

The molecular mechanisms underlying biological differences between two Leishmania species that cause cutaneous disease, L. major and L. amazonensis, are poorly understood. In L. amazonensis, reactive oxygen species (ROS) signaling drives differentiation of nonvirulent promastigotes into forms capable of infecting host macrophages.
KEYWORDS: ascorbate-dependent peroxidase, mitochondria, redox signaling, Leishmania, differentiation
ABSTRACT
The molecular mechanisms underlying biological differences between two Leishmania species that cause cutaneous disease, L. major and L. amazonensis, are poorly understood. In L. amazonensis, reactive oxygen species (ROS) signaling drives differentiation of nonvirulent promastigotes into forms capable of infecting host macrophages. Tight spatial and temporal regulation of H2O2 is key to this signaling mechanism, suggesting a role for ascorbate-dependent peroxidase (APX), which degrades mitochondrial H2O2. Earlier studies showed that APX-null L. major parasites are viable, accumulate higher levels of H2O2, generate a greater yield of infective metacyclic promastigotes, and have increased virulence. In contrast, we found that in L. amazonensis, the ROS-inducible APX is essential for survival of all life cycle stages. APX-null promastigotes could not be generated, and parasites carrying a single APX allele were impaired in their ability to infect macrophages and induce cutaneous lesions in mice. Similar to what was reported for L. major, APX depletion in L. amazonensis enhanced differentiation of metacyclic promastigotes and amastigotes, but the parasites failed to replicate after infecting macrophages. APX expression restored APX single-knockout infectivity, while expression of catalytically inactive APX drastically reduced virulence. APX overexpression in wild-type promastigotes reduced metacyclogenesis, but enhanced intracellular survival following macrophage infection or inoculation into mice. Collectively, our data support a role for APX-regulated mitochondrial H2O2 in promoting differentiation of virulent forms in both L. major and L. amazonensis. Our results also uncover a unique requirement for APX-mediated control of ROS levels for survival and successful intracellular replication of L. amazonensis.
INTRODUCTION
Parasitic protozoa from the genus Leishmania are the causative agents of leishmaniasis, a leading global health problem affecting more than 12 million people worldwide (1). Depending on the Leishmania species, disease symptoms range from relatively benign skin lesions (cutaneous form) to more severe ulcerating lesions that can cause mucosal tissue disfigurement (mucocutaneous form), to infection of internal organs such as the liver and spleen that can be lethal in the absence of medical intervention (visceral form). Even among species that cause cutaneous disease and share extensive genome sequence identity, such as L. major (Old World species) and L. amazonensis (New World species), there are numerous biological differences that are still poorly understood. The intracellular stages of both species replicate inside lysosome-like parasitophorous vacuoles (PVs) of macrophages, but L. major amastigotes reside in individual PVs, while L. amazonensis amastigotes replicate in large communal PVs (2, 3). In addition, there is evidence that L. amazonensis is more resistant to macrophage microbicidal mechanisms compared to L. major, a property that may be related to the ability of L. amazonensis to cause more severe mucocutaneous skin lesions (3–6).
In spite of this variation in biological properties and disease symptoms, all Leishmania species that are pathogenic to vertebrates have life cycles alternating between insect and vertebrate hosts, with transmission occurring via sand fly (such as Lutzomyia sp. and Phlebotomus sp.) bites (7). Marked changes in metabolism and morphology occur upon differentiation of procyclic into metacyclic promastigotes in insect vector and of metacyclic promastigotes into intracellular amastigotes inside mammalian macrophages. These differentiation processes involve genome-wide changes in gene expression orchestrated at the posttranscriptional and posttranslational levels, enabling the parasites to rapidly adapt to environmental changes between host and vectors (8–13). The signaling cascades that bring about differentiation have not been completely elucidated, but recent studies implicate H2O2-mediated signaling in the development of Leishmania virulence (14–16).
An important role for H2O2 as a regulator of cellular redox sensing, signaling, and cell fate has emerged over the last decade. Initially considered mostly as a source of the extremely toxic hydroxyl ions (OH·−) and perinitrite through the Fenton reaction (17, 18), H2O2 is now recognized as a membrane-diffusible second messenger that can initiate changes in cell proliferation and differentiation by modulating the activity of redox-sensitive proteins (19–21). This extensive class of “redox switch” proteins includes phosphatases, kinases, and transcription factors that contain redox-sensitive metal centers or cysteine residues, and they are thus able to alter their oxidative state in response to H2O2. Thereby, subtle changes in local concentrations of H2O2 can directly modulate the activity of target proteins, which propagate signaling cascades through posttranslational modifications or directly influence gene expression (22). While moderate, transient or spatially localized changes in H2O2 concentration play important physiological roles, large-scale, sustained or widely distributed H2O2 increases can have serious deleterious effects (19). Hence, maintenance of tightly controlled, steady-state H2O2 levels is critical for the normal physiological functioning of eukaryotic cells.
Increasing evidence supports a specific role for mitochondria-generated reactive oxygen species (mROS) in determining cell fate (23–25). This is particularly well demonstrated in vertebrate stem cells, whose characteristic self-renewing capacity is preserved at low mROS levels, but readily lost when mROS elevations inhibit their ability to proliferate and commit to differentiation (26). Mitochondrion-generated H2O2 promotes differentiation of a wide range of specialized cells, including mammalian muscle fibers and Arabidopsis root hair cells (27–29). It also plays a critical role in cell fate determination in cancer cells and in tumor development (30). H2O2 is generated in mitochondria or the cytosol as part of ROS cascades that start with the generation of superoxide ion (O2−) through mitochondrial electron transport chain (mETC) complexes or plasma membrane-associated NAD(P)H oxidases (NOXs). Superoxide is rapidly converted to H2O2 by superoxide dismutase (SOD) enzymes present in mitochondria or the cytosol. H2O2 is more stable than superoxide and is membrane diffusible, properties that make it suitable for propagating signals across organelles. Intracellular steady-state H2O2 levels are tightly controlled by the coordinated action of antioxidant enzymes such as catalase, peroxidases and peroxiredoxins, and even subtle changes in H2O2 levels are known to be sufficient to trigger cellular responses (31).
The H2O2-mediated signaling cascade promoting virulence development in L. amazonensis was proposed to start with increased generation of O2−, either through elevated mETC activity under stress conditions (such as low pH or high temperature) or via electron leakage from iron-sulfur (Fe-S) clusters in mETC complexes during iron deprivation (15, 16). Superoxide then gets converted into H2O2 by the mitochondrial iron-dependent superoxide dismutase (SODA) (15). Studies involving genetic and chemical approaches showed that H2O2 can promote development of infective Leishmania forms by stimulating two axenic differentiation processes: (i) generation of infective metacyclic promastigotes from noninfective promastigotes and (ii) differentiation of avirulent promastigotes into virulent amastigotes (15, 16). The tight control of steady-state H2O2 levels, thought to be essential for these differentiation processes, is likely to be achieved through regulation of H2O2 generation by iron-dependent mETC and SODA, in conjunction with H2O2-degrading enzymes that prevent toxic build-up. Of the three peroxide degrading mechanisms identified in Leishmania (32), previous work suggests that heme-containing ascorbate peroxidase (APX), a mitochondrial enzyme that catalyzes H2O2-dependent ascorbate oxidation through peroxidative one-electron transfers (33), is an important regulator of peroxide-induced differentiation in these parasites. Promastigotes from an APX-null mutant line of L. major showed elevated intracellular H2O2 levels and increased differentiation into metacyclic promastigotes compared to the wild type (WT), which resulted in hypervirulence in macrophage and mouse infections (34).
A role for H2O2-mediated oxidative stress in promoting differentiation and virulence in L. major was suggested (34, 35) but not directly demonstrated, as previously done for L. amazonensis (14–16). In this study, we used genetic and biochemical manipulations to directly investigate APX function in L. amazonensis promastigotes. Unlike L. major, L. amazonensis promastigotes can be efficiently induced to differentiate axenically into infective amastigotes by simple changes in growth conditions (from pH 7.4 and 26°C to pH 4.5 and 32°C). Surprisingly, we found that APX plays a dual role in L. amazonensis: while regulating H2O2-dependent differentiation of virulent forms, it is also essential for the survival of all life cycle stages and for amastigote intracellular replication.
RESULTS
APX is a mitochondrial protein whose expression is induced by increasing ROS concentrations.
Our previous studies suggested a role for mitochondrial APX in the axenic differentiation of L. amazonensis from insect promastigotes into virulent amastigotes (15). In that study, utilizing a low-pH, high-temperature protocol (36) to induce >98% of the promastigotes to differentiate into amastigotes in 48 h, we observed steady upregulation of both APX and superoxide dismutase (SOD) activity in mitochondrion-enriched subcellular fractions. Less APX activity was detected in mitochondria of parasites incapable of upregulating mitochondrial SODA, suggesting that APX expression might be dependent on mitochondrial H2O2 production (15).
Immunofluorescence with polyclonal antibodies raised against purified recombinant L. amazonensis APX revealed a punctate pattern within promastigotes. The APX puncta partially colocalized with the mitochondria-specific dye MitoTracker Red (Fig. 1A), in agreement with our previous detection of the APX protein in mitochondrial subcellular fractions (15).
FIG 1.

L. amazonensis APX is a mitochondrial protein upregulated by ROS. (A) APX localizes to mitochondria. Immunolocalization of APX in L. amazonensis promastigotes was performed using antibodies against APX (a [green]). Nucleus and kinetoplast DNA was stained with DAPI (b [blue]), and mitochondria were stained with MitoTracker Red (c [red]). Merging the two images (d [merge]) confirmed the mitochondrial localization of APX (Pearson’s correlation coefficients were 0.826, 0.773, 0.49, and 0.641 in cells 1 to 4). Bar = 4 μm. The extent of overlap between APX staining (green) and mitochondrial staining (red) was determined for individual cells. (B) APX expression is induced by ROS exposure. Log-phase L. amazonensis promastigotes (2 × 107/ml) were treated for 8 h with increasing concentrations of H2O2 or menadione, as indicated. Western blots of whole-cell lysates (10 μg protein/lane) were used to compare the amounts of APX protein in different ROS-treated samples. Tubulin expression was used as a loading control. The ratio of APX relative to tubulin in each sample is indicated below each lane and values represent the mean ± SD of three independent experiments.
To test whether APX expression was regulated by H2O2, we exposed wild-type L. amazonensis promastigotes to increasing concentrations of H2O2 or menadione, a mitochondrial superoxide generator. Dose-dependent increases in APX protein levels were observed following both treatments, with 5- to 6-fold increases observed after treatment with 2 and 4 μM menadione and a 1.7-fold increase at 100 μM H2O2, respectively (Fig. 1B). The different upregulation levels are most likely due to variations in steady-state H2O2 concentrations inside mitochondria following the treatments. While menadione treatment directly induces H2O2 generation inside mitochondria by acting on the ETC, H2O2 added extracellularly has to traverse several membranes in order to enter the organelle.
APX is essential for L. amazonensis survival.
To determine the mechanistic role of APX in regulating L. amazonensis differentiation, we applied a genetic approach to generate APX-null L. amazonensis lines. Since L. amazonensis APX is encoded by a single-copy gene, we decided to replace both alleles sequentially with gene cassettes containing hygromycin (HYG) or phleomycin (PHLEO) resistance genes, flanked by APX 5′ and 3′ untranslated regions (UTRs) to aid homologous recombination (see Fig. S1A and B in the supplemental material). The first round of transfection efficiently replaced one APX allele and generated the heterozygote (APX/ΔAPX) line. Both wild-type and transgenic promastigotes with a single APX allele showed a progressive increase in APX expression as they matured in culture, with the highest level of APX expression observed in stationary-phase promastigotes (see Fig. S2A in the supplemental material), and also in axenically grown amastigotes (Fig. S2C). However, in all growth phases the APX protein content in APX/ΔAPX cells was ∼50% lower than that of wild-type cells (Fig. S2B). Our repeated attempts to generate APX-null mutants resulted in failure. Even though we were able to select clones resistant to the second drug marker, PCR and Western blot analyses demonstrated the presence of residual APX (Fig. S1). This phenomenon of maintenance of essential genes extrachromosomally is not uncommon in Leishmania and was shown for the mitochondrial iron transporter gene LMIT1 (16) and mitochondrial superoxide dismutase gene SODA (15). Similar observations were reported for Leishmania chagasi gamma-glutamylcysteine synthetase encoded by the GSH1 gene (37).
Consistent with their 50% lower APX protein content, the APX/ΔAPX single-knockout parasites showed significantly higher sensitivity to H2O2 (50% inhibitory concentration [IC50] = 38 ± 2.3 μM) in viability tests compared to the wild type (IC50 = 62.75 ± 5.4 μM) or an overexpression line (APX/APX +APX-HA) episomally expressing additional APX fused to a C-terminal hemagglutinin (HA) tag (IC50 = 68.39 ± 5.16 μM) (Fig. 2A and B). A small but statistically significant increase in intracellular ROS levels was detected in APX/ΔAPX single-knockout promastigotes compared to wild-type promastigotes in stationary-phase culture. No significant change in fluorescence levels of CM-H2DCFDA (a chloromethyl derivative of 2′,7′-dichlorodihydrofluorescein diacetate) was observed for the other cell lines, collected from either mid-log or stationary phases of culture (Fig. 2C). No significant upregulation in other antioxidant genes with possible roles inside mitochondria, such as the peroxidoxin tryparedoxin reductase (PXN) and non-selenium glutathione peroxidase (ns-GPX) genes, was observed in APX/ΔAPX cells (see Fig. S3 in the supplemental material). Small but statistically significant increases in tryparedoxin peroxidase (TRYP5) transcript levels were detected in both APX/ΔAPX and APX/APX+APX-HA cells (Fig. S3).
FIG 2.
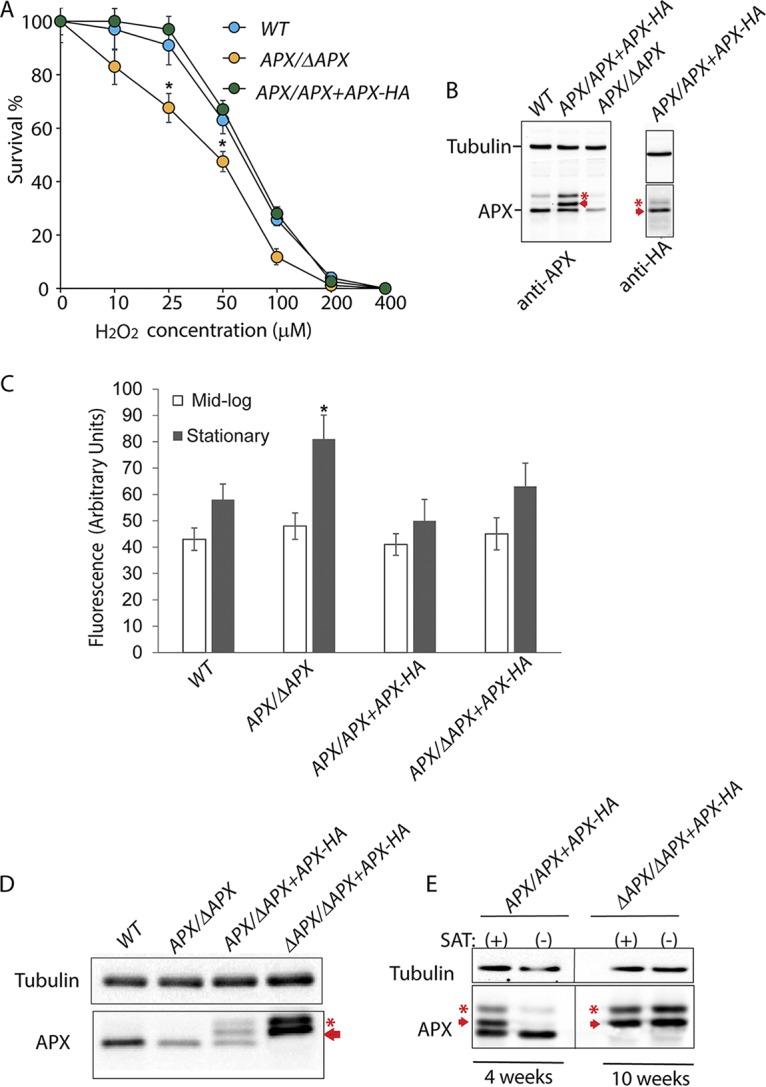
APX is essential for the survival of L. amazonensis promastigotes. (A) Effect of H2O2 on the survival of wild type (WT), APX single-knockout (APX/ΔAPX) or APX-overexpressing (APX/APX+APX-HA) promastigotes. Parasites were cultured in presence of increasing concentrations of H2O2 for 24 h followed by microscopic estimation of viable cell numbers after FDA staining. The data are expressed as percentage of the number of viable cells in cultures without H2O2, represent the mean ± SD of triplicate determinations, and are representative of three independent experiments. One-way ANOVA followed by Dunnett’s post hoc test was performed to compare survival data between cell lines for each H2O2 concentration, using WT intracellular parasite counts as control (*, P ≤ 0.05). (B) Western blot (10 μg protein/lane) using anti-APX rabbit polyclonal antibodies show relative levels of APX in whole-cell extracts of WT, APX/APX+APX-HA or APX/ΔAPX promastigotes from log-phase cultures. Expression of APX protein with C-terminus HA tags was confirmed in APX/APX+APX-HA cell extracts using anti-HA monoclonal antibodies. The arrow indicates the episomally expressed HA-tagged APX protein, the asterisk indicates a possible APX-processing intermediate. (C) Total intracellular ROS levels in WT, APX/APX+APX-HA or APX/ΔAPX promastigotes from mid-log or stationary-phase cultures were measured by determining H2DCFDA fluorescence levels. The data represent the mean ± SD of triplicate determinations of three independent experiments. One-way ANOVA followed by Dunnett’s post hoc test was performed to compare data, using WT as control (*, P ≤ 0.05). (D) Western blot analysis showing APX levels in whole-cell lysates of wild-type (WT), APX single-knockout (APX/ΔAPX), APX single-allelic-knockout complemented (APX/ΔAPX+APX-HA) or APX double-allelic-knockout cells episomally expressing HA-tagged APX (ΔAPX/ΔAPX+APX-HA). The arrow indicates the episomally expressed HA-tagged APX protein. (E) Western blot analysis showing APX in whole-cell lysates of overexpression (APX/APX+APX-HA) and complemented APX double-allelic-knockout (ΔAPX/ΔAPX+APX-HA) promastigotes cultured for 4 and 10 weeks, respectively, with (+) or without (−) the selective drug (SAT) in the culture media. Western blot analysis shows APX levels in whole-cell lysates of APX/APX+APX after 4 weeks of culture and ΔAPX/ΔAPX+APX after 10 weeks of culture. Panels spliced from the same blot for detection with antibodies against tubulin and APX are marked by thin black lines. The arrows indicate the episomally expressed HA-tagged APX protein, asterisks indicate an APX-processing intermediate.
To confirm that APX is an essential gene in L. amazonensis and to verify that our inability to generate L. amazonensis null mutants was not due to technical issues, we attempted to replace the second APX allele in single-knockout cells episomally expressing HA-tagged APX under nourseothricin (SAT) selection. To replace the second APX allele via homologous recombination, complemented single-knockout (APX/APX+APX-HA) cells were subjected to a second round of transfection with the APX-KO-PHLEO construct. Interestingly, under these conditions replacement of the second APX allele with the drug marker cassette was accomplished in 8 out of 10 clones (ΔAPX/ΔAPX+APX-HA) tested. Western blot analysis of whole-cell lysates demonstrated complete absence of native APX in ΔAPX/ΔAPX+APX-HA cells, which now exclusively expressed HA-tagged APX. APX-HA expression levels in these cells were >3-fold higher than endogenous APX detected in WT cells. Interestingly, two distinct isoforms of the C-terminus HA-tagged APX protein were detected, suggesting posttranslational processing (Fig. 2D).
In the absence of selection pressure, episomes expressing drug resistance proteins are usually lost during several rounds of Leishmania replication (38, 39). To examine the extent of episome retention following SAT withdrawal, we performed Western blot analysis on lysates of APX-HA-overexpressing cells in both wild type (APX/APX +APX-HA) and double-knockout backgrounds (ΔAPX/ΔAPX+APX-HA). As expected, within 4 weeks of drug withdrawal, >95% of episomal APX-HA expression was lost in the overexpression line of the wild-type background (APX/APX+APX-HA) and only native APX was detected (Fig. 2E). On the other hand, APX-HA expression in ΔAPX/ΔAPX+APX-HA cells was maintained at constant high levels even after 10 weeks of SAT withdrawal, suggesting that episomal retention was determined by the essentiality of APX and not by the drug selection pressure. Thus, unlike what was previously reported for L. major (34), APX is an essential gene in L. amazonensis.
We found that APX expression is cell cycle dependent and increases over time, with the highest levels of expression occurring during the stationary phase of promastigote growth (Fig. S2A and B). Also, APX expression was higher in amastigotes than in promastigotes (Fig. S2C). This observation is in agreement with the increased resistance to ROS observed in stationary-growth promastigotes and in amastigote stages, as the parasites adapt to elevated ROS levels in their environment and develop virulence (40, 41).
Our inability to generate APX-null lines prompted us to utilize the APX single-knockout L. amazonensis line to investigate the role of APX in regulating virulence development. Comparison of growth rates showed a slight but statistically significant delay in the growth rate of APX single-allele-knockout (APX/ΔAPX) promastigote cultures, which was restored to wild-type levels by episomal expression of APX-HA in the complemented line (APX/ΔAPX+APX-HA) (Fig. 3A). The enhanced sensitivity to H2O2 observed in APX/ΔAPX promastigotes (Fig. 2A) was also restored to wild-type levels in the complemented line (APX/ΔAPX-APX-HA) (IC50 = 60 ± 4.4 μM) (Fig. 3B). In contrast, expression in APX/ΔAPX promastigotes of a mutated APX-HA protein (APX/ΔAPX+mutAPX-HA) with substitutions in the conserved amino acids Trp67 and His68, which are essential for cleaving the O-O bond of its substrate H2O2 (33), failed to restore the defects and caused further decline in growth rates (Fig. 3A) and increased sensitivity to H2O2 (IC50 = 37 ± 1.7 μM) (Fig. 3B). This suggests that the mutated APX-HA protein in APX/ΔAPX +mutAPX-HA cells has a dominant-negative effect.
FIG 3.
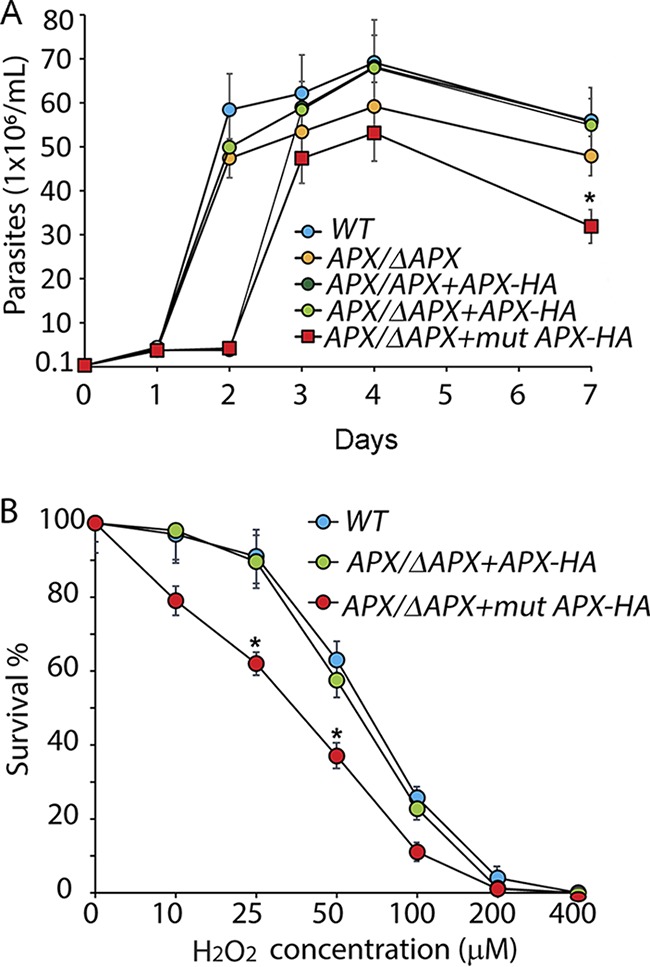
APX depletion results in slower promastigote growth rate and increases sensitivity to H2O2 exposure. (A) Growth curves for wild-type (WT), APX single-knockout (APX/ΔAPX), APX overexpression (APX/APX+APX-HA), APX single-knockout complemented (APX/ΔAPX+APX-HA), or APX single-knockout cells expressing mutated APX-HA (APX/ΔAPX+mutAPX-HA) in regular promastigote growth medium were determined. The data represent the mean ± SD of triplicate determinations and are representative of three independent experiments. A one-way ANOVA with Dunnett’s post hoc test was performed with 7-day WT as control (*, P < 0.05). (B) Effect of H2O2 on the survival of wild-type (WT), APX single-knockout complemented (APX/ΔAPX+APX-HA) or APX single-knockout cells expressing mutated APX-HA protein (APX/ΔAPX+mutAPX-HA). Parasites from the mid-log growth phase were cultured in presence of increasing concentrations of H2O2 for 24 h, followed by microscopic estimation of viable cell numbers after FDA staining. The data are expressed as the percentage of the number of viable cells in control cultures (without H2O2 treatment), represent the mean ± SD of triplicate determinations, and are representative of three independent experiments. One-way ANOVA followed by Dunnett’s post hoc test was performed to compare survival data between cell lines for every H2O2 concentration, using WT intracellular parasite counts as control (*, P ≤ 0.05).
Reduced APX expression promotes generation of metacyclic promastigotes capable of infecting macrophages, but this is followed by a failure to replicate intracellularly.
Our earlier studies suggested that peroxide signaling is involved in two consecutive steps of virulence development: first, during maturation of infective metacyclic forms during the stationary promastigote growth phase, and second, during differentiation into intracellular amastigotes. To assess the role of APX in metacyclogenesis we performed selective agglutination of stationary-phase parasite lines expressing different levels of APX, using the monoclonal antibody m3A.1 (42). This antibody is specific for an L. amazonensis surface epitope only expressed in log-phase promastigotes, allowing quantification of the number of metacyclic promastigotes that do not agglutinate. The APX/ΔAPX or APX/ΔAPX +mutAPX-HA transgenic lines expressing lower levels of active APX yielded 1.5- to 2-fold higher numbers of metacyclic promastigotes compared to the wild-type or complemented lines (APX/ΔAPX +APX-HA). In contrast, the number of metacyclic promastigotes isolated from stationary cultures of the APX overexpression line (APX/APX +APX-HA) was significantly reduced (Fig. 4A). Even though we were not successful in directly quantifying differences in mitochondrial H2O2 concentrations between these parasite lines, our results suggest a role for mitochondrial H2O2 in promoting metacyclogenesis, consistent with the expected increase in H2O2 accumulation in low-APX-expressing cells and the expected lower levels of H2O2 in APX overexpressors.
FIG 4.

APX depletion promotes metacyclogenesis and increases virulence during early stages of infection of BMM. (A) Infective metacyclic forms were isolated from stationary (day 7) cultures of wild type (WT), APX single-knockout (APX/ΔAPX), APX overexpression (APX/APX+APX-HA), APX single-knockout complemented (APX/ΔAPX+APX-HA), or APX single-knockout cells expressing mutated APX-HA (APX/ΔAPX+mutAPX-HA), after agglutinating promastigotes with the m3A.1 monoclonal antibody. The data represent the mean ± SD of the percentage of metacyclic promastigotes in quadruplet determinations and represent data obtained from four independent experiments. One-way ANOVA followed by Dunnett’s post hoc test was performed to compare data, using the percentage of metacyclic promastigotes in the WT as control (*, P ≤ 0.05). (B) The ability of metacyclic promastigotes purified from wild-type (WT), APX single-knockout (APX/ΔAPX), APX overexpression (APX/APX+APX-HA), APX single-knockout complemented (APX/ΔAPX+APX-HA), or APX single-knockout cells expressing mutated APX-HA (APX/ΔAPX+mutAPX-HA) stationary-phase cultures to infect BMMs were compared. BMMs isolated from C57BL/6 mice were infected with metacyclics for 3 h (MOI = 1:5) and either fixed immediately (3 h) or further incubated for 24, 48, or 72 h, and the number of intracellular parasites was determined microscopically. The data represent the mean ± SD of triplicate determinations and are representative of more than five independent experiments. One-way ANOVA followed by Dunnett’s post hoc test was performed to compare data at every time point using WT intracellular parasite counts as control (*, P ≤ 0.05; **, P ≤ 0.005).
Next, we checked the ability of purified metacyclic promastigotes to infect mouse bone marrow-derived macrophages (BMMs) in culture. Intracellular parasite counts 3 h after infection were similar between all tested parasite lines (Fig. 4B). As expected, the numbers of wild-type intracellular parasites started increasing after a 24-h lag period, during which the infective metacyclic promastigotes differentiate into amastigotes. In contrast, APX/ΔAPX parasites did not show the characteristic delay in initiating replication, showing a significantly increased number of intracellular parasites relative to the wild type at 24 h after infection. However, quantifications done at later time points indicated that APX/ΔAPX parasites were impaired in their ability to sustain intracellular replication, as no further growth was observed at subsequent time points. APX single knockouts expressing mutated APX-HA (APX/ΔAPX+mutAPX-HA) also failed to replicate inside macrophages, but their growth ability was restored in complemented lines expressing the functional APX protein (APX/ΔAPX+APX-HA). In contrast, metacyclic promastigotes from the APX overexpression line (APX/APX+APX-HA), even though declining in numbers after 24 h, rebounded strongly to levels comparable to wild type after 72 h.
APX deficiency drastically inhibits infectivity for mice.
To understand how variations in APX expression affect the ability of L. amazonensis to infect mammalian hosts, we injected purified L. amazonensis metacyclic promastigotes into the footpads of C57BL/6 mice and monitored cutaneous lesion development (Fig. 5A). By the eighth week, large lesions with signs of ulceration were observed in wild-type infections, which prompted us terminate the experiment in accordance with IACUC guidelines. In contrast, very little or no lesion development was noted during this period in mice infected with the APX-deficient APX/ΔAPX and APX/ΔAPX+mutAPX-HA lines. Both APX-overexpressing (APX/APX+APX-HA) and complemented (APX/ΔAPX+APX-HA) lines were able to induce lesions, which were not statistically distinguishable from the wild type. The larger variation in lesion size observed within these two groups of infected mice was probably due to different levels of episomal APX expression.
FIG 5.

APX depletion drastically reduces the ability of L. amazonensis metacyclics to produce footpad lesions in mice. (A) C57BL/6 female mice were inoculated with 1 × 106 wild type (WT), APX single-knockout (APX/ΔAPX), APX overexpression (APX/APX+APX-HA), APX single-knockout complemented (APX/ΔAPX+APX-HA), or APX single-knockout cells expressing mutated APX-HA (APX/ΔAPX+mutAPX-HA) purified metacyclic promastigotes in the left hind footpad, and lesion development was measured weekly for 8 weeks. The data represent the mean ± SD of 5 mice. One-way ANOVA followed by Dunnett’s post hoc tests were performed using WT lesion size as control. **, P ≤ 0.005. (B) Graphical representation of estimated parasite load log10 in footpad tissues, 8 weeks after infection. The numbers shown are relative to that assessed for APX single-knockout (APX/ΔAPX) cells (n = 5). One-way ANOVA with Dunnett’s post hoc test was performed with WT as control. **, P ≤ 0.005; ***, P ≤ 0.0005. Relative levels of APX protein was determined in Western blots using 10 μg of whole-cell extracts prepared from all five parasite lines recovered from tissue lesions. Panels spliced from the same blot for detection with antibodies against tubulin and APX are marked by boxes.
Parasite load estimation in mouse footpad tissues at 8 weeks after infection showed significantly lower values for the single-allele APX knockout (APX/ΔAPX) or the single-APX allele line expressing mutated APX (APX/ΔAPX+mutAPX-HA), than what was observed with wild type, overexpressing, or complemented lines (Fig. 5B). Western blot analysis of whole-cell lysates of parasites recovered from lesions confirmed episomal APX expression in all transgenic lines, with slightly elevated levels (∼1.5-fold higher) of APX-HA expression observed in the complemented line (APX/ΔAPX+APX-HA) compared to the overexpression line (APX/APX+APX-HA) or single-knockout-expressing mutated APX (APX/ΔAPX+mutAPX-HA) line (Fig. 5B, top). The reduced expression of episomal APX-HA observed in the APX/APX+APX-HA and APX/ΔAPX-mutAPX cells is most likely due to plasmid loss due to absence of any drug selection in vivo. On the other hand, the higher APX-HA protein expression observed in the complemented APX/APX+APX-HA cells may reflect an increased demand for APX activity during intracellular growth. This in vivo experiment further validates the results of the BMM infection assays and confirms that APX levels are tightly regulated. These results also suggest that APX expression above a certain critical level is required for the survival and successful intracellular replication of L. amazonensis.
APX deficiency promotes promastigote to amastigote differentiation in axenic culture, but these amastigotes are impaired in their ability to replicate.
To assess how APX ablation affects promastigote to amastigote differentiation and to quantify the ability of APX-depleted cells to replicate in axenic culture, we transferred log-phase wild-type or transgenic L. amazonensis promastigotes expressing various levels of APX from promastigote culture conditions (pH of the medium, 7.4; 26°C) to amastigote culture conditions (pH of the medium, 4.5; 32°C) (Fig. 6A). While the wild type (WT), APX overexpression (APX/APX+APX-HA), and complemented APX single-knockout cells (APX/ΔAPX+APX-HA) started replicating after 48 h, growth of APX single-knockout (APX/ΔAPX) cells or cells expressing mutated APX (APX/ΔAPX+mutAPX-HA) was significantly delayed (Fig. 6A).
FIG 6.
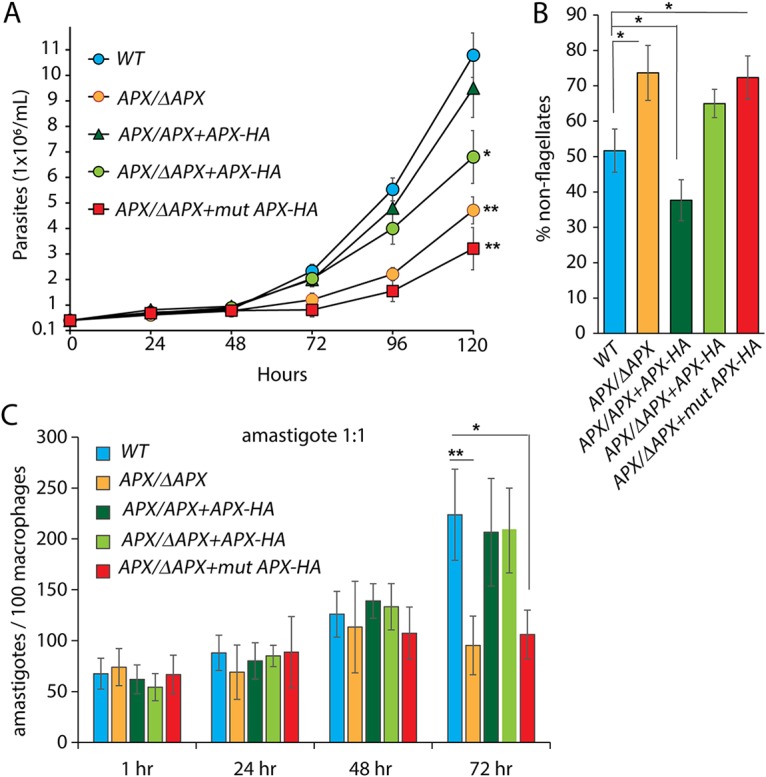
APX depletion facilitates rapid pH/temperature-induced axenic differentiation of promastigotes to amastigotes, but resulting amastigotes are replication-impaired. Log-phase promastigotes were washed, resuspended in pH 4.5 at 2 × 105 parasites/ml, and cultured at 32°C. (A) Numbers of FDA-stained viable wild-type (WT), APX single-knockout (APX/ΔAPX), APX overexpression (APX/APX+APX-HA), APX single-knockout complemented (APX/ΔAPX+APX-HA), or APX single-knockout cells expressing mutated APX-HA (APX/ΔAPX+mutAPX-HA) parasites were estimated at indicated times following shift to amastigote medium. The data represent the mean ± SD of triplicate determinations and are representative of three independent experiments. One-way ANOVA with Dunnett’s post hoc tests were performed to determine significance of growth differences between cell lines at the 120-h time point using WT cell counts as a control. *, P ≤ 0.05. (B) Percentage of viable rounded forms with nondiscernible flagellum in WT, APX/ΔAPX, APX/APX+APX-HA, APX/ΔAPX+APX-HA, or APX/ΔAPX+mutAPX-HA parasites cultured for 24 h in amastigote growth conditions. At least 200 FDA-stained parasites were counted in each sample. The data represent the mean ± SD of triplicate determinations and are representative of three independent experiments. One-way ANOVA with Dunnett’s post hoc tests were performed using the WT percentage of nonflagellate cell count as a control. *, P ≤ 0.05. (C) Viable amastigotes obtained by temperature/pH-induced axenic differentiation of WT, APX/ΔAPX, APX/APX+APX-HA, APX/ΔAPX+APX-HA, or APX/ΔAPX+mutAPX-HA cultures were tested for their ability to infect BMMs. BMMs were infected (MOI of 1:1) and either fixed immediately (1 h) or after further incubation for 24, 48, or 72 h, and the number of intracellular parasites was determined microscopically. The data represent the mean ± SD of triplicate determinations and are representative of five independent experiments. One-way ANOVA followed by Dunnett’s post hoc test performed to compare data at every time point using WT intracellular parasite counts as control (*, P ≤ 0.05; **, P ≤ 0.005).
This low-pH, high-temperature shift efficiently induces wild-type L. amazonensis to differentiate axenically from promastigotes to amastigotes, with >98% of the flagellated promastigotes losing visible flagella within 48 h and assuming a rounded amastigote-like morphology (16, 36). Since >98% of the population for all lines tested showed absence of a visible flagellar structure at 48 h (data not shown), we quantified the appearance of nonflagellated/rounded morphology at an earlier time point. At 24 h after the combined pH-temperature shift, ∼75% of the APX-deficient line populations (APX/ΔAPX and APX/ΔAPX+mutAPX-HA) became nonflagellated, which was almost 1.5- and 2-fold higher than the wild-type and APX overexpression (APX/APX+APX-HA) lines, respectively (Fig. 6B). This suggests that a rapid H2O2 accumulation in low-APX-expressing cells induces promastigote to amastigote differentiation more efficiently, a conclusion that is further corroborated by the lower number of nonflagellated parasites observed in the overexpression line (APX/APX+APX-HA) at the same time point.
Parasites with >98% nonflagellated forms, obtained from all five cultures 48 h after shifting to amastigote growth conditions, were assayed for their ability to infect and replicate inside BMMs (Fig. 6C). The number of intracellular parasites determined 1 h after infection showed no significant variation, suggesting that all tested lines were equally capable of invading BMMs. However, similar to the mouse infection results, APX/ΔAPX and APX/ΔAPX+mutAPX-HA parasites failed to replicate inside macrophages, as evident from the unchanged intracellular parasite counts between 24 and 72 h. Wild-type (WT), APX overexpression (APX/APX+APX-HA), and APX complemented (APX/ΔAPX+APX-HA) lines, on the other hand, grew normally. Together, our data suggest that even though H2O2 accumulation facilitated by low APX levels promotes promastigote to amastigote differentiation, expression of APX above a certain critical level is required for the subsequent replication of intracellular amastigotes.
DISCUSSION
In this study, we characterized the role of APX in Leishmania amazonensis. Protection against oxidative stress by APX overexpression was previously demonstrated in multiple Leishmania strains (43–45), and a role for APX activity in promoting cell differentiation and virulence was reported in L. major (34). However, the L. major studies revealed that APX function is dispensable for the promastigote stage of that species, as APX-null mutant promastigotes cultured in vitro showed little or no growth defects. In contrast, we were only able to generate L. amazonensis APX-null mutants in the presence of an episome driving APX expression, indicating that APX is an essential gene for the survival of L. amazonensis promastigotes. This species-specific variation in APX dependence adds to the evidence that these two cutaneous disease-associated Leishmania species differ in their mechanisms for development of infectivity and pathogenesis (6). While L. amazonensis promastigotes can be reproducibly differentiated into amastigotes by shifting cultures to low-pH/high-temperature conditions or by direct exposure to ROS (14–16, 36), axenic differentiation of L. major is still extremely challenging.
Previously we showed that H2O2-mediated signals act in two distinct steps to promote L. amazonensis infectivity (15). Initially it promotes development of infective metacyclic promastigotes, and subsequently it also triggers differentiation of promastigotes into amastigotes capable of replicating within PVs of host macrophages. Our focus in this study was to understand the role of APX in regulating these two important virulence development steps. To overcome our inability to study APX function in a null genetic background, we generated multiple recombinant L. amazonensis lines expressing different levels of the protein and were able to establish a correlation between levels of APX expression and the ability of the parasites to differentiate into infective forms. We were unable to directly quantify mitochondrial H2O2 content in these cell lines, primarily due to the lack of reliable commercially available reagents. Even though Amplex-Red is highly specific for H2O2, it is poorly permeable for membranes, so is mainly used to detect extracellular H2O2. Using the membrane-permeant fluorescent probe 2′,7′-dichlorodihydrofluorescein ester, we could show the predicted increase in total ROS in APX single-knockout lines, but since this probe reacts with multiple ROS (46), it is not suitable for specifically detecting mitochondrion-generated H2O2.
APX expression varied during different stages of the parasite’s life cycle, with the highest protein expression observed in the infective forms, metacyclic promastigotes and axenic amastigotes. While lower APX levels facilitated maturation of metacyclic promastigotes and provided an early advantage during macrophage infections, similar to what was reported for L. major (34), we found that inability to upregulate APX expression above a critical level negatively impacts the parasite’s ability to survive intracellularly in host macrophages. Further evidence for such a stringent APX requirement in L. amazonensis amastigotes was obtained from pH/temperature shift axenic differentiation experiments, where promastigotes expressing low levels of APX (the APX/ΔAPX and APX/ΔAPX +mutAPX-HA cell lines) efficiently differentiated into amastigotes but failed to establish productive infections in BMMs and were unable to cause lesions when injected into mice. Thus, the lack of virulence observed in APX-deficient L. amazonensis is in stark contrast with what was reported for L. major, where purified APX-null metacyclic promastigotes showed increased infectivity for cultured BMMs and in BALB/c mice (34). The increased overall susceptibility of BALB/c mice to Leishmania infections (47) does not allow direct comparisons with the lesion sizes we observed in C57BL/6 mice, but the more than 2-fold larger lesion size produced by L. major APX-null cells compared to the wild-type or complemented lines after 8 weeks of infection indicates increased virulence resulting from loss of APX. While findings in L. major APX-null cells suggested that other antioxidants may partially compensate for elimination of the peroxide-scavenging gene APX in this species (34), we found that APX is absolutely required for the survival of all L. amazonensis life cycle stages and for infection of mammalian hosts. Our results are consistent with earlier observations that H2O2-mediated signaling promotes L. amazonensis infectivity (14–16) and indicate that APX-mediated H2O2 breakdown is particularly critical for the intracellular stages of L. amazonensis, highlighting a novel functional role for this antioxidant enzyme in regulating amastigote differentiation and replication.
APX is the only heme-containing peroxidase identified to date in Leishmania. It shares 36% sequence identity with plant APX and is a unique hybrid of cytochrome c and ascorbate peroxidase-like proteins. The enzyme contains a 22-amino-acid transmembrane domain at its N terminus and is extremely sensitive to H2O2-mediated inactivation, when the availability of ascorbate as an electron donor is limited. Removal of the N-terminal transmembrane domain enhanced solubility and resistance to H2O2, suggesting that posttranslational processing might play a role in regulating APX activity (33). Our observation of two distinct APX isoforms tagged with HA at the C terminus suggests that N-terminal processing of APX may be involved in regulating enzymatic activity in L. amazonensis, an interesting possibility to be addressed in future studies.
MATERIALS AND METHODS
Leishmania culture.
In vitro cultures of L. amazonensis (IFLA/BR/67/PH8) promastigote forms were maintained at 26°C in M199 medium (pH 7.4) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 0.1% hemin (Frontier Scientific; 25 mg/ml in 50% triethanolamine), 10 mM adenine (pH 7.5), 5 mM l-glutamine, and 5% penicillin-streptomycin (36). Axenic differentiation into amastigote forms was induced mixing promastigote cultures (∼2 × 107 to 4 × 107/ml) with equal volumes of acidic amastigote medium (M199 containing 0.25% glucose, 0.5% Trypticase, and 40 mM sodium succinate, pH 4.5) and further incubating at 32°C. Differentiated amastigotes were maintained in amastigote medium at 32°C. Parasite viability was determined by fluorescence microscopy after staining with fluorescein diacetate (FDA; Sigma-Aldrich) a membrane-permeable molecule that develops green fluorescence after being cleaved intracellularly in metabolically active cells (48). Viable cells were counted microscopically using a 40× (0.75) objective in a Nikon Eclipse 200 microscope.
The parasite’s ability to differentiate was quantitated as the percentage of undifferentiated promastigotes with long flagella versus differentiated parasites with short flagella and rounded shape via phase-contrast microscopy. At least 200 viable cells were scored per sample.
Parasite viability estimation following ROS treatment.
Sensitivity to ROS was determined by seeding promastigotes in log-phase culture (∼2 × 107/ml) at 4 × 105/ml with or without increasing concentrations of H2O2 or menadione. Water and dimethyl sulfoxide (DMSO) were used to dilute H2O2 and menadione, respectively, and were as used as treatment controls. Following 24 h of incubation at 26°C, parasites were stained with FDA and cell viability was quantified with a hemocytometer.
Generation of L. amazonensis recombinant cell lines expressing different APX levels.
The L. amazonensis APX open reading frame (ORF) was targeted for homologous recombination mediated replacement with constructs containing the hygromycin (HYG) or phleomycin (PHLEO) resistance gene through homologous recombination as described earlier (16, 49). Upstream and downstream sequences of the APX ORF were cloned using the following primers that contained sequences for the SfiI restriction enzyme (underlined): LamAPX 5′SfiI-A:FD (GAGGCCACCTAGGCCGTGGGCGGTGG) and LamAPX 5′SfiI B:RV (GAGGCCACGCAGGCTGAACCCGAG) to amplify 5′ UTR sequence and LamAPX 3′SfiI-C:FD (GAGGCCTCTGTGGCCTCTTTGCGTGCC) and LamAPX 3′SfiI-D:RV (GAGGCCTGACTGGCCCTGCGCGATGC) for 3′ UTR. Four-part ligation was performed using the PCR-amplified 5′ and 3′ flanking sequences, drug resistance cassettes and the plasmid backbone and positive clones were identified by analyzing SfiI restriction digests of plasmid DNA samples, and confirmed by sequencing with specific primers as described previously (49). The APX gene-targeting fragment was released by PacI digestion, gel purified and used to transfect L. amazonensis promastigotes by electroporation. Clones with a single APX allele deletion (APX/ΔAPX) were selected on agar plates containing hygromycin (100 μg/ml) or neomycin (50 μg/ml) and analyzed by PCR to verify integration of the drug cassette in the desired location using primers FD5′APX-INT (TCTCCCCGCCCCTTCTGAC) and RV5′APX-INT (AACGATTGAAGTACGCAAGCACG) for confirming 5′ end integration and FD3′APX-INT (AACAGAGCGACACTGCGGGC) and RV3′APX-INT (CTTTCAAGGCTTCCCGAACGCACA) for 3′ end integration.
The rescue plasmid expressing the APX gene tagged with hemagglutinin (HA) at the C terminus was generated as described before (16) with amplification carried out in two steps. Initially, the 912-bp APX ORF was amplified with primers FD-APX HA (AACCCGGGACATATGTTCGGCACCTCG [SmaI site underlined]) and RV-APX HA (CTGGGACGTCGTATGGGTAAAGCTTGCTCCCC), which removed the endogenous stop codon and introduced an in-frame HA tag. The PCR product was reamplified in a second round using the FD-SODA-HA as forward and RV:HA TAG2 (TTGGATCCTTAAGCGTAGTCTGGGACGTCGTATGG [BamHI site underlined]) as reverse primers. The final amplified gene product was cloned into the BamHI and SmaI sites of Leishmania expression plasmid pXG-SAT (courtesy of S. Beverley, Washington University) to generate the plasmid pXG-APX-SAT. APX-HA-expressing Leishmania clones were selected in plates containing 50 μg/ml nourseothricin (Jena Biosciences) and identified via Western blot.
For episomal expression of catalytically dead APX protein, the critically conserved amino acid residues Trp67 and His68 were converted to alanines according to the manufacturer’s protocol using the QuikChange II site-directed mutagenesis kit (Agilent) and mutation-specific primers Sense:LamAPXmut (GAGCCGGCCTCGGCCGCTGCTAGGCGAATCAGCGAAGGT) and Antisense:LamAPXmut (ACCTTCGCTGATTCGCCTAGCAGCGGCCGAGGCCGGCTC) designed using Agilent’s online design tool (https://www.genomics.agilent.com/primerDesignProgram.jsp). Plasmids were sequenced to confirm intended mutations before transfection into L. amazonensis.
Expression of recombinant APX protein and generation of antibodies.
To produce recombinant histidine (6×His)-tagged APX proteins, the APX-encoding gene was PCR amplified using L. amazonensis genomic DNA as the template with primers FD:APXexp (CCACACATGTTCGGCACCTCGCGG) and RV:APXexp (TTCAAAGCTTGCTCCCCGACGCGG). The forward primer contained a PciI site (underlined) contiguous with the start codon, and the reverse primer was engineered to include a HindIII restriction site (underlined) to remove the endogenous stop codon and allow for contiguous synthesis of a 6×-His tag. The amplified PCR product was cloned in the pET28b(+) expression plasmid (Novagen) using NcoI and HindIII restriction sites and used to transform Escherichia coli BL21(DE3)(pLysS) (Novagen). The 6×His-tagged APX protein was expressed in soluble form following induction with 0.1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) (overnight at room temperature in medium supplemented with 2% ethanol) and purified on a nickel column with His60 Ni Superflow resin (Clontech), according to the manufacturer’s protocol. Homogeneity of the purified proteins was confirmed by SDS-PAGE.
Polyclonal antibodies against APX was raised by periodic injection of rabbits with purified protein samples (Pocono Rabbit Farm and Laboratory). Specificity of the antisera was assessed by Western blot.
Immunolocalization of APX.
Localization of APX by immunofluorescence microscopy was performed as described previously (16). To confirm the mitochondrial localization of APX, promastigotes were first incubated with MitoTracker Red CMXRos (Invitrogen) followed by fixation with 4% paraformaldehyde (PFA) and attachment to poly-l-lysine-coated slides (multitest 8-well; MP Biomedicals). After treatment with 50 mM NH4Cl, the cells were permeabilized with 0.1% Triton in phosphate-buffered saline (PBS), blocked with PBS–5% horse serum and 1% bovine serum albumin (BSA) for 1 h at room temperature and incubated with anti-APX rabbit polyclonal antibodies (1:10,000 dilution in PBS–1% BSA) for 1 h followed by anti-rabbit IgG Alexa Fluor 488 (Thermo Fisher) 1:5,000 dilution in PBS–1% BSA for 1 h and staining with 2 μg/ml DAPI (4′,6-diamidino-2-phenylindole) for 1 h. Slides were mounted with ProLong Gold antifade reagent (Thermo Fisher), images were acquired through a Deltavision Elite deconvolution microscope (GE Healthcare) and processed using Volocity Suite (PerkinElmer). Colocalization of APX staining (green channel) and MitoTracker Red (red channel) were analyzed for each cell by performing Costes’ Pearson’s correlation calculations using the Volocity Suite.
Determination of total intracellular ROS content.
The total ROS content of L. amazonensis promastigotes was measured using a chloromethyl derivative of 2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA) (Thermo Fisher), a cell-permeable probe for ROS detection (50). Promastigotes (5 × 107) were collected, washed once with PBS, resuspended in M199, and then incubated with 50 μM CM-H2DCFDA for 15 min at 27°C in the dark. Fluorescence was determined using excitation and emission wavelengths of 480 nm and 520 nm, respectively, and expressed as fluorescence intensity (arbitrary units).
Quantification of Leishmania intracellular growth in macrophages.
A total of 1 × 105 BMMs from C57/BL6 mice (Charles River Laboratories) prepared as previously described were plated on glass coverslips in 3-cm dishes 24 h prior to experiments. Infective metacyclic forms were purified from stationary promastigote cultures (7 days old) using the m3A.1b monoclonal antibody as described earlier (42). Attached BMMs were washed with fresh RPMI 1640 and infected at 1:5 multiplicity of infection (MOI) with purified metacyclics, or at 1:1 MOI with axenically transformed amastigotes in RPMI supplemented with 10% FBS. Following initial incubation to allow for invasion (1 h for amastigotes and 3 h for metacyclics) macrophages were washed three times in PBS and further incubated at 34°C for the indicated times. Coverslips were fixed in 4% PFA after 1 or 3 h of incubation for baseline infection, followed by 24, 48, and 72 h of incubation, permeabilized with 0.1% Triton X-100 for 10 min, and stained with 10 μg/ml DAPI for 1 h. Intracellular parasite numbers were quantified by scoring the total number of macrophages and the total number of intracellular parasites per microscopic field (100× NA 1.3 oil immersion objective; Nikon E200 epifluorescence microscope), and the results were expressed as intracellular parasites per 100 macrophages. At least 300 host cells, in triplicate, were analyzed for each time point.
In vivo virulence and parasite load estimation.
Six-week-old female C57BL/6 mice (n = 5 per group) were inoculated in the left hind footpad with 1 × 106 purified metacyclics (36) from WT, APX/ΔAPX, APX/APX+APX-HA, APX/ΔAPX+APX-HA, and APX/ΔAPX+mutAPX-HA parasites from stationary-phase cultures in a volume of 50 μl PBS. Footpads were measured weekly with a caliper (Mitutoyo Corp., Japan), and lesion progression was expressed as the difference of measurements between the left and right hind footpads. Parasite load was estimated in infected tissue collected from footpads of sacrificed mice 8 weeks postinfection using a limiting dilution assay (51). The Institute of Animal Care and Use Committee at the University of Maryland approved the animal study protocol.
Determination of gene expression by qPCR.
A total of 5 × 107 cells per biological replicate were used to extract total cellular RNA using the RNeasy kit (Qiagen) as per the manufacturer’s protocol. Equal amounts of RNA from each biological set were used to synthesize cDNA (Bio-Rad iScript), The cDNA samples were diluted to 1-ng/μl concentrations and amplified with gene-specific primers FD-PXN (TTGCTCGTGACTATGGTGTG) and RV-PXN (CCGCCTTTGTGGTGTCCAG), FD-TRYP5 (GTGGGTCGTGCTCTTCTTCTAC) and RV-TRYP5 (GCTCTTGGTCTTGTCGGCTA), and FD-nsGPX (TGGCATCCATCTTCACCT) and RV-ns-GPX (ACCTCGTTCAGCATCTCG) using the SYBR green master mix as described earlier (gene expression was normalized to ubiquitin hydrolase [14], which is known to be expressed constitutively).
Statistical analysis.
Statistical analysis and IC50 determinations were performed using GraphPad Prism 5 (GraphPad Software, Inc., San Diego, CA, USA). Data are presented as the mean ± of standard deviation (SD) of determinations in biological replicates. Comparisons between two groups were made using the Student's t test. One-way analysis of variance (ANOVA) followed by Dunnett’s post hoc test were performed to compare multiple experimental groups using one group as control. P < 0.05 was taken as evidence of statistical significance.
Supplementary Material
ACKNOWLEDGMENTS
We thank P. Yates (Oregon Health & Science University) and S. M. Beverley (Washington University) for their generous gifts of plasmids. L. amazonensis (IFLA/BR/67/PH8) was a kind gift from David Sacks (Laboratory of Parasitic Diseases, NIAID, NIH). We are also thankful to Amrita Sarkar for assistance with BMM infection assays.
This work was supported by National Institutes of Health grant R01 AI067979 to N.W.A.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00193-19.
REFERENCES
- 1.WHO. 2010. Control of the leishmaniases. World Health Organization Technical Report Series. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.Antoine JC, Prina E, Lang T, Courret N. 1998. The biogenesis and properties of the parasitophorous vacuoles that harbour Leishmania in murine macrophages. Trends Microbiol 6:392–401. doi: 10.1016/S0966-842X(98)01324-9. [DOI] [PubMed] [Google Scholar]
- 3.Veras PS, de Chastellier C, Rabinovitch M. 1992. Transfer of zymosan (yeast cell walls) to the parasitophorous vacuoles of macrophages infected with Leishmania amazonensis. J Exp Med 176:639–646. doi: 10.1084/jem.176.3.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gomes IN, Calabrich AF, Tavares RS, Wietzerbin J, de Freitas LA, Veras PS. 2003. Differential properties of CBA/J mononuclear phagocytes recovered from an inflammatory site and probed with two different species of Leishmania. Microbes Infect 5:251–260. doi: 10.1016/S1286-4579(03)00025-X. [DOI] [PubMed] [Google Scholar]
- 5.Sacks D, Sher A. 2002. Evasion of innate immunity by parasitic protozoa. Nat Immunol 3:1041–1047. doi: 10.1038/ni1102-1041. [DOI] [PubMed] [Google Scholar]
- 6.Soong L. 2012. Subversion and utilization of host innate defense by Leishmania amazonensis. Front Immunol 3:58. doi: 10.3389/fimmu.2012.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bates PA. 2007. Transmission of Leishmania metacyclic promastigotes by phlebotomine sand flies. Int J Parasitol 37:1097–1106. doi: 10.1016/j.ijpara.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palmeri A, Gherardini PF, Tsigankov P, Ausiello G, Spath GF, Zilberstein D, Helmer-Citterich M. 2011. PhosTryp: a phosphorylation site predictor specific for parasitic protozoa of the family trypanosomatidae. BMC Genomics 12:614. doi: 10.1186/1471-2164-12-614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosenzweig D, Smith D, Opperdoes F, Stern S, Olafson RW, Zilberstein D. 2008. Retooling Leishmania metabolism: from sand fly gut to human macrophage. FASEB J 22:590–602. doi: 10.1096/fj.07-9254com. [DOI] [PubMed] [Google Scholar]
- 10.Rosenzweig D, Smith D, Myler PJ, Olafson RW, Zilberstein D. 2008. Post-translational modification of cellular proteins during Leishmania donovani differentiation. Proteomics 8:1843–1850. doi: 10.1002/pmic.200701043. [DOI] [PubMed] [Google Scholar]
- 11.Saar Y, Ransford A, Waldman E, Mazareb S, Amin-Spector S, Plumblee J, Turco SJ, Zilberstein D. 1998. Characterization of developmentally-regulated activities in axenic amastigotes of Leishmania donovani. Mol Biochem Parasitol 95:9–20. doi: 10.1016/s0166-6851(98)00062-0. [DOI] [PubMed] [Google Scholar]
- 12.Saxena A, Lahav T, Holland N, Aggarwal G, Anupama A, Huang Y, Volpin H, Myler PJ, Zilberstein D. 2007. Analysis of the Leishmania donovani transcriptome reveals an ordered progression of transient and permanent changes in gene expression during differentiation. Mol Biochem Parasitol 152:53–65. doi: 10.1016/j.molbiopara.2006.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsigankov P, Gherardini PF, Helmer-Citterich M, Zilberstein D. 2012. What has proteomics taught us about Leishmania development? Parasitology 139:1146–1157. doi: 10.1017/S0031182012000157. [DOI] [PubMed] [Google Scholar]
- 14.Mittra B, Cortez M, Haydock A, Ramasamy G, Myler PJ, Andrews NW. 2013. Iron uptake controls the generation of Leishmania infective forms through regulation of ROS levels. J Exp Med 210:401–416. doi: 10.1084/jem.20121368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mittra B, Laranjeira-Silva MF, Miguel DC, Perrone Bezerra de Menezes J, Andrews NW. 2017. The iron-dependent mitochondrial superoxide dismutase SODA promotes Leishmania virulence. J Biol Chem 292:12324–12338. doi: 10.1074/jbc.M116.772624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mittra B, Laranjeira-Silva MF, Perrone Bezerra de Menezes J, Jensen J, Michailowsky V, Andrews NW. 2016. A trypanosomatid iron transporter that regulates mitochondrial function is required for Leishmania amazonensis virulence. PLoS Pathog 12:e1005340. doi: 10.1371/journal.ppat.1005340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Long LH, Evans PJ, Halliwell B. 1999. Hydrogen peroxide in human urine: implications for antioxidant defense and redox regulation. Biochem Biophys Res Commun 262:605–609. doi: 10.1006/bbrc.1999.1263. [DOI] [PubMed] [Google Scholar]
- 18.Cuypers A, Hendrix S, Amaral Dos Reis R, De Smet S, Deckers J, Gielen H, Jozefczak M, Loix C, Vercampt H, Vangronsveld J, Keunen E. 2016. Hydrogen peroxide, signaling in disguise during metal phytotoxicity. Front Plant Sci 7:470. doi: 10.3389/fpls.2016.00470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sies H. 2017. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: oxidative eustress. Redox Biol 11:613–619. doi: 10.1016/j.redox.2016.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marinho HS, Real C, Cyrne L, Soares H, Antunes F. 2014. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol 2:535–562. doi: 10.1016/j.redox.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lennicke C, Rahn J, Lichtenfels R, Wessjohann LA, Seliger B. 2015. Hydrogen peroxide—production, fate and role in redox signaling of tumor cells. Cell Commun Signal 13:39. doi: 10.1186/s12964-015-0118-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sies H. 2014. Role of metabolic H2O2 generation: redox signaling and oxidative stress. J Biol Chem 289:8735–8741. doi: 10.1074/jbc.R113.544635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Finkel T. 2012. Signal transduction by mitochondrial oxidants. J Biol Chem 287:4434–4440. doi: 10.1074/jbc.R111.271999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reczek CR, Chandel NS. 2015. ROS-dependent signal transduction. Curr Opin Cell Biol 33:8–13. doi: 10.1016/j.ceb.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hamanaka RB, Chandel NS. 2010. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci 35:505–513. doi: 10.1016/j.tibs.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khacho M, Slack RS. 2017. Mitochondrial and reactive oxygen species signaling coordinate stem cell fate decisions and life long maintenance. Antioxid Redox Signal doi: 10.1089/ars.2017.7228. [DOI] [PubMed] [Google Scholar]
- 27.Lee S, Tak E, Lee J, Rashid MA, Murphy MP, Ha J, Kim SS. 2011. Mitochondrial H2O2 generated from electron transport chain complex I stimulates muscle differentiation. Cell Res 21:817–834. doi: 10.1038/cr.2011.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsukagoshi H, Busch W, Benfey PN. 2010. Transcriptional regulation of ROS controls transition from proliferation to differentiation in the root. Cell 143:606–616. doi: 10.1016/j.cell.2010.10.020. [DOI] [PubMed] [Google Scholar]
- 29.Chandel NS. 2015. Evolution of mitochondria as signaling organelles. Cell Metab 22:204–206. doi: 10.1016/j.cmet.2015.05.013. [DOI] [PubMed] [Google Scholar]
- 30.Kong H, Chandel NS. 2018. Regulation of redox balance in cancer and T cells. J Biol Chem 293:7499–7507. doi: 10.1074/jbc.TM117.000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Antunes F, Brito PM. 2017. Quantitative biology of hydrogen peroxide signaling. Redox Biol 13:1–7. doi: 10.1016/j.redox.2017.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Castro H, Tomas AM. 2008. Peroxidases of trypanosomatids. Antioxid Redox Signal 10:1593–1606. doi: 10.1089/ars.2008.2050. [DOI] [PubMed] [Google Scholar]
- 33.Adak S, Datta AK. 2005. Leishmania major encodes an unusual peroxidase that is a close homologue of plant ascorbate peroxidase: a novel role of the transmembrane domain. Biochem J 390:465–474. doi: 10.1042/BJ20050311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pal S, Dolai S, Yadav RK, Adak S. 2010. Ascorbate peroxidase from Leishmania major controls the virulence of infective stage of promastigotes by regulating oxidative stress. PLoS One 5:e11271. doi: 10.1371/journal.pone.0011271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adak S, Pal S. 2013. Ascorbate peroxidase acts as a novel determiner of redox homeostasis in Leishmania. Antioxid Redox Signal 19:746–754. doi: 10.1089/ars.2012.4745. [DOI] [PubMed] [Google Scholar]
- 36.Huynh C, Sacks DL, Andrews NW. 2006. A Leishmania amazonensis ZIP family iron transporter is essential for parasite replication within macrophage phagolysosomes. J Exp Med 203:2363–2375. doi: 10.1084/jem.20060559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mukherjee A, Langston LD, Ouellette M. 2011. Intrachromosomal tandem duplication and repeat expansion during attempts to inactivate the subtelomeric essential gene GSH1 in Leishmania. Nucleic Acids Res 39:7499–7511. doi: 10.1093/nar/gkr494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mukherjee A, Roy G, Guimond C, Ouellette M. 2009. The gamma-glutamylcysteine synthetase gene of Leishmania is essential and involved in response to oxidants. Mol Microbiol 74:914–927. doi: 10.1111/j.1365-2958.2009.06907.x. [DOI] [PubMed] [Google Scholar]
- 39.Chawla B, Jhingran A, Singh S, Tyagi N, Park MH, Srinivasan N, Roberts SC, Madhubala R. 2010. Identification and characterization of a novel deoxyhypusine synthase in Leishmania donovani. J Biol Chem 285:453–463. doi: 10.1074/jbc.M109.048850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wilson ME, Andersen KA, Britigan BE. 1994. Response of Leishmania chagasi promastigotes to oxidant stress. Infect Immun 62:5133–5141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zarley JH, Britigan BE, Wilson ME. 1991. Hydrogen peroxide-mediated toxicity for Leishmania donovani chagasi promastigotes. Role of hydroxyl radical and protection by heat shock. J Clin Invest 88:1511–1521. doi: 10.1172/JCI115461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pinto-da-Silva LH, Fampa P, Soares DC, Oliveira SM, Souto-Padron T, Saraiva EM. 2005. The 3A1-La monoclonal antibody reveals key features of Leishmania (L) amazonensis metacyclic promastigotes and inhibits procyclics attachment to the sand fly midgut. Int J Parasitol 35:757–764. doi: 10.1016/j.ijpara.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 43.Mukherjee A, Boisvert S, Monte-Neto RL, Coelho AC, Raymond F, Mukhopadhyay R, Corbeil J, Ouellette M. 2013. Telomeric gene deletion and intrachromosomal amplification in antimony-resistant Leishmania. Mol Microbiol 88:189–202. doi: 10.1111/mmi.12178. [DOI] [PubMed] [Google Scholar]
- 44.Kumar A, Das S, Purkait B, Sardar AH, Ghosh AK, Dikhit MR, Abhishek K, Das P. 2014. Ascorbate peroxidase, a key molecule regulating amphotericin B resistance in clinical isolates of Leishmania donovani. Antimicrob Agents Chemother 58:6172–6184. doi: 10.1128/AAC.02834-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moreira DS, Xavier MV, Murta S. 2018. Ascorbate peroxidase overexpression protects Leishmania braziliensis against trivalent antimony effects. Mem Inst Oswaldo Cruz 113:e180377. doi: 10.1590/0074-02760180377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dikalov SI, Harrison DG. 2014. Methods for detection of mitochondrial and cellular reactive oxygen species. Antioxid Redox Signal 20:372–382. doi: 10.1089/ars.2012.4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Loeuillet C, Banuls AL, Hide M. 2016. Study of Leishmania pathogenesis in mice: experimental considerations. Parasit Vectors 9:144. doi: 10.1186/s13071-016-1413-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sacks DL, Melby PC. 2001. Animal models for the analysis of immune responses to leishmaniasis. Curr Protoc Immunol Chapter 19:Unit 19.2. doi: 10.1002/0471142735.im1902s28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fulwiler AL, Soysa DR, Ullman B, Yates PA. 2011. A rapid, efficient and economical method for generating leishmanial gene targeting constructs. Mol Biochem Parasitol 175:209–212. doi: 10.1016/j.molbiopara.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mukherjee SB, Das M, Sudhandiran G, Shaha C. 2002. Increase in cytosolic Ca2+ levels through the activation of non-selective cation channels induced by oxidative stress causes mitochondrial depolarization leading to apoptosis-like death in Leishmania donovani promastigotes. J Biol Chem 277:24717–24727. doi: 10.1074/jbc.M201961200. [DOI] [PubMed] [Google Scholar]
- 51.Lima HC, Bleyenberg JA, Titus RG. 1997. A simple method for quantifying Leishmania in tissues of infected animals. Parasitol Today 13:80–82. doi: 10.1016/s0169-4758(96)40010-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.