

Abstract
Fragile-X syndrome (FXS) is characterized by neurological and psychiatric problems symptomatic of cortical hyperexcitability. Recent animal studies identified deficient γ-aminobutyricacid (GABA) inhibition as a key mechanism for hyperexcitability in FXS, but the GABA system remains largely unexplored in humans with the disorder. The primary objective of this study was to assess GABA-mediated inhibition and its relationship with hyperexcitability in patients with FXS. Transcranial magnetic stimulation (TMS) was used to assess cortical and corticospinal inhibitory and excitatory mechanisms in 18 patients with a molecular diagnosis of FXS and 18 healthy controls. GABA-mediated inhibition was measured with short-interval intracortical inhibition (GABAA), long-interval intracortical inhibition (GABAB), and the corticospinal silent period (GABAA+B). Net intracortical facilitation involving glutamate was assessed with intracortical facilitation, and corticospinal excitability was measured with the resting motor threshold. Results showed that FXS patients had significantly reduced short-interval intracortical inhibition, increased long-interval intracortical inhibition, and increased intracortical facilitation compared to healthy controls. In the FXS group, reduced short-interval intracortical inhibition was associated with heightened intracortical facilitation. Taken together, these results suggest that reduced GABAA inhibition is a plausible mechanism underlying cortical hyperexcitability in patients with FXS. These findings closely match those observed in animal models, supporting the translational validity of these markers for clinical research.
Subject terms: Autism spectrum disorders, Physiology
Introduction
Fragile-X syndrome (FXS) is a rare disorder, but the leading monogenic cause of autism spectrum disorder and the first hereditary cause of intellectual disability. FXS results from the methylation of the Fmr1 gene, which leads to a marked reduction or absence of the fragile-X mental retardation protein (FMRP), an important regulator of protein synthesis involved in brain development and synaptic function1. While the clinical phenotype may vary considerably between individuals affected by the disorder, it typically involves psychiatric and neurological manifestations indicative of neuronal hyperexcitability, including seizures, anxiety, hyperactivity, hypersensitivity, and hyperarousal2,3. Accumulating EEG evidence supports the existence of cortical hyperexcitability in patients with FXS4–7. However, the physiological mechanisms involved in hyperexcitability in FXS remain poorly understood. Research in this field is of prime clinical importance since correcting the alterations involved in neuronal and circuit excitability could alleviate several core symptoms of the disorder2.
Hyperexcitability is a consistent observation across animal models of the disorder3,8,9, and is believed to result from an imbalance between excitatory and inhibitory drives in intracortical circuits3,10. The dominant view over the past 15 years has been that the absence of FMRP in FXS induces an overactivation of metabotropic glutamate receptors (mGluRs), thereby increasing neuronal excitability11. Although experimental data from Fmr1-ko animals support the idea that mGluRs play a role in the process12,13, clinical trials using mGlurR antagonist for FXS have been unsuccessful14,15. Moreover, there is evidence suggesting that mGluR overactivity alone is insufficient to fully explain hyperexcitability in FXS16–18. In that regard, several recent studies show the presence of dysfunctions affecting diverse elements of the GABAergic system in various animal models of FXS17–21, suggesting that reduced inhibition is a key mechanism for circuit hyperexcitability. This theory is further supported by in vivo evidence confirming that defective inhibition from GABAergic interneurons is causally involved in the behavioral and sensory phenotype of the fly and mouse FXS models17,18. These findings suggest that the GABAergic system may be a promising therapeutic target to correct circuit hyperexcitability in FXS2.
The GABA system remains surprisingly unexplored in humans with FXS, despite several past and ongoing clinical trials aiming to modulate it22,23 (NCT03697161; NCT01911455). To date, the most significant evidence of a GABA dysfunction in humans with FXS comes from Hulst and colleagues (2015), who showed diminished GABAA receptor binding throughout the brain in a small group of patients using positron-emission tomography (PET) (n = 10). Yet, it is unknown if this reduction in receptor availability translates into functional alterations at the cortical level. Transcranial magnetic stimulation (TMS) is valuable tool to answer this question, as this technique is capable of probing the excitatory and inhibitory mechanisms in the cortex of awake humans24–26. TMS is a painless and non-invasive neurostimulation technique that uses magnetic fields to stimulate a restricted part of the brain. TMS acts on the cortex through the recruitment of interneurons in upper cortical layers mediating GABAergic inhibition onto pyramidal neurons27, whose resulting activity closely match what is seen non-invasively and at a macroscopic level in humans following stimulation of the primary motor cortex28. Abundant evidence demonstrates the sensitivity of TMS to GABAergic mechanisms in healthy and clinical populations24,25,29,30. Because the technique is safe, well tolerated, and does not require extensive training or sedation to be used with people with intellectual disability, TMS is well suited for the study of the GABA system in FXS.
In the present study, we used TMS to investigate intracortical inhibition and excitation in a cohort of individuals with a molecular diagnosis of FXS (n = 18). We assessed three measures of GABAergic inhibition, including short-interval intracortical inhibition (SICI; GABAA mediated), long-interval intracortical inhibition (LICI, GABAB mediated), and the corticospinal silent period (CSP; GABAA+B). Resting motor threshold (rMT) was used to assess corticospinal excitability, and net intracortical excitation involving glutamate was measured with intracortical facilitation (ICF). We hypothesized that GABA-mediated inhibition would be reduced, while corticospinal excitability and ICF would be enhanced in individuals with FXS compared to healthy controls.
Material and methods
Participants
Individuals aged 13–50-year old with a molecular diagnosis of FXS were eligible to participate in the study. Age and sex-matched control participants were eligible to take part in the study if they reported being in good general health, exempt of genetic or chronic conditions, with no history of neurological, neurodevelopmental, or psychiatric disorders. All control participants were alcohol, caffeine, and medication free at the moment of testing. Exclusion criteria comprised: any absolute contraindication for TMS, untreated hypothyroidism, being pregnant, or breastfeeding. Additional exclusion criteria for FXS patients comprised: change in medication in the previous 3 months, being diagnosed with epilepsy, taking more than three psychoactive drugs, and being unable to comply to simple verbal commands.
Twenty patients with a molecular diagnosis of FXS (two females), and 20 age and sex-matched healthy controls were recruited. Data from one male FXS patient were rejected due to technical EMG recording issues, and multivariate outliers were identified and removed using the Mahalanobis distance (p < 0.05), resulting in 18 patients in each group (one FXS, two controls, all males). Power analysis showed that this sample size allowed the detection of an effect size of f = 0.5 (large effect) in 83% of the cases with an alpha level of 0.05 (two-tails). Seven FXS patients were taking psychoactive medication, four of them taking two types of medications or more (Tables 1 and 2); none of them had a diagnosis of epilepsy. The legal guardian of each participant provided signed informed consent. The study was approved by the research ethics board from the Sherbrooke Hospital Research Center and conducted in accordance with the 1964 Declaration of Helsinki.
Table 1.
Sample characteristics.
| Characteristics | FXS N = 18 | Controls N = 18 |
|---|---|---|
| Age in years (mean, interquartile range) | 24.88 (7.5) | 23.88 (5.3) |
| IQ (mean, interquartile range) | 49.64 (17.5) | – |
| Intellectual disability (n, %) | 18 (100%) | – |
| Sex | ||
| Male | 16 (89%) | 16 (89%) |
| Female | 2 (11%) | 2 (11%) |
| Mutation (n, %) | ||
| Full | 16 (89%) | – |
| Mosaic | 2 (11%) | – |
| Medication status (n, %) | ||
| None | 11 (61.1%) | 18 (100%) |
| One | 3 (16.6%) | – |
| Two or more | 4 (22.2%) | – |
IQ Intellectual quotient
Table 2.
Sex, age, and medication at time of testing for FXS patients.
| Patient | Sex, age y | Psychoactive medication mg/d |
|---|---|---|
| FXS 1 | M, 20 | Abilify 2, Olanzapine 2.5, Zoloft 50 |
| FXS 2 | M, 40 | None |
| FXS 3 | M, 26 | None |
| FXS 4 | M, 18 | None |
| FXS 5 | F, 18 | None |
| FXS 6 | M, 35 | Venlafaxine 75, |
| FXS 7 | M, 28 | Quetiapine 50, Risperidone 2, Sertraline 50 |
| FXS 8 | M, 21 | None |
| FXS 9 | M, 26 | Sertraline 50 |
| FXS 10 | M, 26 | None |
| FXS 11 | M, 20 | None |
| FXS 12 | M, 27 | None |
| FXS 13 | M, 14 | Abilify 2, Adderal XR 40 |
| FXS 14 | M, 26 | Citalopram 40 |
| FXS 15 | M, 25 | None |
| FXS 16 | M, 24 | Sertraline 25, Quetiapine 25, Clonidine 0.1 |
| FXS 17 | F, 19 | None |
| FXS 18 | M, 35 | None |
TMS procedure
TMS was performed with a Magstim BiStim-2 using a 70 mm figure-of-eight coil (Magstim Compagny Limited, UK). To identify the optimal stimulation site, the coil was initially positioned over the C3 location of the International 10–20 EEG system at an intensity of 35% of maximum stimulator output (MSO). Single pulse stimulations were administered at a 1 cm interval covering a 16 cm2 region centered at C3, and the process was repeated while increasing the intensity (5% MSO steps) until a motor-evoked potential was obtained in the contralateral first dorsal interosseus (FDI) muscle. The location was then entered in the neuronavigation system (BrainSight, Rogue Research, Canada), and stimulations of decreasing intensity (2% MSO steps) were administered in the vicinity (±1 cm) to identify the most sensitive location. Electromyographic signals (EMG) were recorded with surface electrodes, amplified using a Powerlab 4/30 system, digitized at 4 KHz, and recorded with Scope v4.0 for offline analysis.
Standard TMS procedures were used31 and administered following the guidelines from the International Federation of Clinical Neurophysiology32. Briefly, resting motor threshold (rMT) was established as the minimal intensity to induce MEPs of at least 50 µV amplitude in five out of ten consecutive trials using the relative frequency criterion33, and intracortical facilitation (ICF, 12 ms interpulse interval (IPI)) was acquired to measure net intracortical excitability. Tests for intracortical inhibitory functions included short-interval intracortical inhibition (SICI; 3 ms IPI; GABAA) and long-interval intracortical inhibition (LICI, 100 ms IPI; GABAB). Corticospinal silent period (CSP; GABAA+B) was acquired while participants performed an isometric voluntary contraction of the FDI corresponding to 20% of maximal muscular contraction. In a subset of participants (n = 20; 10 FXS), short-interval intracortical facilitation (SICF) was acquired using the same interval as SICI (3 ms IPI) to assess the potential contribution of I-wave facilitatory components on SICI34. Conditioning stimuli (CS) for SICI, ICF, and the test stimulus for SICF, were set to an intensity corresponding to 80% of rMT; baseline MEPs and all other stimulations were administered using a test stimulus (TS) intensity set to induce MEPs of 1 mV peak-to-peak amplitude (≈125% of rMT). Fifteen trials were acquired for each measure, except for CSP (n = 5). A frameless neuronavigation system (BrainSight, Rogue Research) was used to ensure stable coil positioning during testing. The procedure was well tolerated by all participants and there was no serious adverse event. Typical EMG responses for each TMS protocol are depicted in Fig. 1.
Fig. 1. Typical EMG responses to the TMS stimulation protocols.

For SICI, ICF, and SICF, the average peak-to-peak MEP amplitude resulting from the CS-TS combination is compared to the baseline response induced by the TS alone in order to compute the degree of inhibition or facilitation. For LICI, the MEP amplitude induced by the second TS is compared to the amplitude of the MEP evoked by the first TS. The CSP is the duration between the onset of the MEP to the return of baseline EMG activity. For paired-pulse measures, the interval between TMS pulses is specified in milliseconds (ms). CS conditioning stimulus, CSP corticospinal silent period, ICF intracortical facilitation, LICI long-interval intracortical inhibition, SICI short-interval intracortical inhibition, SICF short-interval intracortical facilitation, TS test stimulus.
Data processing and statistical analysis
Individual EMG epochs were visually inspected and rejected if visible muscular activity was present in the 100 ms time window preceding the TMS pulse. Peak-to-peak MEP amplitude was calculated and averaged for baseline, SICI, ICF, and SICF independently. Ratios were then computed for SICI, ICF, and SICF, using the mean baseline MEP as the denominator. For LICI, the amplitude of the MEP evoked by the TS was divided by the one induced by the CS within each trial, and the resulting ratios were averaged35. CSP was measured independently by two raters as the duration from the MEP onset to the return to baseline EMG activity (inter rater agreement, r = 0.950). For each measure, a minimum of 10 valid trials were kept, resulting in Cronbach’s alpha being superior to 0.9036. Between-group differences were assessed using independent samples two-tailed t-tests (adjusted for unequal variance when Levene’s test p < 0.05) and multiple comparisons were handled with false-discovery rate (adjusted q-values). Data are available from the authors upon request.
Results
FXS patients and controls did not differ regarding rMT (T32.4 = 1.97, q = 0.43), amplitude of baseline MEP (T29.4 = 0.68, q = 0.44), or CSP length (T33 = 0.77, q = 0.43) (Fig. 2a, b). However, FXS patients showed increased ICF (T22.8 = 2.60, q = 0.017), increased LICI (T22.7 = 2.90, q = 0.011), and reduced SICI (T21.7 2.41, q = 0.021) compared with controls. Groups did not differ significantly on SICF (T18 = 0.37, q = 0.43) (Fig. 2c). Since deficient GABAA inhibition has been associated with circuit hyperexcitability in animal models of FXS17,37, we explored the relationship between SICI and ICF. Correlation analysis showed that FXS patients with less SICI had larger ICF (r = 0.648, q = 0.011), indicating a positive relationship between intracortical hypo-inhibition and hyperexcitability. This relationship was significantly different from the one observed in the control group (r = −0.222 in controls; Fisher’s z = 2.73; q = 0.011) (Fig. 2d). Because TMS measures are sensitive to several pharmacological agents29, we ran exploratory analyses (non-corrected) comparing only non-medicated FXS with healthy controls. Results corroborated observations made at the whole-group level, with non-medicated patients differing significantly from controls on SICI (T26 = 3.71, p = 0.001) and ICF (T26 = 2.38, p = 0.025), and LICI showing a similar trend (T26 = 1.96, p = 0.060). The correlation between SICI and ICF remained significant in the non-medicated FXS group (r = 0.737; p = 0.010), and significantly different from the control group (Fisher’s z = 2.67; p = 0.007). Comparing medicated FXS patients with non-medicated FXS patients and controls did not show significant differences on any TMS measures (Mann–Whitney–Wilcoxon tests), possibly due to the small number of medicated FXS patients (n = 7). Excluding FXS females from the analysis did not change the overall pattern of results. LICI and ICF remained significant (T32 > 2.31, <0.05), the relationship between SICI and ICF remained significant in the FXS group (r = 0.635, p = 0.008), and significantly different from the control group (Fisher’s z = 2.57, p = 0.010). The most notable difference was that the p value for SICI was now slightly above the significance threshold (T32 = 1.98, p = 0.057).
Fig. 2. TMS results.

a Amplitude of baseline motor-evoked potentials (MEP) for each group (in mV). b Corticospinal silent period duration (CSP) duration; c Results of paired-pulse stimulation protocols expressed in ratio from baseline. Significant differences were found on intracortical facilitation (ICF), short-intracortical inhibition (SICI), and long-interval cortical inhibition (LICI). Short-interval cortical facilitation (SICF) did not differ between groups. d Association between SICI and ICF in each group. A significant positive correlation was present in the FXS group, and significantly different from the one in the control group. Error bars indicate standard error of the mean.
Discussion
Recent evidence from animal studies showed the pivotal role of GABAergic inhibitory interneurons in the neurophysiological phenotype of FXS17,18,37. However, it remained unclear how these abnormalities translated to the human brain. The current study bridges this gap by providing evidence that GABAA-mediated intracortical inhibition is reduced in humans with FXS, and that this alteration co-occurs with increased intracortical circuit excitability.
It is recognized that SICI reflects the activity of cortical interneurons on post-synaptic GABAA receptor29. Importantly, since groups did not differ on SICF, it is unlikely that between-group differences in SICI level result from a contamination by indirect facilitatory waves34. The presence of altered GABAA mediated inhibition concurs with the only previous study directly assessing GABAergic function in FXS patients, where [11C]-flumazenil PET was used to show a reduction in GABAA receptor binding38. These results are in line with preclinical evidence showing a reduction of GABA synthesizing enzyme and GABAA receptor mRNA levels in animal models of the disorder2. Similarly, the number of parvalbumin expressing interneurons, the most abundant type of GABAergic interneurons in the cortex39,40, is reduced in Fmr1 KO animals41. Considering the fact that TMS is believed to interact predominantly with parvalbumin interneurons42,43, a reduction in this cell population could also contribute to the aberrant intracortical inhibition observed in FXS patients. Interestingly, recent evidence suggests a causal role between parvalbumin dysfunctions and impairments in sensory processing in FXS, a behavioral phenotype that is common to both animals and humans18. Sensory processing was not evaluated in the present study, thus the association between TMS measures of inhibition and the magnitude of sensory abnormalities in FXS patients remains to be investigated.
The between-group difference observed on ICF confirms the presence of circuit hyperexcitability in FXS patients, as was previously inferred by EEG studies4,5,7,44. Because ICF reflects, at least in part, the activity of NMDA receptors45, one explanation for this observation is that the increase in ICF is directly linked to the exacerbation of glutamate signaling described in animal models of the disorder3,11. However, considering that it is also modulated by other neurotransmitter systems, ICF can more accurately be conceived as “glutamatergic facilitation tempered by GABAergic inhibition”46, namely because the time interval at which ICF can be observed (6–20 ms) encompasses the tail of the GABAA-mediated inhibition measured by SICI47. Hence, while it is established that ICF and SICI depend on distinct neuronal populations, it is their interaction that ultimately controls the corticospinal output29,48. Further supporting a relationship between GABAA-mediated inhibition and intracortical excitability, pharmacological manipulations that reduce or enhance SICI have the opposite effect on ICF (reviewed in29), and changes in SICI can mediate variations in ICF49. Here, the fact that reduced SICI is associated with increased circuit excitability in FXS patients, as expressed by ICF, concurs with recent evidence obtained from in vivo animals pointing to faulty GABAA inhibition as a primary cause for circuit hyperexcitablity in the absence of FMRP17,18. Although the present data cannot ascertain that the alteration in GABAA signaling is a causative factor of hyperexcitability in FXS, it nonetheless supports the significance of the GABAA system as a potential target to correct a core neurophysiological feature of FXS.
Surprisingly, FXS patients showed enhanced LICI, which is primarily mediated through the activity of post-synaptic GABAB receptors50–52. The increase in LICI may explain why CSP values did not differ between both groups despite reduced SICI, as CSP amalgamates both GABAA and GABAB inhibition29. Although the mechanism responsible for the increase in LICI observed in FXS patients is elusive, it could reflect a compensatory mechanism. Indeed, there is cross talk between GABAA and GABAB receptors at the post-synaptic site in such a way that activation of GABAB receptors increases GABAA inhibition53. Whether such cross talk is at play in FXS remains to be established. However, the preservation of LICI can be related to evidence from mice models showing that post-synaptic GABAB receptors are spared in FXS, in contrast with what is observed at the presynaptic level19,20. Specifically, reduced presynaptic GABAB signaling has been reported at glutamatergic synapses19, while excessive presynaptic GABAB signaling appears to be present at inhibitory synapses20. Since presynaptic GABAB receptors are involved in auto-inhibition and regulate neurotransmitter release, both alterations are susceptible to contribute to circuit hyperexcitability. The presence of increased GABAB mediated inhibition described here, coupled with the apparent heterogeneity in terms of GABAB receptor function may explain, at least in part, the limited efficacy of selective GABAB agonists for treating FXS in recent clinical trials22,23.
The lack of difference in rMT between controls and FXS patients is intriguing considering that FMRP influences the activity of ion channels54, including voltage gated sodium channels which are believed to play a role in the cortical hyperexcitability of FXS55,56. It is however known that rMT is normal in several disorders associated with cortical hyperexcitability30,57, including in patients with Dravet syndrome, which is caused by a mutation of a gene directly involved in the proper function of voltage gated sodium channels24. It is possible that variables unrelated to neuronal function, such as the scalp-to-cortex distance58 and cerebral tissue structure59,60, may have obscured potential between-group differences. These evidence, coupled with abundant TMS data from epilepsy patients30, suggest that the rMT is less sensitive to hyperexcitability than paired-pulses protocols. Figure 3 recapitulates the main findings and the presumed underlying neurotransmitter dysfunctions of FXS.
Fig. 3. Schematic representation of the main findings.
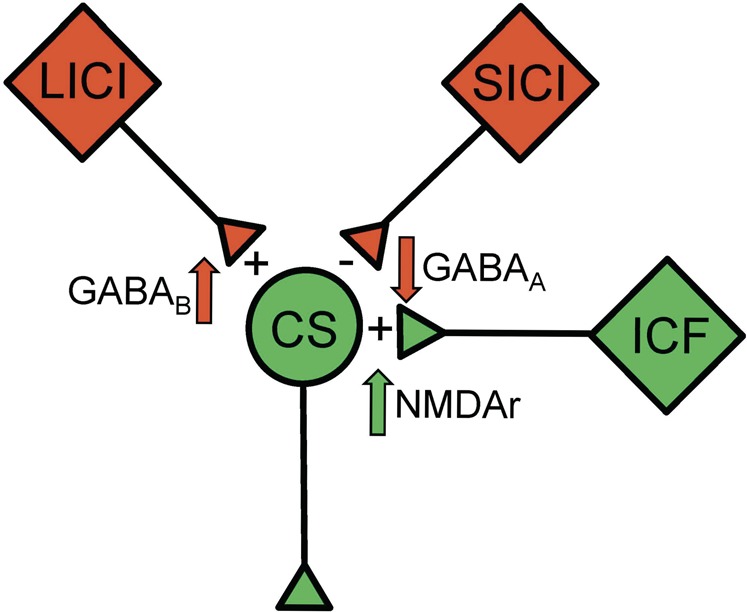
Inhibitory circuits are in red and excitatory circuits in green. The “plus” and “minus” signs indicate the observed effect compared to controls, and the nearby arrows specify the presumed mechanisms involved. CS corticospinal neuron, ICF intracortical facilitation, NMDAr N-methyl-d-aspartate receptors, LICI long-interval intracortical inhibition, SICI short-interval intracortical inhibition.
Interestingly, the overall pattern of TMS abnormalities of FXS patients is strikingly similar to what is seen in patients with a missense mutation of the γ2 subunit of the GABAA receptor (GABRG2(R43Q)), which is associated with an hereditary form of generalized epilepsy57. These patients exhibit normal rMT and CSP, but display reduced SICI, and an increase in ICF of similar magnitude to the one observed in FXS patients57. Furthermore, patients with a missense mutation of the γ2 subunit show reduced [11C]-flumazenil binding61, like FXS patients38. Given that [11C]-flumazenil interacts with benzodiazepine binding sites, it is possible that the reduction in binding observed in FXS patients does not result from an under expression of the γ2 subunit per se, but from the diminished expression of other GABAA receptor subunits composing this binding site, including α1,3, β1,2 and γ2 subunits, which are all under expressed in animal models of FXS62,63. It thus remains to be seen if the same alterations are responsible for the reduction in SICI and [11C]-flumazenil binding in FXS. It is also noteworthy that the TMS alterations common to FXS and GABRG2(R43Q) patients differ markedly from those seen in disorders involving other aspects of the GABAergic system, including patients with succinic semialdehyde dehydrogenase (SSADH) deficiency25, who lack an enzyme involved in GABA metabolism, and Prader-Willi syndrome, caused by deletion or imprinting defect of genes encoding α5, β3, and γ3 subunits of GABAA receptors64. Considering that all of these disorders show a ubiquitous reduction of [11C]-flumazenil binding38,61,65,66, the sensitivity of TMS to specific elements of the GABAergic system emerges as being particularly enlightening for clinical research in neurogenetic disorders associated with inhibitory dysfunctions.
The lack of objective outcome measures and biomarkers sensitive to the mechanisms targeted by experimental drugs is a major hurdle for clinical trials in FXS67–69. Biomarkers would benefit clinical trials by allowing a stratification of the clinical population and providing non-biased outcome measures. Eventually, they could be used to predict response to treatment and design personalized therapies. Because the consequences of FXS are primarily cognitive, emotional, and behavioral, there is tremendous interest in objectively assessing the impact of interventions on brain function. In that regards, interesting advances have been made with EEG70. Namely, auditory evoked EEG responses appeared partially normalized in FXS children treated with minocycline71, and recent data showed remarkably similar electrocortical responses in terms of neural synchronization between FXS patients and Fmr1 KO mice72,73, an important step towards the implementation of translational-relevant biomarkers in clinical trials. While additional research is required to assess the translational value of TMS markers for FXS, the upcoming results from two ongoing clinical trials including TMS as a secondary outcome could provide some answers (NCT02680379; NCT03722290). The unique features of TMS provide an interesting complement to the widely available EEG, notably by allowing for the discrimination of which inhibitory and excitatory processes aggregated in the EEG response are being modulated by an intervention. This is particularly relevant considering that all EEG markers put forth for FXS are hypothesized to involve, directly or indirectly, faulty GABAergic inhibition as an underlying mechanism6,71,73.
Limitations
Although large for a study involving FXS patients, the sample size remains limited, which might have induced type II errors. The heterogeneity of the sample in terms of medication status can also be considered as a limitation. However, more than half of our sample was medication free and still displayed alterations identical to those observed at the whole-group level, showing that our results were not due to the presence of psychoactive drugs. Due to time constraint and to maximize collaboration from low functioning patients, relevant measures such as input-output recruitment curves and late cortical disinhibition74, could not be performed. As it is usually the case in clinical studies with TMS, measures were obtained from stimulation of the primary motor cortex. It thus remains to be seen whether the alterations reported here extend to other cortical areas. This might well be the case, as the decrease in GABAA receptor binding reported by Hulst and colleagues38 seems fairly homogenous across the brain of FXS patients. The use of simultaneous TMS-EEG protocols could help assess the functional impact of this decrease in regions beyond the primary motor cortex. Indeed, using EEG to measure the brain response to TMS stimulations, recent studies have shown that paired-pulse TMS applied to the dorsolateral prefrontal cortex of healthy individuals induces EEG evoked potentials that are consistent with the electrophysiological responses typically derived from MEPs using SICI, ICF, and LICI protocols75,76. Although technically challenging, this approach could provide valuable insight on the inhibition-excitation imbalance across the cerebral cortex of patients with FXS.
Conclusion
This study shows the presence of aberrant inhibitory mechanisms in patients with FXS. These alterations offer a plausible explanation for the cortical hyperexcitability typical of this disorder, and suggest new avenues for pharmacological interventions targeting the GABAergic system. Further research is required to elucidate the precise neurochemical mechanisms responsible for the physiological alterations reported here. While the GABAergic system holds promises as a therapeutic target, the increase in GABAB inhibition illustrates the complex dynamics that are likely to be at play in terms of inhibitory mechanisms in FXS. These findings illustrate the usefulness of TMS in assessing intracortical excitability in FXS and, possibly, in monitoring it following treatment. These results also highlight the crucial need to better characterize the neurophysiology of humans with the disorder in order to accelerate the discovery of new treatment avenues.
Acknowledgements
The authors thank the participants and their family. This study was funded by the FRAXA Foundation (FC) and the Fonds de la Recherche du Québec-Santé (FRQS) (JL). FM is supported by a graduate scholarship from the Fondation du Grand Défi Pierre-Lavoie, CC is supported by a graduate scholarship from the New Brunswick Health Research Foundation, AL is supported by a graduate scholarship from the FRQS.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Greenblatt EJ, Spradling AC. Fragile X mental retardation 1 gene enhances the translation of large autism-related proteins. Science. 2018;361:709–712. doi: 10.1126/science.aas9963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Braat S, Kooy RF. The GABA-A receptor as a therapeutic target for neurodevelopmental disorders. Neuron. 2015;86:1119–1130. doi: 10.1016/j.neuron.2015.03.042. [DOI] [PubMed] [Google Scholar]
- 3.Contractor A, Klyachko VA, Portera-Cailliau C. Altered neuronal and circuit excitability in fragile X syndrome. Neuron. 2015;87:699–715. doi: 10.1016/j.neuron.2015.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferri R, et al. BIT-mapped somatosensory evoked potentials in the fragile X syndrome. Clin. Neurophysiol. 1994;24:413–426. doi: 10.1016/S0987-7053(05)80074-7. [DOI] [PubMed] [Google Scholar]
- 5.Knoth IS, Vannasing P, Major P, Michaud JL, Lippe S. Alterations of visual and auditory evoked potentials in fragile X syndrome. Int J. Dev. Neurosci. 2014;36:90–97. doi: 10.1016/j.ijdevneu.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 6.Ethridge LE, et al. Reduced habituation of auditory evoked potentials indicate cortical hyper-excitability in Fragile X Syndrome. Transl. Psychiatry. 2016;6:e787–8. doi: 10.1038/tp.2016.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van der Molen MJW, et al. Auditory change detection in fragile X syndrome males: a brain potential study. Clin. Neurophysiol. 2012;123:1309–1318. doi: 10.1016/j.clinph.2011.11.039. [DOI] [PubMed] [Google Scholar]
- 8.Gibson JR, Bartley AF, Hays SA, Huber KM. Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model of fragile X syndrome. J. Neurophysiol. 2008;100:2615–2626. doi: 10.1152/jn.90752.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garcia-Pino E, Gessele N, Koch U. Enhanced excitatory connectivity and disturbed sound processing in the auditory brainstem of fragile X mice. J. Neurosci. 2017;37:7403–7419. doi: 10.1523/JNEUROSCI.2310-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nelson SB, Valakh V. Excitatory/inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron. 2015;87:684–698. doi: 10.1016/j.neuron.2015.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27:370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 12.Westmark PR, Dekundy A, Gravius A, Danysz W, Westmark CJ. Rescue of Fmr1KO phenotypes with mGluR5 inhibitors MRZ-8456 versus AFQ-056. Neurobiol. Dis. 2018;119:190–198. doi: 10.1016/j.nbd.2018.08.008. [DOI] [PubMed] [Google Scholar]
- 13.Dölen G, et al. Correction of fragile X syndrome in mice. Neuron. 2007;56:955–962. doi: 10.1016/j.neuron.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bailey DB, et al. Mavoglurant in adolescents with fragile X syndrome: analysis of Clinical Global Impression-Improvement source data from a double-blind therapeutic study followed by an open-label, long-term extension study. J. Neurodev. Disord. 2016;8:1–10. doi: 10.1186/s11689-015-9134-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berry-Kravis E, et al. Mavoglurant in fragile X syndrome: Results of two randomized, double-blind, placebo-controlled trials. Sci. Transl. Med. 2016;8:321ra5. doi: 10.1126/scitranslmed.aab4109. [DOI] [PubMed] [Google Scholar]
- 16.Moskalyuk A., Kooy F. R., Giugliano M. Single-cell and neuronal network alterations in an in vitro model of Fragile X syndrome. Cereb. Cortex 2019, 10.1093/cercor/bhz068 (2019). [DOI] [PubMed]
- 17.Franco LM, Okray Z, Linneweber GA, Hassan BA, Yaksi E. Reduced lateral inhibition impairs olfactory computations and behaviors in a drosophila model of fragile X syndrome. Curr. Biol. 2017;27:1111–1123. doi: 10.1016/j.cub.2017.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goel A, et al. Impaired perceptual learning in a mouse model of fragile X syndrome is mediated by parvalbumin neuron dysfunction and is reversible. Nat. Neurosci. 2018;21:1404–1411. doi: 10.1038/s41593-018-0231-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kang JY, et al. Deficits in the activity of presynaptic γ-Aminobutyric acid type B receptors contribute to altered neuronal excitability in fragile X syndrome. J. Biol. Chem. 2017;292:6621–6632. doi: 10.1074/jbc.M116.772541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wahlstrom-Helgren S, Klyachko VA. GABA-B receptor-mediated feed-forward circuit dysfunction in the mouse model of fragile X syndrome. J. Physiol. 2015;593:5009–5024. doi: 10.1113/JP271190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.D’Hulst C, et al. Expression of the GABAergic system in animal models for fragile X syndrome and fragile X associated tremor/ataxia syndrome (FXTAS) Brain Res. 2009;1253:176–183. doi: 10.1016/j.brainres.2008.11.075. [DOI] [PubMed] [Google Scholar]
- 22.Berry-Kravis E, et al. Arbaclofen in fragile X syndrome: results of phase 3 trials. J. Neurodev. Disord. 2017;9:3. doi: 10.1186/s11689-016-9181-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berry-Kravis E, et al. Effects of STX209 (arbaclofen) on neurobehavioral function in children and adults with fragile X syndrome: a randomized, controlled, phase 2 trial. Sci. Transl. Med. 2012;4:152ra127. doi: 10.1126/scitranslmed.3004214. [DOI] [PubMed] [Google Scholar]
- 24.Stern WM, Sander JW, Rothwell JC, Sisodiya SM. Impaired intracortical inhibition demonstrated in vivo in people with Dravet syndrome. Neurology. 2017;88:1659–1665. doi: 10.1212/WNL.0000000000003868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reis J, et al. GABAB-ergic motor cortex dysfunction in SSADH deficiency. Neurology. 2012;79:47–54. doi: 10.1212/WNL.0b013e31825dcf71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Camprodon JA, Pascual-Leone A. Multimodal applications of transcranial magnetic stimulation for circuit-based psychiatry. JAMA Psychiatry. 2016;73:407–408. doi: 10.1001/jamapsychiatry.2015.3127. [DOI] [PubMed] [Google Scholar]
- 27.Murphy, S. C., Palmer, L. M., Nyffeler, T., Müri, R. M. & Larkum, M. E. Transcranial magnetic stimulation (TMS) inhibits cortical dendrites. Elife5, 735 (2016). [DOI] [PMC free article] [PubMed]
- 28.Li B, et al. Lifting the veil on the dynamics of neuronal activities evoked by transcranial magnetic stimulation. Elife. 2017;6:1918. doi: 10.7554/eLife.30552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ziemann U, et al. TMS and drugs revisited 2014. Clin. Neurophysiol. 2015;126:1–22. doi: 10.1016/j.clinph.2014.08.028. [DOI] [PubMed] [Google Scholar]
- 30.de Goede AA, Braack ter EM, van Putten MJAM. Single and paired pulse transcranial magnetic stimulation in drug naive epilepsy. Clin. Neurophysiol. 2016;127:3140–3155. doi: 10.1016/j.clinph.2016.06.025. [DOI] [PubMed] [Google Scholar]
- 31.Chen R, et al. The clinical diagnostic utility of transcranial magnetic stimulation: report of an IFCN committee. Clin. Neurophysiol. 2008;119:504–532. doi: 10.1016/j.clinph.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 32.Rossi S, et al. Safety, ethical considerations, and application guidelines for the use of transcranial magnetic stimulation in clinical practice and research. Clin. Neurophysiol. 2009;120:2008–2039. doi: 10.1016/j.clinph.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Groppa S, et al. A practical guide to diagnostic transcranial magnetic stimulation: Report of an IFCN committee. Clin. Neurophysiol. 2012;123:858–882. doi: 10.1016/j.clinph.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peurala SH, Müller-Dahlhaus JF, Arai N, Ziemann U. Interference of short-interval intracortical inhibition (SICI) and short-interval intracortical facilitation (SICF) Clin. Neurophysiol. 2008;119:2291–2297. doi: 10.1016/j.clinph.2008.05.031. [DOI] [PubMed] [Google Scholar]
- 35.Lepage JF, et al. Abnormal motor cortex excitability is associated with reduced cortical thickness in X monosomy. Hum. Brain Mapp. 2011;34:936–944. doi: 10.1002/hbm.21481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang, et al. Optimal number of pulses as outcome measures of neuronavigated transcranial magnetic stimulation. Clin. Neurophysiol. 2016;127:2892–2897. doi: 10.1016/j.clinph.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Antoine MW, Langberg T, Schnepel P, Feldman DE. Increased excitation-inhibition ratio stabilizes synapse and circuit excitability in four autism mouse models. Neuron. 2019;101:648–661. doi: 10.1016/j.neuron.2018.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.D’Hulst C. et al. Positron emission tomography (PET) quantification of GABAA receptors in the brain of fragile X patients. PLoS ONE10, e0131486–12 (2015). [DOI] [PMC free article] [PubMed]
- 39.Rudy B, Fishell G, Lee S, Hjerling-Leffler J. Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev. Neurobiol. 2010;71:45–61. doi: 10.1002/dneu.20853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kubota Y, Hattori R, Yui Y. Three distinct subpopulations of GABAergic neurons in rat frontal agranular cortex. Brain Res. 1994;649:159–173. doi: 10.1016/0006-8993(94)91060-X. [DOI] [PubMed] [Google Scholar]
- 41.Selby L, Zhang C, Sun Q-Q. Major defects in neocortical GABAergic inhibitory circuits in mice lacking the fragile X mental retardation protein. Neurosci. Lett. 2007;412:227–232. doi: 10.1016/j.neulet.2006.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Benali A, et al. Theta-burst transcranial magnetic stimulation alters cortical inhibition. J. Neurosci. 2011;31:1193–1203. doi: 10.1523/JNEUROSCI.1379-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kozyrev V, Eysel UT, Jancke D. Voltage-sensitive dye imaging of transcranial magnetic stimulation-induced intracortical dynamics. Proc. Natl Acad. Sci. USA. 2014;111:13553–13558. doi: 10.1073/pnas.1405508111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Castren M, Paakkonen A, Tarkka IM, Ryynanen M, Partanen J. Augmentation of auditory N1 in children with fragile X syndrome. Brain Topogr. 2003;15:165–171. doi: 10.1023/A:1022606200636. [DOI] [PubMed] [Google Scholar]
- 45.Ziemann U, Chen R, Cohen LG, Hallett M. Dextromethorphan decreases the excitability of the human motor cortex. Neurology. 1998;51:1320–1324. doi: 10.1212/WNL.51.5.1320. [DOI] [PubMed] [Google Scholar]
- 46.Reis J, et al. Contribution of transcranial magnetic stimulation to the understanding of cortical mechanisms involved in motor control. J. Physiol. 2008;586:325–351. doi: 10.1113/jphysiol.2007.144824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hanajima R, et al. Paired-pulse magnetic stimulation of the human motor cortex: differences among I waves. J. Physiol. 1998;509:607–618. doi: 10.1111/j.1469-7793.1998.607bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ziemann U, Rothwell JC, Ridding MC. Interaction between intracortical inhibition and facilitation in human motor cortex. J. Physiol. 1996;496:873–881. doi: 10.1113/jphysiol.1996.sp021734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Daskalakis ZJ, et al. Exploring the connectivity between the cerebellum and motor cortex in humans. J. Physiol. 2004;557:689–700. doi: 10.1113/jphysiol.2003.059808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McDonnell MN, Orekhov Y, Ziemann U. The role of GABAB receptors in intracortical inhibition in the human motor cortex. Exp. Brain Res. 2006;173:86–93. doi: 10.1007/s00221-006-0365-2. [DOI] [PubMed] [Google Scholar]
- 51.Werhahn KJ, Kunesch E, Noachtar S, Benecke R, Classen J. Differential effects on motorcortical inhibition induced by blockade of GABA uptake in humans. J. Physiol. 1999;517:591–597. doi: 10.1111/j.1469-7793.1999.0591t.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pierantozzi M, et al. Effect of Vigabatrin on motor responses to transcranial magnetic stimulation. Brain Res. 2004;1028:1–8. doi: 10.1016/j.brainres.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 53.Connelly WM, et al. GABAB receptors regulate extrasynaptic GABAA receptors. J. Neurosci. 2013;33:3780–3785. doi: 10.1523/JNEUROSCI.4989-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brown MR, et al. Fragile X mental retardation protein controls gating of the sodium-activated potassium channel Slack. Nat. Neurosci. 2010;13:819–821. doi: 10.1038/nn.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Deng P-Y, Klyachko VA. Increased persistent sodium current causes neuronal hyperexcitability in the entorhinal cortex of Fmr1 knockout mice. Cell Rep. 2016;16:3157–3166. doi: 10.1016/j.celrep.2016.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Routh BN, et al. Increased transient Na+ conductance and action potential output in layer 2/3 prefrontal cortex neurons of the fmr1−/ymouse. J. Physiol. 2017;595:4431–4448. doi: 10.1113/JP274258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fedi M, et al. Intracortical hyperexcitability in humans with a GABAA receptor mutation. Cereb. Cortex. 2008;18:664–669. doi: 10.1093/cercor/bhm100. [DOI] [PubMed] [Google Scholar]
- 58.Stokes MG, et al. Biophysical determinants of transcranial magnetic stimulation: effects of excitability and depth of targeted area. J. Neurophysiol. 2013;109:437–444. doi: 10.1152/jn.00510.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.List J, et al. Relationship between excitability, plasticity and thickness of the motor cortex in older adults. NeuroImage. 2013;83:809–816. doi: 10.1016/j.neuroimage.2013.07.033. [DOI] [PubMed] [Google Scholar]
- 60.Klöppel S, et al. The cortical motor threshold reflects microstructural properties of cerebral white matter. NeuroImage. 2008;40:1782–1791. doi: 10.1016/j.neuroimage.2008.01.019. [DOI] [PubMed] [Google Scholar]
- 61.Fedi M, et al. A GABAA receptor mutation causing generalized epilepsy reduces benzodiazepine receptor binding. NeuroImage. 2006;32:995–1000. doi: 10.1016/j.neuroimage.2006.05.059. [DOI] [PubMed] [Google Scholar]
- 62.D’Hulst C, et al. Decreased expression of the GABAA receptor in fragile X syndrome. Brain Res. 2006;1121:238–245. doi: 10.1016/j.brainres.2006.08.115. [DOI] [PubMed] [Google Scholar]
- 63.Braat S, et al. The GABA Areceptor is an FMRP target with therapeutic potential in fragile X syndrome. Cell Cycle. 2015;14:2985–2995. doi: 10.4161/15384101.2014.989114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Civardi C, Vicentini R, Grugni G, Cantello R. Corticospinal physiology in patients with Prader–Willi syndrome: a transcranial magnetic stimulation study. Arch. Neurol. 2004;61:1585–1589. doi: 10.1001/archneur.61.10.1585. [DOI] [PubMed] [Google Scholar]
- 65.Pearl PL, et al. Decreased GABA-A binding on FMZ-PET in succinic semialdehyde dehydrogenase deficiency. Neurology. 2009;73:423–429. doi: 10.1212/WNL.0b013e3181b163a5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lucignani G, et al. GABAA receptor abnormalities in Prader–Willi syndrome assessed with positron emission tomography and [11C]flumazenil. NeuroImage. 2004;22:22–28. doi: 10.1016/j.neuroimage.2003.10.050. [DOI] [PubMed] [Google Scholar]
- 67.Riley C, Mailick M, Berry-Kravis E, Bolen J. The future of fragile X syndrome: CDC stakeholder meeting summary. Pediatrics. 2017;139:S147–S152. doi: 10.1542/peds.2016-1159B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Berry-Kravis EM, et al. Drug development for neurodevelopmental disorders: lessons learned from fragile X syndrome. Nat. Rev. Drug Disco. 2018;17:280–299. doi: 10.1038/nrd.2017.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jacquemont S, et al. The challenges of clinical trials in fragile X syndrome. Psychopharmacol. (Berl.) 2013;231:1237–1250. doi: 10.1007/s00213-013-3289-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sinclair D, Oranje B, Razak KA, Siegel SJ, Schmid S. Sensory processing in autism spectrum disorders and Fragile X syndrome—from the clinic to animal models. Neurosci. Biobehav Rev. 2016;76:235–253. doi: 10.1016/j.neubiorev.2016.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schneider A, et al. Electrocortical changes associated with minocycline treatment in fragile X syndrome. J. Psychopharmacol. (Oxf.) 2013;27:956–963. doi: 10.1177/0269881113494105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lovelace JW, Ethell IM, Binder DK, Razak KA. Translation-relevant EEG phenotypes in a mouse model of fragile X syndrome. Neurobiol. Dis. 2018;115:39–48. doi: 10.1016/j.nbd.2018.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ethridge LE, et al. Neural synchronization deficits linked to cortical hyper-excitability and auditory hypersensitivity in fragile X syndrome. Mol. Autism. 2017;8:22. doi: 10.1186/s13229-017-0140-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cash RFH, Ziemann U, Murray K, Thickbroom GW. Late cortical disinhibition in human motor cortex: a triple-pulse transcranial magnetic stimulation study. J. Neurophysiol. 2010;103:511–518. doi: 10.1152/jn.00782.2009. [DOI] [PubMed] [Google Scholar]
- 75.Premoli I, et al. Characterization of GABAB-receptor mediated neurotransmission in the human cortex by paired-pulse TMS–EEG. NeuroImage. 2014;103:152–162. doi: 10.1016/j.neuroimage.2014.09.028. [DOI] [PubMed] [Google Scholar]
- 76.Cash RFH, et al. Characterization of glutamatergic and GABAA-mediated neurotransmission in motor and dorsolateral prefrontal cortex using paired-pulse TMS&EEG. Neuropsychopharmacology. 2016;42:502–511. doi: 10.1038/npp.2016.133. [DOI] [PMC free article] [PubMed] [Google Scholar]