ (2011b) Accelerating axonal growth promotes motor recovery after peripheral nerve injury in mice. J Clin Invest 121:4332–4347. 10.1172/JCI58675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsick BM, Flynn KC, Santiago-Medina M, Bamburg JR, Letourneau PC (2010) Activation of ADF/cofilin mediates attractive growth cone turning toward nerve growth factor and netrin-1. Dev Neurobiol 70:565–588. 10.1002/dneu.20800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meberg PJ, Bamburg JR (2000) Increase in neurite outgrowth mediated by overexpression of actin depolymerizing factor. J Neurosci 20:2459–2469. 10.1523/JNEUROSCI.20-07-02459.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Y, Zhang Y, Tregoubov V, Janus C, Cruz L, Jackson M, Lu WY, MacDonald JF, Wang JY, Falls DL, Jia Z (2002) Abnormal spine morphology and enhanced LTP in LIMK-1 knockout mice. Neuron 35:121–133. 10.1016/S0896-6273(02)00758-4 [DOI] [PubMed] [Google Scholar]
- Mizuno K. (2013) Signaling mechanisms and functional roles of cofilin phosphorylation and dephosphorylation. Cell Signal 25:457–469. 10.1016/j.cellsig.2012.11.001 [DOI] [PubMed] [Google Scholar]
- Mullen RJ, Buck CR, Smith AM (1992) NeuN, a neuronal specific nuclear protein in vertebrates. Development 116:201–211. [DOI] [PubMed] [Google Scholar]
- Nishita M, Tomizawa C, Yamamoto M, Horita Y, Ohashi K, Mizuno K (2005) Spatial and temporal regulation of cofilin activity by LIM kinase and slingshot is critical for directional cell migration. J Cell Biol 171:349–359. 10.1083/jcb.200504029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perron JC, Dodd J (2011) Inductive specification and axonal orientation of spinal neurons mediated by divergent bone morphogenetic protein signaling pathways. Neural Dev 6:36. 10.1186/1749-8104-6-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan KD, Hazen VM, Frendo M, Jia Z, Butler SJ (2010) The bone morphogenetic protein roof plate chemorepellent regulates the rate of commissural axonal growth. J Neurosci 30:15430–15440. 10.1523/JNEUROSCI.4117-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper M, Anderson R, Dwivedy A, Weinl C, van Horck F, Leung KM, Cogill E, Holt C (2006) Signaling mechanisms underlying Slit2-induced collapse of Xenopus retinal growth cones. Neuron 49:215–228. 10.1016/j.neuron.2005.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richner M, Jager SB, Siupka P, Vaegter CB (2017) Hydraulic extrusion of the spinal cord and isolation of dorsal root ganglia in rodents. J Vis Exp 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousso DL, Gaber ZB, Wellik D, Morrisey EE, Novitch BG (2008) Coordinated actions of the forkhead protein Foxp1 and Hox proteins in the columnar organization of spinal motor neurons. Neuron 59:226–240. 10.1016/j.neuron.2008.06.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarmiere PD, Bamburg JR (2004) Regulation of the neuronal actin cytoskeleton by ADF/cofilin. J Neurobiol 58:103–117. 10.1002/neu.10267 [DOI] [PubMed] [Google Scholar]
- Scott RW, Olson MF (2007) LIM kinases: function, regulation and association with human disease. J Mol Med 85:555–568. 10.1007/s00109-007-0165-6 [DOI] [PubMed] [Google Scholar]
- Serafini T, Colamarino SA, Leonardo ED, Wang H, Beddington R, Skarnes WC, Tessier-Lavigne M (1996) Netrin-1 is required for commissural axon guidance in the developing vertebrate nervous system. Cell 87:1001–1014. 10.1016/S0092-8674(00)81795-X [DOI] [PubMed] [Google Scholar]
- Shin JE, Geisler S, DiAntonio A (2014) Dynamic regulation of SCG10 in regenerating axons after injury. Exp Neurol 252:1–11. 10.1016/j.expneurol.2013.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaeth CS, Boydston EA, Figard LR, Zuzek A, Bittner GD (2010) A model for sealing plasmalemmal damage in neurons and other eukaryotic cells. J Neurosci 30:15790–15800. 10.1523/JNEUROSCI.4155-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosney KW, Landmesser LT (1985) Development of the major pathways for neurite outgrowth in the chick hindlimb. Dev Biol 109:193–214. 10.1016/0012-1606(85)90360-4 [DOI] [PubMed] [Google Scholar]
- Trachtenberg JT, Chen BE, Knott GW, Feng G, Sanes JR, Welker E, Svoboda K (2002) Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature 420:788–794. 10.1038/nature01273 [DOI] [PubMed] [Google Scholar]
- Tursun B, Schlüter A, Peters MA, Viehweger B, Ostendorff HP, Soosairajah J, Drung A, Bossenz M, Johnsen SA, Schweizer M, Bernard O, Bach I (2005) The ubiquitin ligase Rnf6 regulates local LIM kinase 1 levels in axonal growth cones. Genes Dev 19:2307–2319. 10.1101/gad.1340605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Niekerk EA, Tuszynski MH, Lu P, Dulin JN (2016) Molecular and cellular mechanisms of axonal regeneration after spinal cord injury. Mol Cell Proteomics 15:394–408. 10.1074/mcp.R115.053751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson C, Paxinos G, Kayalioglu G, Christopher and Dana Reeve Foundation, ScienceDirect (2009) The spinal cord: a Christopher and Dana Reeve Foundation text and atlas (online service), Ed 1, pp xv, 387 Amsterdam: Elsevier/Academic. [Google Scholar]
- Wen Z, Han L, Bamburg JR, Shim S, Ming GL, Zheng JQ (2007) BMP gradients steer nerve growth cones by a balancing act of LIM kinase and slingshot phosphatase on ADF/cofilin. J Cell Biol 178:107–119. 10.1083/jcb.200703055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichterle H, Lieberam I, Porter JA, Jessell TM (2002) Directed differentiation of embryonic stem cells into motor neurons. Cell 110:385–397. 10.1016/S0092-8674(02)00835-8 [DOI] [PubMed] [Google Scholar]
- Yamauchi K, Varadarajan SG, Li JE, Butler SJ (2013) Type Ib BMP receptors mediate the rate of commissural axon extension through inhibition of cofilin activity. Development 140:333–342. 10.1242/dev.089524 [DOI] [PMC free article] [PubMed] [Google Scholar]