


Abstract
The momentum of scRNA-seq datasets prompts for simple and powerful tools exploring their meaningful signatures. Here we present Single-Cell_Signature_Explorer (https://sites.google.com/site/fredsoftwares/products/single-cell-signature-explorer), the first method for qualitative and high-throughput scoring of any gene set-based signature at the single cell level and its visualization using t-SNE or UMAP. By scanning datasets for single or combined signatures, it rapidly maps any multi-gene feature, exemplified here with signatures of cell lineages, biological hallmarks and metabolic pathways in large scRNAseq datasets of human PBMC, melanoma, lung cancer and adult testis.
INTRODUCTION
The development of single cell RNA sequencing (scRNA-seq) techniques yields an increasing number of datasets available to the scientific community. These comprise reference samples of human tissues and organs from healthy individuals as in the Human Cell Atlas repository (1), or published studies of samples from dysfunctional or pathological tissues. Therefore, it becomes important to assess in each single cell from such large data sets any kind of hallmark possibly defined by a gene set, and to visualize this feature across the dimensionality reduction plots such as those produced by t-distributed stochastic neighborhood embedding (t-SNE) (2) or Uniform Manifold Approximation and Projection (UMAP) (3) in a global and comprehensive viewpoint. This is conceptually similar to gene set enrichment analysis (GSEA) (4), which finds the functional significance of genes differentially expressed between bulk transcriptomes of two series of samples. Currently however, there is no method for performing this at a single cell level and to compare all the dataset’s cells, in a reasonable time scale. Here we report Single Cell Signature Explorer, an algorithm which rapidly scores many gene set-defined signatures at the single cell level as qualitative measures for their straightforward and interactive visualization using t-SNE or UMAP plots of scRNA-seq data sets, together with highly informative examples of its applications.
MATERIALS AND METHODS
Single cell experiments
Data for 4k and 8k PBMC obtained by 10× Genomics 3′ chemistry V2 were downloaded from the 10× Genomics website (https://support.10xgenomics.com/single-cell-gene-expression/datasets/2.1.0/pbmc8k, https://support.10xgenomics.com/single-cell-gene-expression/datasets/2.1.0/pbmc4k).
Data for 10k PBMC obtained by 10× Genomics 3′ chemistry V3 were downloaded from the 10× Genomics website (https://support.10xgenomics.com/single-cell-gene-expression/datasets/3.0.0/pbmc_10k_protein_v3).
Data for 33k PBMC obtained by 10× Genomics 3′ chemistry V1 were downloaded from the 10× Genomics website (https://support.10xgenomics.com/single-cell-gene-expression/datasets/1.1.0/pbmc33k)
Data for 19k of lung tumor and normal adjacent cells (5) obtained by 10× Genomics 3′ chemistry V2 were downloaded from Array Express E-MAAB-6149 and E-MTAB-6653.
The human spermatogenesis data (6) obtained by 10× Genomics 3′ chemistry V2 were downloaded from the GEO data set GSE120508.
The 64k melanoma dataset obtained by MARS platform (7) were downloaded from GEO data set GSE12313.
Noise reduction of datasets
Single cell transcriptomic technology is powerful to explore gene expression at the single cell level, but may yield datasets in which technical noise can hinder biological variability. This technical noise from gene sampling fluctuations and cell-to-cell variations in sequencing efficiency (8) has motivated the development of various methods performing pre-processing of raw count normalizations (9,10,11,12,13,14). These either apply a single per-cell scaling factor to all gene counts, or deploy statistical models estimating the per-gene errors of UMI counts. However, a single scaling factor cannot optimally normalize both lowly and highly expressed genes, while standard statistical modeling by negative binomial regression overfit the UMI data (15). A novel normalization procedure that addresses both issues by using regularized negative binomial regression was recently developed. This pre-processing tool, named sctransform (15), normalizes and stabilizes the technical noise variance of UMI counts prior to downstream analyses by principal component analysis (PCA), dimensionality reduction and further exploration of results. Unless stated otherwise, the data sets analyzed below using Single-Cell Signature Explorer were UMI counts pre-processed by sctransform normalization.
t-SNE/UMAP plots
The t-SNE or UMAP plots were produced by Seurat (16) as follows: the raw data (FastQ files) were computed with CellRanger 3.0 and then loaded in an R session with the Seurat 3.0 toolkit package involving the normalization and variance stabilization package sctransform (15). Samples were individually filtered using UMI and percentage of mitochondrial genes criteria. Samples were then merged using correction to align datasets as described in (16). t-SNE or UMAP coordinates were then calculated using the 11 first PCA and exported in a table.
Single-Cell Signature Explorer
Single-Cell Signature Explorer is a package of four successive tools dedicated to high throughput signature exploration in single-cell analysis:
Single-Cell Signature Scorer computes a signature score for each cell.
Single-Cell Signature Merger collates the signature score table with t-SNE and UMAP coordinates.
Single-Cell Signature Viewer displays signature scores on a t-SNE or UMAP plot.
Single-Cell Signature Combiner displays arithmetic combinations of two signature scores on a t-SNE or UMAP plot.
These four softwares were developed with usability, low memory usage and performance in mind. They require no complex command lines or computing skills, they can be used on a laptop but also scale up well on powerful workstations for fast computations. Results are obtained within minutes as a function of the number of explored gene sets (n) and size of each gene set (s): O(n*s).
Single-Cell Signature Scorer
This algorithm computes in each single cell transcriptome a score for any gene set from a database. More than 17 000 curated gene sets from MSigDB (4,17) and Reactome (18), as well as additional user-defined gene sets can be computed by the software. Each gene set is a list of HUGO gene symbols in a text file format labeled by the name of the gene set. The database can also be implemented by additional custom pathways composed of text files for the user-defined list of HUGO gene symbols. Scores were calculated as follows : The score of the GSx gene set in the Cj cell was computed as the sum of all UMI for all the GSx genes expressed in Cj, divided by the sum of all UMI expressed by Cj:
score of Cj cell for GSx gene set 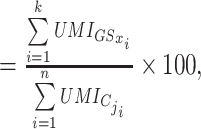
where 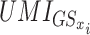 are the UMI of the k genes from GSx found in the Cj cell . We also implemented the possibility to subtract the contribution of a gene when it is preceded by a ‘-’ sign in the gene list. Although single cell signature scores can be computed from raw UMI counts, their relative variance improves when computed from UMI normalized by Seurat’s sctransform (see below). The signature scores primarily represent a qualitative measure for further visualization on a t-SNE or other dimensionality reduced map, rather than a quantitative measure. In addition, as the number of genes in each signature is highly variable, a single cell score for a signature cannot be compared to that of another signature. The Single-Cell Signature Scorer was developed in Go language v1.11.5 (https://golang.org/). The software is multi-threaded to take full advantage of multicore processors. Single-Cell Signature Scorer can compute 13 million (1000 gene sets for 13 300 cells) scores in <4′30″ on a bi-Xeon E5-2687w-v3 workstation. The Single-Cell Signature Scorer was compiled for GNU Linux and Microsoft©Windows™64 bits, and can be compiled for any platform using cross-compilation by Go.
are the UMI of the k genes from GSx found in the Cj cell . We also implemented the possibility to subtract the contribution of a gene when it is preceded by a ‘-’ sign in the gene list. Although single cell signature scores can be computed from raw UMI counts, their relative variance improves when computed from UMI normalized by Seurat’s sctransform (see below). The signature scores primarily represent a qualitative measure for further visualization on a t-SNE or other dimensionality reduced map, rather than a quantitative measure. In addition, as the number of genes in each signature is highly variable, a single cell score for a signature cannot be compared to that of another signature. The Single-Cell Signature Scorer was developed in Go language v1.11.5 (https://golang.org/). The software is multi-threaded to take full advantage of multicore processors. Single-Cell Signature Scorer can compute 13 million (1000 gene sets for 13 300 cells) scores in <4′30″ on a bi-Xeon E5-2687w-v3 workstation. The Single-Cell Signature Scorer was compiled for GNU Linux and Microsoft©Windows™64 bits, and can be compiled for any platform using cross-compilation by Go.
Single-Cell Signature Merger
This algorithm merges scores from Single-Cell Signature Scorer and t-SNE coordinates produced by Seurat (16) to produce tables compatible with the Single-Cell Signature Viewer (see below). This software is multi-threaded and can merge in parallel a set of score tables with a t-SNE or UMAP coordinate table.
Single-Cell Signature Viewer
This tool was developed to display the gene set scores computed by Single-Cell Signature Scorer on a t-SNE map. It takes as input a table score merged with t-SNE coordinates. Single-Cell Signature Viewer was written in R (19) with the Shiny package (20) to draw in real time a colored score scale of signatures selected from a drop-down list on t-SNE or UMAP maps. Since this scaling is sensitive to outliers, the viewer draws a density distribution of scores and provides a color scale cursor allowing users to prune such potential outliers.
Single-Cell Signature Combiner
This tool was developed to display the combination of two gene set scores previously computed by Single-Cell Signature Scorer on a t-SNE map. The user must select two signatures from two drop-down lists. As stated above, the highly variable number of genes in signatures does not allow the comparison of a single cell score for a signature to that of another signature. Therefore prior to combining the two corresponding score sets, they are normalized between [0-1]. Operators that are available to compute the combination are [− + *], thus the user selects to add, subtract or multiply two signatures prior to visualizing the resulting score on a map. Importantly, those combined scores should only be interpreted in the context of the changes observed across multiple cells. Single-Cell Signature Combiner takes as input a table score merged with t-SNE or UMAP coordinates. Single-Cell Signature Combiner was written in R (19) with the Shiny package (20) to draw in real time on a t-SNE or UMAP plot a colored score scale of two signatures selected from the drop-down lists. As above, since this scaling is sensitive to potential outliers, the viewer draws a density distribution of scores and provides a color scale cursor allowing the user to prune such outliers.
Code availability
Single-Cell Signature Explorer was developed using the high performance cross-platform Go language v1.11.1 (https://golang.org/) and the map viewers were developed using the cross-platform R (19) language with the Shiny package (20). Files can be accessed at (https://sites.google.com/site/fredsoftwares/products/single-cell-signature-explorer)
RESULTS AND DISCUSSION
Scoring of gene signature across single cells
Although GSEA measures the relative enrichment of functionally defined sets of genes, it cannot characterize the gene signature from an isolated transcriptome, while the related ssGSEA (21), GSVA (22) and AutoCompare_SES (23) achieve this by scoring the intrinsic enrichment of pre-defined gene sets in any single sample transcriptome. Nevertheless, both algorithms rely on statistics that are inappropriate for single cell transcriptomes mostly composed of zero values, and where the missing genes of each cell vary extensively across the entire scRNA-seq dataset. To address these issues with minimal computing time, here we introduce Single-Cell Signature Explorer, a package of four softwares dedicated to high-throughput signature exploration in single-cell transcriptome analysis. The raw data are pre-processed with CellRanger 3.0 and then with the Seurat 3.0 and sctransform to normalize and stabilize variance of UMI counts across the data set. The score of the GSi gene set in the Cj cell is then computed as the sum of all UMI for all the GSi genes expressed by Cj, divided by the sum of all UMI expressed by Cj. Using this, the scores of each single cell for thousands of gene sets (e.g. 17 000 gene sets from the Molecular Signatures Database (MSigDB) (http://www.broad.mit.edu/gsea/msigdb/index.jsp) are computed within a few minutes, and these scores are then visualized across entire scRNA-seq datasets.
B-cell signature score and relevance
As a first example, the scRNA-seq datasets of 4k and 8k PBMC from one healthy donor were downloaded (https://support.10xgenomics.com/single-cell-gene-expression/datasets), integrated, processed and the resulting t-SNE was plotted. In parallel, each of these 12k single cells was individually scored for each of the ∼15 000 MSIgDB gene sets. As negative controls, these cells were also scored likewise for 1000 random gene sets, each composed of n randomly sampled genes without replacement (RGS1-RGS1000). The PBMC data set comprises a cluster of (n = 1731) B lymphocytes which can be defined either as cells expressing the B-cell identifying CD19 gene alone (n = 883 cells), or as (n = 1826) cells scoring for a ‘B cell’ signature composed of the four genes CD79A, CD79B, CD19, MS4A11. Both cell counts and ROC curves indicated that the B-cell signature outperformed the single gene in spotting all B lymphocytes from the dataset, including the few (n = 109) plasmocytes which were mapped outside of the B-cell cluster (Figure 1). Single-Cell Signature Explorer may process datasets of raw or normalized UMI counts, yielding similar B-cell signature scores from these different data. Nevertheless the relative variability of B-cell signature scores was decreased within all B-cell clusters when computed from normalized UMI counts instead of raw UMI, and this decrease was even better with sctransform-normalized data than with log-normalized data (Supplementary Figure S1). By contrast, no such improvement was seen for the other clusters of non-B cells, for which this signature is not relevant. Hence analyses such as those performed with Single-Cell Signature Explorer benefit from prior data normalization and variance stabilization by Pearson residual as recently reported (15). We then questioned the relevance of the above result by searching for other signatures which might overlay with the B-cell signature. Although random signatures were not enriched across the t-SNE, 57 MSigDB signatures correlated (Pearson r > 0.75) with -and superimposed to- the above ‘B cell’ signature. These signatures comprised relevant gene sets such as ‘B-cell activation’, ‘CD22-mediated BCR regulation’, ‘GO-Immunoglobulin complex’, as well as less relevant signatures that were nevertheless fully consistent with B-cell biology such as ‘de novo protein folding’ and ‘somatic mutation’, (Supplementary Table S1 and Figure S1). Importantly by contrast, no signature defining other non-B-cell lineages was enriched in this cluster (not shown), suggesting that the transcriptomic B-cell signature was specifically mapped in this complex dataset.
Figure 1.

Visualization of gene and signatures in the t-SNE map of 12k PBMC from an healthy individual. Top left: expression of the CD19 gene. Top and bottom right: scores for ‘B-cell signature’, ‘random gene set’, ‘de novo post-translational protein folding’ and ‘adaptive immune response based on somatic recombination of immune receptors built from immunoglobulin superfamily domains’ (both from Gene Ontology) displayed on the t-SNE map. ROC curve (Bottom left) of B cell identification performance by using either the CD19 gene alone (violet curve) or the B-cell signature score (red curve).
Signature score specificity and robustness
To definitively validate the specificity of scores, we reasoned that PBMC formally identified as B lymphocytes by their cell surface expression of the CD19 antigen should match with cells identified by the above B-cell signature. We thus downloaded another data set comprising 10k PBMC from a healthy donor analyzed for both gene and cell surface protein expression by the Feature-Seq method (24). This dataset was processed as above, its UMAP was plotted and the PBMC were visualized for expression of the CD19 cell surface marker and for the B-cell signature score. Both criteria appeared overlaid (Figure 2A), supporting the specificity of signature mapping. Concurrent plots of each B-cell signature gene, taken individually, versus the cell surface CD19 expression displayed frequent CD19+ B cells lacking expression of some B-cell signature genes (Figure 2B), confirming that the signature outperformed its single genes in identifying B lymphocytes.
Figure 2.

Visualization of antibody, genes and signature on UMAP of 10k PBMC from an healthy individual. (A) UMAP with the CD19 cell surface marker (antibody detection, left) and B-cell signature score (right). (B) Data from 10k PBMC chemistry 3 were used to compare the CD19 cell surface (antibody detection, y-axis) to CD19, CD79A, CD79B and MS4A1 genes or B-cell score (x-axis). (C) Violin plots of B-cell signature scores within B cells, PC or others cell clusters computed by Single Cell Signature Scorer from three different datasets: 10k PBMC from 10x Genomics chemistry 3, 8k PBMC from chemistry 2 and 33k from chemistry 1. Red dotted line indicates the mean of the dataset.
The robustness of the B-cell signature was then analyzed in three datasets from PBMC’s of healthy donors with the V1, V2 and V3 generations of the 10× Genomics 3′ chemistry platforms. The B-cell signature scores and their relative intracluster variances were then compared for the clusters of B cell, plasma cells (PC) and the non-B-cell clusters from each generation of platform. Although as expected, all the relative intra-cluster variances decreased with higher generations of platform, the B-cell signature scores consistently differentiated the B cells from the non-B-cell clusters with both platforms (Wilcoxon P < 10−300 for V1, V2 and V3) (Figure 2C). The same conclusion was reached for a further dataset of 64K melanoma tumor cells obtained with the MARS platform, processed as above and plotted as UMAP (7) (GSE12313, Figure 3). The scores therefore consistently discriminated B from non-B cells in various datasets, chemistries and platforms.
Figure 3.

Visualization of B cell and PC signatures on the UMAP plot of 64k cells from melanoma biopsy produced by the MARS-Seq technology (7) (GSE123139). left: Signature scores calculated by Single-Cell Signature Explorer right: Expression level of single genes defining these respective populations as described in (7).
Multipurpose applications of Single-Cell Signature Explorer
Owing to the versatility of signatures from user-defined gene sets or curated databases, Single-Cell Signature Explorer allows not only to visualize relative enrichment of any gene set-based hallmark, but also to combine several of these, such as cell lineage, metabolic pathway and or biological function, as exemplified below. Of note however, the highly variable number of genes in signatures precludes comparisons of a single cell score for two different signatures. Instead, combined signature scores allow straightforward visualization of their variation across the dataset.
In the above-depicted 12k PBMC dataset (Figure 1), myeloid cells were identified by a myeloid-specific signature composed of 31 genes (Supplementary Table S2 and Figure S2a). Then, visualization of metabolic pathway signatures showed that in these resting PBMC, myeloid cells display higher scores than lymphocytes for glycolysis, oxidative phosphorylation (OXPHOS) and tricarboxylic acid (TCA) cycle, but not for fatty acid synthesis (Supplementary Figure S2b), consistent with (25).
In cancer, the metabolic reprogramming is even more pronounced within immune cells from the intra-tumoral microenvironment (26). Using Single-Cell Signature Explorer, this feature was explored in a lung cancer dataset comprising matched samples of malignant and adjacent non-malignant tissues. The scRNA-seq datasets of 19k cells from 20 tumors and 4 matched normal adjacent biopsies from 6 lung cancer patients (5) were downloaded from Array Express E-MTAB-6149 and E-MTAB-6653, processed, integrated, sctransform-normalized and the resulting t-SNE was plotted. Each of these 19k single cells was then scored for all the ∼15 000 human gene sets from MSigDB. Visualizing a series of lineage-defining signatures identified the cell types of each cluster (Supplementary Figure S3) which were consistent with their previous study (5). Myeloid cells from either the tumor or adjacent tissue were then gated in parallel analyses, and their respective scores for the metabolic signatures were visualized as above. Their comparison showed the higher glycolytic scores of some intra-tumoral myeloid cells compared to adjacent myeloid cells, while all cells scored similarly for the other pathways. Interestingly, lung cancer cells also displayed this higher glycolytic signature, consistent with the glycolytic bias of lung cancer (27,28) (Figure 4).
Figure 4.

Metabolic signatures of myeloid cells in 19k cells from lung adenocarcinoma tumors and normal adjacent tissue (5). (A) Myeloid cell signature in the lung adenocarcinoma tumors and normal adjacent samples. (B) Single cell scores for the specified metabolic pathways in myeloid cells (gated as shown in panel A) from either the tumor (middle panels, red) or adjacent normal tissue (right panels, blue). Left panel shows the comparative distributions of scores for the specified pathway in intra-tumoral (red) versus normal adjacent (blue) myeloid cells. (C) Same pathways visualized across the entire t-SNE featuring cancer cell clusters (dotted circles).
These examples showed that Single-Cell Signature Explorer enables users to visualize two distinct hallmarks such as cell identities and biological hallmarks. We then reasoned that this strategy enabled the visualization of two signatures combined through simple subtraction or addition of their normalized scores, and projection of the result on a dimensionality reduction map. This was illustrated using the human testis cell atlas scRNASeq dataset of 6.5 k cells involved in adult spermatogenesis (6). This publicly available data set was downloaded, processed as above and its partitioning into 17 clusters was projected onto a UMAP featuring the germ cell maturation trajectory reported earlier (6). Both cluster identities and positions on the trajectory (Figure 5A) were consistent with the molecular transitions underlying this development (6) . Of note, the ‘GO_embryonic organ development’ signature was observed in spermatogonial stem cells (SSC) and their cell partners (testicular macrophages, Leydig, Sertoli, myoid and endothelial cells) (Supplementary Figure S4a), while ‘GO_male meiosis’ and ‘Hallmark_spermatogenesis’ signatures hit only the early primary spermatocytes and mature sperm cells, respectively. The ‘GO_mitochondrion’ signature gradually decreased along the spermatocyte maturation, contrasting with other micro-environmental cells.
Figure 5.

Single and combined signatures of metabolic pathways in human adult testis cells. SSC: spermatogonial stem cells. (A): Human adult testis cell types and their maturation trajectory UMAP. (B): (bottom panels) Single signatures of either ‘glycolysis glucogenesis’ (KEGG, 62 genes), or ‘oxidative phosphorylation’ (KEGG, 136 genes), and (top panel) their combined signature using the ‘minus’ operator.
At last, there are debates as to whether the spermatogenesis energy is fueled by mitochondrial oxidative phosphorylation, or by glycolysis, or both continuously, or even by their stage-specific complementation. Here, displaying across the same UMAP the combined score for the ‘GO_Glycolysis’ gene set minus the ‘GO_OXPHOS’ gene set shed light on the contrasted glycolytic imbalance which progressively settles along sperm cell maturation (Figure 5B). This result was also consistent with the above mentioned decrease of mitochondria in maturing spermatozoids (Supplementary Figure S4a). Furthermore, the progressive catabolic switch of the glucose metabolism could be evidenced by the combined score for the ‘GO_Glucose metabolic process’ gene set minus the ‘GO_Glucose catabolic process’ gene set (Supplementary Figure S4b).
These results illustrated the versatility of the Single-Cell Signature Explorer for visualizing combinations of distinct signatures able to produce newer displays complementing those from single signatures.
Software benchmarks
The recently published algorithms Seurat’s Cell CycleScore module (16), AUCell (29) and GSVA/ssGSEA (21) can also compute the enrichment scores of gene set-based signatures from single cell transcriptomes. We thus compared Single-Cell Signature Explorer with these algorithms for their respective computation efficiency and displayed the results on a t-SNE map. Benchmarks were performed on a Linux Xubuntu 18.10 workstation with two processors Xeon E5-2687w-v3 and 128Gb RAM. The KEGG database was downloaded from MSigDB v6.2 (http://www.broad.mit.edu/gsea/msigdb/index.jsp). For this benchmark of Single-Cell Signature Scorer, the KEGG database was downloaded from the software web site (https://sites.google.com/site/fredsoftwares/products/databases) where additional, ready-to-use databases of ∼17 000 gene sets for Human, Mouse, Rat, Zebra Fish and Macaccus obtained from MSigDB v6.2 are also available .
For the ssGSEA speed test, both software issues and the excessive computation time (>12 h) for the entire KEGG database did not permit this comparative evaluation. Hence this speed test was only performed on two gene sets and its result was extrapolated. Since Seurat function CellCycleScore can only compute gene sets with anti-correlated expression levels, Seurat computation time for all gene sets of the KEGG database could not be performed.
Performance, computing time and display of the benchmark results are shown in Supplementary Table S3 and Figure S5.
While Seurat CellCycleScoring function computes almost as quickly, and yields similar results, as Single-Cell Signature Explorer (Supplementary Figure S6), it only scores few gene sets pairs with anti-correlated expression and does not display t-SNE plots or UMAP of the results. GSVA/ssGSEA, incepted for scoring gene sets from bulk transcriptomes but not zero-inflated single cell transcriptomes, requires such tedious computing times for a single signature that it cannot perform massive parallel scoring of large collections of gene sets, and does not display t-SNE results. Although the gene rank-based AUCell algorithm computes scores five-times faster than GSVA/ssGSEA, it remains 30-times slower than Signature Explorer and does not display interactive t-SNE maps of the resulting scores.
In addition to its computing performances, the versatility of Single-Cell Signature Explorer permits analysis of data from various scRNA-Seq and sequencing platforms (Figure 3). Beyond the above B-cell signature of 64K human melanoma cells from MARS-Seq, our algorithm allows likewise exploration of non-human samples using gene set databases for macaccus, mouse, rat and zebra fish (https://sites.google.com/site/fredsoftwares/products/databases). Thus Single-Cell Signature Explorer represents the reference tool for general exploration of scRNASeq datasets.
CONCLUSION
Functionally meaningful displays of gene set-based signature enrichment are essential to understand t-SNE or UMAP maps of complex cell samples, but so far, no current method perform this rapidly for a plethora of signatures at the single cell level across large scRNAseq datasets. By quickly delineating multi-gene features such as cell lineage or metabolic pathways with Single-Cell Signature Explorer in lung tumors and normal human blood and testis, we showed that gene set-based signatures outperform single genes and provide a straightforward visualization of the sample’s hallmarks. Within a few minutes from any computer, this new method provides users with thousands of gene set-based signatures for thousands of single cells, and the immediate visualization in t-SNE or UMAP of any of these single signatures or combination of signatures. The compatibility of Single-Cell Signature Explorer to any scRNASeq platform, sequencing technology and used-defined or curated gene set renders its applications as broad as the scRNAseq technology itself.
Supplementary Material
ACKNOWLEDGEMENTS
Aviv Regev and Asaf Madi (Broad Institute) are acknowledged for kindly sharing R scripts for density plots, Eric Espinosa (CRCT) for providing the mastocyte gene set and Cathy Greenland for english editing of the manuscript. This work was granted access to the HPC resources of CALMIP supercomputing center under the allocation 2019-T19001. We are also grateful to the Genotoul bioinformatics platform (Bioinfo Genotoul, Toulouse Midi-Pyrenees) for providing computing resources.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Centre National de la Recherche Scientifique; Université de Toulouse; Institut National de laSanté et de la Recherche Médicale; Labex Toucan 2 Funding for open access charge: INSERM.
Conflict of interest statement. None declared.
REFERENCES
- 1. Regev A., Teichmann S.A., Lander E.S., Amit I., Benoist C., Birney E., Bodenmiller B., Campbell P., Carninci P., Clatworthy M. et al.. The human cell atlas. Elife. 2017; 6:e27041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Maaten L.v.d., Hinton G.. Visualizing data using t-SNE. J. Machi. Learn. Res. 2008; 9:2579–2605. [Google Scholar]
- 3. Becht E., McInnes L., Healy J., Dutertre C.A., Kwok I.W., Ng L.G., Ginhoux F., Newell E.W.. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2019; 37:38–44. [DOI] [PubMed] [Google Scholar]
- 4. Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. et al.. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lambrechts D., Wauters E., Boeckx B., Aibar S., Nittner D., Burton O., Bassez A., Decaluw H., Pircher A., Van den Eynde K. et al.. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med. 2018; 24:1277–1289. [DOI] [PubMed] [Google Scholar]
- 6. Guo J., Grow E.J., Mlcochova H., Maher G.J., Lindskog C., Nie X., Guo Y., Takei Y., Yun J., Cai L. et al.. The adult human testis transcriptional cell atlas. Cell Res. 2018; 28:1141–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li H., van der Leun A.M., Yofe I., Lubling Y., Gelbard-Solodkin D., van Akkooi A.C., van den Braber M., Rozeman E.A., Haanen J.B., Blank C.U. et al.. Dysfunctional CD8 T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell. 2018; 176:775–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Grün D., Kester L., Van Oudenaarden A.. Validation of noise models for single-cell transcriptomics. Nat. Methods. 2014; 11:637–640. [DOI] [PubMed] [Google Scholar]
- 9. Bacher R., Chu L.-F., Leng N., Gasch A.P., Thomson J.A., Stewart R.M., Newton M., Kendziorski C.. SCnorm: robust normalization of single-cell RNA-seq data. Nat. Methods. 2017; 14:584–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vallejos C.A., Marioni J.C., Richardson S.. BASiCS: Bayesian analysis of single-cell sequencing data. PLoS Comput. Biol. 2015; 11:e1004333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lun A.T., McCarthy D.J., Marioni J.C.. A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Research. 2016; 5:2122–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Risso D., Perraudeau F., Gribkova S., Dudoit S., Vert J.-P.. A general and flexible method for signal extraction from single-cell RNA-seq data. Nat. Commun. 2018; 9:284–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lopez R., Regier J., Cole M.B., Jordan M.I., Yosef N.. Deep generative modeling for single-cell transcriptomics. Nat. Methods. 2018; 15:1053–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Qiu X., Hill A., Packer J., Lin D., Ma Y.-A., Trapnell C.. Single-cell mRNA quantification and differential analysis with Census. Nat. Methods. 2017; 14:309–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hafemeister C., Satija R.. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. 2019; bioRxiv doi: 10.1101/576827, 14 March 2019, preprint: not peer reviewed. [DOI] [PMC free article] [PubMed]
- 16. Butler A., Hoffman P., Smibert P., Papalexi E., Satija R.. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018; 36:411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J.P., Tamayo P.. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015; 1:417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fabregat A., Jupe S., Matthews L., Sidiropoulos K., Gillespie M., Garapati P., Haw R., Jassal B., Korninger F., May B. et al.. The reactome pathway knowledgebase. Nucleic Acids Res. 2018; 46:D649–D655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Team R.C. R: A Language and Environment for Statistical Computing. 2017; Vienna: R Foundation for Statistical Computing. [Google Scholar]
- 20. Chang W., Cheng J., Allaire J., Xie Y., McPherson J.. shiny: Web Application Framework for R. 2016. 2017; R package version 0.13. 2https://shiny.rstudio.com/.
- 21. Barbie D.A., Tamayo P., Boehm J.S., Kim S.Y., Moody S.E., Dunn I.F., Schinzel A.C., Sandy P., Meylan E., Scholl C. et al.. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009; 462:108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hänzelmann S., Castelo R., Guinney J.. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013; 14:7–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tosolini M., Algans C., Pont F., Ycart B., Fournié J.-J.. Large-scale microarray profiling reveals four stages of immune escape in non-Hodgkin lymphomas. Oncoimmunology. 2016; 5:e1188246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stoeckius M., Hafemeister C., Stephenson W., Houck-Loomis B., Chattopadhyay P.K., Swerdlow H., Satija R., Smibert P.. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods. 2017; 14:865–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pearce E.L., Poffenberger M.C., Chang C.-H., Jones R.G.. Fueling immunity: insights into metabolism and lymphocyte function. Science. 2013; 342:e1242454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kishton R.J., Sukumar M., Restifo N.P.. Metabolic regulation of T cell longevity and function in tumor immunotherapy. Cell Metab. 2017; 26:94–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gong Y., Yao E., Shen R., Goel A., Arcila M., Teruya-Feldstein J., Zakowski M.F., Frankel S., Peifer M., Thomas R.K. et al.. High expression levels of total IGF-1R and sensitivity of NSCLC cells in vitro to an anti-IGF-1R antibody (R1507). PLoS One. 2009; 4:e7273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nomura M., Morita M., Tanuma N.. A metabolic vulnerability of small-cell lung cancer. Oncotarget. 2018; 9:32278–32279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Aibar S., González-Blas C.B., Moerman T., Imrichova H., Hulselmans G., Rambow F., Marine J.-C., Geurts P., Aerts J., van den Oord J. et al.. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017; 14:1083–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.