


Abstract
The addition of poly(ADP-ribose) (PAR) chains along the chromatin fiber due to PARP1 activity regulates the recruitment of multiple factors to sites of DNA damage. In this manuscript, we investigated how, besides direct binding to PAR, early chromatin unfolding events controlled by PAR signaling contribute to recruitment to DNA lesions. We observed that different DNA-binding, but not histone-binding, domains accumulate at damaged chromatin in a PAR-dependent manner, and that this recruitment correlates with their affinity for DNA. Our findings indicate that this recruitment is promoted by early PAR-dependent chromatin remodeling rather than direct interaction with PAR. Moreover, recruitment is not the consequence of reduced molecular crowding at unfolded damaged chromatin but instead originates from facilitated binding to more exposed DNA. These findings are further substantiated by the observation that PAR-dependent chromatin remodeling at DNA lesions underlies increased DNAse hypersensitivity. Finally, the relevance of this new mode of PAR-dependent recruitment to DNA lesions is demonstrated by the observation that reducing the affinity for DNA of both CHD4 and HP1α, two proteins shown to be involved in the DNA-damage response, strongly impairs their recruitment to DNA lesions.
INTRODUCTION
In eukaryotic cells, the tight packing of chromatin acts as a physical barrier, hindering the accessibility of the genomic material. This poses a challenge for DNA repair factors, which need to bind directly to DNA lesions in order to restore genomic integrity. As part of the DNA damage response (DDR), early chromatin remodeling events triggered by the activation of different pathways help ensure timely access of repair proteins to DNA lesions. Among these different pathways, poly(ADP-ribose) (PAR) signaling is one of the first to be activated upon DNA damage (1). PAR signaling relies on the activity of PAR polymerases and in particular, on the founding member of this family of enzymes, the protein PARP1 which recognizes and binds to DNA lesions leading to a dramatic increase of its catalytic activity (2,3). Activated PARP1 then catalyzes the formation of PAR chains on its target proteins, mainly PARP1 itself and histones (4), in the vicinity of the breaks. These PAR chains act as a binding platform for PAR-binding effector proteins including chromatin remodelers, such as ALC1 or CHD2 (5,6), and also modulate the activity of these effectors by allosteric regulation (7,8). The action of the remodelers combined with electrostatic repulsion between DNA and PAR chains decorating the chromatin fiber contribute to the rapid relaxation of the chromatin structure at DNA damage sites (9,10).
The most likely function of PAR-dependent chromatin unfolding is to increase the accessibility to DNA at the lesions. In line with this hypothesis, a study by Izhar et al. showed that a number of transcription factors with no known role in DNA repair were recruited to sites of DNA damage in a PAR-dependent manner (11). However, this work did not discriminate between recruitment due to an affinity for PAR chains, a property reported for some DNA-binding proteins (12), or from increased local accessibility to chromatin as a result of relaxation. Recently, we also observed that early chromatin relaxation, and not direct PAR binding, was responsible for the PAR-dependent recruitment of some chromatin remodelers, namely CHD3 and CHD4 (13).
In the present study, we aimed to more precisely delineate the contribution of PAR-dependent chromatin remodeling mechanisms in modulating the accessibility of chromatin at sites of DNA damage. We show that PAR-signaling promotes the recruitment of DNA-binding but not histone-binding domains to DNA lesions. We attribute this recruitment to affinity for DNA rather than interaction with PAR chains. Furthermore, we find that the recruitment of DNA-binding domains to regions of relaxed damaged chromatin cannot be explained by a decrease of the macromolecular crowding conditions in this area but rather rely on an increase in the binding rates of the DNA-binding domains controlled by chromatin unfolding. Finally, we show that this mechanism of recruitment also holds true for CHD4 and HP1α, two proteins known to be involved in DNA repair.
MATERIALS AND METHODS
Plasmids
Plasmids encoding YFP-macroH2A1.1 macrodomain (14), PATagRFP-H2B, GFP-WWE (from RNF146) (10), PARP1-mCherry (5), GFP nanobody-LacI fusion, iRFP670-ALC1(WT), iRFP670-ALC1(E175Q) (13), the FRET-crowding sensor (15), GFP-LANA-1-32 (16), pH3-paGFP (17), YFP-HMGN2-NBD (HMGN2-ΔC43) (18), GFP-5 (19), GFP-C/EBPalpha (20), paGFP-H2B (21) GFP-CHD4 (22), GFP-CHD4 (4A) (23), and GFP-ZF45 and GFP-ZF345 (24) have been previously described. Monomeric GFP was expressed from pmEGFP-N1. iRFP670-N1 (25) was digested with BamHI and NotI to release the coding sequence of iRFP670 which was ligated into reciprocal sites on pH2B-mCherry (26) to make pH2B-iRFP670. GFP-Sox5 and GFP-HMG (amino acid 556–624 of Sox5) were made using Gibson assembly (New England Biolabs) with pmEGFP-N1 and and 5′-GATCTCGAGCTCAAGCTTCGAATTCATGCTTACTGACCCTGATTTACCTC-3′ and 5′-GGATCCCGGGCCCGCGGTACCTTGTTGGCTTGTCCCGCAATG-3′, to amplify full-length Sox5 and 5′-GATCTCGAGCTCAAGCTTCGAATTCATGCTTACTGACCCTGATTTACCTC-3′ and 5′-GGATCCCGGGCCCGCGGTACCTTGTTGGCTTGTCCCGCAATG-3′ to amplify the HMG domain from CBIG-Sox5 variant 3, a gift from Jeffrey Macklis (Addgene plasmid # 48707). The BZIP domain from C/EBPa was cut from pEBG-BZIPa (gift from Dong-Er Zhang, Addgene plasmid # 12436 (27)) and ligated into pmEGFP-C1 using BspEI and EcoRI restriction sites on both plamids, to obtain the construct GFP-BZIP. GFP-LZIP was made by amplifying LZIP from BZIP using 5′-AATGAATTCACGCGCAGTTGCC-3′ and 5′-TTATCCGGACTGACCAGTGACAATGACCG-3′ and ligated into pmEGFP-C1 between BspEI and EcoRI restriction sites. GFP-HOXc10 and GFP-Homeo (amino acid 268–327 of HOXc10) was made using Gibson assembly with pmEGFP-N1 and pCMV-SPORT6-HOXc10 from the Mammalian Gene Collection (IMAGE ID 3458115) using 5′-GATCTCGAGCTCAAGCTTCGAATTCATGACATGCCCTCGCAATG-3′ and 5′ GGATCCCGGGCCCGCGGTACCTTGGTGAAATTAAAATTGGAGGTCAG-3′ (for full-length HOXc10) and 5′-GATCTCGAGCTCAAGCTTCGAATTCATGGGAAGGAAGAAGAGGTGC-3′ and 5′- GGATCCCGGGCCCGCGGTACCTTGTTCATTTTCTTGAGTTTCATTC-3′ (for Homeo). The tandem ZF345 construct (GFP-(ZF345)2) was made using Gibson assembly with GFP-ZF345 (24) and ZF345 amplified using 5′-TGCCCTTCCCATGCCCCTTCCCGGGCTGCGGGAAGATCTTTGC-3′ and 5′-GGGCAAAGATCTTCCCGCAGCCCGGGAAGGGGCAAATCACTAGTGACCCTTGAGATTC-3′. To make GFP-CHD4 (W508E) and GFP-CHD4 (W644E), GFP-CHD4 was digested with BamHI to remove a 2.7 kb fragment and exchanged with the same region from pBluescript-CHD4 (W508E) or pBluescript-CHD4 (W644E) (28). HP1α was amplified from cDNA using 5′- ATATAAGATCTGGAAAGAAAACCAAGCGGAC-3′ and 5′-TATATAGAATTCAGCTCTTTGCTGTTTCTTTC-3′ and ligated into pmEGFP-C1 between BglII and EcoRI restriction sites to make GFP-HP1α. The chromodomain of HP1α was amplified using 5′- CGCAGATCTGTCGTGGAGAAGGTGCTAGAC-3′ and 5′- AACCCGAATTCAGGGTTTATTATTTTCAC-3′ and ligated into pmEGFP-C1 between BglII and EcoRI restriction sites to make GFP-HP1α-Chromodomain. The N-terminal region of HP1α was amplified using 5′- GCTGTACAAGTCCGGACTCAGATCTGGAAAGAAAACCAAGCGG-3′ and 5′-CTGGTTCCAGTTTATTATTTTCACCCTCCTTCATC-3′ and the C-terminal region of HP1α was amplified using 5′- AAATAATAAACTGGAACCAGAAAAGATCATTG-3′ and 5′-CGCGGTACCGTCGACTGCAGAATTCTTAGCTCTTTGCTGTTTCTTTC-3′. These fragments, together with pmEGFP-C1, were used to make GFP-HP1α-Δhinge using Gibson assembly. The macrodomain of macroH2A1.1 was amplified using 5′-GGAGATCTCAGGGTGAAGTCAGTAA-3′ and 5′-CCGGAATTCCTAGTTGGCGTCCAGCTT-3′ and cloned into pmEGFP-C1 between BglII and EcoRI restriction sites to make pmEGFP-macroH2A1.1 macrodomain.
Cell culture, Hoechst presensitization, osmotic shocks and PARP inhibition
U2OS cells were cultured in DMEM (4.5 g/l glucose, Sigma) supplemented with 10% fetal bovine serum (Life Technologies), 2 mM glutamine (Sigma), 100 μg/ml penicillin and 100 U/ml streptomycin (Sigma) in a humidified incubator at 37°C with 5% CO2. For transient expression of plasmids, cells were transfected 12–24 h after seeding into eight-well Lab-Tek II chambered coverglass (Thermo Scientific) with XtremeGENE HP (Sigma) or Xfect (Clontech) according to the manufacturer's instructions and incubated for 48 h prior to imaging. For Hoechst presensitization, growth medium was aspirated from the Lab-Tek and replaced with fresh medium containing 0.3 μg/ml Hoechst 33342 for 1 h at 37°C. Immediately prior to imaging, growth medium was replaced with CO2-independent imaging medium (Phenol Red-free Leibovitz's L-15 medium (Life Technologies) supplemented with 20% fetal bovine serum, 2 mM glutamine, 100 μg/ml penicillin and 100 U/ml streptomycin). For hypotonic treatment, the imaging medium was replaced with distilled water 10 min prior to imaging. For hypertonic treatment, cells were bathed with imaging medium supplemented by 160 mM sucrose 5 min prior to imaging (29). For PARP inhibition, cells were treated with 30 μM AG14361 (Euromedex) for 30 min prior to imaging. All experiments were completed with unsynchronized cells.
Confocal imaging and laser microirradiation
Live-cell imaging experiments were completed on a Ti-E inverted microscope from Nikon equipped with a CSU-X1 spinning-disk head from Yokogawa, a Plan APO 60×/1.4 NA oil-immersion objective lens and a sCMOS ORCA Flash 4.0 camera. The fluorescence of EGFP and the activated form of PATagRFP were excited with lasers at 490 and 561 nm, respectively. For fluorescence detection, we used bandpass filters adapted to the fluorophore emission spectra. Laser microirradiation and local photoactivation at 405 nm was performed along a 16 μm-line through the nucleus using a single-point scanning head (iLas2 from Roper Scientific) coupled to the epifluorescence backboard of the microscope. To ensure reproducibility laser power at 405 nm was measured at the beginning of each experiment and set to 125 μW at the sample level. Cells were maintained at 37°C with a heating chamber.
Fluorescence correlation spectroscopy
Fluorescence correlation spectroscopy (FCS) experiments were performed on a Leica SP8 or a Zeiss LSM880 confocal setup. The Leica system was equipped with a Plan APO 63×/1.2 NA water immersion objective. GFP fluorescence was excited with a 488 nm laser and single emitted photons, selected by a bandpass filter at 500–550 nm, were detected and counted using a single photon avalanche photodiode and a PicoHarp module from PicoQuant. On the Zeiss system, a C-Apo 40×/1.2 NA water immersion objective was used. GFP fluorescence was excited with a 488 nm laser and single emitted photons at wavelength ranging between 500 and 550 nm were detected and counted on the GaAsP spectral detector. On both systems, laser power used for FCS measurements were adjusted to minimize photobleaching and avoid the induction of photodamage in Hoechst-sensitized cells. Each FCS acquisition lasted 30–45 s to reduce the noise in the autocorrelation curves. For each nucleus, FCS acquisitions were performed before and ∼1–2 min after damage induction. The power of the 405 nm laser used for microirradiation was set to 160 μW at the sample level. Irradiation was performed within nuclear regions of 20 × 1 μm (Leica SP8) or 12 × 2 μm (Zeiss LSM880). Cells were maintained at 37°C with a heating chamber. Raw photon traces obtained for GFP-tagged DNA-binding domains were detrended for slow fluctuations using the Fluctuation Analyzer 4G software (30).
Fluorescence lifetime imaging
Fluorescence lifetime imaging (FLIM) experiments were performed on a Leica SP8 confocal setup equipped with a Plan APO 63×/1.4 NA oil immersion objective. CFP fluorescence was excited with a pulsed 440 nm laser and emitted photons were selected by a bandpass filter at 467–499 nm. Time-correlated photon counting images were acquired using a single photon avalanche photodiode and a PicoHarp module from PicoQuant. After manual segmentation of the irradiated area on the acquired images, fluorescence lifetimes were estimated by fitting the fluorescence decay curves with a single-exponential decay model using the Symphotime software (PicoQuant). For each nucleus, FLIM acquisitions were performed before and ∼120 s after damage induction. The power of the 405 nm laser used for microirradiation was set to 160 μW at the sample level and irradiation was performed within nuclear regions of 20 × 1 μm. Cells were maintained at 37°C with a heating chamber.
Quantification of protein recruitment and changes in chromatin compaction
We used an assay described previously by Lebeaupin et al. to quantify recruitment kinetics and chromatin relaxation at DNA damage sites (31). Briefly, we used U2OS cells co-expressing the protein of interest tagged with GFP together with core histone H2B tagged with the photoactivatable fluorescent protein PATagRFP. Irradiation at 405 nm along a line of 16 μm through Hoechst-sensitized nuclei allowed simultaneously infliction of DNA damage and photoactivation of PATagRFP to highlight the damaged area. Using a custom MATLAB routine, this highlighted area was segmented automatically and the segmentation mask was applied to the image of the GFP channel to quantify protein accumulation at sites of DNA damage. The mask of the damaged chromatin area was also fitted with an ellipsoid and its minor axis length was used to measure the thickness of the damaged line area. By calculating the ratio between this thickness at a given time point and the thickness immediately post-microirradiation, we could then assess changes in the chromatin compaction state at DNA damage sites.
To measure change in chromatin compaction upon hypertonic treatment, we acquired 3D stacks of the nuclei before and after addition of the hypertonic medium and performed automatic 3D thresholding to estimate the volume of the photoactivated chromatin region.
Fitting of the FCS curves
FCS correlation curves were fitted either with an effective diffusion or a reaction dominant model (32). The effective diffusion model is as follows:
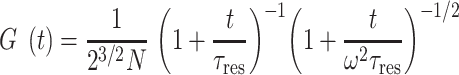 |
(1) |
where N is the number of tagged molecules in the focal volume, τres is the residence time in the focal volume, and ω is the structural parameter of the focal volume, which was fixed to 6. For the fluorescently tagged DNA-binding motifs assessed below, we assumed that the increase in residence time compared to purely diffusible GFP could be attributed mainly to binding to chromatin rather than a change in the gyration radius. Under this assumption, according to (32), one can estimate:
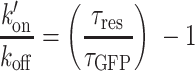 |
(2) |
where k′on is the pseudo first-order association rate with chromatin, koff is the dissociation rate and τGFP is the residence time in the focal volume measured for GFP. The values of the 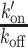 ratio post-irradiation were corrected as follows for changes in chromatin concentration compared to the pre-damage conditions. Cells expressing H2B-PATagRFP were irradiated at 405 nm with the same settings as the ones used for the FCS acquisitions and the area of the photoactivated damaged chromatin region was measured by automatic segmentation immediately after irradiation (A(0)) and 60 s post-irradiation (A(60)). The corrected
ratio post-irradiation were corrected as follows for changes in chromatin concentration compared to the pre-damage conditions. Cells expressing H2B-PATagRFP were irradiated at 405 nm with the same settings as the ones used for the FCS acquisitions and the area of the photoactivated damaged chromatin region was measured by automatic segmentation immediately after irradiation (A(0)) and 60 s post-irradiation (A(60)). The corrected 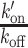 ratio was then calculated as:
ratio was then calculated as:
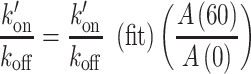 |
(3) |
with 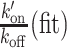 the value obtained from the fit.
the value obtained from the fit.
When using the reaction-dominant model, the correlation curves were first normalized as follows: 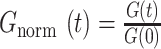 with G(0) calculated as the mean of the correlation curve for times ranging between 10 and 90 μs.
with G(0) calculated as the mean of the correlation curve for times ranging between 10 and 90 μs.
The expression of the reaction-dominant model is then given by:
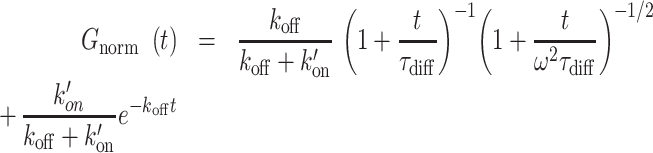 |
(4) |
where τdiff is the diffusion time in the focal volume, ω is the structural parameter of the focal volume, which was fixed to 6, k′on is the pseudo first-order association rate with chromatin and koff is the dissociation rate. The correlation curves obtained pre- and post-irradiation for a given nucleus were fitted together. Since diffusion is not affected by chromatin remodeling at DNA damage sites (see Results), the parameter τdiff was shared for the fits pre- and post-irradiation. Similar to the 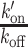 ratio, the values of the association rate post-irradiation were corrected for changes in chromatin concentration compared to the pre-damage conditions according to:
ratio, the values of the association rate post-irradiation were corrected for changes in chromatin concentration compared to the pre-damage conditions according to:
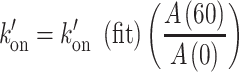 |
(5) |
with k′on(fit) the value obtained from the fit.
Micropore irradiation assay
A modified protocol previously described by Suzuki et al. was established to quantify PARylation induced chromatin relaxation at sites of microirradiation (33). U2OS cells were cultured as described above and labeled with 50 μM 5-bromo-2′-deoxyuridine (BrdU; Sigma) for 24 h followed by 1 h treatment with 10 μM Olaparib (Selleckchem) or were left untreated prior to UV irradiation. For UV irradiation, the medium was removed and the cells were covered with polycarbonate membranes with a 5 μm pore size (Millipore) and exposed to 100 J/m2 of UVC light. After UV irradiation, fresh cell medium was added to the cells and the polycarbonate membrane was removed. Cells were fixed with 3% PFA at different time points post-irradiation, washed with 0.5% Triton X-100 in PBS for 10 min and then treated with 0.005 U/μl DNase I (Thermo Fisher Scientific) for 5 min. The digested DNA ends were labeled with 200 μM 5-iodo-2′-deoxyuridine (IdU; Sigma) by terminal deoxynucleotidyl transferase (Thermo Fisher Scientific) for 2 h. Cells were immunostained with anti-IdU (Abcam, ab187742 at 1:100) or anti–poly-ADPr (10H, ascites at 1:300) and anti-γH2AX (Abcam, ab81299 at 1:2000) antibodies and counterstained with Hoechst. Primary antibodies were detected using Alexa Fluor 488 conjugated goat anti-mouse IgG (Invitrogen, A21422 at 1:1000) and Alexa Fluor 555 conjugated goat anti-rabbit IgG (Invitrogen, A21428 at 1:1000). z-stacks of images were acquired using a Visitron spinning disk confocal system (Visitron systems GmBH) equipped with Yokogawa CSU-W1 spinning disk unit, Olympus IX83 inverted microscope (60× oil objective, NA 1.42), Andor Zyla 4.2 Plus camera, with 405, 488 and 561 nm lasers. The raw images were analyzed in CellProfiler (34) after generating the maximum intensity projections of the z-stacks in Fiji (35). In CellProfiler, the Hoechst stained DNA was used to segment the nuclei and the γH2AX signal was used to identify the irradiated areas. The average signal intensity of the background-subtracted IdU channel was measured in the identified nuclei and irradiated areas. The signal intensities in the irradiated areas were divided by the signal intensities of the corresponding nuclei and plotted using Prism.
Statistics
For the recruitment and chromatin relaxation curves, means ± SEM are shown. For the FCS correlation curves, mean curves are shown. Boxplots were generated using a web-tool developed by the Tyers and Rappsilber labs (http://boxplot.tyerslab.com/). The box limits correspond to the 25th and 75th percentiles and the bold line indicates the median value. The whiskers extend 1.5 times the interquartile range and outliers are shown by dots. Mean curves as well as boxplots were obtained from measurements in at least 15 individual nuclei for each condition. P values were calculated using two-sided unpaired Student's t-test assuming unequal variances except for comparing measurements performed in the same nuclei before and after irradiation, for which two-sided paired Student's t-test assuming unequal variances were used. On the boxplots, * refers to P < 0.05, ** to P < 0.01, *** to P < 0.001, **** to P < 0.0001 and n.s. to non significant.
RESULTS
DNA-binding but not histone-binding domains are recruited to sites of DNA damage in a PAR-dependent manner
The most basic, repeating unit of chromatin is composed of two main components, the DNA double-helix and histone octamers, together forming the nucleosome core-particle (36). To assess whether the local changes in chromatin structure regulated by PAR signaling upon DNA damage could modulate the accessibility to either DNA or histones, we analyzed the behavior of different DNA- and histone-binding domains at sites of DNA damage.
We chose to study three different DNA-binding domains: the HMG domain from SOX5, the BZIP domain from C/EBPa and the Homeobox (Homeo) domain from HOXC10 (27,37,38). These transcription factors have no reported role in the DDR thus their DNA-binding domains are not expected to show specificity for DNA lesions. Here, as well as throughout all this work, we induced DNA damage by microirradiating Hoechst-sensitized nuclei of U2OS cells with a continuous laser at 405 nm. To quantitatively assess the level of DNA-binding domain recruitment to sites of damage over time, the GFP-tagged DNA-binding domains were co-expressed with core histone H2B fused to the photoactivatable protein PATagRFP (Figure 1A and Supplementary Figure S1A). The mask of the microirradiated line highlighted by the photoconverted PATagRFP was used both (i) to measure the accumulation of the GFP-tagged DNA-binding domain at DNA lesions and (ii) to assess changes in the chromatin compaction state at the DNA damage sites by measuring the thickness of the mask over time (31). Using this assay, we found that all the tested DNA-binding domains were recruited to sites of DNA damage with kinetics closely matching the ones characterizing chromatin relaxation (Figure 1B and Supplementary Figure S1A).
Figure 1.

DNA-binding but not histone-binding domains are recruited to sites of DNA damage in a PAR-dependent manner. (A) Recruitment of the DNA-binding domain BZIP from C/EBPa tagged with GFP to sites of DNA damage induced by laser microirradiation. In addition to BZIP-GFP, cells expressed the core histone H2B tagged with the photoactivatable PATagRFP, which allowed highlighting of the irradiated area. Cells were left untreated or treated with 30 μM of the PARP inhibitor AG14361 (PARPi). (B) Automatic segmentation of the photoconverted chromatin area by image analysis allowed quantitative measurement of BZIP recruitment kinetics at DNA damage sites (left) and the assessment of chromatin relaxation in this area (right). (C) Recruitment intensity of GFP-tagged DNA-binding domains at sites of DNA damage in the presence or absence of PARP inhibitor (PARPi) measured 80 s after DNA damage induction. (D) Recruitment intensity of GFP-tagged DNA-binding domains at sites of DNA damage 80 s after damage induction plotted against their relative residence time as measured by FCS. (E–G) Absence of recruitment to sites of DNA damage of (E) the N-terminal (1-32) histone-binding domain of LANA, (F) the nucleosome-binding domain (NBD) of HMGN2 and (G) the chromodomain (CD) of HP1α. Photoactivated H2B-PATagRFP indicate regions of damage. Scale bars = 4 μm.
Next, cells were treated with a PARP inhibitor (PARPi) to block PAR-signaling (Supplementary Figure S1B). In agreement with previous findings (10), we observed that PARPi treatment not only fully abolished chromatin relaxation but even induced a slight over-compaction of the chromatin structure at DNA lesions (Figure 1B). Regarding recruitment, we observed that the stronger the accumulation of DNA-binding domains at DNA lesions, the less PAR-dependent this accumulation was. Indeed, while the weak recruitment of the HMG domain was fully suppressed upon PARPi treatment, the moderate accumulation of the BZIP domain was only partially abolished and the strong recruitment of the Homeo domain appeared to be independent of PARylation (Figure 1C). This PAR-dependent recruitment of DNA binding motifs at DNA lesions was observed not only upon PARPi treatment but also in cells lacking PARP1 (Supplementary Figure S1C–E). Moreover, we observed that full-length SOX5, C/EBPa and HOXC10 accumulated to irradiated areas similarly to the isolated DNA-binding domains indicating that these domains are sufficient to recapitulate the behavior of the full-length proteins in terms of recruitment to DNA lesions (Supplementary Figure S1F).
A plausible explanation of the differences in the levels of DNA-binding domains recruited to sites of DNA damage is that their recruitment is dependent on their affinity for DNA. In agreement with this hypothesis, we found that removing the DNA-binding stretch of BZIP to only keep the leucine-zipper area (LZIP) abolished recruitment to sites of DNA damage (Supplementary Figure S1G). Furthermore, we correlated DNA affinity of each DNA-binding domain with their level of accumulation at DNA lesions. To semi-quantitatively assess the binding affinity of the chosen DNA-binding domains directly in living nuclei in the absence of DNA damage, we measured the residence time of each DNA-binding domain within the focal volume of the confocal microscope by fluorescence correlation spectroscopy (FCS) and normalized it to the residence time measured for GFP (Supplementary Figure S1H and I). Since each of these GFP-tagged DNA-binding domains have relatively similar molecular weights and given that diffusion only varies as the cube root of the molecular weight, we assumed that a larger relative residency time, corresponding to slower mobility, was mostly due to stronger binding to undamaged DNA. We found a clear correlation between the level of recruitment of the DNA-binding domain at DNA lesions and relative residence time, indicating higher levels of recruitment for stronger DNA binding motifs (Figure 1D).
Finally, we assessed potential changes in the accessibility of the histone octamer upon DNA damage. Similar to the rationale behind the choice of the DNA-binding domains, we analyzed nucleosome accessibility by monitoring the recruitment of three histone-binding domains with no reported roles in the DDR. Neither the N-terminal domain of the Latency-Associated Nuclear Antigen (LANA-1–32) nor the nucleosome-binding domain of HMGN2 (HMGN2-NBD), which both bind to the folded domain of H2A-H2B (16,39), recruited to DNA lesions (Figure 1E and F). In fact, we even observed a slight eviction of these two domains from the damaged area which may originate from the local dilution of nucleosome concentration due to chromatin relaxation. HMGN2-NBD reportedly also binds DNA at the entry/exit site of the nucleosome, although with a much weaker affinity than for the H2A-H2B dimer (39). However, this weak DNA affinity does not appear sufficient to promote recruitment of HMGN2-NBD at DNA damage sites. We also monitored the recruitment of the chromodomain of HP1α (HP1α-CD) which recognizes the tail of histone H3 when it is trimethylated on lysine 9, a post-transcriptional modification that has not been reported to be involved in early steps of the DDR. Similar to the domains binding to the folded nucleosome core, we did not observe any accumulation of HP1α-CD to sites of DNA-damage (Figure 1G), in line with what has been reported for the chromodomain of HP1β (40).
Altogether, these data show that an affinity for DNA, but not for histones, promotes recruitment of proteins to DNA lesions. Significantly, PAR-signaling appears to be required for the recruitment of weak DNA-binding domains while being dispensable for stronger ones.
PAR-independent recruitment of DNA-binding domains to sites of DNA damage correlates with a high affinity for DNA
To further investigate the impact of the affinity for DNA on PAR-dependent recruitment to sites of DNA damage, we analyzed the behavior of different combinations of the zinc-finger domains 3–5 from the transcription factor ZIC3 (24). Similar to the DNA-binding domains studied in the previous section, these zinc-finger domains have no reported specificity for DNA lesions and ZIC3 has not been shown to be part of the DDR. First, we compared two GFP-tagged constructs comprising two (ZF45) or three (ZF345) zinc-finger domains. This three zinc-finger construct showed a higher affinity for DNA than ZF45 when comparing their relative residence times measured by FCS (Figure 2A). Correlating with this difference in DNA affinity, ZF345 recruited to sites of DNA damage at higher levels compared to ZF45 (Figure 2B,C).
Figure 2.
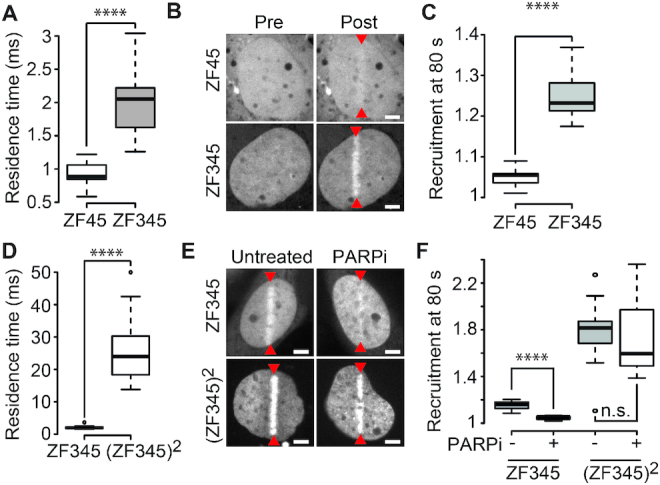
PAR-independent recruitment of DNA-binding domains to sites of DNA damage correlates with a high affinity for DNA. (A) Residence time measured by FCS for GFP-tagged zinc-finger domains 4 and 5 (ZF45) or 3, 4 and 5 (ZF345) of ZIC3. (B) Localization of GFP-ZF45 and GFP-ZF345 before (Pre) and 80 s after (Post) DNA damage induced by laser microirradiation (red arrowheads). (C) Recruitment intensity of GFP-ZF45 and GFP-ZF345 in the irradiated area measured 80 s after DNA damage induction. (D) Residence time for GFP-ZF345 and the tandem repeat of the three zinc-finger domains ((ZF345)2) tagged with GFP as determined by FCS. (E) Localization of GFP-ZF345 and GFP-(ZF345)2 80 s after DNA damage induced by laser microirradiation in untreated cells or cells treated with PARP inhibitor (PARPi). (F) Recruitment intensity of GFP-ZF345 and GFP-(ZF345)2 in the irradiated area measured 80 s after DNA damage induction in untreated cells or cells treated with PARP inhibitor (PARPi). Scale bars = 4 μm.
To assess the impact of further increasing binding affinity to DNA, we compared ZF345 with a tandem repeat of the three zinc-finger domains referred to as (ZF345)2. As expected, (ZF345)2 showed higher affinity for DNA and displayed higher levels of recruitment to DNA lesions than ZF345 (Figure 2D–F). Importantly, we also observed that, while the accumulation of ZF345 to sites of DNA damage relied on PAR-signaling, (ZF345)2 was recruited to sites of damage in a PAR-independent manner (Figure 2F).
These observations fully corroborate our observations with the different classes of DNA-binding domains, indicating that the affinity for DNA is the major factor controlling the accumulation of DNA-binding domains at sites of DNA damage and that, above a certain level of affinity, PARylation becomes dispensable for recruitment.
Recruitment of DNA-binding domains to DNA lesions is not triggered through direct PAR binding but rather relies on PAR-dependent chromatin unfolding
PARylation triggers two main events at sites of DNA damage: the recruitment of PAR-binding effector proteins and chromatin relaxation. Since certain DNA-binding domains of some repair proteins have been shown to bind PAR (12), it is possible that the PAR-dependent recruitment to sites of DNA damage we observed for the different DNA-binding domains presented above occurs through direct binding of PAR chains.
To test for this possibility, we first assessed the ability of the different DNA-binding domains to bind DNA and PAR in vitro. By gel shift assays, we could confirm that the four DNA-binding domains are able to bind DNA in contrast to the macrodomain of macroH2A1.1, which is a well-known PAR binder (41) (Supplementary Figure S2A). Next, we performed an in vitro PAR binding assay and saw that the four DNA-binding domains are able to bind PAR chains (Supplementary Figure S2B). This ability of the DNA-binding domains to bind PAR was still observed under denaturing conditions in contrast to the macrodomain of macroH2A1.1.
Despite the in vitro data showing interactions with PAR chains, the observation that the normalized recruitment kinetics of the different DNA-binding domains to the irradiation area are much slower than the ones of well-known PAR binders such as the WWE domain of RNF146 (42) or the macrodomain of macroH2A1.1 (Figure 3A) prompted us to question the involvement of direct PAR-binding in the accumulation of the DNA-binding domains at sites of damage. To further assess PAR binding in living cells, we used a PAR-3H assay recently developed in our lab (13). Briefly, GFP-tagged proteins of interest are tethered to the LacO array using a LacI-GFP nanobody fusion and co-expressed with mCherry-PARP1 (Supplementary Figure S2C). Upon DNA damage, PARP1 is rapidly PARylated at the site of damage that facilitates its release from the damaged DNA and diffusion through the nucleus in a PARylated state (13,43). If the GFP-tagged tethered protein is able to bind PAR, such as the macrodomain of macroH2A1.1, PARylated PARP1 enriches at the LacO array (Supplementary Figure S2D). Using this assay, we show that the HMG, BZIP or ZF345 domains, all of which show PAR-dependent recruitment to sites of DNA damage, were unable to recruit PARylated PARP1 (Figure 3B and Supplementary Figure S2E and F). Nevertheless, one could still argue that the PAR chains found in the vicinity of the sites of damage are different in terms of quality and quantity from the ones present on PARP1 released from the lesions, and, as such, are able to trigger the recruitment of the DNA-binding domains by direct interaction.
Figure 3.

Recruitment of DNA-binding domains to sites of DNA damage is not triggered by PAR-binding. (A) Normalized recruitment kinetics of PAR binding domains GFP-WWE and YFP-macroH2A1.1 macrodomain (black) and DNA-binding domains GFP-BZIP, GFP-HMG and GFP-Homeo (red) in addition to normalized chromatin relaxation kinetics (blue). (B) GFP-BZIP tethered to the LacO array is unable to recruit PARylated mCherry-PARP1 after DNA damage induction. Insets, pseudocolored according to the look-up table displayed below panel B, show magnification of the LacO array. Boxplots show mCherry-PARP1 intensity at the LacO array quantified pre- and 60 s post-damage. The average intensity at the spot is corrected for background and normalized to average intensity of the nucleus. (C, D) Confocal images showing accumulation of GFP-WWE (C) or GFP-BZIP (D) at sites of DNA damage. Cells were left untreated or treated with PARPi 60 s after DNA damage induction. Boxplots show levels of GFP-WWE, GFP-BZIP or ZF345 accumulation at sites of damage 360 s after microirradiation. (E) Recruitment of the GFP-BZIP to sites of DNA damage induced by laser microirradiation in cells overexpressing WT ALC1 (red) or ATPase dead ALC1 (E175Q, black). (F) Recruitment intensity of GFP-tagged DNA-binding domains at sites of DNA damage in cells overexpressing WT ALC1 or ATPase dead ALC1 (E175Q) measured 80 s after DNA damage induction. For each domain, the recruitment has been normalized to the median recruitment in the E175Q expressing cells. Scale bars = 4 μm
We therefore used a second independent assay to more directly analyze the involvement of the interactions with PAR chains localized at the sites of damage in the recruitment of the DNA-binding domains. Cells were irradiated and, 60 s post-irradiation, i.e. after the completion of the PAR-dependent chromatin relaxation process, we added PARPi to acutely block PARP1 enzymatic activity. Under these conditions, the WWE domain of RNF146 was rapidly released from the irradiated area and, 360 s post-irradiation, the accumulation of WWE from the lesions was nearly completely reversed (Figure 3C), due to the rapid degradation of PAR by the sustained PAR glycohydrolase activity. In contrast, PARPi treatment did not revert the accumulation of BZIP and ZF345 at the irradiation area (Figure 3D). HMG was already released from the DNA lesions at 360 s post-irradiation in the absence of PARPi. Consequently, for this domain, we tested the effect of PARPi treatment over a shorter time frame. At 180 s post-irradiation, we observed no impact of the addition of PARPi on HMG accumulation at sites of damage while WWE was already partially released compared to the control conditions (Supplementary Figure S2G). These results, together with the ones obtained with the PAR-3H assay, argue against the involvement of direct interactions between the DNA-binding domains and PAR chains during the recruitment of these domains to DNA lesions.
In lieu of direct binding to PAR chains, the accumulation of the DNA-binding domains to sites of damage may be promoted by the rapid PAR-dependent unfolding of the chromatin. To assess this possibility, we analyzed the impact of modulating the chromatin relaxation process on the recruitment of the DNA-binding domains. In agreement with our previous results (10,13), we confirmed that chromatin relaxation was enhanced in cells overexpressing the chromatin remodeler ALC1 compared to those overexpressing its catalytically inactive mutant (Supplementary Figure S2H). We found that the four DNA-binding domains, but not the PAR binding macrodomain of macroH2A1.1, displayed enhanced recruitment to DNA lesions in cells overexpressing wild-type ALC1 (Figure 3E, F and Supplementary Figure S2I). This was even true for Homeo, for which the recruitment to DNA lesions did not appear to be PAR-dependent in normal conditions (Figure 1C).
Based on the different assays presented in this section, it is unlikely that the recruitment of the DNA-binding domains to DNA lesions rely on direct interactions with PAR chains. Instead, recruitment appears promoted by the early chromatin remodeling processes occurring upon damage.
Recruitment of DNA-binding domains to relaxed damaged chromatin cannot be attributed to reduced molecular crowding conditions
Chromatin is the major crowding agent in the nucleus and its compaction state modulates diffusion-reaction kinetics of DNA-binding proteins within the nucleus (19). Given the similarities between the kinetics of the recruitment of DNA-binding domains and chromatin relaxation at sites of DNA damage, it is possible that PAR-dependent chromatin relaxation leads to reduced crowding conditions at DNA lesions, thus facilitating the accumulation of DNA-binding domains in this area.
Crowding affects diffusion-reaction dynamics in three different ways: (i) it leads to volume exclusion since the volume occupied by the background molecules, in this case the chromatin, is not accessible to diffusible proteins, (ii) it hinders diffusion, and (iii) it favors compact molecular conformations which, in the case of a protein complex, shifts the binding equilibrium towards the bound state (44). These three consequences of crowding are observed in dense chromatin areas (19) and can be reversed by relaxing chromatin via hypotonic treatment which leads to global nuclear swelling (29). In the following experiments, we quantitatively assessed crowding conditions at sites of DNA damage to distinguish between two alternative scenarios (Supplementary Figure S3): (Model 1) PAR-dependent chromatin unfolding leads to a local increase of the accessible volume fraction and thus, reduced crowding conditions or (Model 2) PAR-dependent chromatin unfolding occurs concomitantly to fiber ‘swelling’, potentially due to the addition of a large number of PAR chains, which keeps the level of crowding unchanged at sites of DNA damage.
We first assessed volume exclusion by measuring the concentration of diffusible probes in and out the damaged area after chromatin relaxation. If chromatin relaxation at sites of DNA damage leads to reduced crowding, it is expected that a higher proportion of the volume is accessible to a diffusible tracer within the relaxed chromatin area compared to the undamaged region, leading to a local increase in the concentration of this tracer (Supplementary Figure S3, Model 1). We observed an increase in the concentration of monomeric GFP in the DNA-damage sites, which allowed us to estimate a local change in accessible volume (Figure 4A,B). The increase in accessible volume measured for GFP was nevertheless modest compared to the change in the volume of the irradiated area, measured by the size of the photoactivated H2B area (Figure 4A and B). In PARPi treated cells, no local increase of GFP concentration was observed in the damaged area, showing that the slight change in accessible volume observed in untreated cells was the consequence of PAR-dependent chromatin remodeling at DNA lesions (Figure 4C). This first experiment suggests a slight reduction in the volume exclusion effect at relaxed damaged chromatin, indicative of decreased crowding conditions compared to undamaged chromatin. To confirm this finding, we also measured changes in volume exclusion for a larger diffusible probe—a GFP pentamer (GFP-5)—which are more sensitive to changes in molecular crowding (45). However, no change in accessible volume at the site of DNA damage was observed for GFP-5 suggesting that this larger probe, in contrast to a single GFP, does not experience reduced molecular crowding in this area (Figure 4C). It is worth noting that the molecular size of many of the repair complexes more closely matches that of GFP-5 rather than a single GFP molecule.
Figure 4.
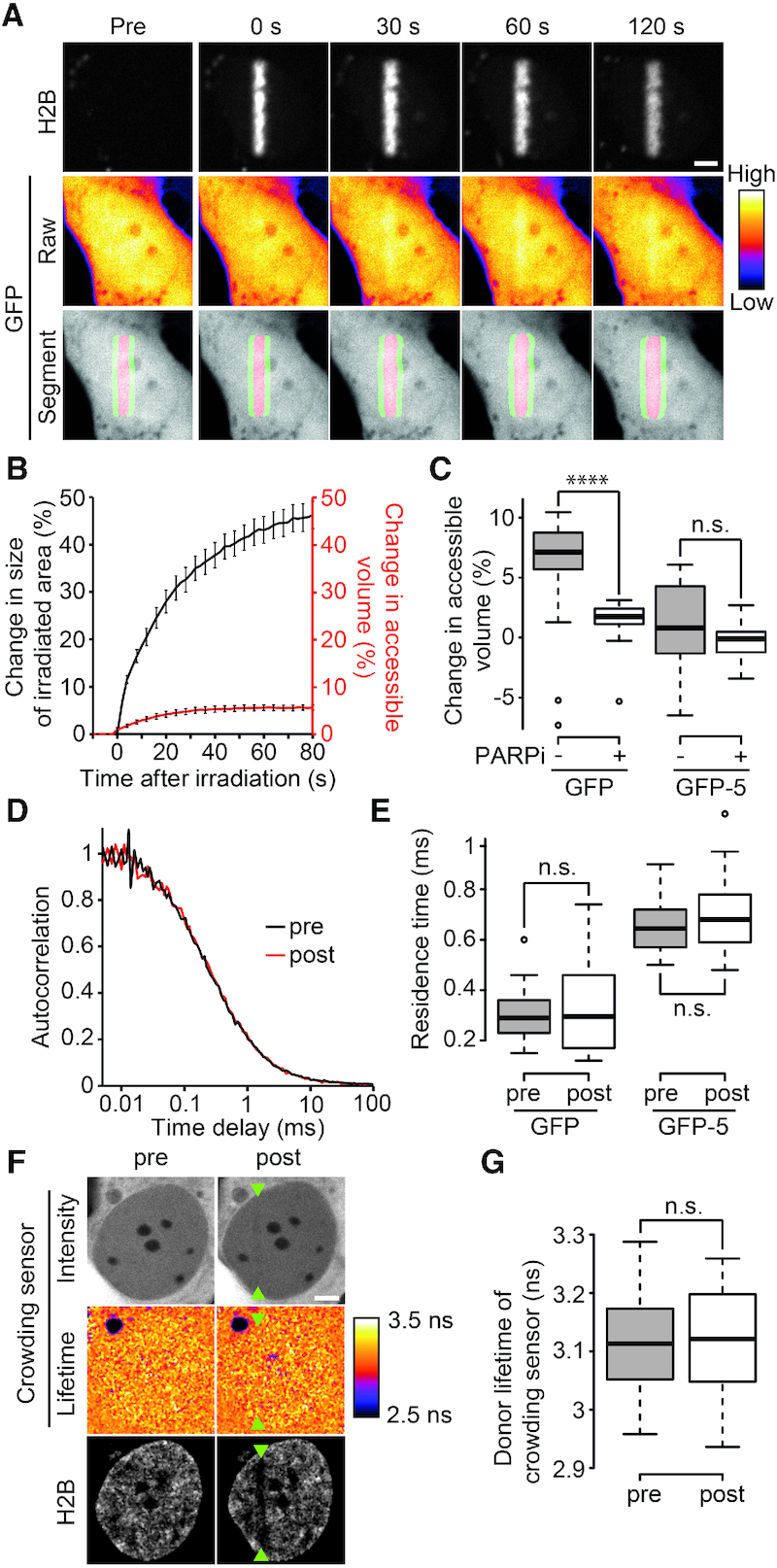
PAR-dependent chromatin relaxation does not reduce macromolecular crowding at DNA damage sites. (A) Raw images to assess changes in volume exclusion at DNA damage sites for freely diffusible GFP. Cells co-express GFP and H2B-PATagRFP. The volume of the damaged area is determined by measuring the area of photoconverted chromatin area. The segmentation of this area is also used as a mask to measure GFP intensities inside (Red area in segment panel) and outside (Green area in segment panel) the irradiated area, whose ratio is used to determine changes in accessible volume for this probe. Raw GFP images are pseudocolored according to the look-up table displayed on the right of panel A. (B) Quantification of changes in size of irradiated area (Black) and changes in accessible volume for GFP (Red). (C) Quantification of changes in accessible volume for GFP and for the GFP-5 array in the absence and presence of PARP inhibitor (PARPi) 80 s post-microirradiation. (D) Normalized FCS correlation curves obtained for GFP at DNA damage sites before (black) and ∼1–2 min after (red) microirradiation. (E) Residence time measured by FCS for GFP and GFP-5 pre- and post-DNA-damage induction. (F) Intensity and donor-lifetime images of molecular crowding sensor before (pre) and ∼1–2 min after (post) DNA damage induction. Microirradiation area is highlighted with green arrowheads. Bleaching of H2B-iRFP670 shows the irradiated area. Donor-lifetime images are pseudocolored according to the look-up table displayed on the right of panel F. (G) Measurement of donor lifetime of crowding sensor in microirradiated area pre- and post-DNA damage induction. Scale bars = 4 μm.
Decreasing crowding should also reduce diffusion hindrance. However, using FCS, we observed no change in the residence time of GFP or GFP-5 before and after DNA damage (Figure 4D and E). The absence of an effect on diffusion hindrance could be explained by a poor sensitivity of the assay. To test this possibility, we induced chromatin over-compaction by bathing cells with hypertonic medium (29) and then triggered relaxation of the over-condensed chromatin by inflicting DNA damage by laser irradiation (10). We estimated that, after this unfolding process, the chromatin compaction state in the irradiated area was similar that in the isotonic medium (Supplementary Figure S4A–C). We assessed the diffusion speed of GFP by FCS in nuclei in isotonic medium, after hypertonic treatment and ∼60 s after laser irradiation. In agreement with enhanced crowding conditions (29), we observed an increase of the residence time of GFP upon hypertonic treatment (Supplementary Figure S4D). In contrast, relaxation of the hyper-compacted chromatin in response to DNA damage had no effect on GFP diffusion. This experiment indicates that the assay is sensitive enough to probe diffusion hindrance in the irradiated area and further supports the fact that chromatin relaxation does not facilitate diffusion in the vicinity of the DNA lesions.
Finally, to test the predicted consequence of crowding on protein conformation, we used a FRET crowding-sensor whose extension varies with the amount of macromolecular crowding (15). To confirm the functionality of this probe, we induced nuclear swelling by bathing cells with hypotonic medium, which reduces intranuclear crowding conditions and leads to chromatin relaxation (Supplementary Figure S4E). As expected, we observed an increase of the donor lifetime in hypotonic conditions compared to isotonic conditions, which reflects a decrease in FRET efficiency due to larger extension of the crowding-sensor (Supplementary Figure S4F and G). In contrast, when examined before and after DNA damage induction, no change in the donor lifetime was detected within the irradiated area suggesting that chromatin relaxation does not lead to reduced macromolecular crowding at sites of DNA damage (Figure 4F and G). We also performed the experiment using precompacted chromatin as described above for diffusion hindrance measurements to assess the sensitivity of the crowding sensor. While we detected a decrease in the lifetime of the crowding-sensor upon hypertonic treatment, no change in lifetime was measured after relaxation of the hyper-compacted chromatin in response to laser irradiation (Supplementary Figure S4H). This tends to rule-out a false negative response of the crowding sensor when probing the crowding state at DNA lesions.
Overall, although we cannot formally exclude a false-negative response of our different crowding reporter assays, the quantitative analysis of crowding conditions at the DNA lesions supports the idea that the PAR-dependent recruitment of DNA-binding domains to sites of DNA damage cannot be attributed to increased accessibility of DNA due to reduced crowding conditions at the sites of DNA damage. Nevertheless, by analyzing crowding conditions, we only assessed the compaction state of the chromatin in terms of the impact of the fraction of occupied nuclear volume without considering consequences arising from changes affecting its spatial conformation. The way chromatin is organized in three dimensions is thought to affect the ability of diffusible DNA-binding proteins navigating through the nucleus to bind DNA (46). We therefore proceeded to assess how PAR-dependent changes in chromatin conformation impacts dynamic binding of DNA-binding domains to DNA at damage sites.
PAR-dependent chromatin remodeling increases the association rates of DNA-binding domains to sites of DNA damage
To better understand the mechanisms underlying the recruitment to DNA lesions of the DNA-binding domains, their local dynamics was assessed by FCS before and after laser microirradiation (Figure 5A). The association between the DNA-binding domains and DNA was modeled by the following reaction:
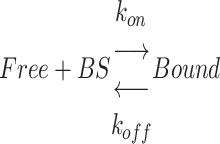 |
Figure 5.

PAR-dependent chromatin remodeling leads to an increase in the association rates of DNA-binding domains at sites of DNA damage. (A) Representative image of a nucleus in which the FCS measurements are performed to assess the local dynamics of GFP-BZIP before (pre) and after (post) DNA-damage induction. Microirradiation area is highlighted with green arrowheads. FCS measurement points are shown in blue. Images are pseudocolored according to the look-up table displayed on the right. Scale bar = 4 μm. (B) 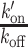 ratios estimated from FCS curves fitted with an effective diffusion model pre- and ∼1–2 min after (post) DNA damage induction in the presence or absence of PARP inhibitor (PARPi) for GFP-BZIP. (C)
ratios estimated from FCS curves fitted with an effective diffusion model pre- and ∼1–2 min after (post) DNA damage induction in the presence or absence of PARP inhibitor (PARPi) for GFP-BZIP. (C) 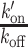 ratios estimated from FCS curves fitted with an effective diffusion model pre- and ∼1–2 min after (post) DNA damage induction for GFP-LANA-1–32. (D) Normalized FCS correlation curves obtained for GFP-BZIP in undamaged nuclei. The experimental curve (continuous black line) is fitted with an effective-diffusion model (dotted black line) or a reaction-dominant model (dotted red line). (E and F) Association (k′on) and dissociation rates (koff) measured by FCS for GFP-BZIP pre- and post-DNA damage and in untreated cells or cells treated with PARP inhibitor (PARPi).
ratios estimated from FCS curves fitted with an effective diffusion model pre- and ∼1–2 min after (post) DNA damage induction for GFP-LANA-1–32. (D) Normalized FCS correlation curves obtained for GFP-BZIP in undamaged nuclei. The experimental curve (continuous black line) is fitted with an effective-diffusion model (dotted black line) or a reaction-dominant model (dotted red line). (E and F) Association (k′on) and dissociation rates (koff) measured by FCS for GFP-BZIP pre- and post-DNA damage and in untreated cells or cells treated with PARP inhibitor (PARPi).
with Free and Bound referring to the two states of the DNA-binding domain and BS to the binding sites along the DNA. FCS curves were fitted with an effective diffusion model to estimate the ratio 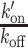 according to Equation (2). k′on is a pseudo first-order association rate corresponding to the product of the association rate kon by the concentration of binding sites [BS]. Thus, k′on depends on the local chromatin concentration which varies upon irradiation due to PAR-dependent remodeling. To assess changes in the binding kinetics which are not due to variations in chromatin concentration,
according to Equation (2). k′on is a pseudo first-order association rate corresponding to the product of the association rate kon by the concentration of binding sites [BS]. Thus, k′on depends on the local chromatin concentration which varies upon irradiation due to PAR-dependent remodeling. To assess changes in the binding kinetics which are not due to variations in chromatin concentration, 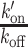 ratios measured post-irradiation were corrected for chromatin relaxation according to Equation (3). Using this analysis framework, we found that all DNA-binding domains displayed an increase in the
ratios measured post-irradiation were corrected for chromatin relaxation according to Equation (3). Using this analysis framework, we found that all DNA-binding domains displayed an increase in the 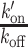 ratio after DNA damage induction (Figure 5B and Supplementary Figure S5A–C), showing a shift of the binding equilibrium towards the DNA-bound state in the damaged area. The increase in the
ratio after DNA damage induction (Figure 5B and Supplementary Figure S5A–C), showing a shift of the binding equilibrium towards the DNA-bound state in the damaged area. The increase in the 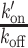 ratio upon laser irradiation was impaired by PARPi treatment for HMG, BZIP and ZF345, but not for Homeo, which mirrors the results obtained regarding recruitment (Figures 1 and 2). In contrast to the DNA-binding domains, the
ratio upon laser irradiation was impaired by PARPi treatment for HMG, BZIP and ZF345, but not for Homeo, which mirrors the results obtained regarding recruitment (Figures 1 and 2). In contrast to the DNA-binding domains, the 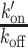 ratio measured for the histone-binding domain of LANA remained unchanged after DNA damage induction (Figure 5C), in agreement with the absence of recruitment to the lesions.
ratio measured for the histone-binding domain of LANA remained unchanged after DNA damage induction (Figure 5C), in agreement with the absence of recruitment to the lesions.
The estimation of the 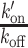 ratios demonstrate an increased binding at sites of DNA damage for the DNA-binding domains. However, this analysis cannot distinguish between an increase in the association rate k′on, indicative of facilitated binding, or a decrease in the dissociation rate koff, implying a longer lifetime of the bound state. Estimating the association and dissociation rates separately can only be achieved for probes displaying reaction-dominant kinetics (32). In such regime, the FCS curves cannot be properly fitted with the one-component effective diffusion model (Equation (1)) but require the use of a two-components reaction-dominant model (Equation (4)). Among the three DNA-binding domains displaying PAR-dependent recruitment to DNA lesions, BZIP was the only one showing significant deviation from the effective diffusion model (Figure 5D and Supplementary Figure S5D–F). Using a reaction-dominant model for BZIP yielded improved fits of the FCS curves and allowed the estimation of the pseudo first-order association rate k′on and the dissociation rate koff of the binding reaction before and after damage. The k′on rates post-irradiation were calculated according to Equation (5) to assess changes in association rates that are not due to variations in chromatin concentration. In the undamaged situation we recovered values for k′on and koff, which are in the same range as those previously reported for BZIP (32). Upon irradiation, we found that k′on was enhanced by ∼5-fold (Figure 5E) while koff underwent a less prominent increase (Figure 5F). This result implies that the accumulation of BZIP at DNA-damage sites is due to facilitated binding to DNA in this area rather than a longer lifetime of the bound state (which should have led to a decreased koff). Furthermore, PARPi treatment led to a significant reduction of k′on post-damage while not affecting koff (Figure 5E and F), indicating that PAR-dependent chromatin remodeling specifically facilitates binding to DNA at sites of damage.
ratios demonstrate an increased binding at sites of DNA damage for the DNA-binding domains. However, this analysis cannot distinguish between an increase in the association rate k′on, indicative of facilitated binding, or a decrease in the dissociation rate koff, implying a longer lifetime of the bound state. Estimating the association and dissociation rates separately can only be achieved for probes displaying reaction-dominant kinetics (32). In such regime, the FCS curves cannot be properly fitted with the one-component effective diffusion model (Equation (1)) but require the use of a two-components reaction-dominant model (Equation (4)). Among the three DNA-binding domains displaying PAR-dependent recruitment to DNA lesions, BZIP was the only one showing significant deviation from the effective diffusion model (Figure 5D and Supplementary Figure S5D–F). Using a reaction-dominant model for BZIP yielded improved fits of the FCS curves and allowed the estimation of the pseudo first-order association rate k′on and the dissociation rate koff of the binding reaction before and after damage. The k′on rates post-irradiation were calculated according to Equation (5) to assess changes in association rates that are not due to variations in chromatin concentration. In the undamaged situation we recovered values for k′on and koff, which are in the same range as those previously reported for BZIP (32). Upon irradiation, we found that k′on was enhanced by ∼5-fold (Figure 5E) while koff underwent a less prominent increase (Figure 5F). This result implies that the accumulation of BZIP at DNA-damage sites is due to facilitated binding to DNA in this area rather than a longer lifetime of the bound state (which should have led to a decreased koff). Furthermore, PARPi treatment led to a significant reduction of k′on post-damage while not affecting koff (Figure 5E and F), indicating that PAR-dependent chromatin remodeling specifically facilitates binding to DNA at sites of damage.
Finally, to compare the behavior of the DNA-binding domains with the one of a motif recruited to DNA lesions via direct PAR binding, we probed the dynamics of the macrodomain of macroH2A1.1 at the sites of damage by FCS. While, prior to damage, the macrodomain was following an effective diffusion kinetic, a reaction-dominant model had to be used to fit the FCS curves acquired after laser irradiation (Supplementary Figure S5G). This switch from an effective diffusion to a reaction-dominant dynamic implies a massive reduction of the dissociation rate koff upon DNA damage (32), in agreement with a strong binding to PAR chains. This behavior is in sharp contrast with the results obtained for BZIP for which we observed a slight increase of the dissociation rate after laser irradiation (Figure 5F).
Increased accessibility to DNase I at sites of DNA damage is promoted by PAR-signaling
The sensitivity of chromatin to nuclease digestion, such as DNase I that non specifically cleaves DNA, is broadly used in genomics to evaluate DNA accessibility (47,48). Previous studies have demonstrated the nuclease sensitive rearrangement of chromatin after UV irradiation in [3H]thymidine labeled human cells indicating that chromatin remodeling mechanisms can tune DNA accessibility during DNA damage repair (49). While it has also been shown that PARP-dependent relaxation of in vitro reconstituted chromatin leads to increased micrococcal nuclease sensitivity (50), it is unknown whether this also holds true in living cells subjected to DNA damage.
To test this possibility, we assessed DNAse sensitivity at DNA lesions inflicted using micropore UV irradiation (33). With this irradiation method we could detect robust activation of PAR signaling as well as γH2AX signaling, although with slower kinetics (Supplementary Figure S6A). The irradiated cells were treated with DNAse and free DNA ends were labeled by the Terminal Deoxynucleotidyl Transferase-based incorporation of IdU (Figure 6A). While no IdU incorporation was detected in the absence of DNase treatment (Supplementary Figure S6B and C), we observed a strong local IdU signal at sites of DNA damage in the presence of DNase I, demonstrating an increased DNase sensitivity of the chromatin at the irradiated areas as compared to the undamaged DNA (Figure 6B and C). Furthermore, inhibiting PARylation strongly reduced the incorporation of IdU at the sites of DNA damage suggesting that PAR-signaling, likely via its role in chromatin remodeling, is responsible for the increased DNase sensitivity displayed by the damaged chromatin (Figure 6B and C). Combined with the results described in the previous sections, these findings indicate that the same PAR-dependent chromatin remodeling mechanisms underlie the recruitment of DNA-binding motifs at the sites of DNA damage observed by live-cell imaging and the increased DNase hypersensitivity used for mapping ‘open’ damaged chromatin regions in genomics.
Figure 6.

The accessibility of DNase I at sites of micropore irradiation is regulated by PAR-signaling. (A) Schematic representation of micropore irradiation assay. Cells are labeled with BrdU to photosensitize the DNA. After micropore irradiation (100 J/m2 of UVC light) and fixation, chromatin is digested by DNase I. The generated free DNA-ends are labeled with IdU by Terminal Deoxynucleotidyl Transferase (TdT) and visualized by immunofluorescence. (B) Representative images of the IdU-signal 30 minutes post-micropore-irradiation to analyze the chromatin accessibility to DNase I in the presence or absence of PARP inhibitor (Olaparib). The microirradiation sites are visualized with anti-γH2AX and anti-rabbit Alexa Fluor 555 antibodies. The free DNA-ends are visualized by anti-IdU and anti-mouse Alexa Fluor 488 antibodies. Scale bar = 10 μm. (C) Quantification of the IdU-signal intensity in the presence or absence of PARP inhibitor (PARPi). The irradiated areas are segmented by γH2AX signal in Hoechst positive nuclei and the average signal intensities of the background-subtracted IdU channel are measured. These average signal intensities are divided by the signal intensities of the corresponding nuclei and plotted.
The PAR-dependent recruitment of CHD4 and HP1α to DNA lesions relies on their ability to bind DNA
Our findings indicate that early PAR-dependent chromatin remodeling promotes the recruitment of DNA-binding proteins to sites of damage due to facilitated association with DNA. To assess the relevance of this recruitment mechanism for DNA repair, we tested whether the accumulation of CHD4 and HP1, two proteins contributing to DNA repair potentially via their known functions in the regulation of the chromatin architecture (13,40), could be due to binding to DNA at unfolded damaged chromatin.
The chromatin remodeler CHD4 has been shown to be important for the repair of DNA double-strand break (DSB) by homologous recombination (HR) (51) and we recently reported that its PAR-dependent recruitment to DNA lesions is not triggered by direct PAR-binding but rather promoted by chromatin relaxation (13). Different domains of CHD4 were shown to bind DNA in vitro, the two chromo-domains (CD1 and CD2) (52) as well as an HMG box-like domain localized at the N-terminus (23). We analyzed the behavior of point mutations localized in each of these domains and which were suspected to affect the ability of CHD4 to bind DNA (23,28). While mutation in the HMG box-like (4A) domain did not affect CHD4 dynamics assessed by FCS, mutations W508E and W644E, localized respectively in CD1 and CD2, displayed a strong reduction of the residence time, suggesting reduced affinity for DNA in living cells (Figure 7A and B). Concomitantly, monitoring the accumulation of these mutants to sites of damage showed that the mutations W508E and W644E, but not the 4A, impaired recruitment, correlating with the reduced affinity of these mutants for DNA (Figure 7C and D).
Figure 7.

Recruitment of CHD4 and HP1α to sites of DNA damage is driven by DNA binding. (A) Schematic representation of the domains of CHD4. Mutations in the HMG-box-like domain (4A) and in the chromodomains (W508E and W644E) are indicated. (B) Residence times measured by FCS in the absence of DNA damage for wild-type and mutant GFP-CHD4. (C) Confocal images showing recruitment of WT and mutant GFP-CHD4 to sites of damage 80 s post-microirradiation. Microirradiation sites are indicated by red arrowheads. (D) Recruitment intensity of wild-type and mutant GFP-CHD4 80 s post-DNA-damage induction. (E) Schematic representation of the domains of HP1α. (F) Confocal images of GFP-HP1α pre- and 60 s post-DNA damage induction in the presence and absence of PARP inhibitor (PARPi). (G) Recruitment kinetics of HP1α at sites of DNA damage in the presence (red) and absence (black) of PARPi. (H) Residence times measured by FCS in the absence of DNA damage for GFP tagged wild-type HP1α and a mutant lacking the hinge domain (ΔHinge). (I, J) Confocal images (I) and recruitment intensity (J) showing recruitment of wild-type HP1α-GFP and the Δhinge mutant 80 s post-damage induction. Microirradiation sites are indicated with red arrowheads. Scale bars = 4 μm.
The chromatin scaffolding protein HP1 has roles in DNA repair (40) in addition to its central function in transcription repression. Indeed, the three isoforms (HP1α, HP1β and HP1γ) were shown to be recruited to DNA lesions (40) and depletion of any of them dramatically impairs efficiency of DSB repair by HR (53). Here, we focused on HP1α (Figure 7E) and observed that its recruitment to DNA lesions was strongly impaired upon PARP inhibition (Figure 7F,G). The fact that we could not detect direct interactions between HP1α and PARylated PARP1 using the PAR-3H assay described previously (Supplementary Figure S7), prompted us to test the involvement of DNA binding in the recruitment of HP1α to DNA lesions. It was previously reported that HP1α binds to DNA via its hinge domain (54) and in agreement with these findings, we measured by FCS a strong reduction of the residence time for a mutated version of HP1α lacking the hinge domain (Δhinge) compared to wild-type HP1α (WT) (Figure 7H). Furthermore, deleting the hinge domain was sufficient to significantly reduce accumulation to sites of damage (Figure 7I and J). These results suggest that, similar to CHD4, the PAR-dependent recruitment of HP1α relies on the ability of this protein to bind DNA.
DISCUSSION
The removal of PARP1, ALC1 or CHD2, all of which are key players of the PAR-dependent chromatin remodeling mechanisms characterizing the early stages of the DDR, leads to cell hypersensitivity to DNA damage (6,10,55). This suggests that the open chromatin organization established by these different actors at sites of DNA damage is essential for repair efficiency, yet its exact function remains unclear. In this current study, we report that different protein domains with affinity for DNA are recruited to sites of damage in a PAR-dependent manner and we propose that this recruitment relies on chromatin unfolding rather than direct binding to PAR chains. A more systematic analysis of the behavior of a larger spectrum of DNA as well as histone-binding domains at sites of damage would be necessary to further establish the generic nature of our findings. Nevertheless, the fact that such recruitment is observed for DNA-binding but not for histone-binding domains and that macromolecular crowding conditions remain mainly unchanged at the irradiated site, suggest that the early PAR-dependent chromatin remodeling events do not simply unfold the chromatin structure to facilitate access to damaged chromatin for any kind of nuclear protein. Rather, they promote the establishment of a particular chromatin conformation in which DNA becomes more exposed while leaving the accessibility of histones unaffected (Figure 8). This specific chromatin conformation is likely to contribute to DNA repair efficiency not only by facilitating access to DNA lesions but also by influencing the choice of the repair pathway, as was shown for the PAR-dependent remodeler CHD2 that specifically affects double-strand break repair through non-homologous end-joining (6).
Figure 8.
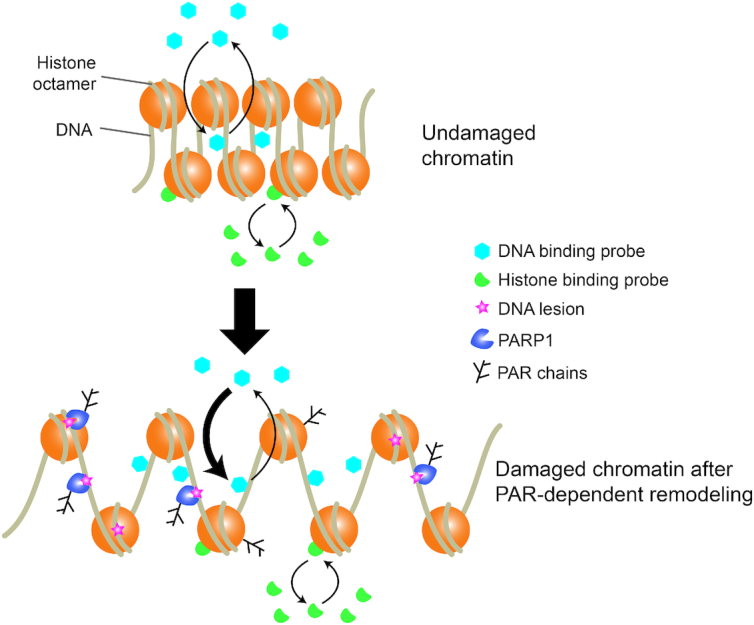
Model of the impact of PAR-dependent chromatin remodeling at DNA-damage sites. In undamaged chromatin, DNA-binding and histone-binding domains interact dynamically with chromatin at equilibrated rates. Upon DNA damage, PARP1 is activated and induces chromatin remodeling. The newly established chromatin architecture is characterized by facilitated association with DNA, as evidenced by increased binding rates of DNA-binding protein, while interaction dynamics with histones remains unchanged.
Our results show a PAR-dependent increase of the pseudo first-order association rate k′on of the DNA-binding domain BZIP at sites of DNA damage, in support of the increased DNA accessibility characterizing the chromatin architecture established by PAR-dependent remodeling mechanisms at DNA lesions. In future work, it would be important to confirm these findings obtained for BZIP by analyzing the behavior of other DNA-binding domains displaying interaction dynamics allowing for the estimation of the k′on rates specifically. Nevertheless, our results still provide insight regarding the mechanisms by which PAR-dependent chromatin remodeling could promote recruitment to DNA lesions. Since the k′on measured for BZIP is the product of the binding rate, kon, and the concentration of binding sites, [BS], the increase in k′on could be attributed to a modulation of either of the two parameters. On the one hand, it has been reported that chromatin remodeling mechanisms that occur at DNA lesions is associated with the release of core histones (56), a process that would enhance the amount of free DNA at sites of DNA damage leading to an increase in [BS]. However, when we monitored core histone turnover within the 2 min time frame in which we observed recruitment of the DNA-binding domains, we found no significant release of either H3 or H2B from DNA damage sites compared to undamaged conditions (Supplementary Figure S8). In contrast to the absence of release of core histone within the studied time frame, it has been shown that the histone linker H1 is quickly displaced from the chromatin upon damage (57). Such eviction of H1 as well as partial unwrapping of nucleosomes or changes in the fiber conformation triggered by PARylation, might be sufficient to increase [BS] due to the uncovering of DNA stretches that were inaccessible in the undamaged chromatin. The PAR-dependent increase in DNase I accessibility observed at DNA lesions is in line with this hypothesis, since DNase I cleavage activity is inhibited by the nucleosomes (48). On the other hand, early chromatin remodeling may also increase the association rate, kon. Indeed, kon is not only a function of the ‘chemical’ affinity of the DNA-binding protein for the DNA, which is probably not affected by chromatin remodeling, but also depends on how fast this protein finds a binding site on the DNA (44). It has been proposed by several groups that the search process in the nucleus is influenced by chromatin conformation (58–60), switching from local and systematic exploration in compact heterochromatin to more long-range sparse exploration in the less-compact euchromatin (19). In the context of DNA repair, the PAR-dependent increase in DNA exposure due to the uncovering of previously hidden DNA stretches or the reduction of the target-search time could thus ensure the timely detection of the DNA lesions by later actors of the repair machinery (61). Noteworthy, such PAR-dependent facilitated binding to DNA observed in the context of the DDR might also hold true during transcription regulation. Such a mechanism may contribute, for example, to the recruitment of CHD4 at heat-shock genes undergoing PAR-dependent puffing in Drosophila (62). It may also explain why mutating CHD4 at residues W508E and W644E, which reduces its affinity for DNA (Figure 7B), impairs the ability of this remodeler to inhibit transcription (28). This recruitment mechanism may also be at play in the context of the PAR-dependent binding of the heat-shock transcription factor 1 to DNA (63).
We also found that, while weak DNA-binding domains require PAR-dependent chromatin remodeling to recruit efficiently to sites of DNA damage, domains displaying a stronger affinity for DNA were recruited independently of PARylation (Figures 1 and 2). This result suggests that, for these strong DNA-binding domains, chromatin remodeling processes controlled by signaling pathways other than PARylation, such as the mobilization of linker histone H1 (10) or core histone acetylation (64,65), might be sufficient to promote recruitment at DNA lesions. Moreover, the recruitment of high affinity DNA-binding domains at DNA lesions observed even after PARP inhibition may provide a mechanism for PARP-independent repair, such as certain base-excision repair pathways (66) or double-strand break repair via Ku-dependent non-homologous end joining (67).
A recent screen monitoring the accumulation of 70 different proteins to sites of DNA damage demonstrated that PAR-signaling regulates the recruitment kinetics of a majority of these proteins (68). Our findings, together with previous work (11,13), indicate that such PAR-dependent recruitment may not solely rely on direct PAR-binding but likely also involves enhanced binding to DNA at damaged chromatin remodeled by PAR-dependent mechanisms. Our present results, together with our previous work (13), indicate that this PAR-dependent recruitment mechanism can account for the accumulation of CHD4, and also probably CHD3, at the sites of damage. Future work will be necessary to establish whether this recruitment mode also holds true for other DNA-binding repair proteins or for regulatory factors modulating the activity of these repair proteins such as the recently identified regulation of the Ataxia-telangiectasia mutated (ATM) kinase by homeobox transcription factors (69). Furthermore, the recruitment via enhanced DNA binding of proteins displaying functions in transcription modulation may contribute to the early repression of transcription at the sites of DNA damage (70). In line with this hypothesis, we show that the PAR-dependent recruitment of HP1α, a chromatin-scaffolding protein involved in transcription silencing, relies on its ability to bind DNA. Conversely, this recruitment mechanism may also explain the PAR-dependent accumulation of transcription factors to DNA lesions ((11) and this study), a potential early step of transcription-coupled repair mechanisms (71). Nevertheless, the potentially generic nature of the PAR-dependent recruitment via enhanced DNA association may also drive the accumulation at DNA lesions of DNA-binding proteins with no functional role in DNA repair. Therefore, after an initial generic protein accumulation phase, one might expect ‘filtering’ by specific retention mechanisms to keep only factors with roles in DDR at the damage sites. It was shown that PAR chains can drive liquid demixing at DNA lesions, allowing the filtering of proteins based on electrostatic interactions and their degree of structural disorder (72). In addition to phase separation, this filtering step might also involve later chromatin reorganization events, including further relaxation and recondensation (13,73) as well as post-translational modifications (74). This hypothesis then suggests that in the future it will be important to address which proteins are retained in the damaged area and what the underlying ‘filtering’ mechanisms are.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Microscopy Rennes Imaging Center (BIOSIT, Université Rennes 1) for technical assistance. This work would not have been possible without the generous donation of plasmids from a number of people. We would like to specifically thank Jeffrey Macklis for the Sox5 plasmid, Arnold Boersma for the crowding FRET sensor, Kenneth Kaye for the GFP-LANA-1-32 plasmid, Michael Bustin for the YFP-HMGN2-NBD (HMGN2-ΔC43) plasmid, Jun Aruga for the ZIC3 plasmids, Dong-Er Zhang for the C/EBPa plasmid, Richard Day for the GFP-tagged full length C/EBPa plasmid, Sophie Polo for GFP-CHD4, James Hagman for pBluescript-CHD4 domain mutants, Ana Silva for GFP-CHD4 (4A), Vladislav Verkhusha for iRFP670-N1, Jan Ellenberg for paGFP-H2B and Johnathan Chubb for pH3-paGFP. We also thank Ivan Ahel for his gift of purified PARP1, James McNally and Davide Mazza for sharing MatLab codes used for fitting the FCS correlation curves and Malte Wachsmuth for his advice when using the Fluctuation Analyzer 4G software.
Author contributions: R.S., T.L., S.J., O.D., S.D., P.B. and S.H. completed the experiments within the manuscript. C.C. and C.B. generated cell lines and DNA constructs. G.T. and S.H. conceived and supervised this study. R.S., S.H. and G.T. wrote the manuscript. All authors read and commented on the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
People Programme (Marie Curie Actions) of the European Union's Seventh Framework Programme (FP7/ 2007–2013) under REA grant agreement [PCOFUND-GA-2013-609102]; PRESTIGE program coordinated by Campus France [PRESTIGE-2017-2-0042]; Université Bretagne-Loire; Fondation ARC pour la recherche sur le cancer [PDF20181208405 to R.S.]; Hungarian Academy of Sciences [LP2017-11/2017 to G.T.]; Ligue contre le Cancer du Grand-Ouest (committees 35, 22 and 72); Fondation ARC pour la recherche sur le cancer [20161204883]; Agence Nationale de la Recherche [PRC-2018 REPAIRCHROM]; Institut Universitaire de France (to S.H.); Max Planck Research Group Leader Program, Max Planck Society (to C.B.); National Research Development and Innovation Office [NKFIH K-119361 to P.B.]; the collaboration between S.H. and G.T. benefited from funding from the CNRS via the PICS program. Funding for open access charge: Agence Nationale de la Recherche.
Conflict of interest statement. None declared.
REFERENCES
- 1. Liu C., Vyas A., Kassab M.A., Singh A.K., Yu X.. The role of poly ADP-ribosylation in the first wave of DNA damage response. Nucleic Acids Res. 2017; 45:8129–8141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ohgushi H., Yoshihara K., Kamiya T.. Bovine thymus poly(adenosine diphosphate ribose) polymerase. Physical properties and binding to DNA. J. Biol. Chem. 1980; 255:6205–6211. [PubMed] [Google Scholar]
- 3. Langelier M.-F., Planck J.L., Roy S., Pascal J.M.. Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science. 2012; 336:728–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gibbs-Seymour I., Fontana P., Rack J.G.M., Ahel I.. HPF1/C4orf27 is a PARP-1-interacting protein that regulates PARP-1 ADP-ribosylation activity. Mol. Cell. 2016; 62:432–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gottschalk A.J., Timinszky G., Kong S.E., Jin J., Cai Y., Swanson S.K., Washburn M.P., Florens L., Ladurner A.G., Conaway J.W. et al.. Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:13770–13774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Luijsterburg M.S., de Krijger I., Wiegant W.W., Shah R.G., Smeenk G., de Groot A.J.L., Pines A., Vertegaal A.C.O., Jacobs J.J.L., Shah G.M. et al.. PARP1 links CHD2-mediated chromatin expansion and H3.3 deposition to DNA repair by Non-homologous End-Joining. Mol. Cell. 2016; 61:547–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lehmann L.C., Hewitt G., Aibara S., Leitner A., Marklund E., Maslen S.L., Maturi V., Chen Y., van der Spoel D., Skehel J.M. et al.. Mechanistic insights into autoinhibition of the oncogenic chromatin remodeler ALC1. Mol. Cell. 2017; 68:847–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. DaRosa P.A., Wang Z., Jiang X., Pruneda J.N., Cong F., Klevit R.E., Xu W.. Allosteric activation of the RNF146 ubiquitin ligase by a poly(ADP-ribosyl)ation signal. Nature. 2015; 517:223–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Poirier G.G., de Murcia G., Jongstra-Bilen J., Niedergang C., Mandel P.. Poly(ADP-ribosyl)ation of polynucleosomes causes relaxation of chromatin structure. Proc. Natl. Acad. Sci. U.S.A. 1982; 79:3423–3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sellou H., Lebeaupin T., Chapuis C., Smith R., Hegele A., Singh H.R., Kozlowski M., Bultmann S., Ladurner A.G., Timinszky G. et al.. The poly(ADP-ribose)-dependent chromatin remodeler Alc1 induces local chromatin relaxation upon DNA damage. Mol. Biol. Cell. 2016; 27:3791–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Izhar L., Adamson B., Ciccia A., Lewis J., Pontano-Vaites L., Leng Y., Liang A.C., Westbrook T.F., Harper J.W., Elledge S.J.. A systematic analysis of factors localized to damaged chromatin reveals PARP-Dependent recruitment of transcription factors. Cell Rep. 2015; 11:1486–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pleschke J.M., Kleczkowska H.E., Strohm M., Althaus F.R.. Poly(ADP-ribose) binds to specific domains in DNA damage checkpoint proteins. J. Biol. Chem. 2000; 275:40974–40980. [DOI] [PubMed] [Google Scholar]
- 13. Smith R., Sellou H., Chapuis C., Huet S., Timinszky G.. CHD3 and CHD4 recruitment and chromatin remodeling activity at DNA breaks is promoted by early poly(ADP-ribose)-dependent chromatin relaxation. Nucleic Acids Res. 2018; 46:6087–6098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Timinszky G., Till S., Hassa P.O., Hothorn M., Kustatscher G., Nijmeijer B., Colombelli J., Altmeyer M., Stelzer E.H.K., Scheffzek K. et al.. A macrodomain-containing histone rearranges chromatin upon sensing PARP1 activation. Nat. Struct. Mol. Biol. 2009; 16:923–929. [DOI] [PubMed] [Google Scholar]
- 15. Boersma A.J., Zuhorn I.S., Poolman B.. A sensor for quantification of macromolecular crowding in living cells. Nat. Methods. 2015; 12:227–229. [DOI] [PubMed] [Google Scholar]
- 16. Barbera A.J., Chodaparambil J.V., Kelley-Clarke B., Joukov V., Walter J.C., Luger K., Kaye K.M.. The nucleosomal surface as a docking station for Kaposi's sarcoma herpesvirus LANA. Science. 2006; 311:856–861. [DOI] [PubMed] [Google Scholar]
- 17. Müller I., Boyle S., Singer R.H., Bickmore W.A., Chubb J.R.. Stable morphology, but dynamic internal reorganisation, of interphase human chromosomes in living cells. PLoS One. 2010; 5:e11560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ueda T., Catez F., Gerlitz G., Bustin M.. Delineation of the protein module that anchors HMGN proteins to nucleosomes in the chromatin of living cells. Mol. Cell Biol. 2008; 28:2872–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bancaud A., Huet S., Daigle N., Mozziconacci J., Beaudouin J., Ellenberg J.. Molecular crowding affects diffusion and binding of nuclear proteins in heterochromatin and reveals the fractal organization of chromatin. EMBO J. 2009; 28:3785–3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schaufele F., Enwright J.F., Wang X., Teoh C., Srihari R., Erickson R., MacDougald O.A., Day R.N.. CCAAT/enhancer binding protein alpha assembles essential cooperating factors in common subnuclear domains. Mol. Endocrinol. 2001; 15:1665–1676. [DOI] [PubMed] [Google Scholar]
- 21. Beaudouin J., Mora-Bermúdez F., Klee T., Daigle N., Ellenberg J.. Dissecting the contribution of diffusion and interactions to the mobility of nuclear proteins. Biophys. J. 2006; 90:1878–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Polo S.E., Kaidi A., Baskcomb L., Galanty Y., Jackson S.P.. Regulation of DNA-damage responses and cell-cycle progression by the chromatin remodelling factor CHD4. EMBO J. 2010; 29:3130–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Silva A.P.G., Ryan D.P., Galanty Y., Low J.K.K., Vandevenne M., Jackson S.P., Mackay J.P.. The N-terminal region of chromodomain helicase DNA-binding Protein 4 (CHD4) is essential for activity and contains a high mobility group (HMG) Box-like-domain that can bind Poly(ADP-ribose). J. Biol. Chem. 2016; 291:924–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hatayama M., Tomizawa T., Sakai-Kato K., Bouvagnet P., Kose S., Imamoto N., Yokoyama S., Utsunomiya-Tate N., Mikoshiba K., Kigawa T. et al.. Functional and structural basis of the nuclear localization signal in the ZIC3 zinc finger domain. Hum. Mol. Genet. 2008; 17:3459–3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shcherbakova D.M., Verkhusha V.V.. Near-infrared fluorescent proteins for multicolor in vivo imaging. Nat. Methods. 2013; 10:751–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Neumann B., Walter T., Hériché J.-K., Bulkescher J., Erfle H., Conrad C., Rogers P., Poser I., Held M., Liebel U. et al.. Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature. 2010; 464:721–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang D.E., Hetherington C.J., Meyers S., Rhoades K.L., Larson C.J., Chen H.M., Hiebert S.W., Tenen D.G.. CCAAT enhancer-binding protein (C/EBP) and AML1 (CBF alpha2) synergistically activate the macrophage colony-stimulating factor receptor promoter. Mol. Cell Biol. 1996; 16:1231–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ramírez J., Dege C., Kutateladze T.G., Hagman J.. MBD2 and multiple domains of CHD4 are required for transcriptional repression by Mi-2/NuRD complexes. Mol. Cell Biol. 2012; 32:5078–5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Walter A., Chapuis C., Huet S., Ellenberg J.. Crowded chromatin is not sufficient for heterochromatin formation and not required for its maintenance. J. Struct. Biol. 2013; 184:445–453. [DOI] [PubMed] [Google Scholar]
- 30. Wachsmuth M., Conrad C., Bulkescher J., Koch B., Mahen R., Isokane M., Pepperkok R., Ellenberg J.. High-throughput fluorescence correlation spectroscopy enables analysis of proteome dynamics in living cells. Nat. Biotechnol. 2015; 33:384–389. [DOI] [PubMed] [Google Scholar]
- 31. Lebeaupin T., Smith R., Huet S., Timinszky G.. Poly(ADP-ribose)-dependent chromatin remodeling in DNA repair. Methods Mol. Biol. 2017; 1608:165–183. [DOI] [PubMed] [Google Scholar]
- 32. Michelman-Ribeiro A., Mazza D., Rosales T., Stasevich T.J., Boukari H., Rishi V., Vinson C., Knutson J.R., McNally J.G.. Direct measurement of association and dissociation rates of DNA binding in live cells by fluorescence correlation spectroscopy. Biophys. J. 2009; 97:337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Suzuki K., Yamauchi M., Oka Y., Suzuki M., Yamashita S.. Creating localized DNA double-strand breaks with microirradiation. Nat. Protoc. 2011; 6:134–139. [DOI] [PubMed] [Google Scholar]
- 34. Kamentsky L., Jones T.R., Fraser A., Bray M.-A., Logan D.J., Madden K.L., Ljosa V., Rueden C., Eliceiri K.W., Carpenter A.E.. Improved structure, function and compatibility for CellProfiler: modular high-throughput image analysis software. Bioinforma. Oxf. Engl. 2011; 27:1179–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. et al.. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012; 9:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Luger K., Mäder A.W., Richmond R.K., Sargent D.F., Richmond T.J.. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997; 389:251–260. [DOI] [PubMed] [Google Scholar]
- 37. Privalov P.L., Jelesarov I., Read C.M., Dragan A.I., Crane-Robinson C.. The energetics of HMG box interactions with DNA: thermodynamics of the DNA binding of the HMG box from mouse sox-5. J. Mol. Biol. 1999; 294:997–1013. [DOI] [PubMed] [Google Scholar]
- 38. LaRonde-LeBlanc NA, Wolberger C.. Structure of HoxA9 and Pbx1 bound to DNA: Hox hexapeptide and DNA recognition anterior to posterior. Genes Dev. 2003; 17:2060–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kato H., van Ingen H., Zhou B.-R., Feng H., Bustin M., Kay L.E., Bai Y.. Architecture of the high mobility group nucleosomal protein 2-nucleosome complex as revealed by methyl-based NMR. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:12283–12288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Luijsterburg M.S., Dinant C., Lans H., Stap J., Wiernasz E., Lagerwerf S., Warmerdam D.O., Lindh M., Brink M.C., Dobrucki J.W. et al.. Heterochromatin protein 1 is recruited to various types of DNA damage. J. Cell Biol. 2009; 185:577–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kustatscher G., Hothorn M., Pugieux C., Scheffzek K., Ladurner A.G.. Splicing regulates NAD metabolite binding to histone macroH2A. Nat. Struct. Mol. Biol. 2005; 12:624–625. [DOI] [PubMed] [Google Scholar]
- 42. Wang Z., Michaud G.A., Cheng Z., Zhang Y., Hinds T.R., Fan E., Cong F., Xu W.. Recognition of the iso-ADP-ribose moiety in poly(ADP-ribose) by WWE domains suggests a general mechanism for poly(ADP-ribosyl)ation-dependent ubiquitination. Genes Dev. 2012; 26:235–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Satoh M.S., Lindahl T.. Role of poly(ADP-ribose) formation in DNA repair. Nature. 1992; 356:356–358. [DOI] [PubMed] [Google Scholar]
- 44. Lebeaupin T., Smith R., Huet S.. Lavelle C, Victor J-M. 9 - The multiple effects of molecular crowding in the cell nucleus: From molecular dynamics to the regulation of nuclear architecture. Nuclear Architecture and Dynamics. 2018; 2:Boston: Translational Epigenetics. Academic Press; 209–232. [Google Scholar]
- 45. Hall D., Minton A.P.. Macromolecular crowding: qualitative and semiquantitative successes, quantitative challenges. Biochim. Biophys. Acta. 2003; 1649:127–139. [DOI] [PubMed] [Google Scholar]
- 46. Cortini R., Filion G.J.. Theoretical principles of transcription factor traffic on folded chromatin. Nat. Commun. 2018; 9:1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Burkholder G.D., Weaver M.G.. DNA-protein interactions and chromosome banding. Exp. Cell Res. 1977; 110:251–262. [DOI] [PubMed] [Google Scholar]
- 48. Boyle A.P., Davis S., Shulha H.P., Meltzer P., Margulies E.H., Weng Z., Furey T.S., Crawford G.E.. High-resolution mapping and characterization of open chromatin across the genome. Cell. 2008; 132:311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Smerdon M.J., Lieberman M.W.. Nucleosome rearrangement in human chromatin during UV-induced DNA- reapir synthesis. Proc. Natl. Acad. Sci. U.S.A. 1978; 75:4238–4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kim M.Y., Mauro S., Gévry N., Lis J.T., Kraus W.L.. NAD+-dependent modulation of chromatin structure and transcription by nucleosome binding properties of PARP-1. Cell. 2004; 119:803–814. [DOI] [PubMed] [Google Scholar]
- 51. Qi W., Chen H., Xiao T., Wang R., Li T., Han L., Zeng X.. Acetyltransferase p300 collaborates with chromodomain helicase DNA-binding protein 4 (CHD4) to facilitate DNA double-strand break repair. Mutagenesis. 2016; 31:193–203. [DOI] [PubMed] [Google Scholar]
- 52. Bouazoune K., Mitterweger A., Längst G., Imhof A., Akhtar A., Becker P.B., Brehm A.. The dMi-2 chromodomains are DNA binding modules important for ATP-dependent nucleosome mobilization. EMBO J. 2002; 21:2430–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lee Y.-H., Kuo C.-Y., Stark J.M., Shih H.-M., Ann D.K.. HP1 promotes tumor suppressor BRCA1 functions during the DNA damage response. Nucleic Acids Res. 2013; 41:5784–5798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bryan L.C., Weilandt D.R., Bachmann A.L., Kilic S., Lechner C.C., Odermatt P.D., Fantner G.E., Georgeon S., Hantschel O., Hatzimanikatis V. et al.. Single-molecule kinetic analysis of HP1-chromatin binding reveals a dynamic network of histone modification and DNA interactions. Nucleic Acids Res. 2017; 45:10504–10517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ahel D., Horejsí Z., Wiechens N., Polo S.E., Garcia-Wilson E., Ahel I., Flynn H., Skehel M., West S.C., Jackson S.P. et al.. Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science. 2009; 325:1240–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Adam S., Dabin J., Chevallier O., Leroy O., Baldeyron C., Corpet A., Lomonte P., Renaud O., Almouzni G., Polo S.E.. Real-time tracking of parental histones reveals their contribution to chromatin integrity following DNA damage. Mol. Cell. 2016; 64:65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Strickfaden H., McDonald D., Kruhlak M.J., Haince J.-F., Th’ng J.P.H., Rouleau M., Ishibashi T., Corry G.N., Ausio J., Underhill D.A. et al.. Poly(ADP-ribosyl)ation-dependent transient chromatin decondensation and histone displacement following laser microirradiation. J. Biol. Chem. 2016; 291:1789–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Erdel F., Schubert T., Marth C., Längst G., Rippe K.. Human ISWI chromatin-remodeling complexes sample nucleosomes via transient binding reactions and become immobilized at active sites. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:19873–19878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Erdel F., Krug J., Längst G., Rippe K.. Targeting chromatin remodelers: signals and search mechanisms. Biochim. Biophys. Acta. 2011; 1809:497–508. [DOI] [PubMed] [Google Scholar]
- 60. Knight S.C., Xie L., Deng W., Guglielmi B., Witkowsky L.B., Bosanac L., Zhang E.T., El Beheiry M., Masson J.-B., Dahan M. et al.. Dynamics of CRISPR-Cas9 genome interrogation in living cells. Science. 2015; 350:823–826. [DOI] [PubMed] [Google Scholar]
- 61. Normanno D., Boudarène L., Dugast-Darzacq C., Chen J., Richter C., Proux F., Bénichou O., Voituriez R., Darzacq X., Dahan M.. Probing the target search of DNA-binding proteins in mammalian cells using TetR as model searcher. Nat. Commun. 2015; 6:7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tulin A., Spradling A.. Chromatin loosening by poly(ADP)-ribose polymerase (PARP) at Drosophila puff loci. Science. 2003; 299:560–562. [DOI] [PubMed] [Google Scholar]
- 63. Fujimoto M., Takii R., Katiyar A., Srivastava P., Nakai A.. Poly(ADP-Ribose) Polymerase 1 Promotes the human heat shock response by facilitating heat shock transcription factor 1 Binding to DNA. Mol. Cell Biol. 2018; 38:e00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ogiwara H., Ui A., Otsuka A., Satoh H., Yokomi I., Nakajima S., Yasui A., Yokota J., Kohno T.. Histone acetylation by CBP and p300 at double-strand break sites facilitates SWI/SNF chromatin remodeling and the recruitment of non-homologous end joining factors. Oncogene. 2011; 30:2135–2146. [DOI] [PubMed] [Google Scholar]
- 65. Toiber D., Erdel F., Bouazoune K., Silberman D.M., Zhong L., Mulligan P., Sebastian C., Cosentino C., Martinez-Pastor B., Giacosa S. et al.. SIRT6 recruits SNF2H to DNA break sites, preventing genomic instability through chromatin remodeling. Mol. Cell. 2013; 51:454–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Campalans A., Kortulewski T., Amouroux R., Menoni H., Vermeulen W., Radicella J.P.. Distinct spatiotemporal patterns and PARP dependence of XRCC1 recruitment to single-strand break and base excision repair. Nucleic Acids Res. 2013; 41:3115–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wang M., Wu W., Wu W., Rosidi B., Zhang L., Wang H., Iliakis G.. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006; 34:6170–6182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Aleksandrov R., Dotchev A., Poser I., Krastev D., Georgiev G., Panova G., Babukov Y., Danovski G., Dyankova T., Hubatsch L. et al.. Protein dynamics in complex DNA lesions. Mol. Cell. 2018; 69:1046–1061. [DOI] [PubMed] [Google Scholar]
- 69. Johnson T.E., Lee J.-H., Myler L.R., Zhou Y., Mosley T.J., Yang S.-H., Uprety N., Kim J., Paull T.T. Bio-Bricks for Molecular Machines FRI Stream . Homeodomain proteins directly regulate ATM kinase activity. Cell Rep. 2018; 24:1471–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ayrapetov M.K., Gursoy-Yuzugullu O., Xu C., Xu Y., Price B.D.. DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:9169–9174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Fousteri M., Mullenders L.H.F.. Transcription-coupled nucleotide excision repair in mammalian cells: molecular mechanisms and biological effects. Cell Res. 2008; 18:73–84. [DOI] [PubMed] [Google Scholar]
- 72. Altmeyer M., Neelsen K.J., Teloni F., Pozdnyakova I., Pellegrino S., Grøfte M., Rask M.-B.D., Streicher W., Jungmichel S., Nielsen M.L. et al.. Liquid demixing of intrinsically disordered proteins is seeded by poly(ADP-ribose). Nat. Commun. 2015; 6:8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Burgess R.C., Burman B., Kruhlak M.J., Misteli T.. Activation of DNA damage response signaling by condensed chromatin. Cell Rep. 2014; 9:1703–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Khurana S., Kruhlak M.J., Kim J., Tran A.D., Liu J., Nyswaner K., Shi L., Jailwala P., Sung M.-H., Hakim O. et al.. A macrohistone variant links dynamic chromatin compaction to BRCA1-dependent genome maintenance. Cell Rep. 2014; 8:1049–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.