

Abstract
Predicting the risk of liver metastasis can have important prognostic and therapeutic implications, given the availability of liver-directed therapy. Uveal melanoma has a striking predisposition for liver metastasis despite the absence of anatomical proximity. Understanding its biology may uncover factors promoting liver metastasis in other malignancies. We quantified gene expression by RNAseq in 76 uveal melanomas and combined with public data in a meta-analysis of 196 patients. The meta-analysis of uveal melanoma gene expression identified 63 genes which remained prognostic after adjustment for chromosome 3 status. Two genes, PTP4A3 and JPH1, were selected by L1-penalized regression and combined in a prognostic score. The score predicted liver-specific relapse in a public pan-cancer dataset and in two public colorectal cancer datasets. The score varied between colorectal consensus molecular subtypes (CMS), as did the risk of liver relapse, which was lowest in CMS1. Additional prospective validation was done by real-time PCR in 463 breast cancer patients. The score was significantly correlated with liver relapse in hormone receptor positive tumors. In conclusion, the expression of PTP4A3 and JPH1 correlates with risk of liver metastasis in colorectal cancer and breast cancer. The underlying biological mechanism is an interesting area for further research.
Subject terms: Metastasis, Tumour biomarkers, Cancer genomics, Tumour biomarkers
Introduction
Almost all patients with metastatic uveal melanoma present liver metastases1, making this rare disease an ideal model for the identification of biological features predisposing to liver metastasis. Our objective was to use the gene expression profile of uveal melanoma to identify genes that would predict the risk of liver metastasis in epithelial malignancies, especially colorectal cancer. Surgical resection of liver metastases from colorectal cancer can be performed with curative intent and is recognized as standard of care under certain conditions2,3. The optimal management of oligometastatic liver spread from other tumor types is not as clearly defined but curative treatment can be contemplated on a case-by-case basis. In addition to surgery, less invasive modalities such as radiofrequency ablation4, hepatic arterial infusion chemotherapy5 or local immunotherapy (trials NCT02509507 and NCT03256344) are being explored for patients with metastases limited to the liver. In conclusion, the identification of patients at high risk of liver metastasis could justify more aggressive treatment up-front (for example, adjuvant chemotherapy) and more intensive liver-specific follow-up with the intention to detect and treat liver metastases as early as possible with an appropriate treatment modality.
Several prognostic features have been identified in uveal melanoma and are associated with the development of distal metastasis, typically in the liver. Monosomy of chromosome 3 often co-occurs with mutation of the BAP1 gene in 3p21.1, results in bi-allelic inactivation and is a hallmark of uveal melanoma with unfavorable prognosis6. However, the BAP1 mutation is rare in other cancer types (less than 5%), with the exception of kidney cancer7, mesothelioma8 and cholangiocarcinoma9. Gain of chromosome arm 8q is also associated with unfavorable prognosis in uveal melanoma10–12, as is overexpression of PTP4A3, which resides in this arm, specifically in 8q24.313. In contrast with the BAP1 mutation, gain of 8q is a frequent event in many cancer types, including colorectal14, gastric15 and breast cancer16.
A previous gene expression study has subdivided uveal melanoma in two distinct molecular classes with prognostic implications, as class 1 tumors are associated with a favorable prognosis17. A recent integrative study of uveal melanoma has further characterized four distinct classes, primarily based on chromosome 3 and chromosome 8 copy number alterations and secondarily based on gene alterations (EIF1AX, SF3B1), gene expression and methylation patterns18.
Unfortunately, many gene expression studies of uveal melanoma are small, limited by the rarity of the disease. Here, in addition to an analysis of 76 primary uveal melanoma samples using RNAseq, we present a meta-analysis of 196 patients with uveal melanoma which served as a training set for the discovery of gene expression patterns favoring liver metastasis in other tumor types.
Results
We pooled three gene expression studies into a training dataset with 196 uveal melanoma patients and 13560 genes. For each gene, we fitted a univariable Cox survival model of relapse-free survival against expression separately in each dataset and computed a meta-analysis summary. This approach identified 656 genes negatively associated and 810 genes positively associated with relapse (FDR < 0.05). The Q-test for heterogeneity and visual inspection of forest plots did not show discrepancies between datasets. The top genes, ordered by FDR, are shown in Table 1 (detailed results in Table S1).
Table 1.
Genes associated with relapse in uveal melanoma.
| Gene | HR | 95% CI | FDR | Location |
|---|---|---|---|---|
| PTP4A3 | 2.54 | (2.01 to 3.20) | <0.001 | 8q24.3 |
| JPH1 | 2.34 | (1.87 to 2.94) | <0.001 | 8q21.11 |
| ID2 | 0.48 | (0.39 to 0.6) | <0.001 | 2p25.1 |
| ANG | 0.42 | (0.33 to 0.55) | <0.001 | 14q11.2 |
| PRKDC | 1.97 | (1.61 to 2.41) | <0.001 | 8q11.21 |
| POP1 | 1.91 | (1.57 to 2.33) | <0.001 | 8q22.2 |
| RAB11FIP1 | 0.52 | (0.42 to 0.63) | <0.001 | 8p11.23 |
| CDC25B | 2.06 | (1.65 to 2.57) | <0.001 | 20p13 |
| CHAC1 | 1.97 | (1.59 to 2.43) | <0.001 | 15q15.1 |
| LPIN1 | 0.51 | (0.42 to 0.64) | <0.001 | 2p25.1 |
| CHD7 | 1.97 | (1.59 to 2.45) | <0.001 | 8q12.2 |
| VCPIP1 | 1.99 | (1.60 to 2.48) | <0.001 | 8q13.1 |
| ROPN1B | 0.50 | (0.40 to 0.62) | <0.001 | 3q21.2 |
| MTFR1 | 1.86 | (1.52 to 2.28) | <0.001 | 8q13.1 |
| MCM4 | 1.81 | (1.49 to 2.19) | <0.001 | 8q11.21 |
| COL9A3 | 1.88 | (1.53 to 2.31) | <0.001 | 20q13.33 |
| ASAP1 | 2.02 | (1.61 to 2.55) | <0.001 | 8q24.22 |
| SPIRE1 | 2.02 | (1.60 to 2.55) | <0.001 | 18p11.21 |
| TLCD1 | 1.83 | (1.50 to 2.24) | <0.001 | 17q11.2 |
| RAB2A | 1.80 | (1.48 to 2.18) | <0.001 | 8q12.1 |
Top 20 genes with lowest FDR shown (full results in the supplement) in a meta-analysis of univariable survival models against the expression of each individual gene.
A recent study of uveal melanoma19 identified 12 genes that have prognostic value. Eleven of these genes were available in the training dataset and for nine of them we confirmed the expected statistically significant positive (EIF1B, ID2, LMCD1, MTUS1, ROBO1, SATB1) respectively negative (ECM1, HTR2B, RAB31) association with relapse-free survival (all FDR < 0.05) in our meta-analysis. High expression of BAP1, suggestive of chromosome 3 disomy, was associated with a low risk of relapse (HR = 0.64, 95% 0.54–0.78, FDR < 0.001).
Many of the statistically significant genes in our meta-analysis resided either on chromosome 3 (N = 185) or on chromosome 8 (N = 183), together accounting for one fourth of all significant genes (25.1%, 369/1466). The log hazard ratio (HR) of genes from 3p and 3q was almost always negative, therefore higher expression, presumably in patients with chromosome 3 disomy, predicted longer relapse-free survival (mean log HR estimate −0.020, 95% −0.029 to −0.011, t-test P < 0.0001). The same was observed for genes from 6p (log HR −0.018, 95% −0.0305 to −0.006, P = 0.0033) and 6q (log HR −0.101, 95% −0.115 to −0.087, P < 0.0001). Finally, high expression of genes from 8q was associated with shorter relapse-free survival (P < 0.0001) while the inverse was observed for genes from 8p (P < 0.0001).
The training data were pooled by computing standardized gene expression values (mean-centered and scaled to a variance of one). In Fig. 1, a heatmap of prognostic genes (FDR < 0.05) delineates four major gene clusters (I-IV). Gene expression within these clusters is roughly associated with molecular class, chromosome 3 status and relapse. The heatmap and tumor similarity tree also suggest the presence of three patient groups (a, b and c). All tumors from the top tier of the tree (subtree a) belong to class 1 (72 of 72 = 100%), shown in grey, and demonstrate rare relapses, shown in black. Inversely, the tumors from the high-risk group (subtree c) almost always belong to class 1 (74 of 80 = 92.5%)17, which is shown in black. In the middle, we see a group (subtree b) that is associated with intermediate risk of relapse (Fig. S1). This group is probably not adequately captured by the traditional two classes and appears to be biologically distinct. Tumors from the intermediate and high-risk groups mostly seem to differ on genes from cluster I and III, such as CHD7, JPH1, ASAP1, POP1 and PRKD, which are highly inter-correlated (Pearson’s correlation P < 0.0001 for all pairs). An additional heatmap of all available genes can be found in the supplement (Fig. S2).
Figure 1.

Gene expression heatmap. Hierarchical clustering with Pearson’s correlation similarity and average linkage. Heatmap showing the 1466 genes associated with prognosis in the meta-analysis of Cox models (FDR < 0.05). We perceive four main clusters (I-IV). In addition to favorable (a) and unfavorable (c) patient groups, there seems to be an intermediate group (b), characterized by a distinct pattern of low expression in cluster I and III and high expression in clusters II and IV. Chromosome 3 loss, class 2 tumors and relapse before 3 years of follow-up are shown in black.
Derivation of a liver metastasis score
Given the propensity of uveal melanoma for metastasis to the liver, we assumed that gene expression analysis could reveal features that predispose to liver metastasis in other cancer types. We further reasoned that it would be necessary to adjust for chromosome 3 loss, which is known to be associated with BAP1 biallelic inactivation and metastatic risk in uveal melanoma. BAP1 inactivation (mutation or loss) is a rare event in most cancer types and would be unlikely to be a universal predictor of liver metastasis. Therefore, we performed an additional meta-analysis. Within each dataset and for each gene we fitted multivariable Cox model of relapse-free survival that included gene expression and chromosome 3 status. In this approach, 63 genes were significantly correlated with relapse (FDR < 0.05). Again, the Q-test for heterogeneity and visual inspection of forest plots did not show discrepancies between datasets. The top 10 genes are shown in Table 2 (full results in Table S2). After adjustment for chromosome 3 status, the relation of genes residing on chromosome 3 with relapse was attenuated. Indeed, the expression of most chromosome 3 genes (777/782), including BAP1 (HR = 0.779, 95% 0.620–0.977, FDR = 0.374), was not significantly associated with relapse-free survival when chromosome 3 status was included in the model. Twenty-eight significant genes (44% of all significant genes) were located on chromosome 8 and seven thereof in 8q24.3, the distal end of the chromosome 8.
Table 2.
Genes associated with relapse in uveal melanoma.
| Gene | HR | 95% CI | FDR | Location |
|---|---|---|---|---|
| JPH1 | 2.00 | (1.56 to 2.57) | <0.001 | 8q21.11 |
| PTP4A3 | 2.13 | (1.62 to 2.80) | <0.001 | 8q24.3 |
| ID2 | 0.56 | (0.44 to 0.70) | 0.002 | 2p25.1 |
| POP1 | 1.68 | (1.37 to 2.06) | 0.002 | 8q22.2 |
| CYP4X1 | 1.62 | (1.33 to 1.97) | 0.004 | 1p33 |
| HPN | 1.65 | (1.33 to 2.05) | 0.009 | 19q13.11 |
| FYN | 0.61 | (0.50 to 0.75) | 0.009 | 6q21 |
| LPIN1 | 0.58 | (0.46 to 0.73) | 0.009 | 2p25.1 |
| ETNPPL | 1.69 | (1.34 to 2.12) | 0.011 | 4q25 |
| MARC2 | 0.63 | (0.52 to 0.78) | 0.012 | 1q41 |
| TLCD1 | 1.68 | (1.33 to 2.10) | 0.012 | 17q11.2 |
| TSC22D1 | 1.61 | (1.30 to 1.99) | 0.013 | 13q14.11 |
| VCPIP1 | 1.69 | (1.33 to 2.15) | 0.014 | 8q13.1 |
| ANG | 0.52 | (0.39 to 0.70) | 0.014 | 14q11.2 |
| RAB11FIP1 | 0.62 | (0.50 to 0.77) | 0.014 | 8p11.23 |
| DNAAF3 | 0.56 | (0.43 to 0.73) | 0.014 | 19q13.42 |
| GALM | 0.67 | (0.55 to 0.80) | 0.014 | 2p22.1 |
| ASAP1 | 1.70 | (1.33 to 2.17) | 0.015 | 8q24.22 |
| CBS | 1.48 | (1.23 to 1.77) | 0.016 | 21q22.3 |
| MTFR1 | 1.61 | (1.29 to 2.01) | 0.018 | 8q13.1 |
Top 20 genes by lowest FDR shown (full results in the supplement) in a meta-analysis of bivariable survival models including chromosome 3 and the expression of each individual gene.
The pooled data of the training dataset were further used to build a prognostic scoring formula with the L1-penalized likelihood algorithm of the GLMnet package (supplemental methods) limited to the genes that were prognostic after adjustment for chromosome status 3 at FDR < 0.1 (Table S2). The procedure converged on a linear score based on the expression of only the two most significant genes, namely JPH1 and PTP4A3 (coefficients 0.147 and 0.249 respectively). In the training data, the two-gene prognostic score was significantly associated with progression-free survival in a multivariable survival model that included tumor class and chromosome 3 status (P < 0.0001). The prognostic score was also correlated with the expression of class genes (R2 = 0.66 against the mean of class 1 minus the mean of class 2 genes, P < 0.0001).
Validation
Uveal melanoma
The prognostic performance of the two-gene score was validated in independent public uveal melanoma data (GSE39717, N = 30 and the TCGA, N = 60). Meta-analysis of univariable Cox survival regression confirmed a statistically significant association with prognosis (Figs 2A and S3, log HR = 3.63, 95% 1.07–6.19, P = 0.0055). As expected, the score remained prognostic in a bivariable model including chromosome 3 status (Figs 2B and S3, log HRscore = 3.75, 95% 1.00–6.50, P = 0.0075).
Figure 2.

Forest plot of univariable (A) and bivariable (B) Cox regression of the continuous risk score in uveal melanoma. Log hazard ratio estimates with standard error bars are shown horizontally for both datasets (upper part) and the meta-analysis (RE model, bottom part). The lack of association is at zero (dotted vertical line). There was a positive association in both the univariable and bivariable model, both statistically significant in the meta-analysis.
Pan-cancer
Our main objective was to examine whether the predisposition of uveal melanoma to liver relapse can be used to estimate the risk of liver metastasis in other malignancies, especially of epithelial origin. To this end, we examined the two-gene score values in a pan-cancer dataset (GSE2109) including 1181 primary tumors (1036 non-metastatic, 57 with liver metastases, 89 with other metastases). Most samples were obtained from colorectal (N = 274), breast (N = 248), endometrial (N = 147), ovarian (N = 132) and lung (N = 98) cancer. The risk score was significantly higher in tumors that developed liver metastases (t-test P < 0.0001, Fig. S4), a difference that persisted when we restricted the analysis to tumors that had metastasized (N = 146, t-test P < 0.0001, Fig. S4). The difference between tumors with and without liver metastasis also remained significant in a bivariable logistic model including the primary origin (logOR = 0.26, 95% 0.005–0.41, P = 0.043). A similar result is obtained when the score values are standardized per primary site (logOR = 0.29, 95% 0.03–0.55, P = 0.028). Looking at individual metastatic sites, the risk score was highest in the group with liver metastases and was statistically significant in pairwise comparisons with non-metastatic tumors but also against tumors harboring metastases in bone and lung (Fig. 3).
Figure 3.
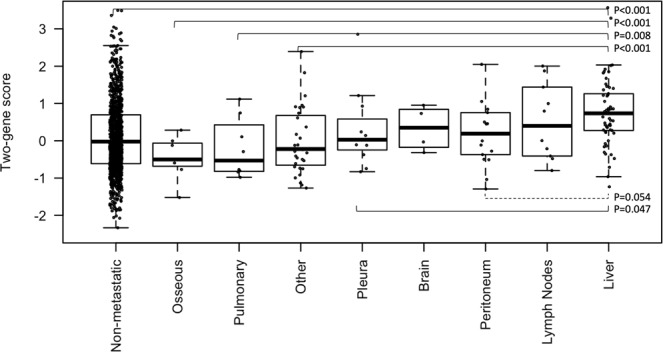
The two-gene score is highest in tumors with liver metastases. Data from the GSE2109 pan-cancer dataset. Statistically significant pairwise Wilcoxon’s comparisons are shown in solid lines (all values are FDR).
Breast cancer
We measured the expression of PTP4A3 and JPH1 in a breast cancer cohort by qPCR and used it to calculate the risk score in a series of 463 tumors for which the time and site of relapse were known (271 hormone receptor-positive, 86 triple-negative and 106 HER2-positive). The proportion of patients with liver-first relapses did not differ significantly (Fisher’s test P = 0.53) between HER2-positive tumors (10%), hormone receptor-positive (8%) and triple-negative tumor (6%). In hormone receptor-positive tumors, the two-gene prognostic score was associated with relapse (HR = 1.24, 1.04–1.47, P = 0.016) and specifically with first relapse to the liver (Fig. 4, HR = 1.50, 1.10–2.05, P = 0.0098) but not with relapse to non-liver sites (HR = 1.15, 0.94–1.41, P = 0.183). In triple-negative tumors, the two-gene score was not associated with relapse at any site (HR = 1.25, 0.77–22.4, P = 0.88) or liver-specific relapse (HR = 0.78, 0.29–2.08, P = 0.62). Similarly, in Her2 + tumors the score was not associated with relapse at any site (HR = 0.96, 0.70–1.32, P = 0.789, 38 events) or liver-specific relapse (HR = 0.99, 0.55–1.78, P = 0.981, 11 events).
Figure 4.
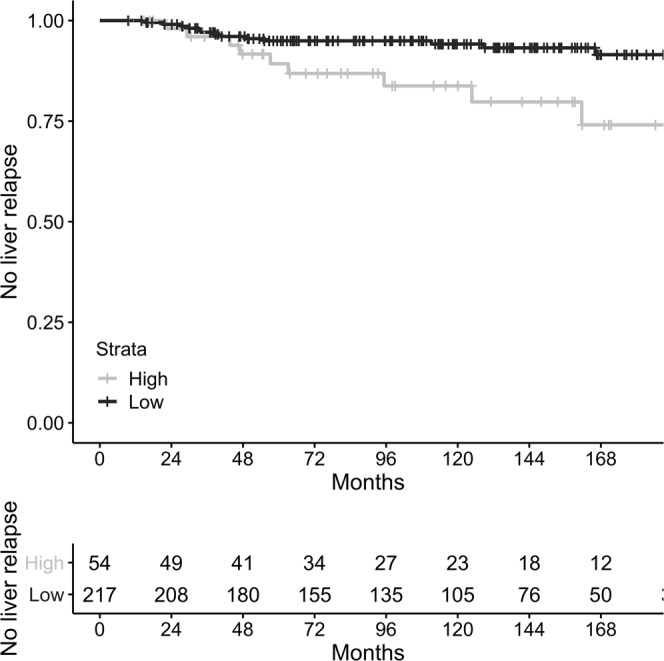
Kaplan-Meier curve of liver-first relapse in hormone-receptor positive breast cancer. Liver-first relapse occurred more often in the high-score group (P = 0.0098 for the continuous risk score). Data plotted using the top quintile as cutoff value (P = 0.011 at the specified cutoff).
Colorectal cancer
Finally, we used colorectal cancer data from the PETACC-3 trial (N = 604, 63 liver relapses) and a public retrospective series (GSE14095, N = 189, 53 liver relapses) to validate the risk score in colorectal cancer. In both datasets the risk score was significantly higher in patients with liver relapse (Wilcoxon’s test P = 0.047 and P = 0.011). In a meta-analysis of logistic regression models, a high-risk score was significantly associated with liver metastasis (Fig. 5A, logOR = 0.54, 95% 0.24–0.85, P = 0.0005). Interestingly, in the PETACC data, which also included non-liver relapses, the score was not associated with relapse in general (logOR = 0.00, 95% −0.57 to 0.57, P = 0.98), while relapse to non-liver sites was a negative trend (logOR = −0.65, 95% −1.39 to 0.06, P = 0.078).
Figure 5.

The risk score was specifically associated with liver relapse in colorectal cancer. Log hazard ratio estimates with standard error bars are shown horizontally for both datasets (upper part) and the meta-analysis (RE model, bottom part). The lack of association is at zero (dotted vertical line). In a meta-analysis (RE model) of (A) univariable and (B) bivariable logistic regression, the risk score was consistently and significantly associated with liver relapse, even after adjustment for CMS groups. Data from PETACC-3 and GSE14095. (C) In the prospective PETACC data, the proportion of total relapse was similar in CMS1-3, as expected. Liver relapse was lower in CMS1.
The distribution of the risk score was visibly bimodal in both colorectal cancer datasets (Fig. S5), suggesting the presence of distinct sub-populations. Indeed, when tumors were separated by the consensus molecular subtype (CMS) classifier, the average risk score was highest in CMS2 and 4, and lowest in CMS1 and 3 (Fig. S5). The association between liver metastasis and the risk score remained significant in a bivariable model that included the CMS subtype (Fig. 5B, logOR = 0.40, 95% 0.09–0.71, P = 0.011). In the prospective PETACC data, the distribution of the risk score agreed with the proportion of liver relapses, which was lowest in CMS1 (Fig. 5C, logOR = −1.04 vs non-CMS1, 95% −2.11 to −0.19, P = 0.03). Liver relapse was only 13% of all relapse in CMS1, against 34%, 31% and 30% in CMS2–4 respectively. In the pooled colorectal cancer data, prediction of liver metastasis was improved by including both CMS and the risk score (ANOVA Chi2 test P = 0.0013 against a model including CMS only, P < 0.001 against the score only).
Discussion
Different tumors have different patterns of metastatic spread20,21. Predicting the most likely site of relapse can help determine the most appropriate treatment and follow-up, such as scintigraphy for bone metastases and liver MRI for liver metastases. Early work has demonstrated that it is possible to distinguish tumors with metastatic potential from tumors that do not metastasize from their gene expression profile22. However, despite advances in our understanding of the cellular adaptations that occur in every distant site23, efforts to predict organ-specific metastasis are limited in size and scope. A small study of 20 colorectal tumors resulted in the identification of 37 genes that differed between tumors with and without liver metastases24. Another study of 123 colorectal tumors found 46 genes associated with liver metastasis25. Several of the genes, including MMP1, MMP2, TIMP1 and HIF1A, are known cancer genes and facilitate stromal invasion. In a study of over 1000 breast tumors the most important risk factor for liver metastasis was the HER2-enriched subtype, but without more specific information at the gene level26.
A predictor of liver metastasis can be derived from any gene expression dataset with adequate follow-up. The advantage of using uveal melanoma is that its proclivity for liver metastasis is highly specific (~90%), meaning that it rarely metastasizes to other organs, and this proclivity cannot be explained by anatomical or vascular proximity. In that sense, tumors that behave biologically like uveal melanoma may be better candidates for liver-specific follow-up and interventions than tumors with a general tendency to colonize multiple organs, which would be at risk of extrahepatic recurrence.
We derived and validated a prognostic score that predicted relapse in uveal melanoma based on the weighted expression of only two genes (PTP4A3 and JPH1) and applied it to the prediction of liver relapse in other malignancies. The observation that PTP4A3 expression strongly correlates with metastatic risk in uveal melanoma is in agreement with previous studies of uveal melanoma13. In colorectal cancer, the PTP4A3 gene regulates cancer cell adhesion and the expression of key ECM and adhesion genes27. PTP4A3 also seems to facilitate metastasis through accumulation of MMP1428 and regulation of integrin β129. Interestingly, copy number gains of PTP4A3 at 8q are more frequent in colorectal tumors with liver metastases30 although this observation also includes broader alterations such as loss of 8p and gain of centromeric 8q31.
The JPH1 gene codes junctophilin-1, a component of the junctional complex that binds the plasma membrane with the endoplasmic reticulum and regulates intracellular calcium32. Although JPH1 has been found to be differentially expressed in metastatic versus non-metastatic uveal melanoma33 and thymoma34, its biological significance in cancer remains to be elucidated. Even in the absence of a direct causal relation, JPH1 upregulation may reflect other upstream events, such as driver mutations in other genes or copy number gains in 8q21 involving nearby regulatory regions.
Our hypothesis was that the gene expression of uveal melanoma could identify biological predictors of liver-specific metastasis. In agreement with our initial hypothesis, the risk score developed in uveal melanoma was associated specifically with the risk of liver relapse. In the pan-cancer dataset (GSE2109) the score values were highest in tumors which developed liver metastasis, compared with tumors which metastasized to other sites. Although the number of events (liver metastases) is not sufficient to characterize individual tumor types, the relation between the two-gene score and liver metastasis remained significant in a multivariable model after adjustment for the site of the primary.
Metastasis to the liver is common in breast cancer20, but locoregional treatment such as surgical resection is only beneficial for carefully selected patients35,36, making their early identification essential. In hormone receptor positive breast cancer, the score was associated with metastatic liver relapse but not with relapse to other sites. The score was not predictive of liver relapse in triple negative or Her2+ breast cancer, a fact that could result from profound biological differences, significant heterogeneity in the case of triple negative tumors, or simply a lack of statistical power due to a small number of liver events in these cohorts. Her2+ breast tumors have a higher risk of liver metastasis26, but did not have higher score values in our study and may depend on the contribution of alternative biological pathways. For example, previous studies have shown that the expression of Claudin-2 is associated with early liver relapse in triple negative and hormone receptor positive breast cancer37.
The score was consistently associated with liver metastasis in a meta-analysis of two different colorectal cancer datasets. Other genes, such as EREG, AREG and LCK have also been associated with liver metastasis in a previous gene expression study of 160 colorectal cancer samples38 although it is unclear whether these genes are generally related with liver metastasis or are specific to colorectal cancer. CMS was also predictive of liver relapse in colorectal cancer, with CMS1 having a significantly lower proportion of liver relapses than CMS2–4. Given that the overwhelming majority of CMS1 tumors are right-sided, the influence of anatomical factors cannot be excluded, although a causal link between right-sidedness and low risk of liver metastasis is not obvious and cannot be inferred from our data. A lower incidence of liver metastases in right-sided tumors has also been observed previously31. Since both CMS and the risk score were independently predictive of liver metastasis in a bivariable model, integrating both in a clinically-oriented algorithm should further improve predictive performance.
Judging the reliability and, generally, the clinical performance (precision, accuracy etc) of the score at this stage is probably premature. First, the practical utility depends on the prior risk of liver metastasis, which varies considerably between cancer types20. It would be very hard to show a meaningful absolute increase in the probability of liver metastasis in cancer types which very rarely metastasize to the liver. Second, the overall risk of metastasis must also be estimated according to the known prognostic variables of each cancer type. For example, a T1N0 tumor would be very unlikely to spread to any organ. Finally, a specific cut-off would have to be chosen per tumor type but also depending on the clinical consequences. For example, even a modest increase in the estimated risk of metastasis could justify a liver MRI, while an invasive test or a therapeutic intervention would require a higher degree of confidence. A more detailed characterization of the score’s performance would be useful in a subsequent work that examines a precisely defined clinical context (disease type, risk factors, threshold for intervention etc).
Based on the above, our work provides proof-of-concept for predicting the risk of liver metastasis in hormone-sensitive breast cancer and colorectal cancer, based on a simple two-gene score. The performance could further be improved with the integration of type-specific information, such as the CMS classification of colorectal cancer. The results from a pan-cancer analysis indicate that the score could be useful in other tumor types and that it is highest in tumors with liver metastases, compared with tumors which colonize other sites. This suggests the existence of a common underlying biological process of liver metastasis that could be exploited for prognostic or therapeutic purposes. The elucidation of this mechanism presents an opportunity for further study.
Methods
Origin of samples
Untreated human samples from uveal melanoma and breast cancer were collected by the unit of surgical oncology from patients who provided written informed consent. The samples were stored in the Biological Resource Center (BRC) of the Institut Curie, France, in accordance with French regulations. The study was approved by the ethics committee and the institutional review board of the Curie Institute with approval number A10–024. All research was performed in accordance with relevant guidelines and regulations.
We searched for gene expression data from human primary samples of uveal melanoma in the Gene Expression Omnibus39 and manually curated datasets with adequate clinical annotation. At the time of retrieval, 18 data sets were screened and three were retained for analysis: GSE44295 (N = 57), GSE22138 (N = 63) and GSE39717 (N = 39, 30 with follow-up). The two largest datasets were used in conjunction with our own RNAseq data for model development (training dataset). We also downloaded publicly available RNAseq data from the TCGA (accessed on November 2017) for 80 patients, of which 60 had follow-up information and were retained for analysis. GSE39717 and TCGA data were used for validation (see Supplement Sections 1–3 and Fig. S6).
Gene expression data for colorectal cancer were obtained from the PETACC-3 study of adjuvant chemotherapy in colon cancer40, available in ArrayExpress (E-MTAB-990) and from a series of colorectal patients41 that were followed for the occurrence of liver metastases (GSE14095, N = 187). Finally, we also obtained a pan-cancer dataset which contained information on the site of relapse (GSE2109, N = 2158, 1181 of which with metastatic relapse).
We considered the use of the PanCancer TCGA data, based on the recent publication of curated clinical annotation42 (see Supplementary Table 1 in that article, columns T and U). Unfortunately, despite the inclusion of over 10,000 patients, the occurrence of liver events was recorded for only 262 patients, raising concerns about the quality of the follow-up with respect to that specific outcome. For example, none were documented for patients with colorectal cancer (N = 618), a fact that limited the usefulness of this resource for our purposes.
Inactivation of BAP1 is common in uveal melanoma but can also occur in kidney cancer7, mesothelioma8 and cholangiocarcinoma9. However, mesothelioma and cholangiocarcinoma are rare and we could not find gene expression datasets documenting liver metastasis, not just overall survival or relapse-free survival, which could be used our purposes. In addition, cholangiocarcinoma is in immediate proximity to the liver and would not be a useful model of liver metastasis, more likely reflective of local invasion instead. Out of 33 patients with renal cancer in the pan-cancer GSE2109 dataset only 10 had distant metastases and none had liver metastases, making any kind of statistical analysis impossible.
Total RNA extraction
Total RNA was extracted from fresh frozen uveal melanoma and breast cancer samples with the acid-phenol guanidium method. The quantity of RNA was assessed by using an ND-1000 NanoDrop Spectrophotometer with corresponding software (Thermo Fisher Scientific Inc., Wilmington, DE). RNA quality was determined by agarose gel electrophoresis.
RNAseq
Library preparation was done using the Illumina TruSeq kit according to manufacturer’s instructions. Libraries were sequenced to produce paired-end reads. We used STAR v2.543 to align against the GRCh37 DNA primary assembly and the Ensembl annotation r75 (available from ftp://ftp.ensembl.org/pub/release-75/). Quality control was done with RNA-SeQC (v1.1.8)44 and custom scripts.
The raw counts were estimated with the Rsubread package (v1.2) and then imported by edgeR (v3.12)45. Raw counts were filtered by only keeping genes with at least 1 count-per-million in at least 5 samples, normalized with the trimmed mean of M-values (TMM) and log-transformed, according to the edgeR recommendations.
Real-time PCR
The RT-qPCR method, including cDNA synthesis and PCR conditions, has been previously described in detail46. We quantified transcripts of an endogenous RNA control gene involved in cellular metabolic pathway, namely TBP (Genbank NM_003194), which encodes the TATA box-binding protein (a component of the DNA-binding protein complex TFIID)47. Each sample was normalized on the basis of its TBP content and against a median of 10 normal breast tissue samples, with the ΔCt method.
Data analysis and statistics
Statistical analyses were done in R version 3.5. All statistical tests were two-sided and considered significant if P < 0.05. P-value adjustment for multiple comparisons was used to control the false discovery rate48. Survival models were built with the survival R package49 and the coxph() function. By default, this function fits a Cox proportional hazards regression model with the Efron approximation for tied event times.
We used the metafor package with the random effects model and the default restricted maximum-likelihood estimator of residual heterogeneity (τ2) to perform meta-analyses50. Specifically, for each dataset and each gene the log-transformed hazard ratio estimate (yi) and its associated standard error (sei) were extracted from a univariable and bivariable (with chromosome 3 status) model. The rma() function from the metafor package was then called with rma(yi, sei). The Q-test was used to test for significant heterogeneity.
The nearest-neighbor chain algorithm was used for hierarchical clustering (reviewed in51). Pearson’s correlation was used as a similarity metric with average linkage. We used the nclust package (v2.0.1) for this purpose, available from https://bcf.isb-sib.ch/nclust/.
The coxnet method52 in the GLMnet package was used to derive a linear prognostic score. The development of the prognostic score was in accordance with the REMARK recommendations.
Gene expression estimate of chromosome 3 status
Over all uveal melanoma datasets, the chromosome 3 status had been estimated by dedicated methods (aCGH or FISH) for 119 patients. For the remaining patients, we inferred the chromosome 3 status from gene expression data. Using patients with known chromosome 3 status, we trained a binomial (loss/no loss) GLMnet model on 95 patients and validated it on the remaining 24 patients. Data from the 24 patients were not considered during the derivation of the model. Only the expression of genes residing on chromosome 3 was used, as these genes would be directly affected by a loss of chromosome 3.
Validation on the remaining 24 patients demonstrated a very high concordance (23/24 = 96%) of gene expression estimates with other methods (FISH, aCGH). The same model was subsequently applied to 77 patients for whom the chromosome 3 status was missing. The gene expression prediction of chromosome 3 status was significantly associated with relapse-free survival (HRmonosomy 4.30, 95% 2.34–7.88, P < 0.0001). The proportion of samples with monosomy did not differ significantly by estimation method (FISH, aCGH or gene expression, Fisher’s test P = 0.519).
Supplementary information
Acknowledgements
The work of Petros Tsantoulis was supported by the “Ligue Genevoise contre le Cancer”. The work of Wulfran Cacheux was supported by the Nuovo Soldati Foundation (2014–2015). The authors gratefully acknowledge the technical assistance of Pratyaksha Wirapati (Swiss Institute of Bioinformatics).
Author contributions
P.T. and M.D. performed the data analysis, contributed to its interpretation and drafted the initial manuscript. I.B. and S.V. acquired material and data from breast cancer patients and revised the manuscript. P.M., N.C., A.H., M.H.S. and S.R.-R. acquired material and clinical data from uveal melanoma patients and did a critical revision of the manuscript. P.Y.D. helped interpret the results and did a critical revision of the manuscript. A.R. and W.C. proposed the study design, helped interpret the results and contributed to the initial manuscript draft. All authors approved the final manuscript.
Data availability
The RNAseq data produced by the study can be found in the European Genome-phenome Archive (accession EGAS00001002932).
Public data used in this work:
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE44295
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE22138
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE39717
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE2109
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-019-52841-y.
References
- 1.The Collaborative Ocular Melanoma Study Assessment of Metastatic Disease Status at Death in 435 Patients With Large Choroidal Melanoma in the Collaborative Ocular Melanoma Study (COMS) Arch. Ophthalmol. 2001;119:670. doi: 10.1001/archopht.119.5.670. [DOI] [PubMed] [Google Scholar]
- 2.Tomlinson JS, et al. Actual 10-Year Survival After Resection of Colorectal Liver Metastases Defines Cure. J. Clin. Oncol. 2007;25:4575–4580. doi: 10.1200/JCO.2007.11.0833. [DOI] [PubMed] [Google Scholar]
- 3.Van Cutsem E, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann. Oncol. 2016;27:1386–1422. doi: 10.1093/annonc/mdw235. [DOI] [PubMed] [Google Scholar]
- 4.Van Tilborg AAJM, et al. Long-term results of radiofrequency ablation for unresectable colorectal liver metastases: a potentially curative intervention. Br. J. Radiol. 2011;84:556–565. doi: 10.1259/bjr/78268814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Groot Koerkamp B, et al. Perioperative Hepatic Arterial Infusion Pump Chemotherapy Is Associated With Longer Survival After Resection of Colorectal Liver Metastases: A Propensity Score Analysis. J. Clin. Oncol. 2017;35:1938–1944. doi: 10.1200/JCO.2016.71.8346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harbour JW, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010;330:1410–3. doi: 10.1126/science.1194472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hakimi AA, et al. Adverse Outcomes in Clear Cell Renal Cell Carcinoma with Mutations of 3p21 Epigenetic Regulators BAP1 and SETD2: A Report by MSKCC and the KIRC TCGA Research Network. Clin. Cancer Res. 2013;19:3259–3267. doi: 10.1158/1078-0432.CCR-12-3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Joseph NM, et al. Genomic profiling of malignant peritoneal mesothelioma reveals recurrent alterations in epigenetic regulatory genes BAP1, SETD2, and DDX3X. Mod. Pathol. 2017;30:246–254. doi: 10.1038/modpathol.2016.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Al-Shamsi HO, et al. BRCA-associated protein 1 mutant cholangiocarcinoma: an aggressive disease subtype. J. Gastrointest. Oncol. 2016;7:556–561. doi: 10.21037/jgo.2016.03.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Royer-Bertrand B, et al. Comprehensive Genetic Landscape of Uveal Melanoma by Whole-Genome Sequencing. Am. J. Hum. Genet. 2016;99:1190–1198. doi: 10.1016/j.ajhg.2016.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cassoux N, et al. Genome-wide profiling is a clinically relevant and affordable prognostic test in posterior uveal melanoma. Br. J. Ophthalmol. 2014;98:769–774. doi: 10.1136/bjophthalmol-2013-303867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Damato B, et al. Multiplex ligation-dependent probe amplification of uveal melanoma: correlation with metastatic death. Invest. Ophthalmol. Vis. Sci. 2009;50:3048–55. doi: 10.1167/iovs.08-3165. [DOI] [PubMed] [Google Scholar]
- 13.Laurent C, et al. High PTP4A3 phosphatase expression correlates with metastatic risk in uveal melanoma patients. Cancer Res. 2011;71:666–74. doi: 10.1158/0008-5472.CAN-10-0605. [DOI] [PubMed] [Google Scholar]
- 14.Haan JC, et al. Genomic landscape of metastatic colorectal cancer. Nat. Commun. 2014;5:5457. doi: 10.1038/ncomms6457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bass AJ, et al. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–9. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walker LC, et al. Dual-Color Fluorescence In Situ Hybridization Reveals an Association of Chromosome 8q22 but Not 8p21 Imbalance with High Grade Invasive Breast Carcinoma. PLoS One. 2013;8:e70790. doi: 10.1371/journal.pone.0070790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Onken MD, Worley LA, Ehlers JP, Harbour JW. Gene expression profiling in uveal melanoma reveals two molecular classes and predicts metastatic death. Cancer Res. 2004;64:7205–9. doi: 10.1158/0008-5472.CAN-04-1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robertson AG, et al. Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma. Cancer Cell. 2017;32:204–220.e15. doi: 10.1016/j.ccell.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Onken MD, Worley LA, Tuscan MD, Harbour JW. An Accurate, Clinically Feasible Multi-Gene Expression Assay for Predicting Metastasis in Uveal Melanoma. J. Mol. Diagnostics. 2010;12:461–468. doi: 10.2353/jmoldx.2010.090220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hess KR, et al. Metastatic patterns in adenocarcinoma. Cancer. 2006;106:1624–1633. doi: 10.1002/cncr.21778. [DOI] [PubMed] [Google Scholar]
- 21.Nguyen DX, Bos PD, Massagué J. Metastasis: from dissemination to organ-specific colonization. Nat. Rev. Cancer. 2009;9:274–284. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- 22.Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat. Genet. 2003;33:49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- 23.Obenauf AC, Massagué J. Surviving at a distance: organ specific metastasis. Trends in cancer. 2015;1:76–91. doi: 10.1016/j.trecan.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.D’Arrigo A, et al. Metastatic transcriptional pattern revealed by gene expression profiling in primary colorectal carcinoma. Int. J. Cancer. 2005;115:256–262. doi: 10.1002/ijc.20883. [DOI] [PubMed] [Google Scholar]
- 25.Ki DH, et al. Whole genome analysis for liver metastasis gene signatures in colorectal cancer. Int. J. Cancer. 2007;121:2005–2012. doi: 10.1002/ijc.22975. [DOI] [PubMed] [Google Scholar]
- 26.Harrell JC, et al. Genomic analysis identifies unique signatures predictive of brain, lung, and liver relapse. Breast Cancer Res. Treat. 2012;132:523–535. doi: 10.1007/s10549-011-1619-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McQueeney Kelley E., Salamoun Joseph M., Ahn Jennifer G., Pekic Paula, Blanco Isabella K., Struckman Heather L., Sharlow Elizabeth R., Wipf Peter, Lazo John S. A chemical genetics approach identifies PTP4A3 as a regulator of colon cancer cell adhesion. The FASEB Journal. 2018;32(10):5661–5673. doi: 10.1096/fj.201701446R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maacha S, et al. Protein Tyrosine Phosphatase 4A3 (PTP4A3) Promotes Human Uveal Melanoma Aggressiveness Through Membrane Accumulation of Matrix Metalloproteinase 14 (MMP14) Investig. Opthalmology Vis. Sci. 2016;57:1982. doi: 10.1167/iovs.15-18780. [DOI] [PubMed] [Google Scholar]
- 29.Foy M, Anézo O, Saule S, Planque N. PRL-3/PTP4A3 phosphatase regulates integrin β1 in adhesion structures during migration of human ocular melanoma cells. Exp. Cell Res. 2017;353:88–99. doi: 10.1016/j.yexcr.2017.03.012. [DOI] [PubMed] [Google Scholar]
- 30.Nakayama N., Yamashita K., Tanaka T., Kawamata H., Ooki A., Sato T., Nakamura T., Watanabe M. Genomic gain of the PRL-3 gene may represent poor prognosis of primary colorectal cancer, and associate with liver metastasis. Clinical & Experimental Metastasis. 2015;33(1):3–13. doi: 10.1007/s10585-015-9749-7. [DOI] [PubMed] [Google Scholar]
- 31.Bruin SC, et al. Specific genomic aberrations in primary colorectal cancer are associated with liver metastases. BMC Cancer. 2010;10:662. doi: 10.1186/1471-2407-10-662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takeshima H, et al. Junctophilins. Mol. Cell. 2000;6:11–22. doi: 10.1016/s1097-2765(00)00003-4. [DOI] [PubMed] [Google Scholar]
- 33.Fagone P, et al. Identification of novel chemotherapeutic strategies for metastatic uveal melanoma. Sci. Rep. 2017;7:44564. doi: 10.1038/srep44564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Badve S, et al. Molecular Analysis of Thymoma. PLoS One. 2012;7:e42669. doi: 10.1371/journal.pone.0042669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Selzner M, Morse MA, Vredenburgh JJ, Meyers WC, Clavien PA. Liver metastases from breast cancer: long-term survival after curative resection. Surgery. 2000;127:383–9. doi: 10.1067/msy.2000.103883. [DOI] [PubMed] [Google Scholar]
- 36.Maksan S-M, Lehnert T, Bastert G, Herfarth C. Curative liver resection for metastatic breast cancer. Eur. J. Surg. Oncol. 2000;26:209–212. doi: 10.1053/ejso.1999.0778. [DOI] [PubMed] [Google Scholar]
- 37.Kimbung S, et al. Claudin-2 is an independent negative prognostic factor in breast cancer and specifically predicts early liver recurrences. Mol. Oncol. 2014;8:119–128. doi: 10.1016/j.molonc.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Watanabe T, et al. Prediction of liver metastasis after colorectal cancer using reverse transcription-polymerase chain reaction analysis of 10 genes. Eur. J. Cancer. 2010;46:2119–2126. doi: 10.1016/j.ejca.2010.04.019. [DOI] [PubMed] [Google Scholar]
- 39.Barrett T, et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 2013;41:D991–D995. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Budinska E, et al. Gene expression patterns unveil a new level of molecular heterogeneity in colorectal cancer. J. Pathol. 2013;231:63–76. doi: 10.1002/path.4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Watanabe T, et al. Gene expression signature and response to the use of leucovorin, fluorouracil and oxaliplatin in colorectal cancer patients. Clin. Transl. Oncol. 2011;13:419–425. doi: 10.1007/s12094-011-0676-z. [DOI] [PubMed] [Google Scholar]
- 42.Liu J, et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell. 2018;173:400–416.e11. doi: 10.1016/j.cell.2018.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dobin A, et al. Bioinformatics. 2013. STAR: ultrafast universal RNA-seq aligner; pp. 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.DeLuca DS, et al. RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics. 2012;28:1530–1532. doi: 10.1093/bioinformatics/bts196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40:4288–4297. doi: 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bièche I, et al. Quantification of estrogen receptor α and β expression in sporadic breast cancer. Oncogene. 2001;20:8109–8115. doi: 10.1038/sj.onc.1204917. [DOI] [PubMed] [Google Scholar]
- 47.Bièche I, et al. Real-time reverse transcription-PCR assay for future management of ERBB2-based clinical applications. Clin. Chem. 1999;45:1148–56. [PubMed] [Google Scholar]
- 48.Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 289–300 (1995).
- 49.Therneau Terry M., Grambsch Patricia M. Modeling Survival Data: Extending the Cox Model. New York, NY: Springer New York; 2000. [Google Scholar]
- 50.Viechtbauer W. Conducting Meta-Analyses in R with the metafor Package. J. Stat. Softw. 2010;36:1–48. doi: 10.18637/jss.v036.i03. [DOI] [Google Scholar]
- 51.Murtagh F. A Survey of Recent Advances in Hierarchical Clustering Algorithms. Comput. J. 1983;26:354–359. doi: 10.1093/comjnl/26.4.354. [DOI] [Google Scholar]
- 52.Simon N, Friedman J, Hastie T, Tibshirani R. Regularization Paths for Cox’s Proportional Hazards Model via Coordinate Descent. J. Stat. Softw. 2011;39:1–13. doi: 10.18637/jss.v039.i05. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNAseq data produced by the study can be found in the European Genome-phenome Archive (accession EGAS00001002932).
Public data used in this work:
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE44295
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE22138
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE39717
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE2109