


Abstract
Bloom helicase (BLM) and its orthologs are essential for the maintenance of genome integrity. BLM defects represent the underlying cause of Bloom Syndrome, a rare genetic disorder that is marked by strong cancer predisposition. BLM deficient cells accumulate extensive chromosomal aberrations stemming from dysfunctions in homologous recombination (HR). BLM participates in several HR stages and helps dismantle potentially harmful HR intermediates. However, much remains to be learned about the molecular mechanisms of these BLM-mediated regulatory effects. Here, we use DNA curtains to directly visualize the activity of BLM helicase on single molecules of DNA. Our data show that BLM is a robust helicase capable of rapidly (∼70–80 base pairs per second) unwinding extensive tracts (∼8–10 kilobases) of double-stranded DNA (dsDNA). Importantly, we find no evidence for BLM activity on single-stranded DNA (ssDNA) that is bound by replication protein A (RPA). Likewise, our results show that BLM can neither associate with nor translocate on ssDNA that is bound by the recombinase protein RAD51. Moreover, our data reveal that the presence of RAD51 also blocks BLM translocation on dsDNA substrates. We discuss our findings within the context of potential regulator roles for BLM helicase during DNA replication and repair.
INTRODUCTION
RecQ helicases constitute a highly conserved subgroup of the SF2 helicases (super–family 2) and they play essential roles in maintaining genome integrity in species ranging from bacteria to man (1–5). Humans possess five RecQ homologs, namely WRN, BLM, RECQ1, RECQ4 and RECQ5 (1,3–5). These helicases play diverse roles in transcription, DNA replication and DNA repair, and are broadly associated with functions that contribute to the maintenance of genome stability (1–5). Their importance is reflected in the fact that mutations in BLM, WRN and RECQ4 cause Bloom, Werner, and Rothmund–Thompson syndromes, respectively (1–5). These diseases are associated with profound developmental abnormalities and increased cancer risk, and the latter two syndromes are also characterized by premature ageing (1–5).
Bloom syndrome (BS) is an autosomal recessive disorder characterized by severe developmental defects and strong cancer predisposition (6–9). Homozygous null BLM mutations are embryonic lethal in mice, whereas hypomorphic BLM mutations result in growth defects, severe anemia, chromosomal instability and cancer (1–5). Cell lines derived from BLM-deficient mice and patient-derived BS cells exhibit extensive genome rearrangements (3,4). BLM has been implicated in several distinct aspects of genome maintenance, including DSB end processing (10–12), disruption of strand invasion intermediates (13), Holliday junction (HJ) dissolution (14), recognition and processing of ultra-fine anaphase bridges (15,16), replication fork progression through its involvement in the rescue of stalled or collapsed replication forks and by unwinding difficult to replicate DNA secondary structures, such as G-quartets (17–20). Given their diverse roles in genome maintenance, BLM and other RecQ helicases have emerged as potential targets for novel anticancer chemotherapeutics (5,21).
In addition to its helicase core, BLM (1417 amino acids) harbors a RecQ C-terminal (RQC) domain that confers high-affinity structure-specific DNA-binding activity, and a helicase and RNase D–like C-terminal (HRDC) domain that is necessary to promote BLM recruitment to DNA damage (1,4,14). BLM is a ssDNA translocase that moves along one strand of DNA to unwind a duplex. BLM exhibits ATP-dependent 3′→5′ helicase activity and is capable of unwinding DNA structures mimicking a variety of DNA replication and repair intermediates, including 3′ tailed duplexes, forked duplexes, G-quartets, D-loops and four-way Holliday junctions (1–4,22,23). In vitro studies have also recapitulated a number of BLM-related reactions that contribute to homologous recombination (10,14). For instance, BLM promotes DNA end resection in combination with the DNA2 helicase/nuclease, to yield 3′ ssDNA overhangs that serve as a binding platform for the RAD51 recombinase during the initial stage of homologous recombination (10,24,25). BLM has also been suggested to act as an anti-recombinase, by disrupting ssDNA-bound RAD51 filaments (13,26,27). However, unlike other known anti-recombinases (13), BLM appears to act only upon the unstable, ADP-bound form of the RAD51–ssDNA filament, which is inactive for recombination (13,26,27). BLM can also disrupt D-loops formed by RAD51, but again, this activity is contingent upon inactivation of the RAD51 filament (26). Finally, BLM associates with Topoisomerase IIIα (TOPO IIIα) and the RecQ-mediated genome instability protein 1 and protein 2 (RMI1/2) to form the BTR complex, which promotes the dissolution of the double Holliday junction (HJ), a HR intermediate and the precursor to crossover recombinants that entail the exchange of chromosome arms (10,14). As such, dHJ dissolution helps ensure that non-crossover recombination products are made, BTR-mediated dHJ dissolution helps minimize the potential for aberrant recombination-dependent chromosomal rearrangements, such as chromosome arm translocations (10,14).
Our current understanding of the mechanistic basis of BLM function(s) in maintaining genome integrity remains relatively limited. Since BLM participates in a multiplicity of processes, it remains difficult to fully define how BLM mutations impact any one particular process. Similarly, BLM interacts with both WRN and RECQ5, and there may be partial functional redundancy between these helicases (3–5). In our effort to delineate the roles of helicases in genome maintenance, we have begun to apply our DNA curtain methodology to examine helicase/translocase interactions with DNA substrates in real-time by total internal reflection fluorescence microscopy (TIRFM) (28–32). Here, we report the use of these DNA curtain assays to analyze the basic properties of BLM helicase as it acts upon naked and protein-bound ssDNA and dsDNA substrates. This work relies upon two strategies, either by the use of mCherry-tagged RPA to label the ssDNA strands generated as a result of BLM-mediated dsDNA unwinding, or by directly visualizing GFP-tagged BLM as it acts upon dsDNA. The results reveal that BLM is a remarkably robust dsDNA helicase capable of unwinding DNA strands at a rate of ∼70 bp per second (bp/sec) with an average processivity of ∼8000 bp. Surprisingly, we have been unable to find any activity of BLM on RPA-bound ssDNA, RAD51-bound ssDNA, RAD51-bound dsDNA or even RAD51-bound heteroduplex DNA joints. These observations argue against BLM acting to strip DNA of bound RPA or RAD51. Instead, our data are most consistent with a model with the action of BLM helicase being restricted to the DNA end resection stage of HR, and acting after the completion of DNA strand exchange when RAD51 has already dissociated from the heteroduplex DNA joint and also during dHJ dissolution.
MATERIALS AND METHODS
Plasmid construction
The GFP gene was inserted in front of the N-terminus of the BLM gene with a C-terminal hexahistidine tag in a pYES2 vector using Gibson assembly and the following four primers: 5′-GGA TCC CTA ACC ATG TCA ACT GAA CCG ATG AGC AAA GGA GAA GAA CTT TTC ACT GG–3′; 5′-GAT TAT TTT GAG GAA CAG CAG CCA TTC CTC CAG GTC CAC CCA TGC CAT GTG TAA TCC CAG-3′; 5′-CTG GGA TTA CAC ATG GCA TGG GTG GAC CTG GAG GAA TGG CTG CTG TTC CTC AAA ATA ATC–3′; 5′-CCA GTG AAA AGT TCT TCT CCT TTG CTC ATC GGT TCA GTT GAC ATG GTT AGG GAT CC-3′.
Protein expression
RPA, RPA-mCherry, RPA-GFP, RAD51, TOPO IIIα, RMI1/2 and BLM were purified as previously described (33–36). GFP–BLM was purified similarly to untagged BLM with the following modifications (35). GFP–BLM-pYES2 was transformed into a protease-deficient yeast expression strain (JEL-1). Six liters of cells in basic medium minus uracil (0.17% yeast nitrogen base, 0.5% ammonium sulfate, 2% sodium lactate, 3% glycerol, 0.87 g/L amino acid mix without uracil) were grown at 30°C until OD reached ∼1.0 and were induced by the addition of 2% galactose for 24 h at 25°C. Then, cells were harvested by centrifugation at 4°C. All subsequent steps were performed at 4°C and care was taken to minimize sample exposure to light. The cells were resuspended in 40 ml cell lysis buffer (50 mM Tris–HCl [pH 7.0], 1 M NaCl, and 10% glycerol, 1 mM TCEP, protease inhibitor cocktail (Roche, Cat. No. 05892988001), 2 mM EDTA and mixed with 40 ml glass beads (425–600 μm, Sigma). Cells were lysed by vortexing 10 times for 30 s each time and the samples were chilled on ice for 2 min between each cycle of vortexing. The lysate was collected and centrifuged for 1 h at 40 000 rpm at 4°C using Ti–45 rotor. The supernatant was precipitated with 20% ammonium sulfate (10 g per 50 ml supernatant) for 1 h. The pellet was recovered by centrifugation at 10 000 rpm at 4°C for 10 min. Then, the pellet was dissolved in cell lysis buffer plus 15 mM imidazole and without EDTA. The resuspension was passed through a 0.45 μm filter (Millex; Cat No. SLHV033RS) to remove any undissolved precipitate. The protein solution was then purified using Ni-NTA (Qiagen) resin and eluted with an imidazole step gradient (with successive washes buffer containing 20, 30, 40, 50, 60, 70, 80, 100, 120, 140, 160, 180, 200 and 250 mM imidazole) and fractions containing GFP–BLM were identified by SDS-PAGE and Coomassie staining. The purified GFP–BLM fraction was concentrated by dialysis into 50% PEG 20,000 for 48 hours. The concentrated protein was aliquoted and stored at –80°C.
ATP hydrolysis assays
Comparison of unlabeled BLM and GFP–BLM ATP hydrolysis activities was performed in BLM buffer (20 mM Tris–HCl [pH 7.5], 1 mM MgCl2, 2 mM ATP, 1 mM DTT, 0.2 mg/ml BSA) at 37°C. All reactions contained either M13 ssDNA (2 μM nucleotides; NEB, Cat. No. N4040S), pUC19 (2 μM nucleotides; NEB, Cat No. N3041A) or ΦX174 (2 μM nucleotides, RF I or RF II; NEB, Cat No's. N3021L and N3022L), as indicated. Reactions were initiated by the addition of 10 nM (Figure 2A) or 5 nM (Figures 5B, C, 6B, C and Supplementary Figure S3) BLM or GFP–BLM, as indicated; note, that protein levels were reduced in the later experiments to conserve material. Aliquots were removed at the indicated time points and quenched by addition of 50 mM EDTA. The quenched reactions were quantified by the ATPase/GTPase Activity Assay Kit as per the manufacturer's instructions (Sigma, Cat. No. MAK113).
Figure 2.
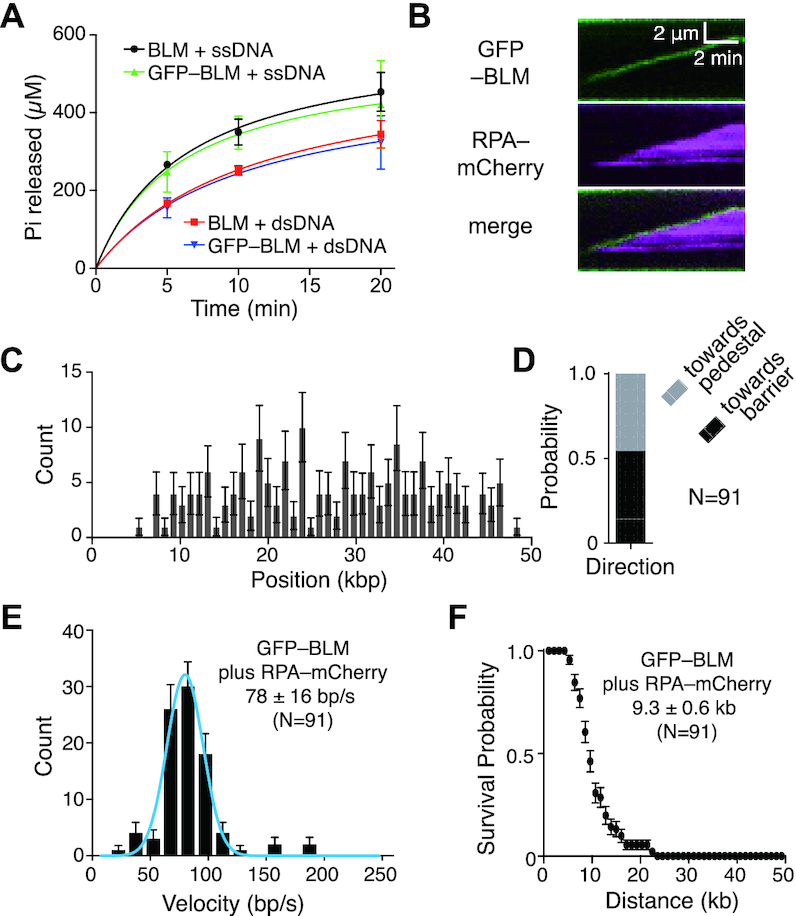
GFP-tagged BLM is active for dsDNA translocation and unwinding. (A) ATP hydrolysis assays comparing unlabeled BLM (10 nM) to GFP-tagged BLM (10 nM) in the presence of either ssDNA (M13) or dsDNA (pUC19). Data points represent the mean and standard deviation of three independent experiments. (B) Kymograph showing GFP–BLM (green) unwinding dsDNA in the presence of 1 nM RPA-mCherry (magenta); note that buffer flow was OFF during data collection. Reactions contained 0.2 nM GFP–BLM and 1 nM RPA-mCherry. (C) Distribution of initiation sites for GFP–BLM unwinding in the presence of RPA-mCherry (N = 175). Error bars represent 95% confidence intervals calculated from bootstrap analysis. (D) Quantification of GFP–BLM translocation direction in the presence of 1 nM RPA-mCherry. (E) Velocity distribution of GFP–BLM unwinding rates with 1 nM RPA-mCherry on double-tethered dsDNA. The solid blue line represents a Gaussian fit to the data. Error bars represent 95% confidence intervals calculated from bootstrap analysis. (F) Survival probability plot of GFP–BLM translocation with 1 nM RPA-mCherry on double-tethered dsDNA. Error bars represent 95% confidence intervals calculated from bootstrap analysis.
Figure 5.
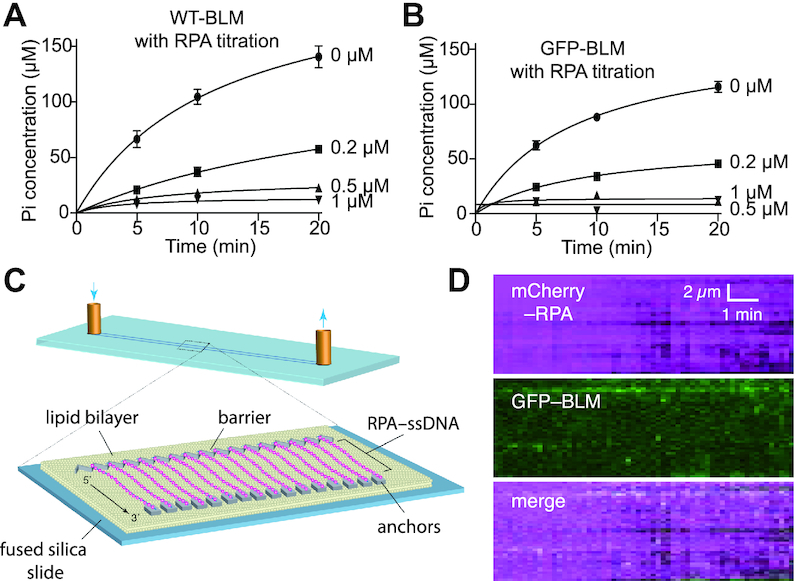
RPA blocks BLM interactions with ssDNA. (A) ATPase assays containing 5 nM unlabeled BLM with either 0, 0.2, 0.5 or 1 μM RPA in the presence of ssDNA (M13). Data points represent the mean and standard deviation of three independent experiments; note that the lower total Pi concentration levels in the minus RPA control assays in comparison to Figure 2A is due the difference in BLM concentration (5 nM versus 10 nM). (B) ATPase assays containing 5 nM GFP–BLM with 0, 0.2, 0.5, or 1 μM RPA in the presence of ssDNA. Data points represent the mean and standard deviation of three independent experiments; note that the lower total Pi concentration levels in the minus RPA control assays in comparison to Figure 2A is due the difference in GFP–BLM concentration (5 nM versus 10 nM). (C) Schematic illustration of a ssDNA curtain bound by RPA-mCherry. (D) Kymograph showing that ssDNA-bound RPA-mCherry blocks GFP–BLM (5 nM) interactions with ssDNA; note that buffer flow was OFF during data collection and unbound RPA-mCherry was flushed out of the sample chamber prior to the injection of GFP–BLM.
Figure 6.
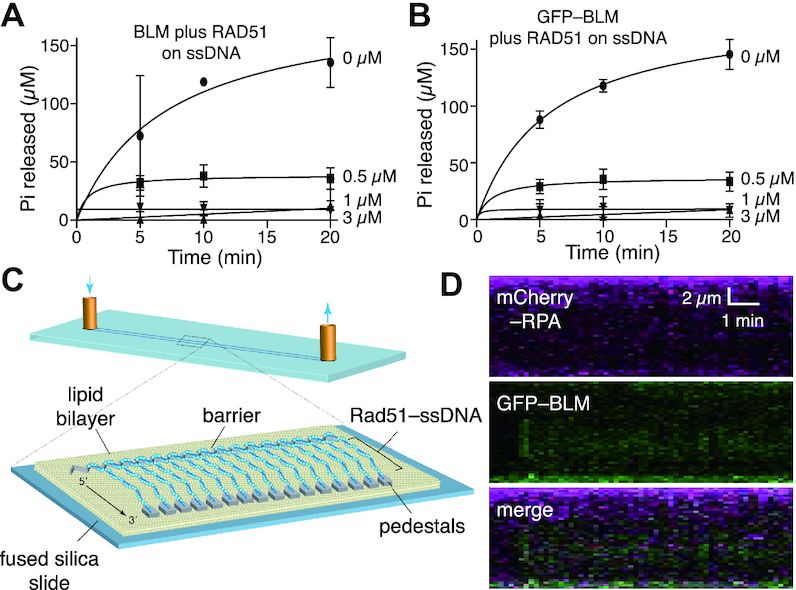
RAD51 blocks BLM activity on ssDNA. (A) ATPase assays containing 5 nM unlabeled BLM in the presence of 0, 0.5, 1 or 3 μM RAD51 and ssDNA (M13). Data points represent the mean and standard deviation of three independent experiments; note that the lower total Pi concentration levels in the minus RPA control assays in comparison to Figure 2A is due the difference in BLM concentration (5 nM versus 10 nM). (B) ATPase assays containing 5 nM GFP–BLM in the presence of 0, 0.5, 1 or 3 μM RAD51 and ssDNA (M13). Data points represent the mean and standard deviation of three independent experiments; note that the lower total Pi concentration levels in the minus RPA control assays in comparison to Figure 2A is due the difference in GFP–BLM concentration (5 nM versus 10 nM). (C) Schematic illustration of a ssDNA curtain bound by unlabeled RAD51-mCherry. (D) Kymograph showing that unlabeled RAD51 prevents GFP–BLM (5 nM) from interacting with ssDNA; note that buffer flow was OFF during data collection, unbound RAD51 was flushed from the sample chamber prior to the injection of GFP–BLM and the reactions contained 1 nM RPA-Cherry.
ATP hydrolysis assays testing for the effects of RPA and RAD51 were performed in BLM buffer plus calcium (20 mM Tris–HCl [pH 7.5], 1 mM MgCl2, 5 mM CaCl2, 2 mM ATP, 1 mM DTT, 0.2 mg/ml BSA) at 37°C. RPA or RAD51 with indicated concentrations were incubated with either M13 ssDNA (2 μM nucleotides) or pUC19 dsDNA (2 μM nucleotides) for 10 mins at 37°C before the addition of 5 nM BLM or GFP–BLM. Aliquots were removed at the indicated time points and quenched by addition of 50 mM EDTA and were quantified as described above.
Single- and double-strand DNA curtains
All experiments were conducted with a prism–type total internal reflection fluorescence (TIRF) microscope (Nikon) equipped with a 488-nm laser (Coherent Sapphire, 200 mW), a 561-nm laser (Coherent Sapphire, 200 mW), and two Andor iXon EMCCD cameras (29,37). Flow cells and ssDNA curtains were prepared as previously described (29,37,38). In brief, lipid bilayers were prepared with 91.5% DOPC, 0.5% biotinylated-PE, and 8% mPEG 2000-DOPE. The ssDNA substrate was generated using rolling circle replication with a biotinylated primer, a circular M13 ssDNA template, and phi29 DNA polymerase, as described (29,37,38). For assays with dsDNA, the substrates were prepared using λ-phage DNA (48.5 kb; NEB Cat No. N3011S) that was biotinylated at one end and labeled with digoxigenin (DIG) at the other end, as previously described (39,40). The biotinylated ssDNA or dsDNA was injected into the sample chamber and attached to the bilayer through a biotin–streptavidin linkage. The downstream ends of the ssDNA or dsDNA were then anchored to exposed Cr pedestals either through non-specific adsorption (for ssDNA) or through an antibody-DIG interaction (for dsDNA), as described (29,37–40). The flow cells were then attached to a microfluidic system and sample delivery was controlled using a syringe pump (Kd Scientific). For all two–color images, we used a custom–built shuttering system to avoid signal bleed–through during image acquisition. With this system, images from the green (GFP) and the red (mCherry) channels are recorded independently, these recordings are offset by 100 milliseconds such that when one camera records the red channel image, the green laser is shuttered off, and vice versa (29).
For experiments with RPA–ssDNA curtains, biotinylated ssDNA was aligned at the barriers by application of flow in BSA buffer (40 mM Tris–HCl [pH 8.0], 1 mM MgCl2, 1 mM DTT, 0.2 mg/ml BSA) at 37°C with a flow rate of 1 ml/min. Secondary structure was reduced with a single 500 μl injection of 7 M urea, immediately followed by 5–10 ml of BSA buffer containing 100 pM RPA-mCherry. Then buffer was changed to BLM buffer (20 mM Tris–HCl [pH 7.5], 1 mM MgCl2, 2 mM ATP, 1 mM DTT, 0.2 mg/ml BSA). 5 nM GFP–BLM in BLM buffer was injected into the flow cell through a 150 μl loop under the flow of BLM buffer. Buffer flow was stopped when GFP–BLM entered the sample chamber.
For experiments with RAD51–ssDNA curtains, 1 μM RAD51 was injected into a sample chamber containing pre-assembled RAD51–ssDNA filaments in the presence of HR buffer (30 mM Tris–HCl [pH 7.5], 1 mM MgCl2, 5 mM CaCl2, 100 mM KCl, 2 mM ATP, 1 mM DTT, 0.2 mg/ml BSA). Buffer flow was then terminated and the samples were incubated for 15 min in the absence of buffer flow at 37°C to allow RAD51 filament formation. Flow was resumed with HR buffer at 0.5 ml/min for 5 min to flush away unbound RAD51. The buffer was then switched to BLM plus calcium buffer and the 5 nM GFP–BLM and 1 nM RPA-mCherry in BLM plus calcium buffer were injected through a 150 μl loop into the sample chamber. Buffer flow was stopped when GFP–BLM entered the flow cell. For D-loop experiments, before the injection of GFP–BLM, 2 nM Atto565–labelled dsDNA (70 bp) with 15 nt of homology to the M13 ssDNA was injected through a 150 μl loop under the flow of HR buffer, followed by a 10-min incubation at 37°C, as described (41,42). Flow was resumed with HR buffer at 1 ml/min for 2 min to flush away any remaining Atto565–labelled dsDNA. Then buffer was changed to BLM plus calcium buffer. 15 nM GFP–BLM and 1 nM WT RPA in BLM plus calcium buffer were injected through a 150 μl loop under the flow of BLM plus calcium buffer. Buffer flow was stopped when GFP–BLM entered the sample chamber.
For experiments with RAD51–dsDNA curtains, double-tethered dsDNA curtains were prepared as described previously (39,40). RAD51 (1 μM) was injected into flow cell under the flow of BLM plus calcium buffer, followed by a 15-min incubation in the absence of buffer flow at 37°C. Flow was resumed with BLM plus calcium buffer at 0.5 ml/min for 5 min to flush any remaining RAD51. 0.4 nM GFP–BLM and 1 nM RPA-mCherry were injected through a 150 μl loop under the flow of BLM plus calcium buffer. Buffer flow was stopped when GFP–BLM entered the sample chamber.
Single-molecule data analysis
All data were collected at one frame per 10 s with 100-ms integration time, and the laser was shuttered between each acquired image to minimize photo-bleaching. Raw TIFF images were imported as image stacks into ImageJ. Images were corrected for drift using the StackReg function in ImageJ. Kymographs were then generated from the corrected image stacks by defining a one-pixel wide region-of-interest along the long axis of the individual ssDNA or dsDNA molecules and these kymographs were used for analysis of BLM or GFP–BLM processivity and velocity as described (29). Note, that all processivity and velocity measurements only included analysis of actively translocating molecules; inactive complexes were excluded from analysis.
The size of the DNA-bound GFP–BLM complexes was estimated by comparison to the signal intensity of a single GFP molecule, as follows. First, flow cells were prepared with a lambda-DNA substrate containing a 30-nt 3′ ssDNA, which serves as the binding site for a single molecule of GFP-tagged RPA, as described (43). GFP-RPA (1 nM) was injected into the flow cell in the presence of BSA buffer (40 mM Tris–HCl [pH 8.0], 1 mM MgCl2, 1 mM DTT, 0.2 mg/ml BSA). Buffer flow was then terminated and incubated for 10 min. Flow was resumed with BSA buffer at 0.5 ml/min for 5 min to flush away free GFP-RPA. The fluorescence signal intensity of the end-bound GFP-RPA molecules was then recorded and the fluorescent intensity of GFP-RPA was plotted and fitted to a Gaussian distribution. Photobleaching of the end-bound RPA-GFP was performed at 200 mM laser power (488 nm) under continuous laser illumination to confirm that the observed complexes reflected single molecules of GFP-RPA. Experiments using GFP–BLM on dsDNA in ATP-containing buffer were then performed as described above, and the fluorescence signal intensity of actively translocating molecules was then recorded and plotted with a bin size corresponding to the signal intensity of a single GFP based upon the measurements of single RPA-GFP molecules.
RESULTS
Single-molecule dsDNA curtain assays for BLM helicase activity
For visualizing BLM activity, we prepared double-tethered dsDNA curtains using λ-DNA (48.5-kb) as a substrate (39,40). In brief, the DNA molecules were anchored to a lipid bilayer deposited onto a flow-cell surface and aligned at nanofabricated chromium (Cr) barriers, allowing the DNA molecules to be visualized by total internal reflection fluorescence microscopy (Figure 1A). DNA unwinding by BLM could be detected by monitoring the association of mCherry-tagged RPA with the resulting strands of ssDNA (Figure 1B). This approach is similar to a strategy previously used for E. coli RecQ and SSB (44), and it leverages our expertise in the use of RPA-mCherry or RPA-GFP in single molecule DNA curtain assays (29,37). Incubation of unlabeled BLM and RPA-mCherry with the dsDNA curtains resulted in the formation of long tracts of RPA-mCherry fluorescence bound to the DNA, consistent with the expectation that BLM unwound the dsDNA to allow RPA-mCherry binding to the ssDNA strands (Figure 1C and Supplementary Figure S1).
Figure 1.
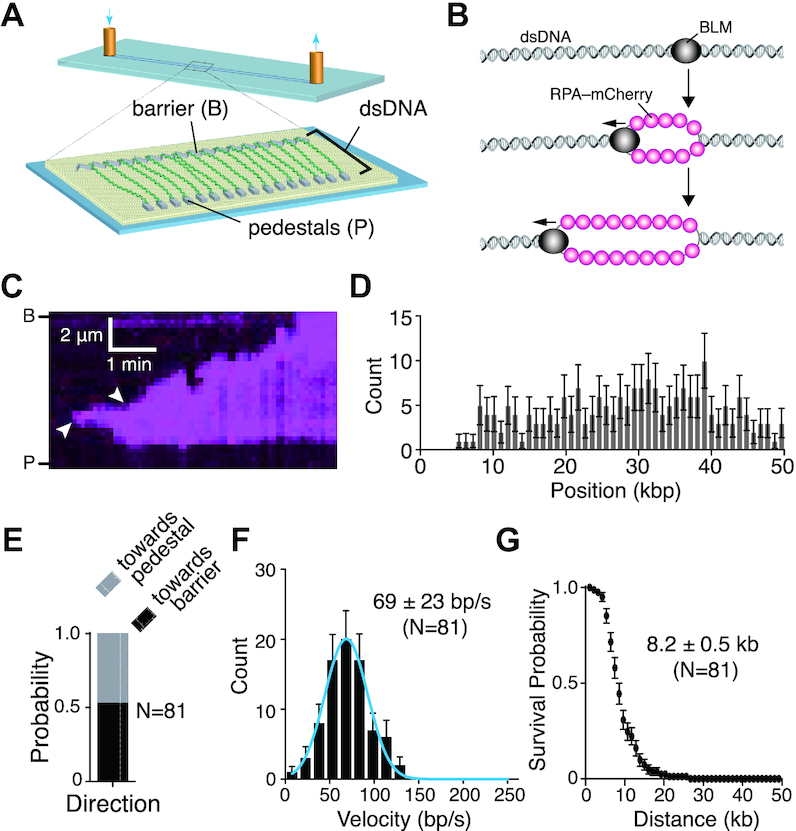
BLM is a fast and highly processive dsDNA helicase. (A) Schematic illustration of the double-tethered DNA curtains assay. (B) Schematic showing experimental rational for the detection of dsDNA unwinding activity for unlabeled BLM as revealed by the binding of RPA-mCherry to the resulting ssDNA products. (C) Kymograph showing BLM (unlabeled) unwinding dsDNA (unlabeled) as revealed by the binding of RPA-mCherry (magenta); note that buffer flow was OFF during data collection. Arrowheads indicate the sites where BLM initiated dsDNA unwinding based upon the appearance of RPA-mCherry. Reactions contained 0.2 nM unlabeled BLM and 1 nM RPA-mCherry. (D) Distribution of sites where BLM initiated dsDNA unwinding; error bars represent 95% confidence intervals calculated from bootstrap analysis. (E) Quantification of BLM translocation direction in reactions with 1 nM RPA-mCherry. ‘Towards pedestal’ indicates BLM movement in the direction from the barrier to the pedestal, and ‘towards barrier’ indicates movement in the opposite direction. (F) Velocity distribution of BLM unwinding rates in reactions with 1 nM RPA-mCherry on double-tethered dsDNA. The solid blue line represents a Gaussian fit to the data. Error bars represent 95% confidence intervals calculated from bootstrap analysis. (G) Survival probability plot of BLM translocation with 1 nM RPA-mCherry on double-tethered dsDNA. Error bars represent 95% confidence intervals calculated from bootstrap analysis.
Inspection of the resulting kymographs indicated that the RPA-mCherry signal typically appeared at random locations along the dsDNA (Figure 1C, D) and then spread outward from these sites (Figure 1C and Supplementary Figure S1). The observed patterns of time-dependent RPA-mCherry accumulation suggested that BLM was located at the leading edges of the spreading RPA-mCherry signal (see below). Approximately 50% of the BLM trajectories proceeded towards the Cr barriers and the remaining trajectories proceeded towards the Cr pedestals, indicating that there was no preferred direction for translocation along the dsDNA molecules (Figure 1E). The vast majority (N = 80/81) of events gave the appearance of unidirectional translocation (as depicted in Figure 1B), based upon the progressive unidirectional spreading of the RPA–mCherry signal. Analysis of the resulting trajectories suggested that BLM could unwind the dsDNA at a rate of 69 ± 23 bp/s (mean ± sd, N = 81; Figure 1F) and exhibited an average processivity of 8.2 ± 0.5 kb (mean ± sd, N = 81; Figure 1G). These results show that BLM is a fast and highly processive helicase capable of unwinding long tracts of dsDNA.
Direct visualization of GFP-tagged BLM during dsDNA unwinding
In the assays described above, we could only indirectly infer the location of unlabeled BLM relative to the RPA-mCherry signal on the DNA. To directly detect BLM, we expressed and purified a GFP–BLM fusion protein. BLM exhibits robust DNA-dependent ATP hydrolysis activity with both ssDNA and dsDNA substrates (22,45). Accordingly, bulk biochemical assays confirmed that GFP–BLM has ATPase activity comparable to that of unlabeled BLM with either ssDNA or dsDNA as a cofactor (Figure 2A). Moreover, previous studies have shown that GFP–BLM is functional in vivo (46), again suggesting that GFP–BLM has functional properties similar to untagged BLM.
We could easily detect dsDNA unwinding activity of GFP–BLM in assays with either RPA-mCherry or unlabeled RPA. In assays with RPA-mCherry, the GFP–BLM co-localized with the leading edges of the spreading RPA-mCherry signal, consistent with the interpretation that BLM generated the ssDNA strands that were then bound by RPA (Figure 2B and Supplementary Figure S2). Data analysis showed that GFP–BLM initially bound to random locations on the dsDNA (Figure 2C) and could then translocate in either direction along the dsDNA (Figure 2D). In assays with 0.2 nM GFP–BLM and 2 mM ATP there were ∼2–3 GFP–BLM complexes bound per dsDNA molecule (Supplementary Figure S3A, B). Approximately 47% of the molecules underwent translocation (Supplementary Figure S3C-S3D), revealing a translocation velocity of 78 ± 16 bp/sec (mean ± sd, N = 91) and an average translocation distance of 9.3 ± 0.6 kb (mean ± sd, N = 91) (Figure 2E, F). Control experiments using GFP–BLM with either no nucleotide co-factor, ADP or the nonhydrolyzable ATP analog ATPγS revealed no evidence for DNA translocation activity (Supplementary Figure S3A, B). GFP–BLM bound at comparable levels to dsDNA in buffer with ATP, ADP or ATPγ, however, binding activity was significantly reduced (∼10-fold) in buffer that lacked any nucleotide co-factor (Supplementary Figure S3A, B). The observation that GFP–BLM dsDNA-binding activity was compromised in the absence of nucleotide co-factor was consistent with prior reports of poor dsDNA binding activity in the absence of ATP (15). Assays with the ATP hydrolysis deficient mutant GFP–BLM-K695A revealed DNA-binding activity, but showed no evidence for translocation (Supplementary Figure S3C). Interestingly, this mutant should be deficient in ATP binding, which given our results with GFP–BLM in the absence of nucleotide co-factor (Supplementary Figure S3A, B), might suggest that GFP–BLM-K695A would not bind to DNA. We speculate that GFP–BLM-K695A may have residue ATP-binding activity sufficient to support DNA-binding activity, although further studies would be necessary to conclusively test this possibility. Regardless, our data show that ATP hydrolysis is essential for BLM movement on DNA in our assays.
We could also document GFP–BLM activity in reactions with unlabeled RPA (Figure 3A, Supplementary Figures S3D and S4). In this case, GFP–BLM bound to random locations on the dsDNA (Figure 3B) and could translocate in either direction (Figure 3C) at an average velocity of 101 ± 34 bp/sec (mean ± sd, N = 99) (Figure 3D) for an average distance of 8.1 ± 0.7 kb (mean ± sd, N = 99) (Figure 3E). The velocity of GFP–BLM in reactions with unlabeled RPA is higher than that seen with unlabeled BLM plus RPA-mCherry or GFP–BLM plus RPA-mCherry. This suggests that the mCherry tag on RPA may affect the translocation velocity of BLM slightly.
Figure 3.
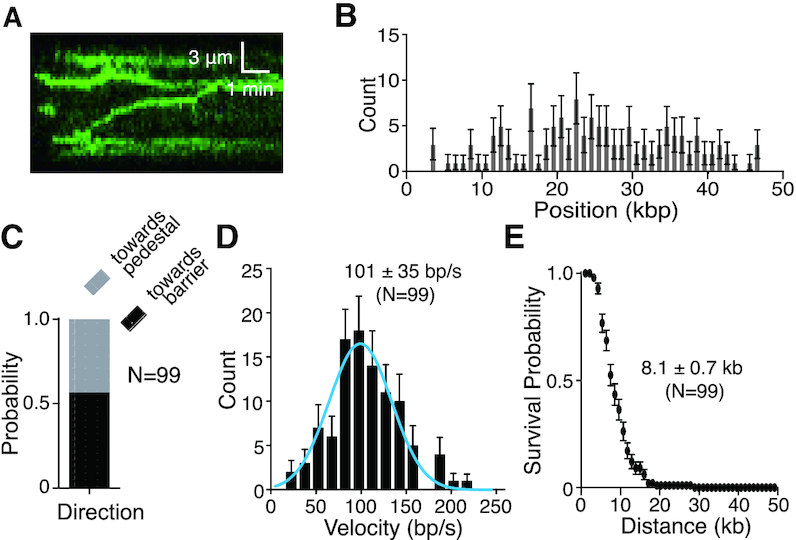
GFP–BLM translocation on dsDNA with unlabeled RPA. (A) Kymograph showing GFP–BLM (0.2 nM; green) translocation on dsDNA (unlabeled) in the presence of 1 nM unlabeled RPA; note that buffer flow was OFF during data collection. (B) Distribution of initiation sites for GFP–BLM in the presence of 1 nM unlabeled RPA (N = 132). Error bars represent 95% confidence intervals calculated from bootstrap analysis. (C) Quantification of GFP–BLM translocation direction in the presence of 1 nM unlabeled RPA. (D) Velocity distribution of GFP–BLM translocation in reactions with 1 nM unlabeled RPA on double-tethered dsDNA. The solid blue line represents a Gaussian fit to the data. Error bars represent 95% confidence intervals calculated from bootstrap analysis. (E) Survival probability plot of GFP–BLM translocation with 1 nM unlabeled RPA on double-tethered dsDNA. Error bars represent 95% confidence intervals calculated from bootstrap analysis.
RPA is not necessary for BLM translocase activity on dsDNA
BLM exhibited robust ATP hydrolysis activity on dsDNA (pUC19, 2686 bp) in the absence of RPA (Figure 2A), consistent with prior studies (22,45). We could also detect long-range translocation of GFP–BLM on dsDNA molecules even in the absence of RPA (Figure 4A, Supplementary Figures S3D and S5). Analysis of the resulting data revealed that GFP–BLM bound to seemingly random sites on the dsDNA in the absence of RPA (Figure 4B) and could translocate in either direction (Figure 4C), yielding an average translocation velocity of 94 ± 37 bp/sec (mean ± sd, N = 95) and an average processivity of 9.4 ± 0.5 kb (mean ± sd, N = 95; Figure 4D, E). From these results, we conclude that BLM can translocate on dsDNA without RPA being present. As shown schematically in Figure 1B, the DNA curtain assays can only be used to detect strand unwinding when fluorescent RPA is present in the buffer because we require fluorescent RPA as a tool to detect ssDNA production. In the absence of fluorescent RPA, we cannot detect any ssDNA that might be produced as a consequence of strand unwinding. Therefore, these assays performed in the absence of RPA cannot be used to infer whether or not the translocating BLM complexes unwind the dsDNA. However, previous studies have shown that dsDNA unwinding by BLM is compromised in the absence of RPA (11,12,47,48). Therefore, we speculate that the BLM translocation activity observed in the absence of RPA in our assays may not be coupled to extensive strand unwinding. One possibility is that under these conditions, BLM only transiently opens the dsDNA strands, but the ssDNA strands might quickly re-anneal when RPA is absent from the buffer. Alternatively, it is also possible that BLM could be moving along the dsDNA and not unwinding the strands
Figure 4.
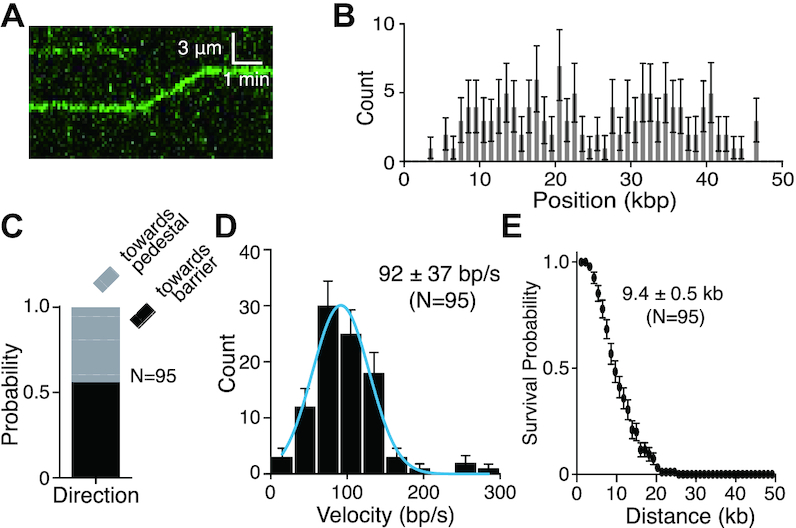
RPA is not necessary for GFP–BLM translocation on dsDNA. (A) Kymograph showing GFP–BLM (0.2 nM; green) translocation on dsDNA (unlabeled) without RPA; buffer flow was OFF during data collection. (B) Distribution of initiation sites for GFP–BLM in the absence of RPA (N = 134). Error bars represent 95% confidence intervals calculated from bootstrap analysis. (C) Quantification of GFP–BLM translocation direction in the absence of RPA. (D) Velocity distribution of GFP–BLM translocation rates without RPA on double-tethered dsDNA. The solid blue line represents a Gaussian fit to the data. Error bars represent 95% confidence intervals calculated from bootstrap analysis. (E) Survival probability plot of GFP–BLM translocation without RPA on double-tethered dsDNA. Error bars represent 95% confidence intervals calculated from bootstrap analysis.
GFP–BLM acts as a multimeric complex while translocating on dsDNA
Previous studies have suggested that BLM can exist in a number of different oligomeric states, although the biological implications of this ability to adopt different states remains uncertain [reviewed in ref. (4)]. Electron-microscopy studies revealed that full-length BLM in solution formed a six-fold symmetrical structure consistent with hexameric ring and also formed a four-fold symmetrical structure consistent with an alternative oligomeric state (49). The N-terminal domain of BLM is necessary for oligomerization (50) and studies of the monomeric BLM core lacking this oligomerization domain show that the monomer still retains ATP hydrolysis and helicase activities (45,51). Interestingly, one study suggested that in the presence of ATP, the BLM hexamer dissociated into either dimer or monomeric forms (52). To estimate the size of the actively translocating GFP–BLM complexes observed in the dsDNA curtain assays, we compared their fluorescence signal intensity to the signal intensity of a single RPA-GFP molecule (see Materials and Methods and Supplementary Figure S6A–D) (43). This analysis revealed a range of signal intensities with the average fluorescence signal intensity for the actively translocation GFP–BLM complexes corresponded to expectations for 3.1 ± 1.6 molecules of GFP–BLM per complex (mean ± s.d., N = 69; Supplementary Figure S6E). Notably, there was no correlation between signal intensity and translocation velocity or signal intensity and processivity, suggesting that although the complexes might contain different numbers of GFP–BLM molecules, as evidenced by the range of GFP–BLM signal intensity values, the general dsDNA translocation characteristics remained constant (Supplementary Figure S6F and G). Importantly, although it is safe to conclude that the complexes we observe translocating on the DNA are comprised of multiple BLM molecules, we caution that the values we report are only an estimate. Exact details of the BLM oligomeric state will be better revealed through high-resolution structural studies of the active DNA-bound BLM complex
BLM is unable to act upon RPA–ssDNA
The experiments described above show that BLM is a potent helicase capable of processively unwinding long tracts of dsDNA. We next sought to determine the translocation properties of BLM on ssDNA. Given that BLM hydrolyzes ATP avidly when ssDNA is present, we surmise that it also translocates on ssDNA (Figure 2A) (47). In the physiological setting, ssDNA is coated by RPA (53,54). Therefore, we asked whether we could detect the activity of GFP–BLM on RPA–ssDNA. Bulk biochemical assays revealed that RPA strongly inhibited the ssDNA-dependent ATPase activity of BLM (M13 ssDNA, 6407 nt; Figure 5A, B), which is in agreement with a previous report (47). This suggested to us that RPA either blocks BLM association with ssDNA, interferes with the DNA translocation of bound BLM, or both. To help distinguish between these possibilities, we examined the behavior of GFP–BLM in DNA curtain assays using RPA-coated ssDNA as a substrate (29,37). Here, we found limited evidence of GFP–BLM binding to ssDNA even when the concentration of GFP–BLM was increased 12.5-fold relative to that used with the dsDNA substrate (5 nM versus 0.4 nM; cf. Figures 2B, 5D and Supplementary Figure S7). This result suggests that BLM is unable to readily associate with RPA–ssDNA. Moreover, in no instance did we observe evidence for GFP–BLM translocation on RPA-bound ssDNA molecules. Taken together, our bulk biochemical and single molecule assays strongly suggest that RPA prevents BLM from binding and translocating on ssDNA.
BLM is unable to act upon active Rad51–ssDNA filaments
RAD51 forms an extended helical filament on ssDNA derived from DNA end resection (10,55,56). The resulting nucleoprotein filament is referred to as the presynaptic complex, which catalyzes the ATP-dependent pairing of the bound ssDNA with a homologous dsDNA target and DNA strand exchange between the paired DNA molecules (10,55,56). BLM is capable of removing RAD51 from ssDNA when RAD51 is in its inactive ADP-bound state (26,27). In contrast, BLM does not act upon the active ATP-bound form of the RAD51 filaments (26,27). Consistent with these results, bulk biochemical assays demonstrated that the ssDNA-dependent ATPase activity of BLM was strongly inhibited by RAD51 (Figure 6A, B). Note, assembly of active RAD51 filaments on ssDNA requires the presence of both ATP and Ca2+ in the reaction buffer (33,57), therefore, we also conducted control ATP hydrolysis reactions in which we compared the ATPase activity of BLM on either naked ssDNA or naked dsDNA in the presence or absence of 5 mM Ca2+. These control assays confirmed that the DNA-dependent ATP hydrolysis activity of BLM was not drastically inhibited by Ca2+ (Supplementary Figure S8). Moreover, we were unable to detect evidence for BLM having any capacity to disrupt the active form of the RAD51–ssDNA nucleoprotein filament in DNA curtain assays (Figure 6C). We were unable to detect significant binding or any translocation of GFP–BLM on the RAD51–ssDNA nucleoprotein filaments even when the concentration of GFP–BLM was 12.5-fold of what was necessary to observe activity on naked dsDNA (c.f. Figures 2B and 6D, Supplementary Figures S3A, B and S9). Notably, addition of the BLM–interacting protein complex Topo IIIα/RMI1/RMI2 (TRR) modestly enhanced the binding of BLM to the RAD51–ssDNA, yielding ∼2 binding events per ssDNA, but only when the GFP–BLM was 12.5-fold of what was necessary to observe activity on naked dsDNA (Supplementary Figure S3A, B). However, these TRR-containing BLM complexes still exhibited no evidence of translocation activity (Supplementary Figure S3C). We conclude that the active form of the RAD51 filament prevents BLM from associating with ssDNA, and although TRR can promote BLM binding on RAD51–ssDNA, the bound BLM complexes still do not strip the active RAD51 filaments from the ssDNA.
RAD51 blocks BLM activity on dsDNA
As shown above, both RPA and RAD51 attenuate the ssDNA-dependent ATPase activity of BLM by inhibiting the association of BLM with ssDNA. We next asked whether BLM would act on RAD51-bound dsDNA, as, following D-loop formation, RAD51 would remain on the heteroduplex dsDNA product (10,55,56). Our bulk biochemical assays confirmed that BLM exhibits robust ATP hydrolysis activity in assays with dsDNA (Figure 7A, B, Supplementary Figures S8B and S10), consistent with prior studies (22,45), however, we find that the addition of RAD51 to these assays inhibited the ATP hydrolysis activity of BLM (Figure 7A,B). This observation suggests that BLM is not active when the dsDNA substrate is bound by RAD51. Consistent with this observation, using double-tethered dsDNA curtains coated with RAD51 (Figure 7C), we were able to detect association of GFP–BLM with the RAD51-bound dsDNA molecules (Figure 7D, Supplementary Figures S3A, B and S11). However, we did not observe any evidence for the highly processive dsDNA unwinding trajectories that are characteristic of BLM activity on naked dsDNA (cf. Figures 2B and 7D, Supplementary Figures S3C and S11). Note, in these assays, the BLM could in principle bind to either (i) the dsDNA (for instance, in small gaps between RAD51 filaments), (ii) directly to the RAD51 proteins or (iii) perhaps both RAD51 and the dsDNA. Regardless of the binding mechanism, these results indicate that RAD51 greatly restricts the ability of BLM to act on dsDNA.
Figure 7.

BLM cannot unwind dsDNA bound by RAD51. (A) ATPase assays containing 5 nM unlabeled BLM in the presence of 0, 0.5, 1 or 3 μM RAD51 and dsDNA (pUC19). Data points represent the mean and standard deviation of three independent experiments. (B) ATPase assays containing 5 nM GFP–BLM in the presence of 0, 0.5, 1 or 3 μM RAD51 and dsDNA (pUC19). Data points represent the mean and standard deviation of three independent experiments. (C) Schematic illustration of a double-tethered dsDNA curtain coated with RAD51. (D) Kymograph showing that GFP–BLM (0.4 nM) can bind to RAD51-bound dsDNA, but is unable to unwind the dsDNA; note that buffer flow was OFF during data collection, unbound RAD51 was flushed from the sample chamber prior to the injection of GFP–BLM and the reactions contained 1 nM RPA-Cherry.
BLM is recruited to RAD51-bound heteroduplex DNA intermediates
BLM exhibits highly active binding activity for a variety of DNA structures, including G-quadraplexes, Holliday junctions, DNA forks and DNA bubbles (4,23,45). Moreover, BLM exhibits robust helicase activity on deproteinized D-loops prepared with bacterial recombinase RecA (58). BLM can also prevent DNA loop formation by RAD51 when the RAD51 filaments are inactivated by removal of Ca2+ (26,27), but BLM is unable to disrupt D-loops bound by the active ATP-bound form of RAD51 (13,26,27). Therefore, we next tested whether BLM could bind to and dismantle heteroduplex DNA joints prepared in the presence of active RAD51 in the DNA curtain assays. For these assays, we prepared the RAD51–ssDNA filaments using ssDNA curtains, as described above. The RAD51–ssDNA filaments were then incubated with a fluorescently-tagged (Atto565) 70-bp oligonucleotide substrate bearing a 15-nucleotide tract of sequence homologous to the M13 ssDNA bound by RAD51 to allow for the formation of heteroduplex DNA joints (Figure 8A), as previously described (41,42). Remarkably, the majority of the observed GFP–BLM molecules (∼85%) co-localized with Atto565 labelled heteroduplex DNA joints (Figure 8B), demonstrating that BLM has a high affinity for these recombination intermediates. The remain smaller population of GFP–BLM molecules (∼15%) were targeted to regions that were not co-localized with a visible Atto565-labeled heteroduplex joint, although we cannot rule out the possibility that these molecules may have been bound at dark (unlabeled) heteroduplex joints that had been photobleached (Figure 8B). Importantly, as with the other protein-bound recombination intermediates, there was no evidence that the bound GFP–BLM could disrupt the heteroduplex intermediates or translocate along the RAD51 filaments (Figure 8C and Supplementary Figure S12). These results are in good agreement with prior studies showing that BLM is unable to act upon D-loops prepared with active ATP-bound RAD51 (26,27) and extend these findings by showing that BLM can bind to these structures even though they remain resistant to disrupt. Given that RAD51 blocks the binding of BLM to ssDNA (see Figure 6) we speculate that in assays with the heteroduplex DNA joints, the BLM complexes are targeted to the short dsDNA segments that flank the heteroduplex joint or the displaced ssDNA strand (depicted in Figure 8A).
Figure 8.

BLM cannot dismantle RAD51-bound heteroduplex DNA joints. (A) Schematic illustration of heteroduplex joints prepared with RAD51–ssDNA curtains and an Atto565-labeled dsDNA oligonucleotide (70-bp) bearing 15-nts of sequence homologous to the M13-derived ssDNA substrate. (B) Percent of GFP–BLM (15 nM) molecules bound the Atto565-labeled heteroduplex DNA joints or bound elsewhere on the RAD51–ssDNA (N = 116). (C) Kymograph showing outcomes of reactions containing GFP–BLM (15 nM) with RAD51–ssDNA curtains preassembled with the Atto565-labeled heteroduplex DNA joints; note that buffer flow was OFF during data collection. The Asterix highlights an example of bound GFP–BLM that did not co-localize with the heteroduplex DNA joint.
DISCUSSION
Our work shows that human BLM is a fast and highly processive helicase and is capable of completely unwinding long regions of naked dsDNA, leaving the unwound ssDNA strands coated with RPA. In striking contrast to its robust interactions with naked dsDNA, we are unable to detect evidence for efficient BLM interactions with either RPA-coated ssDNA or RAD51-coated ssDNA. Similarly, the ATP-bound form of RAD51 also blocks BLM activity on dsDNA and heteroduplex DNA joints. These findings are consistent with prior studies showing that BLM cannot disrupt the active ATP-bound form of the RAD51 filament and cannot dismantle D-loops in the presence of active RAD51 (13,26,27). As discussed below, the differential responses of BLM towards potential substrates may have important implications with respect to the roles that BLM plays in DNA replication and repair.
BLM is a robust dsDNA helicase
Our data show that BLM can translocate at a rate of ∼70–100 bp/s and travel an average distance of ∼8–10 kb while unwinding naked dsDNA, allowing the unwound single strands to become quickly coated with RPA. This activity is likely relevant to the numerous functions that BLM fulfills in DNA replication and repair. In particular, we speculate that robust BLM translocation activity could contribute to processive DNA end resection (11,59,60) and may help compensate for the possible need to disrupt nucleosomes as it acts upon chromatin during DNA end resection in vivo (24,61–63). Interestingly, previous single molecule fluorescence resonance energy transfer (smFRET) studies of the BLM core domain, comprised of amino acids 642–1290 (45), indicated that BLM could undergo repetitive cycles of dsDNA unwinding, suggesting a strand switching model where BLM moved back and forth along a short section of dsDNA while unwinding <34 bp (48). Bulk biochemical studies of this same BLM core domain suggested that it could translocate on ssDNA, but could only travel ∼50 nucleotides before dissociating from the ssDNA substrate (64). Similarly, repetitive unwinding and/or low processivity have been reported for BLM acting upon short G-quartet containing substrates (20,65). We do not see evidence for repetitive cycling by BLM in our assays with longer dsDNA substrates, but we would not be able to spatially resolve back and forth motions over a short distance, so we cannot rule out this type of behavior for the fraction of GFP-tagged BLM molecules that appear to remain stationary.
We find clear evidence for very long-range dsDNA translocation (∼8–10 kb) by both unlabeled BLM and GFP-tagged BLM on naked dsDNA substrates. In contrast, earlier bulk biochemical studies of BLM suggested that it was a relatively slow helicase with low processivity and was poor at acting upon fully dsDNA substrates (23,45,66). However, these findings are in contrast to studies showing that BLM can unwind both short (50- to 80-bp) and longer (2.0- to 2.7-kb) substrates (11,12). One key difference between these studies is that the more recent works used lower concentrations of MgCl2 (2 mM) (11,12), whereas the earlier studies used a higher concentration of MgCl2 (5 nM), which was later found to inhibit BLM activity (11). Similarly, single molecule smFRET and bulk biochemical studies performed at higher concentrations of MgCl2 also reported low BLM processivity (48,64,65). Our experiments were all conducted at lower MgCl2 (1 mM) and our results are in general agreement with studies showing that BLM can act upon both short and longer dsDNA substrates (11,12). Importantly, the bulk biochemical studies did not report processivity values for BLM in these assays with the longer dsDNA substrates, so we are unable to directly compare our processivity results to theirs (11,12). But, one might infer a lower bound on the order of 2.7 kb for BLM processivity on dsDNA, since a substantial portion of the substrates in these studies were completely unwound (11,12). There is another important difference between our work and the bulk biochemical studies, namely, we see a large proportion of inactive GFP–BLM complexes that fail to translocate on naked dsDNA (Supplementary Figure S3C). Accordingly, these inactive complexes are not included in our calculations of BLM translocation velocity and processivity, whereas it would not be possible to distinguish between active and inactive complexes in ensemble assays. If a large proportion of BLM complexes are inactive in the ensemble biochemical measurements, then one might anticipate that these measurements would underestimate kinetic parameters (i.e. velocity and processivity) for DNA unwinding activity.
Notably, in the absence of RPA, BLM is unable to unwind short blunt ended 25- to 50-bp blunt ended DNA fragments, suggesting that BLM required a ssDNA overhang or bubble or other DNA structural elements for efficient strand unwinding (23,45). However, other studies showed that BLM could efficiently unwind blunt ended 50- and 80-bp substrates so long as RPA is present (11,12). Our work showing that BLM can act upon naked dsDNA indicates that BLM can initiate translocation and unwinding at random positions throughout the dsDNA molecules. Moreover, assuming that ATP hydrolysis activity serves as a good proxy for translocation/helicase activity, then our ATP hydrolysis assays with closed circular DNA, as well as other published ATPase assays using closed circular dsDNA (22,45), also support the conclusion that BLM can bind to internal locations on dsDNA molecules and can initiate translocation and strand unwinding from these internal sites. However, we do not precisely what structural features within the dsDNA are recognized by BLM, and given that our assays use long dsDNA substrates, either pUC19 (2.7 kb), ΦX174 (5.4 kb), or lambda phage DNA (48.5 kb), it is possible that BLM binds to transient bubbles within the dsDNA. Finally, our data demonstrate that BLM can bind to and translocate on dsDNA in the absence of RPA, but as indicated above in the Results section, our assays conducted in the absence of RPA should not be interpreted as suggesting BLM helicase activity is independent of RPA. Instead, given the prior works showing that RPA is necessary for BLM helicase activity (11,12,47), it is possible that BLM translocation and strand unwinding (i.e. helicase) activities are decoupled from one another when RPA is absent. One simple explanation would be that BLM can perhaps transiently separate the dsDNA strands, but then they immediately reanneal because there is no RPA to keep the strands apart. Future studies using an alternative fluorescently tagged ssDNA-binding protein, such as E. coli SSB, might allow one to overcome this technical limitation.
BLM interactions with ssDNA are blocked by RPA
BLM interacts with RPA based upon ELISA and Far Western assays (47). However, ATP hydrolysis by BLM is greatly attenuated in reactions with RPA and ssDNA (this study and (47)) and we are unable to detect evidence of BLM interactions with RPA-bound ssDNA. Our data strongly suggest that RPA inhibits BLM by blocking its association with ssDNA. These findings are also in good agreement with a recent study showing that RPA blocked BLM association with ssDNA (15). RPA is a highly abundant with an extremely high affinity for ssDNA, so all ssDNA is likely to become coated by RPA in the physiological setting (53,54). Thus, these findings predict that RPA may prevent BLM from binding to ssDNA intermediates present during DNA recombination and repair, and may instead restrict BLM activity to nearby regions dsDNA or ssDNA/dsDNA junctions. Interestingly, although conventional microscopy studies show that BLM and RPA overlap in DNA repair foci during meiosis (67), higher resolution SIM-microscopy studies suggest that BLM and RPA are actually slightly offset from one another, suggesting that they do not directly interact but instead occupy distinct positions within spatially juxtaposed DNA intermediates (68).
We have previously characterized the anti-recombinase activity of the UvrD-like helicase Srs2 and the RecQ helicase Sgs1 from Saccharomyces cerevisiae (28,30–32). These helicases are initially loaded onto small clusters of RPA present at the ends of Rad51 filaments, but do not appear to load at internal positions within the Rad51 filaments (28,31,32). They then undergo rapid 3′→5′ translocation while stripping Rad51 from the ssDNA (28,31,32). In addition, Srs2 and Sgs1 can both bind to and translocate along RPA-coated ssDNA molecules (28,30). Thus, the properties of these yeast anti-recombinases with respect to interactions with ssDNA-containing recombination intermediates appear to be very different from human BLM.
Is BLM a bona fide anti-recombinase?
Previous studies have suggested that BLM can act as an anti-recombinase to dismantle RAD51 filaments both in vitro and in vivo (26,69). This type of activity would require BLM to act upon protein-bound ssDNA intermediates. However, while BLM acts upon the ADP-bound form of the RAD51 filament, which is highly unstable (33,70), it cannot dissociate the catalytically active, ATP-bound form of the RAD51 filament (13,26,27). Consistent with these prior studies (26,27), we find no evidence for BLM interactions with the ATP-bound RAD51 filament in our assays (Figure 7). It should be noted that human RAD51 forms active ATP-bound filaments in vitro only in the presence of the Ca2+ ion, which restricts ATP hydrolysis by RAD51 (33,57). Prior reports indicate that RAD51 can form inactive filaments on DNA in the ADP-bound state in vitro (26,71,72). However, we are unable to detect the formation of inactive RAD51 filaments in our assays, and the active RAD51 filaments quickly dissociate from ssDNA when inactivated by the removal of Ca2+ (33,70). Our inability to detect inactive ADP-bound RAD51 filaments is likely due to a technical limitation in that our ssDNA curtain assays require the presence of RPA; we cannot make ssDNA curtains without RPA due to the formation of extensive ssDNA secondary structure (which is removed by RPA) and because we need fluorescent RPA to see the ssDNA (29,37,38). RPA has an extremely high affinity for ssDNA (53,54) and we anticipate that the inactive ADP-bound form of the RAD51 filaments cannot assemble onto ssDNA when RPA is present.
The inability of BLM to dismantle active RAD51 filaments in vitro raises the question of what form(s) of RAD51 are most prevalent in vivo and how these forms may change over the course of a repair reaction. Fluorescence recovery after photobleaching (FRAP) studies show that subnuclear RAD51 foci are highly stable in vivo at sites of DNA damage (73), suggesting that these foci reflect the active form of RAD51. However, it is not yet possible to correlate a microscopically observable RAD51 focus with underlying mechanistic events, thus any given RAD51 focus could in principle reflect any stage of recombination beginning with early presynaptic complex intermediates to post-strand invasion intermediates. As such, it is possible that RAD51 foci measurements do not reflect the earliest and latest stages of recombination when RAD51 may be unstable and undergoing substantial turnover, but may instead only reflect the most stable intermediates that exist after presynaptic complex assembly and perhaps the earliest stages of strand invasion. Interestingly, a recent study has shown that RAD51 focus formation is regulated by BLM (69,74). Cells lacking BRCA1 show decreased RAD51 foci formation upon exposure to ionizing radiation, but deletion of BLM restores RAD51 focus formation in these BRCA1-deficient cells (69). Conversely, BLM overexpression reduces RAD51 focus formation (69). These findings are consistent with a model where BLM prevents RAD51 filament from being made in BRCA1-deficient cells (69,74). However, the exact molecular mechanism underlying these observations remains unknown (69,74). If BLM really removes RAD51 from ssDNA in cells, then one would need to invoke a model in which the in vivo activity of BLM is markedly different from its in vitro characteristics. One possibility is that there exists an important aspect of these in vivo reactions that is not fully recapitulated in vitro. For instance, it is possible that a post-translational modification (e.g. phosphorylation, acetylation, and SUMOylation) or perhaps an interaction with an unknown partner protein might promote the recruitment and activity of BLM on active RAD51 filaments. Alternatively, something may inactivate the RAD51 filaments, allowing for their disruption by BLM. This later possibility is intriguing in that we show BLM is specifically targeted to the RAD51 bound heteroduplex DNA joints, and might be poised to quickly dismantle these structures if RAD51 were inactivated. Nevertheless, the inability of BLM to dismantle active RAD51 filaments is in sharp contrast with known anti-recombinases including human RECQ5 (75), and yeast Srs2 and Sgs1 (28,31,76,77), all of which are adept at disrupting active RAD51 filaments (13). Moreover, the inability of BLM to act upon RPA- and RAD51–ssDNA complexes manifests at the level of binding suggests that BLM would not be readily targeted to these ssDNA-containing recombination intermediates.
Finally, our results with BLM are in direct contrast to findings with the human helicase RECQ5, which can efficiently dismantle the active form of the RAD51–ssDNA (13,75,78). We speculate that the different behaviors of RECQ5 and BLM toward these types of nucleoprotein substrates may reflect a division of labor for these two human RECQ helicases with respect to substrate specificity during DNA replication and DNA repair. We anticipate that the establishment of DNA curtain assays for studying the properties of BLM will allow for experiments designed to address the roles of BLM in processes such as DNA end resection or Holliday junction dissolution, and these assays may also be adapted for other human RECQ helicases.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health grants [R01CA236606 to E.C.G., R35GM118026 to E.C.G., R01ES007061 to P.S., P30CA054174 to P.S., P01CA92584 to E.C.G. and P.S.]; NSF grant [MCB-1154511 to E.C.G.]; Cancer Prevention & Research Institute of Texas (CPRIT) Recruitment of Established Investigators (REI) Award [RR180029 to P.S.]. Funding for open access charge: National Institutes of Health Grants.
Conflict of interest statement. None declared.
REFERENCES
- 1. Bernstein K., Gangloff S., Rothstein R.. The RecQ DNA helicases in DNA repair. Annu. Rev. Genet. 2010; 44:393–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Branzei D., Foiani M.. RecQ helicases queuing with Srs2 to disrupt Rad51 filaments and suppress recombination. Genes Dev. 2007; 21:3019–3026. [DOI] [PubMed] [Google Scholar]
- 3. Chu W., Hickson I.. RecQ helicases: multifunctional genome caretakers. Nat. Rev. Cancer. 2009; 9:644–654. [DOI] [PubMed] [Google Scholar]
- 4. Croteau D., Popuri V., Opresko P., Bohr V.. Human RecQ helicases in DNA repair, recombination, and replication. Annu. Rev. Biochem. 2014; 83:519–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brosh R., Jr. DNA helicases involved in DNA repair and their roles in cancer. Nat. Rev. Cancer. 2013; 13:542–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Arora H., Chacon A., Choudhary S., McLeod M., Meshkov L., Nouri K., Izakovic J.. Bloom syndrome. Int. J. Dermatol. 2014; 53:798–802. [DOI] [PubMed] [Google Scholar]
- 7. Campbell M., Campbell W., Rogers J., Rogers N., Rogers Z., van den Hurk A., Webb A., Webb T., Zaslaw P.. Bloom syndrome: research and data priorities for the development of precision medicine as identified by some affected families. Cold Spring Harb. Mol. Case Stud. 2018; 4:a002816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ellis N., Groden J., Ye T., Straughen J., Lennon D., Ciocci S., Proytcheva M., German J.. The Bloom's syndrome gene product is homologous to RecQ helicases. Cell. 1995; 83:655–666. [DOI] [PubMed] [Google Scholar]
- 9. German J. Bloom's syndrome. XX. The first 100 cancers. Cancer Genet. Cyogenet. 1997; 93:100–106. [DOI] [PubMed] [Google Scholar]
- 10. Kowalczykowski S. An overview of the molecular mechanisms of recombinational dna repair. Cold Spring Harbor Perspect. Biol. 2015; 7:a016410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nimonkar A., Genschel J., Kinoshita E., Polaczek P., Campbell J., Wyman C., Modrich P., Kowalczykowski S.. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011; 25:350–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Daley J., Chiba T., Xue X., Niu H., Sung P.. Multifaceted role of the Topo IIIalpha-RMI1-RMI2 complex and DNA2 in the BLM-dependent pathway of DNA break end resection. Nucleic Acids Res. 2014; 42:11083–11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Branzei D., Szakal B.. Building up and breaking down: mechanisms controlling recombination during replication. Crit. Rev. Biochem. Mol. Biol. 2017; 52:381–394. [DOI] [PubMed] [Google Scholar]
- 14. Bizard A., Hickson I.. The dissolution of double Holliday junctions. Cold Spring Harbor Perspect. Biol. 2014; 6:a016477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sarlos K., Biebricher A., Bizard A., Bakx J., Ferrete-Bonastre A., Modesti M., Paramasivam M., Yao Q., Peterman E., Wuite G. et al.. Reconstitution of anaphase DNA bridge recognition and disjunction. Nat. Struct. Mol. Biol. 2018; 25:868–876. [DOI] [PubMed] [Google Scholar]
- 16. Chan Y., Fugger K., West S.. Unresolved recombination intermediates lead to ultra-fine anaphase bridges, chromosome breaks and aberrations. Nat Cell Biol. 2018; 20:92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Davies S., North P., Hickson I.. Role for BLM in replication-fork restart and suppression of origin firing after replicative stress. Nat. Struct. Mol. Biol. 2007; 14:677–679. [DOI] [PubMed] [Google Scholar]
- 18. van Wietmarschen N., Merzouk S., Halsema N., Spierings D., Guryev V., Lansdorp P.. BLM helicase suppresses recombination at G-quadruplex motifs in transcribed genes. Nat. Commun. 2018; 9:271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cortez D. Preventing replication fork collapse to maintain genome integrity. DNA Repair (Amst.). 2015; 32:149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chatterjee S., Zagelbaum J., Savitsky P., Sturzenegger A., Huttner D., Janscak P., Hickson I., Gileadi O., Rothenberg E.. Mechanistic insight into the interaction of BLM helicase with intra-strand G-quadruplex structures. Nat. Commun. 2014; 5:5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Datta A., Brosh R. Jr.. New insights into DNA helicases as druggable targets for cancer therapy. Front. Mol. Biosci. 2018; 5:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Karow J., Chakraverty R., Hickson I.. The Bloom's syndrome gene product is a 3′-5′ DNA helicase. J. Biol. Chem. 1997; 272:30611–30614. [DOI] [PubMed] [Google Scholar]
- 23. Mohaghegh P., Karow J., Brosh R. Jr., Bohr V., Hickson I.. The Bloom's and Werner's syndrome proteins are DNA structure-specific helicases. Nucleic Acids Res. 2001; 29:2843–2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Daley J., Niu H., Miller A., Sung P.. Biochemical mechanism of DSB end resection and its regulation. DNA Repair (Amst.). 2015; 32:66–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Symington L. End resection at double-strand breaks: mechanism and regulation. Cold Spring Harbor Perspect. Biol. 2014; 6:a016436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bugreev D., Yu X., Egelman E., Mazin A.. Novel pro- and anti-recombination activities of the Bloom's syndrome helicase. Genes Dev. 2007; 21:3085–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bugreev D., Mazina O., Mazin A.. Bloom syndrome helicase stimulates RAD51 DNA strand exchange activity through a novel mechanism. J. Biol. Chem. 2009; 284:26349–26359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Crickard J., Xue C., Wang W., Kwon Y., Sung P., Greene E.. The RecQ helicase Sgs1 drives ATP-dependent disruption of Rad51 filaments. Nucleic Acids Res. 2019; 47:4694–4706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. De Tullio L., Kaniecki K., Greene E.. Single-stranded DNA curtains for studying the srs2 helicase using total internal reflection fluorescence microscopy. Methods Enzymol. 2018; 600:407–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. De Tullio L., Kaniecki K., Kwon Y., Crickard J., Sung P., Greene E.. Yeast Srs2 Helicase Promotes Redistribution of Single-Stranded DNA-Bound RPA and Rad52 in Homologous Recombination Regulation. Cell Rep. 2017; 21:570–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kaniecki K., De Tullio L., Gibb B., Kwon Y., Sung P., Greene E.. Dissociation of rad51 presynaptic complexes and heteroduplex DNA joints by tandem assemblies of srs2. Cell Rep. 2017; 21:3166–3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Crickard J., Kaniecki K., Kwon Y., Sung P., Greene E.. Meiosis-specific recombinase Dmc1 is a potent inhibitor of the Srs2 antirecombinase. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:E10041–E10048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ma C., Gibb B., Kwon Y., Sung P., Greene E.. Protein dynamics of human RPA and RAD51 on ssDNA during assembly and disassembly of the RAD51 filament. Nucleic Acids Res. 2017; 45:749–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ma C., Kwon Y., Sung P., Greene E.. Human RAD52 interactions with replication protein A and the RAD51 presynaptic complex. J. Biol. Chem. 2017; 292:11702–11713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bussen W., Raynard S., Busygina V., Singh A., Sung P.. Holliday junction processing activity of the BLM-Topo IIIalpha-BLAP75 complex. J. Biol. Chem. 2007; 282:31484–31492. [DOI] [PubMed] [Google Scholar]
- 36. Sigurdsson S., Trujillo K., Song B., Stratton S., Sung P.. Basis for avid homologous DNA strand exchange by human Rad51 and RPA. J. Biol. Chem. 2001; 276:8798–8806. [DOI] [PubMed] [Google Scholar]
- 37. Ma C., Steinfeld J., Greene E.. Single-stranded DNA curtains for studying homologous recombination. Methods Enzymol. 2017; 582:193–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gibb B., Silverstein T., Finkelstein I., Greene E.. Single-stranded DNA curtains for real-time single-molecule visualization of protein-nucleic acid interactions. Anal. Chem. 2012; 84:7607–7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gorman J., Fazio T., Wang F., Wind S., Greene E.. Nanofabricated racks of aligned and anchored DNA substrates for single-molecule imaging. Langmuir. 2010; 26:1372–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Greene E., Wind S., Fazio T., Gorman J., Visnapuu M.. DNA curtains for high-throughput single-molecule optical imaging. Methods Enzymol. 2010; 472:293–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Qi Z., Redding S., Lee J., Gibb B., Kwon Y., Niu H., Gaines W., Sung P., Greene E.. DNA sequence alignment by microhomology sampling during homologous recombination. Cell. 2015; 160:856–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee J., Terakawa T., Qi Z., Steinfeld J., Redding S., Kwon Y., Gaines W., Zhao W., Sung P., Greene E.. DNA RECOMBINATION. Base triplet stepping by the Rad51/RecA family of recombinases. Science. 2015; 349:977–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xue C., Wang W., Crickard J., Moevus C., Kwon Y., Sung P., Greene E.. Regulatory control of Sgs1 and Dna2 during eukaryotic DNA end resection. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:6091–6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rad B., Forget A., Baskin R., Kowalczykowski S.. Single-molecule visualization of RecQ helicase reveals DNA melting, nucleation, and assembly are required for processive DNA unwinding. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:E6852–E6861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Janscak P., Garcia P., Hamburger F., Makuta Y., Shiraishi K., Imai Y., Ikeda H., Bickle T.. Characterization and mutational analysis of the RecQ core of the bloom syndrome protein. J. Mol. Biol. 2003; 330:29–42. [DOI] [PubMed] [Google Scholar]
- 46. Hu P., Beresten S., van Brabant A., Ye T., Pandolfi P., Johnson F., Guarente L., Ellis N.. Evidence for BLM and Topoisomerase IIIalpha interaction in genomic stability. Hum. Mol. Genet. 2001; 10:1287–1298. [DOI] [PubMed] [Google Scholar]
- 47. Brosh R. Jr., Li J., Kenny M., Karow J., Cooper M., Kureekattil R., Hickson I., Bohr V.. Replication protein A physically interacts with the Bloom's syndrome protein and stimulates its helicase activity. J. Biol. Chem. 2000; 275:23500–23508. [DOI] [PubMed] [Google Scholar]
- 48. Yodh J., Stevens B., Kanagaraj R., Janscak P., Ha T.. BLM helicase measures DNA unwound before switching strands and hRPA promotes unwinding reinitiation. EMBO J. 2009; 28:405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Karow J., Newman R., Freemont P., Hickson I.. Oligomeric ring structure of the Bloom's syndrome helicase. Curr. Biol. 1999; 9:597–600. [DOI] [PubMed] [Google Scholar]
- 50. Beresten S., Stan R., van Brabant A., Ye T., Naureckiene S., Ellis N.. Purification of overexpressed hexahistidine-tagged BLM N431 as oligomeric complexes. Protein Expr. Purif. 1999; 17:239–248. [DOI] [PubMed] [Google Scholar]
- 51. Yang Y., Dou S., Xu Y., Bazeille N., Wang P., Rigolet P., Xu H., Xi X.. Kinetic mechanism of DNA unwinding by the BLM helicase core and molecular basis for its low processivity. Biochemistry. 2010; 49:656–668. [DOI] [PubMed] [Google Scholar]
- 52. Xu Y., Bazeille N., Ding X., Lu X., Wang P., Bugnard E., Grondin V., Dou S., Xi X.. Multimeric BLM is dissociated upon ATP hydrolysis and functions as monomers in resolving DNA structures. Nucleic Acids Res. 2012; 40:9802–9814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wold M. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem. 1997; 66:61–92. [DOI] [PubMed] [Google Scholar]
- 54. Chen R., Wold M.. Replication protein A: single-stranded DNA’s first responder: dynamic DNA-interactions allow replication protein A to direct single-strand DNA intermediates into different pathways for synthesis or repair. Bioessays. 2014; 36:1156–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. San Filippo J., Sung P., Klein H.. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 2008; 77:229–257. [DOI] [PubMed] [Google Scholar]
- 56. Heyer W., Ehmsen K., Liu J.. Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 2010; 44:113–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bugreev D., Mazin A.. Ca2+ activates human homologous recombination protein Rad51 by modulating its ATPase activity. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:9988–9993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bachrati C., Borts R., Hickson I.. Mobile D-loops are a preferred substrate for the Bloom's syndrome helicase. Nucleic Acids Res. 2006; 34:2269–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Daley J., Jimenez-Sainz J., Wang W., Miller A., Xue X., Nguyen K., Jensen R., Sung P.. Enhancement of BLM-DNA2-mediated long-range DNA end resection by CtIP. Cell Rep. 2017; 21:324–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Niu H., Chung W., Zhu Z., Kwon Y., Zhao W., Chi P., Prakash R., Seong C., Liu D., Lu L. et al.. Mechanism of the ATP-dependent DNA end-resection machinery from Saccharomycescerevisiae. Nature. 2010; 467:108–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Adkins N., Niu H., Sung P., Peterson C.. Nucleosome dynamics regulates DNA processing. Nat. Struct. Mol. Biol. 2013; 20:836–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Peterson C., Almouzni G.. Nucleosome dynamics as modular systems that integrate DNA damage and repair. Cold Spring Harbor Perspect. Biol. 2013; 5:a012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Symington L. Mechanism and regulation of DNA end resection in eukaryotes. Crit. Rev. Biochem. Mol. Biol. 2016; 51:195–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gyimesi M., Sarlos K., Kovacs M.. Processive translocation mechanism of the human Bloom's syndrome helicase along single-stranded DNA. Nucleic Acids Res. 2010; 38:4404–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Budhathoki J., Stafford E., Yodh J., Balci H.. ATP-dependent G-quadruplex unfolding by Bloom helicase exhibits low processivity. Nucleic Acids Res. 2015; 43:5961–5970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Karow J., Constantinou A., Li J., West S., Hickson I.. The Bloom's syndrome gene product promotes branch migration of holliday junctions. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:6504–6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Walpita D., Plug A., Neff N., German J., Ashley T.. Bloom's syndrome protein, BLM, colocalizes with replication protein A in meiotic prophase nuclei of mammalian spermatocytes. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:5622–5627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Woglar A., Villeneuve A.. Dynamic Architecture of DNA Repair Complexes and the Synaptonemal Complex at Sites of Meiotic Recombination. Cell. 2018; 173:1678–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Patel D., Misenko S., Her J., Bunting S.. BLM helicase regulates DNA repair by counteracting RAD51 loading at DNA double-strand break sites. J. Cell Biol. 2017; 216:3521–3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Brouwer I., Moschetti T., Candelli A., Garcin E., Modesti M., Pellegrini L., Wuite G., Peterman E.. Two distinct conformational states define the interaction of human RAD51-ATP with single-stranded DNA. EMBO J. 2018; 37:e98162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hilario J., Amitani I., Baskin R., Kowalczykowski S.. Direct imaging of human Rad51 nucleoprotein dynamics on individual DNA molecules. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kim H., Morimatsu K., Norden B., Ardhammar M., Takahashi M.. ADP stabilizes the human Rad51-single stranded DNA complex and promotes its DNA annealing activity. Genes Cells. 2002; 7:1125–1134. [DOI] [PubMed] [Google Scholar]
- 73. Essers J., Houtsmuller A., van Veelen L., Paulusma C., Nigg A., Pastink A., Vermeulen W., Hoeijmakers J., Kanaar R.. Nuclear dynamics of RAD52 group homologous recombination proteins in response to DNA damage. EMBO J. 2002; 21:2030–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Her J., Ray C., Bunting S.. The BLM Helicase: Keeping recombination honest. Cell Cycle. 2018; 17:401–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hu Y., Raynard S., Sehorn M., Lu X., Bussen W., Zheng L., Stark J., Barnes E., Chi P., Janscak P. et al.. RECQL5/Recql5 helicase regulates homologous recombination and suppresses tumor formation via disruption of Rad51 presynaptic filaments. Genes Dev. 2007; 21:3073–3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Antony E., Tomko E., Xiao Q., Krejci L., Lohman T., Ellenberger T.. Srs2 disassembles Rad51 filaments by a protein-protein interaction triggering ATP turnover and dissociation of Rad51 from DNA. Mol. Cell. 2009; 35:105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Krejci L., Van Komen S., Li Y., Villemain J., Reddy M., Klein H., Ellenberger T., Sung P.. DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature. 2003; 423:305–309. [DOI] [PubMed] [Google Scholar]
- 78. Schwendener S., Raynard S., Paliwal S., Cheng A., Kanagaraj R., Shevelev I., Stark J., Sung P., Janscak P.. Physical interaction of RECQ5 helicase with RAD51 facilitates its anti-recombinase activity. J. Biol. Chem. 2010; 285:15739–15745. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.