

Group B Streptococcus (GBS) is an opportunistic bacterial pathogen that contributes to miscarriage, preterm birth, and serious neonatal infections. Studies have indicated that some multilocus sequence types (STs) of GBS are more likely to cause severe disease than others. We hypothesized that the ability of GBS to elicit varying host responses in maternal decidual tissue during pregnancy is an important factor regulating infection and disease severity.
KEYWORDS: group B Streptococcus, Streptococcus agalactiae, cell death, cytokines, host response, inflammatory responses, macrophage
ABSTRACT
Group B Streptococcus (GBS) is an opportunistic bacterial pathogen that contributes to miscarriage, preterm birth, and serious neonatal infections. Studies have indicated that some multilocus sequence types (STs) of GBS are more likely to cause severe disease than others. We hypothesized that the ability of GBS to elicit varying host responses in maternal decidual tissue during pregnancy is an important factor regulating infection and disease severity. To address this hypothesis, we utilized an antibody microarray to compare changes in production and activation of host signaling proteins in decidualized telomerase-immortalized human endometrial stromal cells (dT-HESCs) following infection with GBS strains from septic neonates or colonized mothers. GBS infection increased levels of total and phosphorylated mitogen-activated protein kinase (MAPK) family members such as p38 and JNK and induced nuclear factor kappa B (NF-κB) pathway activation. Infection also altered the regulation of additional proteins that mediate cell death and inflammation in a strain-specific manner, which could be due to the observed variation in attachment to and invasion of the decidual stromal cells and ability to lyse red blood cells. Further analyses confirmed array results and revealed that p38 promotes programmed necrosis in dT-HESCs. Together, the observed signaling changes may contribute to deregulation of critical developmental signaling cascades and inflammatory responses following infection, both of which could trigger GBS-associated pregnancy complications.
INTRODUCTION
Group B Streptococcus (GBS), also known as Streptococcus agalactiae, frequently colonizes the gastrointestinal and genitourinary tracts of up to 35% of pregnant women worldwide (1). Although GBS colonization is often asymptomatic, it can cause serious complications during pregnancy, including preterm birth and stillbirth, and result in neonatal sepsis and meningitis (2). As maternal GBS colonization is the primary risk factor for prenatal and neonatal disease, women are routinely screened for GBS during pregnancy. In colonized mothers, GBS can induce pregnancy complications by ascending through the vaginal tract and cervix, crossing the extraplacental membranes surrounding the fetus, and initiating a fetal infection in utero (3, 4). It is also possible for infants to become infected as they pass through the birth canal during delivery by aspirating vaginal fluid containing GBS (3, 4). Newborns exposed to GBS may develop early-onset disease (EOD), which occurs during the first week after birth and typically presents as pneumonia and sepsis, or late-onset disease (LOD), which presents as meningitis and sepsis and occurs from 1 week to 3 months of age (2). To lower the probability of developing EOD, it is recommended that colonized mothers receive intrapartum antibiotic prophylaxis (IAP) during delivery; unfortunately, this intervention is not effective at reducing the risk of GBS-related pregnancy complications or LOD in neonates (2).
Dysregulated inflammatory signaling has been shown to contribute to adverse pregnancy outcomes such as extraplacental membrane weakening, which can lead to miscarriage, preterm birth, or neonatal sepsis (3, 5–7). To develop novel prevention and diagnostic strategies capable of combating GBS infection during pregnancy, a more complete understanding of how GBS modulates host signaling at the maternal-fetal interface is of critical importance. When GBS ascends the vaginal tract, passes through the cervix, and enters the uterus, it encounters the extraplacental membranes, which protect and surround the developing fetus in utero (7). The decidua, or outermost layer of the extraplacental membranes, is the first layer of cells with which GBS must interact to gain access to the fetus (7). This tissue layer is composed primarily of decidual stromal cells (DSCs), though it is also interspersed with macrophages (7).
Results from our group and others have shown that GBS strains belonging to specific multilocus sequence types (STs), such as ST-17 and ST-19, are more likely to cause neonatal infections (8–10) and persist in women following childbirth and antibiotic prophylaxis (11). ST-17 strains specifically have been linked to late-onset disease and meningitis (10) and were found to possess unique virulence genes and virulence characteristics (12–14). Select ST-17 strains evaluated in our prior studies have been shown to have an enhanced ability to attach to and invade DSCs, express key virulence genes (15), and persist in macrophages (16, 17). Strains representing other genotypes, such as ST-12, for instance, were more commonly found in pregnant women than in infected neonates and were more likely to be lost after antibiotic prophylaxis during childbirth (11). A select ST-12 strain also survived poorly inside macrophages compared to a ST-17 strain (16). Moreover, we have recently evaluated a larger panel of 15 GBS strains representing four STs (e.g., STs 17, 19, 12, and 1) and three capsule (CPS) types (e.g., CPS III, II, and V) and have found that strains of the same ST and/or CPS type induce similar cytokine responses from macrophages in vitro (18).
Because of this phenotypic and genotypic variation in clinical GBS strains as well as the epidemiological relevance of specific strain types, we sought to investigate how DSC responses vary following exposure to GBS of different genotypes. We hypothesized that distinct GBS strains would vary in their ability to elicit programmed cell death and induce inflammatory signaling cascades in infected DSCs. While some death and inflammatory responses were induced by all GBS strains, others were unique to specific strains or genotypes. These findings shed light on why certain STs may induce more severe pathology and provide new insights for the development of novel therapeutic intervention strategies for the prevention and treatment of GBS disease.
RESULTS
Antibody-based protein array reveals general and strain-specific responses to GBS infection by DSCs.
The antibody array identified many proteins with significantly altered total or phosphorylated levels in the presence of at least one of the four GBS strains examined. A subset of these responses varied at 2 h (see Table S1 in the supplemental material) and 3 h (see Table S2) following infection of decidualized human endometrial stromal cells (dT-HESCs). Pooling the response targets for all four strains allowed us to elucidate general DSC responses to GBS infection. When the targets were grouped according to function, signaling pathways for the regulation of inflammation and the immune response, cell survival and metabolism, rearrangement of the cytoskeleton and cell-cell contacts, hormone signaling, and angiogenesis were most affected (Table 1). Due to the interconnectedness of these pathways, many proteins fell into multiple categories; the most dramatic responses were observed after 3 h. Notably, a set of 34 proteins had 10-fold increases in phosphorylated or total protein levels relative to those with mock infection for at least one GBS strain (see Table S3). Arrestin B, G-protein-coupled receptor kinase 2 (GRK-2), myosin light chain (MLC), platelet-derived growth factor receptor alpha (PDGFRa), and protein kinase cAMP-activated catalytic subunit alpha (PRKACA) were increased by >15-fold, while signal transducer and activator of transcription 2 (STAT2) had a 142- to 318-fold increase.
TABLE 1.
Proteins with significantly altered total or phosphorylated levels following infection with at least one of the four GBS strains at 2 or 3 ha

Color coding corresponds to the number of array hits for each pathway at the two time points tested.
GBS induces MAPK signaling in DSCs.
Because pathways affecting stress responses, inflammatory signaling, and cell death regulation were impacted by GBS infection, key members involved in these processes were selected for follow-up analysis. Members of the mitogen-activated protein kinase (MAPK) family, which aid in the regulation of cellular responses and play roles in death and inflammatory signaling (19), had the greatest number of significant changes (data not shown). Indeed, significant changes in protein activity and level were observed in response to GBS infection at both 2 and 3 h postinfection, particularly for proteins involved in the stress-activated p38 and Jun N-terminal kinase (JNK) MAPK pathways (data not shown). For example, significantly increased phosphorylated (active) p38 levels were observed following infection with all four GBS strains after 3 h (Fig. 1A) even though no difference was observed for total p38 (Fig. 1B). An increase in phosphorylated (phospho)-JNK was also observed at 3 h; however, the increase was only significant for the two non-ST-17 strains, GB590 and GB653 (Fig. 1C). No difference in total JNK levels was observed (Fig. 1D), and the variation in protein levels was confirmed using Western blotting (Fig. 1E).
FIG 1.

GBS induces strain-specific activation of stress-responsive MAPKs. T-HESCs were infected with GBS for 2 or 3 h (MOI = 10), and host cell lysates were collected for Western blotting. Lysates were assessed for phosphorylated (active) or total protein levels of p38 (A and B) and JNK (C and D), and densitometry was used to compare differences between infection conditions. Densitometry values represent pooled results from at least three independent biological replicates, and error bars represent standard deviations of the means. Significance was determined by ANOVA (p-p38, P < 0.0001; p38, P = 0.8053; p-JNK, P < 0.0001; JNK, P = 0.4604; p-ERK1/2, P = 0.9963; ERK1/2, P = 0.6838) with post hoc Dunnett’s testing to compare each infection condition to the mock infection. *, P < 0.05; **, P < 0.01; ****, P < 0.0001. (E) Representative Western blots from one biological replicate with its corresponding loading control (GAPDH) are shown. Equal amounts of the same protein lysate preparations were loaded onto the gels for each protein.
A significant increase in total but not phosphorylated levels of Fos was observed for the ST-17 GB411 invasive strain as well as the ST-19 GB590 colonizing strain (Fig. 2A and B), both of which possess the capsule type (CPS) III. In contrast, the level of phospho-Jun but not total Jun was increased at 3 h for only the two ST-17 CPS III strains, GB112 and GB411 (Fig. 2C and D). Phosphorylated crystallin αB was also increased by 3 h (Fig. 2E) for all four GBS strains, though the highest levels were seen in GB411. Interestingly, a decline in total levels of crystallin αB was observed for all infection conditions but was only significant for the ST-12 CPS II strain, GB653, at 3 h postinfection (Fig. 2F); both GB411 and GB590 were not quite significant (P = 0.06). As this protein is activated in response to stress and normally functions to inhibit programmed cell death, enhanced levels of phospho-crystallin αB along with declining levels of total protein suggest that the stress of GBS infection induces a shift that can lead to programmed cell death. All protein levels and differences were confirmed by Western blotting (Fig. 2G).
FIG 2.

GBS induces strain-specific activation of downstream MAPK transcription factors and effectors. Phosphorylated (active) and total protein levels of Fos (A and B), Jun (C and D), and crystallin αB (CRYaB; E and F) are shown and differences were calculated based on the densitometry values. These values represent pooled results from at least three independent biological replicates, and error bars represent standard deviations of the means. Significance was determined by ANOVA (p-Fos, P = 0.1976; Fos, P = 0.0384; p-Jun, P = 0.0003; Jun, P < 0.0001; p-CRYaB, P < 0.0001; CRYaB, P = 0.0114) with post hoc Dunnett’s testing to compare each infection condition to the mock infection. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. (G) Representative Western blots from one biological replicate of each protein target with its corresponding loading control (GAPDH) are shown. Equal amounts of the same protein lysate preparations were loaded onto the gels for each protein target shown.
GBS induces nuclear factor kappa B signaling in DSCs downstream of JNK MAPK activation.
Members of the nuclear factor kappa B (NF-κB) proinflammatory pathway, which is frequently activated downstream of MAPK signaling, were also activated following GBS infection (data not shown). To confirm activation of this pathway, immunofluorescence microscopy was used to examine nuclear localization of NF-κB in response to mock or GBS infection with each of the four strains at 2 through 5 h postinfection (data not shown). A significant increase in NF-κB activation was observed for all four strains by 5 h postinfection (Fig. 3A). Intriguingly, activation occurred more rapidly and was more widespread in response to the two non-ST-17 strains than in response to the two ST-17 strains. These results correlated with decreasing levels of the NF-κB inhibitory protein, IκB, which was significantly reduced for all four strains by 3 h postinfection (Fig. 3B). Similarly, depletion of IκB was most pronounced in response to the two non-ST-17 strains. Protein levels were confirmed by Western blotting (Fig. 3C).
FIG 3.
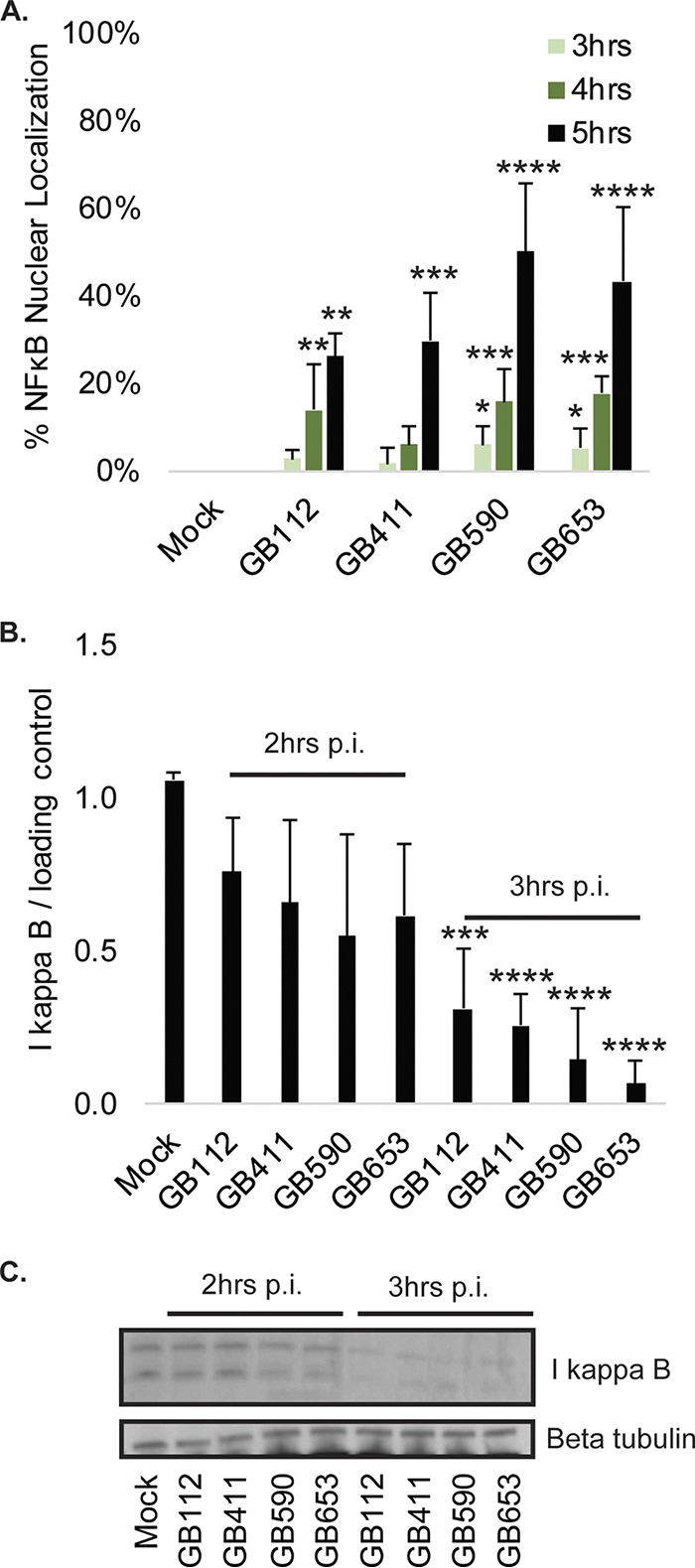
GBS induces NF-κB pathway activation. (A) T-HESCs were infected with GBS for 3, 4, or 5 h at an MOI of 10 prior to fixation, nuclear staining (DAPI), and detection of NF-κB p65 (Alexa Fluor 488) by immunofluorescence microscopy. Percent NF-κB nuclear localization compares the number of cells with positive nuclear localization (Alexa Fluor 488) to the total cell number in a given field (DAPI) using ImageJ. Results from multiple fields from three independent biological replicates were pooled and graphed for each time point, with error bars representing standard deviations. Significance was determined by ANOVA (3 h, P = 0.025; 4 h, P < 0.0001; 5 h, P < 0.0001) with post hoc Dunnett’s testing to compare each infection condition to the mock infection. *, P < 0.05; **, P < 0.01; ***, P< 0.001; ****, P < 0.0001. (B and C) Total levels of IκB were assessed 2 and 3 h postinfection (p.i.) by Western blotting. IκB was normalized to a beta-tubulin loading control, and densitometry values represent pooled results from four independent biological replicates. Significance was determined by ANOVA (P < 0.0001) with post hoc Dunnett’s testing to compare each infection condition to the mock infection. ***, P < 0.001; ****, P < 0.0001.
To determine whether the increase in NF-κB activity was occurring downstream of p38 and JNK, we treated DSCs with the p38 inhibitor SB203580 as well as the JNK inhibitor SP600125 (data not shown) for comparison to vehicle controls prior to mock infection or infection with each strain. Although inhibition of p38 did not significantly decrease NF-κB nuclear localization (Fig. 4A), JNK inhibition contributed to a significant reduction in activation of NF-κB for each strain at 5 h postinfection (Fig. 4B). Together, these data suggest that JNK activation contributes to NF-κB pathway activation in the presence of GBS and that this occurs independently of p38 activity.
FIG 4.
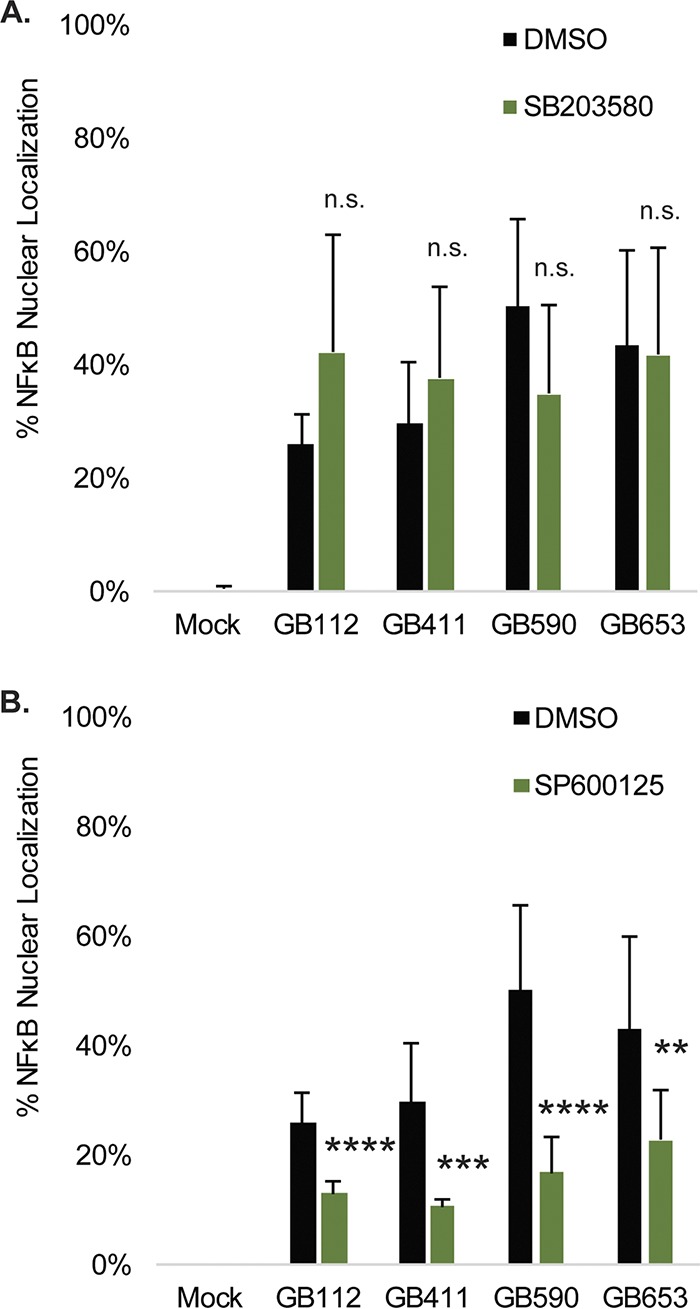
Effects of p38 and JNK inhibition on GBS-mediated NF-κB nuclear localization. T-HESCs were infected with GBS for 5 h at an MOI of 10 prior to fixation, nuclear staining (DAPI), and detection of NF-κB p65 (Alexa Fluor 488) by immunofluorescence microscopy. Prior to infection, cells were treated for 1.25 h with 10 μM SB203580 (p38 inhibitor) (A), 25 μM SP600125 (JNK inhibitor) (B) or a vehicle control (DMSO). Percent NF-κB nuclear localization was determined by comparing the number of cells with positive nuclear localization (Alexa Fluor 488) to the total cell number in a given field (DAPI) using ImageJ. Results from at least two fields (>300 individual cells per condition per time point) from each of three independent biological replicates were pooled and graphed for each time point, with error bars representing standard deviations. Significance was determined by t tests to compare differences between the inhibitor treatment for each infection condition with the corresponding vehicle control for the same infection condition. ns, not significant; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
GBS induces programmed cell death in DSCs downstream of p38 MAPK activation.
In addition to regulating the induction of many proinflammatory responses, stress-activated MAPKs often aid in the regulation of programmed cell death (19). The array data indicated changes in the activation and total protein levels of proteins such as p53, Akt, and mammalian Ste20-like kinases 1, 2, and 3 (MST 1, 2, and 3, respectively), which are involved in the regulation of various programmed cell death pathways (data not shown). To address how DSC signaling responses contribute to the induction of programmed cell death, we compared changes in cell death during infection using an ethidium homodimer membrane permeabilization assay. Significantly enhanced cell death was observed by 4 h in response to all four strains and occurred most rapidly in response to the ST-12 GB653 strain (Fig. 5A). Inhibition of p38 resulted in a modest but significant reduction in cell death in response to all four strains (Fig. 5B). In contrast, inhibition of JNK caused a significant increase in cell death in response to the GB112, GB411, and GB653 strains, with a trend toward increased cell death in the GB590 strain (P = 0.06) (Fig. 5C). These data indicate that p38 activation contributes to the induction of programmed cell death in response to GBS infection, while JNK activation has a prosurvival effect, perhaps through its contribution to NF-κB pathway activation.
FIG 5.
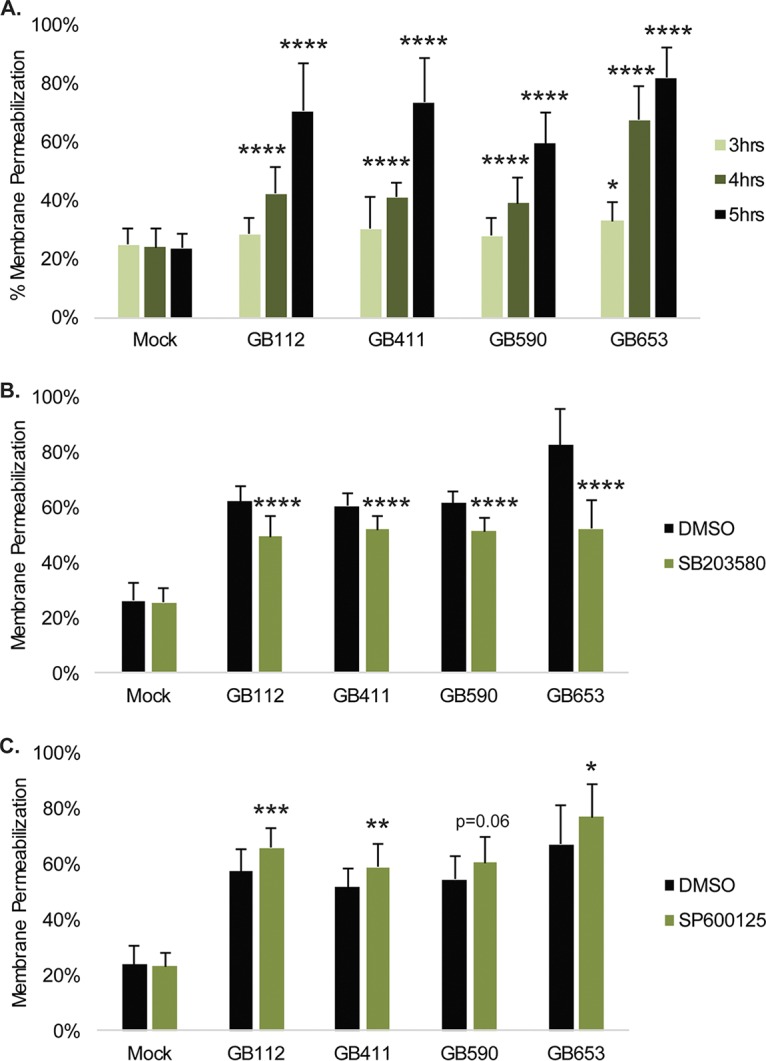
GBS-mediated cytotoxicity in DSCs is influenced by p38 and JNK MAPK signaling. (A) T-HESCs were infected with GBS for 4, 4, or 5 h at an MOI of 10, and cell death was assessed using an ethidium homodimer membrane permeabilization assay. Significance was determined by ANOVA (3 h, P = 0.0449; 4 h, P < 0.0001; 5 h, P < 0.0001) with post hoc Dunnett’s testing to compare each infection condition to the mock infection. *, P < 0.05; ****, P < 0.0001. Effects of p38 inhibition on cell death were assessed by treating cells for 1.25 h prior to infection with 10 μM SB203580 (B), and effects of JNK inhibition were assessed by treating cells for 1.25 h prior to infection with 25 μM SP600125 (C). Cell death for p38 and JNK inhibition compared to corresponding vehicle controls for each infection condition was assessed 4 h postinfection by ethidium homodimer assay, and significance was determined by t tests. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
GBS-mediated cell death in DSCs occurs through RIPK1- and RIPK3-dependent programmed necrosis.
To determine whether cell death was dependent on caspases, cells were treated with a general caspase inhibitor prior to infection with GBS. The caspase inhibitor significantly reduced cell death by 10% to 20% under the GB112, GB411, and GB590 infection conditions (Fig. 6A). To confirm that GBS-induced programmed cell death was dependent on caspases, we assessed caspase 3/7 activation at 2 through 5 h postinfection (Fig. 6B). Although we saw a slight increase in caspase 3/7 activation at 3 h postinfection, we observed a dramatic loss of caspase 3/7 activity (1.4- to 2.9-fold) by 4 h postinfection. Activity was further decreased (1.7- to 3.2-fold) at 5 h. In the presence of staurosporine, however, GBS reduced caspase 3/7 activation to levels that were 4-fold lower than those observed under mock infection conditions containing staurosporine (Fig. 6C). These results suggest that GBS infection may be actively degrading or inhibiting the activation of caspases.
FIG 6.
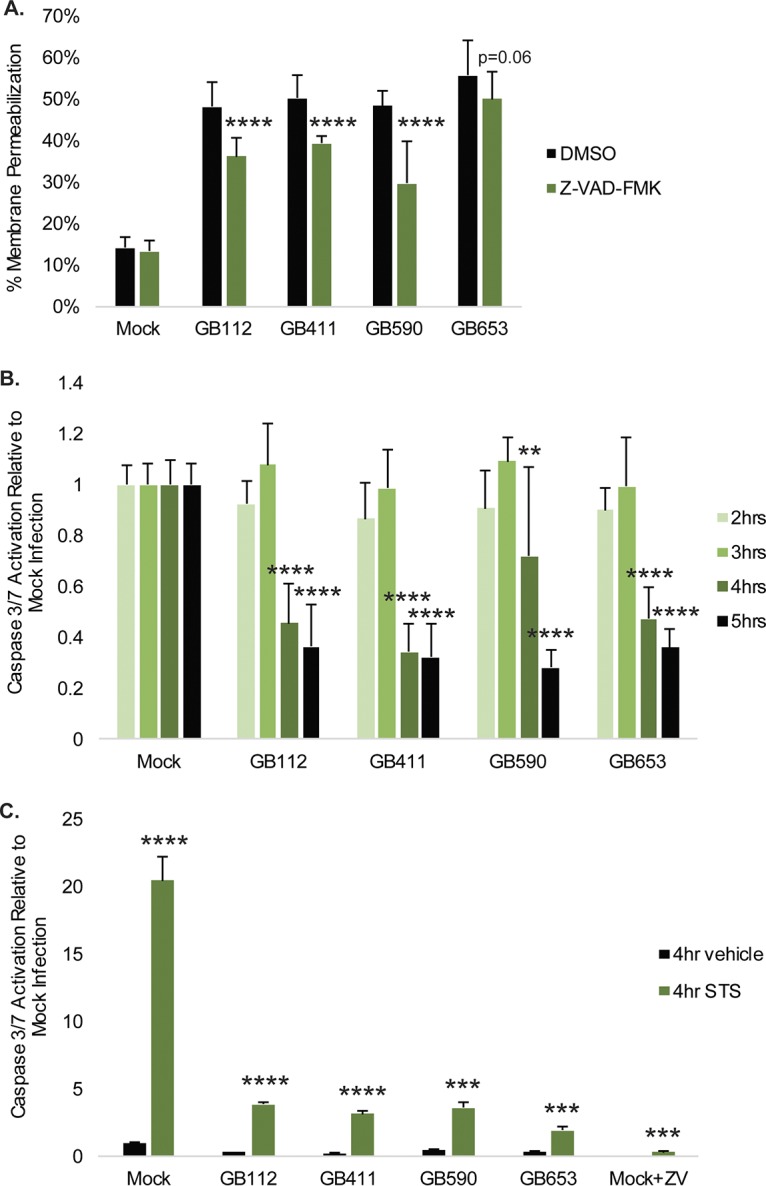
Classic apoptosis is not the primary mechanism by which GBS mediates cytotoxicity in DSCs. (A) T-HESCs were treated with 50 μM Z-VAD-fmk for 1.25 h prior be being infected with GBS for 4 h at an MOI of 10. Cell death was assessed using an ethidium homodimer membrane permeabilization assay. Results from at least three independent biological replicates were pooled, and significance was determined by t tests to compare each treatment condition to the corresponding vehicle control for the same strain. ****, P < 0.0001. (B) Activity levels of the executioner caspases 3 and 7 were measured 2 through 5 h postinfection at an MOI of10. Results from at least three independent biological replicates were pooled, and significance was determined by ANOVA (2 h, P = 0.1615; 3 h, P = 0.3582; 4 h, P < 0.0001; 5 h, P < 0.0001) with post hoc Dunnett’s testing to compare each infection condition to the mock infection. **, P < 0.01; ****, P < 0.0001. (C) Caspase 3/7 activation 4 h postinfection was assessed in the presence of 2 μM staurosporine (STS) or DMSO vehicle control. Results from at least three independent biological replicates were pooled, and significance was determined by t tests to compare each STS treatment condition to the corresponding vehicle control for the same strain. ***, P < 0.001; ****, P < 0.0001.
As GBS-infected DSCs did not appear to be undergoing caspase-dependent apoptosis, we explored whether they might be undergoing programmed necrosis, or necroptosis. Necroptosis is one of the best-characterized forms of programmed necrosis and relies on the activation of receptor-interacting protein kinase 1 (RIPK1) and RIPK3 to form a complex called the necrosome (20–22). Necrosome formation leads to activation of the protein mixed lineage kinase domain-like (MLKL) and ultimately to cell rupture and release of inflammatory factors (20–22). To determine whether GBS was inducing necroptosis in DSCs, cells were treated with RIPK1 and RIPK3 inhibitors prior to infection (Fig. 7A). Both inhibitors significantly reduced programmed cell death by 10% to 15% in response to all four strains of GBS. Significantly increased levels of phospho-MLKL (4- to 8-fold) were also observed by 3 h postinfection in response to all four strains, though no difference was noted for total MLKL levels (Fig. 7B). Protein levels were confirmed by Western blotting (Fig. 7C).
FIG 7.
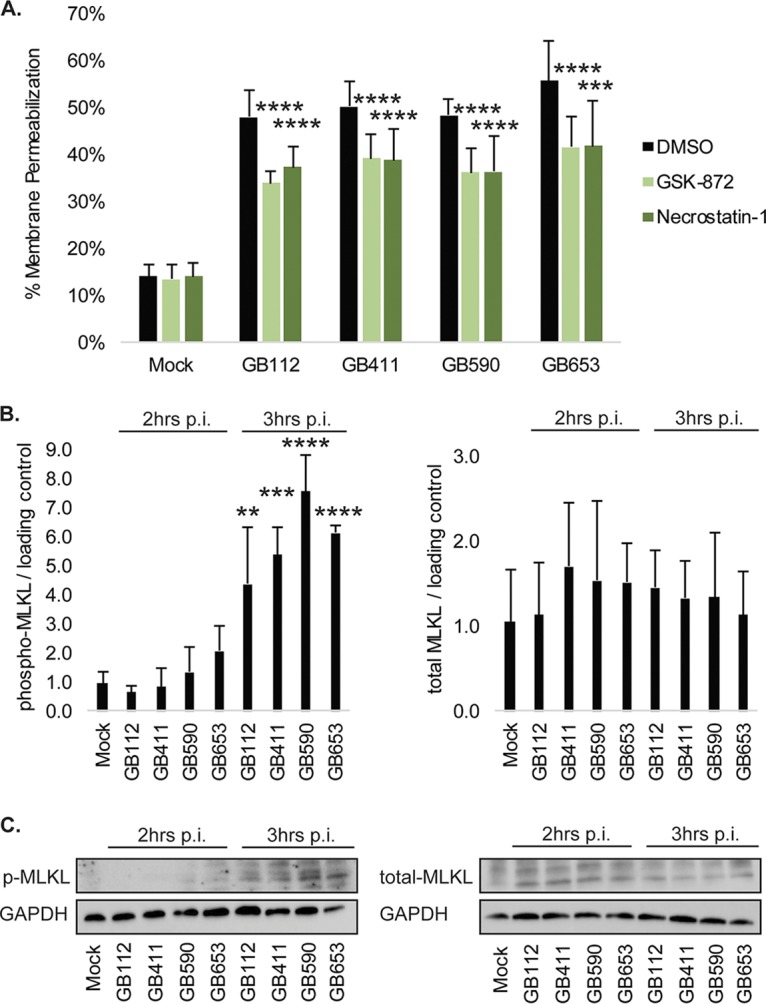
GBS induces necroptosis in infected DSCs. (A) T-HESCs were treated with 15 μM GSK-872 or 50 μM necrostatin-1 for 1.25 h prior be being infected with GBS for 4 h at an MOI of 10. Cell death was assessed using an ethidium homodimer membrane permeabilization assay. Results from at least three independent biological replicates were pooled, and significance was determined by t tests to compare each treatment condition to the corresponding vehicle control for the same strain. ***, P < 0.001; ****, P < 0.0001. Levels of phosphorylated (active) and total MLKL were assessed by Western Blotting in T-HESC lysates 2 and 3 h post-GBS infection at an MOI of 10. (B) Phosphorylated and total levels of MLKL were normalized to GAPDH, and densitometry values represent pooled results from at least three independent biological replicates. Significance was determined by ANOVA (p-MLKL, P < 0.0001; MLKL, P = 0.871) with post hoc Dunnett’s testing to compare each infection condition to the mock infection. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001. (C) Representative Western blots from one biological replicate of each protein target with its corresponding loading control (GAPDH) are shown. Equal amounts of the same protein lysate preparations were loaded onto the gels for each protein target shown.
Morphologically, the GBS-infected cells had a swollen appearance consistent with the onset of the membrane permeabilization that occurs during necrotic cell death (data not shown). Interestingly, by 4 to 5 h postinfection, infected cells underwent a noticeable reduction in nuclear size with an accompanying shriveled appearance of the cell body. This reduction was most noticeable following exposure to the GB653 strain and in the presence of the JNK inhibitor (data not shown), which could be due to continued breakdown of the ruptured membranes as the infection progressed.
GBS induces general and strain-specific changes in decidual cytokine signaling.
Because GBS infection increased both proinflammatory and prodeath signaling pathways, we also investigated how GBS infection impacts cytokine production in DSCs via cytokine array (data not shown). Despite significant activation of p38, JNK, and NF-κB, only minimal production of most proinflammatory cytokines was observed in DSCs in response to all four GBS strains (Fig. 8A). Cytokine array analyses indicated modest GBS-induced cytokine elevation for leukocyte activators such as granulocyte colony-stimulating factor (G-CSF) and interleukin 4 (IL-4) as well as for multifunctional proinflammatory cytokines such as I-309 and IL-6 and the angiogenesis regulator angiogenin. In contrast, many leukocyte activators and chemokines, such as epithelial-neutrophil activating peptide 78 (ENA-78), IL-8, and granulocyte-macrophage colony-stimulating factor (GM-CSF), were markedly downregulated during all GBS infections compared to those with mock infection. The same was true for several multifunctional proinflammatory cytokines, including IL-1β and tumor necrosis factor alpha (TNF-α) as well as regulators of cell growth and embryonic and placental development (e.g., fibroblast growth factor 4 [FGF-4], Flt-3 ligand, placental growth factor [PLGF], and transforming growth factor β1 [TGF-β1]).
FIG 8.

DSC cytokine responses to specific GBS strains or genotypes. Values are expressed as a ratio of the densitometry value of each infection condition compared to that for mock infection. Values of >1 indicate an increase, and values of <1 indicate a decrease. (A) Responses with similar trends for all four infection conditions are shown if an increase or decrease of >15% from mock infection was observed for at least three of the four strains. (B) Responses that were unique to specific GBS strains or sequence types (STs) are shown. Pink cells indicate increased fold changes and blue cells indicate decreased fold changes, while bolded values are ≥1.5-fold higher or lower than that for the mock infection. Underlined values indicate cases where the mock infection and the infection condition being compared both had values that were similar to or less than the internal negative-control values; such values are likely to be below the range of accurate detection.
It is interesting to note that several strain-specific cytokine responses were also observed (Fig. 8B). The ST-17 GB411 strain isolated from a neonate with sepsis induced more widespread reductions in inflammatory and growth and development-related cytokines than the other three GBS strains isolated from colonized mothers. This invasive strain also induced lower levels of the anti-inflammatory cytokine IL-13.
GBS strains exhibit variation in attachment to and invasion of DSCs and lysis of RBCs.
The ability of each of the strains to attach to and invade DSCs under conditions matching those used in the signaling analyses was evaluated. Though only a small percentage of the total bacterial population attaches to and invades DSCs under these conditions, our results demonstrate that the two ST-17 strains attached and invaded the cells better than the two non-ST-17 strains (Fig. 9A to C). In addition, we evaluated variation in the production of β-hemolysin/cytolysin (βH/C), which has been shown to be cytolytic against multiple host cell types and can cause damage to maternal and fetal tissues during pregnancy (23). Our results indicate that the ST-12 strain (GB653) induced more rapid red blood cell (RBC) lysis than the ST-17 and ST-19 strains as well as control strain GB37 (Fig. 10), which was previously shown to lack βH/C production (24). These findings were consistent with the cytotoxicity trends observed in DSCs.
FIG 9.

GBS strains exhibit various attachment and invasion capabilities in DSCs. T-HESCs were infected with GBS for 2 h at an MOI of 10. The number of bacteria for each condition is expressed relative to the total number of bacteria in each well at the end of the infection period (2 h). (A) The percentages of cell-associated bacteria include both attached and invaded bacteria relative to the final inoculum. (B) The percent invaded values represent bacteria obtained from lysed DSCs 1 h post-antibiotic treatment (to kill extracellular bacteria) and normalized to the final inoculum. (C) The percent attached values represent the percent invaded values subtracted from the percent associated values. Results were pooled from at least three independent biological replicates, each of which was performed in triplicates. Error bars represent standard deviations of the means. Significance was determined by ANOVA (P < 0.0001 for all) followed by post hoc Tukey’s tests to compare the mean of each condition to the means of each of the other conditions. *, P < 0.05; ****, P < 0.0001.
FIG 10.
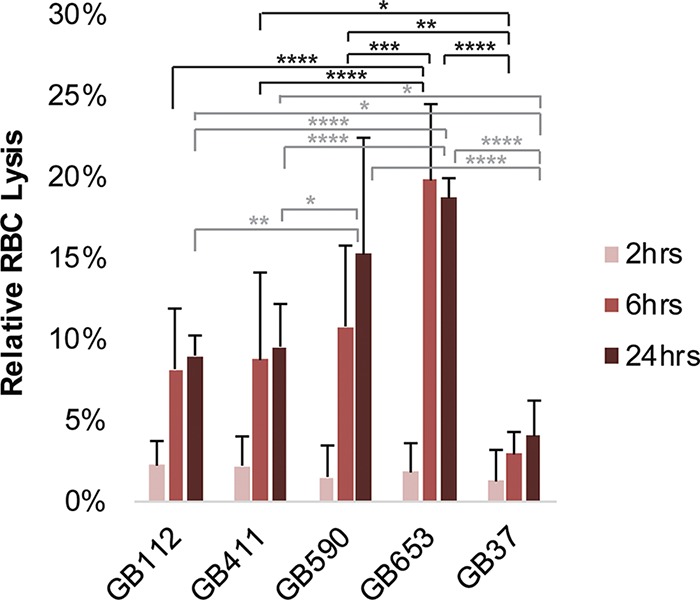
GBS strains exhibit differential hemolytic activity. The upper and lower limits of RBC lysis were determined by using the detergent Triton X-100 as a positive control and PBS as a negative control; percent RBC lysis was determined relative to these values. Three biological replicates were averaged for each condition (each performed in technical triplicates), and error bars represent standard deviations of the means. Significance was determined by ANOVA for each time point (2 h, P = 0.7442; 6 h, P < 0.0001; 24 h, P < 0.0001), and Tukey’s tests were performed post hoc to determine differences in RBC lysis between treatments for each time point. No significant differences were observed at 2 h; significant differences at 6 h are indicated in black, while significant differences at 24 h are indicated in gray. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
DISCUSSION
The goal of this study was to characterize general and strain-specific responses induced in DSCs following infection with four unique GBS clinical isolates. The use of an antibody-based protein array enabled the identification of major pathways involved in the regulation of inflammation and immune responses, cell survival and metabolism, cytoskeletal and cell-cell contact regulation, hormone signaling, and angiogenesis that are modulated in response to GBS infection. We anticipate that GBS-mediated alteration of specific inflammatory and immune responses impacts the ability of infected mothers and neonates to appropriately recognize and respond to GBS infection. Changes in the regulation of cell survival, cytoskeleton rearrangements, and cell-cell contacts are likely to influence GBS colonization and invasion of host cells and negatively impact the integrity of infected tissue, which may also contribute to extraplacental membrane weakening as was discussed in a prior study (25). Additionally, GBS-mediated modulation of cellular metabolism, hormone signaling, and angiogenesis could have severe impacts on fetal development, perhaps contributing to outcomes such as miscarriage or stillbirth.
The MAPK family, particularly, members of the p38 and JNK pathways, had the greatest number of pathway members whose total or phosphorylated levels were altered in the presence of GBS infection. Strain-specific differences were also observed. Our data also suggest that JNK contributes to NF-κB pathway activation, while p38 promotes the induction of programmed cell death in DSCs. The connection between p38 activation and enhanced cytotoxicity was further supported by the increase in phospho-crystallin αB with an accompanying decline in total protein levels. As this protein is activated in response to stress and normally functions to inhibit programmed cell death, the observed changes suggest that the stress of GBS infection induces a signaling shift that ultimately leads to programmed cell death.
Activations of p38, JNK, and NF-κB have been observed in macrophage cell lines in response to GBS infection, where they were associated with cytokine activation and neonatal sepsis. Activation of these key host pathways, however, was not previously reported in DSCs in response to GBS (26–29). It has been observed, however, that increased activation of NF-κB and downstream cytokine signaling in decidual tissue are associated with miscarriage (30) and that other bacterial pathogens could activate these pathways to promote inflammation, contractions, and extraplacental membrane weakening (7, 31–34).Therefore, it is possible that activation of the proinflammatory and pro-cell death pathways identified in this study could have serious impacts on fetal and maternal health during pregnancy.
Because bacterial pathogens have been reported to induce cell death in multiple host cell types through both caspase-dependent and caspase-independent mechanisms (35–42), we investigated whether GBS infection of DSCs induced caspase activation. Although a reduction in cell death was observed in the presence of a pan-caspase inhibitor, significant reductions in the levels of the executioner caspases 3 and 7 were seen in the presence of GBS. These results differ from those described for endothelial cells and macrophages, where GBS induced classic apoptosis following infection (43, 44). Our data also suggest that GBS induces activation of other caspases, particularly caspase 1, which promotes programmed cell death through nonapoptotic mechanisms (45). These results also indicate that GBS has a mechanism to actively degrade proapoptotic caspases or inhibit their activation, as GBS dramatically reduced caspase 3 and 7 activity in the presence of staurosporine. Although caspase inhibition has been observed during infection with other bacterial pathogens (46), this mechanism has yet to be described in response to GBS.
Our results provide convincing evidence that the primary mechanism of programmed cell death in DSCs in response to GBS is necroptosis. Indeed, inhibitors of necrosome components significantly reduced cell death in response to GBS, and consistent with this, significant increases in the activity of MLKL, a key downstream protein of this cell death cascade, were observed. Furthermore, the cells underwent visible morphological changes that were consistent with this form of cell death (47). These results, however, do not rule out the possibility that other nonapoptotic forms of cell death, such as caspase 1-dependent pyroptosis, may also be occurring in the infected cell population. One study, for instance, demonstrated that GBS induces membrane permeabilization and fetal injury through both nucleotide-binding domain and leucine-rich repeat containing protein (NLRP) inflammasome-dependent and -independent mechanisms in a murine model of in utero infection (48). Such a scenario could explain the modest reduction in cell death that we observed in the presence of the general caspase inhibitor Z-VAD-fmk. Although necroptosis was not previously reported in response to GBS infection, it has been observed in other cell types in response to related streptococcal species (49, 50). Indeed, necrosis and general cytotoxicity following GBS infection have been observed in a variety of other cell types and tissues in studies where the specific mechanism of cell death was not determined (51–54). It is possible that further evaluation of such models will reveal GBS-mediated necroptosis in some of these cell types as well.
Despite our observations that proinflammatory pathways are activated in response to GBS and evidence for the induction of a highly proinflammatory form of programmed cell death in GBS-infected DSCs, we failed to observe high levels of proinflammatory cytokines by cytokine array. This apparent contradiction may be due to the cell culture model itself or the timing of NF-κB activation in infected cells. High levels of nuclear localized NF-κB, which drives the production of many proinflammatory cytokines, were not observed in response to most of the GBS strains until 4 to 5 h postinfection. Importantly, our cytotoxicity studies indicated that by this time, a large percentage of the cell population was already undergoing programmed cell death. Thus, by the time the cells receive the signal to produce proinflammatory cytokines, there is little time to do so before they are killed. However, once these decidual cells rupture at the culmination of necroptosis, it is likely that a variety of damage-associated molecular patterns (DAMPs) and various inflammatory mediators are released (20). In vivo, we anticipate that such factors would signal to other nearby cells and surrounding tissues to induce a variety of proinflammatory responses that lead to immune activation, local tissue destruction, extraplacental membrane weakening, and dissemination of GBS into deeper tissues. Nonetheless, these outcomes would be dependent on the timing and intensity of the response and will require further investigation using more quantitative approaches and different model systems. Indeed, one limitation of this work is the loss of biological context when host-microbe interactions are modeled in vitro, as the situation is likely more complicated in vivo, given the potential input of other cell types and soluble paracrine mediators such as sex hormones. The new knowledge revealed by the present in vitro work will need to be confirmed in tissue models or, optimally, in vivo.
Future work will also explore changes to some of the other major signaling pathways identified in the array. Such pathways include those that affect fetal development, including hormone signaling and angiogenesis, as well as pathways involved in colonization of maternal and fetal tissues. It will also be interesting to assess host signaling responses across a larger panel of GBS clinical isolates to determine whether factors such as ST, capsule type, or clinical source (colonizing versus invasive) impact the key host responses that were identified. Indeed, GBS strains of various genetic backgrounds have been shown to vary in their ability to attach to and invade DSCs (15), which was confirmed for the strains evaluated in this study. It is therefore possible that these differences partly contribute to the variation in host cell signaling responses that were observed. Intracellular bacterial populations could impact death and inflammatory signaling pathways differently than extracellular populations, perhaps accounting for some of the differences in the timing and intensity of the observed responses. The GBS strains examined also varied in βH/C production, and enhanced lysis of RBCs correlated with enhanced inflammatory responses by DSCs. This trend suggests that the hemolysin may be a contributing factor to the strain-specific differences in host cell death and inflammatory signaling pathways that were observed.
Though we primarily investigated the role of MAPK signaling in inflammation and programmed cell death responses, many of the other pathways identified through the KEGG and STRING array analyses also interface with the MAPK pathway. Together, these pathways form extensive and complex signaling networks that influence a diverse array of cellular functions. Extensive analysis will be required to piece together how these pathways interact during GBS infection and to understand how GBS-induced changes in host signaling contribute to specific pregnancy complications and various aspects of neonatal disease. Further evaluation of the differences in host responses between strains may also shed light on why certain sequence types tend to cause more severe cases of disease and may reveal potential therapeutic targets to combat GBS neonatal infections.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
Four previously characterized GBS strains were used in this study; three strains (GB00112 [capsule {CPS} type III, ST-17], GB00590 [CPS III, ST-19], and GB00653 [CPS II, ST-12]) were isolated from colonized mothers (11) and one (GB00411 [CPS III, ST-17]) was from a neonate with EOD (10). Strains were grown in Todd-Hewitt broth (THB) at 37°C for 16 to 20 h, washed in sterile phosphate-buffered saline (PBS), and resuspended in cell culture medium prior to infection of host cells.
Cell culture and infection.
Telomerase-immortalized human endometrial stromal cells (T-HESCs; ATCC CRL-4003) (55) were cultured in Dulbecco’s modified Eagle medium (DMEM)/nutrient mixture Ham’s F-12 with l-glutamine (Sigma) containing 1.5 g/liter sodium bicarbonate (J.T.Baker), 1% BD Biosciences ITS+ universal culture supplement premix containing insulin, human transferrin, and selenous acid (Corning), 10% charcoal-treated fetal bovine serum (FBS; Atlanta Biologicals), and 2% antibiotic-antimycotic (Gibco). Cells were maintained at 37°C with 5% carbon dioxide, and seeded into 24-well, 6-well, or 10-cm dishes (MidSci) for infection experiments. Confluent cells were decidualized (dT-HESCs) as previously described (15) by incubation with 0.5 mM 8-bromo-cyclic AMP (cAMP) (Sigma) for 3 to 5 days in T-HESC medium.
Prior to bacterial infection, dT-HESCs were washed with PBS and resuspended in T-HESC infection medium. Overnight GBS cultures were centrifuged, washed in PBS, and resuspended in T-HESC infection medium. The optical density at 600 nm (OD600) of each culture was normalized to calculate the multiplicity of infection (MOI), which was verified using standard mammalian cell counting and CFU quantification. dT-HESCs were infected with the normalized cultures of GBS at an MOI of 10 bacteria per host cell and incubated at 37°C with 5% CO2 for 2 to 5 h.
Antibody array.
The KAM-900P antibody microarray kit (Kinexus, Vancouver, Canada) includes antibodies to 386 different human proteins with 613 phosphorylation site-specific antibodies and 265 pan-specific antibodies. Phosphorylation of specific sites provides information about a protein’s activity or regulatory state, while pan-specific sites provide information regarding total protein level within the cell. Following GBS infection of dT-HESCs, media and nonadherent bacteria were aspirated, and the cells were washed twice with PBS, lysed with Kinexus lysis buffer, and disrupted using microprobe sonication (Branson Sonifier 250). The protocols for protein collection, labeling, and antibody array incubation were according to the manufacturer’s instructions. Data were submitted to Kinexus for signal detection and statistical analysis to compare each infection condition to the corresponding mock infection control for each time point.
Proteins with z-score ratios greater than 1.0 or less than −1.0 were considered significant. Additionally, proteins identified as “priority” or “possible leads” (referred to as “leads”) were included in follow-up annotations and pathway groupings. A priority lead was defined as a protein with phosphorylated or total level percent changes from control (%CFC; control refers to mock infection) of ≥300% (equivalent to a fold change of 6), a sum of percent error ranges of <0.75 × %CFC value, and at least one globally normalized intensity value ≥1,500, as described in the Kinexus KAM-900P instructions. A possible lead was defined as a %CFC of ≥150 (equivalent to a fold change of 3), a sum of percent error ranges of <0.85 × %CFC value, and at least one globally normalized intensity value of ≥1,000. Following this statistical analysis, proteins with phosphorylated or total levels that were significantly altered compared to those with mock infection or were identified as possible leads were functionally annotated and grouped into major signaling pathways using the KEGG and STRING software programs and the UniProt protein database (49).
SDS-PAGE and Western blotting.
The protein concentration of each dT-HESC sample lysate was determined by bicinchoninic acid (BCA) assay (Pierce) using bovine serum albumin (BSA) protein standards and normalized prior to loading the samples on a 4% to 15% polyacrylamide gel (Bio-Rad). Samples were transferred to a polyvinylidene difluoride (PVDF) membrane and blocked in 5% BSA (Fisher Scientific) plus 0.1% Tween 20 (Sigma) in Tris-buffered saline (TBS); membranes were incubated with primary antibodies overnight at 4°C. Primary antibodies obtained from Cell Signaling were used at a dilution of 1:1,000, and antibodies obtained from Santa Cruz Biotechnology were used at a dilution of 1:500. The membranes were washed for 1.5 h in TBS plus 0.1% Tween 20 and incubated with goat anti-mouse or goat anti-rabbit IgG-horseradish peroxidase (HRP) secondary antibodies (Life Technologies) at a dilution of 1:5,000 for 1.5 h at room temperature. Membranes were then washed for 1.5 h in TBS plus 0.1% Tween 20, incubated with ECL chemiluminescence reagent (Pierce), and developed using an Amersham Imager 600 (GE Life Sciences). Relative protein levels were quantified by densitometry using ImageJ, and values from at least 3 independent biological replicates were pooled for each protein target. To account for possible differences in loading or protein transfer, GAPDH (glyceraldehyde-3-phosphate dehydrogenase) or beta-tubulin was used as the loading control to determine the normalized protein levels that are reported for each protein analyzed. Graphed densitometry data represent averaged values from 3 to 5 biological replicates and error bars indicate the standard deviations from the means.
Immunofluorescence staining and imaging.
Cells were plated in sterile 24-well cell culture-treated plates, and GBS was added as described by Flaherty et al. for group A Streptococcus (49). Following infection, cells were washed in PBS and fixed overnight in a 4% paraformaldehyde solution in PBS (wt/vol). Treated cells were washed in PBS and blocked for 2 h at room temperature in PBS with 1% (wt/vol) normal goat serum, 2% (vol/vol) Triton, and 0.5% (vol/vol) Tween 20. The cells were washed with PBS for 1.5 h, incubated with primary antibody (1:400) in blocking solution overnight at 4°C, washed in PBS for 1.5 h, and incubated for 2 h at room temperature in secondary antibody (goat anti-rabbit IgG Alexa Fluor 488) at a 1:400 ratio of antibody to blocking solution. Cells were washed for 1 h and added to DAPI (4′,6-diamidino-2-phenylindole) nuclear stain (1:500) in blocking solution for incubation at room temperature (30 min) prior to a final wash in PBS for 30 min at room temperature. The plates were stored in PBS at 4°C prior to imaging, which was performed using a BioTek Cytation 3 Imager (20× objective). ImageJ and Adobe Photoshop Lightroom 6 were used to process the captured images. At least three biological replicates were performed per condition for all immunofluorescence experiments, with multiple fields captured and counted per condition to obtain the reported data. Statistics were calculated from at least 275 cells per condition, which were pooled from the three biological replicates. Averaged values from each replicate were graphed with error bars representing the standard deviations from the means.
Antibodies and stains.
Antibodies to NF-κB p65 (8242S), IκBα (9242S), beta-tubulin (2128S), GAPDH (5174S), phospho-MAPK p38 (T180+Y182; 4511S), total MAPK p38 (8690S), phospho-extracellular signal-regulated kinase 1 and 2 (ERK1/2) (T202+Y204; 4370T), total ERK1/2 (4695T), total crystallin αB (45844S), total stress-activated protein kinase (SAPK)/JNK (9252T), phospho-MLKL (S358), and total MLKL (98110T) were obtained from Cell Signaling Technology. Antibodies to phospho-JNK (T183+Y185; 6254), phospho-Fos (S374; 81485), total c-Fos (166940), phospho-Jun (S63; 822), total c-Jun (74543), and phospho-crystallin αB (S59; 365884) were obtained from Santa Cruz Biotechnology. Goat anti-rabbit and anti-mouse IgG-HRP secondary antibodies (31460 and 31430) were obtained from Thermo Fisher Scientific. DAPI nuclear stain was obtained from Cell Signaling, and goat anti-rabbit IgG Alexa Fluor 488 was obtained from Molecular Probes (Life Technologies).
Vehicle controls and chemical inhibitors.
For each experiment involving chemical inhibitors, sterile dimethyl sulfoxide (DMSO; Sigma) was used as the chemical solvent and experimental vehicle control. SB203580 (Cell Signaling) was used to inhibit p38 MAPK activity, and SP600125 (Cell Signaling) was used to inhibit JNK activity. Z-VAD-fmk (ApexBio) was used as a general caspase inhibitor, and staurosporine (Cell Signaling), which is a potent protein kinase inhibitor that induces apoptosis, was used as a positive control for caspase 3/7 activation and induction of apoptosis. Necrostatin-1 (Sigma-Aldrich) was used to inhibit RIPK1 kinase, and GSK872 (Calbiochem) was used to inhibit RIPK3. Each inhibitor (except staurosporine) was incubated with each of the GBS strains used in this study at time points and drug concentrations matching experimental conditions to determine effects on GBS growth and viability (data not shown). Following incubation in liquid media, bacterial samples were collected from each sample/condition, plated on Todd-Hewitt agar, and grown overnight at 37°C in 5% CO2 to quantify CFU/ml from the liquid cultures as conducted previously (49).
Ethidium homodimer cell death and caspase 3/7 activity assays.
dT-HESCs were plated in 24-well tissue culture plates and infected with GBS using the conditions described above (MOI = 10). Cell death was determined using the membrane permeabilization method described by Flaherty et al. (49). Briefly, after infection, the cells were washed with sterile PBS. Cells were then covered and incubated at room temperature for 30 min with 4 μM ethidium homodimer 1 (Fisher Scientific) in PBS. The level of fluorescence was determined using a BioTek Cytation 3 plate reader set to 528 nm excitation and 617 nm emission. The percentage of dead cells was determined by adding 0.1% (wt/vol) saponin (Sigma) to each well following the initial reading and allowing the plate to incubate for an additional 20 min at room temperature before reading the plate a second time at the same settings. Percent membrane permeabilization values were obtained individually for each well by dividing the initial fluorescence reading (posttreatment) by the second fluorescence reading (postsaponin). For each biological replicate, treatment conditions were performed in technical triplicates (at minimum), and at least 3 independent biological replicates were performed and pooled per condition. The pooled average and standard deviation from each condition were plotted for comparison.
To detect caspase 3/7 activity, the dT-HESC cells were infected at an MOI of 10 for 2 to 5 h and treated with Caspase-Glo 3/7 reagent (Promega), which contains both cell lysing agents and the luminescent caspase substrate to promote caspases 3 and 7 are executioner caspases that mediate activation of classic apoptosis (47). Luminescence values were determined using a BioTek Cytation 3 plate reader.
Cytokine arrays.
Supernatant collection, processing, and analysis methods were adapted from those described previously (18, 49); modifications included the time point of sample collection, the human cell type analyzed, and the bacterial species tested. Briefly, the supernatants containing bacteria and secreted proteins were collected and centrifuged (2,400 relative centrifugal force [rcf] for 10 min) after a 5 h infection and stored at −20°C. Following centrifugation (16,000 rcf) for 5 min to remove debris, the Abcam Human Cytokine Antibody Array kit (ab133998) was used according to the manufacturer’s instructions to quantify relative cytokine levels per sample using an Amersham Imager 600 (GE Life Sciences). Cytokine functions were defined by UniProt (http://www.uniprot.org/). Densitometry was performed using ImageJ to determine relative protein levels; values were corrected using internal positive controls for each array, and the normalized values were graphed. Each infection condition was compared to the corresponding mock infection condition to calculate fold changes. A fold change of ≥1.5 was considered biologically significant.
Association assays.
As described previously (15), dT-HESCs were infected for 2 h at an MOI of 10 bacteria per host cell. Briefly, wells were washed with PBS after a 2-h incubation at 37°C with 5% CO2 to remove nonadherent bacteria. The number of associated bacteria (attached and invaded) was quantified following lysis of the dT-HESCs with 0.1% Triton X-100 (Sigma), plating on Todd-Hewitt agar, and incubating overnight at 37°C. The percentage of bacterial cells that had invaded host cells was quantified after adding 100 μg/ml of gentamicin (Gibco) and 5 μg/ml of penicillin G (Sigma) and incubating at 37°C for 1 h to kill extracellular bacteria. The number of attached bacteria was calculated by subtracting the number of invaded bacteria from the number of associated bacteria. All data were expressed as percentages of the total number of bacteria per well at the end of the 2-h infection period. Assays were run in triplicates at least three times.
RBC lysis.
Whole sheep blood was washed three times in PBS and diluted to a final concentration of 1:20 (vol/vol) in PBS. GBS cultures were diluted to an OD600 value of 0.1 in cell culture infection medium and combined with the blood/PBS mixture at a 1:5 ratio (bacteria to blood) and incubated at 37°C for 2 h, 6 h, or 24 h. A mixture of 1% Triton in PBS and blood was used as a positive control, representing 100% RBC lysis, and PBS was used as a negative control for RBC lysis. Each test condition was performed in triplicates for each of three biological replicates. After the incubation period, the samples were centrifuged at 275 rcf for 10 min, and an aliquot (200 μl) of the supernatant was collected from each sample and transferred to a clear, flat-bottom 96-well plate for analysis. A microplate reader was used to determine sample absorbance at 450 nm, a wavelength that allows for detection of the heme released from the lysed RBCs. Percent lysis was determined by normalizing absorbance values to the Triton positive control (100% lysis) and PBS buffer as the negative control. GBS strain GB00037, which was previously shown to exhibit decreased hemolysis and pigmentation (24), was also examined for comparison.
Data analysis.
All statistical analyses were carried out using GraphPad Prism 6.0 or Microsoft Excel. Normal distribution was assumed for all data based on the similarity of standard deviations between conditions within each data set and on the absence of outlier values. Significant differences for individual pairs of means were determined using Student’s t tests, and P values of <0.05 were considered significant. For larger data sets comparing 3 or more groups, analysis of variance (ANOVA) was used to calculate P values followed by post hoc Dunnett’s tests for comparing each mean to a control mean for each experiment. When the mean of each condition was to be compared to the mean of all other conditions, post hoc Tukey’s tests were used after the ANOVA. P values of <0.05 were considered significant. Individual P values from t tests, post hoc Dunnett’s tests, and post hoc Tukey’s tests are reported in the figure legends.
Supplementary Material
ACKNOWLEDGMENTS
Funding for this work was provided by the Michigan State University (MSU) Foundation and the National Institutes of Health (AI134036 and HD090061). M.M. was supported by the MSU Molecular Biology Study Abroad Program.
We declare no conflicts of interest.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00729-19.
REFERENCES
- 1.Russell NJ, Seale AC, O’Driscoll M, O’Sullivan C, Bianchi-Jassir F, Gonzalez-Guarin J, Lawn JE, Baker CJ, Bartlett L, Cutland C, Gravett MG, Heath PT, Le Doare K, Madhi SA, Rubens CE, Schrag S, Sobanjo-Ter Meulen A, Vekemans J, Saha SK, Ip M. 2017. Maternal colonization with group B Streptococcus and serotype distribution worldwide: systematic review and meta-analyses. Clin Infect Dis 6(Suppl 2):S100–S111. doi: 10.1093/cid/cix658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Verani JR, McGee L, Schrag SJ. 2010. Prevention of perinatal group B streptococcal disease–revised guidelines from CDC, 2010. MMWR Recomm Rep 59:1–36. [PubMed] [Google Scholar]
- 3.Vornhagen J, Adams Waldorf KM, Rajagopal L. 2017. Perinatal group B streptococcal infections: virulence factors, immunity, and prevention strategies. Trends Microbiol 25:919–931. doi: 10.1016/j.tim.2017.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patras KA, Nizet V. 2018. Group B streptococcal maternal colonization and neonatal disease: molecular mechanisms and preventative approaches. Front Pediatr 6:27. doi: 10.3389/fped.2018.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wennekamp J, Henneke P. 2008. Induction and termination of inflammatory signaling in group B streptococcal sepsis. Immunol Rev 225:114–127. doi: 10.1111/j.1600-065X.2008.00673.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vanderhoeven JP, Bierle CJ, Kapur RP, McAdams RM, Beyer RP, Bammler TK, Farin FM, Bansal A, Spencer M, Deng M, Gravett MG, Rubens CE, Rajagopal L, Adams Waldorf KM. 2014. Group B streptococcal infection of the choriodecidua induces dysfunction of the cytokeratin network in amniotic epithelium: a pathway to membrane weakening. PLoS Pathog 10:e1003920. doi: 10.1371/journal.ppat.1003920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anders AP, Gaddy JA, Doster RS, Aronoff DM. 2017. Current concepts in maternal-fetal immunology: recognition and response to microbial pathogens by decidual stromal cells. Am J Repro Immunol 77:e12623. doi: 10.1111/aji.12623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones N, Bohnsack JF, Takahashi S, Oliver KA, Chan MS, Kunst F, Glaser P, Rusniok C, Crook DWM, Harding RM, Bisharat N, Spratt BG. 2003. Multilocus sequence typing system for group B Streptococcus. J Clin Microbiol 41:2530–2536. doi: 10.1128/JCM.41.6.2530-2536.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luan S-L, Granlund M, Sellin M, Lagergård T, Spratt BG, Norgren M. 2005. Multilocus sequence typing of Swedish invasive group B Streptococcus isolates indicates a neonatally associated genetic lineage and capsule switching. J Clin Microbiol 43:3727–3733. doi: 10.1128/JCM.43.8.3727-3733.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Manning SD, Springman AC, Lehotzky E, Lewis MA, Whittam TS, Davies HD. 2009. Multilocus sequence types associated with neonatal group B streptococcal sepsis and meningitis in Canada. J Clin Microbiol 47:1143–1148. doi: 10.1128/JCM.01424-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manning SD, Lewis MA, Springman AC, Lehotzky E, Whittam TS, Davies HD. 2008. Genotypic diversity and serotype distribution of group B Streptococcus isolated from women before and after delivery. Clin Infect Dis 46:1829–1837. doi: 10.1086/588296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brochet M, Couvé E, Zouine M, Vallaeys T, Rusniok C, Lamy MC, Buchrieser C, Trieu-Cuot P, Kunst F, Poyart C, Glaser P. 2006. Genomic diversity and evolution within the species Streptococcus agalactiae. Microbes Infect 8:1227–1243. doi: 10.1016/j.micinf.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 13.Springman AC, Lacher DW, Wu G, Milton N, Whittam TS, Davies HD, Manning SD. 2009. Selection, recombination, and virulence gene diversity among group B streptococcal genotypes. J Bacteriol 191:5419–5427. doi: 10.1128/JB.00369-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parker RE, Laut C, Gaddy JA, Zadoks RN, Davies HD, Manning SD. 2016. Association between genotypic diversity and biofilm production in group B Streptococcus. BMC Microbiol 16:86. doi: 10.1186/s12866-016-0704-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Korir ML, Knupp D, LeMerise K, Boldenow E, Loch-Caruso R, Aronoff DM, Manning SD. 2014. Association and virulence gene expression vary among serotype III group B Streptococcus isolates following exposure to decidual and lung epithelial cells. Infect Immun 82:4587–4595. doi: 10.1128/IAI.02181-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Korir ML, Laut C, Rogers LM, Plemmons JA, Aronoff DM, Manning SD. 2017. Differing mechanisms of surviving phagosomal stress among group B Streptococcus strains of varying genotypes. Virulence 8:924–937. doi: 10.1080/21505594.2016.1252016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Korir ML, Flaherty RA, Rogers LM, Gaddy JA, Aronoff DM, Manning SD. 2018. Investigation of the role that NADH peroxidase plays in oxidative stress survival in group B Streptococcus. Front Microbiol 9:2786. doi: 10.3389/fmicb.2018.02786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flaherty RA, Borges EC, Sutton JA, Aronoff DM, Gaddy JA, Petroff MG, Manning SD. 2019. Genetically distinct group B Streptococcus strains induce varying macrophage cytokine responses. PLoS One 14:e0222910. doi: 10.1371/journal.pone.0222910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cargnello M, Roux P. 2011. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev 75:50–83. doi: 10.1128/MMBR.00031-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaczmarek A, Vandenabeele P, Krysko DV. 2013. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity 38:209–223. doi: 10.1016/j.immuni.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 21.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. 2010. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 22.Degterev A, Hitomi J, Germscheid M, Ch'en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J. 2008. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Randis TM, Gelber SE, Hooven TA, Abellar RG, Akabas LH, Lewis EL, Walker LB, Byland LM, Nizet V, Ratner AJ. 2014. Group B Streptococcus hemolysin/cytolysin breaches maternal-fetal barriers to cause preterm birth and intrauterine fetal demise in vivo. J Infect Dis 210:265–273. doi: 10.1093/infdis/jiu067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gendrin C, Vornhagen J, Armistead B, Singh P, Whidbey C, Merillat S, Knupp D, Parker R, Rogers LM, Quach P, Iyer LM, Aravind L, Manning SD, Aronoff DM, Rajagopal L. 2018. A nonhemolytic group B Streptococcus strain exhibits hypervirulence. J Infect Dis 217:983–987. doi: 10.1093/infdis/jix646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park H-R, Harris SM, Boldenow E, McEachin RC, Sartor M, Chames M, Loch-Caruso R. 2018. Group B Streptococcus activates transcriptomic pathways related to premature birth in human extraplacental membranes in vitro. Biol Reprod 98:396–407. doi: 10.1093/biolre/iox147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ali SR, Fong JJ, Carlin AF, Busch TD, Linden R, Angata T, Areschoug T, Parast M, Varki N, Murray J, Nizet V, Varki A. 2014. Siglec-5 and Siglec-14 are polymorphic paired receptors that modulate neutrophil and amnion signaling responses to group B Streptococcus. J Exp Med 211:1231–1242. doi: 10.1084/jem.20131853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kenzel S, Santos-Sierra S, Deshmukh SD, Moeller I, Ergin B, Fitzgerald KA, Lien E, Akira S, Golenbock DT, Henneke P. 2009. Role of p38 and early growth response factor 1 in the macrophage response to group B Streptococcus. Infect Immun 77:2474–2481. doi: 10.1128/IAI.01343-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kenzel S, Mancuso G, Malley R, Teti G, Golenbock DT, Henneke P. 2006. c-Jun kinase is a critical signaling molecule in a neonatal model of group B streptococcal sepsis. J Immunol 176:3181–3188. doi: 10.4049/jimmunol.176.5.3181. [DOI] [PubMed] [Google Scholar]
- 29.Mancuso G, Midiri A, Beninati C, Piraino G, Valenti A, Nicocia G, Teti D, Cook J, Teti G. 2002. Mitogen-activated protein kinases and NF-kappa B are involved in TNF-alpha responses to group B streptococci. J Immunol 169:1401–1409. doi: 10.4049/jimmunol.169.3.1401. [DOI] [PubMed] [Google Scholar]
- 30.Wang L, Yu X, Yan C, Wang X, Wang L, Yu X, Yan C, Wang X. 2010. Nuclear translocation of nuclear factor kappa B in first trimester deciduas and chorionic villi in early spontaneous miscarriage women. Int J Mol Sci 11:521–531. doi: 10.3390/ijms11020521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Agrawal V, Hirsch E. 2012. Intrauterine infection and preterm labor. Semin Fetal Neonatal Med 17:12–19. doi: 10.1016/j.siny.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lappas M. 2013. NOD1 and NOD2 regulate proinflammatory and prolabor mediators in human fetal membranes and myometrium via nuclear factor-kappa B. Biol Reprod 89:14. doi: 10.1095/biolreprod.113.110056. [DOI] [PubMed] [Google Scholar]
- 33.Romero R, Wu YK, Brody DT, Oyarzun E, Duff GW, Durum SK. 1989. Human decidua: a source of interleukin-1. Obstet Gynecol 73:31–34. [PubMed] [Google Scholar]
- 34.Romero R, Espinoza J, Gonçalves L, Kusanovic J, Friel L, Hassan S. 2007. The role of inflammation and infection in preterm birth. Semin Reprod Med 25:21–39. doi: 10.1055/s-2006-956773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.N’Guessan PD, Schmeck B, Ayim A, Hocke AC, Brell B, Hammerschmidt S, Rosseau S, Suttorp N, Hippenstiel S. 2005. Streptococcus pneumoniae R6x induced p38 MAPK and JNK-mediated caspase-dependent apoptosis in human endothelial cells. Thromb Haemost 94:295–303. doi: 10.1160/TH04-12-0822. [DOI] [PubMed] [Google Scholar]
- 36.Stringaris AK, Geisenhainer J, Bergmann F, Balshüsemann C, Lee U, Zysk G, Mitchell TJ, Keller BU, Kuhnt U, Gerber J, Spreer A, Bähr M, Michel U, Nau R. 2002. Neurotoxicity of pneumolysin, a major pneumococcal virulence factor, involves calcium influx and depends on activation of p38 mitogen-activated protein kinase. Neurobiol Dis 11:355–368. doi: 10.1006/nbdi.2002.0561. [DOI] [PubMed] [Google Scholar]
- 37.Krzymińska S, Frąckowiak H, Kaznowski A. 2012. Acinetobacter calcoaceticus-baumannii complex strains induce caspase-dependent and caspase-independent death of human epithelial cells. Curr Microbiol 65:319–329. doi: 10.1007/s00284-012-0159-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robinson N, McComb S, Mulligan R, Dudani R, Krishnan L, Sad S. 2012. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat Immunol 13:954–962. doi: 10.1038/ni.2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morinaga Y, Yanagihara K, Nakamura S, Hasegawa H, Seki M, Izumikawa K, Kakeya H, Yamamoto Y, Yamada Y, Kohno S, Kamihira S. 2010. Legionella pneumophila induces cathepsin B-dependent necrotic cell death with releasing high mobility group box1 in macrophages. Respir Res 11:158. doi: 10.1186/1465-9921-11-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kennedy CL, Smith DJ, Lyras D, Chakravorty A, Rood JI. 2009. Programmed cellular necrosis mediated by the pore-forming alpha-toxin from Clostridium septicum. PLoS Pathog 5:e1000516. doi: 10.1371/journal.ppat.1000516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Autheman D, Wyder M, Popoff M, D’Herde K, Christen S, Posthaus H. 2013. Clostridium perfringens beta-toxin induces necrostatin-inhibitable, calpain-dependent necrosis in primary porcine endothelial cells. PLoS One 8:e64644. doi: 10.1371/journal.pone.0064644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kling DE, Tsvang I, Murphy MP, Newburg DS. 2013. Group B Streptococcus induces a caspase-dependent apoptosis in fetal rat lung interstitium. Microb Pathog 61–62:1–10. doi: 10.1016/j.micpath.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beyrich C, Löffler J, Kobsar A, Speer CP, Kneitz S, Eigenthaler M. 2011. Infection of human coronary artery endothelial cells by group B Streptococcus contributes to dysregulation of apoptosis, hemostasis, and innate immune responses. Mediators Inflamm 2011:971502. doi: 10.1155/2011/971502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fettucciari K, Rosati E, Scaringi L, Cornacchione P, Migliorati G, Sabatini R, Fetriconi I, Rossi R, Marconi P. 2000. Group B Streptococcus induces apoptosis in macrophages. J Immunol 165:3923–3933. doi: 10.4049/jimmunol.165.7.3923. [DOI] [PubMed] [Google Scholar]
- 45.LaRock CN, Nizet V. 2015. Inflammasome/IL-1β responses to streptococcal pathogens. Front Immunol 6:518. doi: 10.3389/fimmu.2015.00518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wall DM, McCormick BA. 2014. Bacterial secreted effectors and caspase-3 interactions. Cell Microbiol 16:1746–1756. doi: 10.1111/cmi.12368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Van Cruchten S, Van Den Broeck W. 2002. Morphological and biochemical aspects of apoptosis, oncosis and necrosis. Anat Histol Embryol 31:214–223. doi: 10.1046/j.1439-0264.2002.00398.x. [DOI] [PubMed] [Google Scholar]
- 48.Whidbey C, Vornhagen J, Gendrin C, Boldenow E, Samson JM, Doering K, Ngo L, Ezekwe EAD, Gundlach JH, Elovitz MA, Liggitt D, Duncan JA, Adams Waldorf KM, Rajagopal L. 2015. A streptococcal lipid toxin induces membrane permeabilization and pyroptosis leading to fetal injury. EMBO Mol Med 7:488–505. doi: 10.15252/emmm.201404883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Flaherty RA, Puricelli JM, Higashi DL, Park CJ, Lee SW. 2015. Streptolysin S promotes programmed cell death and enhances inflammatory signaling in epithelial keratinocytes during group A Streptococcus infection. Infect Immun 83:4118–4133. doi: 10.1128/IAI.00611-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.González-Juarbe N, Gilley RP, Hinojosa CA, Bradley KM, Kamei A, Gao G, Dube PH, Bergman MA, Orihuela CJ. 2015. Pore-forming toxins induce macrophage necroptosis during acute bacterial pneumonia. PLoS Pathog 11:e1005337. doi: 10.1371/journal.ppat.1005337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Da Costa AFE, Pereira CS, Santos Gda S, Carvalho TMU, Hirata R Jr, De Mattos-Guaraldi AL, Rosa A, Nagao PE. 2011. Group B Streptococcus serotypes III and V induce apoptosis and necrosis of human epithelial A549 cells. Int J Mol Med 27:739–744. doi: 10.3892/ijmm.2011.635. [DOI] [PubMed] [Google Scholar]
- 52.Alkuwaity K, Taylor A, Heckels JE, Doran KS, Christodoulides M. 2012. Group B Streptococcus interactions with human meningeal cells and astrocytes in vitro. PLoS One 7:e42660. doi: 10.1371/journal.pone.0042660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kaplan A, Chung K, Kocak H, Bertolotto C, Uh A, Hobel CJ, Simmons CF, Doran K, Liu GY, Equils O. 2008. Group B Streptococcus induces trophoblast death. Microb Pathog 45:231–235. doi: 10.1016/j.micpath.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bifrare Y-D, Gianinazzi C, Imboden H, Leib SL, Täuber MG. 2003. Bacterial meningitis causes two distinct forms of cellular damage in the hippocampal dentate gyrus in infant rats. Hippocampus 13:481–488. doi: 10.1002/hipo.10142. [DOI] [PubMed] [Google Scholar]
- 55.Krikun G, Mor G, Alvero A, Guller S, Schatz F, Sapi E, Rahman M, Caze R, Qumsiyeh M, Lockwood CJ. 2004. A novel immortalized human endometrial stromal cell line with normal progestational response. Endocrinology 145:2291–2296. doi: 10.1210/en.2003-1606. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.